
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGallium nanoparticles as novel inhibitors of Aβ40 aggregation
Kyabeth M.
Torres
a,
Ambar S.
Delgado
a,
Erika R.
Serrano
b,
Nitza V.
Falcón-Cruz
b,
Anamaris
Meléndez
c,
Idalia
Ramos
 c,
Deguo
Du
*d and
Rolando
Oyola
*b
c,
Deguo
Du
*d and
Rolando
Oyola
*b
aUniversity of Puerto Rico-Humacao, Department of Biology, Call Box 860, Humacao 00792 PR, USA
bUniversity of Puerto Rico-Humacao, Department of Chemistry, Call Box 860, Humacao 00792 PR, USA. E-mail: rolando.oyola@upr.edu
cUniversity of Puerto Rico-Humacao, Department of Physics & Electronics, Call Box 860, Humacao 00792 PR, USA
dFlorida Atlantic University, Department of Chemistry & Biochemistry, Boca Raton, 33431, FL, USA. E-mail: ddu@fau.edu
First published on 9th July 2021
Abstract
Alzheimer's disease (AD) has been consistently related to the formation of senile amyloid plaques mainly composed of amyloid β (Aβ) peptides. The toxicity of Aβ aggregates has been indicated to be responsible for AD pathology. One scenario to decrease Aβ toxicity is the development of effective inhibitors against Aβ amyloid formation. In this study, we investigate the effect of gallium nitride nanoparticles (GaN NPs) as inhibitors of Aβ40 amyloid formation using a combination of biophysical approaches. Our results show that the lag phase of Aβ40 aggregation kinetics is significantly retarded by GaN NPs in a concentration dependent manner, implying the activity of GaN NPs in interfering with the formation of the crucial nucleus during Aβ aggregation. Our results also show that GaN NPs can reduce the amyloid fibril elongation rate in the course of the aggregation kinetics. It is speculated that the high polarization characteristics of GaN NPs may provoke a strong interaction between the particles and Aβ40 peptide and in this way decrease self-association of the peptide monomers to form amyloids.
Introduction
Alzheimer's disease (AD) is a progressive, irreversible, and currently incurable neurodegenerative disorder characterized by neuronal loss, memory impairment, and cognitive decline.1,2 As the leading cause of dementia in a rapidly aging population, AD is a growing threat to global health, economies, and society in general. One possible pathway to develop AD has been linked to the formation of senile plaques due to the aggregation of a group of key amyloid-β (Aβ) peptides, e.g., Aβ40 and Aβ42, that are formed from the cleavage of the transmembrane amyloid precursor protein (APP) by β- and γ-secretases.2–4 Amyloid formation of Aβ is typically viewed as a nucleated polymerization process, manifested by an initial lag phase in which the oligomeric nuclei are formed, followed by a rapid growth phase during which the nuclei grow by the addition of monomers to form fibrillar structures.5,6 Moreover, amyloid oligomers have also been shown to form hetero-oligomers by interacting with its isoforms or other proteins.5,7 Therefore great effort has been devoted to characterize oligomers to better understand the amyloid formation mechanism and conceive plausible therapeutic alternatives.4,6One of the plausible therapeutic strategies for treating the disease is to inhibit the formation of toxic Aβ aggregate species. As such, a wide variety of amyloid inhibitors that can prevent the formation of Aβ aggregates, e.g., small organic compounds and designed peptides, have been reported.2,3,8–10 Also, recent studies have demonstrated that various nanoparticles (NPs) can either completely or partially inhibit Aβ fibril formation, due to their interactions with Aβ.11–13 NPs have been used as an important build block to construct new Aβ inhibitors using surface modification and changes in morphology of the particles.14
Different substrates have been used to establish NP cores for the development of efficient NPs for therapeutic applications. The common NP formulations employed in the diagnosis and therapeutic studies of AD include polymeric NPs, gold NPs (Au NPs), gadolinium NPs, selenium NPs, protein-based NPs, and polysaccharide-based NPs.15 For example, Liao et al. showed that bare Au NPs inhibited Aβ fibrillation, leading to the formation of fragmented fibrils and spherical oligomers.16 These authors reasoned that the negative surface potential of Au NPs is an important factor for the inhibition of Aβ amyloid fibril formation. Similarly, Yin et al. demonstrated that selenium NPs not only effectively inhibit Aβ aggregation but could disaggregate preformed Aβ fibrils into non-toxic amorphous oligomers.17 Family III nitrides, particularly gallium, aluminium, and indium nitrides, have gained considerable interest for use as transducing materials in biosensors.18 Specifically, gallium nitride (GaN) is a semiconductor material with wurtzite crystal structure that has a wide (3.4 eV) band gap and is widely used in ultraviolet–blue light emitting diodes, photodetectors and lasers.19 In addition, GaN demonstrates good biocompatibility and aqueous stability in conditions relevant to biological applications.20–22 Therefore, GaN has also been used in various biological applications. Braniste et al. reported the uptake of GaN NPs by porcine endothelial cells,23 and their results suggested that the uptake of the particles by the cells is strongly dependent upon whether the GaN NPs are fixed to a substrate surface or free floating in the medium. The endothelial cells growing on surfaces functionalized with GaN NPs demonstrated excellent adhesion and proliferation, suggesting good biocompatibility of the nanoparticles. The same research group expanded their studies and demonstrated that GaN/Fe-based nanoparticles exhibiting magnetic properties can be taken up by endothelial cells and stored within vesicles.24 In addition, Hofstetter et al. performed an investigation of the effects of differently treated GaN surfaces on mouse fibroblast cell proliferation and growth dynamics.25 Their results suggest that GaN does not require specific surface treatments to be biocompatible and that cell signalling is not affected by GaN–cell interactions. Jewett et al. demonstrated that functionalized and etched GaN surfaces are stable in aqueous environments and leach a negligible amount of Ga in solution even in the presence of hydrogen peroxide.26 Also, GaN surfaces in cell culture did not interfere with nearby cell growth, and the etched GaN promoted the adhesion of cells better than the etched silicon surfaces. On the other hand, Ga3+ can interfere with the iron metabolism in cells leading to apoptosis.27 This was attributed to the similar ionic radius of Ga3+ and Fe3+ and Ga3+ being less electroactive than Fe3+ in the iron metabolic processes.28
While GaN has emerged as a promising candidate for bio-interfacing,29 to the best of our knowledge, the potential use of GaN as an inhibitor of aggregation of amyloidogenic proteins including Aβ, has not been explored. Since previous studies have shown that GaN can interact with various cellular components, it is possible that GaN NPs are able to interact with Aβ peptide and in consequence affect the aggregation behaviour. In this study, we investigate the effect of GaN NPs on the aggregation of Aβ40 in vitro using a combination of biophysical approaches. In comparison to Aβ42, Aβ40 is less toxic to neurons and exerts a more moderate aggregation propensity and thus is appropriate for a sophisticated investigation of the properties of aggregation kinetics. Our results demonstrate that GaN NPs markedly affects the characteristics of Aβ40 oligomerization and fibrillization. It is expected that further molecular modification of the surface characteristics such as the charge properties of GaN NPs may be an approach for designing potent NP inhibitors against protein amyloidogenesis.
