
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synergistic reinforcement of a reversible Diels–Alder type network with nanocellulose†
Anna
Peterson
a,
Manta
Roy
 a,
Julia
Fagerlund
a,
Giada
Lo Re
b and
Christian
Müller
*ac
a,
Julia
Fagerlund
a,
Giada
Lo Re
b and
Christian
Müller
*ac
aDepartment of Chemistry and Chemical Engineering, Chalmers University of Technology, 412 96 Göteborg, Sweden. E-mail: christian.muller@chalmers.se
bDepartment of Industrial and Materials Science, Chalmers University of Technology, 412 96 Göteborg, Sweden
cWallenberg Wood Science Center, Chalmers University of Technology, 412 96 Göteborg, Sweden
First published on 7th July 2021
Abstract
Covalent adaptable networks are attractive intermediates between thermosets and thermoplastics. To achieve an optimal combination of dimensional stability at the temperature of use and macroscopic flow at elevated temperatures, materials that combine two reversible networks are highly sought after. We demonstrate that such a material can be created through the addition of cellulose microfibrils to a polymer matrix that can undergo thermoreversible Diels–Alder reactions. The cellulose microfibrils and crosslinked polymer form two independent reversible networks that display clear synergistic effects on the thermomechanical properties of the nanocomposite. Above the glass transition temperature of the polymer matrix the two networks work in tandem to reduce tensile creep by a factor of 40 at 80 °C, while increasing the storage modulus by a factor of 60 at the same temperature. The adaptability of the Diels–Alder network is not compromised by the addition of cellulose microfibrils, as shown by kinetic studies and repeated reprocessing. Further, the cellulose network significantly improves the dimensional stability at elevated temperatures where the Diels–Alder network dissociates.
Introduction
Covalent adaptable networks (CANs), currently receive considerable interest as next generation plastic materials.1–3 They display a previously unattainable combination of material properties that lie in between thermosets and thermoplastics, obtained via reversible crosslinking. The resulting polymer networks typically show reduced creep rates, increased stiffness and increased solvent and thermal resistance, characteristic for a thermoset. Simultaneously, the materials retain their ability for macroscopic flow, classically only displayed by thermoplastics.3 CANs can be divided into two categories based on the mechanism that underlies the reversible crosslinking chemistry: dissociative networks formed by reversible addition or condensation reactions4 and associative networks formed by reversible bond exchange processes.2 Dissociative networks comprise crosslinks that open up at elevated temperature, leading to a complete loss of network integrity in the state of macroscopic flow. The dissociation of crosslinks allows the use of reprocessing methods such as extrusion or fused filament fabrication type 3D printing, which are otherwise reserved for thermoplastic materials.5,6One widely used thermoreversible crosslink exploits the Diels–Alder (DA) reaction between furan (F) and maleimide (MI).7 The popularity of the furan/maleimide coupling reaction stems from the relatively low temperature (100–120 °C) of the retro Diels–Alder (rDA) decoupling reaction8–10 and the click-chemistry type nature and high chemoselectivity of the reaction.7,11 The DA reaction is an equilibrium reaction between a forward coupling reaction, forming the adduct (A), and the reverse decoupling reaction that regenerates the reactants (eqn (1)).
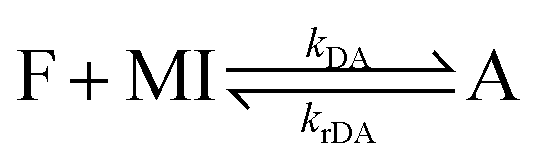 | (1) |
At low temperatures (association temperature range Tassociation ∼ 50–60 °C) the equilibrium is shifted towards the crosslinked state while at elevated temperatures (dissociation temperature range Tdiss ∼ 100–120 °C) the reverse reaction is dominating, leading to a loss of network integrity.12 Since 2002, when Wudl et al.8 published their pioneering work using the DA coupling reaction to form reversibly crosslinked networks, various furan-modified molecules, ranging from oligomers8,9,13 to elastomers14–18 and other macromolecules5,19–21 have been coupled with bismaleimides or maleimide functionalized polymers, rendering the DA reaction the (probably) most well studied coupling reaction within CANs.
The dimensional stability and resistance to flow, even at elevated temperatures are important characteristics of thermosets.3 It is characterized by the creep of the material, i.e. the slow deformation in response to a persistent mechanical stress. The glass transition temperature (Tg) as well as the density of crosslinks in a plastic material will dictate the relaxation kinetics of the polymer chains and, hence, the creep behavior. Design of new CANs must find balance between the temperature range in which macroscopic flow is restricted, imparting dimensional stability, and the temperature range in which macroscopic flow is needed to permit reprocessability.
Apart from a sufficiently high Tg and crosslink density the introduction of dually crosslinked dynamic networks has recently been shown beneficial to arrest creep22e.g. by using a combination of irreversible and dynamic crosslinks23,24 or introduction of metal complexes in dynamic networks.25,26 The network forming properties of nanocellulose within a polymer matrix, however, has not been addressed in this regard. Nanocelluloses are stiff, elongated particles of cellulose with at least one dimension in the nanometer range. The numerous hydroxyl groups on the cellulose surface allow the particles to interact via hydrogen bonding.27 Composites containing a nanocellulose network are, hence, effectively dissociative networks, where hydrogen bonds between nanocellulose particles constitute the crosslinking points.
The formation of a nanocellulose network in polymer composites has been shown beneficial to arrest creep, especially in the high temperature regime.28 Composites of CANs reinforced by nanocellulose have recently been demonstrated, where the nanocellulose imparts a higher toughness, stiffness and resistance to macroscopic flow. Impregnation of a cellulose nanopaper with oligomers29 or a polymer latex30,31 of a CAN prepolymer have been shown to produce reshapable and reweldable composites. Such composites have been suggested as recyclable replacements for traditional fiber reinforced thermosetting composites.29,32 Functionalized cellulose crystals have been used as crosslinkers33–35 or reinforcement36 in CAN matrices, providing an increase in the crosslinking density but a decrease in reaction rate of the reversible reaction.36 The impact of a nanocellulose reinforcing agent on the reaction rate of a dissociative CAN, however, has not yet been shown.
Here, we show a dually crosslinked dynamic network, where synergistic effects between DA reversible covalent interactions and secondary interactions between nanocellulose increase the stiffness and resistance to macroscopic flow. We synthesized a DA type dissociative CAN, which is soluble in dimethylformamide (DMF), to facilitate compounding of microfibrillated cellulose (MFC) together with the CAN. Nanocellulose materials can only be processed from polar solvents, such as water and DMF, which limits the type of CAN matrix that can be used. The commercially available copolymer poly(ethylene-alt-maleic anhydride), p(E-alt-MA) was modified with pendant furan groups enabled by the abundance of reactive maleic anhydride groups in the polymer chain. Apart from the ease of functionalization due to the numerous anhydrides along the polymer backbone, the synthetic product was confirmed to be soluble in DMF. The resulting polymers exhibit a Tg between 60–80 °C, depending on the furan content. By varying the amount of the furan functionality, the crosslink density of the obtained DA network could be controlled. Via crosslinking with bismaleimide (BMI) we obtain fully thermoreversible DA networks. An oligomeric BMI that contains flexible alkyl segments was chosen, which plasticizes the network, as vitrification at too high temperatures would decrease the rate of the DA and rDA reactions. Below Tg = 77 °C, creep is almost completely arrested while, above Tg, a higher density of crosslinks and the presence of MFC reduces creep, resulting in a combined decrease of creep by almost two orders of magnitude at 80 °C. Interestingly, we find that the addition of MFC allows to drastically reduce the creep at temperatures above Tdiss. While the neat CAN rapidly elongates and fractures, the addition of MFC reduces the creep strain to 20%. This is attributed to the formation of a MFC network, which governs the dimensional stability of the CAN even in the dissociated state.
