
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSilver quasi-nanoparticles: bridging the gap between molecule-like clusters and plasmonic nanoparticles†
Fatima
Douma
a,
Louwanda
Lakiss
 a,
Oleg I.
Lebedev
b,
Julien
Cardin
c,
Krassimir L.
Kostov
d,
Jaafar
El Fallah
a,
Valentin
Valtchev
a and
Mohamad
El-Roz
*a
a,
Oleg I.
Lebedev
b,
Julien
Cardin
c,
Krassimir L.
Kostov
d,
Jaafar
El Fallah
a,
Valentin
Valtchev
a and
Mohamad
El-Roz
*a
aNormandie Université, ENSICAEN, UNICAEN, CNRS, Laboratoire Catalyse et Spectrochimie, 14050 Caen, France. E-mail: mohamad.elroz@ensicaen.fr
bLaboratoire CRISMAT, ENSICAEN, CNRS UMR 6508, 14050 Caen, France
cLaboratoire CIMAP, Normandie Université, ENSICAEN, UNICAEN, CNRS UMR 6252, CEA, 14000 Caen, France
dBulgarian Academy of Sciences, Institute of General and Inorganic Chemistry, 1113 Sofia, Bulgaria
First published on 30th June 2021
Abstract
Herein, we report a new strategy for preparing connected silver sub-nanoparticles with unique optical behavior via the selective photo-assisted electrochemical reduction of silver cations in FAU-type zeolite X (FAUX) cages. Bi2+/Bi3+-doped zeolite nanoparticles (ZX-Bi) were prepared by one-pot hydrothermal synthesis and stabilized as a colloidal water suspension. A ZX-Bi suspension, containing silver nitrate, was subjected to UV irradiation resulting in the reduction of silver cations and the generation of Agn&+ (with n > &) clusters (Ag@ZX-Bi). The physicochemical characterization of the samples, using XRD, TG, N2 sorption, NMR, HRTEM-STEM, ICP, EDX and XPS analyses, provided comprehensive information on the textural and structural properties, the chemical compositions and the metal oxidation states of the samples. Their optical behavior was investigated using UV-visible and photoluminescence spectroscopies. The IR-operando analysis under visible-light revealed local heating of Ag@ZX-Bi up to 400 K. Theoretical calculation of the absorption, scattering, and extinction cross-sections, σabs, σsca and σext, respectively, of the different silver models prepared in this study was in agreement with the experimental data, elucidating the unique optical behavior of the silver particles. The set of analyses shows that quasi-nanoparticles of Ag are formed from bridged Ag clusters (AgCLs) through zeolite channels closing the gap between clusters and plasmonic nanoparticles for the first time.
1 Introduction
Downsizing the dimension of metallic particles to the nanometer, sub-nanometer and quasi-atomic level provides a new approach for achieving novel structural, electronic and optical properties. One of the consequences of reducing the size of metallic particles to less than the Fermi wavelength is their molecule-like behavior with the intralevel energy exceeding the thermal activation energy (kBT), which gives rise to a size-dependent HOMO–LUMO band gap comparable to that of semiconductors. Thus, the layer-dependent transitions of silver clusters render them optically active emissive centers.1,2 However, the interaction between the incident light, at a specific wavelength, and metal nanoparticles (MNPs) could induce collective oscillation of the conduction electrons, producing local surface plasmonic resonance (LSPR) with no direct luminescence properties.3 This behavior makes them very attractive for various applications in solar-cells,4 imaging,5 biomedecine,6–8 (bio)sensing,9,10etc.Several recent research studies have aimed at preparing M-NPs and metal clusters (M-CLs) with an ordered shape and size.1–10 They focused on tuning the metal clusters’ physicochemical properties (size, morphology, and charge) to improve their performance. The crucial key to reaching this objective is to control the self-assembly process of the metal atomsand to stabilize their nucleation at a distinct stage.
In the case of M-CLs, one of the main challenges is their stability, as they are susceptible to agglomerate into larger particles due to their high surface energy. Typically, organic stabilizing agents (micelle formation with M-NP cores) or porous host supports are used to ensure their stability.11,12 Several recent works have shown that the use of zeolite as a host environment is one of the most convenient strategies for promoting the formation of well-defined M-CLs. For instance, embedding a few silver atoms and clusters in the well-defined dielectric cavities of a zeolite reveals novel optical properties with respect to plasmonic silver nanoparticles (Ag-NPs). This behavior is dependent on multiple factors, including the size, geometry, degree of hydration, and the electrostatic interaction between the metal and the local host environment.13,14
Silver clusters (Ag-CLs) in zeolite pores are usually prepared by thermal treatment of a zeolite exchanged with silver cations at a relatively high temperature (>450 °C). The optical properties of the final materials depend on the framework topology and charge density of the zeolite, its counter-ions, the degree of silver exchange, and its hydration state.15,16 However, a very low concentration of silver is required in order to minimize the migration of the silver clusters and their agglomeration into large particles. Therefore, only a few isolated clusters can be obtained with interesting emission behavior in visible light.
Recently, we demonstrated that Ag-CLs can be prepared under soft irradiation conditions at RT.17 We showed that UV irradiation of a suspension of faujasite X (FAUX) doped with vanadium oxide clusters (ZX-V) promotes a local reduction of silver nitrate into Ag-CLs, via a photocatalytic-like process in the presence of ethanol as an electron donor. Due to the limited loading of the highly dispersed vanadium oxide clusters in the zeolite, only isolated Ag-CLs and some external Ag-NPs were obtained.
Herein, we report an innovative and selective strategy for preparing Ag-CLs in FAUX doped with Bi2+/Bi3+ cations via the local photo-assisted electrochemical reduction of Ag cations. The successful preparation of highly crystalline zeolites with abundant Bi active sites and their subsequent localization in sodalite and double six-membered ring (6MR) prisms offered a unique opportunity for bridging the reduced silver clusters through the zeolite channel instead of the isolated clusters obtained previously. Contrary to our previous photocatalytic process, the new process is a photo-assisted electrochemical approach that can even be performed in the dark. With this new approach, the optical behavior of Ag-CLs can be easily tuned by adjusting the amount of Bi without changing the host environment.
2 Experimental
Materials
The following chemical products were purchased and used without further purification: Al powder (325 mesh, 99.5%, Alfa Aesar), NaOH (VWR Chemicals-Prolabo, 99%), SiO2 (Ludox-AS 30, 30 wt% SiO2, pH = 9.8, Aldrich), and Bi(NO3)3·5H2O (99.9%, Aldrich), Ag(NO3) (99.9%, Aldrich).The Ag@ZX-Bi sample was prepared using the synthesis procedure that was previously reported in our recent work for the preparation of Ag@ZX-V.17 First, ZX-Bi is prepared by the in situ incorporation of bismuth as a metal precursor for photoactive species in the synthesis mixtures.
Synthesis of ZX-Bi
As a first step, the nanosized ZX-Bi-x samples were synthesized according to a sol–gel method without employing an organic template (x represents the weight percentage, which varies between 0.5 and 10 wt%, relative to the weight of alumina and silica). In a typical synthetic procedure, sodium aluminate and sodium silicate are prepared separately (denoted as solutions A and B, respectively). The sodium aluminate was prepared by adding ∼0.5 g of sodium hydroxide to 3 g of water in a plastic bottle. Then aluminum powder was slowly dispersed under vigorous stirring until it had completely dissolved. After that bismuth(III) nitrate pentahydrate (Bi(NO3)3·5H2O) was introduced into the solution, which was sonicated for 30 min to completely dissolve the Bi(NO3)3. Sodium silicate was prepared concurrently by adding 1 g of NaOH to 1 g of water and 10 g of a colloidal silica suspension at room temperature. The obtained sol was heated for 7 min at 90 °C until a clear sol was formed and kept under constant stirring. Solution A was added dropwise into solution B under intensive stirring after placing the latter in an ice bath in order to control the exothermicity of the reaction. The formed sol–gel mixture was aged for 14–24 h at room temperature. The water content was adjusted using a freeze dryer to avoid aggregation. The solution was then crystallized at 50 °C for 24 h. The molar composition of the clear gel is given as: 10SiO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.1Al2O3:xBi:9Na2O:122H2O (x = 0.066, 0.20, 0.40, and 0.66 atm% equivalent to 0.5, 1.5, 3.0 and 5.0 wt% of Bi with respect to (Si + Al), respectively). Then, very stable suspensions were obtained. The solid was recovered by centrifugation (25 min, 20000 rpm) followed by re-dispersion in double-distilled water. The procedure was repeated until a pH of ∼8 was reached. It has been observed that the crystallinity and the purity of ZX-Bi are strongly affected by the amount of bismuth precursor added to the initial synthesis mixture. In order to obtain a highly crystalline and pure ZX-Bi sample, the synthesis parameters such as the aging and crystallization times, the amount of water eliminated by freeze-drying and the Bi content were optimized. The optimization processes and the results are detailed in the ESI.† To estimate the crystallinity of the zeolite samples, a Bi-free high ZX sample is used as the reference. Then the crystallinity of the as-synthesized samples was estimated by comparing the integrated area of the reflection peaks of ZX-Bi relative to their corresponding peaks in the pure ZX.
:1.1Al2O3:xBi:9Na2O:122H2O (x = 0.066, 0.20, 0.40, and 0.66 atm% equivalent to 0.5, 1.5, 3.0 and 5.0 wt% of Bi with respect to (Si + Al), respectively). Then, very stable suspensions were obtained. The solid was recovered by centrifugation (25 min, 20000 rpm) followed by re-dispersion in double-distilled water. The procedure was repeated until a pH of ∼8 was reached. It has been observed that the crystallinity and the purity of ZX-Bi are strongly affected by the amount of bismuth precursor added to the initial synthesis mixture. In order to obtain a highly crystalline and pure ZX-Bi sample, the synthesis parameters such as the aging and crystallization times, the amount of water eliminated by freeze-drying and the Bi content were optimized. The optimization processes and the results are detailed in the ESI.† To estimate the crystallinity of the zeolite samples, a Bi-free high ZX sample is used as the reference. Then the crystallinity of the as-synthesized samples was estimated by comparing the integrated area of the reflection peaks of ZX-Bi relative to their corresponding peaks in the pure ZX.
Preparation of Ag@ZX-Bi
Zeolite samples with different contents of Bi were prepared to investigate the effect of the Bi content on the evolution of silver species formation. Ag@ZX-Bi samples were prepared by dispersing ZX-Bi (2.4 g l−1) in pure water or in aqueous solutions containing ethanol and silver nitrate (10 mM) with a ratio of ethanol/water = 1/3. Upon UV irradiation (Hg–Xe lamp; polychromatic irradiance = 205 mW cm−2; infrared cut filter), the color of the suspension changed from white to pink, confirming the reduction of silver Agnn+ to Agnδ+ clusters (with δ < n). The optical penetration depth of zeolite-based structures was evaluated, and indicated that UV radiation is transmitted at least partially through the walls of these structures. Thanks to the known refractive indices of the zeolite material's constituents (H2O, SiO2, and Al2O3), we estimate that the optical penetration depth, defined as the depth at which the intensity of the radiation inside the material falls to 1/e at different UV wavelengths ranging from 240 to 365 nm. The penetration depths were found to range from 7.8 nm in pure Al2O3 to 6 m in pure water at 365 nm. These extreme values frame the depth of penetration into the walls of the zeolite-based structures. In view of the dimensions of the zeolite-based structures, it can therefore be concluded that UV radiation is transmitted at least partially through the walls of these structures. In addition, the concentration of the zeolite suspension (2 mg ml−1) and the optical pathway (10–20 cm) were optimized (based on our previous study) in order to obtain highly reproducible results (many grams of Ag@ZXBi-3 were prepared, corresponding to the treatment of tens of samples).Characterization techniques
UV-vis spectroscopy measurements were performed using a Cary 4000 UV-vis spectrophotometer at room temperature in transmission mode using a quartz cuvette with a 1 cm path length. The absorbance spectra were corrected by subtracting the spectrum of pure zeolite (ZX). The DR-UV-vis spectra of the dried powder samples were recorded using a RSA-CU40 diffuse reflectance cell. The spectrum of the non-irradiated sample was subtracted from the spectra of the irradiated samples. The chemical composition of the samples was measured using a Varian Vista AX CCD inductively coupled plasma atomic emission spectrometer (ICP-AES).Continuous wave (CW) photoluminescence (PL) measurements were carried out at room temperature using two excitation lasers: a Crylas FQCW266 emitting at 266 nm with an average power of 27 mW, and a coherent Innova 90C argon laser with an average power of 150 mW at 488 nm. The excitation was at an incident angle of 45° on a beam spot size of about 1 mm2 and chopped at 3.00 Hz for samples. Emitted photons were collected by means of a set of lenses using a Horiba Jobin-Yvon Triax 180 monochromator. At the exit of this monochromator, an R5108 Hamamatsu photomultiplier tube ensured photon detection. This detected signal was amplified by means of a Femto DLPCA-200 trans-impedance amplifier and sent to an SR830 lock-in amplifier referenced at the excitation laser chopper frequency. Different high pass filter cutting at 500 nm or 700 nm was used, respectively, to prevent collection of the excitation line by the detection system and to prevent eventual second-order contributions. The PL spectra were measured on sample powder stacked between two quartz Suprasil (TM) glass slides from Hellma Analytics.
The water content and stability of the samples were investigated by thermogravimetric analysis (TGA) using a SETSYS analyzer instrument (SETARAM) (at a heating rate of 5 °C min−1 under a 40 ml min−1 flow of air).
The structural and morphological features of the samples were investigated by transmission electron microscopy (TEM) using an aberration probe and image corrected JEM ARM200F cold FEG microscope operated at 200 kV equipped with a CENTURIO EDX detector and GIF Quantum spectrometer. Specimens for transmission electron microscopy studies were dispersed in ethanol and drop cast onto a Cu holey carbon grid.
The crystallinity and phase purity of zeolite samples were studied by powder X-ray diffraction analysis (PXRD) using a PANalytical X'Pert Pro diffractometer with CuKα wavelength (λ = 1.5418 Å). The PXRD diagrams were recorded at room temperature between 3 and 50° (2θ) with a step size of ∼0.014° (2θ). Variable divergent slits with a constant illuminated sample length of 6 mm were used. Phase identification was performed using the PANalytical HighScore plus program. Nitrogen sorption analyses were performed using Model ASAP 2020 apparatus. After degassing at 200 °C under vacuum for 12 h, the samples were cooled, and nitrogen adsorption was performed. Specific surface areas were determined from the BET equation. The total pore volume is the total volume adsorbed at P/P0 = 0.99. The t-plot method was used to distinguish the micropores from the mesopores in the samples. The pore size distribution (PSD) was derived from the desorption branch of the N2 isotherm using the Barrett–Joyner–Halenda (BJH) algorithm.
Aluminum MAS NMR experiments were performed using a 500 MHz Bruker AVANCE III spectrometer operating at 130.32 MHz for 27Al, and equipped with a double-resonance 4 mm probe head.
X-Ray photoelectron spectroscopy (XPS) measurements were carried out using an AXIS Supra electron spectrometer (Kratos Analyical Ltd) with a base vacuum in the analysis chamber of the order of 10−8 Pa. The samples were irradiated with monochromatized Al Kα radiation with a photon energy of 1486.6 eV. The photoemitted electrons were separated, according to their kinetic energy, by 180° – a hemispherical analyzer with a total instrumental resolution of 0.6 eV (as measured by the FWHM of Ag 3d5/2 line) at a pass energy of 20 eV. Due to the charging effect, a resolution of ∼1.0 eV was measured on the selected samples. Energy calibration was performed by normalizing the C 1s line of adventitiously adsorbed hydrocarbons on silver folio 284.6 eV. The diameter of the analysis area was 700 × 300 μm2.
Operando IR experiments were performed using a ‘sandwich like’ IR cell-reactor.18 The FITR spectra of the samples (shaped as a pellet of ∼20 mg with 65 μm of thickness) were recorded under Ar and at different temperatures. The employed ThermoNicolet NEXUS 670 FTIR instrument was equipped with an MCT detector with a spectral resolution of 4 cm−1 and a time resolution of 1 spectrum per s. The irradiation of the samples with visible light was performed using a Xe-lamp (LC8 Hamamatsu, 200 W) equipped with a cut-UV filter (λ > 390 nm).
For CO-adsorption, the sample, activated in an in situ IR cell, was first cooled to −173 °C under vacuum. Then, small doses of 0.5–10 μmol of CO were introduced into the cell. IR spectra were recorded after each dose. IR spectra were recorded using a Nicolet Magna 550-FT-IR spectrometer at 4 cm−1 optical resolution. Prior to the measurements, the samples were pressed into self-supporting discs (diameter: 1.6 cm, 20 mg) and pretreated in the IR cell attached to a vacuum line (10−6 torr), at 100 °C for 18 h.
The boundary element method (BEM) approach presented by F. J. Garcia de Abajo et al.19 and developed by U. Hohenester et al.20 in the MNPBEM MatLab toolbox was used in order to model the far-field response of singular silver objects. Silver nanostructure models were built based on the TEM data, which help to localize and determine the particle sizes of the Ag clusters. Then, the absorption, scattering, and extinction cross-sections, σabs, σsca, and σext, respectively, of the structure models were calculated. The silver nanostructures in the faujasite structure were reproduced by the alternation of large-diameter spherical silver nanoparticles (LNPs; 1.1 nm) for the sodalite cages, and small diameter silver nanoparticles (SNPs; 0.7 nm) for the hexagonal prisms. The effect of NP interconnection was investigated by comparing the response of the silver nanostructure with isolated and interconnected nanoparticles. The silver nanostructures were also simulated with an increasing number of faujasite unit cells of 1, 2, 5 and 10 to reproduce the different structures with various lengths observed by TEM. The surrounding medium was considered as water with a constant permittivity (ε = 1.332) and the silver permittivity was taken from ref. 21. It should be underlined that the simulations simplified our experimental system. For example, the global charge of the clusters, the presence of Bi3+, the zeolite–Ag interaction, and the silver NP size may change the silver permittivity with respect to that of bulk silver in the standard. Moreover, supplementary orientation effects and overlapping of many objects (silver nanostructures) may complicate the plasmon response. Although this simulation will not give a response strictly identical to that observed in complex experimental systems, its contribution is representative and significant for the interpretation.
