
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effect of kaolin pre-treatment method and NaOH levels on the structure and properties of kaolin-derived faujasite zeolites
Stephen
Otieno
a,
Chrispin
Kowenje
a,
Fredrick
Kengara
b and
Robert
Mokaya
 *c
*c
aDepartment of Chemistry, Maseno University, P. O. Box, 333-40105, Maseno, Kenya
bSchool of Pure and Applied Sciences, Bomet University College, P. O. Box 701-20400, Bomet, Kenya
cSchool of Chemistry, University of Nottingham, University Park, Nottingham NG7 2RD, UK. E-mail: r.mokaya@nottingham.ac.uk
First published on 16th August 2021
Abstract
Fusion with NaOH is considered effective in the conversion of kaolin to soluble aluminosilicates. However, to date, there is no consistency in the nature and type of zeolite structures prepared from kaolin via this approach, suggesting a lack of clarity on the effect of varying the fusion method. In this report, we have explored the effect of NaOH as an activator in the fusion step during the synthesis of kaolin-derived zeolites by performing partial or full fusion of the kaolin before the hydrothermal crystallization step. The content of NaOH was adjusted to maintain similar hydrogel composition, and the subsequent hydrothermal synthesis conditions were identical for the partial and full fusion protocols. The effect of the kaolin pre-treatment prior to full fusion was also explored by mixing the clay with NaOH under varied conditions. Zeolite Y and zeolite X structures were obtained from partial and full fusion, respectively. For full fusion, pre-treatment of kaolin by dry mixing before dry fusion resulted in zeolite X with some zeolite A impurity. On the other hand, wet mixing promoted homogeneity in the hydrogel with no formation of undesirable zeolite A. Wet mixing before dry fusion generated pure zeolite X with porosity (645 m2 g−1 and 0.24 cm3 g−1) that is comparable to that of commercial zeolite 13X. However, wet mixing with subsequent wet fusion resulted in poorly crystalline zeolite X along with some small pore hydroxysodalite impurity. Our findings demonstrate the crucial role of NaOH concentration in the fusion of kaolin when used as starting material in determining the zeolite structure. We also show the need for an efficient pre-treatment method that allows the complete conversion of kaolin into suitable ingredients before the hydrothermal crystallization step. We, therefore, identify synthesis protocols that enable use of sustainable and cheap kaolin as a raw material for high quality faujasite zeolites.
1. Introduction
Zeolites are important materials for various applications, including catalysis, detergents, water purification, and agriculture. The hydrothermal synthesis of zeolites is known to be influenced by several factors. Key synthesis variables, which determine the structure type, elemental composition, porosity, and crystallinity of the resulting zeolites include the hydrogel silica (SiO2) to alumina (Al2O3) molar ratio, concentration of alkaline activator, ageing time, and crystallization time and temperature.1–3 The multiplicity of the variables means that the synthesis of zeolites can be a complex process.The industrial production of zeolites involves the use of costly commercially available sources of silica and alumina. For this reason, there is ongoing research on finding cost-effective and sustainable alternatives. Naturally available kaolin, rich in kaolinite (Kaol) minerals, is currently being extensively explored as a raw material for zeolite synthesis due to its abundance and high silica and alumina content.4 Kaol is a hydrous aluminium silicate formed by the decomposition of feldspar minerals. Before the hydrothermal synthesis of zeolites, kaolin is usually treated at high temperature by either calcination to form metakaolinite (MK) or fusing with an alkaline activator to form fused-metakaolinite (F-MK). The advantage of using F-MK over MK in pre-treating kaolin is that it enables complete breakdown of the kaolinite structure and also promotes dry reactions of other thermally stable mineral phases, such as quartz and muscovite, that are usually present in the raw materials.5 The alkaline activation method therefore improves the dissolution and reactivity of the kaolin components. This ensures that a large amount of aluminosilicate structural building units are subsequently dissolved in the hydrogel solution6 prior to hydrothermal crystallisation. The amount of alkaline activator used in the fusion step is also crucial to the extent that the proportion of kaolin constituents, as indicated by intensity of their powder X-ray diffraction peaks, effectively decreases with increasing concentration of alkaline activator in the fusion step.7
Although alkaline fusion is considered effective in the conversion of kaolin to structural building units, the manner and steps via which it is achieved and the post modifications carried out on the synthesis hydrogels also play a role in determining the resulting zeolite structure types and their properties. A review of studies reported in the literature to date (Table 1) shows that different zeolite structures are formed from the two methods of kaolin treatment. Bahgaat and co-workers recently reported synthesis of zeolite Y from impure Egyptian kaolin prepared via MK without modification of the synthesis hydrogel.8 However, such hydrogels derived via calcination of kaolin to MK and without further modification of their chemical compositions in most cases favour the formation of zeolite A.3,5,9–12 On the other hand, the fusion of kaolin to F-MK without further modification of the hydrogel composition, but with the same hydrogel composition as in the MK hydrogels, favours the formation of low silica faujasite structures.7,9,13–16 Some studies have however reported synthesis of hydroxysodalite17 and zeolite A with quartz and hydroxysodalite impurities3,5,6,15,18 from F-MKs derived from the same fusion protocols and without further modification of the hydrogel composition.
| Kaolin source | Kaolin treatment | Hydrogel modification | Hydrogel composition | Zeolite product | % Cryst | A BET | V μ pore | Ref. |
|---|---|---|---|---|---|---|---|---|
| FAU = faujasite zeolite, ANA = analcime, SDA = structure directing agent, A = zeolite A, X = zeolite X, Y = zeolite Y, Q = quartz, S = hydroxysodalite, P = philipsite, M = muscovite, I = illite, K = kaolin, n.d = not donea Identified impurity.b Unidentified impurity. | ||||||||
| Brazil | MK at 850 °C/2 h | Na2SiO3 | 10 g MK & 15 g NaOH in 100 g of H2O | Y, Pa | 78% | 555 | n.d | 21 |
| Sigma | MK at 900 °C/0.5 h | Na2SiO3·5H2O | Na2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Al2O3:SiO2:H2O = 3.75:1.0:2.5:243.7 :Al2O3:SiO2:H2O = 3.75:1.0:2.5:243.7 |
X, A, P, S | n.d | 436 | n.d | 24 |
| Iraq | F-MK at 850 °C/3 h | Na2SiO3 | 50 g F-MK & 63 g Na2SiO3in 500 ml H2O | Y, Ib | n.d | 390 | 0.11 | 25 |
| Egypt | Patented | Na2SiO3 | Patented | Y, Q, MK | n.d | n.d | n.d | 23 |
| Nigeria | MK at 600 °C/50 min | Na2SiO3 | Na2O:Al2O3:SiO2:H2O = 15:1:15:450 |
Y, Ib | n.d | n.d | n.d | 19 |
| Cameroon | MK at 750 °C/8 h | — | SiO2:Al2O3:Na2O:H2O = 3.5:2.1:1:143.2 |
A, Q | 88.1% | 18 | — | 9 |
| Fumed silica | SiO2:Al2O3:Na2O:H2O = 3.5:2.1:1:143.2 |
A, X, Y, P, Q | n.d | n.d | n.d | |||
| F-MK at 750 °C/8 h | — | SiO2:Al2O3:Na2O:H2O = 10:1:7.98:120 |
X, A | 95.9% | 579 | 0.18 | ||
| Fumed silica | SiO2:Al2O3:Na2O:H2O = 10:1:7.98:120 |
Y | n.d | 600 | 0.18 | |||
| South Africa | F-MK at 550 °C/1.5 h | — | Al2O3:Na2O:SiO2:H2O = 1:9.70:7.79:403.20 |
S | n.d | n.d | n.d | 17 |
| Al(OH)3 | Al2O3:Na2O:SiO2:H2O = 1:7.11:4.71:292.35 |
X | n.d | n.d | n.d | |||
| Poland | MK at 800 °C/6 h | Silica gel | Na2O:SiO2:Al2O3:H2O = 10:10:1:200 |
Y, Q, M, Pa | n.d | n.d | n.d | 29 |
| Poland | MK at 800 °C/6 h | — | Na2O:SiO2:Al2O3:H2O = 10:10:1:200 |
Y, Q | n.d | 686 | 0.26 | 30 |
| Ethiopia | MK at 600 °C/3 h | — | SiO2:Al2O3:Na2O:H2O = 2:1:1:37 |
A, Q | 75% | n.d | n.d | 5 |
| F-MK at 600 °C/1 h | — | SiO2:Al2O3:Na2O:H2O = 2:1:1:37 |
A, S | 84% | n.d | n.d | ||
| Tunisia | — | Al(OH)3 | SiO2:Al2O3:Na2O:H2O = 3.4:1:10.4:319.8 |
S, Q, K, I | n.d | 62 | - | 14 |
| Fusion at 550 °C/2 h | Al(OH)3 | SiO2:Al2O3:Na2O:H2O = 3.4:1:10.4:319.8 |
X, Aa, Sa | n.d | 293 | 0.12 | ||
| Brazil | MK at 700 °C/2 h | — | Na/Al & Si/Al of 1.64 & 1, H2O = 15 ml | A | n.d | n.d | n.d | 10 |
| Algeria | F-MK at 800 °C/2 h | Ludox (40 wt% SiO2) | Na2O:Al2O3:SiO2:H2O = 4.01:1:9.37:156 |
Y | n.d | 626 | 0.26 | 26 |
| India | MK at 900 °C/1 h | Na2SiO3 | SiO2/Al2O3 = 7.5–15; Na2O/SiO2 = 0.5–1 and H2O/Na2O = 20–30 | Y, P, Pa | n.d | 490 | n.d | 20 |
| Jordan | MK at 650 °C/2 h | — | MK/NaOH solution of 1.0 g/25 ml | A, Q, S | n.d | n.d | n.d | 31 |
| England | F-MK at 600 °C/1 h | — | 4.40 g F-MK in 21.5 ml water | A | n.d | n.d | n.d | 6 |
| Egypt | MK at 800 °C/6 h | — | 10 g MK in 100 ml of 1 M NaOH | Y, P | n.d | n.d | n.d | 8 |
| Ghana | F-MK at 600 °C/2 h | — | F-MK/water in 1:5 w/w |
X, Q, Aa | n.d | 389 | 0.84 | 16 |
| Brazil | F-MK at 700 °C/2 h | Na2SiO3 | 8.72 g MK, 16.63 g Na2SiO3, 6.28 g NaOH | Y | n.d | n.d | n.d | 27 |
The post-modification of the synthesis hydrogels in both MK and F-MK methods, for example by addition of an extra source of silica, results in high silica products. Again, the reported structures, and their properties, from the two treatment methods when extra silica was used are inconsistent. More importantly, the structure types vary from one study to another where high silica faujasite X or faujasite Y structures were reported to have been formed when MK,9,19–24 or F-MK5,7,9,14–16,25–27 methods were used. Despite the attraction and huge potential of kaolinite rich clays as a sustainable alternative source of aluminosilicates in zeolite synthesis, the apparent inconsistency in the products generated and their properties are an on-going challenge.9,19–25
The review in Table 1 and the inconsistencies therein of the synthesised zeolite structures suggests that current practice, which involves pre-treatment of clays via F-MK, may be ineffective. The homogeneity of the starting synthesis hydrogels and their transformation into crystalline zeolite material are an important area of research.28 It is therefore important to fully explore the conversion of aluminosilicates in kaolin, into a form that ensures subsequent complete dissolution to a homogeneous hydrogel. In this work, the effects of NaOH concentration in the alkaline fusion step have been investigated by comparing partial fusion with full fusion of kaolin before hydrothermal crystallization. In addition, the effects of pre-treatment of kaolin with alkaline activator prior to the fusion step have been explored by subjecting the raw clay to two regimes, namely, dry mixing with NaOH followed by dry fusion, or wet mixing with NaOH followed by wet/dry fusion. The fusion and synthesis conditions, i.e., alkaline fusion, hydrogel composition, ageing and hydrothermal synthesis were identical for all protocols. The findings of this study will inform on the choice of pre-treatment processes that may be applied to clay materials before fusion and hydrothermal synthesis of zeolites.