Experimental
Materials
Aβ40 was obtained from Life Tein Company (NJ, USA). Certified analysis of the peptide established that the purity was higher than >95% with a formula weight of 4329.9. The peptide was used as received. Gallium nitrate hydrate was obtained from Sigma-Aldrich (USA) with >99.9% purity trace metals basis. Congo red (CR) with high purity was obtained from Calbiochem (UK). Thioflavin T (ThT) was obtained from Sigma-Aldrich (USA). Other solvents and reagents were obtained from either Sigma-Aldrich (USA) or Fisher Scientific (Cayey, PR) and used without further purification.Synthesis of GaN NPs
The GaN NPs were synthesized according to previous reports.30–34 Briefly, Ga(NO3)3 was dissolved in nitric acid, the pH was adjusted to 8.2 with ammonium hydroxide and citric acid was added until the solution became clear. The resulting solution was dried and heated in a burnout furnace under air at 400 °C for 4 hours producing a Ga2O3 powder that was converted to GaN in a tube furnace at 900 °C under an NH3 gas flow for 3 hours to ensure complete conversion.31X-Ray diffraction (XRD) measurements
The phase structures of the nanoparticle samples were identified using X-ray diffraction techniques using a Bruker D2 Phaser with Cu Kα radiation (wavelength of the radiation, λ = 1.54 Å) operating at 30 kV and 10 mA. The incident beam passed through a 2.5° Soller slit, a 0.6° (1.0 mm) divergence slit, a secondary 2.5° Soller slit, a Ni filter, and a receiving slit of 0.2 mm. After interaction with the sample, the diffracted beam was detected using a scintillation counter. The samples were analyzed utilizing a Bragg–Brentano geometry over a range of 5–85° 2θ with a step size of 0.02° 2θ, with each step measured for one second. For all XRD measurements, the samples were prepared as self-supported pressed powders over PMMA specimen holders from Bruker. Data analysis was performed using the instrument software, which includes the spectral database. In addition, XRD is a convenient method for determining the mean size of single-crystal nanoparticles or crystallites in nanocrystalline bulk materials.35 The size of the synthesized nanoparticles was calculated using the Scherrer formula, D = Kλ/βcos![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ where D is the average grain size, K is the shape factor (taken as 0.9), λ is the X-ray wavelength, β is the peak width measured in radians as the full width at half of the maximum intensity, and the Bragg angle is 21.485°.
θ where D is the average grain size, K is the shape factor (taken as 0.9), λ is the X-ray wavelength, β is the peak width measured in radians as the full width at half of the maximum intensity, and the Bragg angle is 21.485°.
Aβ40 aggregation kinetics followed by thioflavin T (ThT) emission
The Aβ40 peptide utilized in the kinetic aggregation assay was monomerized as described previously.36,37 Briefly, lyophilized Aβ40 powder was dissolved in aqueous NaOH solution (2 mM) and the pH was adjusted to 11 using a 100 mM NaOH solution followed by sonicating the solution for 1 hour in an ice-cold water bath. The resulting solution was filtered through a 0.22 μm filter and kept on ice before use. The concentration of the Aβ40 stock solution was determined using tyrosine UV absorbance at 280 nm [ε280 = 1280 (M cm)−1] using an Agilent 8453 photodiode array spectrometer. The fluorescence kinetics were measured using a Fluoroskan Ascent microplate reader (Thermo-Nicolet, USA). Fibril formation of Aβ40 was followed using the ThT fluorescence emission using filters (bandpass of 15 nm) with an excitation of 435 and emission at 485 nm, respectively. Each well contained 20 μM Aβ40, 20 μM ThT, 150 mM NaCl in 50 mM sodium phosphate buffer saline (PBS pH = 7.4) with or without GaN NPs in a total volume of 300 μL. The emission was measured through the bottom of the plate every five minutes with five seconds of gentle shaking (100 rpm) before readings at 37 °C. The Aβ40 aggregation kinetic parameters were obtained from the average of at least three replicates. Fitting analysis was performed according to eqn (1);38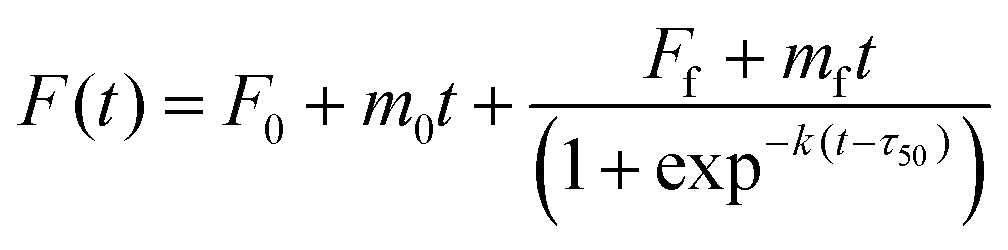 | (1) |
Congo red binding assay
