DOI:
10.1039/D1MA00391G
(Paper)
Mater. Adv., 2021,
2, 5160-5170
Non-doped OLEDs based on tetraphenylethylene phenanthroimidazoles with negligible efficiency roll-off: effects of end group regulated stimulus responsive AIE luminogens†
Received
29th April 2021
, Accepted 22nd June 2021
First published on 23rd June 2021
Abstract
The positional isomers of phenanthroimidazoles, TPE-NPPB and TPE-APPB, have been designed to understand the positional effects of triphenylamine (TPA) and tetraphenylethylene (TPE) units on the aggregation-induced emission and mechanochromism. The phenanthroimidazoles TPE-NPPB and TPE-APPB show reversible mechanochromism between blue and green colours and were analysed via powder X-ray diffraction (PXRD). The PXRD analysis reveals that the phase transition of TPE-NPPB and TPE-APPB from crystalline to amorphous state is associated with colour changes. The non-doped OLEDs based on TPE-NPPB and TPE-APPB exhibit high external quantum efficiency (EQE) of 3.2 and 5.3%, current efficiency (CE) of 4.32 and 5.28 cd A−1 and power efficiency (PE) of 4.01 and 4.92 lm W−1, respectively. The improvement of mechanochromic materials with aggregation-induced emission (AIE) behaviour play a vital role in optoelectronics.
1. Introduction
Luminescent materials are commonly used in organic light-emitting diodes (OLEDs) and are used as chemosensors as well as mechano-sensors.1–3 Organic materials with tunable solid state emission are of broad and current interest owing to their promising applications in due to their promising applications in security systems, memory devices, data storage and fluorescent probes.1,2 Mechanochromic materials alter their colours on applying external mechanical stimuli such as grinding, rubbing and crushing.1,3 A prerequisite for reversible mechanochromism is high solid-state emission, which depends on planarity, effective conjugation and interaction with the surrounding environment.4–6 However, conventional organic materials suffer from aggregation caused quenching (ACQ), which decreases the luminance efficiency.7–9 Aggregation-induced emission (AIE) is a promising solution to ACQ.10–13 Mechanochromic materials with the AIE behaviour become highly emissive in the solid state and can be employed as emissive materials in OLEDs.14
The luminescence properties of materials in solid state depend on the molecular arrangement as well as intermolecular interactions.14 The mode of molecular packing of the materials will affect molecular stacking as well as conjugation and tune the luminescence properties.15 Thus, monitoring the molecular packing is significant in altering the photophysical and electroluminescent properties.15–17 Phenanthroimidazole exhibits near-ultraviolet emission with electron-transport ability. Its twisted and rigid molecular structure can enhance the stability and carrier transport ability.18–20 To shift the emission maximum to the visible region, an electron-donating triphenylamine (TPA) group was incorporated into the molecule. Organic emitters containing the TPA group are used as hole transport materials because of their unique optoelectronic properties, which include electron-donating ability and high hole mobility. Although TPA can be used as a complexing agent in the high temperature superconducting materials, it causes ACQ effect in solids. Therefore, introducing an aggregation-induced emission (AIE) group such as triphenylethylene (TPE) into the molecule would improve the solid state luminous efficiency.21 TPE is highly emissive in the solid state due to the restriction of intramolecular rotation (RIR) due to the twisted propeller shaped structure and is utilized for the development of AIE active mechanochromic materials.22–26 The combination of the phenanthroimidazole core with TPA and TPE units produced intriguing mechanochromic and OLED properties. Another strategy such as an asymmetrical structure has been introduced to strengthen the amorphous morphological stability and efficiency by slightly changing the substituents.26 Recently, some progress has been made to obtain highly stable and efficient deep-blue anthracene derivatives by introducing a non-planar rotational molecular structure, which efficiently interrupts the π-conjugation.26
Herein, we report N-phenyl-N-(4-(1-(1-(4-(1,2,2-triphenylvinyl)phenyl)naphthalen-4-yl)-1H-phenanthro[9,10-d]imidazol-2-yl)phenyl)benzenamine (TPE-NPPB) and N-phenyl-N-(4-(2-(10-(4-(1,2,2-triphenylethyl)phenyl)anthracen-9-yl)-1H-phenanthro[9,10-d]imidazol-1yl)phenyl)benzenamine (TPE-APPB) consisting of TPA and TPE units. The AIE and mechanochromic properties of TPE-NPPB and TPE-APPB have been investigated. These excellent solid state emitters, TPE-NPPB and TPE-APPB show reversible mechanochromism between blue and green colours. The non-doped OLEDs based on TPE-NPPB and TPE-APPB fluorophores exhibit high external quantum efficiency (EQE) of 3.2 and 5.3%, current efficiency (CE) of 4.32 and 5.28 cd A−1 and power efficiency (PE) of 4.01 and 4.92 lm W−1, respectively.
2. Experimental section
2.1. Materials
The chemicals used in the Experimental section were obtained from Sigma-Aldrich. The synthetic route of the emissive materials has been outlined in Scheme S1 (ESI†).
2.2. Synthesis of mechanochromic materials
(i)
N-(4-(1-(1-Bromonaphthalen-4-yl)-1H-phenanthro[9,10-d]imidazole-2-yl)phenyl)-N-phenylbenzamine (BrNPPB).
A mixture of 9,10-phenanthrenequinone (2.54 mmol), 4-(diphenylamino)benzaldehyde (2.80 mmol), ammonium acetate (25.4 mmol) and 4-bromonaphthalen-1-amine (3.04 mmol) in acetic acid (10 mL) was refluxed under continuous stirring for 24 h (120 °C; N2). The resulting solid was filtered, washed with water and dried. The intermediate BrNPPB was collected as a pale yellow solid. Yield: 70%. 1H NMR (400 MHz; CDCl3; δ, ppm): 6.68–6.54 (m, 6H), 6.58 (d, 2H), 7.03 (s, 4H), 7.21–7.24 (d, 4H), 7.41–7.50 (t, 2H), 7.69 (d, 2H), 7.85–7.89 (t, 3H), 8.12–8.20 (d, 2H), 8.91 (d, 2H). 13C NMR (100 MHz; CDCl3; δ, ppm): 121.2, 122.4, 122.5, 122.6, 122.7, 122.8, 125.1, 125.2, 126.0, 126.2, 126.3, 126.4, 126.5, 126.7, 126.8, 127.0, 127.3, 127.6, 129.6, 129.8, 130.1, 131.0, 131.1, 131.3, 132.7, 141.0, 141.3, 141.4, 149.6. MS: m/z. 666.60 [M+]; calcd 666.51 (Scheme S2, ESI†).
(ii)
N-(4-(2-(10-Bromoanthracen-9-yl)-1H-phenanthro[9,10-d]imidazol-1-yl)phenyl)-N-phenylbenzenamine (BrAPPB).
A mixture of 9,10-phenanthrenequinone (2.54 mmol), 9-bromoanthracene-10-carbaldehyde (2.80 mmol), ammonium acetate (25.4 mmol) and N′,N′-diphenylbenzene-1,4-diamine (4.32 mmol) in acetic acid (10 mL) was refluxed with continuous stirring for 42 h (120 °C; N2). The resulting solid was filtered, washed with water and dried. The intermediate BrAPPB was collected as a grey coloured solid. Yield: 68%. 1H NMR (400 MHz; CDCl3; δ, ppm): 6.42–6.49 (m, 4H), 6.53 (d, 4H), 701–7.10 (m, 5H), 7.38–7.42 (m, 5H), 7.60 (t, 2H), 7.70–7.78 (m, 6H), 7.93 (d, 2H), 8.03 (d, 2H). 13C NMR (100 MHz; CDCl3; δ, ppm): 118.0, 120.3, 122.6, 124.0, 125.3, 126.2, 127.2, 128.3, 129.4, 130.6, 134.8, 135.0, 140.3, 141.2, 142.6, 144.8, 148.4, 151.6, 156.4. MS: m/z. 716.66 [M+]; calcd 715.79 (Scheme S2, ESI†).
(iii) N-Phenyl-N-(4-(1-(1-(4-(1,2,2-triphenylvinyl)phenyl) naphthalen-4-yl)-1H-phenanthro[9,10-d]imidazol-2-yl)phenyl)benzenamine (TPE-NPPB).