2. Experimental
2.1. Materials
The clay material was supplied by the Department of Inorganic Chemistry, University of Yaoundé 1 – Cameroon. Sodium hydroxide, ≥99%, pellets, and molecular sieve 13X (4–8 mesh) were obtained from Thermo Fisher Scientific while sodium metasilicate pentahydrate (Na2SiO3·5H2O), ≥97%, was obtained from Sigma Aldrich. Deionized water was via an Elga PURELAB Option 4463 deionizer.2.2. Synthesis of zeolites
The zeolites synthesis was carried out in two steps, the first step being the alkaline fusion of kaolin with NaOH while the second step was the hydrothermal reaction step. The alkaline fusion step was carried out via two protocols utilizing different quantities of NaOH in the fusion. The NaOH content was subsequently adjusted in the hydrogel for the partial fusion protocol to match that of the full fusion protocol. Fig. 1 shows a schematic of the two protocols used in this study and their modifications.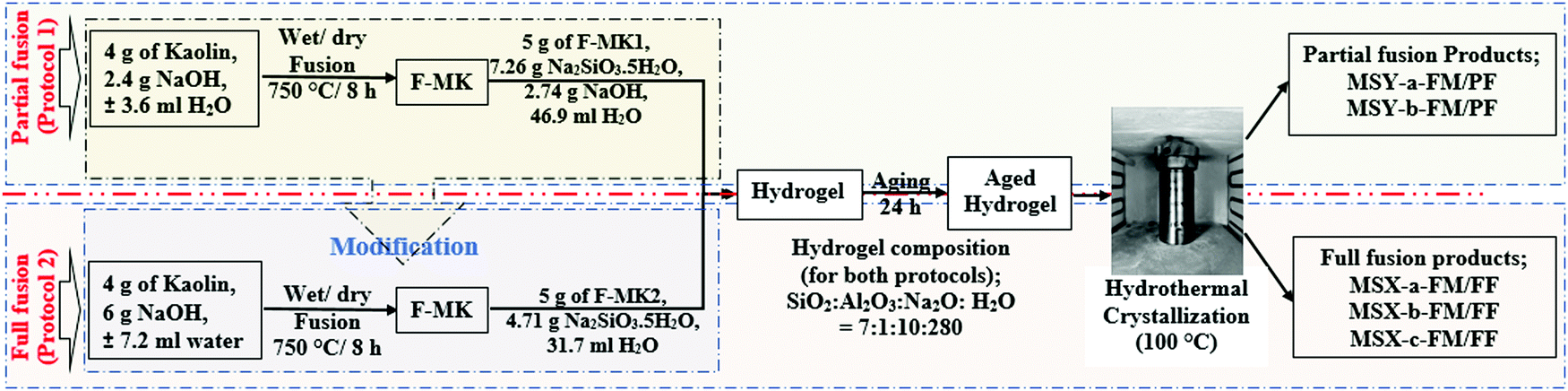 | ||
| Fig. 1 Schematic for partial and full fusion protocols used for the synthesis of zeolites. | ||
The first synthesis method (protocol 1) utilizing lower quantity of NaOH in the fusion step, (hereinafter referred to as partial fusion protocol), was a modified version of the method reported by Doyle and co-workers.25 Specifically, fused-metakaolinite (F-MK1) was prepared by grinding 4 g of kaolin with 2.4 g of NaOH pellets in agate motor before fusing the mixture at 750 °C for 8 h. The resulting F-MK1 was then ground to powder before preparing a synthesis hydrogel by dissolving 5 g of the F-MK1, 7.26 g of Na2SiO3·5H2O and 2.74 g of residual NaOH in 46.9 ml of deionized water. Based on the elemental composition of the kaolin, the final molar composition of the synthesis hydrogel was SiO2:Al2O3:Na2O:H2O = 7:1:10:282. The hydrogel was then magnetically stirred (600 rpm) at 25 °C for 4 h before ageing without stirring at 25 °C for 24 h. The aged hydrogel was placed in an oven to crystallize at 100 °C for 48 h leading to products denoted as MSY zeolites. The products were washed with deionized water to pH < 8 followed by drying at 100 °C for 24 h.
To match protocol 1 with our previous work,9 and without altering the synthesis hydrogel composition, the fusion step in protocol 1 was modified in protocol 2 by carefully calculating and grinding all the required NaOH activator with kaolin before fusion. Specifically, 4.0 g of kaolin was mixed with 6 g of NaOH by grinding in agate motor followed by fusion as described above. The resulting F-MK2 was then ground to powder before preparing a synthesis hydrogel with a chemical composition of SiO2:Al2O3:Na2O:H2O = 7:1:10:282, similar to protocol 1, by dissolving 5 g of the F-MK2 powder and 4.71 g of Na2SiO3·5H2O in 31.7 ml of deionized water. The resulting hydrogel was then stirred, aged and crystallized as described in protocol 1 and resulted in products denoted as MSX zeolites.
2.3. Pre-treatment of kaolin before fusion step
To investigate the effects of pre-treatment before the fusion step, three pre-treatment methods of kaolin were performed by either dry mixing or wet mixing of kaolin with NaOH before either wet or dry fusion step followed by hydrothermal crystallization step at conditions similar those described in Section 2.2. The first pre-treatment (also referred to as dry mixing before dry fusion) was achieved by dry mixing the kaolin with NaOH pellets through grinding in a mortar before dry fusing the mixture as described above. The second pre-treatment method (wet mixing before wet fusion) was achieved by grinding kaolin with 3.6 ml of 16.7 M NaOH and 7.2 ml of 20.8 M NaOH solution, for partial and full fusion protocols, respectively, in agate motor before fusing the wet mixture. In the third pre-treatment (wet mixing before dry fusion), the mixing was achieved by wet mixing of kaolin with NaOH solution, as described for wet mixing above, after which the water content was removed by drying the mixture at 100 °C for 48 h in an oven before dry fusing the resulting mixture. Subsequent hydrogel preparation and hydrothermal crystallization were performed as described in Section 2.2.The products obtained through the partial fusion in protocol 1 and the full fusion in protocol 2 are denoted as MSY-(a/b)-FM/PF and MSX-(a/b/c)-FM/FF respectively. Where MSX and MSY denotes zeolites related to the zeolite Y and the zeolite X families, respectively, FM denotes fused metakaolinite (F-MK) while PF and FF denotes products obtained in the partial fusion and full fusion protocols respectively. The letter a, b or c denotes products obtained via the pre-treatment of kaolin by dry mixing with NaOH before dry fusion, wet mixing with NaOH before wet fusion or wet mixing with NaOH followed by evaporation of the water solvent before dry fusion, respectively.
For further investigations, the three pre-treatment procedures were performed on our previously reported F-MK protocol, which reported the formation of zeolite Y when fumed silica modified hydrogel was used.9 F-MK was prepared by fusing a mixture of 6 g of kaolin and 7.2 g of NaOH pellets at 750 °C for 8 h before dissolving 10.0 g of the F-MK and 8 g of fumed silica in 35.1 ml of H2O water to give a synthesis hydrogel composition of SiO2:Al2O3:Na2O:H2O molar ratio of 14.08:1:7.13:150.18. The products are denoted FSY-(a,b/c)-FM/FF where FSY is the zeolite Y obtained via this method.