Results and discussion
Synthesis and characterization of furan functionalized p(E-alt-MA)
We modified the copolymer p(E-alt-MA) with furfuryl amine and heptyl amine through a thermally initiated addition–substitution reaction (Fig. 1, cf. ESI† for detailed synthetic protocol). The ratio of furfuryl to alkyl functionality was varied, rendering two samples, 50f having 50 mol% of the MA units substituted with furfuryl and 50 mol% with heptyl, and 10f having 10 mol% of the MA units substituted with furfuryl and 90 mol% with heptyl (Table 1). The difference in furan content between the two samples later allows us to obtain CANs with different crosslinking densities. The presence of furfuryl functional groups was confirmed by 1H nuclear magnetic resonance spectroscopy (1H NMR) and Fourier-transform infrared spectroscopy (FTIR). 1H NMR peaks at δ = 7.96, 7.46, 6.31 and 6.21 ppm are ascribed to the protons of furan rings, while the peak at δ = 4.49 ppm is attributed to the presence of methylene protons (CH2) of the furfuryl (Fig. S1a, ESI†). A comparison between the 1H NMR spectra of 50f and 10f (Fig. S1, ESI†) shows the drastically higher content of alkyl protons for 10f, attributed to the higher grafting density of heptyl instead of furfuryl. The CH3 proton from the heptyl chain is observed at δ = 0.83 ppm, where the integral value of 10f is about 8 times as high compared to 50f. A quantitative assignment of the relative grafting densities from 1H NMR, however, is not possible due to overlap of the alkyl proton peaks. | ||
| Fig. 1 (a) Modification of p(E-alt-MA) with furfuryl and heptyl amine rendering imide formation via an addition–elimination reaction, (b) FTIR absorbance spectra of p(E-alt-MA) and furan modified p(E-alt-MA). | ||
| Sample | Composition of furan modified p(E-alt-MA) | Furan:maleimide ratio | Microfibrillated cellulose | |
|---|---|---|---|---|
| 1a | 1b | |||
| 50f | 0.5 | 0.5 | — | — |
| 10f | 0.1 | 0.9 | — | — |
| XL50f | 0.5 | 0.5 | 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
— |
| XL10f | 0.1 | 0.9 | 2:1 |
— |
| XL50f-MFC | 0.5 | 0.5 | 2:1 |
10 wt% |
| XL10f-MFC | 0.1 | 0.9 | 2:1 |
10 wt% |
FTIR spectroscopy was used to further confirm grafting of furan groups and imide formation (Fig. 1b), as well as quantification of the furan graft density. Characteristic peaks are the furan ring asymmetric and symmetric vibration of COC (ϑCOC = 1070 cm−1 and 1008 cm−1), the CH in-plane deformation (ϑCH = 1138 cm−1), the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond stretching vibration (ϑCC = 1503 cm−1) and the significant CH out of plane deformation (ϑCH = 754 cm−1). Moreover, the disappearance of the anhydride peaks (ϑCO = 1856 cm−1 and 1776 cm−1) and the simultaneous appearance of imide peaks (ϑCO = 1687 cm−1 and 1773 cm−1) confirms grafting of p(E-alt-MA) with alkyl and furfuryl. The maleic anhydride groups are prone to moisture absorption and thereby hydrolyze into dicarboxylic acid, a conversion that reduces the reactivity of the carbonyl group and hence the efficiency of nucleophilic substitution. However, the acid peak (ϑCO = 1710 cm−1) cannot be spotted in pristine p(E-alt-MA) while the presence of anhydride characteristic peaks (ϑCO = 1856 cm−1 and 1776 cm−1)37 is evident (Fig. 1b). Quantification of the degree of furan substitution was done based on the area of the furan peak, centered around ϑCH = 754 cm−1 (Table 2, applied equations and raw data are found in Fig. S2, ESI†). Further, elemental analysis was used to confirm grafting of furfuryl and heptyl to the p(E-alt-MA) backbone.38 The experimental results are in excellent agreement with the predicted mass fraction of the repeating units in case of both 50f and 10f, where especially the amount of nitrogen, stemming from imide formation between anhydride and amine confirms a high yield of the grafting reaction (Table S1, ESI†).
C double bond stretching vibration (ϑCC = 1503 cm−1) and the significant CH out of plane deformation (ϑCH = 754 cm−1). Moreover, the disappearance of the anhydride peaks (ϑCO = 1856 cm−1 and 1776 cm−1) and the simultaneous appearance of imide peaks (ϑCO = 1687 cm−1 and 1773 cm−1) confirms grafting of p(E-alt-MA) with alkyl and furfuryl. The maleic anhydride groups are prone to moisture absorption and thereby hydrolyze into dicarboxylic acid, a conversion that reduces the reactivity of the carbonyl group and hence the efficiency of nucleophilic substitution. However, the acid peak (ϑCO = 1710 cm−1) cannot be spotted in pristine p(E-alt-MA) while the presence of anhydride characteristic peaks (ϑCO = 1856 cm−1 and 1776 cm−1)37 is evident (Fig. 1b). Quantification of the degree of furan substitution was done based on the area of the furan peak, centered around ϑCH = 754 cm−1 (Table 2, applied equations and raw data are found in Fig. S2, ESI†). Further, elemental analysis was used to confirm grafting of furfuryl and heptyl to the p(E-alt-MA) backbone.38 The experimental results are in excellent agreement with the predicted mass fraction of the repeating units in case of both 50f and 10f, where especially the amount of nitrogen, stemming from imide formation between anhydride and amine confirms a high yield of the grafting reaction (Table S1, ESI†).
The number-average molar mass (Mn), from gel permeation chromatography (GPC) analysis, is increased from 40 kg mol−1 for neat p(E-alt-MA) to 70 and 100 kg mol−1 for 50f and 10f, respectively (Table 2 and Fig. S3, ESI†). The increase in molar mass of the repeat unit obtained after functionalization is expected to be about 70%, with a slightly higher mass for the 10f repeat unit due to the abundance of heptyl chains. GPC analysis hence confirms successful functionalization of p(E-alt-MA). The discrepancy between expected mass increase and the experimental results is ascribed to differences in the hydrodynamic radii of the polymers compared to the polystyrene standard used, while also bearing in mind that the samples show a high polydispersity (PDI).
CAN preparation via Diels–Alder crosslinking
We crosslinked the functionalized p(E-alt-MA) polymers (10f and 50f) with oligomeric bismaleimide (oligo-BMI) using a solvent casting procedure (cf. Experimental) to form dissociative CANs, denoted XL10f and XL50f (Fig. 2a and Table 1). A molar stoichiometry of F/M = 2:1 was chosen due to the incomplete conversion of the equilibrium controlled DA reaction for a stoichiometric ratio (F/M = 1).39 The DA reaction between furan modified p(E-alt-MA) and oligo-BMI renders a thermally reversible polymer network. The reversible nature was confirmed by solubility tests where the initial reaction mixture, containing 50f and oligo-BMI dissolved in DMF, formed a gel after thermal treatment at 60 °C overnight (Fig. 2b). Upon heating the mixture at 140 °C, the gel reverts back to the liquid state, a clear light brown liquid, which again may be turned into a gel by annealing at 60 °C.