A mixture of Pd(PPh3)4 (0.04 mmol), BrNPPB (0.4 mmol), 4-(1,2,2-triphenylvinyl)phenylboronic acid pinacol ester (0.52 mmol) and K2CO3 (2.0 mmol) in a mixture of ethanol (8.0 mL), toluene (24.0 mL) and H2O (4.0 mL) was refluxed (120 °C; N2; 42 h). The crude was purified by column chromatography. TPE-NPPB was separated as a white powder. Yield: 65%. 1H NMR (400 MHz, CDCl3, δ, ppm): 6.23–6.46 (m, 4H), 6.50–6.83 (t, 4H), 6.86–7.16 (m, 6H), 7.20–7.29 (m, 6H), 7.32–7.40 (m, 4H), 7.44 (t, 3H), 7.50 (s, 2H), 7.63 (s, 2H), 7.68 (t, 2H), 7.70–7.81 (m, 5H), 8.22 (s, 4H), 8.32–8.58 (m, 5H). 13C NMR (100 MHz, CDCl3, δ, ppm): 108.1, 122.4, 122.8, 124.7, 125.1, 126.2, 127.6, 127.8, 126.4, 126.6, 128.5, 128.4, 129.7, 131.2, 131.3, 131.5, 131.6, 134.1, 136.2, 135.8, 138.9, 140.0, 149.4. MS: m/z. 918.13 [M+]; calcd 918.08 (Scheme S2, ESI†).
(iv)
N-Phenyl-N-(4-(2-(10-(4-(1,2,2-triphenylethyl)phenyl)anthracen-9-yl)-1H-phenanthro[9,10-d]imidazol-1-yl)phenyl)benzenamine (TPE-APPB).
A mixture of Pd(PPh3)4 (0.04 mmol), BrAPPB (0.4 mmol), 4-(1,2,2-triphenylvinyl)phenylboronic acid pinacol ester (0.52 mmol) and K2CO3 (2.0 mmol) in a mixture of ethanol (8.0 mL), toluene (24.0 mL) and H2O (4.0 mL) was refluxed (120 °C; N2; 42 h). The crude was purified by column chromatography. The TPE-APPB was separated as white powder. Yield: 60%. 1H NMR (400 MHz; CDCl3; δ, ppm): 6.20 (t, 4H), 6.32 (d, 4H), 6.40–6.83 (m, 4H), 6.98 (t, 3H), 7.10–7.28 (m, 6H), 7.33–7.40 (m, 6H), 7.48–7.58 (m, 5H), 7.60 (d, 4H), 7.75 (t, 3H), 7.85–8.00 (m, 4H), 8.06–8.46 (m, 6H). 13C NMR (100 MHz; CDCl3; δ, ppm): 102.0, 122.5, 123.2, 123.4, 125.4, 126.5, 127.4, 128.0, 129.3, 129.7, 131.9, 133.1, 134.9, 135.0, 136.3, 137.0, 139.3, 140.5, 141.6. MS: m/z. 968.19 [M+]; calcd 968.12 (Scheme S2, ESI†).
2.3. Fabrication of devices
An ITO glass (resistance 20 Ω sq−1) was cleaned with acetone, deionized water and isopropanol, dried (120 °C) and transferred into a deposition system after UV-zone treatment (20 min). The devices were fabricated by the multiple source beam deposition method in vacuum at a pressure of 4 × 10−5 mbar. Evaporation rates of 2–4 Å s−1 (organic materials) and 0.1 and 4 Å s−1 for LiF and metal electrodes were applied, respectively. The thickness of each deposition layer was monitored using a quartz crystal thickness monitor. The EL measurement with CIE coordinates was recorded using a USB-650-VIS-NIR spectrometer (Ocean Optics, Inc, USA). The current density–voltage–luminance (J–V–L) characteristics were measured using a source meter (Keithley 2450) equipped with an LS-110 light intensity meter. The external quantum efficiency was determined from luminance, current density and EL spectrum assuming Lambertian distribution.
2.4. Computational details
The ground (S0) (DFT)/excited (Sn*) (TD-DFT) states of emissive materials were analyzed by the Gaussian 09 program.27
3. Results and discussion
3.1. Theoretical calculation
To gain more insight into the structure–activity analysis, molecular optimization and frontier molecular orbital (FMO) analysis for TPE-NPPB and TPE-APPB were carried out by DFT and TD-DFT methods (Fig. 1 and Fig. S1, S2, ESI†).27 The optimized structures and HOMO (highest occupied molecular orbital) and LUMO (lowest unoccupied molecular orbital) for TPE-NPPB and TPE-APPB are shown in Fig. 1 (Table 1). The optimized structures confirm the twisted confirmation of TPE and TPA units and reveal only C–H interactions. Theoretical calculations reveal only C–H interactions and no H-bonding or π–π interactions, which make it loosely packed, making it sensitive to grinding, thereby allowing mechanochromism. The twisted confirmation leads to restraint on the π–π stacking interaction results in intensified emission in the aggregated form of TPE-NPPB and TPE-APPB. The optimized structures of intermediates, BrNPPB and BrAPPB, reveal a propeller orientation of the TPA unit. The TPA unit and bromo-substituted phenyl ring in BrNPPB and BrAPPB are not in the plane of the phenanthroimidazole unit and exhibit a highly twisted geometry. The HOMO and LUMO are well-separated in TPE-NPPB, HOMO being mainly localized on the phenanthroimidazole and TPA units, whereas LUMO is localized on the TPE unit. However, in TPE-APPB, HOMO and LUMO are not well separated and mainly localized on the TPE and imidazole units. The attachment of the TPE unit through the azomethine nitrogen atom of phenanthroimidazole in TPE-NPPB causes disruption in the extended conjugation, resulting in a better separation of molecular orbitals. In TPE-APPB, the TPE unit attached with a carbon atom of the imidazole ring produces highly extended conjugation allowing the molecular orbitals to be delocalized over the whole molecule.
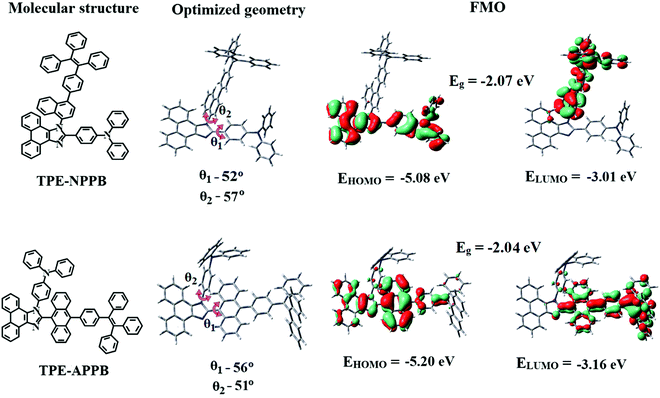 |
| | Fig. 1 Molecular structure, optimized geometry and frontier molecular orbitals (FMO) of TPE-NPPB and TPE-APPB. | |
Table 1 Summary of optical and thermal properties
| Properties |
TPE-NPPB
|
TPE-APPB
|
|
T
d/Tg/Tm (°C) |
449/261/273 |
462/286/320 |
|
λ
ab (nm) |
243, 338 |
249, 346 |
|
λ
em (nm) |
403 |
416 |
|
ϕ (soln) |
0.16 |
0.46 |
| HOMO/LUMO (eV) |
−5.08/−3.01 |
−5.20/−3.16 |
|
E
g (eV) |
−2.07 |
−2.04 |
From the optimized molecular geometry, the π-conjugation between imidazole and anthracene in TPE-APPB is more interrupted that could induce pure blue emission. The theoretical band-gap of TPE-NPPB and TPE-APPB are of 2.12 and 2.10 eV, respectively. The computed vertical transitions, oscillator strength and HONTOs and LUNTOs of TPE-NPPB and TPE-APPB are shown in Fig. S2 (Tables S1 and S2, ESI†).
3.2. Mechanochromic materials
In order to get a clear understanding about the geometry and electronic structures of phenanthroimidazoles, TPE-NPPB and TPE-APPB, density functional theory (DFT) calculations were performed. Based on calculations, the mechano-sensor materials TPE-NPPB and TPE-APPB have been designed and synthesized by simple and efficient two step approach, (Scheme S1, ESI†). The intermediates BrNPPB and BrAPPB were synthesized by the condensation reaction of 9,10-phenanthroquinone using corresponding aldehydes, amines and ammonium acetate. The Suzuki cross-coupling reaction of BrNPPB and BrAPPB with 4-(1,2,2-triphenylvinyl)phenylboronic acid pinacol ester in nitrogen atmosphere using the Pd(PPh3)4 catalyst yielded the target phenanthroimidazoles TPE-NPPB and TPE-APPB with 65% and 60% yield, respectively.28 The luminogens, TPE-NPPB and TPE-APPB, were characterized via NMR and MS techniques.