Congo red (CR) was dissolved in PBS at a concentration of 100 μM and filtered through a 0.22 μm membrane. The solutions were prepared by dilution of stock solutions to obtain the final concentrations of 5.0 μM CR, 150 mM NaCl, 50 mM PBS 7.4, and Aβ40 (15 μM) and incubated at 37 °C in a 10 mm pathlength optical quartz cell. The absorbance spectra were measured at certain times for the samples in triplicate. The incubation was run with gentle agitation.39Turbidity study
The aggregation kinetics of Aβ40 was determined by monitoring the absorbance at 600 nm as reported previously.40,41 Samples were prepared as described in the CR binding studies but without the dye.Scanning electron microscopy (SEM)
SEM images for nanoparticle characterization were acquired on a JEOL JSM-IT-100 with an Energy Dispersive Spectrometer operating at 25 or 30 kV. For amyloid fibrils, SEM images were acquired on an FEI Quanta 600 ESEM operating at 15 kV at the Singh Center for Nanotechnology at the University of Pennsylvania.42 The sample solution was diluted 100-fold with pure water. Subsequently, the diluted samples were placed on silicone and dried under nitrogen for 10 min before measuring the images.Results and discussion
Characterization of the synthesized GaN NPs
The morphology of the synthesized GaN NPs was investigated using SEM. As shown in Fig. 1, the GaN NP samples dispersed in water exhibit a typical crystallite morphology. The XRD spectrum of the GaN NPs resembles that previously reported by Chen et al.,30 as shown in Fig. 2A. Specifically, the XRD pattern of the GaN NPs exhibits peaks at 2θ of 32.80, 35.20, and 37.26 and four other major peaks at 48.8, 58.2, 64.2, and 69.3 degrees, respectively. All these peaks can be indexed to a hexagonal (wurtzite) GaN structure. The GaN NP sizes determined using the Scherrer formula and the (110) peak at 58.2 degrees range from 6 to 12 nm. The energy-dispersive spectrum (Fig. 2B) reveals that the elemental composition of the GaN NPs is 88.53, 10.01, and 1.46 w/w% for gallium, nitrogen, and oxygen, respectively. | ||
| Fig. 1 SEM images of the synthesized GaN NPs. The images were measured at different kilovolts and magnifications: (A) 25 kV, 5000×; (B) 30 kV, 10000×; (C) 30 kV, 20000×; and (D) 30 kV, 50000×. | ||
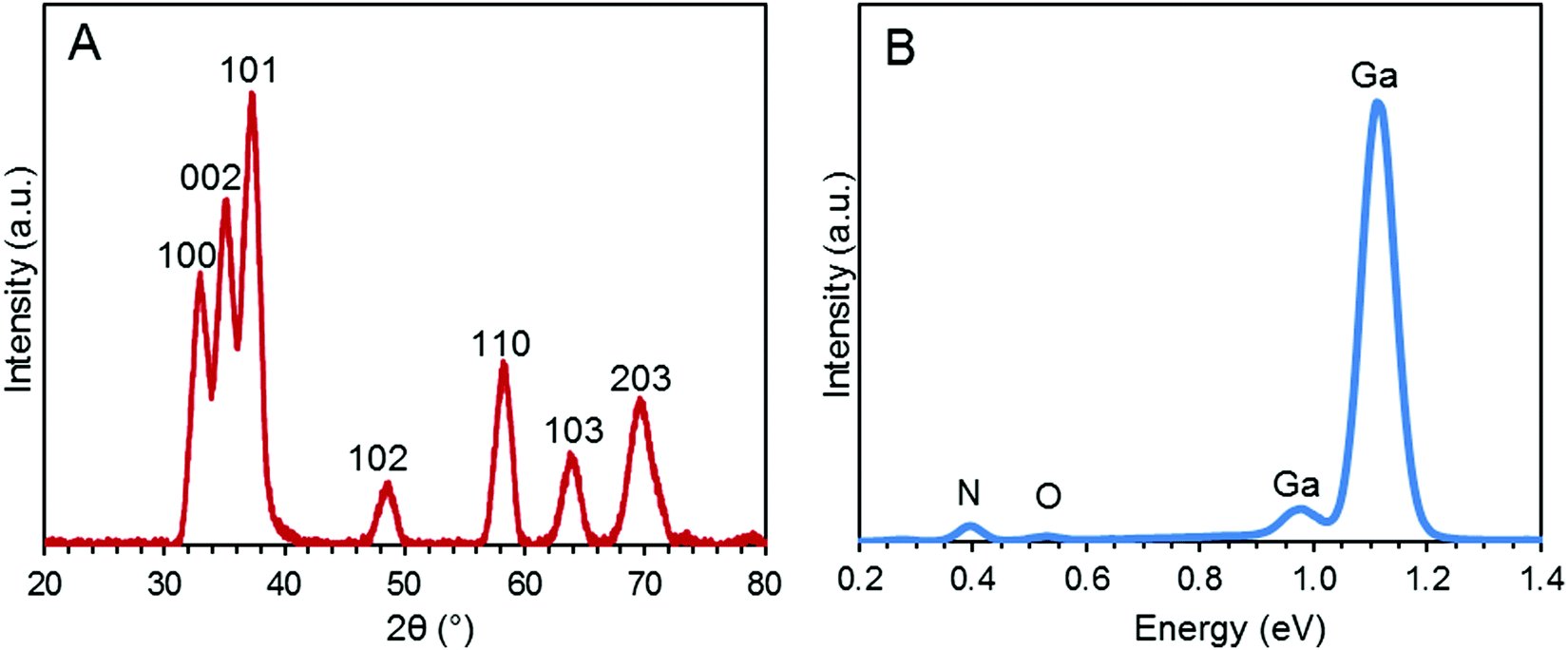 | ||
| Fig. 2 (A) XRD pattern of the GaN NPs. (B) Energy-dispersive spectrum of the GaN NPs. The weight percent elemental data for nitrogen, oxygen, and gallium is 10.01, 1.46, and 88.53%, respectively. | ||
Impact of GaN NPs on Aβ40 aggregation kinetics measured using ThT fluorescence
The formation of Aβ40 amyloid aggregates starting from its monomeric state was studied using ThT fluorescence in the absence or presence of GaN NPs in sodium phosphate buffer (50 mM PBS buffer, 150 mM NaCl, pH 7.4). ThT is an environmentally sensitive fluorophore whose binding to amyloid fibrils dramatically increases its fluorescence quantum yield.43Fig. 3 shows the time dependent ThT fluorescence intensity changes of the Aβ40 sample (20 μM) in the absence or presence of GaN NPs. The kinetic curves are fitted using eqn (1) to obtain aggregation parameters such as τ50 and τlag (summarized in Fig. 4). In the absence of GaN NPs, the Aβ40 aggregation kinetics exhibit a typical sigmoidal shape, characteristic of a nucleation polymerization mechanism,44 which consists of a lag phase associated with nucleation, a fast growth phase related to the propagation of fibrils, and a final stationary phase related to the aggregated state. The kinetics exhibit a τ50 of 1044 minutes (Fig. 4), and a growth phase with a rate constant of 5.45 × 10−3 min−1. To rule out the possibility that ThT can directly interact with the GaN NPs, causing its fluorescence intensity to change, we also performed control experiments where no Aβ40 is present. The results show that the ThT emission in the presence of GaN NPs does not only change with time (Fig. 3, curve f) and that the amplitude overlaps with the initial baseline of the kinetic runs containing Aβ40. This confirms that the observed ThT fluorescence intensity change in the Aβ40 aggregation measurements is not due to direct interactions between ThT and GaN NPs.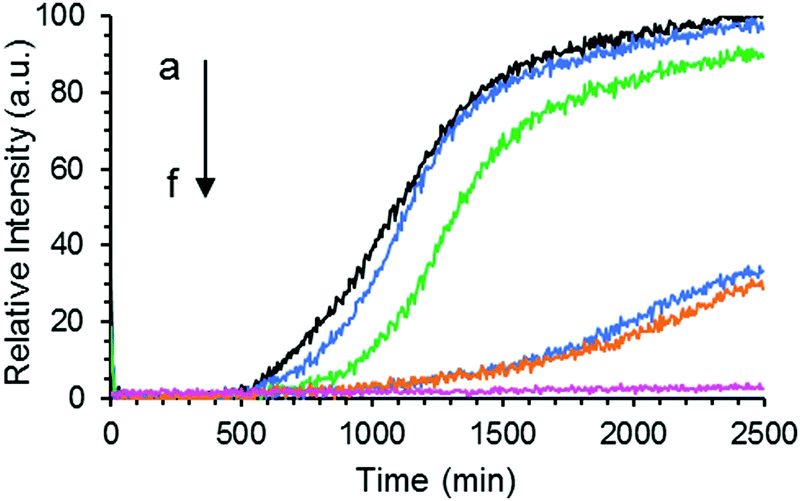 | ||