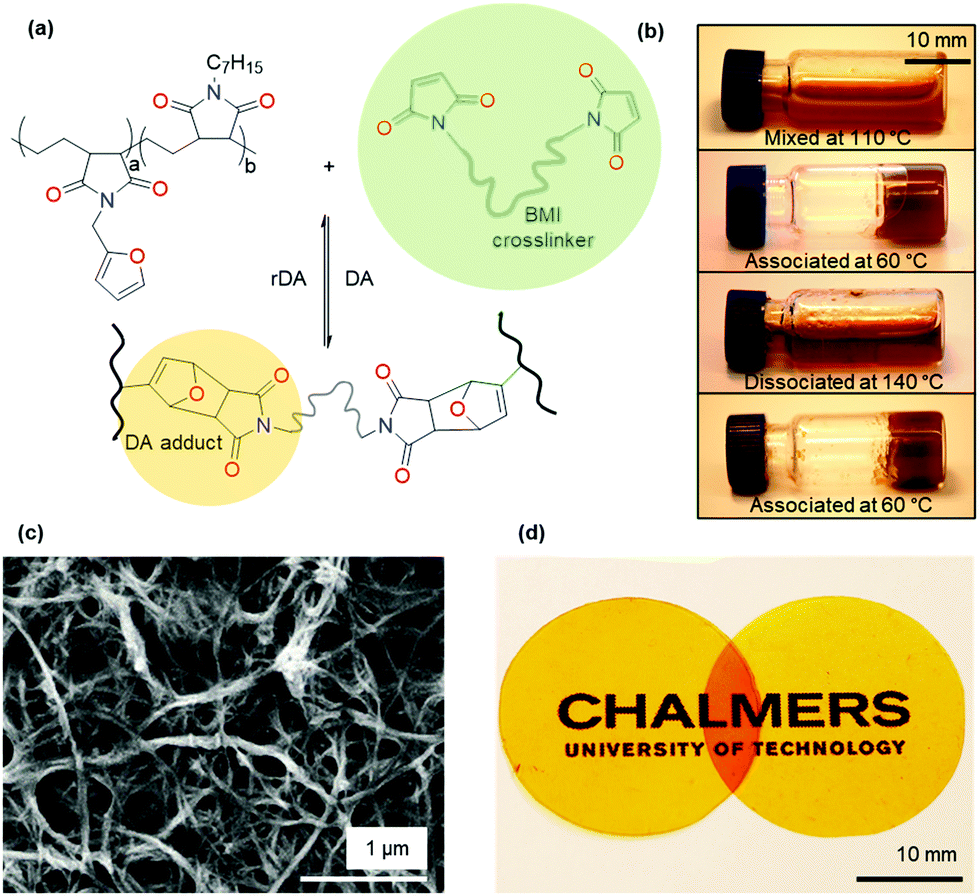 | ||
| Fig. 2 (a) Synthesis of the Diels–Alder type dissociative CAN via bismaleimide crosslinking, (b) the reversible sol–gel transition process of the XL50f sample in DMF, (c) SEM micrograph of MFC, (d) photograph of XL50f (left) and XL50f-MFC (right) showcasing the efficient dispersion of MFC in the XL50f matrix. | ||
As described above, the design of the here presented CAN allows for dissolution in DMF (Table S2 (ESI†), solubility chart). Network formation of nanocellulose in composites processed from DMF has been previously reported,40,41 making our CAN ideal for the study of formation of a second network, based on MFC (Fig. 2c). Samples containing 10 wt% MFC were prepared with the same solvent casting procedure as was used for the neat CANs. Once dried, all samples were cured at 60 °C for at least 60 h to ensure complete crosslinking. The progress of crosslinking was followed by FTIR spectroscopy, specifically the intensity of the absorption peak of the out of plane bending of the olefinic protons of maleimide (ϑCH = 828 cm−1),42 which reaches a stable value after about 20 h at 60 °C (Fig. S4, ESI†). The visual appearance of cured films is equivalent for samples with and without MFC. Films are bright orange, homogenous and transparent (Fig. 2d). The lack of visible aggregates indicates efficient dispersion of MFC in the CAN.
DSC heating thermograms of pristine p(E-alt-MA) reveal a high Tg, around 145 °C. After introduction of furan groups and alkyl chains the Tg is significantly reduced. DSC thermograms of 10f and 50f display a Tg of 60 and 77 °C, respectively (Fig. S5a (ESI†) and Table 1). The decrease in Tg is attributed to the presence of flexible alkyl chains and bulky furan groups, increasing the mobility of the polymer. The oligomeric BMI crosslinker (cf. Fig. S5b (ESI†) for molecular structure) used for CAN formation can be viewed as a simultaneous crosslinker and plasticizer, due to the long flexible alkyl segments. Post crosslinking, the magnitude of the heat flow step, which indicates the Tg, has decreased while new transitions arise from the thermal history of the crosslinked samples (Fig. S5c, ESI†). While the DSC heating thermograms of XL10f and XL50f do not allow for an unambiguous assignment of the Tg of the CANs, they indicate that the Tg did not significantly increase, in agreement with dynamic mechanical analysis (DMA) thermograms (Fig. S6, ESI†). Additionally, we observe two broad endothermic peaks in the DSC heating thermograms of XL50f and XL50f-MFC, which are attributed to the rDA reaction of the two DA adduct stereoisomers. The endothermic peaks centered around 170 °C and 145 °C (Fig. S5c, ESI†) are ascribed to the rDA reaction of the more thermally stable exo adduct and the less stable endo adduct, respectively.43 The peak temperatures of the rDA endotherms are at the higher end of the range of temperatures reported for DA systems, for which the equilibrium is typically shifted towards the dissociated state at temperatures above 120 °C.7 DSC thermograms feature very broad transitions with an onset at about 120 °C (Fig. S5c, ESI†), which also correlates with the softening point observed in DMA thermograms (cf. discussion below). We ascribe the broad endotherms and relatively high peak temperatures of the rDA endotherms to slow relaxation kinetics of the network, as the network points are attached to macromolecular chains.5,44 The endotherms of the rDA reaction for XL10f are less pronounced due to the lower crosslinking density.
Kinetics of the DA and rDA reaction
We studied the impact of the furan content and the addition of nanocellulose on the kinetics of the DA association. The kinetics of the DA and rDA reactions depend on the local mobility of the molecules involved in the reaction, while the overall processability depends on the macroscopic viscosity of the material. While the local mobility and viscosity are similar in some systems, such as oligomer mixtures and many neat polymers, in case of e.g. composites, a material may show a high resistance to melt flow, but still feature a high local reactivity. Decoupling of the local mobility and viscosity was demonstrated for associative CANs reinforced with cellulose nanomaterials, which display reversible crosslink chemistry and at the same time a higher stiffness and resistance to flow.34,36,45 However, the impact of a nanocellulose reinforcing agent on a dissociative CAN is yet to be shown. We used FTIR spectroscopy, and specifically, the maleimide absorption peak (ϑCH = 828 cm−1) to follow the course of the reaction at different temperatures (Fig. 3a). The relative conversion, RIR, was calculated using the peak intensities (MI) according to eqn (2) where the C–H methyl peak (ϑCH = 722 cm−1) was used for normalization.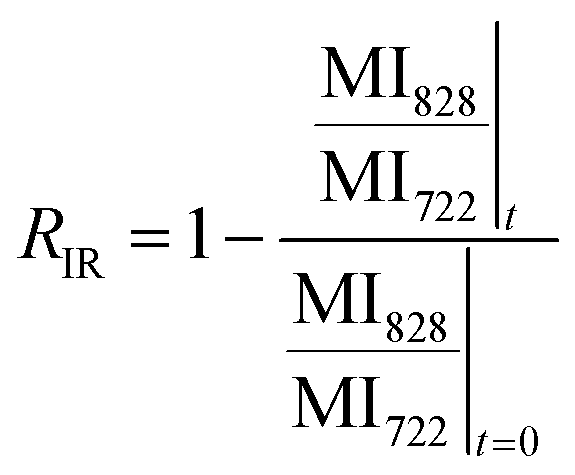 | (2) |
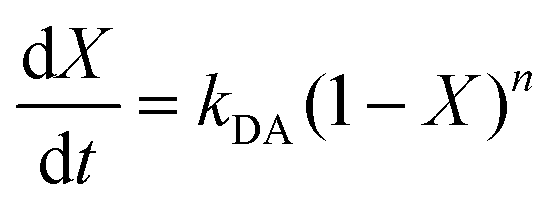 | (3) |
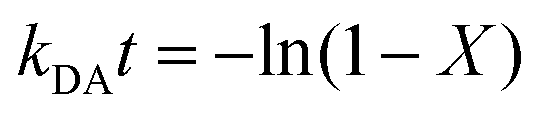 | (4) |
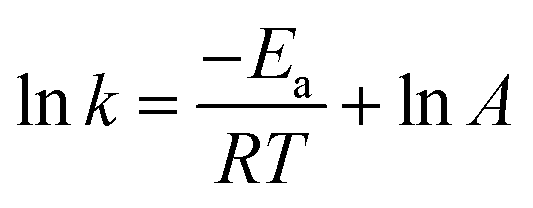 | (5) |
:1.