3.3. Thermal and electrochemical studies
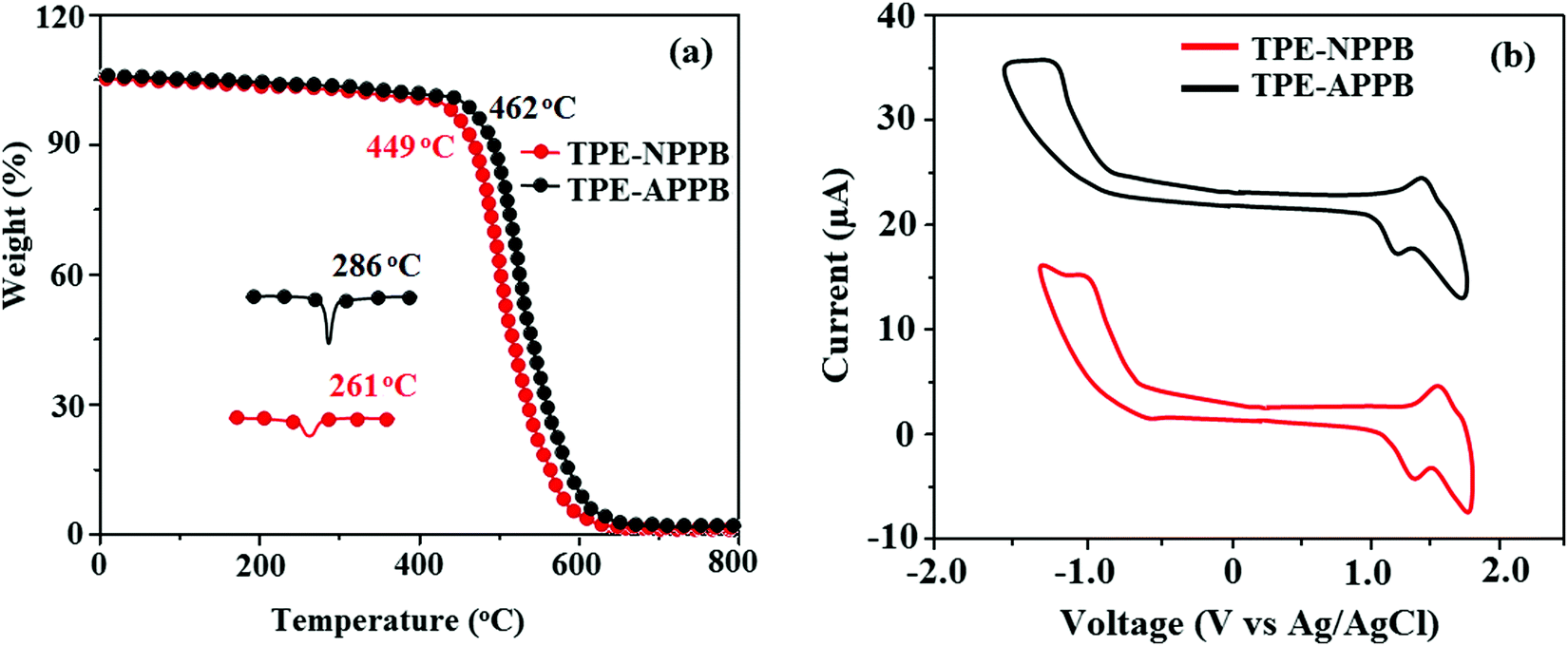
The fabrication of devices required thermally stable materials and hence the materials should be stable at elevated temperatures. The thermal stability of TPE-NPPB and TPE-APPB was analysed by thermogravimetric analysis and differential scanning calorimetry (Fig. 2 and Table 1). The twisted molecular architecture of TPE-NPPB and TPE-APPB with a twist angle of ∼56° between the C2 substituent and the phenanthrimidazole core effectively suppressed the conjugation and intermolecular π–π stacking (Fig. 1). The twisted molecular architecture increased the thermal stability of TPE-NPPB and TPE-APPB. The thermal decomposition temperature (Td) of TPE-NPPB (449 °C) and TPE-APPB (462 °C) implies that these materials are thermally stable and hence they are a suitable choice for material applications. Compared to TPE-NPPB, the more asymmetrically twisted conformations between anthracene and imidazole units in TPE-APPB efficiently interrupt the molecular π-conjugation and inhibit the π–π intermolecular interactions, resulting in high thermal stability, which is important for application in devices. This geometrical morphology tends to maintain certain distance between the anthracene units of adjacent molecules and suppress close packing and π–π intermolecular interactions. The DSC curves reveal an endothermic process for TPE-NPPB and TPE-APPB with high melting temperatures of 273 and 320 °C, respectively. Because of C and N-side coupling with a bulky substituents, the rigid phenanthrimidazole shows high glass transition temperatures (TPE-NPPB – 261 °C; TPE-APPB – 286 °C). The interaction of substituents at C2 with N1 of the phenanthroimidazole core induced more condensed molecular packing and enhanced the thermal stability, which is essential for application in the fabrication of devices.
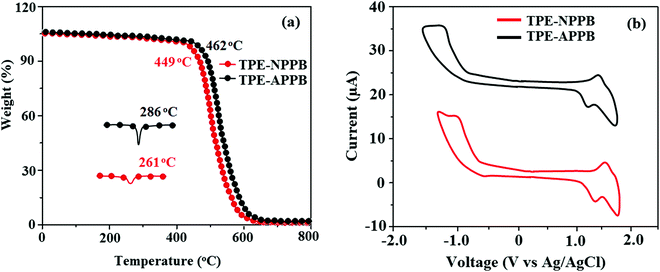 |
| | Fig. 2 (a) TGA and DSC graph and (b) cyclic voltammograms of TPE-NPPB and TPE-APPB. | |
The electrochemical properties and frontier energy level of TPE-NPPB and TPE-APPB were examined by cyclic voltammetry in dichloromethane using tetrabutylammonium hexafluorophosphate (TBAPF6) as the supporting electrolyte. The cyclic voltammograms (Fig. 2) of TPE-NPPB and TPE-APPB reveal quasi-reversible oxidation and reduction waves. The energies for highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) were calculated by the equations: HOMO = −(Eonset + 4.4) eV and LUMO = HOMO−1239/λonset.29 From the calculated HOMO energies of TPE-NPPB (−5.08 eV) and TPE-APPB (−5.20 eV) and LUMO energies of TPE-NPPB (−3.01 eV) and TPE-APPB (−3.16 eV), the electrochemical HOMO–LUMO gap of TPE-NPPB and TPE-APPB are found to be 2.07 eV and 2.04 eV, respectively.