| Fig. 3 Aggregation kinetics of Aβ40 (20 μM) only (a), or in the presence of 47 (b), 60 (c), 67 (d), 73 μg mL−1 (e) of GaN NPs, respectively followed by ThT fluorescence. Curve f is the fluorescence intensity of the sample containing 80 μg mL−1 GaN NPs and 20 μM ThT only without Aβ40. | ||
 | ||
| Fig. 4 (A) τ50 of the aggregation kinetics of Aβ40 (20 μM) in the presence of different amounts of GaN NPs. (B) τlag of the aggregation kinetics of Aβ40 (20 μM) in the presence of different amounts of GaN NPs. | ||
The addition of GaN NPs markedly delays the aggregation kinetics of Aβ40. In the presence of 60 μg mL−1 GaN NPs, the value of τ50 increases to 1227 minutes (Fig. 3 and 4), ca.18% longer than that of Aβ40 alone. Increasing the GaN concentration to 73 μg mL−1 results in a two-fold increase in τ50 relative to that of Aβ40 in the absence of GaN NPs, up to 2197 minutes. Further increase of the GaN concentration shows smaller change in τ50. Furthermore, as shown in Fig. 4B, the τlag of the Aβ40 aggregation kinetics is approximately 700 minutes, and the addition of 67 μg mL−1 GaN increases the τlag value to 1349 minutes. This result suggests that the presence of appropriate amounts of GaN can delay the rate of critical oligomeric nucleus formation of Aβ. It is worth noting that the highest GaN concentration achievable in the experiments is limited by its solubility. Increasing the GaN concentration also affects the apparent growth rate constant (k) of the exponential phase. For instance, the k value of the kinetics in the presence of 67 μg mL−1 GaN NPs is 2.57 × 10−3 min−1, which is less than half that of the kinetics of the peptide only. A larger k value suggests a higher order exponential phase where growth of the fibrils occurs.45–49 The results suggest that the addition of higher amount of GaN NPs can also significantly interfere with the growth process of the amyloid fibrils, thus inhibiting the formation of amyloids.
The maximum of ThT fluorescence amplitude in Aβ aggregation kinetics is influenced by the addition of GaN NPs. In general, ThT fluorescence emission intensity is proportional to the amyloid fibers present, and a decrease in the intensity can be indicative of a decrease in the overall fiber content.50 As depicted in Fig. 5, increasing the GaN NP concentration lowers the ThT maximum fluorescence amplitude. These results suggest the inhibition of Aβ40 fibril formation by GaN NPs. It is interesting that there is also an apparent threshold concentration value of approximately 60 μg mL−1 where the inhibition efficiency starts to increase sharply. Taken together, these results demonstrate that GaN NPs can delay the formation of the crucial nucleus in Aβ aggregation and slow down the rate of Aβ40 amyloid fibril formation. In addition, GaN NPs can also decrease the growth rate constant of Aβ40 amyloid formation.
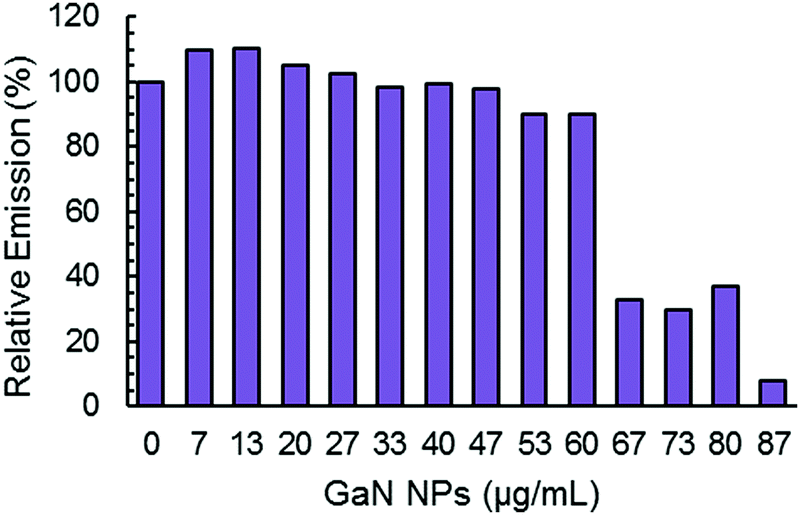 | ||
| Fig. 5 Normalized maximum ThT fluorescence emission intensity during the aggregation of Aβ40 (20 μM) without or with different concentrations of GaN NPs. | ||
Although a general mechanism for the underlying impact of NPs on protein fibril formation remains elusive,14 the structural characteristics of the NPs such as size, shape, and the surface properties have been demonstrated to be crucial in the interactions of the particles and proteins and the inhibition activities of NPs on protein amyloid formation.49,51,52 The inhibition activity of GaN on Aβ40 aggregation likely arises from interactions between the peptide and the NPs in aqueous solution, which may be driven by interactions such as electrostatic and van der Waals interactions. The non-centrosymmetric wurtzite hexagonal structure of GaN NPs has planes consisting of anions and cations that attract each other via electrostatic forces inducing a spontaneous polarization that is larger than other materials.18 It is conceivable that this polarization property may play an important role in the interaction of GaN and Aβ40 peptide, which, as a result, would prevent or decrease peptide–peptide interactions possibly by stabilizing the Aβ40 monomers.