2.4. Characterization of clay and zeolite products
Powder XRD patterns were obtained in continuous mode with 2θ scan step size of 0.026° using a PANalytical X-Pert Pro X-ray powder diffractometer employing Cu-Kα radiation. X-rays were generated from the Cu anode at 40 kV and a current of 40 mA. The diffraction of the sample was recorded over the 2θ range from 5° to 50° at room temperature. Zeolite phases were identified by comparison with the reference XRD patterns in the zeolite database from the International Zeolite Association32 and that of commercial molecular sieve 13X. The presence of functional groups was monitored by infrared spectroscopy between 400–4000 cm−1 using a Bruker Alpha Attenuated Total Reflectance – Fourier Transform Infrared (ATR-FTIR) spectrometer. Thermogravimetric analysis (TGA) was performed using a TA Instruments SDT Q600 thermal analyser by heating samples at ramp rate of 10 °C min−1 under flowing air (100 ml min−1) up to 1000 °C. The morphology of the samples was explored via scanning electron microscopy (SEM) using a FEI Quanta 200 3D Dual Beam FIB microscope.Elemental composition was obtained using an energy dispersive spectrometer (EDS) during SEM analysis and a PerkinElmer Inductively Coupled Plasma Optical Emission Spectroscopy (ICP-OES) Optima 2000 DV fitted with an auto sampler. For the ICP-OES, the samples were prepared using the reported fusion procedure by ref. 33. The textural properties were obtained using a Micromeritics 3FLEX sorptometer employing N2 gas as a sorbate at liquid nitrogen temperature (−196 °C). The samples were outgassed under vacuum at 300 °C for 16 h before analysis. The surface area was calculated using the Brunauer–Emmett–Teller (BET) method. The total pore volume was calculated from the nitrogen uptake at close to saturation pressure (P/P0 ∼ 0.99), micropore surface area, external surface area and micropore volume were determined from t-plot analysis while pore distribution and pore size were determined by the Horvath–Kawazoe model.34
3. Results and discussion
3.1. Characterization of clay material
The FTIR spectra of the raw and pre-treated clay material are shown in Fig. 2a. The spectra for the raw clay reveals four bands between 3600 and 3900 cm−1 characteristic of the triclinic layer structure of kaolinite clays.35 The band at ca. 3620 cm−1 is due to stretching vibrations of inner Al–OH groups, while those at ca. 3654, 3667 and 3687 cm−1 are due to the stretching vibrations of surface –OH groups. The Si–O in plane and out of plane stretching vibrations bands appear at ca. 997 and 1114 cm−1, respectively, while the Al–O stretching of the AlO6 octahedron and the Al–OH inner surface bending vibration bands are at 540 and 911 cm−1, respectively. The doublet bands at ca. 700 and 665 cm−1 due to Si–O from quartz phase have been previously observed.36 Other bands at ca. 525 and 1025 cm−1 are due to Si–O–Si bending and stretching vibrations respectively. The quartz and kaolinite bands were masked in the wet clay treated with NaOH solution in the pre-treatment step. The kaolinite bands were however suppressed after drying out the water solvent leaving the quartz bands and other newly formed bands. Further treatment of the mixture by fusion at 750 °C eliminated all the kaolinite and quartz bands with formation of more new bands in the low wave number region.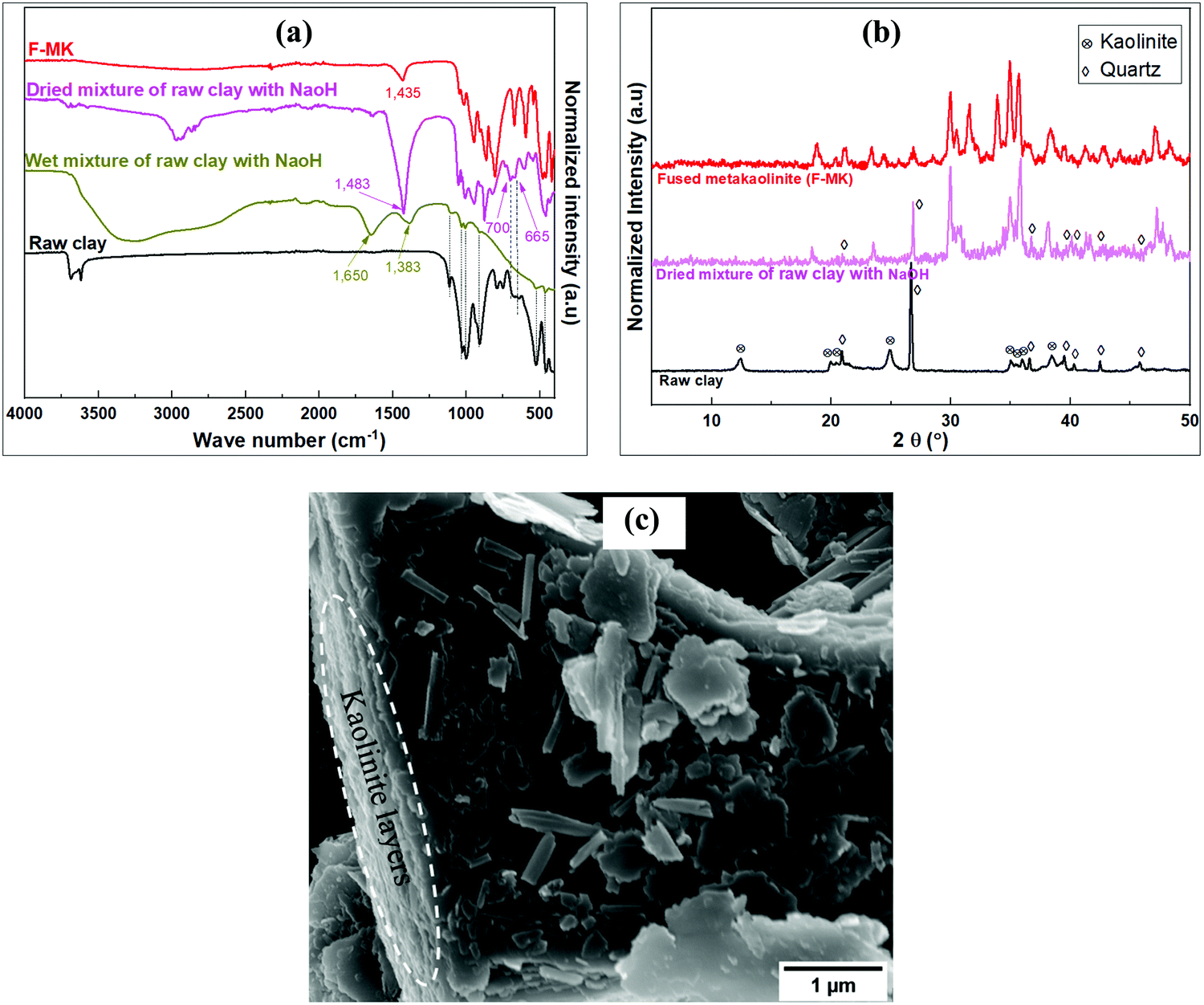 | ||
| Fig. 2 Analysis of raw and pre-treated clay material; (a) FTIR spectra of raw clay, modified and F-MK, (b) XRD patterns of raw clay, modified and F-MK, (c) SEM image of raw clay. | ||
Fig. 2b shows the XRD patterns of the raw and pre-treated clay material. The peaks at 2θ of 12.44° and 24.86° for the of raw clay are characteristic of the kaolinite phase while those at 2θ of 20.87°, 26.67°, 36.60°, 39.49°, 42.49° and 45.82° correspond to quartz phases. Pre-treatment of kaolin by wet mixing with NaOH followed by drying out of the water led to the suppression of kaolinite peaks and emergence of new peaks alongside the initial quartz peaks. The kaolinite and quartz peaks were not observed in the fused-metakaolinite (F-MK) after fusing the kaolin at 750 °C, however, new peaks were observed at higher 2θ values in addition to those observed for the dried mixture of raw clay and NaOH. Fig. 2c shows the SEM image of the raw clay depicting layered structures with pseudohexagonal platy morphology typical of kaolinites.37
The elemental data for kaolin is shown in Table 2, and indicates that SiO2 (56.74 wt%) and Al2O3 (30.34 wt%), with SiO2/Al2O3 molar ratio of 3.17, were the main oxides in the raw clay. Other elements which were detected in trace amounts included Ti, Fe, Na, K and Cu.
| wt% | SiO2 in the Kaol structure | SiO2 in as quartz |
|---|---|---|
| 35.71 | 21.03 | |
| Calculated formula | Fe0.93Cu0.39Ti4.89Na2.69K0.69Al100Si158.70O475.76 | |
Thermal analysis curves of the raw clay and the pre-treated clay are shown in Fig. 3. The raw clay is stable up to 400 °C with subsequent exothermic dehydroxylation occurring from ca. 500 °C accompanied with 10.49% mass loss on ignition (LOI) to form MK (Fig. 3a). The modified clay (wet mixed with NaOH then dried) in Fig. 3b shows multiple steps of exothermic mass loss. In the first step, ca. 5% mass loss occurs between 30 °C and 500 °C, due to the loss of absorbed water. The second step occurs between 500 °C and 720 °C and may be due to dehydroxylation reactions to form Si–O–Si(Al), which could also account for the steps between 720 and 810 °C. The steps above 810 °C may arise from change in sodium aluminosilicate phases to more stable phases.