3 Results and discussion
Highly crystalline ZX-Bi samples were obtained after extensive optimization of the synthesis parameters. It should be noted that ZX-V is used in this work as a reference. Details about the ZX-V sample can be found in ref. 17 and the ESI.† The XRD results demonstrate that the presence of Bi in the synthesis medium significantly affects the crystal-growth kinetics of zeolite X (Fig. 1 and Fig. S1–S3, ESI†). We attribute this effect to the additional charge. Thus, the presence of bismuth reduces the crystallization time from 24 h to 8 h, compared with the Bi-free sample (ZX) prepared under similar synthesis conditions. The maximal amount of bismuth introduced in the synthesis medium without perturbing the crystallization processes is 3.0 wt% (with respect to the amounts of Si and Al) (Fig. 1). For simplification, the “x” value in the ZX-Bi-x samples corresponds to the amount of Bi initially introduced. For a higher bismuth concentration (>5 wt% Bi), the crystallization of faujasite is altered, and sodalite arises as the major crystalline product (Table S1 and Fig. S1–S3, ESI†). Thus, at larger amounts the Bi perturbs the structure-directing role of Na+ and consequently the final zeolite product.22 The XRD pattern of ZX-Bi-3 did not show any evidence of the presence of crystalline Bi oxide (Fig. 1). The broadening of the XRD peaks is due to the nanosized dimensions of the zeolite particles, as shown by DLS analysis. The hydrodynamic diameters of ZX and ZX-Bi particles are 45 and 50 nm, respectively (Fig. S4, ESI†). Hence, ZX-Bi shows similar crystallinity of the parent zeolite ZX with no extra-framework alumina detected (Fig. S5, ESI†), when it is prepared under the optimal conditions cited above. | ||
| Fig. 1 (A) X-Ray diffraction patterns of ZX-Bi samples prepared with different bismuth contents: (a) 0 wt% (ZX or ZX-Bi-0), (b) 0.5 wt% (ZX-Bi-0.5), (c) 1.5 wt% (ZX-Bi-1.5), (d) 3.0 wt% (ZX-Bi-3), and (e) 5.0 wt% (ZX-Bi-5). Samples crystallized at 50 °C for 8 h after aging for 14 h at RT. The weight percentage of bismuth corresponds to the initial quantity added and calculated by considering the quantity of Al and Si. (B) XRD patterns of (a) pure ZX and (b) ZX-Bi-3 crystallized for 24 h and 8 h, respectively. Crystallization temperature: 50 °C; after aging for 24 h and 14 h at RT, respectively. | ||
According to the ICP analysis, the bismuth amounts in the ZX-Bi-0.5, ZX-Bi-1.5 and ZX-Bi-3 samples are 0.38, 1.15 and 2.3 wt%, respectively (equivalent to 0.18, 0.33 and 0.74 atm%). The Si/Al ratio was in the range of 1.3–1.5 in all samples, in agreement with the EDX results (Table S2, ESI†). The loss of Bi during synthesis is relatively low (around 25%) with respect to that previously observed when a vanadium precursor was used (around 90%).17 This is probably due to the counter-ion role that Bin+ plays for compensating the negative charge of the oxygen lattice.
The textural properties of ZX-Bi-3 compared with the reference ZX sample are assembled in Fig. 2. The N2 adsorption data analysis of ZX and the ZX-Bi-3 sample (Fig. 2A) shows a combination of type I and type IV isotherms with a large hysteresis H1 type loop. The latter corresponds to a material with textural porosity originating from packed nanoparticles that are uniform in size.23 The slightly lower micropore volume of ZX-Bi compared with pure ZX (Fig. 2B) might be due to the slightly lower crystallinity of the sample (95% with respect to the pure ZX). It should be noted that the pore size distribution for ZX and ZX-Bi-3 (Fig. 2B) in the mesopore range is identical, indicating a similar particle size and regular textural porosity. Thermal analysis of the ZX sample (Fig. 2C) shows a water weight loss with two endothermic peaks at around 80 °C and 140 °C, which correspond to the dehydration of physisorbed and structural water, respectively. However, the elimination of water in the ZX-Bi sample takes place at a relatively higher temperature (125 °C and 200 °C). This could be related to the higher water affinity of multivalent Bi cations.
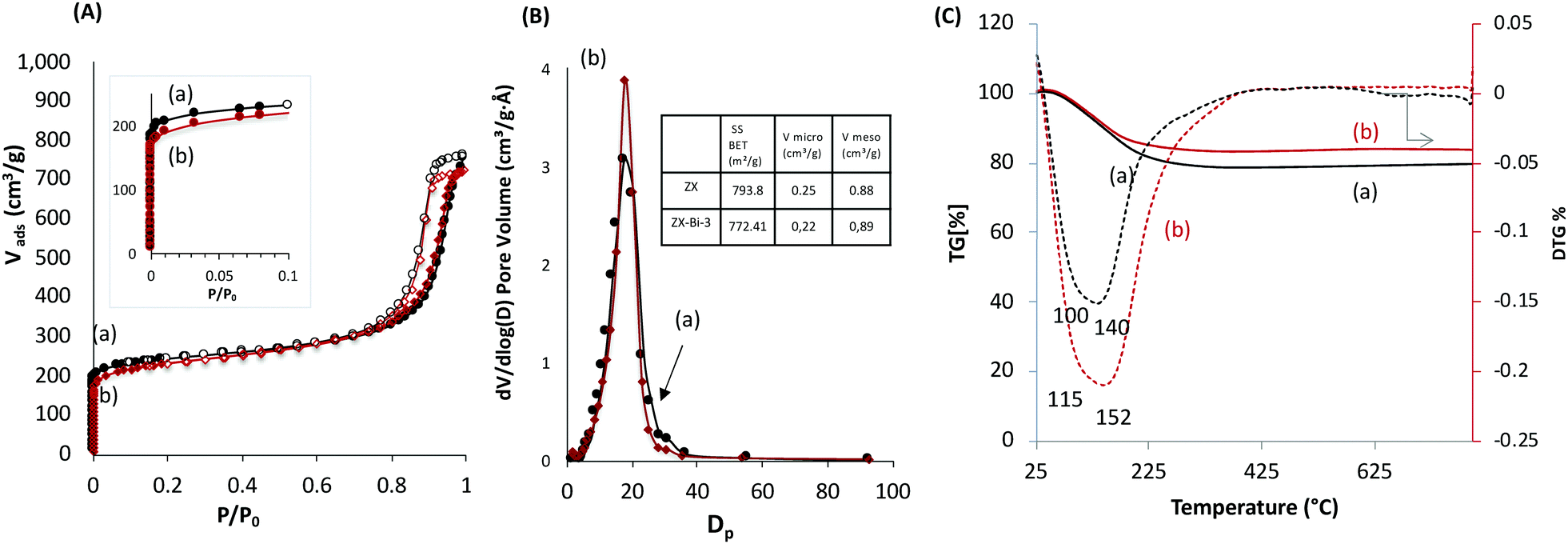 | ||
| Fig. 2 (A) Nitrogen adsorption and desorption isotherms, (B) mesopore size distribution, and (C) TG (solid lines) and DTG (dotted lines) curves of (a) ZX and (b) ZX-Bi-3 samples. The surface area and micropore volume values of both samples are reported in the table in inset of (B). | ||
The electron microscopy study (Fig. 3 and Fig. S6, ESI†) confirmed the presence of highly dispersed Bi in the zeolite. It is challenging to localize the Bi particle with precision in the zeolite structure based on the TEM analysis, which is due to the low Bi concentration and high mobility under the electron-beam. In this respect, HAADF-STEM is considered a more appropriate technique due to its atomic number dependence (∼Z2) and sensitivity to a small amount of material (down to single atoms). The HAADF-STM images reveal the presence of ordered Bi atoms along the [112] plane (Fig. 3a), which are mainly located in the sodalite cage and double six-membered ring (6MR) prism (Fig. 3b–d).
 | ||
| Fig. 3 HAADF-STEM analysis of the ZX-Bi sample: low magnification HAADF-STEM images of different ZX-Bi nanoparticles viewed along the two most informative directions: (a) the [112]FAU and (b) the [011]FAU zone axis; (c) and (d) correspond to the enlargement of the boxed zones in (b) and (c), respectively, overlaid with the corresponding structural model. The images reveal different Bi filling levels mainly in the sodalite cage and the 6MR prism: from high (white contrast) to almost empty (dark contrast). | ||
Ag@ZX-Bi-3 samples were prepared by UV irradiation of ZX-Bi-3 suspensions in the presence of AgNO3. Then the samples were washed with deionized water three times in order to remove the unreacted silver nitrate. XPS analysis performed on ZX-Bi-3 and Ag@ZX-Bi-3 samples shed more light on the nature of bismuth before and after Ag+ reduction. In the case of the parent sample (ZX-Bi), in addition to the main signal observed at 153.3 eV and attributed to silicon (Si 2s), the high-resolution XPS spectrum (Fig. 4) shows characteristic peaks of Bi3+ and Bi2+: (1) the Bi 4f core level spectrum with two spin–orbit doublet peaks (J = 5/2 and 7/2) centered at 165 (Bi 4f5/2) and 159.5 eV (Bi 4f7/2) with a peak separation of 5.5 eV, and (2) the pair of peaks at lower binding energies 163 and 157.3 eV with a peak separation of 5.7 eV, respectively.24–26
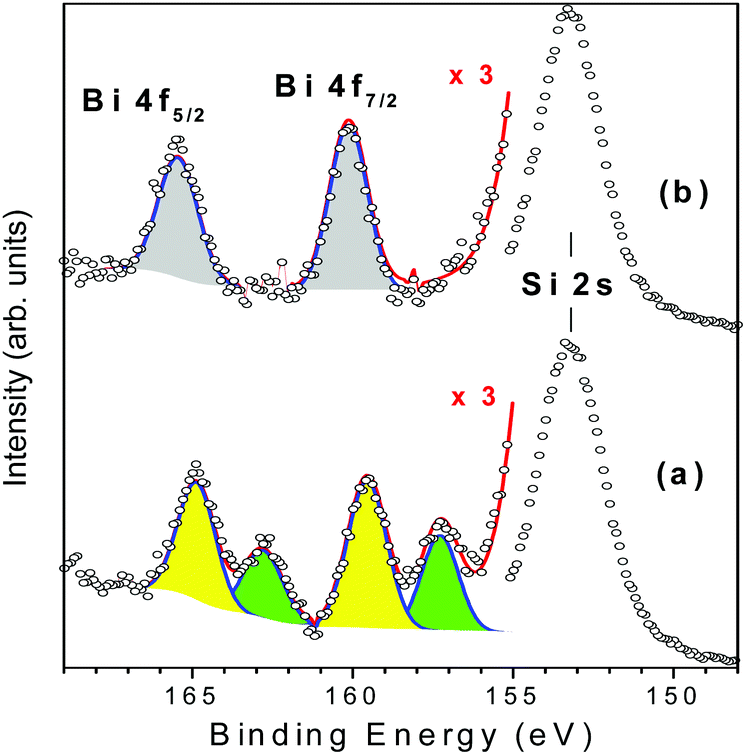 | ||
| Fig. 4 Gaussian deconvolutions of the XPS spectra of ZX-Bi-3 (a) and Ag@ZX-Bi-3 (b) samples. The red lines correspond to the intensity sum of the different peak contributions. | ||
The chemical composition obtained by XPS is very close to that obtained by ICP and EDX analyses, confirming the homogeneous distribution of bismuth cations. The unexpected detection of Bi2+ in the ZX-Bi-3 sample reveals the chemical reduction of the Bi3+ precursor during the synthesis process. The reduction of Bi3+ to its lower-valence bismuth ions usually occurs after thermal treatment at a relatively high temperature under anaerobic conditions.27 The low synthesis temperature (50 °C) excludes the thermal-reduction hypothesis, which is probably due to the in situ loading of Bi in the presence of metallic alumina in the synthesis, characterized with a very low standard potential (−1.67 V SHE, for Al3+/Al at 25 °C) with respect to Bi3+/Bi (+0.317 V SHE at 25 °C). The XPS analysis shows that Bi2+ and Bi3+ coexist with a Bi2+/Bi3+ ratio of 2.3. This result was confirmed on two different ZX-Bi-3 samples (with an error of below 10%). We anticipate two different positions of the Bi2+ and Bi3+ counter cations compensating the (AlOSi)− charges of the zeolite framework. Sun et al.28 demonstrated by high-resolution synchrotron XRD analysis of dehydrated zeolite that Bi+ with an ionic radius of 1.45 Å occupies a single-type site I′ in the sodalite cage of the FAU-type zeolite, where it is coordinated with three oxygen atoms of the 6MR prism. However, the characteristic ionic radii of Bi2+ and Bi3+ are 1.16 and 0.96 Å (vs. 1.0 Å for Na+), respectively.28
Considering the ionic radius, it is reasonable to presume that Bi2+ and Bi3+ ions could occupy the sodalite cage and the hexagonal prism of FAUX, respectively. However, the XPS results of Ag@ZX-Bi reveal the presence of Bi5+ with two characteristic peaks at 160.1 and 166.2 eV (Fig. 4b).29,30 This result indicates that a redox reaction occurs between the Bi2+/B3+ and Ag+. It is important to mention that a similar result was obtained in the absence of ethanol (used as an electron donor in the case of Ag@ZX-V; Fig. S7, ESI†). Also, similar results were recorded after 30 days in the dark (with a lower absorbance intensity), showing a different process of silver reduction in comparison with Ag@ZX-V (Fig. S8, ESI†). Hence, a photo-assisted redox process between Bi2+/Bi3+ and Ag+ is the most credible explanation supported by the selective detection of Bi5+on the Ag@ZX-Bi samples.
Fig. 5 shows the time progression of the Ag@ZX-Bi UV-visible spectrum versus the UV-irradiation time. A distinctive color change from milky white to magenta can be clearly observed (inset Fig. 5). The absorbance band positions indicate the presence of isolated Ag-CLs (Agnδ+) with different nuclearities (≠n) inside FAUX. These Ag-CLs have a characteristic absorbance in the UV region, similar to that observed previously for Ag@ZX-V samples (Fig. S8 and Table S3, ESI†). The broad absorption extended over the 266–310 nm region is usually assigned to the electronic transitions of few-atom-containing silver ionic clusters (Agnδ+), e.g., Ag3+, Ag42+, and ionic Ag8δ+.31,32 The shoulders around 295 and 360 nm are characteristic fingerprints of hydrated Ag42+.33 A distorted symmetry due to the interaction with water molecules explains the Ag42+ absorption spectrum splitting with maxima at 295 and 360 nm.34 A similar effect is most probably the origin of a broad peak at 360 nm.35 The super-atom orbital model suggests that the Ag42+ electronic transitions are s–p transitions resulting from the contribution of 5s-based orbitals, i.e., from a doubly symmetric occupied orbital (s orbital) to an unoccupied orbital (p orbital).36 The well-defined peak at 320 nm is usually assigned to the presence of the atomic Ag80 in the sodalite cage.37 Each of the eight silver atoms is pointing toward one six-membered ring, thus forming a distorted cube inside the sodalite cages. The band at 380 nm is close to the characteristic absorption bands of silver species containing approximately seven nuclei embedded in zeolite cages.38,39 It should be noted that the band intensities reflect the abundance of the different Agnδ+ species (with n > δ), which can have different extinction coefficients.
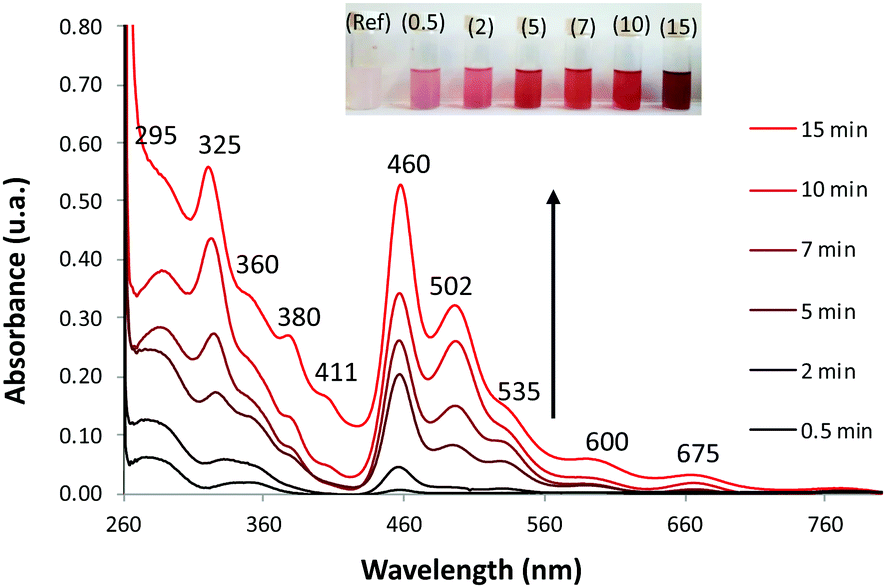 | ||
| Fig. 5 Time evolution of the UV-vis absorption spectra of ZX-Bi suspensions after (a) 0.5, (b) 2, (c) 5, (d) 7, (e) 10 and (f) 15 min of UV irradiation in the presence of 10−2 M of AgNO3. Conditions: lamp Hg–Xe; polychromatic = 205 mW cm−2; filter cut infrared; total volume = 5 ml; [ZX-Bi] = 2.4 g l−1. Inset: Images of the corresponding suspensions. ‘Ref’ corresponds to the sample before irradiation. | ||
Compared with metallic silver, the XPS analysis of Ag@ZX-Bi shows a shift to a higher binding energy (Fig. S9B, ESI†), confirming the cluster/nanoparticle nature of the Ag particles.40 The Auger spectrum (Fig. S9C, ESI†) shows two broad bands with kinetic energy near 349 nm and 354 nm with a shift of ∼2 eV compared with that of metallic silver. This demonstrates the ionic state of the silver clusters.41,42 This result is also confirmed by the IR analysis, using CO as a probe molecule, which demonstrates the presence of both metallic and ionic types of silver (Fig. S10, ESI†). Therefore, based on these results and the TEM data we conclude that silver particles are in the form of reduced Agnδ+ clusters with a partial cationic charge (n > δ). Contrariwise to the UV region, Ag@ZX-Bi illustrates very distinctive optical behavior in the visible range compared with that usually observed for classical Ag-CLs and/or plasmonic Ag-NPs confined/supported in/on the zeolite. Interestingly, the spectrum of the Ag@ZX-Bi sample shows new bands at around 460, 502, 535, 600 and 675 nm, which might be the origin of the magenta color. The shallow pink color has been previously reported for Bi-doped FAU zeolite after intense thermal treatment and the absorbance intensity is generally proportional to the Bi+ concentration.43 Bi+ is characterized with two significant NIR-absorption bands at 500 and 700 nm and promoted by the thermal reduction of Bi3+. However, no Bi+ in Ag@ZX-Bi is detected, as the XPS characterization of the sample reveals and reported previously (Fig. 3). Therefore, we conclude that the appearance of these new bands in the visible region results from a strong interaction of the zeolite ligand with the reduced silver clusters by transitional charge transfer (CT) and/or to a strong connection between the silver clusters through the zeolite channels.32 Similar optical features were observed for the brick-red samples prepared upon dehydration of Ag-containing zeolites A.31,34,44 It is important to note that the UV-visible band positions are not affected by the nature of the sample (powder or suspension). However, the relative intensity of the bands is dramatically modified, showing a solvatochromic effect of the liquid water (Fig. S11A, ESI†). We should also mention that the Ag@ZX-Bi sample, compared to Ag@ZX-V, is very stable. The analysis repeated eight months after the preparation, for both suspension and powder forms, showed no significant alteration in their UV-visible spectra and crystallinity (Fig. S11B, ESI†). To the best of our knowledge, such an exceptional optical stability has not been reported for silver species (clusters or nanoparticles) so far.
A set of experiments was performed to better understand the Ag@ZX-Bi-3 sample behavior. First, the role of Bin+ was investigated to highlight the characteristics of Ag@ZX-Bi. For this purpose, suspensions containing ZX, ZX-Bi-0.5, ZX-Bi-1.5 and ZX-Bi-3 were exposed to UV irradiation for 15 min in the presence of equimolar amounts of silver nitrate (Fig. S12, ESI†). No significant absorbance is observed for the pure zeolite showing the absence of any silver clusters. The UV-visible spectra of Ag@ZX-Bi-3 and Ag@ZX-Bi-1.5 exhibit two main narrow bands at 318 and 408 nm, respectively, and differ from the ZX-Bi-0.5 spectrum (Fig. S12, ESI†). The 318 and 408 nm band intensities are lower in the case of Ag@ZX-Bi-1.5 with the appearance of the characteristic bands in the visible region, which is much more pronounced for Ag@ZX-Bi-3. On the other hand, zeolites prepared under similar conditions but at different crystallization times (tcrystal) are used to prepare a series of Ag-CL-containing ZX-Bi-3 samples. ICP analysis proves that the Bi content and Si/Al ratio is similar for all samples (Table S4, ESI†). As shown in Fig. S13 (ESI†), the increase of the FAUX samples’ crystallinity (tcrystal < 10 h) impacts the abundance of the Ag-CLs but without a significant effect on their nature. However, when sodalite is the major phase (Fig. S4g, ESI,†tcrystal = 48 h), the optical behavior of the sample is dramatically changed. Therefore, there is a direct relationship between the zeolite framework morphology and the optical behavior of the Ag@ZX-Bi-3 samples. The indirect contribution of Bi cations to the Ag@ZX-Bi optical behavior cannot be totally excluded. However, the variation of the optical behavior (and not only with the absorbance band intensities) with the bismuth concentration allows an important impact of bismuth to be excluded, e.g. if Bi is impacting the optical behaviour, the variation of its concentration would affect the intensity instead of the band positions (wavelengths). This conclusion is fully supported by the simulation of the cluster absorbance. Connecting the silver via the prismatic cages shifts the absorbance bands from the near-visible to the red region. The effect of the zeolite topology is also clear. The optical behavior changes between the FAU type and sodalite type frameworks. On the other hand, it is still clear that the optical behavior of the Ag@ZX-Bi sample depends on the specific topology of the faujasite structure. Namely, the FAU supercages are connected through double six-membered rings forming hexagonal prisms. This structural unit is absent in the sodalite structure. In Ag@ZX-Bi, Ag+ has a strong affinity for the sites I and I′ that are located in the hexagonal prism faces between the sodalite cages,13 while Bin+ occupies mainly the sodalite cages. The positioning of Ag in the 6MR prism, in addition to the abundance of the Bi cations in the sodalite cages, contributes to the formation of Ag clusters and thus ensures the interconnection between silver clusters. This connection does not exist in the sodalite structure and, consequently, the silver clusters remain isolated. On the other hand, the relatively low concentration of Bi active sites in FAUX leads solely to the formation of isolated Ag-CLs. Therefore, the increase of the Bi3+/Bi2+ active site concentration might promote the connectivity of the Ag-CLs and the formation of quasi-AgNPs. This is due to the increase of the reduced silver concentration, thus reducing their inter-distance.
The continuous wave (CW) photoluminescence (PL) measurements shed more light on the optical behavior of the Ag@ZX-Bi-x samples (x = 0.5, 1.5 and 3). The measurements were carried out at room temperature using two excitation lasers at 266 nm and 488 nm. For excitation at 266 nm, we observe several PL bands, depending on the Bi content. The most intense band position is at 2.06 eV (600 nm). The less intense bands are observed at 2.22 eV (558 nm) and at 2.35 eV (527 nm). Upon excitation at 488 nm, new PL bands at 2.67 eV (464 nm) and at 1.39 eV (892 nm) are observed with very low intensity; the latter are attributed to the high Bi content (Fig. 6a and b). The bands observed in all spectra, 2.06 eV (600 nm), 2.22 eV (558 nm) and 2.35 eV (527 nm), seem to be independent of the type of active center or its concentration (Fig. S14, ESI†). We attribute these bands to isolated silver clusters in the sodalite cages. For example, the emission bands observed at 527 and 558 nm are similar to the ones associated with the trinuclear and tetranuclear silver species,13,45 respectively. These assignments agree with the UV-visible absorbance analysis of the samples, which indicates the presence of common Ag species with a characteristic UV absorbance and new species with a characteristic visible absorbance for Ag@ZX-Bi samples with a relatively high Bi content. This latter seems to have very low (or no) radiative emission in the visible range (behavior characteristic of plasmonic particles).
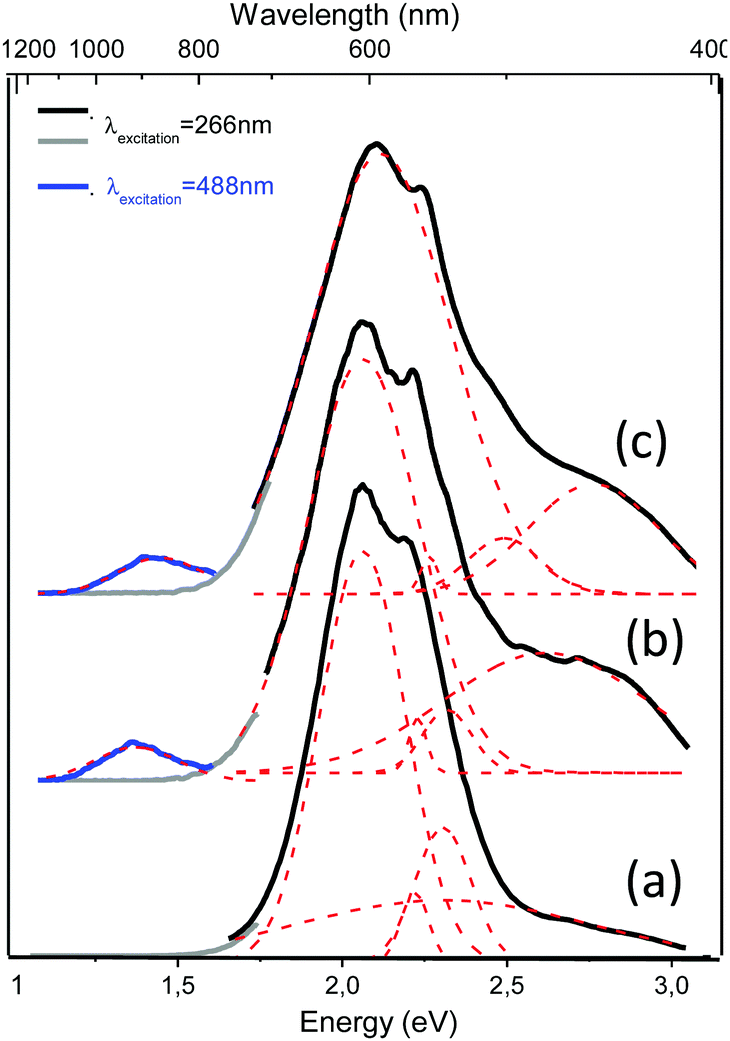 | ||
| Fig. 6 PL spectra of (a) Ag@ZX-Bi-0.5, (b) Ag@ZX-Bi-1.5 and (c) Ag@ZX-Bi-3.0 samples. The excitation laser wavelength is either 266 nm (black and grey) or at 488 nm (blue). PL bands were fitted using the Gaussian function (dashed red lines). Long-pass filters were used with the cut-on wavelength at 700 nm for the grey and blue curves. | ||
This assignment was further supported by the TEM results. Fig. 7 shows the high-resolution TEM images along the most informative [011] FAU zone axis. The morphology of ZX nanoparticles remains intact with clearly expressed faceting with 〈111〉 planes. Ag clusters appear as dark spots in the TEM images (Fig. 7a and b) and as bright white dots (Z = 47) in the HAADF-STEM images (Fig. 7c–e), where the contrast is proportional to atomic number (∼Z2) and thickness.
 | ||
| Fig. 7 TEM analysis of the Ag@ZX-Bi sample: (a) bright-field low-magnification TEM images of Ag@ZX-Bi; (b) HRTEM image of single Ag@ZX-Bi crystallites revealing the resolved Ag lattice planes following the FAU structure. The corresponding FT pattern confirmed the structure and the 3D arrangement of the Ag clusters aligned to the FAU framework (see enlargements of the central FT spots). (c) Low-magnification HAADF-STEM image of Ag@ZX-Bi. Bright-contrast Ag nanostructures are clearly visible. (d) High-resolution HAADF-STEM image of Ag@ZX-Bi. (e) High-resolution HAADF-STEM images of Ag@ZX-Bi structures close to the [112] (left panel) and to the [011] (right panel) FAU zone axis. Ag nanostructures within the ZX-Bi structure exhibiting bright contrast are marked with white arrows. The structural model of FAU is overlaid on the experimental image. | ||
In the latter, the dark spots correspond to the super cages and/or surrounding ZX frameworks, which are not clearly resolved due to the low sum HAADF-STEM contrast of the silica–alumina zeolite framework (ZSi = 14; ZAl = 13; ZO = 8) and large difference with the atomic number of Ag. Despite the high sensitivity of the Ag@ZX-Bi particles to the electron beam and their tendency to agglomerate into larger silver NPs, it was possible to acquire high-resolution images along the [011] and [112] zone axis of the Ag@ZX-Bi crystallite in both modes – HRTEM (Fig. 7b) and HAADF-STEM (Fig. 7e) – using low doses and weak e-beam conditions. Thus, the lattice planes of the Ag clusters were resolved. The corresponding Fourier transform (FT) patterns (inset Fig. 7b) confirm the presence and the 3D arrangement of Ag [111] particles within the pores of the FAU-type structure.
The Ag sub-nanoparticles appear in projection to have close to a ball shape with a typical diameter of about 1.0 nm, interconnected by Ag “nanobridges” and forming a checkerboard pattern reflecting the [011] FAU structure symmetry. The size of Ag is compatible with the 6-membered ring prism of the FAU structure. The Ag in such a position makes a nanobridge between the silver clusters situated in the sodalite cage, as shown in Fig. 7e. The [011] FAU skillet structural model is placed over the experimental [011] HAADF-STEM image (Fig. 7e). The HAADF-STEM image of Ag@ZX-Bi projected on the [112] zone axis reveals the presence of silver nanoparticles situated in the sodalite cages connected through the 6MR prism, thus creating a 3D nanostructure. The needle-like silver particles (Fig. 7e) are more pronounced and longer than those observed in our previous work on Ag@ZX-V. This could explain the characteristic visible absorbance of the silver quasi-nanoparticles. It should be noted that due to the low concentration of Bi, we assume that the white spot corresponds mainly to the silver particles that are clearly much more abundant than Bi (Fig. 3). Taking into account the redox process, the oxidation of Bi2+ and Bi3+ to Bi5+ generates three to two reduced silver atoms, respectively. These reduced silver atoms are surrounded with cationic silver atoms to form the clusters, making silver much more abundant than bismuth. The oxidation of the Bi allows the positive charge of the reduced silver to be recompensated. The IR spectrum of Ag@ZX-Bi (Fig. S17, ESI†) (1600–1300 cm−1) reveals the presence of residual nitrate, indicating that the excess of the positive charge of bismuth (with respect to the zeolite framework) is recompensated by the negative charge of nitrate as a counter ion.
MNPBEM modeling experiments (see the ESI† for more details) were performed to understand better the optical behavior of the nanostructured silver species in the faujasite (Ag-FAU-Rep) pore space (Fig. 8A). We have modelled the far-field response in terms of extinction absorption and scattering cross-section spectra and near-field responses in terms of the field enhancement map (Fig. 8B). The extinction, absorption, and scattering cross-section spectra of isolated and interconnected Ag-FAU nanoparticles with two perpendicular excitation polarizations (E‖xO, and E‖yO) were calculated. The extinction cross-section sums of the absorption were found to be clearly greater than those of the scattering cross-section. A plasmon narrow extinction band centered at 384 nm, independent of the excitation polarization, is observed in the samples with isolated silver nanostructures in the sodalite cages (i.e., Ag-free 6MR prism). This band is similar to that observed for a single Ag cluster (Fig. S15, ESI†) and experimentally observed in the case of Ag@ZX-V and Ag-ZX-Bi-0.5 (ZX-Bi with low Bi content). However, in the case of the Ag-FAU nanostructures with interconnected silver clusters (Ag in the sodalite cage and the 6MR prism), the extinction spectra show new extinction bands distributed in the visible region (370–700 nm).
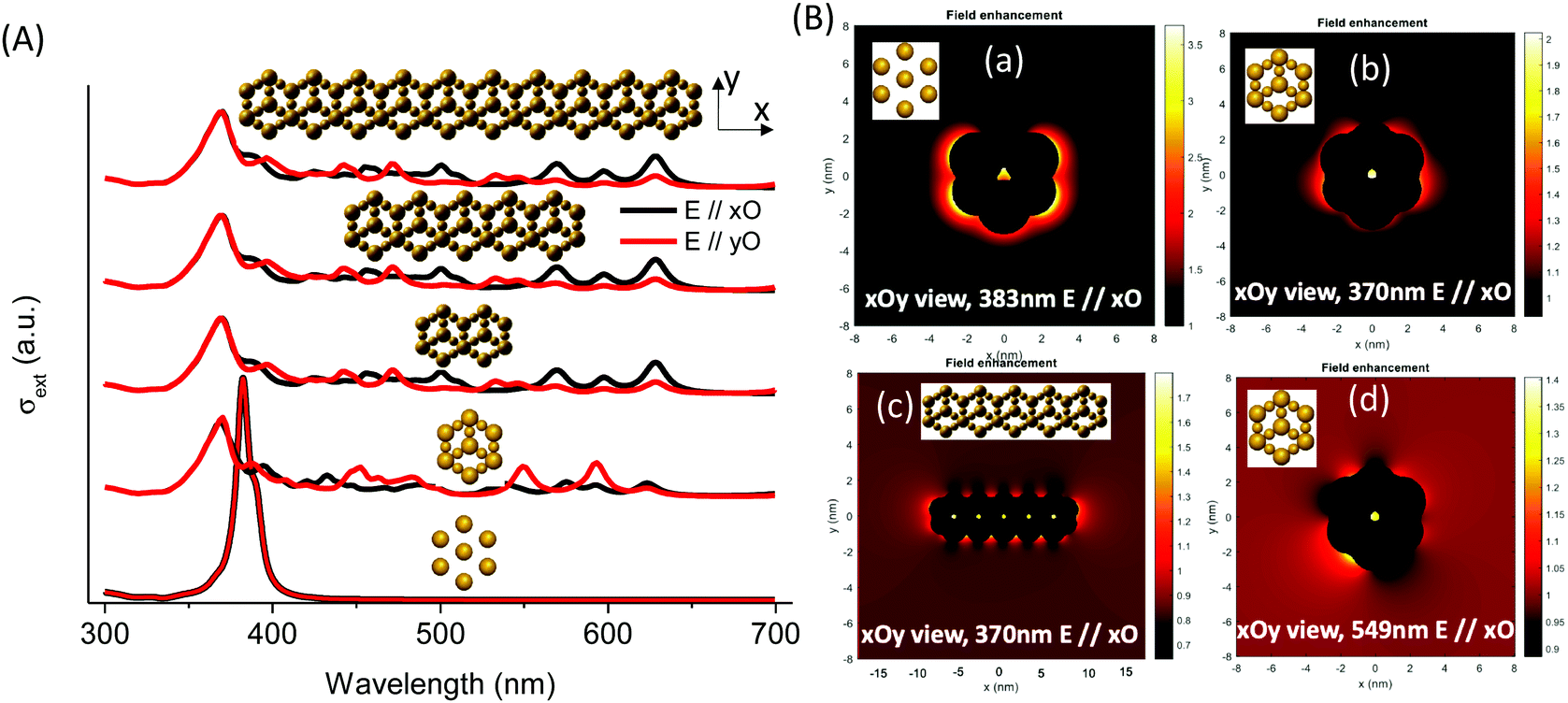 | ||
| Fig. 8 (A) Extinction cross-sections of silver nanostructures with isolated and interconnected nanoparticles in a single unit cell, two interconnected unit cells, five interconnected unit cells, and ten interconnected unit cells with polarization along the xO and yO axes. (B) Near-field enhancement maps for isolated silver (a) and interconnected (b and d) silver-replica nanostructure resonance energies of one (b) and five (c) unit cells at 370 nm (b and c), 383 nm (a), and 549 nm (d). | ||
The simplification of the modelled system is probably the reason for the difference in the band positions between the simulated and the experimental spectra (more details in the Experimental section). Nevertheless, it reveals important information concerning the nanostructure shape effect on the global optical behavior. More significantly, the results confirm the hypothesis that the interconnection of silver clusters through the zeolite channels is responsible for absorbing multiple bands in the visible range. The simulation shows that, with the exception of the 384 nm and 370 nm bands, the intensity depends on the polarization angle (Fig. S16, ESI†). This shows that these resonances arise on structures with less symmetry than the individual particles in the sodalite cage or the 6MR prism, and that they might be due to a delocalized mode over several of these elements. The origin of different bands obtained on single silver faujasite nanostructures was investigated by exploring the plasmon response of elemental building blocks. Large and small individual nanoparticles were considered: rods formed by two large and a small nanoparticle (vertical and oriented at 19.47°/xOy plane); faujasite's basis ring formed by six rods; and the conical top structure formed by three rods oriented at θ = 120° (Fig. S16, ESI†). The plasmon response in terms of the extinction cross-section spectra of those building blocks was calculated and compared with that of the silver faujasite nanostructures. This comparison allowed us to confirm that the resonance centered at 383 nm is due to the individual/isolated response of large and small silver nanoparticles/clusters reported above or to rods parallel to the wavevector (perpendicular to the xOy plane). The resonances between 386 and 500 nm can be attributed to resonances occurring in rods oriented out of the perpendicular axis to the xOy plane. Resonances occurring above 500 nm are attributed to resonance occurring in the whole ring and conical top structures (Fig. 8B). Larger structures created by adding several faujasite unit cells resulting in the linear chain were investigated, leading to comparable cross-section spectra (Fig. 8) with resonance peaks ranging from 383 to 700 nm.
The calculated near-field enhancement maps for isolated and interconnected silver nanostructures with resonance energies at 370, 383 and 549 nm are reported in Fig. 8B. Localized resonance around each constituting nanoparticle was found on isolated silver faujasite nanostructures, while delocalized resonance over several constituting nanoparticles was found on interconnected silver faujasite nanostructures. This result confirms the existence of delocalized plasmon modes on several Ag-FAU building blocks, leading to numerous extinction bands whose positions are red-shifted up to 700 nm. The near-field enhancement map at 549 nm shows that the red-shifted plasmon resonance occurs in a single-cell nanostructure. Considering that the plasmon decay follows three different channel processes with Drude friction-like dissipation, inter-band transitions, and the generation of energetic hot electrons,46 we may have a different kind of behavior depending on the spectral position of excitation. For short wavelengths (high energy), we may have energy transfer to resonance energy levels of luminescent species, leading to enhanced photoluminescence (as observed above under 266 nm of excitation). For larger wavelengths (lower energy), only the dissipation process may occur with Drude friction or electron–phonon interactions leading to heat release in the local nanostructure environment.
Operando IR analysis was used to elucidate the plasmonic behavior of the Ag@ZX-Bi-3 sample. Under visible-light irradiation, it shows an immediate response with fast water desorption for almost 80% in the first three minutes of irradiation (Fig. 9A).
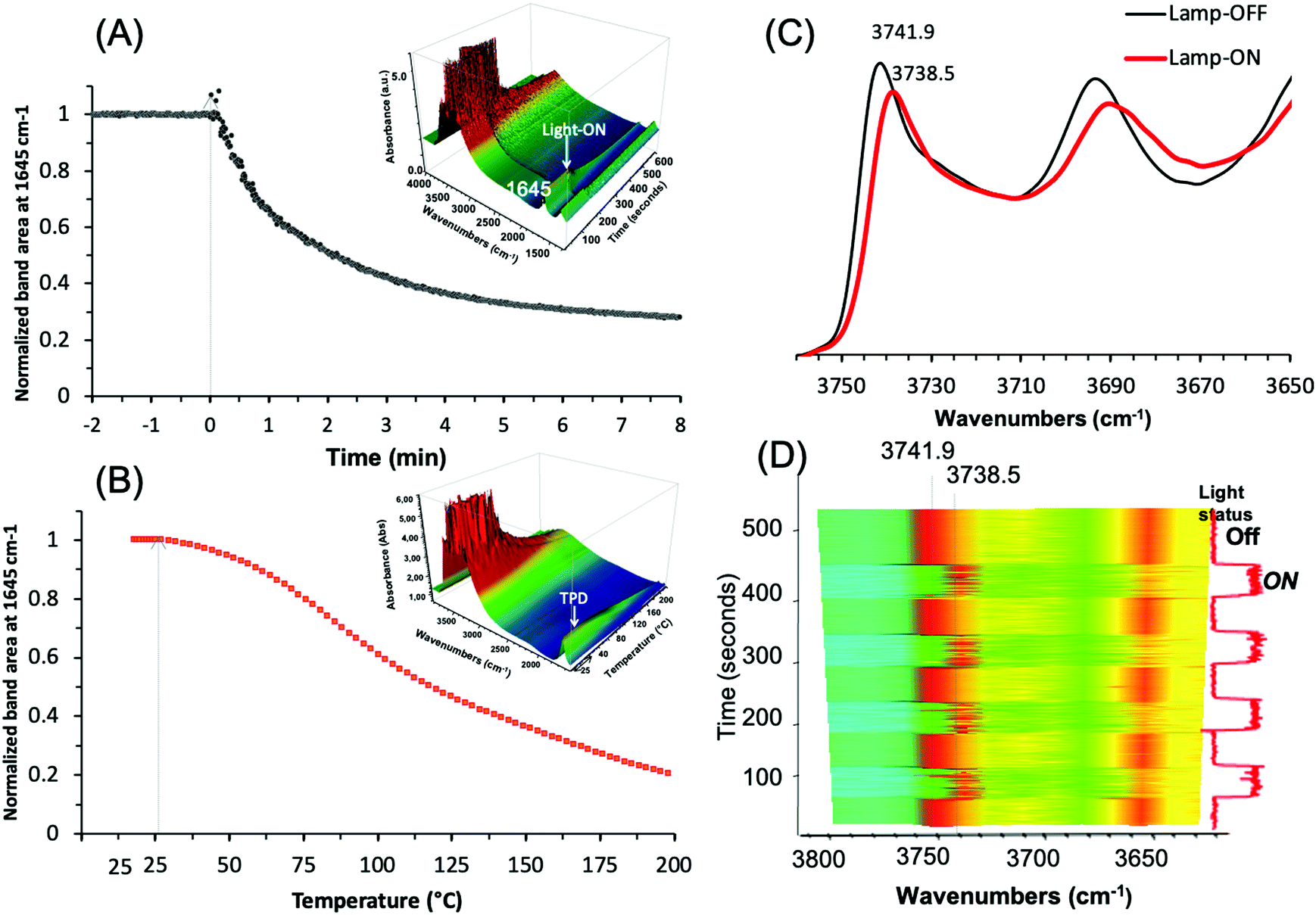 | ||
| Fig. 9 Evolution of the IR band area (centered at 1645 cm−1) of the adsorbed water on the as-prepared Ag@ZX-Bi-3 samples versus (A) the visible-light irradiation time at 25 °C, and (B) the temperature in the dark. Insets: 3D mapping of the corresponding original spectra. The arrows correspond to the turning on of the light (t = 0 s) and the starting point of heating (rate = 1.75 °C min−1), respectively. Experimental conditions: Ar flow (25 cm3 min−1); mcat = 20 mg (pellet of 2 cm2 diameter and 65 μm thickness); light source used in (A) Xe lamp with cut UV filter (λ > 390 nm) and irradiance ∼150 mW cm−2. (C) IR spectra of Ag@ZX-Bi sample in the vibrational region of SiOH in the dark (black; lamp-OFF) and under visible-light irradiation (red; lamp-ON). Sample activated at 28 °C under visible-light irradiation for 30 min. (D) 2D mapping of the band intensity variation in the 3800–3650 cm−1 region for different cycles of lamp-OFF/lamp-ON (red and blue-green correspond to the higher and lower intensities, respectively). (The light relative intensities (0% or 100%): low position: shutter closed; high position: shutter open.) The vertical scale axis corresponds to the time of the experiments. Note: (C) and (D) were measured on the same sample pellet and under the same conditions used in (A). | ||
This loss is usually taking place at around 200 °C (Fig. 9B), and is in agreement with the TG result for ZX-Bi (Fig. 3B). It is important to mention that this effect is more than two times higher than that observed with Ag@ZX-V (Fig. S17, ESI†) and that there are no responses for Ag@ZX-Bi and Ag+@ZX-Bi under monochromatic UV (366 nm) or visible (>390 nm) light irradiation, respectively. Therefore, this behavior is directly related to the presence of the Ag-quasi-NPs (Ag-QNPS). Another noteworthy behavior is observed on the activated samples (after 30 min of visible irradiation at 28 °C). The IR spectrum of this sample shows a reversible and instantaneous shift toward lower wavenumbers (for 3.4 cm−1) in the vibration region of ν(OH) under visible irradiation with respect to that collected in the dark (Fig. 9C and D). Such an effect is usually a sign of a temperature increase of a few hundred degrees.47
The relationship between the ν(OH) band and temperature was measured on a series of Ag@ZX-Bi samples (Fig. S18, ESI†). The results show that the shift of 3.4 cm−1 corresponds to a heating temperature of 190 °C. Under permanent visible irradiation and with the increase of the sample temperature, the variation of ν(OH) (Fig. S18A, ESI†) is almost constant (i.e. Δν(OH) ∼ 3.5 cm−1) until 150 °C. However, the shift is similar (in the dark or under visible light) at a temperature equal to or higher than 200 °C. The HRTEM analysis shows that the activation of Ag@ZX-Bi at T ≥ 200 °C promotes the formation of external AgNPs (Fig. S19, ESI†), probably due to the migration and aggregation of the silver on the zeolite surface. This explains the major drop of the plasmonic behavior of the S-QNPs. It should be noted that the zeolite surface is rich in OH groups, but is without direct contact with the Ag-QNPs. The instantaneous effect on these site vibration frequencies allows us to think that the energy transfer from the Ag-QNPs to the OH is probably radiative and agrees with the water desorption observed during the irradiation of Ag@ZX-Bi under visible light.
4 Conclusion
Bismuth (Bi2+/Bi3+)-doped zeolite nanoparticles (ZX-Bi), successfully prepared using one-pot hydrothermal synthesis and stabilized as colloidal suspensions, were used for the preparation of bridged silver clusters. UV-irradiation of the ZX-Bi suspensions in the presence of silver nitrate promotes the formation of reduced silver particles (Ag@ZX-Bi) in the zeolite micropore space. The set of experimental results revealed that the optical behavior of the confined silver particles depends on the Bi concentration initially introduced into the zeolite. The composite sample's UV-visible absorbance shows narrow bands in the visible range (400–800 nm), which is characteristic of metal nanoparticles with well-defined sizes. The TEM analysis, the photoluminescence results, and the boundary element method (BEM) approach showed that the unique optical behavior of Ag is a consequence of the formation of interconnected silver clusters (silver quasi-nanoparticles) through the zeolite channels. The IR operando analysis of the samples in the dark and under visible-light irradiation confirmed the surprising plasmonic behavior of the Ag@ZX-Bi samples and registered local heating equivalent to 190 °C under visible irradiation, behavior that is characteristic of plasmonic nanoparticles. Therefore, to our knowledge, the gap between clusters and plasmonic nanoparticles with the well-controlled and reproducible size of Ag-QNPs is bridged for the first time. The simplicity of the used method allows easy scale-up for different possible applications (μ-sensors, electronics, optics, etc.).Author contributions
The manuscript was prepared and written through contributions of all the authors. All the authors have approved the final version of the manuscript.Conflicts of interest
The authors declare that no competing interest.Acknowledgements
F. Douma and M. El-Roz acknowledge the PHC Tassili program (17MDU983) and European Regional Development Fund (ERDF), Normandy region (RAPHYD project), for the financial support. The authors acknowledge their colleagues Mama Lafjah, Fatiha Djafri and Jaun Pablo Bolletta for the scientific discussions and support.Notes and references
- Y. Lu and W. Chen, Chem. Soc. Rev., 2012, 41, 3594–3623 RSC.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS PubMed.
- T. Q. Yang, B. Peng, B. Q. Shan, Y. X. Zong, J. G. Jiang, P. Wu and K. Zhang, Nanomaterials, 2020, 10(2), 261 CrossRef CAS PubMed.
- N. Sakai, S. Nakamura and T. Tatsuma, Dalton Trans., 2013, 42, 16162–16165 RSC.
- A. Royon, K. Bourhis, M. Bellec, G. Papon, B. Bousquet, Y. Deshayes and L. Canioni, Adv. Mater., 2010, 22, 5282–5286 CrossRef CAS PubMed.
- Y. Wang, C. Dai and X. P. Yan, Chem. Commun., 2014, 50(92), 14341–14344 RSC.
- Y. Yu, J. Geng, E. Y. X. Ong, V. Chellappan and Y. N. Tan, Adv. Healthcare Mater., 2016, 5, 2528–2535 CrossRef CAS PubMed.
- J. Li, Y. Dai, S. Wang, C. Han and K. Xu, Sens. Actuators, B, 2016, 232, 1–8 CrossRef CAS.
- L. Shang and S. Dong, Biosens. Bioelectron., 2009, 24, 1569–1573 CrossRef CAS PubMed.
- I. Díez, M. Pusa, S. Kulmala, H. Jiang, A. Walther, A. S. Goldmann and R. H. Ras, Angew. Chem., Int. Ed., 2009, 48, 2122–2125 CrossRef PubMed.
- M. Cao, R. Pang, Q. Y. Wang, Z. Han, Z. Y. Wang, X. Y. Dong and T. C. Mak, J. Am. Chem. Soc., 2019, 141, 14505–14509 CrossRef CAS PubMed.
- T. Wu, D. Yin, X. Hu, B. Yang, H. Liu, Y. P. Xie and G. G. Gao, Nanoscale, 2019, 11(35), 16293–16298 RSC.
- T. Altantzis, E. Coutino-Gonzalez, W. Baekelant, G. T. Martinez, A. M. Abakumov, G. V. Tendeloo and J. Hofkens, ACS Nano, 2016, 10, 7604–7611 CrossRef CAS PubMed.
- D. Yao, S. Xu, Y. Wang and H. Li, Mater. Chem. Front., 2019, 3, 1080–1084 RSC.
- S. Aghakhani, D. Grandjean, W. Baekelant, E. Coutiño-Gonzalez, E. Fron, K. Kvashnina and P. Lievens, Nanoscale, 2018, 10, 11467–11476 RSC.
- T. Yumura, A. Oda, H. Torigoe, A. Itadani, Y. Kuroda, T. Wakasugi and H. Kobayashi, J. Phys. Chem. C, 2014, 118, 23874–23887 CrossRef CAS.
- (a) M. El-Roz, I. Telegeiev, N. E. Mordvinova, O. I. Lebedev, N. Barrier, A. Behilil and V. Valtchev, ACS Appl. Mater. Interfaces, 2018, 10(34), 28702–28708 CrossRef CAS PubMed; (b) H. I. Hamoud, M. Lafjah, F. Douma, O. I. Lebedev, F. Djafri, V. Valchev and M. El-Roz, Sol. Energy, 2019, 189, 244–253 CrossRef CAS.
- M. El-Roz, L. Lakiss, I. Telegeiev, O. I. Lebedev, P. Bazin, A. Vicente and V. Valtchev, ACS Appl. Mater. Interfaces, 2017, 9, 17846–17855 CrossRef CAS PubMed.
- F. G. De Abajo and A. Howie, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 115418 CrossRef.
- U. Hohenester and A. Trügler, Comput. Phys. Commun., 2012, 183, 370–381 CrossRef CAS.
- P. B. Johnson and R. W. Christy, Phys. Rev. B: Solid State, 1972, 6, 4370 CrossRef CAS.
- A. M. Kuznetsov, M. S. Shapnik, A. N. Masliy and K. V. Zelenetskaya, Russ. J. Electrochem., 2002, 38, 669–675 CrossRef CAS.
- H. Awala, J. P. Gilson, R. Retoux, P. Boullay, J. M. Goupil, V. Valtchev and S. Mintova, Nat. Mater., 2015, 14, 447–451 CrossRef CAS PubMed.
- X. Qin, Y. Li, D. Wu, Y. Wu, R. Chen, Z. Ma and J. Qiu, RSC Adv., 2015, 5, 101347 RSC.
- A. Yousif, R. M. Jafer, S. Som, M. M. Duvenhage, E. Coetsee and H. C. Swart, RSC Adv., 2015, 5, 54115–54122 RSC.
- H. Li, Z. Yang, J. Zhang, Y. Huang, H. Ji and Y. Tong, Appl. Surf. Sci., 2017, 423, 1188–1197 CrossRef CAS.
- M. Majerová, R. Klement, A. Prnová, J. Kraxner, E. Bruneel and D. Galusek, R. Soc. Open Sci., 2018, 5, 181667 CrossRef PubMed.
- H. T. Sun, Y. Matsushita, Y. Sakka, N. Shirahata, M. Tanaka, Y. Katsuya and K. Kobayashi, J. Am. Chem. Soc., 2012, 134, 2918–2921 CrossRef CAS PubMed.
- M. G. Sales, L. Herweyer, E. Opila and S. McDonnell, Appl. Surf. Sci., 2020, 508, 145256 CrossRef.
- C. Lv, G. Chen, X. Zhou, C. Zhang, Z. Wang, B. Zhao and D. Li, ACS Appl. Mater. Interfaces, 2017, 9, 23748–23755 CrossRef CAS PubMed.
- I. Tuzovskaya, N. Bogdanchikova, A. Pestryakov, V. Gurin, A. Simakov and V. Lunin, Advances in the Understanding and Application of Catalysts, 2003, p. 248 Search PubMed.
- V. S. Gurin, N. E. Bogdanchikova and V. P. Petranovskii, J. Phys. Chem. B, 2000, 104, 12105–12110 CrossRef CAS.
- T. Yumura, A. Oda, H. Torigoe, A. Itadani, Y. Kuroda, T. Wakasugi and H. Kobayashi, J. Phys. Chem. C, 2014, 118, 23874–23887 CrossRef CAS.
- E. Fron, S. Aghakhani, W. Baekelant, D. Grandjean, E. Coutino-Gonzalez, M. Van der Auweraer and J. Hofkens, J. Phys. Chem. C, 2019, 123, 10630–10638 CrossRef CAS.
- N. T. Cuong, H. M. T. Nguyen, M. P. Pham-Ho and M. T. Nguyen, Phys. Chem. Chem. Phys., 2016, 18, 18128–18136 RSC.
- T. Yumura, M. Kumondai, Y. Kuroda, T. Wakasugi and H. Kobayashi, RSC Adv., 2017, 7, 4950–4959 RSC.
- B. Concepción-Rosabal, G. Rodríguez-Fuentes, N. Bogdanchikova, P. Bosch, M. Avalos and V. H. Lara, Microporous Mesoporous Mater., 2005, 86, 249–255 CrossRef.
- R. Kellerman and J. Texter, J. Chem. Phys., 1979, 70, 1562–1563 CrossRef CAS.
- E. Gachard, J. Belloni and M. A. Subramanian, J. Mater. Chem., 1996, 6, 867–870 RSC.
- H. S. Shin, H. C. Choi, Y. Jung, S. B. Kim, H. J. Song and H. J. Shin, Chem. Phys. Lett., 2004, 383, 418–422 CrossRef CAS.
- A. M. Fonseca and I. C. Neves, Microporous Mesoporous Mater., 2013, 181, 83–87 CrossRef CAS.
- O. Fenwick, E. Coutiño-Gonzalez, F. Richard, S. Bonacchi, W. Baekelant, D. de Vos and P. Samorì, Small, 2020, 16, 2002063 CrossRef CAS PubMed.
- Z. Bai, M. Fujii, T. Hasegawa, K. Imakita, Y. Miwa, M. Mizuhata and S. Hayashi, Microporous Mesoporous Mater., 2011, 145, 21–25 CrossRef CAS.
- R. Seifert, R. Rytz and G. Calzaferri, J. Phys. Chem. A, 2000, 104(32), 7473–7483 CrossRef CAS.
- B. Dong, R. Retoux, V. De Waele, S. G. Chiodo, T. Mineva, J. Cardin and S. Mintova, Microporous Mesoporous Mater., 2017, 244, 74–82 CrossRef CAS.
- L. V. Besteiro, X. T. Kong, Z. Wang, G. Hartland and A. O. Govorov, ACS Photonics, 2017, 4, 2759–2781 CrossRef CAS.
- V. Zholobenko, C. Freitas, M. Jendrlin, P. Bazin, A. Travert and F. Thibault-Starzyk, J. Catal., 2020, 385, 52–60 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00382h |
| This journal is © The Royal Society of Chemistry 2021 |