Inhibition of GaN NPs on Aβ40 aggregation measured using CR binding
CR has also been widely used in monitoring peptide aggregation and fibril formation. The absorption spectrum of CR peaks at around 490 nm (π–π* transition of the azo group) in aqueous solution,53 is shifted to ca. 550 nm when Aβ40 fibrils are present. This bathochromic shift is easily seen visually where an orange red to rose change in colour is observed. We conducted an experiment using CR as a probe to further prove that GaN NPs can decrease the aggregation rate of Aβ40 and its fibril quantity. The absorption spectra of different solutions containing CR alone, Aβ40–CR, and Aβ40–CR–GaN were measured at different times of incubation. The solution containing only Aβ40 shows an initial absorption spectrum that resembles the one of CR with a broad band peak with a maximum at 490 nm (Fig. 6A). After ∼5 hours, the formation of Aβ40 amyloids is indicated by the absorbance increase in the 520–550 nm region (Fig. 6A). The absorbance in this region increases with time reaching a plateau after ∼20 hours. When Aβ40 is incubated in the presence of GaN NPs (87 μg mL−1), the intensity change of the absorbance at 541 nm, characteristic for Aβ40 aggregation, is slower than that of Aβ40 alone (Fig. 6B vs. 6A). This result further indicates the inhibition activity of GaN NPs on Aβ40 aggregation. To rule out the possibility that CR can directly interact with GaN NPs and cause a change in the absorption, we also performed control experiments in the absence of Aβ40. The results show that the CR–GaN NPs absorption spectrum does not change with incubation time (Fig. 6C), hence corroborating that the spectral changes are due to the formation of Aβ40 fibrils.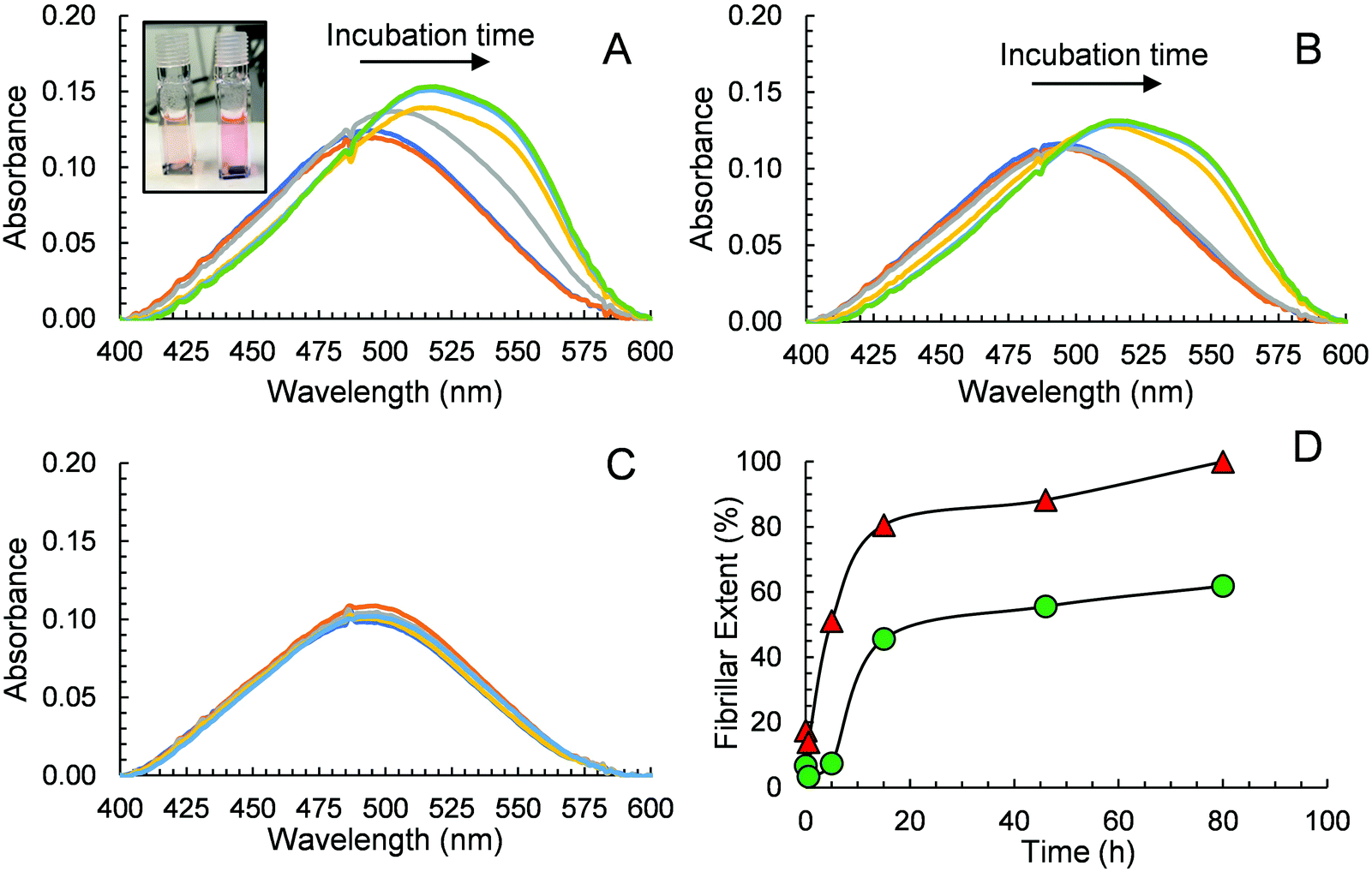 | ||
| Fig. 6 Absorption spectra of Aβ40 (15 μM, containing 5 μM CR) in the absence (A) or in the presence (B) of 87 μg mL−1 GaN NPs. The spectra were measured at the incubation times of 0 h (dark blue), 0.5 h (orange), 5 h (gray), 15 h (yellow), 46 h (light blue), and 80 h (green). Inset in (A): photo of samples containing GaN–Aβ40 (left) and Aβ40 (right) showing the difference in colour after a 5 h incubation. (C) Absorption spectra of CR and GaN NPs only measured at the incubation times of 0 h (dark blue), 0.5 h (orange), 5 h (gray), 15 h (yellow), 46 h (light blue), and 80 h (green). (D) Quantitative evaluation of fibrillar extent during the aggregation of Aβ40 in the absence (red triangles) or in the presence (green circles) of GaN NPs (87 μg mL−1). A baseline correction due to scattering was applied to each spectrum before reading the absorbance values. The fibrillar extent values are relative to the maximum Aβ40 fibrillar concentration at 80 h for an Aβ40 solution in the absence of GaN NPs. | ||
Quantitative fibrillar formation was also studied using CR as a probe.54 The concentration of Aβ fibrils at certain times, in μg mL−1, was estimated from CR binding where [Aβ40fibrils]t = (A541mix/4780) − (A403mix/6830) − (A403CR/8620) where A541mix and A403mix are the absorbances of the solutions containing Aβ40 or Aβ40–GaN at 541 and 403 nm, respectively, and A403CR is the absorbance of the solution containing CR alone under the same experimental incubation time.54 The results are shown in Fig. 6D. The data shows that the lag phase is around 4 hours for Aβ40 in the presence of GaN (87 μg mL−1), which is approximately three times longer than that in the absence of GaN. In addition, the presence of GaN NPs reduces the fibrillar content by up to 62% after 80 hours of incubation. These results are in accord with what observed in the ThT kinetics study.