3.4. Photophysical properties
Phenanthroimidazoles, TPE-NPPB and TPE-APPB are freely soluble in dichloromethane, tetrahydrofuran, chloroform and hexane but they are poorly soluble in water, alcohol and acetonitrile. The photophysical properties of TPE-NPPB and TPE-APPB were studied in tetrahydrofuran via electronic spectroscopy (Fig. 3 and Table 1). TPE-NPPB and TPE-APPB show absorption peaks at 243 and 249 nm, respectively, corresponding to the π–π* transition of aromatic segments. The absorption peaks at 338 and 346 nm are attributed to the π–π* transition of the phenanthroimidazole unit.30 The high extinction coefficients (εmax) of TPE-NPPB and TPE-APPB are due to the increase in the conjugation length [TPE-NPPB: 243 nm (εmax – 41152.26 cm−1 M−1), 338 nm (εmax – 29585.80 cm−1 M−1); TPE-APPB: 249 nm (εmax – 40160.64 cm−1 M−1), 346 nm (εmax – 28901.73 cm−1 M−1)], which confirm the presence of effective charge transfer transitions in these materials.31,32
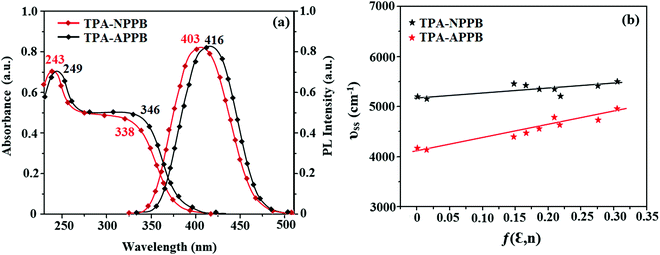 |
| | Fig. 3 (a) Normalised absorption and emission spectra and (b) Lippert–Mataga plot of TPE-NPPB and TPE-APPB. | |
The photoluminance characteristics of these materials were studied in numerous solvents. TPE-NPPB and TPE-APPB show emission maxima at 403 and 416 nm, respectively. Solvatochromism was examined by emission (Fig. S3, ESI†) and absorption spectra (Fig. S4, ESI†) of TPE-NPPB and TPE-APPB (Tables S3 and S4, ESI†). The emission spectra of TPE-NPPB and TPE-APPB in non-polar solvents display a fine vibrational structure and the vibrational structure vanishes in polar solvents. The emission red-shifted in polar solvents. In non-polar solvents, the emission arises from the localised excited (LE) state, whereas in polar solvents, the emission originated from the charge transfer (CT) state. The Lippert–Mataga plot (Fig. 3) of TPE-NPPB show higher slope than the TPE-APPB, which suggests that in TPE-NPPB, the excited state is more polarized than in TPE-APPB. The TPE moiety in these emissive materials is well established for the AIE characteristics due to its propeller shape. The emission behaviour was used to study the AIE properties of TPE-NPPB and TPE-APPB (Fig. 4). TPE-NPPB and TPE-APPB exhibit good solubility in tetrahydrofuran and poor solubility in water. Hence, the gradual increase in the water percentage in the tetrahydrofuran solution initiates the formation of nanoaggregates. TPE-NPPB and TPE-APPB are weekly emissive at low water fractions and the relative PLQYs for TPE-NPPB and TPE-APPB are 0.16 and 0.46, respectively. The non-radiative energy loss of the excited state caused by the free rotation of phenyl rings of the TPE moiety in the solution state leads to non-emissive nature.33 The formation of aggregates at a higher water percentage restricts the free rotation of the phenyl ring, resulting in enhanced intensity with red-shifted emission. TPE-NPPB and TPE-APPB exhibit an increase in the emission intensity with a bathochromic shift for above 70% water fraction due to effective aggregate formation. TPE-APPB has a higher degree of enhancement of intensity as compared to TPE-NPPB due to the positional effects. The superposition of the emission spectra of TPE-NPPB and TPE-APPB at 90% water fraction results in higher extent of intensity enhancement (Fig. S5, ESI†). The AIE behaviour was also studied via absorption spectroscopy (Fig. S6, ESI†). The absorption spectra for TPE-NPPB and TPE-APPB reveal a similar trend of absorption bands up to water fraction of 70%, above which there is scattering of light or Mie effect observed due to the formation of nanoaggregates. The AIE behaviours of TPE-NPPB and TPE-APPB are shown in photographs under UV illumination (Fig. 4).
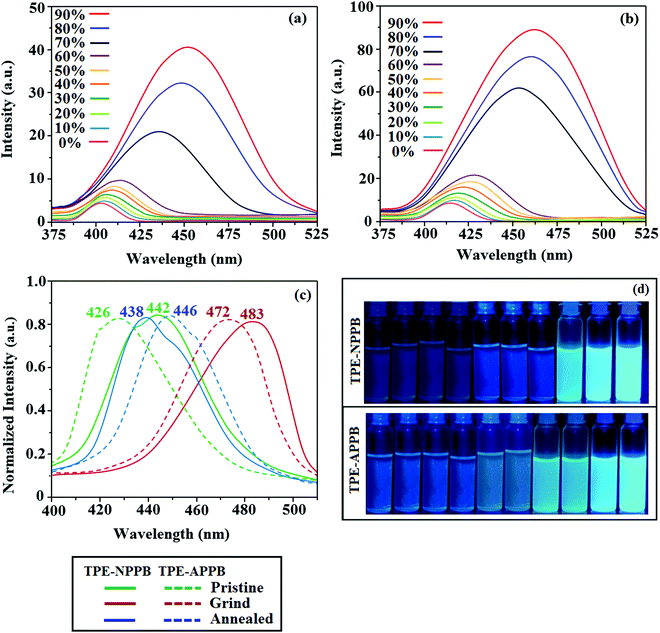 |
| | Fig. 4 Emission spectra of (a) TPE-NPPB and (b) TPE-APPB in THF–H2O with different water fractions; (c) UV illumination of TPE-NPPB and TPE-APPB in THF–Water mixtures with different water fractions (10 μM) (0% to 90% water fraction from L to R) and (d) emission spectra of pristine, grind and annealed solids of TPE-NPPB and TPE-APPB. | |
3.5. Mechanochromism
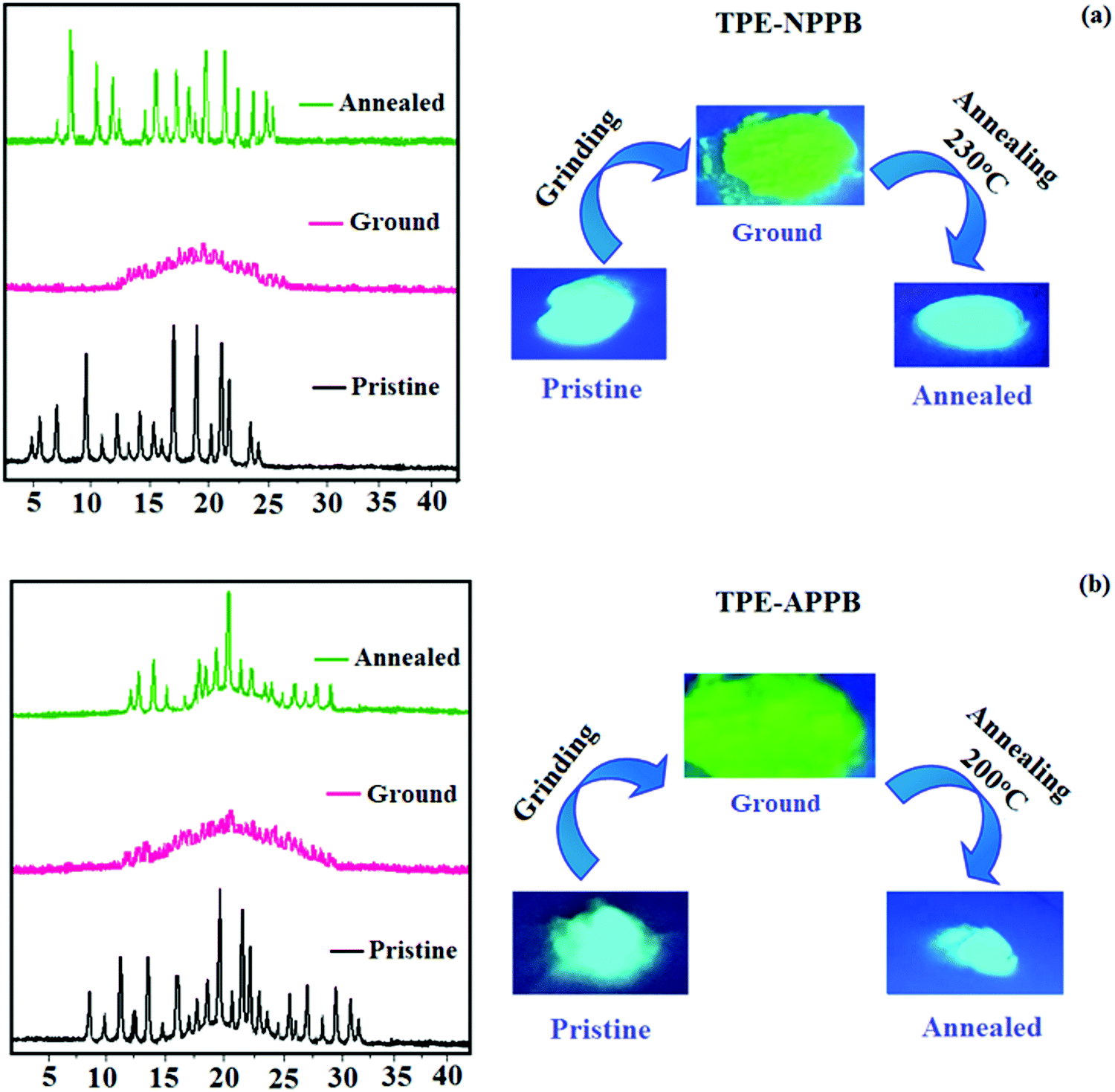
Phenanthroimidazoles, TPE-NPPB and TPE-APPB were determined to exhibit the reversible mechanochromic behaviour on account of the twisted structure of the TPE moiety. The mechanochromic behaviour was studied via fluorescence spectroscopy (Fig. 4 and Table 2). TPE-NPPB and TPE-APPB were mechanically ground with the aid of a mortar and pestle. Pristine TPE-NPPB emits at 442 nm (sky blue), which red-shifted to 483 nm (bluish green) on grinding. The reversibility of colour switching was analysed by annealing TPE-NPPB at 230 °C for five minutes, which leads to blue emission (438 nm) with a higher blueshift. The peak at 476 nm could be ascribed to the intermediate species that arise from the grinding of the pristine sample. These observations indicate the highly sensitive nature of TPE-NPPB towards mechanical stimuli. The high sensitivity of TPE-NPPB could be due to the position of TPA and TPE, TPE being directly attached to the nitrogen atom of the imidazole ring. Similarly, pristine TPE-APPB shows blue emission at 426 nm, which red-shifted to 472 nm (green) on grinding. The colour switching was analysed by annealing at 200 °C for five minutes, which led to blue emission at 446 nm. Due to the more twisted molecular structure, the PL spectra of TPE-APPB in the film exhibit a hypochromatic shift in comparison with those of TPE-NPPB. It should be noted that using 9,10-diphenylanthracene units to substitute at the C-2 position of phenanthrimidazole can efficiently limit the π-conjugation, which induces deep-blue emission. The absolute quantum yield for TPE-NPPB in its pristine, ground and annealed forms were of 48.63%, 51.08% and 66.93%, respectively. The absolute quantum yield for TPE-APPB in its pristine, ground and annealed forms were of 69.03%, 78.92% and 81.03%, respectively. TPE-NPPB and TPE-APPB exhibited grinding-induced spectral shifts of 45 and 26 nm, respectively. The powder XRD (PXRD) studies of TPE-NPPB and TPE-APPB were performed to gain insight into the morphological change that could be a possible reason behind the reversible mechanochromic behaviour (Fig. 5). The pristine TPE-NPPB and TPE-APPB displayed sharp and intense diffraction peaks attributed to crystalline structures. The sharp diffraction peaks disappeared on grinding giving a broad band, which implies the morphological change from the crystalline state to the amorphous state. The crystallinity of TPE-NPPB and TPE-APPB was restored on annealing, which is clearly seen by the sharp diffraction peaks.