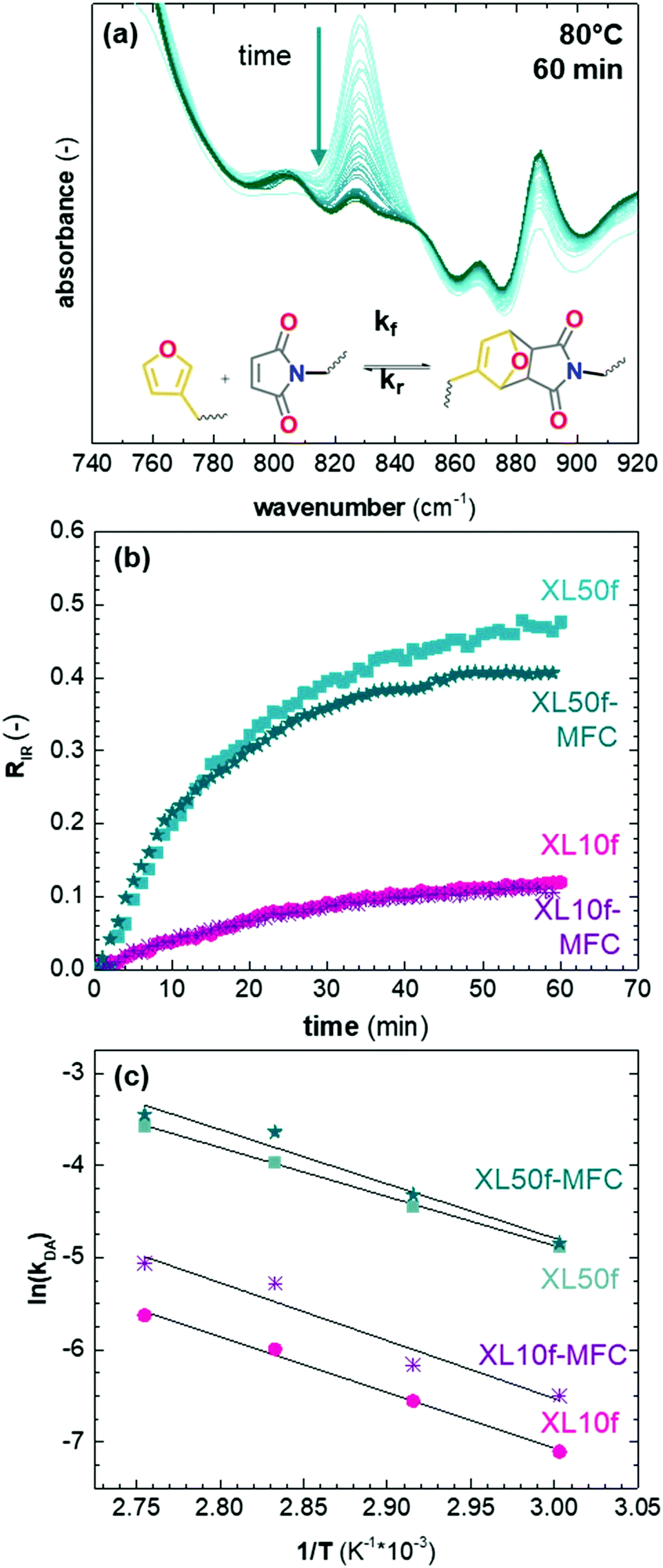 | ||
| Fig. 3 (a) FTIR spectra showing the reduction of the maleimide peak at 828 cm−1 during a 60 min isotherm at 80 °C, (b) the relative conversion, RIR, deduced for XL10f and XL50f with and without MFC at 80 °C, and (c) Arrhenius plots of ln(kDA) versus reciprocal temperature. | ||
The initial slope of RIR(t) was fitted with eqn (4) to determine the apparent rate of the DA association reaction, kDA. The slope is increasing with the isothermal annealing temperature (Fig. S7a, ESI†). The growing impact of the reverse reaction at high temperatures and long reaction times is indicated by the fact that the consumption of maleimide after 60 min is higher at 70 and 80 °C compared to 90 °C even though the initial consumption of maleimide at 90 °C is faster. RIR was also compared for XL10f and XL50f where the maleimide consumption pattern is similar for the samples with and without MFC (Fig. 3b), giving a first indication that the addition of nanocellulose does not drastically affect the kinetics of the association reaction. Next, the rate coefficients were fitted using the linearized version of the Arrhenius equation (eqn (5)) to determine the pre-exponential factor (A) and activation energy (Ea) for the reaction (Fig. 3c). Activation energies of Ea = 49.0 and 44.3 kJ mol−1 were obtained for XL50f with and without MFC, respectively, whereas XL10f featured a slightly higher Ea of 52.2 and 50.3 kJ mol−1 (Table 3). The values obtained are within the range of Ea reported for DA association in polymer melts, which are typically 30–60 kJ mol−1.47–49 The small difference in activation energy between samples with and without MFC suggests that the local mobility and macroscopic viscosity are indeed decoupled, giving rise to CAN type materials which display resistance to macroscopic flow without drastically affecting the reaction kinetics.