Inhibition of GaN NPs on Aβ40 aggregation confirmed using turbidity and imaging studies
Turbidity measurements were also performed to monitor Aβ40 aggregation as it is well-known that protein aggregate formation leads to an increase in the sample absorbance at 600 nm.40 The absorbance intensity at 600 nm was monitored as a function of time for Aβ40 aggregation in the absence and presence of GaN NPs at 87 μg mL−1. As shown in Fig. 7, the presence of GaN NPs significantly decreases the fibril formation kinetics, consistent with both the ThT and CR results. The inhibitory activity of the particles on Aβ40 fibrillogenesis was further confirmed using SEM imaging (Fig. 8). SEM images were acquired using the samples from the Aβ40 aggregation kinetics study in the absence or presence of GaN NPs. In the absence of GaN NPs, Aβ40 aggregated to form fibrils of a few micrometers long and produced large conglomerates. In contrast, in the presence of GaN NPs, the number of fibrils was dramatically reduced and no large clusters were observed. Taken together, these results corroborate that GaN NPs are an effective inhibitor of Aβ40 amyloid fibril formation. | ||
| Fig. 7 Turbidity curves for Aβ40 aggregation in the absence (red triangles) or in the presence of 87 μg mL−1 GaN NPs (green circles) measured by monitoring the absorbance at 600 nm. A curve for the solution containing GaN NP and CR alone (blue squares) incubated in parallel is also shown. Solid lines are to guide the reader. | ||
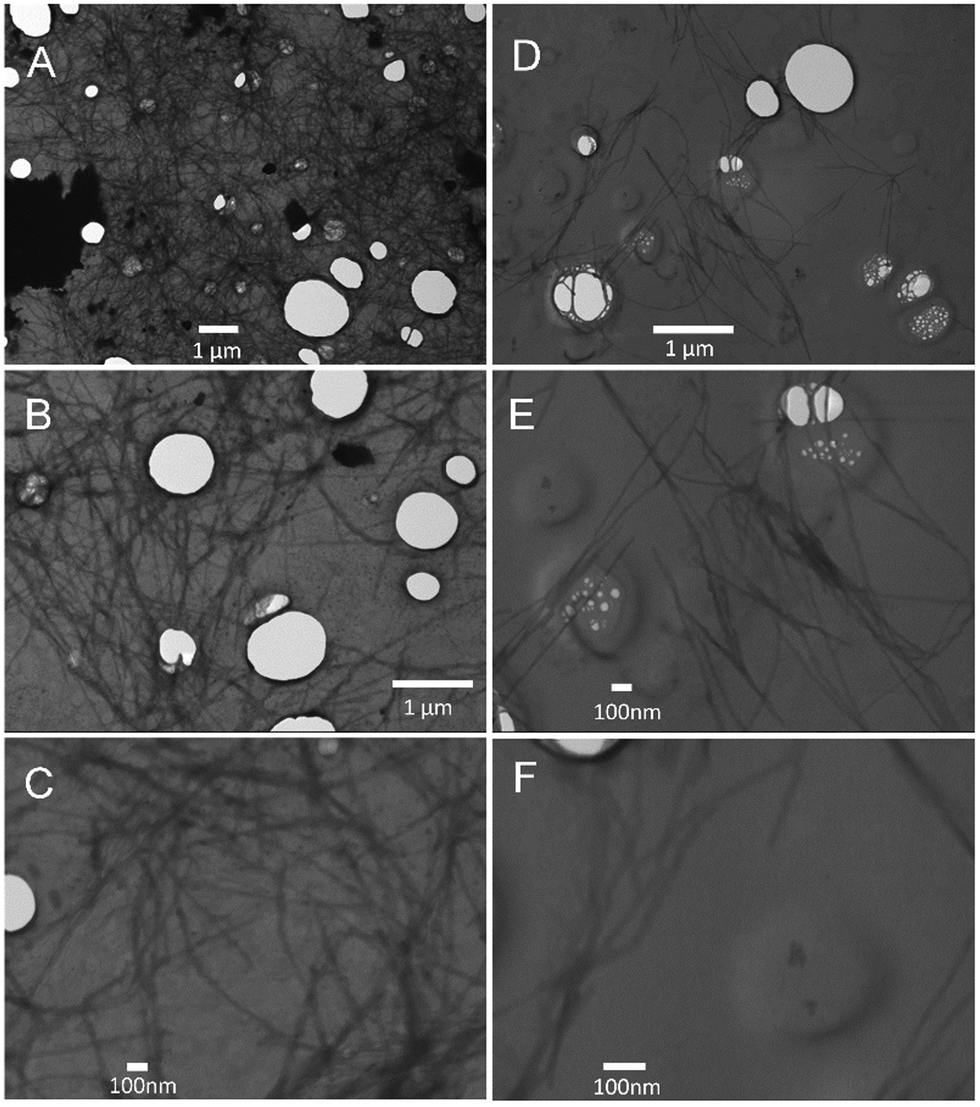 | ||
| Fig. 8 SEM images of Aβ40 (20 μM) acquired after the aggregation kinetics measurement using ThT fluorescence emission. Left: SEM images of Aβ40 only measured at different magnifications: (A) 10000×, (B) 20000×, and (C) 50000×. Right: SEM images of Aβ40 in the presence of 87 μg mL−1 GaN NPs measured at different magnifications: (D) 20000×, (E) 50000×, and (F) 100000×. | ||
Conclusions
In summary, we studied the effect of GaN NPs on Aβ40 amyloid fibril formation using a variety of approaches including ThT fluorescence, CR absorbance, turbidity, and SEM imaging. Our results show that the presence of GaN NPs significantly delays the lag time of Aβ40 aggregation, indicating its inhibiting activity on crucial oligomeric nucleus formation of the peptide. Also, the results show that GaN NPs can slow down the amyloid fibril elongation rate in the amyloid growth phase of the aggregation kinetics. Considering that the non-centrosymmetric wurtzite hexagonal structure of GaN NPs contains planes consisting of anions and cations that induce strong spontaneous polarization, it is plausible that this polarization characteristic of the particle can provoke a stronger interaction between the NPs and Aβ40 peptide and thus decrease intermolecular interactions among the peptide monomers to form amyloids. Future studies on modification of the surface characteristics of gallium nanoparticles to enhance solubility and selectivity may lead to the discovery of new NP inhibitors with high biocompatibility and inhibition efficiency.Author contributions