Table 2 Emission behaviour of TPE-NPPB and TPE-APPB under external stimuli
| Compounds |
λ
pristine
|
λ
ground
|
λ
annealed
|
Δλ |
|
TPE-NPPB
|
442 |
483 |
438 |
45 |
|
TPE-APPB
|
426 |
472 |
446 |
26 |
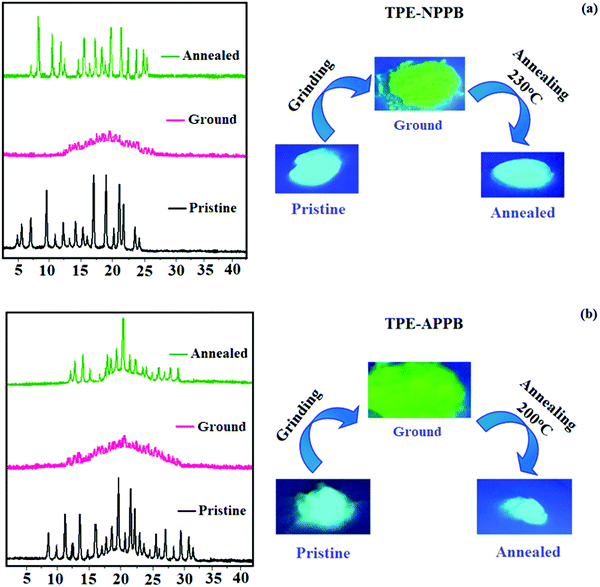 |
| | Fig. 5 Powder X-ray diffraction and mechanochromic behaviour of (a) TPE-NPPB and (b) TPE-APPB. | |
3.6. Electroluminescent studies
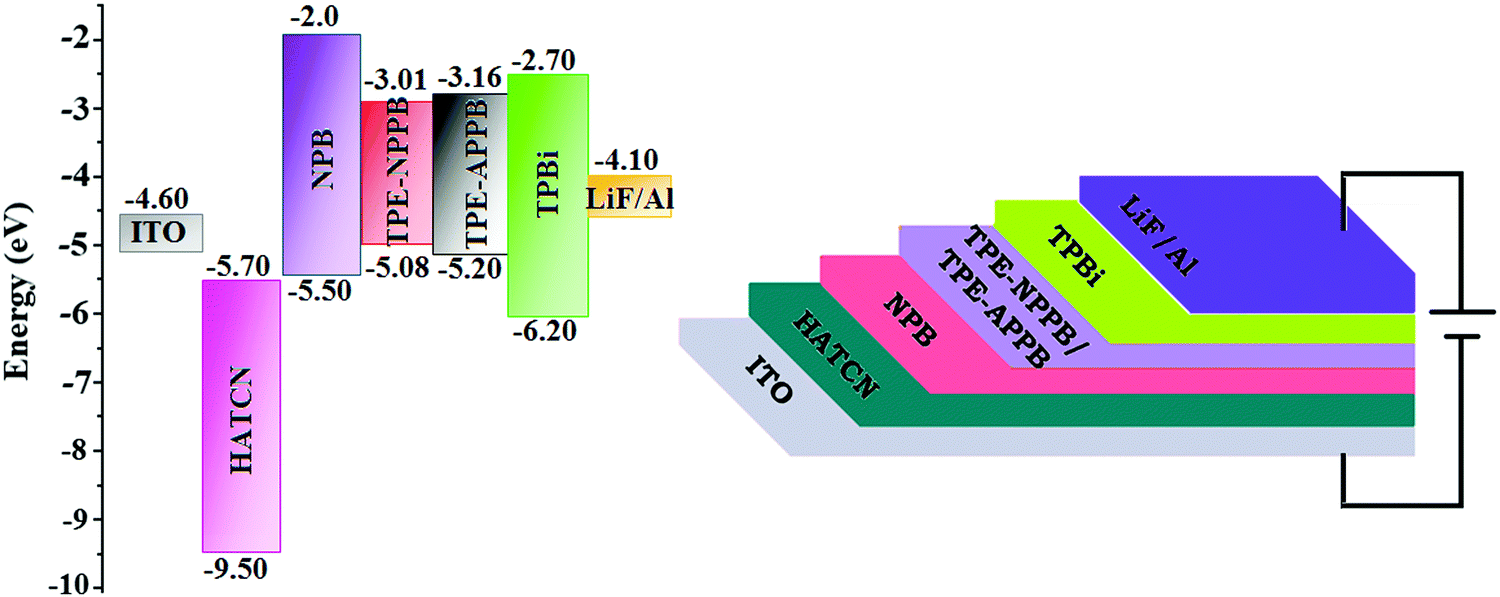
In view of the excellent thermal stability, AIE character and high PLQY in solid state, TPE-NPPB and TPE-APPB were employed as emissive layers to fabricate non-doped OLEDs with the configurations of ITO/HATCN (1,4,5,8,9,11 hexaazatriphen-ylenehexacarbonitrile) (5 nm)/NPB (N,N′-bis(phenyl)-benzidene) (40 nm)/TPE-NPPB and TPE-APPB (20 nm)/TPBi (1,3,5-tris(N-phenylbenzimidazole-2-yl)benzene) (40 nm)/LiF (1 nm)/Al [HATCN-hole injection layer, NPB-hole transporting layer and TPBi-electron transporting layer] (Fig. 6). Fig. 7 shows the current density and luminance of non-doped devices with TPE-NPPB and TPE-APPB as the emissive layer. The small difference in the current density and luminance reveal that the positional change of TPA and TPE units in the backbone structure does not affect the carrier transport properties of the emitters. The turn-on voltages of the devices were same because of the small difference of carrier injection from carrier transport layers to the emissive layer. The non-doped devices based on TPE-NPPB and TPE-APPB exhibited blue emission of 444 and 428 nm, high external quantum efficiency (EQE) of 3.2 and 5.3%, current efficiency (CE) of 4.32 and 5.28 cd A−1, power efficiency (PE) of 4.01 and 4.92 lm W−1 and luminance of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 231 and 15461 cd m−2, respectively. Compared to TPE-NPPB (0.18, 0.21), the device using TPE-APPB as a blue emitter showed a more pure blue emission with CIE coordinates of (0.18, 0.19), which could be attributed to the fully twisted molecular structure. The performance of non-doped devices indicates that the asymmetrically twisted molecular structure can efficiently suppress the π–π intermolecular interaction between the adjacent molecules and result in deep-blue emission.
231 and 15461 cd m−2, respectively. Compared to TPE-NPPB (0.18, 0.21), the device using TPE-APPB as a blue emitter showed a more pure blue emission with CIE coordinates of (0.18, 0.19), which could be attributed to the fully twisted molecular structure. The performance of non-doped devices indicates that the asymmetrically twisted molecular structure can efficiently suppress the π–π intermolecular interaction between the adjacent molecules and result in deep-blue emission.
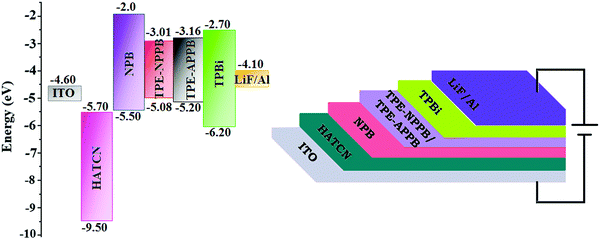 |
| | Fig. 6 Non-doped device configuration with the energy level of TPE-NPPB and TPE-APPB. | |
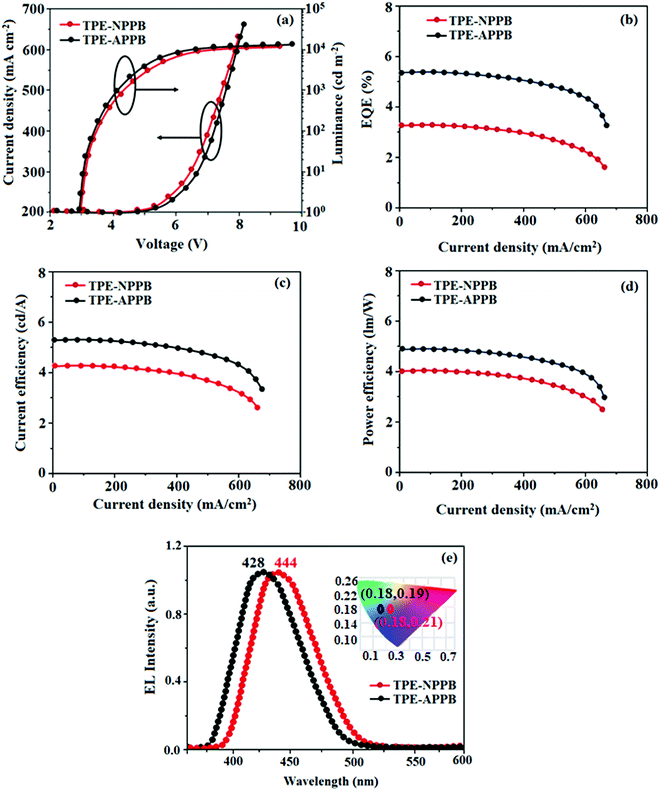 |
| | Fig. 7 Device performances: (a) luminance–current density–voltage; (b) external quantum efficiency–current density (c) current efficiency–current density; (d) power efficiency–current density and (e) normalized EL specta of TPE-NPPB and TPE-APPB. | |
TPE-NPPB- and TPE-APPB-based devices exhibit high EQE because of the AIE character, which suppresses concentration quenching. The EQE of TPE-APPB-based devices was high because of the strong AIE behaviour, as confirmed in the PL intensity change by the addition of water fraction. The efficient AIE property of TPE-APPB contributed to high EQE in the non-doped OLEDs. The non-doped devices based on TPE-NPPB and TPE-APPB exhibited low efficiency roll-off and their EQE still remained 3.15% and 5.25% at a brightness of 1000 cd m−2. All the devices showed negligible roll off efficiency (TPE-NPPB – 1.45% and TPE-APPB – 0.94%) due to the suppression of current-induced exciton quenching and effective h+–e− recombination. The high EQE and low roll-off efficiency further emphasized a great potential of these materials for industrial applications.
The EL spectra of the TPE-APPB-based device were shifted to shorter wavelengths, which is in agreement with the relative PL peak position of the two emitters. Compared with TPE-NPPB, anthracenyl-substituted TPE-APPB showed a bathochromic shift due to conjugation.26 A relatively broad emission modification strategy can be used as a tool for managing the mechanochromism while maintaining EL performances.
4. Conclusion
We have designed positional isomers of phenanthroimidazoles TPE-NPPB and TPE-APPB consisting of TPE and TPA unit and synthesized by the Suzuki cross-coupling reaction. The DFT studies reveal better separation of HOMO and LUMO in TPE-NPPB compared to that in TPE-APPB, which is attributed to the positions of TPE and TPA units. The thermally stable TPE-NPPB and TPE-APPB isomers display strong AIE characteristics and exhibit reversible emission in response to mechanical stimuli with excellent colour contrast. The isomer TPE-NPPB show higher sensitivity towards mechanical stimuli relative to TPE-APPB. The PXRD patterns reveal that the observed morphological change from amorphous state to crystalline state is associated with colour changes. The coupling of TPA and TPE to the phenanthroimidazole backbone provides a new approach for the development of stimulus-responsive solid state emitters. The non-doped device based on TPE-APPB exhibits a high external quantum efficiency (EQE) of 5.3%, current efficiency (CE) of 5.28 cd A−1, power efficiency (PE) of 4.92 lm W−1 and luminance (L) of 15461 cd m−2 (Table 3). The coupling of phenanthroimidazole, TPA and TPE synergistically leads to high solid state emission, which can be further employed in optoelectronic applications.
Table 3 Electroluminescent efficiency of TPE-NPPB- and TPE-APPB-based devices
| Emitters |
CE (cd A−1) |
PE (lm W−1) |
EQE (%) |
L (cd m−2) |
EL (nm) |
CIE (x,y) |
|
TPE-NPPB
|
4.32 |
4.01 |
3.2 |
10231 |
444 |
0.18, 0.21 |
|
TPE-APPB
|
5.28 |
4.92 |
5.3 |
15461 |
428 |
0.18, 0.19 |
Conflicts of interest
There are no conflicts to declare.
References
-
(a) Z. Chi, X. Zhang, B. Xu, X. Zhou, C. Ma, Y. Zhang, S. Liua and J. Xu, Chem. Soc. Rev., 2012, 41, 3878–3896 RSC;
(b) Y. Sagara and T. Kato, Nat. Chem., 2009, 1, 605–610 CrossRef CAS PubMed;
(c) Y. Sagara, S. Yamane, M. Mitani, C. Weder and T. Kato, Adv. Mater., 2016, 28, 1073–1095 CrossRef CAS;
(d)
J. Xu and Z. Chi, Mechanochromic Fluorescent Materials: Phenomena, Materials and Applications, RSC, Cambridge, UK, 2014, 8 RSC;
(e) C. Weder, J. Mater. Chem., 2011, 21, 8235–8236 RSC;
(f) F. Ciardelli, G. Ruggeri and A. Pucci, Chem. Soc. Rev., 2013, 42, 857–870 RSC.
-
(a) T. P. I. Saragi, T. Spehr, A. Siebert, T. Fuhrmann-Lieker and J. Salbeck, Chem. Rev., 2007, 107, 1011–1065 CrossRef CAS PubMed;
(b) J. Liang, B. Z. Tang and B. Liu, Chem. Soc. Rev., 2015, 44, 2798–2811 RSC;
(c) L. Yuan, W. Y. Lin, K. B. Zheng, W. L. He and M. W. Huang, Chem. Soc. Rev., 2013, 42, 622–661 RSC;
(d) Y. X. Zhu, Z. W. Wei, M. Pan, H. P. Wang, J. Y. Zhang and C. Y. Su, Dalton Trans., 2016, 45, 943–950 RSC;
(e) R. Zheng, X. F. Mei, Z. H. Lin, Y. Zhao, H. M. Yao, W. Lv and Q. D. Ling, J. Mater. Chem. C, 2015, 3, 10242–10248 RSC;
(f) S. J. Toal, K. A. Jones, D. Magde and W. C. Trogler, J. Am. Chem. Soc., 2005, 127, 11661–11665 CrossRef CAS PubMed;
(g) B. Xu, M. Xie, J. He, B. Xu, Z. Chi, W. Tian, L. Jiang, F. Zhao, S. Liu, Y. Zhang, Z. Xu and J. Xu, Chem. Commun., 2013, 49, 273–275 RSC;
(h) D. A. Davis, A. Hamilton, J. L. Yang, L. D. Cremar, D. Van Gough, S. L. Potisek, M. T. Ong, P. V. Braun, T. J. Martinez, S. R. White, J. S. Moore and N. R. Sottos, Nature, 2009, 459, 68–72 CrossRef CAS;
(i) Z. Ning, Z. Chen, Q. Zhang, Y. Yan, S. Qian, Y. Cao and H. Tian, Adv. Funct. Mater., 2007, 17, 3799–3807 CrossRef CAS;
(j) A. Kishimura, T. Yamashita, K. Yamaguchi and T. Aida, Nat. Mater., 2005, 4, 546–549 CrossRef CAS PubMed;
(k) B. R. Crenshaw, M. Burnworth, D. Khariwala, A. Hiltner, P. T. Mather, R. Simha and C. Weder, Macromolecules, 2007, 40, 2400–2408 CrossRef CAS;
(l) S. Hirata and T. Watanabe, Adv. Mater., 2006, 18, 2725–2729 CrossRef CAS;
(m) M. S. Kwon, J. Gierschner, S. J. Yoon and S. Y. Park, Adv. Mater., 2012, 24, 5487–5492 CrossRef CAS PubMed;
(n) H. Sun, S. Liu, W. Lin, K. Y. Zhang, W. Lv, X. Huang, F. Huo, H. Yang, G. Jenkins, Q. Zhao and W. Huang, Nat. Commun., 2014, 5, 3601–3609 CrossRef PubMed.
-
(a) X. Zhang, Z. Chi, Y. Zhang, S. Liu and J. Xu, J. Mater. Chem. C, 2013, 1, 3376–3390 RSC;
(b) X. Luo, J. Li, C. Li, L. Heng, Y. Dong, Z. Liu, Z. Bo and B. Z. Tang, Adv. Mater., 2011, 23, 3261–3265 CrossRef CAS PubMed;
(c) S. J. Yoon, J. W. Chung, J. Gierschner, K. S. Kim, M. G. Choi, D. Kim and S. Y. Park, J. Am. Chem. Soc., 2010, 132, 13675–13683 CrossRef CAS PubMed.
-
(a) H. Ito, T. Saito, N. Oshima, N. Kitamura, S. Ishizaka, Y. Hinatsu, M. Wakeshima, M. Kato, K. Tsuge and M. Sawamura, J. Am. Chem. Soc., 2008, 130, 10044–10045 CrossRef CAS PubMed;
(b) C. G. Chandaluri and T. P. Radhakrishnan, Angew. Chem., Int. Ed., 2012, 51, 11849–11852 CrossRef CAS PubMed;
(c) G. Zhang, J. Lu, M. Sabat and C. L. Fraser, J. Am. Chem. Soc., 2010, 132, 2160–2162 CrossRef CAS PubMed;
(d) Y. Dong, B. Xu, J. Zhang, X. Tan, L. Wang, J. Chen, H. Lv, S. Wen, B. Li, L. Ye, B. Zou and W. Tian, Angew. Chem., Int. Ed., 2012, 51, 10782–10785 CrossRef CAS PubMed;
(e) Y. Zhang, F. Gao, X. D. Cao, Y. Q. Li, Y. Z. Xu, W. G. Weng and R. Boulatov, Angew. Chem., Int. Ed., 2016, 55, 3040–3044 CrossRef PubMed.
-
(a) T. Mutai, H. Satou and K. Araki, Nat. Mater., 2005, 4, 685–687 CrossRef CAS PubMed;
(b) W. Z. Yuan, Y. Tan, Y. Gong, P. Lu, J. W. Y. Lam, X. Y. Shen, C. Feng, H. H. Y. Sung, Y. Lu, I. D. Williams, J. Z. Sun, Y. Zhang and B. Z. Tang, Adv. Mater., 2013, 25, 2837–2843 CrossRef CAS PubMed;
(c) X. Zhang, Z. Chi, B. Xu, L. Jiang, X. Zhou, Y. Zhang, S. Liu and J. Xu, Chem. Commun., 2012, 48, 10895–10897 RSC;
(d) S. Xue, W. Liu, X. Qiu, Y. Gao and W. Yang, J. Phys. Chem. C, 2014, 118, 18668–18675 CrossRef CAS.
-
(a) T. Jadhav, B. Dhokale, S. M. Mobin and R. Misra, RSC Adv., 2015, 5, 29878–29884 RSC;
(b) R. Yoshii, A. Hirose, K. Tanaka and Y. Chujo, J. Am. Chem. Soc., 2014, 136, 18131–18139 CrossRef CAS PubMed;
(c) N. Nguyen, G. Zhang, J. Lu, A. Sherman and C. Fraser, J. Mater. Chem., 2011, 21, 8409–8415 RSC.
-
J. B. Birks, Photophysics of Aromatic Molecules, Wiley, London, UK, 1970 Search PubMed.
- S. Jayanty and T. P. Radhakrishnan, Chem. – Eur. J., 2004, 10, 791–797 CrossRef CAS PubMed.
- H. J. Tracy, J. L. Mullin, W. T. Klooster, J. A. Martin, J. Haug, S. Wallace, I. Rudloe and K. Watts, Inorg. Chem., 2005, 44, 2003–2011 CrossRef CAS PubMed.
- J. D. Luo, Z. L. Xie, J. W. Y. Lam, L. Cheng, H. Y. Chen, C. F. Qiu, H. S. Kwok, X. W. Zhan, Y. Q. Liu, D. B. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC.
- Y. Li, T. Liu, H. Liu, M. Z. Tian and Y. Li, Acc. Chem. Res., 2014, 47, 1186–11988 CrossRef CAS PubMed.
- H. Liu, J. Xu and Y. Li, Acc. Chem. Res., 2010, 43, 1496–1508 CrossRef CAS PubMed.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
-
(a) Z. Yang, Z. Mao, T. Yu, Y. Zhang, S. Liu, J. Xu and Z. Chi, ACS Symp. Ser., 2016, 1226, 221–259 CrossRef CAS;
(b) J. Li, T. Shan, M. Yao, Y. Gao, X. Han, B. Yang and P. Lu, J. Mater. Chem. C, 2017, 5, 2552–2558 RSC;
(c) T. Han, X. Gu, J. W. Y. Lam, A. C. S. Leung, R. T. K. Kwok, T. Han, B. Tong, J. Shi, Y. Dong and B. Z. Tang, J. Mater. Chem. C, 2016, 4, 10430–10434 RSC;
(d) K. Matsuoka1, K. Albrecht, K. Yamamoto and K. Fujita, Sci. Rep., 2017, 7, 1–9 CrossRef;
(e) J. Y. Sun, J. Q. Han, Y. Liu, Y. A. Duan, T. Y. Han and J. Yuan, J. Mater. Chem. C, 2016, 4, 8276–8283 RSC.
- Z. B. Henson, K. Müllen and G. C. Bazan, Nat. Chem., 2012, 4, 699–704 CrossRef CAS.
-
(a) R. H. Pawle, T. E. Haas, P. Müller and S. W. Thomas III, Chem. Sci., 2014, 5, 4184–4188 RSC;
(b) Y. Gong, Y. Tan, J. Liu, P. Lu, C. Feng, W. Z. Yuan, Y. Lu, J. Z. Sun, G. He and Y. Zhang, Chem. Commun., 2013, 49, 4009–4011 RSC.
- S. Xue, X. Qiu, Q. Sun and W. Yang, J. Mater. Chem. C, 2016, 4, 1568–1578 RSC.
- T. Jadhav, J. M. Choi, J. Y. Lee, B. Dhokale and R. Misra, Org. Electron., 2016, 37, 448–452 CrossRef CAS.
- H. Liu, Q. Bai, L. Yao, H. Zhang, H. Xu, S. Zhang, W. Li, Y. Gao, J. Li, P. Lu, H. Wang, B. Yang and Y. Ma, Chem. Sci., 2015, 6, 3797–3804 RSC.
- T. Shan, Y. Liu, X. Tang, Q. Bai, Y. Gao, Z. Gao, J. Li, J. Deng, B. Yang, P. Lu and Y. Ma, ACS Appl. Mater. Interfaces, 2016, 8, 28771–28779 CrossRef CAS.
-
(a) Y. Dong, J. W. Y. Lam, A. Qin, J. Liu, Z. Li, B. Z. Tang, J. Sun and H. S. Kwok, Appl. Phys. Lett., 2007, 91, 011111–011114 CrossRef;
(b) J. Huang, X. Yang, J. Wang, C. Zhong, L. Wang, J. Qin and Z. Li, J. Mater. Chem., 2012, 22, 2478–2484 RSC;
(c) S. Wang, W. J. Oldham, R. A. Hudack and G. C. Bazan, J. Am. Chem. Soc., 2000, 122, 5695–5709 CrossRef CAS;
(d) T. Jadhav, B. Dhokale, Y. Patil, S. M. Mobin and R. Misra, J. Phys. Chem. C, 2016, 120, 24030–24040 CrossRef CAS;
(e) T. Jadhav, B. Dhokale, S. M. Mobin and R. Misra, J. Mater. Chem. C, 2015, 3, 9981–9988 RSC.
-
(a) M. Zhu and C. Yang, Chem. Soc. Rev., 2013, 42, 4963–4976 RSC;
(b) G. M. Farinola and R. Ragni, Chem. Soc. Rev., 2011, 40, 3467–3482 RSC;
(c) M. A. Baldo, M. E. Thompson and S. R. Forrest, Nature, 2000, 403, 750–753 CrossRef CAS;
(d) S. Reineke, F. Lindner, G. Schwartz, N. Seidler, K. Walzer, B. Lüssem and K. Leo, Nature, 2009, 459, 234–238 CrossRef CAS;
(e) A. C. Grimsdale, K. L. Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897–1091 CrossRef CAS PubMed;
(f) W. C. H. Choy, W. K. Chan and Y. Yuan, Adv. Mater., 2014, 26, 5368–5399 CrossRef CAS PubMed;
(g) Z. Ge, T. Hayakawa, S. Ando, M. Ueda, T. Akiike, H. Miyamoto, T. Kajita and M. Kakimoto, Chem. Mater., 2008, 20, 2532–2537 CrossRef CAS;
(h) S. Jeong, M. Kim, S. H. Kim and J. Hong, Org. Electron., 2013, 14, 2497–2504 CrossRef CAS.
-
(a) J. Huang, Y. Jiang, J. Yang, R. Tang, N. Xie, Q. Li, H. S. Kwok, B. Z. Tang and Z. Li, J. Mater. Chem. C, 2014, 2, 2028–2036 RSC;
(b) C. Li, J. Wei, J. Han, Z. Li, X. Song, Z. Zhang, J. Zhang and Y. Wang, J. Mater. Chem. C, 2016, 4, 10120–10129 RSC;
(c) J. Yang, J. Huang, Q. Li and Z. Li, J. Mater. Chem. C, 2016, 4, 2663–2684 RSC.
-
(a) C. Jin, J. Liu, Y. Chen, G. Li, R. Guan, P. Zhang, L. Ji and H. Chao, Dalton Trans., 2015, 44, 7538–7547 RSC;
(b) L. Zheng and R. Hua, J. Org. Chem., 2014, 79, 3930–3936 CrossRef CAS PubMed;
(c) Y. Zhang, J. H. Wang, J. Zheng and D. Li, Chem. Commun., 2015, 51, 6350–6353 RSC;
(d) W. C. Chen, C. S. Lee and Q. X. Tong, J. Mater. Chem. C, 2015, 3, 10957–10963 RSC;
(e) X. Tang, Q. Bai, Q. Peng, Y. Gao, J. Li, Y. Liu, L. Yao, P. Lu, B. Yang and Y. Ma, Chem. Mater., 2015, 27, 7050–7057 CrossRef CAS;
(f) S. Zhuang, R. Shangguan, J. Jin, G. Tu, L. Wang, J. Chen, D. Ma and X. Zhu, Org. Electron., 2012, 13, 3050–3059 CrossRef CAS.
-
(a) Y. Zhang, S. L. Lai, Q. X. Tong, M. F. Lo, T. W. Ng, M. Y. Chan, Z. C. Wen, J. He, K. S. Jeff, X. L. Tang, W. M. Liu, C. C. Ko, P. F. Wang and C. S. Lee, Chem. Mater., 2012, 24, 61–70 CrossRef;
(b) S. Zhang, L. Yao, Q. Peng, W. Li, Y. Pan, R. Xiao, Y. Gao, C. Gu, Z. Wang, P. Lu, F. Li, S. Su, B. Yang and Y. Ma, Adv. Funct. Mater., 2015, 25, 1755–1762 CrossRef CAS;
(c) W. Li, Y. Pan, L. Yao, H. Liu, S. Zhang, C. Wang, F. Shen, P. Lu, B. Yang and Y. Ma, Adv. Opt. Mater., 2014, 2, 892–901 CrossRef CAS;
(d) W. Li, D. Liu, F. Shen, D. Ma, Z. Wang, T. Feng, Y. Xu, B. Yang and Y. Ma, Adv. Funct. Mater., 2012, 22, 2797–2803 CrossRef CAS;
(e) Y. Gao, S. Zhang, Y. Pan, L. Yao, H. Liu, Y. Guo, Q. Gu, B. Yang and Y. Ma, Phys. Chem. Chem. Phys., 2016, 18, 24176–24184 RSC;
(f) G. Li, J. Zhao, D. Zhang, Z. Shi, Z. Zhu, H. Song, J. Zhu, S. Tao, F. Lu and Q. Tong, J. Mater. Chem. C, 2016, 4, 8787–8794 RSC.
-
(a) T. Jadhav, J. M. Choi, B. Dhokale, S. M. Mobin, J. Y. Lee and R. Misra, J. Phys. Chem. C, 2016, 120, 18487–18495 CrossRef CAS;
(b) T. Jadhav, J. M. Choi, J. Shinde, J. Y. Lee and R. Misra, J. Mater. Chem. C, 2017, 5, 6014–6020 RSC;
(c) L. Chen, Y. Jiang, H. Nie, R. Hu, H. S. Kwok, F. Huang, A. Qin, Z. Zhao and B. Z. Tang, ACS Appl. Mater. Interfaces, 2014, 6, 17215–17225 CrossRef CAS PubMed;
(d) G. Lin, H. Peng, L. Chen, H. Nie, W. Luo, Y. Li, S. Chen, R. Hu, A. Qin, Z. Zhao and B. Zhong Tang, ACS Appl. Mater. Interfaces, 2016, 26, 16799–16808 CrossRef;
(e) Y. Li, W. Wang, Z. Zhuang, Z. Wang, G. Lin, P. Shen, S. Chen, Z. Zhao and B. Z. Tang, J. Mater. Chem. C, 2018, 6, 5900–5907 RSC;
(f) J. Jayabharathi, S. Panimozhi and V. Thanikachalam, Springer Nature, Sci. Rep., 2019, 9, 17555–17569 Search PubMed.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford CT (Revision A.02), 2009, Gaussian, Inc., Wallingford, CT Search PubMed.
- R. Misra, T. Jadhav, B. Dhokale and S. M. Mobin, Chem. Commun., 2014, 50, 9076–9078 RSC.
- S. Janietz, D. D. C. Bradley, M. Grell, C. Giebeler, M. Inbasekaran and E. P. Woo, Appl. Phys. Lett., 1998, 73, 2453–2455 CrossRef CAS.
-
(a) Y. Yuan, D. Li, X. Zhang, X. Zhao, Y. Liu, J. Zhang and Y. Wang, New J. Chem., 2011, 35, 1534–1540 RSC;
(b) Y. Zhang, S. L. Lai, Q. X. Tong, M. Y. Chan, T. W. Ng, Z. C. Wen, G. Q. Zhang, S. T. Lee, H. L. Kwong and C. S. Lee, J. Mater. Chem., 2011, 21, 8206–8214 RSC;
(c) B. Liu, J. Zhao, C. Luo, F. Lu, S. Tao and Q. Tong, J. Mater. Chem. C, 2016, 4, 2003–2010 RSC.
- J. Jayabharathi, S. Thilagavathy and V. Thanikachalam, RSC Adv., 2021, 11, 8606–8618 RSC.
- J. Jayabharathi, J. Anudeebhana, V. Thanikachalam and S. Sivaraj, RSC Adv., 2020, 10, 8866–8879 RSC.
-
(a) Z. Y. Wanga, J. W. Zhaob, P. Lia, T. Fenga, W. J. Wanga, S. L. Taob and Q. X. Tong, New J. Chem., 2018, 42, 8924–8932 RSC;
(b) R. Misra, T. Jadhav, B. Dhokale and S. M. Mobin, Chem. Commun., 2014, 50, 9076–9078 RSC;
(c) F. Liu, H. Liu, X. Tang, S. Ren, X. He, J. Li, C. Du, Z. Feng and P. Lu, Nano Energy, 2020, 68, 104325–104365 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00391g |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Shanmugam
Thilagavathy
and
Venugopal
Thanikachalam
*,
Shanmugam
Thilagavathy
and
Venugopal
Thanikachalam