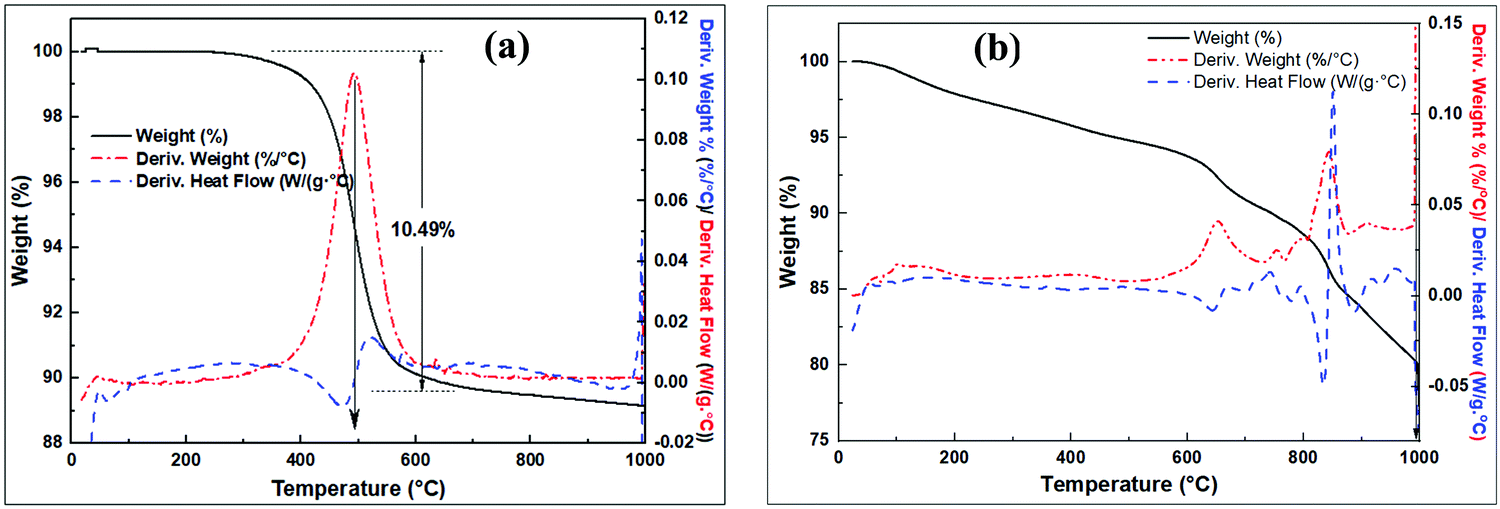 | ||
| Fig. 3 TGA analysis of; (a) raw clay, (b) wet mixed raw clay with NaOH then dried. | ||
3.2. Characterization of zeolite products
The meanings of abbreviations and product codes in this study are shown below in Table 3.| Code/symbol | Description |
|---|---|
| F-MK | Fused-metakaolinite (fused kaolin with NaOH at 750 °C for 8 h) |
| Washed F-MK | Fused-metakaolinite washed with deionized water |
| Partial fusion | Calcining kaolin in the presence of smaller quantity of NaOH compared to full fusion |
| Full fusion | Calcining kaolin in the presence of larger quantity of NaOH |
| SMX | Synthesized zeolite X from hydrogel modified with additional metasilicate |
| FMY | Synthesized zeolite Y from hydrogel modified with additional fumed silica |
| FM | Fused-metakaolinite product |
| PF | Partial fusion product |
| FF | Full fusion product |
| a | Product obtained from dry mixing of kaolin with NaOH followed by dry fusion |
| b | Product obtained from wet mixing of kaolin with NaOH followed by wet fusion |
| c | Product obtained from wet mixing of kaolin with NaOH and then drying out the water before dry fusion |
| MSY-a-FM/PF | Zeolite Na-Y synthesized from partial fusion of dry mixture of kaolin with NaOH (dry fusion of a dry mixture) |
| MSY-b-FM/PF | Zeolite Y synthesized from partial fusion of wet mixture of kaolin with NaOH (wet fusion of a wet mixture) |
| MSX-a-FM/FF | Zeolite X synthesized from full fusion of dry mixture of kaolin with NaOH (dry fusion of a dry mixture) |
| MSX-b-FM/FF | Zeolite X synthesized from full fusion of a wet mixture of kaolin with NaOH (wet fusion of wet mixture) |
| MSX-c-FM/FF | Zeolite X synthesized from full fusion of a dried out wet mixture of kaolin with NaOH (dry fusion of a wet mixture) |
The chemical composition and the calculated chemical formula of the products obtained at varying synthesis durations is shown in Table 4. Both EDX-SEM and ICP-OES methods for elemental analysis indicated relatively similar chemical composition as observed by the analysis of sample MSX-c-FM/FF-48h. Varying the concentration of NaOH in the fusion step, i.e., performing partial or full fusion, did not affect the SiO2/Al2O3 ratio of the resulting zeolite products as observed in the MSY-b-FM/PF and MSX-b-FM/FF products obtained via wet fusion in the partial and full fusion, respectively. In addition, the SiO2/Al2O3 ratio of the resulting products was not significantly affected by the crystallization time (see MSX-c-FM/FF-10h, MSX-c-FM/FF-24h, MSX-c-FM/FF-48h and MSX-c-FM/FF-72h). The SiO2/Al2O3 ratios of the resulting products were, however, affected by the pre-treatment method of the kaolin. The wet fusion products (MSX-b-FM/FF and MSY-b-FM/PF) had low SiO2/Al2O3 ratio of ca. 2.3, while the dry fusion products, in both dry mixing and wet mixing, (MSX-a-FM/FF and MSX-c-FM/FF) had slightly higher SiO2/Al2O3 ratio of ca. 2.5.
| Sample | Percent oxide (wt% MxOy) | Method | Chemical formula | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Al | Si | Na | Ti | Fe | K | Cu | SiO2/Al2O3 | |||
| MSX- a-FM/FF-48h | 30.83 | 49.40 | 16.70 | 1.48 | 0.34 | 0.10 | 0.95 | 2.72 | EDX-SEM | Fe0.7Ti3.1Na89.1K0.6Al100Si136O477 |
| MSX-a-FM/FF-96h | 31.20 | 48.91 | 17.52 | 1.36 | 0.44 | 0.54 | 2.66 | EDX-SEM | Fe0.9Ti2.8Na92.4K0.2Al100Si133O472.1 | |
| MSX-b-FM/FF-48h | 33.01 | 45.75 | 20.08 | 0.78 | 0.14 | 0.26 | 2.35 | EDX-SEM | Fe0.3Ti1.5Na100.1K1.4Al100Si117.6O440.9 | |
| MSX-c-FM/FF-10h | 29.94 | 45.31 | 23.15 | 0.95 | 0.30 | 0.18 | 0.17 | 2.57 | ICP-OES | Fe0.63Cu0.37Ti2.10Na127.2K0.66Al100Si128.42O476.29 |
| MSX-c-FM/FF-24h | 30.33 | 44.71 | 23.23 | 1.01 | 0.30 | 0.18 | 0.23 | 2.50 | ICP-OES | Fe0.63Cu0.48Ti2.22Na125.99K0.65Al100Si125.08O469.32 |
| MSX-c-FM/FF-48h | 29.89 | 44.96 | 23.30 | 1.01 | 0.42 | 0.17 | 0.22 | 2.55 | ICP-OES | Fe0.90Cu0.47Ti2.24Na128.27K0.62Al100Si127.63O476.00 |
| MSX-c-FM/FF-48h | 31.60 | 48.50 | 18.74 | 0.90 | 0.21 | 2.60 | EDX-SEM | Fe0.4Ti1.8Na97.6K0.3Al100Si130.2O465.4 | ||
| MSX-c-FM/FF-72h | 30.02 | 43.79 | 24.58 | 1.00 | 0.30 | 0.13 | 0.17 | 2.48 | ICP-OES | Fe0.63Cu0.36Ti2.20Na134.70K0.48Al100Si123.75O470.81 |
| MSY-b-FM/PF-48h | 31.87 | 42.83 | 18.91 | 1.38 | 0.35 | 2.33 | EDX-SEM | Fe0.7Ti2.8Na97.6Al100Si114O436.2 | ||
The FTIR spectra showing the transformation of F-MK through its modified hydrogel to zeolite products, in both partial fusion and full fusion protocols, and their SEM images are shown in Fig. 4. Two sets of products were obtained depending on the mode of kaolin pre-treatment, i.e., partial fusion products (Fig. 4a) or full fusion products (Fig. 4b). FTIR bands of the F-MKs shift to high wavenumbers as the hydrothermal crystallization products are formed, which shift has been observed in previous studies.9 The FTIR bands at ca. 970 cm−1, 675 cm−1 and 450 cm−1 are due to the asymmetric stretching, symmetric stretching and symmetric bending vibration, respectively, of zeolitic T–O tetrahedral aluminosilicate bonds.38 The band at ca. 556 cm−1 is for symmetric stretching vibrations associated with the six-member rings (D6R) of the faujasite structure.39 The deconvoluted bands of the aged full fusion hydrogel modified with sodium metasilicate pentahydrate in Fig. 4c also show the presences of bands leading to the formation of MSX zeolites products. The bands at ca. 3400 and 1647 cm−1 are associated with the hydroxyl stretching and bending vibrations, respectively, of the absorbed water of hydration.40 The hydroxyl bands are more prominent in the full fusion products obtained in protocol 2 (MSX zeolites) than in the partial fusion products obtained in protocol 1 (MSY zeolites) because the former are more hydrophilic, or have large pores that hold greater amounts of water. The SEM images of the products reveal octahedral-like crystals, which are characteristic of faujasite zeolites, with varying particle size of up to 0.5 μm and 2 μm for MSX-c-FM/FF-48h and MSY-b-FM/PF-48h products, respectively.
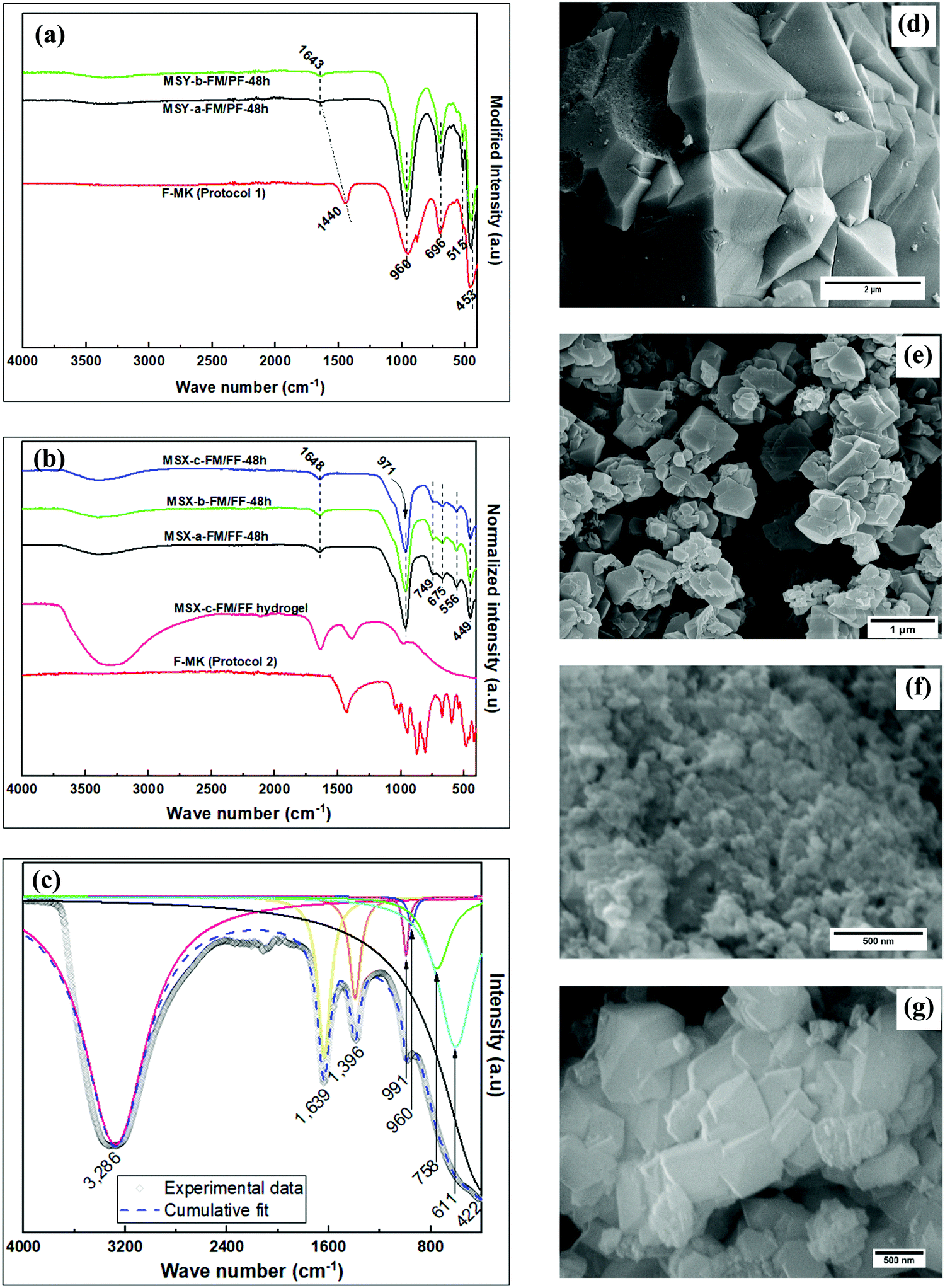 | ||
| Fig. 4 The FTIR spectra of; (a) partial fusion products, (b) full fusion products, (c) deconvoluted FTIR peaks of the aged FF hydrogel modified with Na2SiO3·5H2O, and SEM images of; (d) MSY-b-FM/PF-48h, (e) MSX-a-FM/FF-48h, (f) MSX-b-FM/FF-48h, (g) MSX-c-FM/FF-48h. | ||
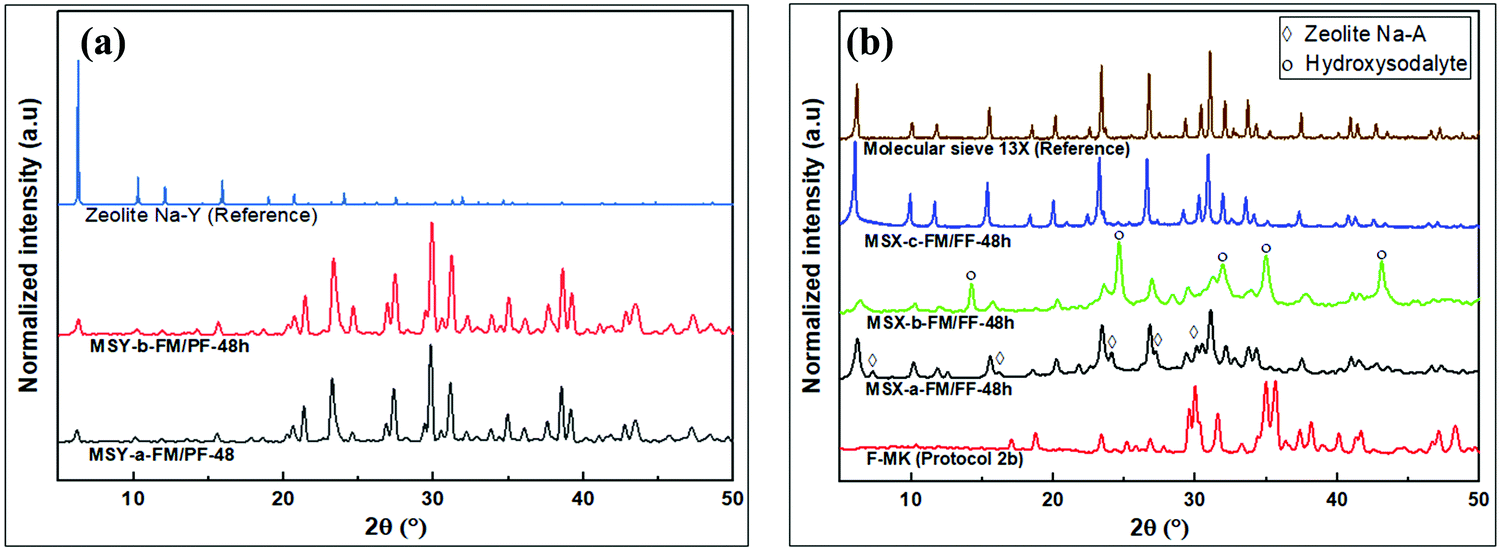
The XRD patterns showing the transformation of kaolin to zeolite products are shown in Fig. 5. Two sets of zeolite structures are observed depending on whether partial or full fusion was applied in the pre-treatment of the kaolin. The intensity of peaks in the low 2θ region for the partial fusion products was lower than that for full fusion products. The XRD peaks of partial fusion products (i.e., MSY-a-FM/PF-48h and MSY-b-FM/PF-48h) at 2θ values of 6.4°, 10.3°, 11.9°, 15.6°, 17.9°, 21.4°, 23.3°, 24.6° and 27.4° in Fig. 5a, are characteristic of zeolite Y32 and are consistent with the zeolite Y product obtained by Doyle et al.25 On the other hand, the XRD patterns of the full fusion products in Fig. 5b (i.e., MSX-a-FM/FF-48h, MSX-b-FM/FF-48h and MSX-c-FM/FF-48h) exhibit peaks at slightly lower 2θ values of 6.2°, 10.1°, 11.7°, 15.4°, 20.1°, 24.0°, 27.2°, 29.3°, and 30.0°, which are consistent with zeolite X.32
 | ||
| Fig. 5 The XRD patterns; (a) partial fusion products and, (b) full fusion products. | ||
The low amount of NaOH in the partial fusion method of protocol 1 may result in incomplete conversion of kaolinite clay components leading to the formation of less reactive aluminosilicates41 alongside the more reactive sodium silicate and sodium aluminate (sodium aluminosilicate) components. The presence of aluminosilicates in the hydrogel may aid the transformation of the faujasite building units to those of faujasite Y during nucleation and/or crystallization. This is consistent with trends observed in the literature review (Table 1) where zeolite Y was the main product when MK (aluminosilicate) hydrogels were used rather than F-MK hydrogels.
In contrast, the high alkaline environment created in the full fusion method of protocol 2 appears to facilitate complete conversion of the kaolin components to sodium aluminosilicates.5,42 The resulting synthesis gel is therefore rich in highly reactive sodium aluminosilicates rather than less reactive aluminosilicates. The reaction of sodium aluminosilicates at high fusion temperatures may also lead to the formation of secondary or tertiary (i.e., dimers, oligomers or polymers) structure building units corresponding to the faujasite X structure9 as observed in the FTIR spectra of the modified hydrogel in Fig. 4c. Alternatively, these faujasite X structure building units may form during the nucleation stage of the hydrogel favoured by the sodium aluminosilicates formed in the fusion step. The form of the final products is therefore significantly influenced by the nature in which the starting kaolin components are present in the synthesis gel. The gels that are rich in aluminosilicates favour formation of faujasite Y, while those rich in sodium aluminosilicates favour formation of faujasite X structures.
In the partial fusion in protocol 1, the mode of kaolin pretreatment did not affect the crystalinity of the resulting zeolite Y as observed in the XRD patterns of MSY-a-FM/PF-48h and MSY-b-FM/PF-48h in Fig. 5a. However, for the full fusion products in protocol 2, the intensity and purity of the resulting zeolite X depended on the mode of kaolin pre-treatment prior to hydrothermal crystalization, Fig. 5b. Apart from the zeolite X, low-intensity peaks at 2θ of 7.3°, 16.2°, 24.1°, 27.2° and 29.9°, which are consistent with zeolite A,32 were also observed for the MSX-a-FM/FF-48h product. The MSX-b-FM/FF-48h product showed poorly crystalline zeolite-X and hydroxysodalite impurity characterized by the peaks at 14.3°, 24.7°, 31.9°, 35.0° and 43.1°.32 The poor-quality of the products in addition to the low silica hydroxisodalite impurity might be the reason for the observed reduction in Si2/Al2O3 molar ratio in the chemical analysis data in Table 4 for the wet fusion products. On the other hand, the MSX-c-FM/FF-48h product showed pure and highly crystalline zeolite X similar to commercially available molecular sieve 13X with all peaks matching those for the reference zeolite.
The presence of zeolite A alongside zeolite X in the MSX-a-FM/FF-48h product, obtained via dry mixing before dry fusion, is therefore an indication of inhomogeneous dry reaction of NaOH with kaolin during the fusion step resulting in F-MK (sodium aluminosilicates) and some burnt kaolin or MK (aluminosilicates). This inhomogeneous dry reaction is a result of inefficient dry mixing of the NaOH with kaolin during the pre-treatment step. The F-MK, which forms the bulk of fused material, is therefore converted to zeolite X during the hydrothermal crystallization step, while the trace MK is transformed to zeolite A. The conversions of F-MK to zeolite X, and MK to zeolite A have previously been reported.9 The product via dry mixing of NaOH with kaolin followed by dry fusion before hydrothermal crystallization step therefore depends on the degree of mixing achieved. Less efficient mixing may therefore explain the presence of zeolite A impurities in previous reports involving this method.7,19–25,28
In both MSX-b-FM/FF-48h and MSX-c-FM/FF-48h products, uniform mixtures of clay and NaOH were obtained in the wet mixing method, implying that the starting kaolin was fully converted to F-MK (sodium aluminosilicates) in the fusion step leading to the absence of zeolite A in the final products. The presence of water in the fusion step for the MSX-b-FM/FF-48h product may aid the formation of non-uniform building blocks consisting of narrow pore zeolite structural building units alongside faujasite structural building units. The narrow pore building units form hydroxysodalite (HS) alongside the poorly crystalline zeolite X product arising from the faujasite building units. This is consistent with the SEM image in Fig. 4f that shows very small particles tending towards being amorphous. Although the fusion process was performed at high temperature of 750 °C, the narrow pore building units which formed HS might have been formed during the temperature ramping stage when the temperature was high enough to lead to chemical reactions of clay with NaOH, and water was still present within the material to influence the reaction to small pore building units. On the other hand, the absence of water in the dry fusion step for MSX-c-FM/FF-48h may yield uniform faujasite building units suitable for conversion to highly crystalline zeolite X (Fig. 5b) with uniform crystal sizes (Fig. 4g). The formation of zeolite X of varying crystallinity and purity arising from differences in the pre-treatment methods of kaolin before the fusion step is further proof that the nature and purity of the final zeolite structure is determined during the high temperature reaction at the fusion step.
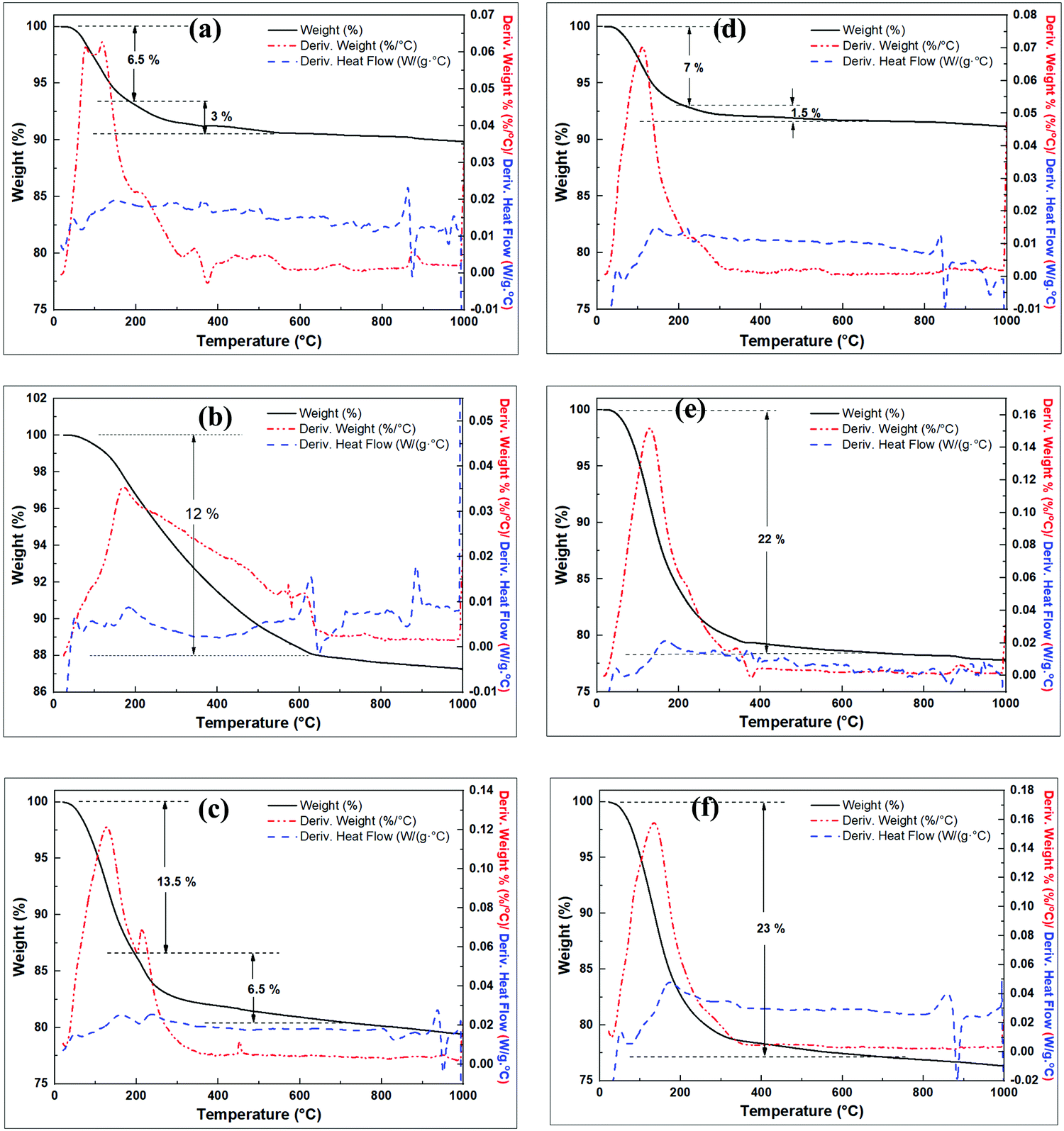 | ||
| Fig. 6 TGA curves of the full fusion derived products, (a). MSY-a-FM/PF-48h, (b) Washed F-MK, (c) MSX-b-FM/FF-48h, (d) MSY-b-FM/PF-48h, (e) MSX-a-FM/FF-48h, (f) MSX-c-FM/FF-48h. | ||
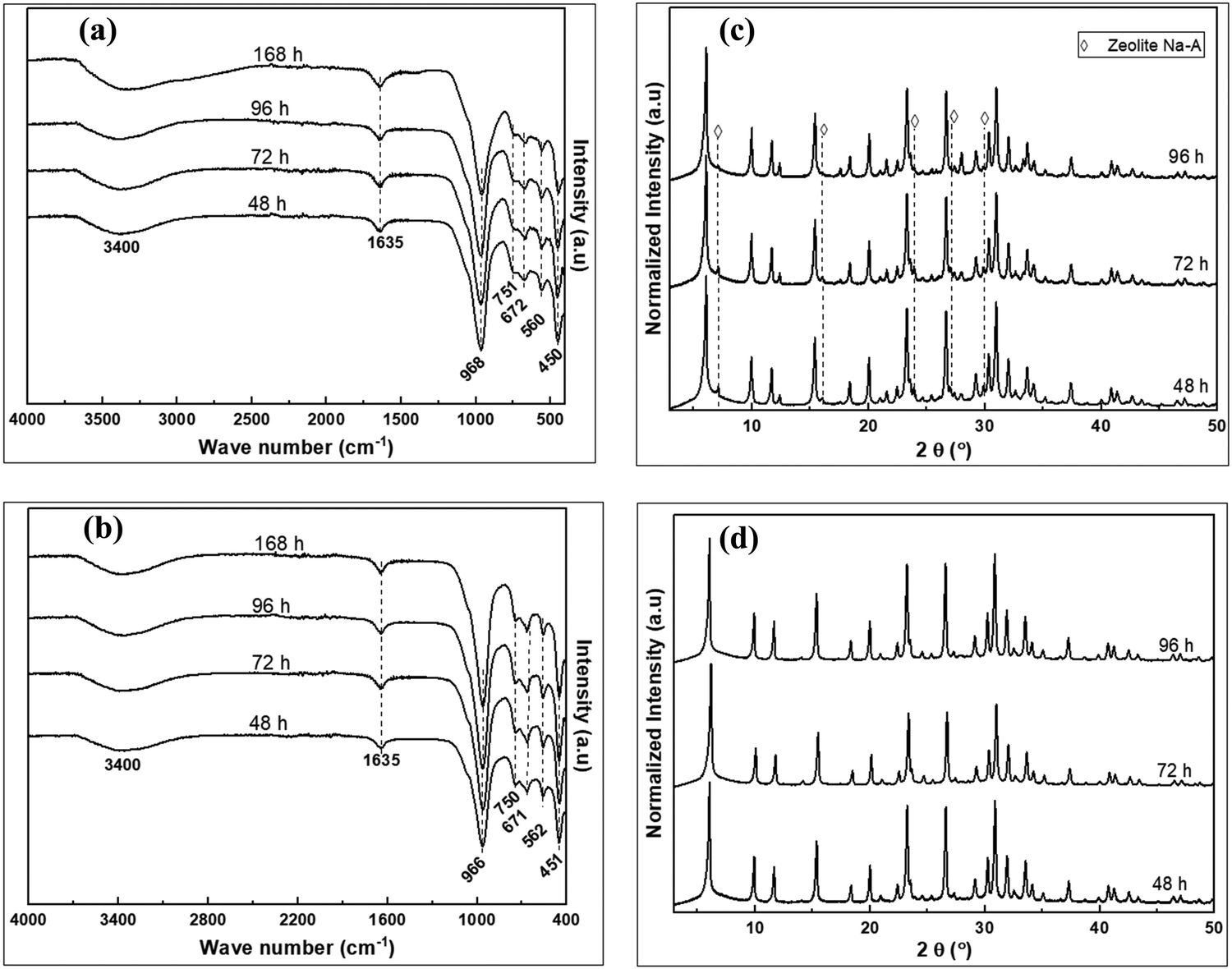 | ||
| Fig. 7 The FTIR spectra of products at different crystallization time; (a) MSX-a-FM/FF, (b) MSX-c-FM/FF, and the XRD patterns of products at different crystallization time; (c) MSX-a-FM/FF, (d) MSX-c-FM/FF. | ||
The FTIR spectra in Fig. 7a and b shows similar functional groups belonging to zeolite X.39 However, the XRD patterns of MSX-a-FM/FF, obtained via dry mixing before dry fusion, in Fig. 7c, shows that zeolite A that formed alongside X at low crystallization time was transformed via dissolution at longer crystallization time to form greater amounts of X via the process of Ostwald's ripening,43 with X being the abundant phase after 96 h. On the other hand, pure zeolite X, with no phase change, even at prolonged reaction time, was obtained in the MSX-c-FM/FF products for wet mixing of kaolin with NaOH before dry fusion. The method leading to the formation of MSX-c-FM/FF is therefore efficient for the conversion of kaolin components and the production of pure and quality zeolite products within a short period of time.
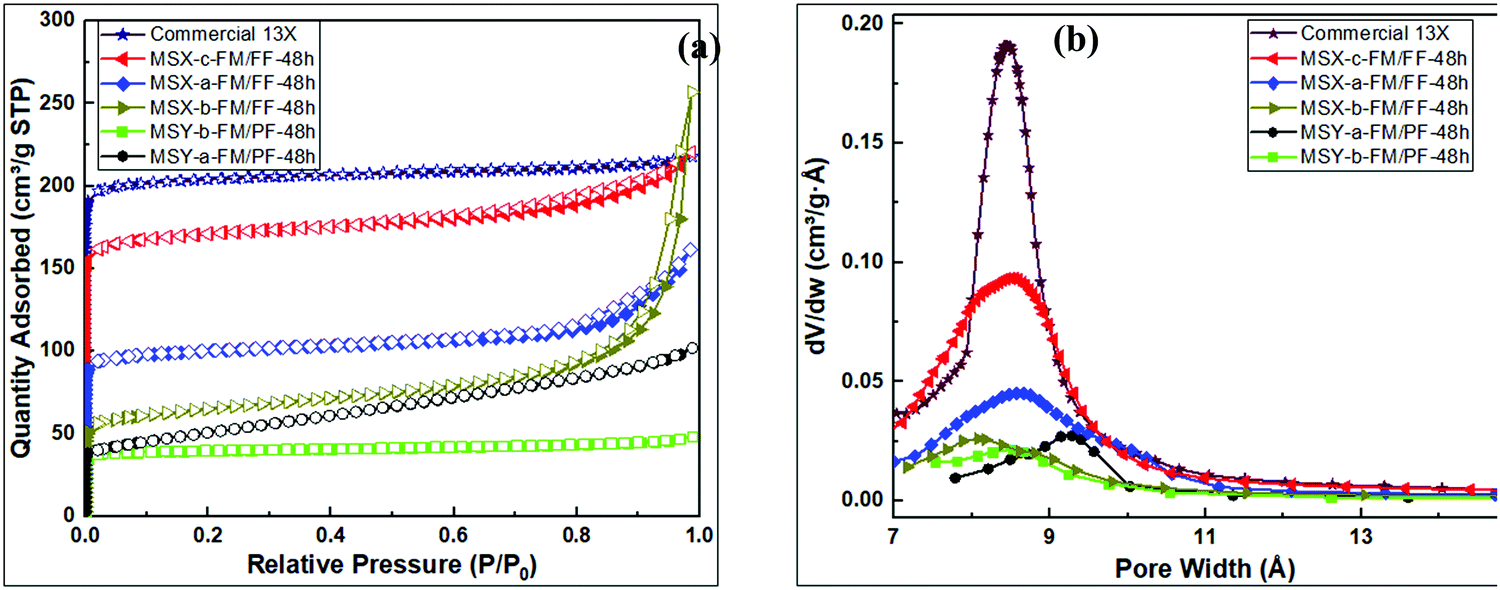 | ||
| Fig. 8 Porosity analysis of synthesized zeolites; (a) N2 sorption isotherms, (b) Horvath–Kawazoe pore size distribution curves. | ||
The textural properties of the zeolite products are summarised in Table 5. The partial fusion product (MSY-b-FM/PF-48h) obtained via protocol 1 had the lowest surface area, micropore surface area and pore volume of 151 m2 g−1, 58 m2 g−1, and 0.03 cm3 g−1, respectively. On the other hand, the MSX-c-FM/FF-48h product obtained via full fusion in protocol 2 had the highest surface area, micropore surface area and pore volume of 645 m2 g−1, 579 m2 g−1 and 0.24 cm3 g−1 respectively.
| Sample | Surface area (m2 g−1) | Micropore surface area (m2 g−1) | External surface area (m2 g−1) | Micropore volume (cm3 g−1) | Total pore volume (cm3 g−1) | Pore size (Å) |
|---|---|---|---|---|---|---|
| MSY-a-FM/PF-48h | 180 | 58 | 122 | 0.03 | 0.14 | 9.3 |
| MSY-b-FM/PF-48h | 151 | 134 | 17 | 0.05 | 0.06 | 8.5 |
| MSX-a-FM/FF-48h | 376 | 327 | 49 | 0.13 | 0.22 | 8.7 |
| MSX-b-FM/FF-48h | 239 | 152 | 86 | 0.06 | 0.32 | 8.0 |
| MSX-c-FM/FF-48h | 645 | 579 | 67 | 0.24 | 0.28 | 8.7 |
| Commercial 13X | 772 | 726 | 46 | 0.30 | 0.27 | 8.7 |
Pore channels of size 8.7 Å are observed for MSY-b-FM/PF-48h, MSX-a-FM/FF-48h and MSX-c-FM/FF-48h products. Product MSY-a-FM/PF-48h had slightly larger pores of size 9.3 Å, while MSX-b-FM/FF-48h had slightly smaller pores of 8.0 Å. The textural properties of the MSX-c-FM/FF-48h product are higher than previously reported values (Table 1) for similar zeolite type and are comparable to those of commercially available molecular 13X (772 m2 g−1 and 0.30 cm3 g−1). The micropore volume was in the ascending order; MSY-a-FM/PF-48h < MSY-b-FM/PF-48h < MSX-b-FM/FF-48h < MSX-a-FM/FF-48h < MSX-c-FM/FF-48h. Although the MSX-b-FM/FF-48h product had similar composition to other protocol 2 products, the presence of water in the wet fusion encouraged reactions or growths of zeolite polymers that lead to narrowing of the zeolite channels and cavities (to 8.0 Å compared to 8.7 Å for other X zeolites), and formation of hydroxysodalite and other mesoporous or amorphous products.
3.3. Effects of the pre-treatment methods
The elemental composition of the products from various pre-treatment methods is shown in Table 6. The products from dry fusion had relatively similar SiO2/Al2O3 molar ratio of ca. 3.8 while that from wet fusion recorded slightly lower ratio of ca. 3.6.| Sample | Percent oxide (wt% MxOy) | Method | Chemical formula | |||||
|---|---|---|---|---|---|---|---|---|
| Al | Si | Na | Ti | Fe | SiO2/Al2O3 | |||
| FSY-a-FM-48h | 25.2 | 53.8 | 15.5 | 0.6 | 0.2 | 3.88 | SEM-EDX | Fe0.2Ti1.5Na101.4K1.2Al100Si181.5O596.7 |
| FSY-b-FM-48h | 25.3 | 57.8 | 15.2 | 0.9 | 0.1 | 3.63 | SEM-EDX | Fe0.3Ti2.2Na98.8K5.4Al100Si193.9O596.9 |
| FSY-c-FM-48h | 25.7 | 57.6 | 15.4 | 0.7 | 0.0 | 3.81 | SEM-EDX | Fe0.1Ti1.8Na98.5K3.9Al100Si190.2O587 |
The SEM images of the FSY-a-FM-48h and FSY-c-FM-48h products in Fig. 9 show octahedral-like crystals typical of faujasite zeolites with average particle size of 1 μm. The FSY-a-FM-48h image shows non-uniform crystals with some amorphous phases (Fig. 9a), in addition to crystals exhibiting fractured fragments (Fig. 9b). On the other hand, FSY-c-FM-48h obtained from wet mixing before dry fusion showed fully formed uniform crystals exhibiting relatively smooth surfaces, Fig. 9c and d.
 | ||
| Fig. 9 The SEM images of the products obtained from fumed silica modified hydrogel; FSY-a-FM-48h (a and b), FSY-c-FM-48h (c and d). | ||
Fig. 10 shows FTIR spectra and XRD patterns of products obtained from various pre-treatment methods. Zeolites related to the Y family, similar to previous reports,9 were the major products. The trend discussed in Section 3.2.2 was observed in the properties of the resulting zeolite Y products obtained from the hydrogels modified with fumed silica. The FSY-a-FM-48h product, obtained through dry mixing of kaolin with NaOH followed by dry fusion before hydrothermal crystallization, resulted in zeolite Y with impurities. The FSY-b-FM-48h product, obtained through wet mixing of kaolin with NaOH followed by wet fusion before hydrothermal crystallization, resulted in zeolite Y with poor crystallinity and impurities similar to those observed for MSX-b-FM/FF-48h. The FSY-c-FM-48h product obtained through wet mixing of kaolin with NaOH followed by dry fusion before hydrothermal crystallization, resulted in pure zeolite Y with improved properties similar to the MSX-b-FM/FF-48h product.
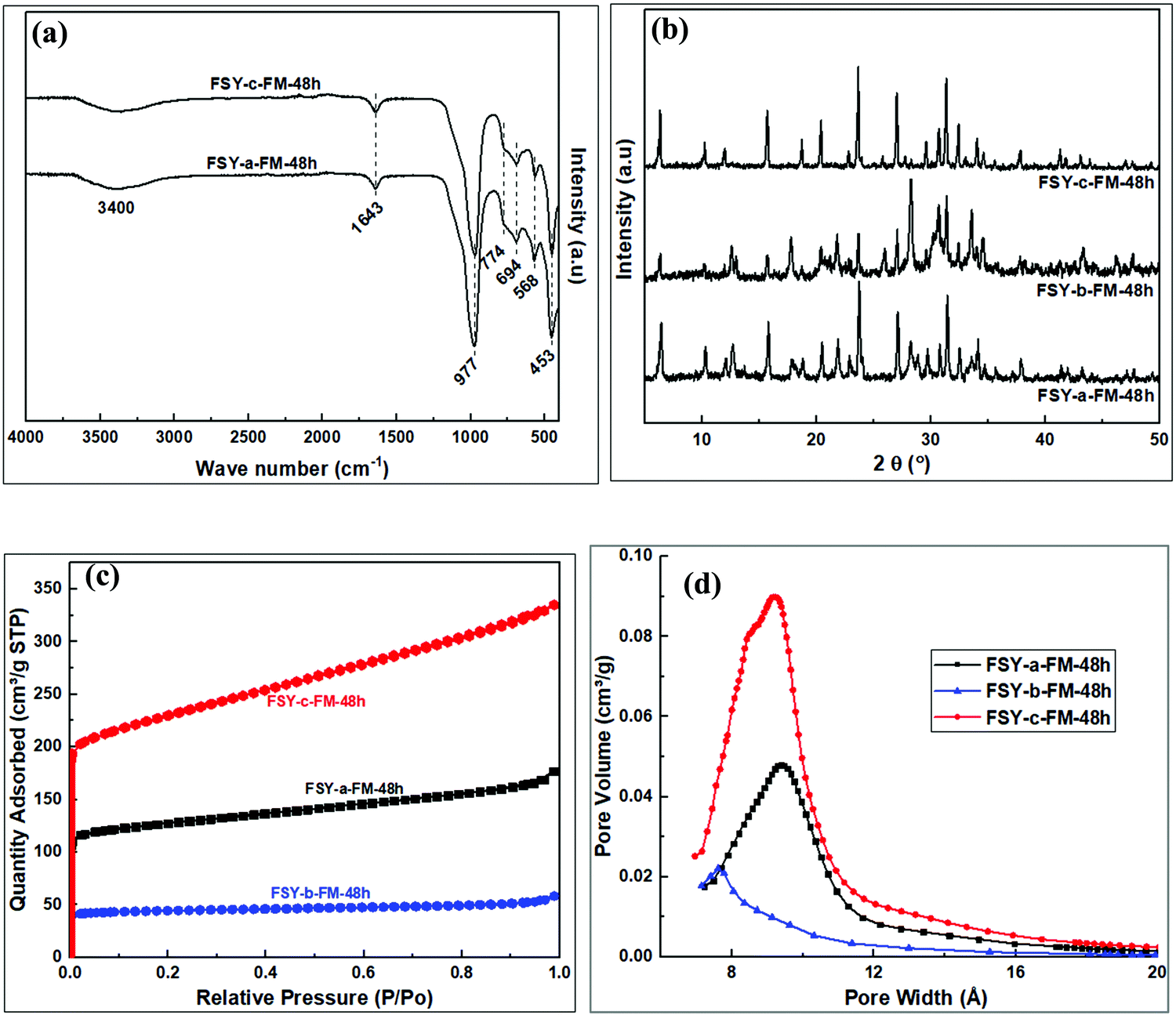 | ||
| Fig. 10 Analysis of products obtained from fumed silica modified hydrogel; (a) FTIR spectra, (b) XRD patterns, (c) N2 sorption isotherms, (d) Horvath–Kawazoe pore size distribution. | ||
The zeolite products exhibited reversible type I isotherms, Fig. 10c, with FSY-b-FM/FF-48h having the lowest porosity (167 m2 g−1 and 0.06 cm3 g−1) as summarised in Table 7. On the other hand, the FSY-c-FM/FF-48h product had high porosity (845 m2 g−1 and 0.23 cm3 g−1). Pore size of 9.9 and 9.6 Å was observed for the FSY-a-FM-48h and FSY-c-FM-48h products, respectively, with slightly lower pore size of 8.9 Å for the FSY-b-FM-48h product.
| Sample | Surface area (m2 g−1) | Micropore surface area (m2 g−1) | External surface area (m2 g−1) | Micropore volume (cm3 g−1) | Total pore volume (cm3 g−1) | Pore size (Å) |
|---|---|---|---|---|---|---|
| FSY-a-FM-48h | 473 | 355 | 118 | 0.15 | 0.27 | 9.9 |
| FSY-b-FM-48h | 167 | 143 | 24 | 0.06 | 0.09 | 8.9 |
| FSY-c-FM-48h | 845 | 549 | 297 | 0.23 | 0.51 | 9.6 |
4. Conclusions
For the synthesis of faujasite zeolites using kaolin as a starting material, the resulting zeolite structure not only depends on the hydrogel composition and the synthesis conditions but also on the fusion conditions, i.e., the amount of NaOH used in the fusion step. For hydrogels with the same concentrations of starting materials, partial fusion of the kaolin, involving limited quantities of NaOH leads to the formation of zeolite Y, while full fusion leads to the formation of zeolite X. The resulting zeolite type depends on the nature of the silica and alumina components of the synthesis hydrogel. Various pre-treatment procedures on kaolin do not affect the nature of zeolite Y products obtained via partial fusion. The influence of pre-treatment procedures on kaolin before the fusion step on the nature of zeolite products was however demonstrated for full fusion. Wet mixing followed by wet fusion results in undesired reactions that lead to the formation of hydroxysodalites as impurities in X and Y products. Dry mixing, on the other hand, results in a non-uniform mixture leading to the formation of impure zeolites. The best pre-treatment method was wet mixing of kaolin followed by removal of the solvent via drying prior to the fusion step, which resulted in high quality X and Y zeolites with textural properties comparable to those of commercially available zeolites. Both NaOH concentration in the fusion step and crystallization time did not affect the SiO2/Al2O3 molar ratio of the resulting products. The ratio was however affected by the mode of kaolin pre-treatment. Our findings show that kaolin can be used as a cheaper and viable alternative to commercial aluminosilicates in the synthesis of high quality zeolites with properties comparable to those of commercially available zeolite analogues.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Royal Society (ACBI programme grant number AQ150029). We thank Dr Justin Kemmegne-Mbouguen at the Department of Chemistry, University of Yaoundé 1 (Cameroon), for the kaolin and Mr Sun Hwi Bang at the Materials Research Institute and Department of Materials Science and Engineering, Pennsylvania State University (USA) for the SEM images.References
- E. B. G. Johnson and S. E. Arshad, Appl. Clay Sci., 2014, 97–98, 215–221 CrossRef CAS.
- T. Abdullahi, Z. Harun and M. H. D. Othman, Adv. Powder Technol., 2017, 28, 1827–1840 CrossRef CAS.
- M. Foroughi, A. Salem and S. Salem, Mater. Chem. Phys., 2021, 258, 123892 CrossRef CAS.
- J. M. Huggett, in Encyclopedia of Geology, ed. R. C. Selley, L. R. M. Cocks and I. R. Plimer, Elsevier, Oxford, 2005, pp. 358–365 Search PubMed.
- L. Ayele, J. Pérez-Pariente, Y. Chebude and I. Díaz, Appl. Clay Sci., 2016, 132–133, 485–490 CrossRef CAS.
- C. A. Ríos, C. D. Williams and O. M. Castellanos, Ingenieria y Competitividad, 2012, 14, 125–137 CrossRef.
- Y. Ma, C. Yan, A. Alshameri, X. Qiu, C. Zhou and D. Li, Adv. Powder Technol., 2014, 25, 495–499 CrossRef CAS.
- A. Bahgaat, M. Mohamed, A. Abdel Karim, A. Melegy and H. Hassan, Egypt. J. Chem., 2020, 63, 3791–3800 Search PubMed.
- S. O. Otieno, F. O. Kengara, J. C. Kemmegne-Mbouguen, H. W. Langmi, C. B. O. Kowenje and R. Mokaya, Microporous Mesoporous Mater., 2019, 290, 109668 CrossRef CAS.
- A. Á. B. Maia, R. N. Dias, R. S. Angélica and R. F. Neves, J. Mater. Res. Technol., 2019, 8, 2924–2929 CrossRef CAS.
- P. Pereira, B. Ferreira, N. Oliveira, E. Nassar, K. Ciuffi, M. Vicente, R. Trujillano, V. Rives, A. Gil, S. Korili and E. De Faria, Appl. Sci., 2018, 8, 608 CrossRef.
- C. A. Rios, C. D. Williams and M. J. Maple, BISTUA, 2007, 5, 15–26 CAS.
- C. A. Ríos, C. D. Williams and M. A. Fullen, Appl. Clay Sci., 2009, 42, 446–454 CrossRef.
- M. Mezni, A. Hamzaoui, N. Hamdi and E. Srasra, Appl. Clay Sci., 2011, 52, 209–218 CrossRef CAS.
- C. Belviso, F. Cavalcante, A. Lettino and S. Fiore, Appl. Clay Sci., 2013, 80–81, 162–168 CrossRef CAS.
- D. Anderson, S. Balapangu, H. N. A. Fleischer, R. A. Viade, F. D. Krampa, P. Kanyong, G. A. Awandare and E. K. Tiburu, Sensors, 2017, 17, 1831 CrossRef PubMed.
- N. M. Musyoka, R. Missengue, M. Kusisakana and L. F. Petrik, Appl. Clay Sci., 2014, 97–98, 182–186 CrossRef CAS.
- A. M. Zayed, A. Q. Selim, E. A. Mohamed, M. S. M. Abdel Wahed, M. K. Seliem and M. Sillanpää, Appl. Clay Sci., 2017, 140, 17–24 CrossRef CAS.
- A. S. Kovo, O. Hernandez and S. M. Holmes, J. Mater. Chem., 2009, 19, 6207–6212 RSC.
- S. Chandrasekhar and P. N. Pramada, Appl. Clay Sci., 2004, 27, 187–198 CrossRef CAS.
- L. B. Bortolatto, R. A. A. Boca Santa, J. C. Moreira, D. B. Machado, M. A. P. M. Martins, M. A. Fiori, N. C. Kuhnen and H. G. Riella, Microporous Mesoporous Mater., 2017, 248, 214–221 CrossRef CAS.
- D. M. El-Mekkawi, F. A. Ibrahim and M. M. Selim, J. Environ. Chem. Eng., 2016, 4, 1417–1422 CrossRef CAS.
- D. M. El-Mekkawi and M. M. Selim, Mater. Charact., 2012, 69, 37–44 CrossRef CAS.
- C. Covarrubias, R. García, R. Arriagada, J. Yánez and M. T. Garland, Microporous Mesoporous Mater., 2006, 88, 220–231 CrossRef CAS.
- A. M. Doyle, T. M. Albayati, A. S. Abbas and Z. T. Alismaeel, Renewable Energy, 2016, 97, 19–23 CrossRef CAS.
- N. Djeffal, M. Benbouzid, B. Boukoussa, H. Sekkiou and A. Bengueddach, Mater. Res. Express, 2017, 4, 035504 CrossRef.
- P. R. D. S. D. Castro, A. Á. B. Maia and R. S. Angélica, Mater. Res., 2019, 22, e20190321 CrossRef.
- N. A. Z. Logar, N. A. N. Tusar, A. Ristic, G. Mali, M. Mazaj and V. E. Kaucic, in Ordered porous solids - recent advances and Prospects, ed. V. Valtchev, S. Mintova and M. Tsapatsis, Elsevier, The Boulevard, Langford lane, Kidlington, Oxford, OX5 1GB, UK, 2009, ch. Five, pp. 101–209 Search PubMed.
- M. Warzybok, A. Chverenhuk and J. Warchoł, Czasopismo Inżynierii Lądowej, Środowiska i Architektury, 2015, z. 62(nr 3/I), 485–495 Search PubMed.
- M. Warzybok and J. Warchoł, Czasopismo Inżynierii Lądowej, Środowiska i Architektury, 2018, z. 65(nr 1), 13–26 Search PubMed.
- M. Gougazeh and J. C. Buhl, J. Assoc. Arab Univ. Basic Appl. Sci., 2014, 15, 35–42 Search PubMed.
- C. Baerlocher and L. B. McCusker, Database of Zeolite Structures, 2017. http://www.iza-structure.org/databases, 2017 ed Access Date 23 March 2020.
- W. Zamechek, in Verified Syntheses of Zeolitic Materials, ed. H. Robson and K. P. Lillerud, Elsevier Science, Amsterdam, 2001, pp. 51–53 Search PubMed.
- L. S. Cheng and Y. Ralph T, Chem. Eng. Sci., 1994, 49, 2599–2609 CrossRef CAS.
- P. Djomgoue and D. Njopwouo, J. Surf. Eng. Mater. Adv. Technol, 2013, 03, 275–282 Search PubMed.
- B. J. Saikia and G. Parthasarathy, J. Mod. Phys, 2010, 01, 206–210 CrossRef CAS.
- H. Hanlie, C. Wang, K. Zeng, K. Zhang, K. Yin and Z. Li, Clay Clay Miner, 2012, 60, 240–253 CrossRef.
- R. Belaabed, S. Elabed, A. Addaou, A. Laajab, M. A. Rodríguez and A. Lahsini, Bol. Soc. Esp. Ceram. Vidrio, 2016, 55, 152–158 CrossRef CAS.
- W. Mozgawa, W. Jastrzębski and M. Handke, J. Mol. Struct., 2005, 744–747, 663–670 CrossRef CAS.
- H. Tounsi, S. Mseddi and S. Djemel, Phys. Procedia, 2009, 2, 1065–1074 CrossRef CAS.
- L. N. Tchadjie and S. O. Ekolu, J. Mater. Sci., 2018, 53, 4709–4733 CrossRef CAS.
- N. Li, T. Li, H. Liu, Y. Yue and X. Bao, Appl. Clay Sci., 2017, 144, 150–156 CrossRef CAS.
- H. Greer, P. S. Wheatley, S. E. Ashbrook, R. E. Morris and W. Zhou, J. Am. Chem. Soc., 2009, 131, 17986–17992 CrossRef CAS PubMed.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CAS.
| This journal is © The Royal Society of Chemistry 2021 |