KMT, ASD, ERS, NVFC, and AM performed the experiments. IR, DD and RO conceived and wrote the paper.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by NSF-DMR-2122102. KT and ES acknowledge support from the NIH-UPRH-MARC-T34GM008156 undergraduate student training in academic research program. The authors acknowledge Dr. Feng Gai (University of Pennsylvania) for valuable discussions. We also acknowledge Jamie Ford for obtaining the SEM images at the Singh Center for Nanotechnology at the University of Pennsylvania, which is supported by the NSF National Nanotechnology Coordinated Infrastructure Program under grant NNCI-1542153. DD acknowledges financial support from the National Institutes of Health (R15GM116006) and the Alzheimer's Association (AARG-17-531423).References
- A. K. Sahoo, J. Dandapat, U. C. Dash and S. Kanhar, J. Ethnopharmacol., 2018, 215, 42–73 CrossRef CAS PubMed.
- J. Cummings, G. Lee, A. Ritter and K. Zhong, Alzheimer's Dementia, 2018, 4, 195–214 CrossRef PubMed.
- S. K. Singh, S. Srivastav, A. K. Yadav, S. Srikrishna and G. Perry, Oxid. Med. Cell. Longevity, 2016, 2016, 7361613 Search PubMed.
- P. H. Nguyen, A. Ramamoorthy, B. R. Sahoo, J. Zheng, P. Faller, J. E. Straub, L. Dominguez, J.-E. Shea, N. V. Dokholyan, A. De Simone, B. Ma, R. Nussinov, S. Najafi, S. T. Ngo, A. Loquet, M. Chiricotto, P. Ganguly, J. McCarty, M. S. Li, C. Hall, Y. Wang, Y. Miller, S. Melchionna, B. Habenstein, S. Timr, J. Chen, B. Hnath, B. Strodel, R. Kayed, S. Lesné, G. Wei, F. Sterpone, A. J. Doig and P. Derreumaux, Chem. Rev., 2021, 121, 2545–2647 CrossRef CAS PubMed.
- B. R. Sahoo, S. J. Cox and A. Ramamoorthy, Chem. Commun., 2020, 56, 4627–4639 RSC.
- E. E. Cawood, T. K. Karamanos, A. J. Wilson and S. E. Radford, Biophys. Chem., 2021, 268, 106505 CrossRef CAS PubMed.
- M. I. Ivanova, Y. Lin, Y.-H. Lee, J. Zheng and A. Ramamoorthy, Biophys. Chem., 2021, 269, 106507 CrossRef CAS PubMed.
- P. Williams, A. Sorribas and M.-J. R. Howes, Nat. Prod. Rep., 2011, 28, 48–77 RSC.
- C. Cabaleiro-Lago, F. Quinlan-Pluck, I. Lynch, S. Lindman, A. M. Minogue, E. Thulin, D. M. Walsh, K. A. Dawson and S. Linse, J. Am. Chem. Soc., 2008, 130, 15437–15443 CrossRef CAS PubMed.
- D. J. Selkoe, Science, 1997, 275, 630–631 CrossRef CAS PubMed.
- R. Liu, J. Yang, L. Liu, Z. Lu, Z. Shi, W. Ji, J. Shen and X. Zhang, Adv. Sci., 2020, 7, 1901555 CrossRef CAS PubMed.
- A. Binda, C. Murano and I. Rivolta, Int. J. Nanomed., 2020, 15, 6113–6135 CrossRef CAS PubMed.
- G. Altinoglu and T. Adali, Curr. Drug Targets, 2020, 21, 628–646 CrossRef CAS PubMed.
- T. John, A. Gladytz, C. Kubeil, L. L. Martin, H. J. Risselada and B. Abel, Nanoscale, 2018, 10, 20894–20913 RSC.
- J. Gupta, M. T. Fatima, Z. Islam, R. H. Khan, V. N. Uversky and P. Salahuddin, Int. J. Biol. Macromol., 2019, 130, 515–526 CrossRef CAS PubMed.
- Y.-H. Liao, Y.-J. Chang, Y. Yoshiike, Y.-C. Chang and Y.-R. Chen, Small, 2012, 8, 3631–3639 CrossRef CAS PubMed.
- T. Yin, L. Yang, Y. Liu, X. Zhou, J. Sun and J. Liu, Acta Biomater., 2015, 25, 172–183 CrossRef CAS PubMed.
- X. Li and X. Liu, Nanoscale, 2017, 9, 7320–7341 RSC.
- D. Franke, A. Luisa da Rosa, M. Lorke and T. Frauenheim, Phys. Status Solidi B, 2019, 256, 1800455 CrossRef.
- C. M. Foster, R. Collazo, Z. Sitar and A. Ivanisevic, Langmuir, 2013, 29, 8377–8384 CrossRef CAS PubMed.
- G. Steinhoff, O. Purrucker, M. Tanaka, M. Stutzmann and M. Eickhoff, Adv. Funct. Mater., 2003, 13, 841–846 CrossRef CAS.