| Sample | E a (kJ mol−1) | A (s−1) | R 2 |
|---|---|---|---|
| XL50f | 44.3 | 11.1 | 0.999 |
| XL50f-MFC | 49.0 | 12.9 | 0.967 |
| XL10f | 50.3 | 11.1 | 0.995 |
| XL10f-MFC | 52.2 | 12.32 | 0.946 |
Crosslinking density
Besides the Tg and the presence of a stiff reinforcement, the crosslinking density has been identified as a key factor affecting the creep resistance of materials. The molecular weight between crosslinks, Mc, of our samples was calculated according to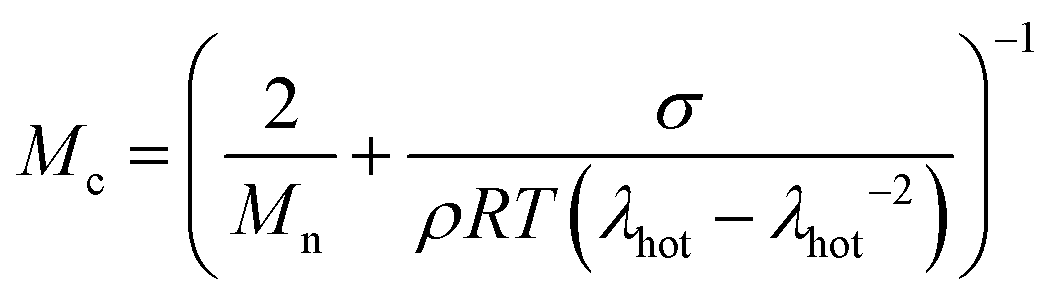 | (6) |
51 and for crosslinked polyethylene around 6 kg mol−1.52
| Sample | M c (kg mol−1) | E′ at 80 °C (MPa) | ||
|---|---|---|---|---|
| Theoretical minimum | Experimental | Associated | Dissociated | |
| XL50f | 5 | 8 | 74 | 4.4 |
| XL10f | 11 | 20 | 49 | 0.13 |
| p(E-alt-MA) | — | — | — | 1560 |
Thermo-mechanical characterization
DMA thermograms show the synergistic effect between formation of a DA type CAN and reinforcement with MFC (Fig. 4). The impact of CAN formation on the storage modulus, E′, below Tdiss is evident in case of XL50f, which was either cured (60 °C, 60 h) or dissociated (160 °C, 15 min with subsequent quick cooling to RT) prior to the DMA experiment. DA crosslinking in XL50f increases E′ by a factor 2 at 40 °C (below Tg) and almost a factor 20 at 80 °C (above Tg) compared to the dissociated sample. Similar results are seen for XL10f, although with a smaller effect from CAN formation due to the lower crosslink density (Fig. S6, ESI†). The addition of MFC to XL50f further increases E′ by a factor 2 at 40 °C, a factor 30 at 80 °C and at least 2 orders of magnitude at 160 °C (above Tdiss). The combined effect of formation of the DA type CAN and MFC addition is a factor 4 at 40 °C and a factor 60 at 80 °C. The increase in E′ is directly related to the dimensional stability and resistance to flow in these CAN composites.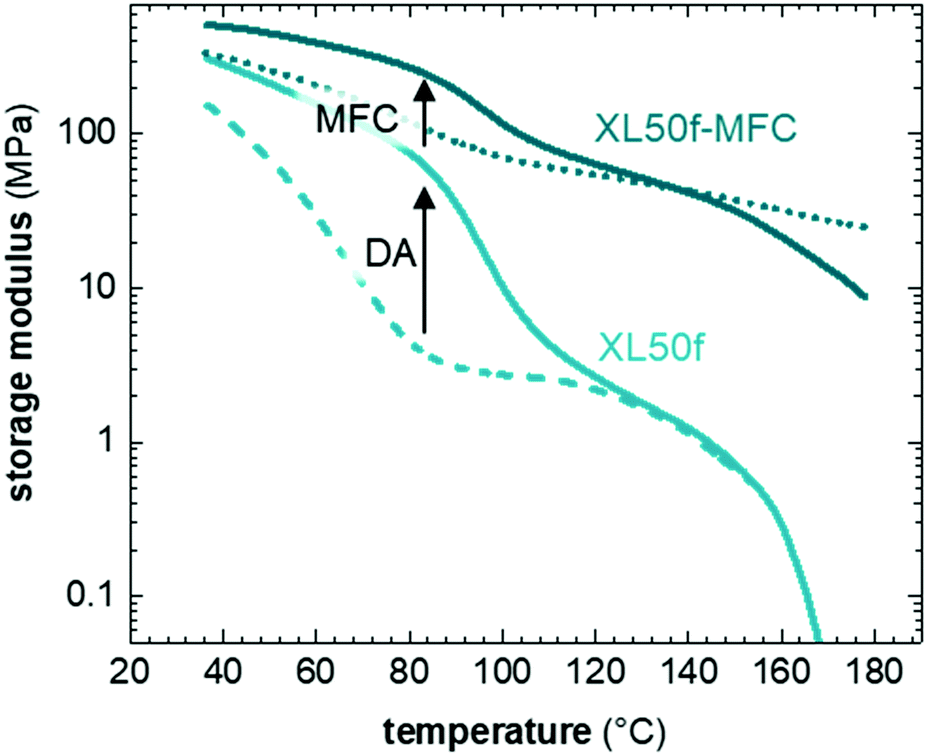 | ||
| Fig. 4 DMA thermograms of XL50f with and without MFC. Dashed and dotted lines are samples of XL50f and XL50f-MFC, respectively, that, prior to analysis, were dissociated at 160 °C and rapidly cooled to prevent crosslink formation. | ||
The set of CANs synthesized in this study allowed us to examine the creep compliance as a function of three different parameters: the crosslink density, the Tg and the presence of a second network of MFC. The impact of crosslink density was probed by creep experiments of XL50f and XL10f at 80 °C, above the Tg of CANs. We find that the crosslink density has a significant impact on the creep compliance, where XL10f and XL50f show creep strains of 20 and 5%, respectively (Fig. 5a). XL10f also shows a much larger instantaneous strain upon subjecting the sample to the applied stress, which correlates with the lower crosslink density, as the long polymer segments between crosslinks can uncoil and align in the direction of stress, a behavior which is suppressed by the higher crosslink density in XL50f. The XL50f sample was additionally probed at 40 °C, e.g. below Tg (Fig. 5b), by which the creep strain is reduced, from 5% to around 1% after 180 min of creep at σ = 50 kPa. A similar reduction of creep strain is seen upon addition of MFC to the XL50f sample (Fig. 5c), again showing the synergistic effect of the CAN and MFC on the thermo-mechanical properties. To probe the effect of MFC only, we carried out creep tests at 140 °C, above Tdiss. At this temperature the DA equilibrium is heavily shifted towards the reactant side and XL50f consequently rapidly elongates and ultimately fractures after reaching a creep strain of more than 200%, whereas the addition of nanocellulose arrests creep and results in a creep strain of not more than 4% after 180 min of creep (Fig. 5d).
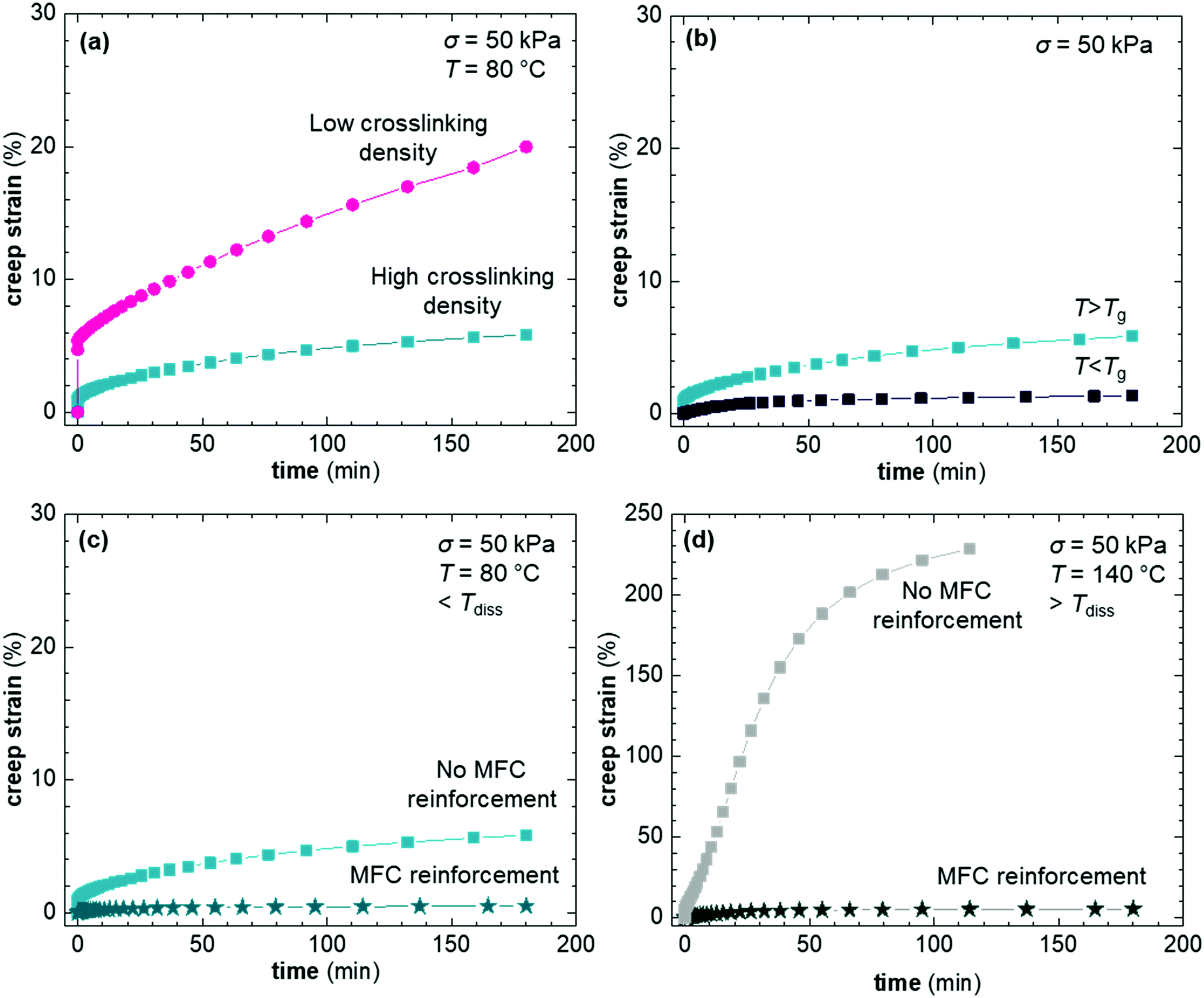 | ||
| Fig. 5 Creep strain as function of time for (a) XL10f and XL50f having different degree of crosslinking, (b) XL50f at temperatures above and below Tg, i.e. 80 and 40 °C, (c) XL50f with and without cellulose addition in the associated state and (d) XL50f with and without cellulose addition in the dissociated state. | ||
Seemingly, MFC arrests the flow of the macromolecular chains in the dissociated state. MFC is a known rheology modifier, increasing the viscosity of melts and suspensions.53 However, this effect alone is not sufficient to explain the complete arrestment of creep. Earlier publications have shown a strong reinforcing effect of nanocellulose networks in low modulus polymers54–56 and network formation has also been shown to effectively arrest creep at high tempeature.28 Numerous hydrogen bonds, as well as entanglements between the individual cellulose fibrils allow formation of a reinforcing network that can transmit mechanical stress, taking load from the dissociated DA network. The MFC fraction at which a continuous network of nanocellulose is predicted to form, i.e. the percolation threshold, is in the range of 3 wt%, based on the aspect ratio of the fibrils (cf. ESI† for calculation of the percolation threshold). The considerably higher MFC content of 10 wt% in our CAN based composites, in combination with solution casting of composites using DMF, is likely to give rise to the formation of a nanocellulose network. A second sample of XL50f reinforced with a lower amount of MFC, 5 wt%, showed a similar creep resistance as the sample containing 10 wt% MFC, where creep is almost completely arrested even at 140 °C and the final creep strain after 180 min is in the range of a few percent (Fig. S9, ESI†). MFC, hence, forms a network even at concentrations close to the theoretical percolation threshold, which indicates efficient dispersion of MFC in the matrix.
Further, the visual appearance of films, where aggregates cannot be spotted, as well as DMA temperature ramps where the rubbery plateau extends into the high temperature region both indicate that a MFC network has indeed formed. The presence of such a network strongly enhances the resistance to creep, especially of the dissociated CAN samples.
Thermal cycling
We examined repeated association and dissociation of the DA adduct of XL50f with and without MFC during 5 temperature cycles. We gathered spectroscopic information, following the intensity of the maleimide FTIR peak (ϑCH = 828 cm−1), and recorded the magnitude of E′ with DMA as a function of temperature (Fig. 6). During each cycle the samples were associated at 60 °C for 240 min and then dissociated for 15 min at 140 °C. | ||
| Fig. 6 Repeated thermal cycling of XL50f with and without MFC, (a) normalized IR absorbance of the maleimide peak of XL50f, (b) E′ of XL50f with and without MFC obtained from DMA, and (c) temperature program during cycling. | ||
We observe a strong response to the repeated temperature cycling. The normalized maleimide peak intensity for XL50f varies between 1 and 0.7 while E′ varies between 1–100 MPa and 20–300 MPa for XL50f and XL50f-MFC, respectively (Fig. 6b). It should be noted that the DMA data show a progressive slight increase in the modulus of the dissociated state for both samples. During every cycle E′ of dissociated XL50f increases with, on average, 5% while dissociated XL50f-MFC displays an average increase of 3%. We argue that a fraction of the crosslinks either do not have time to dissociate during the 140 °C isotherm or that a small fraction of permanent crosslinks is formed during temperature cycling. The spectroscopic analysis (Fig. 6a), however, suggests that the time and temperature for dissociation is sufficient, and that possible covalent bonds that may form do not involve the maleimide group. One plausible side reaction could be attributed to moisture absorption of the anhydrides during synthesis, resulting in the formation of carboxylic acids. These acids could form permanent crosslinks by reaction with secondary amides, resulting from ring opening of the imide. A small trace of secondary amides is visible in the 1HNMR spectra of 50f, while the same peak is not seen in p(E-alt-MA) and 10f (Fig. S10, ESI†). Aromatization of the DA adducts via dehydration is another possible side-reaction, which would yield permanents crosslinks. Aromatization in the bulk has been reported after curing DA adducts at temperatures above 180 °C for several hours.7,57 We argue that aromatization is not the most probable cause of the progressive increase in modulus because of the lower temperature and shorter duration of the thermal cycling steps.
Conclusions
We have observed a synergistic effect of two distinct networks, a DA type CAN and a MFC network, on the thermomechanical properties of the resulting composite material. The macromolecular DA type CAN was synthesized by furan functionalization of the copolymer p(E-alt-MA) and subsequent curing with a bismaleimide crosslinker. The design of the DA network allows dissolution in DMF, a solvent in which MFC can be dispersed, enabling the formation of a second network based on physical interactions between cellulose fibrils. The combined effect of the networks is an increase in storage modulus by a factor of 60 at 80 °C and a decrease in creep strain by a factor of 40 at the same temperature. Further, the MFC network adds mechanical stability to the composite at elevated temperatures, where the DA network dissociates. The presence of a nanocellulose network hence prevents macroscopic flow while, according to the kinetic evaluation, it does not significantly affect the rate of the DA reaction.Finally, the DA type network reinforced with MFC maintains its properties during at least 5 temperature cycles during which the reinforced CAN undergoes repeated association/dissociation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from the Swedish Research Council Formas (grant no. 2018-00886) and the Swedish Foundation for Strategic Research (grant no. GMT14-0036). We thank Anders Mårtensson for help with elemental analysis. We thank Dr Anna I. Hofmann and Dr Mariza Mone for expert help with photography and figure design, respectively.References
- W. Alabiso and S. Schlögl, The impact of vitrimers on the industry of the future: Chemistry, properties and sustainable forward-looking applications, Polymers, 2020, 12, 1660 CrossRef CAS PubMed.
- W. Denissen, J. M. Winne and F. E. Du Prez, Vitrimers: Permanent organic networks with glass-like fluidity, Chem. Sci., 2016, 7, 30–38 RSC.
- D. J. Fortman, J. P. Brutman, G. X. De Hoe, R. L. Snyder, W. R. Dichtel and M. A. Hillmyer, Approaches to sustainable and continually recyclable cross-linked polymers, ACS Sustainable Chem. Eng., 2018, 6, 11145–11159 CrossRef CAS.
- C. J. Kloxin and C. N. Bowman, Covalent adaptable networks: Smart, reconfigurable and responsive network systems, Chem. Soc. Rev., 2013, 42, 7161–7173 RSC.
- K. L. Jin, S. S. Kim, J. Xu, F. S. Bates and C. J. Ellison, Melt-blown cross-linked fibers from thermally reversible Diels-Alder polymer networks, ACS Macro Lett., 2018, 7, 1339–1345 CrossRef CAS.
- B. R. Elling and W. R. Dichtel, Reprocessable cross-linked polymer networks: are associative exchange mechanisms desirable?, ACS Cent. Sci., 2020, 6, 1488–1496 CrossRef CAS PubMed.
- A. Gandini, The furan/maleimide Diels–Alder reaction: A versatile click-unclick tool in macromolecular synthesis, Prog. Polym. Sci., 2013, 38, 1–29 CrossRef CAS.
- X. X. Chen, M. A. Dam, K. Ono, A. Mal, H. B. Shen, S. R. Nutt, K. Sheran and F. Wudl, A thermally re-mendable cross-linked polymeric material, Science, 2002, 295, 1698–1702 CrossRef CAS PubMed.
- A. Cuvellier, R. Verhelle, J. Brancart, B. Vanderborght, G. Van Assche and H. Rahier, The influence of stereochemistry on the reactivity of the Diels–Alder cycloaddition and the implications for reversible network polymerization, Polym. Chem., 2019, 10, 473–485 RSC.
- B. J. Adzima, H. A. Aguirre, C. J. Kloxin, T. F. Scott and C. N. Bowman, Rheological and chemical analysis of reverse gelation in a covalently cross-linked Diels-Alder polymer network, Macromolecules, 2008, 41, 9112–9117 CrossRef CAS PubMed.
- Y. L. Liu and T. W. Chuo, Self-healing polymers based on thermally reversible Diels–Alder chemistry, Polym. Chem., 2013, 4, 2194–2205 RSC.
- J. R. Davidson, G. A. Appuhamillage, C. M. Thompson, W. Voit and R. A. Smaldone, Design paradigm utilizing reversible Diels–Alder reactions to enhance the mechanical properties of 3D printed materials, ACS Appl. Mater. Interfaces, 2016, 8, 16961–16966 CrossRef CAS PubMed.
- L. Rulisek, P. Sebek, Z. Havlas, R. Hrabal, P. Capek and A. Svatos, An experimental and theoretical study of stereoselectivity of furan-maleic anhydride and furan-maleimide Diels–Alder reactions, J. Org. Chem., 2005, 70, 6295–6302 CrossRef CAS PubMed.
- Y. L. Fang, X. S. Du, Y. X. Jiang, Z. L. Du, P. T. Pan, X. Cheng and H. B. Wang, Thermal-driven self-healing and recyclable waterborne polyurethane films based on reversible covalent interaction, ACS Sustainable Chem. Eng., 2018, 6, 14490–14500 CrossRef CAS.
- C. Goussé, A. Gandini and P. Hodge, Application of the Diels-Alder reaction to polymers bearing furan moieties. 2. Diels–Alder and retro-Diels–Alder reactions involving furan rings in some styrene copolymers, Macromolecules, 1998, 31, 314–321 CrossRef.
- R. Gheneim, C. Perez-Berumen and A. Gandini, Diels–Alder reactions with novel polymeric dienes and dienophiles: Synthesis of reversibly cross-linked elastomers, Macromolecules, 2002, 35, 7246–7253 CrossRef CAS.
- P. Berto, A. Pointet, C. Le Coz, S. Grelier and F. Peruch, Recyclable telechelic cross-linked polybutadiene based on reversible Diels–Alder chemistry, Macromolecules, 2018, 51, 651–659 CrossRef CAS.
- L. M. Polgar, A. Kingma, M. Roelfs, M. van Essen, M. van Duin and F. Picchioni, Kinetics of cross-linking and de-cross-linking of EPM rubber with thermoreversible Diels–Alder chemistry, Eur. Polym. J., 2017, 90, 150–161 CrossRef CAS.
- S. A. Canary and M. P. Stevens, Thermally reversible cross-linking of polystyrene via the furan–maleimide Diels–Alder reaction, J. Polym. Sci., Part A: Polym. Chem., 1992, 30, 1755–1760 CrossRef CAS.
- J. M. Raquez, S. Vanderstappen, F. Meyer, P. Verge, M. Alexandre, J. M. Thomassin, C. Jerome and P. Dubois, Design of cross-linked semicrystalline poly(ε-caprolactone)-based networks with one-way and two-way shape-memory properties through Diels–Alder reactions, Chem. – Eur. J., 2011, 17, 10135–10143 CrossRef CAS PubMed.
- K. Inoue, M. Yamashiro and M. Iji, Recyclable shape-memory polymer: Poly(lactic acid) crosslinked by a thermoreversible Diels–Alder reaction, J. Appl. Polym. Sci., 2009, 112, 876–885 CrossRef CAS.
- L. Hammer, N. J. Van Zee and R. Nicolay, Dually crosslinked polymer networks incorporating dynamic covalent bonds, Polymers, 2021, 13, 396 CrossRef CAS PubMed.
- J. J. Cash, T. Kubo, D. J. Dobbins and B. S. Sumerlin, Maximizing the symbiosis of static and dynamic bonds in self-healing boronic ester networks, Polym. Chem., 2018, 9, 2011–2020 RSC.
- L. Q. Li, X. Chen, K. L. Jin and J. M. Torkelson, Vitrimers designed both to strongly suppress creep and to recover original cross-link density after reprocessing: Quantitative theory and experiments, Macromolecules, 2018, 51, 5537–5546 CrossRef CAS.
- S. Wang, S. Q. Ma, O. Li, X. W. Xu, B. B. Wang, K. F. Huang, Y. L. Liu and J. Zhu, Facile preparation of polyimine vitrimers with enhanced creep resistance and thermal and mechanical properties via metal coordination, Macromolecules, 2020, 53, 2919–2931 CrossRef CAS.
- Y. J. Liu, Z. H. Tang, D. Wang, S. W. Wu and B. C. Guo, Biomimetic design of elastomeric vitrimers with unparalleled mechanical properties, improved creep resistance and retained malleability by metal-ligand coordination, J. Mater. Chem. A, 2019, 7, 26867–26876 RSC.
- S. J. Eichhorn, A. Dufresne, M. Aranguren, N. E. Marcovich, J. R. Capadona, S. J. Rowan, C. Weder, W. Thielemans, M. Roman, S. Renneckar, W. Gindl, S. Veigel, J. Keckes, H. Yano, K. Abe, M. Nogi, A. N. Nakagaito, A. Mangalam, J. Simonsen, A. S. Benight, A. Bismarck, L. A. Berglund and T. Peijs, Review: Current international research into cellulose nanofibres and nanocomposites, J. Mater. Sci., 2010, 45, 1–33 CrossRef CAS.
- A. Peterson, I. Östergren, A. Lotsari, A. Venkatesh, J. Thunberg, A. Ström, R. Rojas, M. Andersson, L. A. Berglund, A. Boldizar and C. Müller, Dynamic nanocellulose networks for thermoset-like yet recyclable plastics with a high melt stiffness and creep resistance, Biomacromolecules, 2019, 20, 3924–3932 CrossRef CAS PubMed.
- W. Zhao, Z. H. Feng, Z. H. Liang, Y. F. Lv, F. K. Xiang, C. Y. Xiong, C. Duan, L. Dai and Y. H. Ni, Vitrimer-cellulose paper composites: A new class of strong, smart, green, and sustainable materials, ACS Appl. Mater. Interfaces, 2019, 11, 36090–36099 CrossRef CAS PubMed.
- F. Lossada, J. Q. Guo, D. J. Jiao, S. Groeer, E. Bourgeat-Lami, D. Montarnal and A. Walther, Vitrimer chemistry meets cellulose nanofibrils: Bioinspired nanopapers with high water resistance and strong adhesion, Biomacromolecules, 2019, 20, 1045–1055 CrossRef CAS PubMed.
- S. Sain, D. Åkesson, M. Skrifvars and S. Roy, Hydrophobic shape-memory biocomposites from tung-oil-based bioresin and onion-skin-derived nanocellulose networks, Polymers, 2020, 12, 2470 CrossRef CAS PubMed.
- D. A. Kissounko, P. Taynton and C. Kaffer, New material: Vitrimers promise to impact composites, Reinf. Plast., 2018, 62, 162–166 CrossRef.
- C. H. Xu, Z. J. Zheng, W. C. Wu, L. H. Fu and B. F. Lin, Design of healable epoxy composite based on beta-hydroxyl esters crosslinked networks by using carboxylated cellulose nanocrystals as crosslinker, Compos. Sci. Technol., 2019, 181, 107677 CrossRef CAS.
- E. Trovatti, A. G. Cunha, A. J. F. Carvalho and A. Gandini, Furan-modified natural rubber: A substrate for its reversible crosslinking and for clicking it onto nanocellulose, Int. J. Biol. Macromol., 2017, 95, 762–768 CrossRef CAS PubMed.
- E. Cudjoe, K. M. Herbert and S. J. Rowan, Strong, rebondable, dynamic cross-linked cellulose nanocrystal polymer nanocomposite adhesives, ACS Appl. Mater. Interfaces, 2018, 10, 30723–30731 CrossRef CAS PubMed.
- J. L. Swartz, R. L. Li and W. R. Dichtel, Incorporating functionalized cellulose to increase the toughness of covalent adaptable networks, ACS Appl. Mater. Interfaces, 2020, 12, 44110–44116 CrossRef CAS PubMed.
- Y. Ouyang, M. Mauri, A. M. Pourrahimi, I. Östergren, A. Lund, T. Gkourmpis, O. Prieto, X. Xu, P.-O. Hagstrand and C. Müller, Recyclable polyethylene insulation via reactive compounding with a maleic anhydride-grafted polypropylene, ACS Appl. Polym. Mater, 2020, 2, 2389–2396 CrossRef CAS.
- R. Araya-Hermosilla, G. M. R. Lima, P. Raffa, G. Fortunato, A. Pucci, M. E. Flores, I. Moreno-Villoslada, A. A. Broekhuis and F. Picchioni, Intrinsic self-healing thermoset through covalent and hydrogen bonding interactions, Eur. Polym. J., 2016, 81, 186–197 CrossRef CAS.
- A. Gandini, D. Coelho and A. J. D. Silvestre, Reversible click chemistry at the service of macromolecular materials. Part 1: Kinetics of the Diels–Alder reaction applied to furan-maleimide model compounds and linear polymerizations, Eur. Polym. J., 2008, 44, 4029–4036 CrossRef CAS.
- Y. Zhu, J. Hu, H. Luo, R. J. Young, L. Deng, S. Zhang, Y. Fan and G. Ye, Rapidly switchable water-sensitive shape-memory cellulose/elastomer nano-composites, Soft Matter, 2012, 8, 2509–2517 RSC.
- J. Sapkota, S. Kumar, C. Weder and J. E. Foster, Influence of processing conditions on properties of poly(vinyl acetate)/cellulose nanocrystal nanocomposites, Macromol. Mater. Eng., 2015, 300, 562–571 CrossRef CAS.
- J. Aizpurua, L. Martin, E. Formoso, A. Gonzalez and L. Irusta, One pot stimuli-responsive linear waterborne polyurethanes via Diels–Alder reaction, Prog. Org. Coat., 2019, 130, 31–43 CrossRef CAS.
- E. Dolci, G. Michaud, F. Simon, B. Boutevin, S. Fouquay and S. Caillol, Remendable thermosetting polymers for isocyanate-free adhesives: A preliminary study, Polym. Chem., 2015, 6, 7851–7861 RSC.
- J. Aizpurua, L. Martin, M. Fernández, A. González and L. Irusta, Recyclable, remendable and healing polyurethane/acrylic coatings from UV curable waterborne dispersions containing Diels–Alder moieties, Prog. Org. Coat., 2020, 139 Search PubMed.
- X. Z. Xu, F. Liu, L. Jiang, J. Y. Zhu, D. Haagenson and D. P. Wiesenborn, Cellulose nanocrystals vs. cellulose nanofibrils: A comparative study on their microstructures and effects as polymer reinforcing agents, ACS Appl. Mater. Interfaces, 2013, 5, 2999–3009 CrossRef CAS PubMed.
- X. X. Liu, P. F. Du, L. Liu, Z. Zheng, X. L. Wang, T. Joncheray and Y. F. Zhang, Kinetic study of Diels–Alder reaction involving in maleimide-furan compounds and linear polyurethane, Polym. Bull., 2013, 70, 2319–2335 CrossRef CAS.
- E. Goiti, F. Heatley, M. B. Huglin and J. M. Rego, Kinetic aspects of the Diels–Alder reaction between poly(styrene-co-furfuryl methacrylate) and bismaleimide, Eur. Polym. J., 2004, 40, 1451–1460 CrossRef CAS.
- Y. L. Liu, C. Y. Hsieh and Y. W. Chen, Thermally reversible cross-linked polyamides and thermo-responsive gels by means of Diels–Alder reaction, Polymer, 2006, 47, 2581–2586 CrossRef CAS.
- Y. L. Liu and C. Y. Hsieh, Crosslinked epoxy materials exhibiting thermal remendablility and removability from multifunctional maleimide and furan compounds, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 905–913 CrossRef CAS.
- A. Smedberg, T. Hjertberg and B. Gustafsson, The role of entanglements in network formation in unsaturated low density polyethylene, Polymer, 2004, 45, 4867–4875 CrossRef CAS.
- J. P. Flory, Networks, in Encyclopedia of polymer science and engineering, ed. H. F. B. N. M. Mark, C. G. Overberger and G. Menges, Wiley, New York, 1987, vol. 10, pp. 95–112 Search PubMed.
- M. Mauri, A. Peterson, A. Senol, K. Elamin, A. Gitsas, T. Hjertberg, A. Matic, T. Gkourmpis, O. Prieto and C. Müller, Byproduct-free curing of a highly insulating polyethylene copolymer blend: An alternative to peroxide crosslinking, J. Mater. Chem. C, 2018, 6, 11292–11302 RSC.
- D. Klemm, F. Kramer, S. Moritz, T. Lindström, M. Ankerfors, D. Gray and A. Dorris, Nanocelluloses: A new family of nature-based materials, Angew. Chem., Int. Ed., 2011, 50, 5438–5466 CrossRef CAS PubMed.
- V. Favier, G. R. Canova, J. Y. Cavaille, H. Chanzy, A. Dufresne and C. Gauthier, Nanocomposite materials from latex and cellulose whiskers, Polym. Adv. Technol., 1995, 6, 351–355 CrossRef CAS.
- P. K. Annamalai, K. L. Dagnon, S. Monemian, E. J. Foster, S. J. Rowan and C. Weder, Water-responsive mechanically adaptive nanocomposites based on styrene-butadiene rubber and cellulose nanocrystals: Processing matters, ACS Appl. Mater. Interfaces, 2014, 6, 967–976 CrossRef CAS PubMed.
- J. R. Capadona, K. Shanmuganathan, D. J. Tyler, S. J. Rowan and C. Weder, Stimuli-responsive polymer nanocomposites inspired by the sea cucumber dermis, Science, 2008, 319, 1370–1374 CrossRef CAS PubMed.
- C. D. Diakoumakos and J. A. Mikroyannidis, Polyimides derived from Diels–Alder polymerization of furfuryl-substituted maleamic acids or from the reaction of bismaleamic with bisfurfurylpyromellitamic acids, J. Polym. Sci., Part A: Polym. Chem., 1992, 30, 2559–2567 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00291k |
| This journal is © The Royal Society of Chemistry 2021 |