- T. H. Young and C. R. Chen, Biomaterials, 2006, 27, 3361–3367 CrossRef CAS PubMed.
- T. Braniste, I. Tiginyanu, T. Horvath, S. Raevschi, S. Cebotari, M. Lux, A. Haverich and A. Hilfiker, Beilstein J. Nanotechnol., 2016, 7, 1330–1337 CrossRef CAS PubMed.
- T. Braniste, I. Tiginyanu, T. Horvath, S. Raevschi, B. Andree, S. Cebotari, E. C. Boyle, A. Haverich and A. Hilfiker, Nanoscale Res. Lett., 2017, 12, 486 CrossRef PubMed.
- M. Hofstetter, J. Howgate, M. Schmid, S. Schoell, M. Sachsenhauser, D. Adigüzel, M. Stutzmann, I. D. Sharp and S. Thalhammer, Biochem. Biophys. Res. Commun., 2012, 424, 348–353 CrossRef CAS PubMed.
- S. A. Jewett, M. S. Makowski, B. Andrews, M. J. Manfra and A. Ivanisevic, Acta Biomater., 2012, 8, 728–733 CrossRef CAS PubMed.
- P. Sahoo, P. S. Murthy, S. Dhara, V. P. Venugopalan, A. Das and A. K. Tyagi, J. Nanopart. Res., 2013, 15, 1841 CrossRef.
- S. R. Choi, B. E. Britigan and P. Narayanasamy, Antimicrob. Agents Chemother., 2019, 63, e02643–02618 CAS.
- L. E. Bain, M. P. Hoffmann, I. Bryan, R. Collazo and A. Ivanisevic, Nanoscale, 2015, 7, 2360–2365 RSC.
- Y. Chen, N. Jyoti and J. Kim, Appl. Phys. A, 2011, 102, 517–519 CrossRef CAS.
- H.-L. Li, R.-J. Xie, N. Hirosaki, B. Dierre, T. Sekiguchi and Y. Yajima, J. Am. Ceram. Soc., 2008, 91, 1711–1714 CrossRef CAS.
- J. L. Robles-Garcia, A. Melendez, D. Yates, J. J. Santiago-Aviles, I. Ramos and E. M. Campo, Proc. SPIE, 2011, 8036, 80360F/80361–80360F/80367 CrossRef.
- A. Melendez, K. Morales, I. Ramos, E. Campo and J. J. Santiago-Aviles, Proc. SPIE, 2009, 7402, 740210/740211–740210/740216 CrossRef.
- E. M. Campo, J. Poplawsky, A. Melendez, J. L. Robles-Garcia, I. Ramos, D. Yates, J. E. Fischer and J. J. Santiago-Aviles, RSC Adv., 2014, 4, 2634–2639 RSC.
- U. Holzwarth and N. Gibson, Nat. Nanotechnol., 2011, 6, 534 CrossRef CAS PubMed.
- N. P. Cook, V. Torres, D. Jain and A. A. Martí, J. Am. Chem. Soc., 2011, 133, 11121–11123 CrossRef CAS PubMed.
- H. Liu, R. Lantz, P. Cosme, N. Rivera, C. Andino, W. G. Gonzalez, A. C. Terentis, E. P. Wojcikiewicz, R. Oyola, J. Miksovska and D. Du, Chem. Commun., 2015, 51, 7000–7003 RSC.
- L. Nielsen, R. Khurana, A. Coats, S. Frokjaer, J. Brange, S. Vyas, V. N. Uversky and A. L. Fink, Biochemistry, 2001, 40, 6036–6046 CrossRef CAS PubMed.
- S. Lee, E. J. Fernandez and T. A. Good, Protein Sci., 2007, 16, 723–732 CrossRef CAS PubMed.
- J. Jayamani, G. Shanmugam and E. R. Azhagiya Singam, RSC Adv., 2014, 4, 62326–62336 RSC.
- S.-H. Wang, X.-Y. Dong and Y. Sun, Biochem. Eng. J., 2012, 63, 38–49 CrossRef CAS.
- M. J. Hore, X. Ye, J. Ford, Y. Gao, J. Fei, Q. Wu, S. J. Rowan, R. J. Composto, C. B. Murray and B. Hammouda, Nano Lett., 2015, 15, 5730–5738 CrossRef CAS PubMed.
- E. Arad, H. Green, R. Jelinek and H. Rapaport, J. Colloid Interface Sci., 2020, 573, 87–95 CrossRef CAS PubMed.
- J. T. Pedersen and N. H. H. Heegaard, Anal. Chem., 2013, 85, 4215–4227 CrossRef CAS PubMed.
- L. Hong, X. Qi and Y. Zhang, J. Phys. Chem. B, 2012, 116, 6611–6617 CrossRef CAS PubMed.
- A. K. Buell, Biochem. J., 2019, 476, 2677–2703 CrossRef CAS PubMed.
- T. C. Michaels, H. W. Lazell, P. Arosio and T. P. Knowles, J. Chem. Phys., 2015, 143, 054901 CrossRef PubMed.
- P. Arosio, T. P. Knowles and S. Linse, Phys. Chem. Chem. Phys., 2015, 17, 7606–7618 RSC.
- K. Shezad, K. Zhang, M. Hussain, H. Dong, C. He, X. Gong, X. Xie, J. Zhu and L. Shen, Langmuir, 2016, 32, 8238–8244 CrossRef CAS PubMed.
- S. J. Cox, B. Lam, A. Prasad, H. A. Marietta, N. V. Stander, J. G. Joel, B. R. Sahoo, F. Guo, A. K. Stoddard, M. I. Ivanova and A. Ramamoorthy, Biochemistry, 2020, 59, 2249–2258 CrossRef CAS PubMed.
- G. Gao, M. Zhang, D. Gong, R. Chen, X. Hu and T. Sun, Nanoscale, 2017, 9, 4107–4113 RSC.
- M. Lundqvist, J. Stigler, G. Elia, I. Lynch, T. Cedervall and K. A. Dawson, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14265–14270 CrossRef CAS PubMed.
- K. Yokoyama, A. Fischer, A. R. Amori, D. R. Welchons and R. E. McKnight, J. Biophys. Chem., 2010, 1, 153–163 CrossRef CAS.
- W. E. Klunk, R. F. Jacob and R. P. Mason, Anal. Biochem., 1999, 266, 66–76 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |