
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA ruthenium-inserted hydrotalcite (Ru-HT) heterogeneous catalyst: kinetic studies on the selective hydrogenation of carbon dioxide to formic acid†
Minaxi S.
Maru
 *abc,
Sanwala
Ram
ad,
Noor-ul H.
Khan
ad and
Ram S.
Shukla
*ad
*abc,
Sanwala
Ram
ad,
Noor-ul H.
Khan
ad and
Ram S.
Shukla
*ad
aInorganic Materials and Catalysis Division, Council of Scientific and Industrial Research (CSIR), Central Salt and Marine Chemicals Research Institute, G. B. Marg, Bhavnagar, 364002, Gujarat, India. E-mail: dr.maru139@gmail.com
bInorganic Chemistry Division, Department of Chemistry, Saurashtra University, Rajkot, 360005, Gujarat, India
cDepartment of Chemistry, Indian Institute of Teacher Education (IITE), Gandhinagar, 382016, Gujarat, India
dAcademy of Scientific and Innovative Research (AcSIR), Council of Scientific and Industrial Research (CSIR), Central Salt and Marine Chemicals Research Institute, G. B. Marg, Bhavnagar, 364002, Gujarat, India
First published on 3rd July 2021
Abstract
Kinetic studies have been carried out for the base-free hydrogenation of CO2 to formic acid using a heterogeneous ruthenium-inserted hydrotalcite (Ru-HT) catalyst. An impressive turnover number (TON = 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 389) was achieved for formic acid under the optimized reaction conditions using a methanol:water mixture as the solvent (5:1, v/v) with 60 bar total pressure at 60 °C for 24 hours. The rates were determined directly in terms of the formation of formic acid with time. Kinetics were performed by carrying out experiments concerning the amount of catalyst, the individual partial pressures of CO2 and H2, the total pressure, temperature, agitation speed, reaction volume and v/v ratio of the mixed solvent. The rate of formic acid formation was first order on the amount of catalyst and partial pressures of CO2 and H2. The best reaction conditions achieved from the kinetic parametric optimization were: 100 mg catalyst, 30 bar pCO2, 30 bar pH2, 60 °C temperature, 800 rpm agitation speed and methanol–water (5:1, v/v) solvent. The computed activation parameters obtained from the temperature-dependent rate of formic acid formation were Ea = 34.5 ± 2.5 kJ mol−1, ΔH# = 32 ± 2.5 kJ mol−1 and ΔS# = −384 ± 5 J deg−1 mol−1. The presence of water in the mixed solvent effectively enhanced the reaction rate, which is characteristically observed due to its molecular effect. Two mechanistic routes for CO2 hydrogenation to formic acid are proposed and discussed based on the kinetic and experimental observations. The studied parameters were found to be significantly effective to increase the rate of reaction appreciably.
389) was achieved for formic acid under the optimized reaction conditions using a methanol:water mixture as the solvent (5:1, v/v) with 60 bar total pressure at 60 °C for 24 hours. The rates were determined directly in terms of the formation of formic acid with time. Kinetics were performed by carrying out experiments concerning the amount of catalyst, the individual partial pressures of CO2 and H2, the total pressure, temperature, agitation speed, reaction volume and v/v ratio of the mixed solvent. The rate of formic acid formation was first order on the amount of catalyst and partial pressures of CO2 and H2. The best reaction conditions achieved from the kinetic parametric optimization were: 100 mg catalyst, 30 bar pCO2, 30 bar pH2, 60 °C temperature, 800 rpm agitation speed and methanol–water (5:1, v/v) solvent. The computed activation parameters obtained from the temperature-dependent rate of formic acid formation were Ea = 34.5 ± 2.5 kJ mol−1, ΔH# = 32 ± 2.5 kJ mol−1 and ΔS# = −384 ± 5 J deg−1 mol−1. The presence of water in the mixed solvent effectively enhanced the reaction rate, which is characteristically observed due to its molecular effect. Two mechanistic routes for CO2 hydrogenation to formic acid are proposed and discussed based on the kinetic and experimental observations. The studied parameters were found to be significantly effective to increase the rate of reaction appreciably.
Introduction
Carbon dioxide (CO2), a potent greenhouse gas produced mainly through the combustion of fossil fuels and deforestation, is significantly contributing to global warming. Although many ways are proposed to curtail or damp its emission into the atmosphere, one of attractive way is to convert it into useful chemicals due to its availability as a plentiful, cheap and non-toxic C-1 resource, specifically for formic acid as it is a renewable energy source for H2 gas.1 Formic acid is an essential intermediate and fundamental chemical feedstock in industrial organic chemistry because of its indispensable application in textile-making, dyeing, paper-making, the production of medicinal agents, tanning agents in the leather industry, as a preservative agent in livestock feed and as an important intermediate in chemical synthesis. Formic acid is the foremost chemical for the production of many basic organic compounds like ketones, aldehydes, amides and carboxylic acids via formate esters, which are also essential in the fragrance and perfumery industries.2 The currently established commercial process for the synthesis of formic acid is via a two-step procedure, using toxic carbon monoxide at high pressure and high temperature. In the first step, the formation of methyl formate by the carbonylation of methanol occurs using 2.5% (w/w) sodium methoxide as the catalyst, where only 30% of the methanol is converted into methyl formate, whereas in the second step, the hydrolysis of methyl formate gives formic acid.3 To avoid the use of toxic carbon monoxide, a promising and potential replacement for the production of formic acid is the direct hydrogenation of very abundant, inexpensive and nontoxic CO2 using various transition metal-based homogeneous and heterogeneous catalysts.4–7The greenhouse effect of CO2 is the most environmentally concerning problem in the world; many actions have been taken to counteract the danger of it, and the general reduction of CO2 emissions has been proposed so far. Among the feasible approaches to reducing CO2 emissions, processes based on CO2 recycling have attracted major scientific and technological interest.8 Accordingly, the hydrogenation of CO2 to formic acid is one of the best atom-inexpensive reactions that has engaged the focus of many researchers in the field of catalysis.9,10 The enthalpy change values of the reactions for the direct hydration of CO and the direct hydrogenation of CO2 are ΔH = −28.4 kJ mol−1 and ΔH = −31.6 kJ mol−1, respectively, and these are close enough to suggest the replacement of CO with CO2.3 The direct synthesis of formic acid by CO2 hydrogenation is thermodynamically unfavourable, associated with about  .11 The global demand for formic acid is continuously growing, particularly from the perspective of a renewable energy hydrogen source, and its production via the direct catalytic hydrogenation of CO2 without the use of a base is considerably more sustainable in comparison with the existing reaction routes.
.11 The global demand for formic acid is continuously growing, particularly from the perspective of a renewable energy hydrogen source, and its production via the direct catalytic hydrogenation of CO2 without the use of a base is considerably more sustainable in comparison with the existing reaction routes.
The utilization of CO and CO2 for their conversions into industrially important value-added chemicals has been a focus of our research using heterogeneous catalytic systems since 2006. Recently, we have developed Cu-, Co-, Ru-, and Rh-based heterogeneous catalysts, which were found to be efficient for the selective hydrogenation of CO2 to formic acid/formate and achieved remarkable results for formic acid/formate in terms of the CO2 conversion and product selectivity, with astonishing turnover numbers.12–14 A series of supported Rh complexes as efficient heterogeneous catalysts have been reported for the effective synthesis of alcohols from alkenes using carbon monoxide via hydroformylation, hydroaminomethylation, aldol condensation and stepwise hydrogenation.15–22
Among them, the hydrogenation of CO2 to formic acid has been reported using hydrotalcite (HT) as a support material for the active catalytic system. Hydrotalcite belongs to the class of brucite-like layered double hydroxides with the general formula Mg1−xAlx(CO32−)x/n(OH2)x+·m(H2O) and it is an attractive inorganic base. Its catalytic activity can be improved through the intercalation of appropriate anions in the interlayer space, optimization of the Mg2+ and Al3+ molar ratio and activation of the final material at 450 °C. A number of metals can be grafted with hydrotalcite in its brucite-like layers via a co-precipitation method due to the isomorphic characteristic substitution of Mg2+ or Al3+ ions at the octahedral sites, which are considered to be the active sites for various organic transformations.23,24
Ruthenium-grafted hydrotalcite materials are reported to be highly active catalysts for oxidation, cyclocondensation, alkylation, methanation, and methanolysis reactions.21,22a,25–29 Ru-containing materials and complexes have been the focus of investigations for the catalytic hydrogenation of CO2 by molecular hydrogen, due to their potential favourable activity and selectivity for the formation of formic acid.30 Very few reports are available on hydrotalcite for the hydrogenation of CO2 for the production of formic acid and their kinetic studies. Kinetic studies for this kind of reaction have an important role in understanding the magnitude of the catalyst involvement towards the activation of CO2 and H2 molecules, the formation of a hydride and the insertion of CO2 into such formed metal hydride bonds, thereby forming the formato intermediate resulting in the formation of the hydrogenated product. Investigations on kinetic aspects for the hydrogenation of CO2 to formic acid are quite scarce, and few have been reported using homogeneous catalysts.14,31,32 Further, some authors have reported kinetic studies for CO2 hydrogenation to methanol over heterogeneous catalysts along with theoretical DFT studies.33,34 Kinetic studies of a catalytic reaction are essential for achieving a systematic analysis of the catalytic system, so that improvements in its performance through the optimization of reaction parameters can be achieved to bring the product to a better and higher production level.20
Recently a heterogeneous Ru-hydrotalcite (Ru-HT)-based catalyst for CO2 hydrogenation by molecular hydrogen for the selective synthesis of formic acid with a choice of ruthenium species was reported by our group.13b The catalyst was found to be effective for the hydrogenation and selective formation of formic acid at a moderate temperature without using any additional liquid or solid base, as is usually practised in this reaction. The soft solid base HT remarkably plays the dual role of support and of base. The activity and reusability of the Ru-HT catalyst were appreciable for the production of formic acid at a moderate temperature (60 °C) and 60 bar pressure (CO2 and H2).
The reactivity of the catalyst significantly depends on the kinetic factors in the catalytic process. The role of kinetic factors is very significant to monitor and control the reaction to the stage of the formation of product and optimisation of the reaction parameters, which can promise to bring the process to the commercial level.35 Kinetic studies on the formation of formic acid are also instructive for improving the catalyst system and providing basic information on the design and scale-up of appropriate reactors. In view of the above, and since formic acid is a bulk chemical that is produced at a commercial level, there is a pressing need for detailed kinetic studies on the Ru-HT-catalysed heterogeneous hydrogenation of CO2 to formic acid, so that the reaction parameters can be optimized under the employed reaction conditions. Thermodynamic insight is also substantiated in the present work for formic acid formation by determining activation parameters. The present work discusses the detailed kinetic investigations on the Ru-HT catalysed hydrogenation of CO2 to formic acid according to the influence of the effective operating parameters: the amount of catalyst, the partial pressure of CO2, the partial pressure of H2, total pressure, temperature, agitation speed, the ratio of the methanol/water (v/v) solvent and the reaction volume, directly on the rate of selective formation of formic acid.
Experimental
Materials and methods
Hydrogen gas UHP/zero grade (99.9%) and carbon dioxide UHP/zero grade gas cylinders were purchased from Hydro Gas India Pvt Ltd. Ruthenium trichloride [RuCl3·xH2O] was acquired from Sigma-Aldrich, USA. Magnesium chloride [MgCl2·6H2O; 98%], aluminum chloride [AlCl3·9H2O; 98%], sodium nitrate [NaNO3; 99.9%] and sodium hydroxide [NaOH; 99.9%] were purchased from S.D. Fine Chemicals Ltd, Mumbai, India and used as received. A.R. grade methanol was purchased from LobaChemie and was used without further purification. Double distilled Milli-Q deionized water was used for the synthesis of catalysts. The FT-IR spectra were recorded using a PerkinElmer Spectrum GX FT-IR system using KBr pellets in the mid-IR region. Powder X-ray diffraction (PXRD) patterns were obtained using a Rigaku MINIFLEX-II (FD 41521) powder X-ray diffractometer. The samples were scanned in the 2θ range of 10–80° using CuKα (λ = 1.54178 Å) radiation and a Ni filter. The textural analysis of the samples was carried out by nitrogen adsorption at 77.4 K using a Sorptometer (ASAP-2010, Micromeritics). X-Ray photoelectron spectroscopy (XPS) analysis was performed using an Omicron Nanotechnology system with monochromatic AlKα radiation through 1486.7 eV photon energy. FE-SEM (Field emission scanning electron microscopy) and EDX analysis were carried out using a JEOL JSM 7100F instrument at room temperature. Inductively coupled plasma (ICP) optical emission spectroscopy (ICPS-2000) was performed using a PerkinElmer Optima 2000 for the determination of the concentration of Ru element by using a Ru 240.3 analyte. The formic acid product was analyzed using a UV-vis spectrophotometer-P80+ in the wavelength range of 190–800 nm. These measurements were performed in 1 cm quartz cells using methanol as a standard. The UHPLC analysis was done using a SHIMADZU Nexera UHPLC system. Before HPLC analysis, all the samples and standards were filtered using an Axiva SFNY25XB nylon HPLC syringe filter (25 mm filter diameter/0.45 μm pore size) and left undiluted. Formic acid was separated using a HiQSilC18HS HPLC column (4.6 mm × 250 mm) operated at ambient temperature with an overall flow rate of 0.5 mL per minute of a 0.1% phosphoric acid mobile phase and detected using an SPD-M20A Prominence diode array UV detector set to 190 nm wavelength. The sample injection volume was 20 μL. The analysis was completed within 30 minutes, and data were recorded using the LabSolutions chromatography software.Ru-HT catalyst preparation
The Ru-HT catalyst was prepared using a coprecipitation method at a constant pH, referring to the previously published method.36 Solution A with NaNO3 (0.079 mol) and solution B with RuCl3·xH2O (0.5 mmol), Mg(NO3)2·6H2O (0.0522 mol) and Al(NO3)3·9H2O (0.0144 mol) were each prepared in 50 mL of double distilled deionized water. Solution B was added dropwise to solution A at room temperature over around 45 minutes with continuous stirring and a constant pH of 10 was maintained using 1 M NaOH solution. The resulting mixture was then transferred into a Teflon coated stainless steel autoclave to age at 80 °C for 16 hours under autogenous water vapor pressure. The precipitate was filtered and washed with hot distilled water to remove the unreacted ions present in it. The solid material was dried at 80 °C, then ground and stored under vacuum as Ru-HT with a 3.5/1 molar ratio of Mg/Al in the hydrotalcite (HT). Ru-HT was activated at 450 °C for 4 hours in a muffle furnace. As the catalyst was recycled for up to 7 reaction cycles, the recycled catalyst was named RRu-HT.CO2 hydrogenation and kinetic measurements
The hydrogenation of CO2 was done in a specially made high-pressure laboratory having safety precautions for using hydrogen and conducting reactions at high pressure and temperature, using an autoclave having 100 mL capacity. All operations involving the charging of gases were carried out in a well-ventilated fume hood. The reactor was charged with a known quantity of catalyst and a mixture of methanol and double distilled deionized water in the desired volume ratio (MeOH:H2O, v/v), and the vessel was then sealed and flushed with CO2 three times to remove air and was pressurized up to a final total desired pressure with CO2/H2 (1:1, p/p) at room temperature. The reactor was heated using a temperature controller to the desired temperature. Stirring was started at a fixed agitation speed (200–800 rpm) as the inside temperature of the reactor reached the employed temperature, which was considered to be the start of the reaction. After the desired reaction time, stirring and the heating were stopped and the reactor was allowed to cool to room temperature. The reactor was depressurized at room temperature and the vessel was opened and the reaction solution was collected by simple filtration to separate the catalyst.
The hydrogenation product was analyzed by HPLC and the concentration of the formed product was confirmed using a standard calibration curve plotted using area vs. formic acid concentration at the 190 nm wavelength. The formic acid peak was observed at a 19 minute retention time, which was completely detected as the same as that of the standard formic acid peak (Fig. S1, ESI†). The product was also analyzed spectrophotometrically using a chromotropic acid test and volumetrically by acid–base titration. A UV-vis P80+ spectrophotometer in the wavelength range of 190–800 nm was used for the analysis of the formic acid product. These measurements were performed in 1 cm quartz cells by taking methanol as a standard. Several repeated experiments were done to check the reproducibility of the reaction under identical reaction conditions, and the obtained results were found in the range of 3% variation.
For kinetic measurements, a series of kinetic experiments were carried out as a function of the amount of catalyst, the partial pressure of CO2, partial pressure of H2, total pressure, temperature, reaction volume and agitation speed for observing the effect of each individual parameter directly on the rate of formation of formic acid under identical employed reaction conditions. While varying an individual parameter, all the other parameters were kept constant. Aliquots were withdrawn from the reaction mixture at different desired time intervals and analysed to quantify the conversion in terms of the formation of formic acid in all the performed experiments.16
Results and discussion
Catalyst characterization
BET surface area
Comparative values of the pore size, pore volume and the surface area of HT, Ru-HT and RRu-HT show diminished values for the hydrotalcite-HT structure. The pore size and pore volume of HT, Ru-HT and RRu-HT are 26.18 nm and 0.72 cm3 g−1, 17.4 nm and 0.33 cm3 g−1 and 24 nm and 0.39 cm3 g−1, respectively. The surface areas of HT, Ru-HT and RRu-HT are 101 m2 g−1, 76 m2 g−1 and 65 m2 g−1, respectively. These attenuations in the texture of Ru-HT and RRu-HT propose the superiority of the extent of the size, volume and external surface area of the HT structure in both catalysts by the involvement of Ru. The increase in size and pore volume of RRu-HT and the decrease in surface area after the 7th catalytic cycle lead to the decrease in formic acid production via the weakened connection of the Ru ion to the HT layers.Inductively coupled plasma (ICP)
Inductively coupled plasma (ICP) mass spectrometry analysis was done to find the amount of Ru ion available in the Ru-HT catalyst, followed by the digestion of a known amount of catalyst using aqua regia (a mixture of concentrated HCl and HNO3) and making up the final solution with distilled water to 50 mL in a volumetric flask. The ICP analysis data showed a mean value of 0.487 mg L−1 (0.004 mmol) for the Ru ion present in the Ru-HT catalyst (Table 1). When the catalyst starts to drop the formic acid production after the 7th catalytic cycle, the leaching of the Ru ion was checked by ICP and was found to have a negative mean value of −0.114 mg L−1. The reduction in product formation is maybe due to the morphology and structural destruction of the catalyst after the many catalytic cycles.Kinetic profile
The detailed kinetic study in terms of the formation of the CO2 hydrogenated product formic acid was carried out at 60 °C, at which the maximum catalytic activity with a TON of 11389 for formic acid was achieved under 60 bar of CO2 and H2 (1:1, p/p) with 60 mL of a mixture of methanol and water (5:1, v/v) as the solvent in 24 hours. The kinetic profile for the hydrogenation of CO2 for the production of formic acid is presented in Fig. 1.
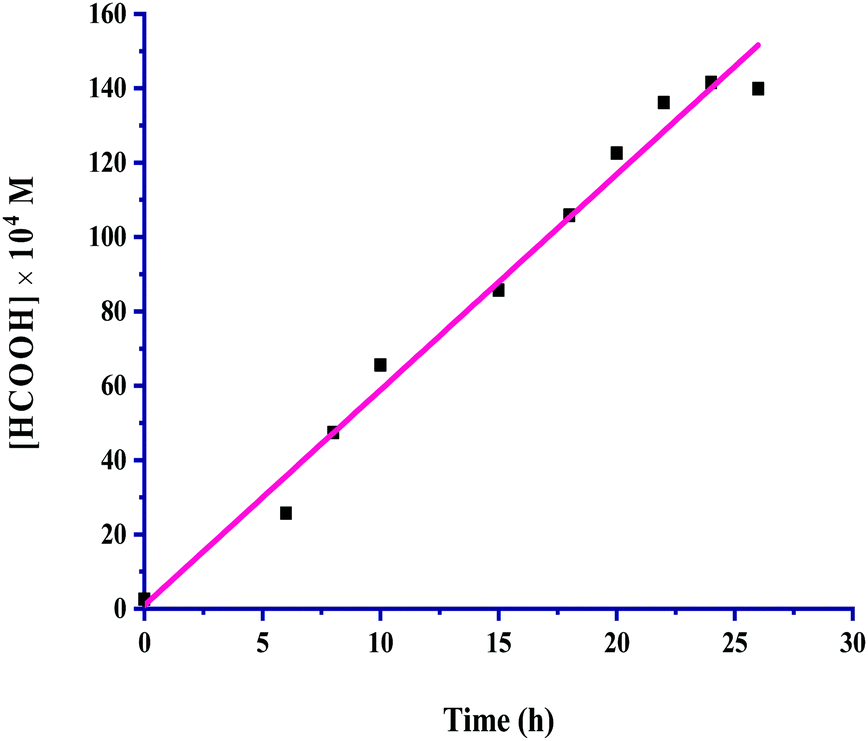 | ||
| Fig. 1 Kinetic profile: catalyst = 100 mg, pCO2 = 30 bar, pH2 = 30 bar, temperature = 60 °C, agitation speed 800 rpm and 60 mL of the solvent of methanol and water (5:1, v/v). | ||
The formic acid formation increased linearly up to 24 hours, and a further increase in the time did not bring about more formation; instead, it started to reach saturation. All other reaction parameters were also taken into account for this kinetic investigation, and the product formation rates were determined appropriately from the slopes of the corresponding plots. The catalyst amount, partial pressure of CO2 and H2, total pressure, temperature, agitation speed, reaction volume and volumetric ratio (v/v) of the solvents methanol and water used were considered in an inclusive range for the kinetic exploration by keeping the other reaction parameters constant, while a particular parameter was being studied under the employed reaction conditions. The range of conditions used for kinetic parametric optimization is given in Table 2.
| Reaction parameter | Range of parameter |
|---|---|
| Amount of catalyst, mg | 20–140 |
| Partial pressure of CO2, bar | 5–40 |
| Partial pressure of H2, bar | 5–40 |
| Total pressure, bar | 40–100 |
| Temperature, °C | 30–100 |
| Agitation speed, rpm | 200–800 |
| Reaction volume, mL | 12–60 |
| Methanol:water ratio (v/v) |
7:1–1:5 |
The rates of the formation of formic acid were examined concerning the effects of all the said reaction parameters and were determined using eqn (1) from the plot given in Fig. 1, drawn by increasing concentration of formic acid with time.
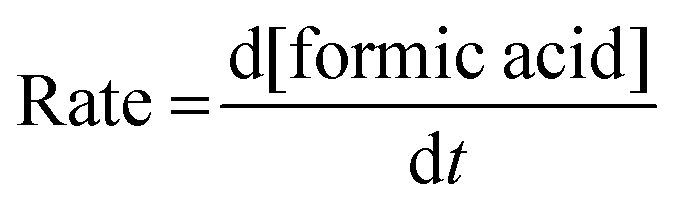 | (1) |
Dependence of the rate on the amount of catalyst
The relationship between the amount of catalyst and the rate of CO2 hydrogenation was considered by taking 20–130 mg of catalyst, and the results are given in Fig. 2. The plot of rate vs. catalyst amount passes through the origin, indicating that the reaction is completely catalytic under the employed reaction conditions. For the increase in the amount of catalyst from 20 mg, the rate of reaction increased steadily as the total available active sites provide more exposure to bind both gases to itself, leading to an effective increment up to 100 mg of catalyst. On further increasing the catalyst amount a linear increase was not seen and the rate reached saturation. The rate of the formation of formic acid was found to have a first-order dependence on the amount of catalyst. | ||
| Fig. 2 Dependence of rate on the amount of catalyst: pCO2 = 30 bar, pH2 = 30 bar, temperature = 60 °C, agitation speed 800 rpm and 60 mL of a solvent of methanol and water (5:1, v/v). | ||
Dependence of the rate on partial pressures of CO2 and H2
The partial pressure of CO2 was varied from 5 to 40 bar to examine its effect on the rate, and the results are portrayed in Fig. 3. It is very clear from the graph that the rate of formic acid formation was very low at a lower pressure of CO2, which suggested that an exact and critical amount of CO2 gas is required along with H2 gas to get itself appreciably hydrogenated to yield formic acid. The rate of formation was increased after an acute pressure of CO2 was provided. The determined product formation first reached a maximum level and then started to remain unaffected on further increasing the pressure above 30 bar. On increasing the partial pressure of CO2 at a constant pressure (30 bar) of H2, the solubility of H2 remained constant, and the solubility of CO2 was increased. The increased solubility of CO2 with pressure increases the concentration of CO2 in the reaction. This causes more availability of CO2 and results in an increased rate of formation of formic acid. First order kinetics were follow in terms of the partial pressure of CO2 gas. The rate of formation of formic acid was increased with increasing the partial pressure of H2 from 5 to 30 bar pressure, and a further increase in H2 pressure provided saturation of the rate. These results, as shown in Fig. 4, indicated that the highest rate was for a 1:1 (p/p) ratio of both gases at a total pressure of 60 bar. Similar to the dependence of partial pressure of CO2, the kinetic dependence in terms of the partial pressure of H2 was also observed to be first order. The experimental observations for the effect of the partial pressures of both gases indicated that the 1:1 (p/p) ratio is highly favourable for the maximum formation of formic acid under the studied chemical and physical conditions. However, the selectivity of formic acid remained effective with other varied ratios of CO2 and H2 gases during partial pressure study.
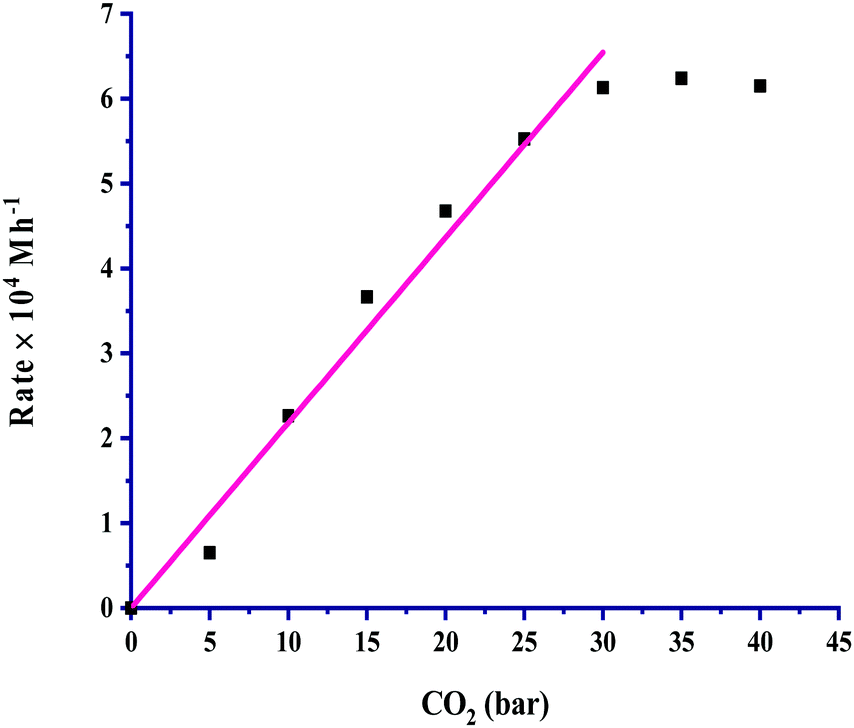 | ||
| Fig. 3 Dependence of rate on pCO2: catalyst = 100 mg, pH2 = 30 bar, temperature = 60 °C, agitation speed 800 rpm and 60 mL of a solvent of methanol and water (5:1, v/v). | ||
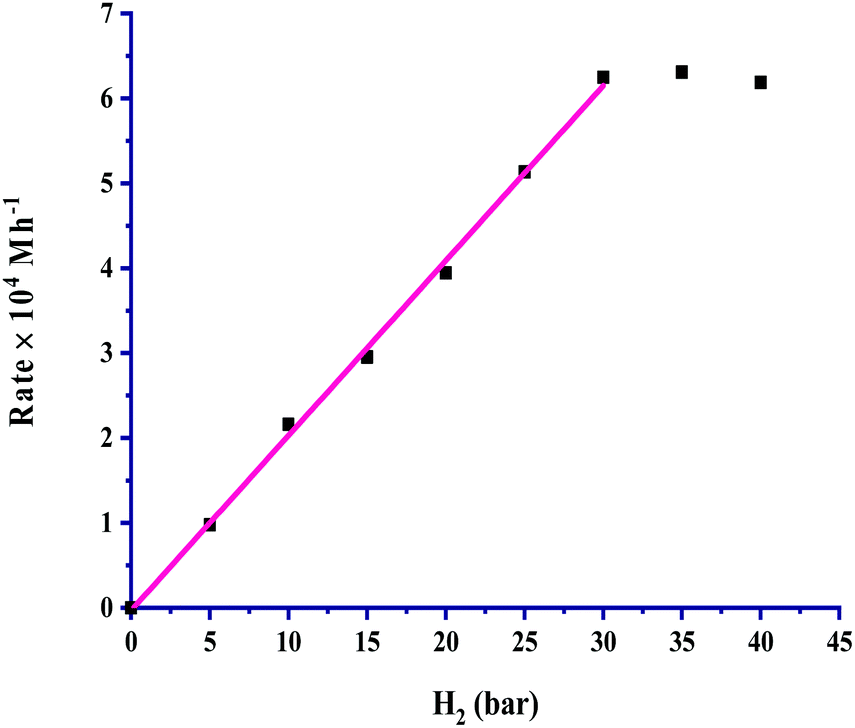 | ||
| Fig. 4 Dependence of rate on pH2: catalyst = 100 mg, pCO2 = 30 bar, temperature = 60 °C, agitation speed 800 rpm and 60 mL of a solvent of methanol and water (5:1, v/v). | ||
Dependence of rate on the total pressure, temperature and agitation speed
The dependence of the rate was investigated to observe the effect of total pressure, temperature and agitation speed, and the corresponding results are listed in Table 3.:1, v/v)
| Entry | pCO2 (bar) | pH2 (bar) | Total pressure (bar) | Temp. (°C) | Agitation speed (rpm) | Rate × 104 (M h−1) |
|---|---|---|---|---|---|---|
| 1 | 20 | 20 | 40 | 60 | 800 | 4.18 |
| 2 | 25 | 25 | 50 | 60 | 800 | 5.15 |
| 3 | 30 | 30 | 60 | 60 | 800 | 6.25 |
| 4 | 35 | 35 | 70 | 60 | 800 | 6.30 |
| 5 | 40 | 40 | 80 | 60 | 800 | 6.32 |
| 6 | 45 | 45 | 90 | 60 | 800 | 6.33 |
| 7 | 50 | 50 | 100 | 60 | 800 | 6.33 |
| 8 | 30 | 30 | 60 | 30 | 800 | 2.01 |
| 9 | 30 | 30 | 60 | 40 | 800 | 3.01 |
| 10 | 30 | 30 | 60 | 50 | 800 | 4.00 |
| 11 | 30 | 30 | 60 | 60 | 800 | 6.25 |
| 12 | 30 | 30 | 60 | 70 | 800 | 6.30 |
| 13 | 30 | 30 | 60 | 80 | 800 | 6.34 |
| 14 | 30 | 30 | 60 | 90 | 800 | 6.35 |
| 15 | 30 | 30 | 60 | 100 | 800 | 6.37 |
| 16 | 30 | 30 | 60 | 60 | 200 | 2.96 |
| 17 | 30 | 30 | 60 | 60 | 300 | 3.80 |
| 18 | 30 | 30 | 60 | 60 | 400 | 4.50 |
| 19 | 30 | 30 | 60 | 60 | 500 | 5.02 |
| 20 | 30 | 30 | 60 | 60 | 600 | 6.20 |
| 21 | 30 | 30 | 60 | 60 | 700 | 6.25 |
| 22 | 30 | 30 | 60 | 60 | 800 | 6.26 |
The total pressure was varied by maintaining the ratio of CO2 and H2 at 1:1 (p/p) in the pressure range of 40–100 bar, and the corresponding results listed in Table 3 (entries, 1–7) showed a significant effect on the rate. The rates were linearly increased with a pressure of up to 60 bar from 4.18 × 10−4 M h−1 to 6.25 × 10−4 M h−1 from 30 to 60 bar, and a further increase in total pressure started to attain saturation. On increasing the pressure, the solubilities of both gases increased, which resulted in an increase in the concentration of the gases. Thus the increased concentration of gases enhanced their availability for the reaction and the rates were increased with pressure. The kinetic trend of the total pressure and partial pressures of the individual gases CO2 and H2 on the rate was in good agreement of being identical, where the rates were linearly increased up to 60 bar pressure, and above that the rates were unaffected. The dependence of the hydrogenation reaction rate for formic acid on the temperature was studied from near room temperature to high temperatures, in the range of 30 to 100 °C. The solubilities of the gases in the solvent tend to decrease with an increase in temperature. The dependence showed a linear increase in the rate with a rise in temperature from 30 to 60 °C, and then with a further increase in the temperature the rates were found to be saturated. The finding indicated that the solubility effect with the rise in temperature from 30 to 60 °C is not very effective towards reducing the available concentrations of the gases to react to form formic acid and the rates were linearly increased with temperature. The saturating effect of the temperature above 60 °C for the formation of formic acid may be considered for the much lower solubility of CO2 and H2 gases at the higher temperature. Under the reaction conditions, lower solubility becomes unable to effectively enhance the available concentration of these gases. The Arrhenius plot drawn in the temperature range of 30 to 60 °C shows a good linear fit and was used for evaluating the activation parameters (Fig. 5). The determined activation parameters of Ea = 34.5 ± 2.5 kJ mol−1, ΔH# = 32 ± 2.5 kJ mol−1 and ΔS# = −384 ± 5 J deg−1 mol−1 indicated that both the activation enthalpy and the highly negative entropy associated with hydrogenation were highly favourable for the activation of CO2 and H2 for the selective formation of formic acid. The reported activation parameters for the Rh-hydrotalcite (Rh-HT)-based catalyst, for the catalyzed hydrogenation of CO2 to formic acid, are Ea = 33.5 ± 2.5 kJ mol−1, ΔH# = 30.9 ± 2.5 kJ mol−1 and ΔS# = −275 ± 5 J deg−1 mol−1.18
 | ||
| Fig. 5 Arrhenius plot of log rate vs. 1/T for the formation of formic acid. | ||
The activation enthalpies are almost comparable for the Ru-HT and Rh-HT catalytic systems; however, the entropy of activation is observed to be remarkably much more favourably associated with Ru-HT. An almost closer activation energy, 31 kJ mol−1, is reported for Ru-based catalysts under homogeneous conditions up to a slightly lower temperature of 40 °C.44 G. Laurenczy et al. investigated hydrogenation of CO2 to formic acid catalyzed by Ru-complexes and obtained an activation enthalpy of 96 kJ mol−1 under homogeneous conditions, studying the reaction in the temperature range of 23–90 °C.31 These observations indicated that heterogeneous catalyst systems performing well towards the activation enthalpies associated with CO2 hydrogenation, and that the enthalpies are effectively reduced thereby making the reaction more favourable.
The dependence of the rate on the agitation speed in the range of 200–800 rpm (Table 3, entries 16–22) showed that the rate significantly increased for 200–500 rpm, from 2.96 × 10−4 M h−1 to 5.02 × 10−4 M h−1, respectively, and on further increasing the agitation speed the rates followed a steady state. At the lower agitation speed, the initial concentration of dissolved gases was less, which indicates a diffusion-controlled reaction; hence, the rate of reaction increases. Furthermore, on increasing the agitation speed, although the initial concentrations of dissolved gases are significantly increased and the concentration of dissolved gases in the reaction mixture becomes higher, the rate remained unaffected. Hence an agitation speed above 500 rpm has a negligible effect upon the reaction rate, which indicated the absence of mass transfer resistance and that the reaction is under the kinetic regime. To ensure a purely kinetic regime, all the experiments were preferentially performed at 800 rpm. The use of finely powdered Ru-HT catalyst eliminated the effect of internal diffusion during the kinetic study. The particle size of the hydrotalcite-based ruthenium catalyst is reported to be in the order of a nanometer.45 Also the pore size of Ru-HT is 17.4 nm, which is bigger than that of general mesoporous materials, enabling easy ingress of the reactants and egress of the products. This is also in line with the observations on the Cu-hydrotalcite catalyst, where the particle size of below 100 micrometers is reported to be in the kinetic regime by eliminating internal diffusion.46 The stability of the product was observed to be well maintained over the entire range of the total applied pressure, temperature and agitation speed.
Dependence of rate on reaction volume and v/v ratio of methanol and water
The hydrogenation of CO2 was carried out by varying the reaction volume fivefold, from 12 to 60 mL by keeping the ratio of methanol/water, 5:1 (v/v), and the corresponding results are given in Table 4. The reaction volume and the respective solubilities of the CO2 and H2 gases in the used solvent have remarkably contributed to influence the rate. The solubilities in mmol L−1 at 1 atm pressure and 25 °C, of H2 and CO2 in water, are 0.68 and 58.8, respectively.47 Under these conditions, the solubilities of H2 and CO2 in methanol are 3.86 and 137.8, respectively. Solubilities of both gases are appreciably higher in methanol. In a comparison of hydrogen, the solubility of CO2 is much higher in both the solvents water and methanol. CO2 is a polar solute with a partial positive charge on the carbon and a partial negative charge on the oxygen, and water and methanol are polar solvents that greatly favour the solubility.
| Entry | Methanol, mL | Water, mL | Rean volume, mL | Methanol/water ratio (v/v) | Rate × 104 M h−1 |
|---|---|---|---|---|---|
| 1 | 10 | 2 | 12 | 5:1 |
1.22 |
| 2 | 15 | 3 | 18 | 5:1 |
1.85 |
| 3 | 20 | 4 | 24 | 5:1 |
2.50 |
| 4 | 25 | 5 | 30 | 5:1 |
3.12 |
| 5 | 30 | 6 | 36 | 5:1 |
3.66 |
| 6 | 35 | 7 | 42 | 5:1 |
4.37 |
| 7 | 40 | 8 | 48 | 5:1 |
4.83 |
| 8 | 45 | 9 | 54 | 5:1 |
5.47 |
| 9 | 52.5 | 7.5 | 60 | 7:1 |
6.26 |
| 10 | 51.5 | 8.5 | 60 | 6:1 |
6.27 |
| 11 | 50 | 10 | 60 | 5:1 |
6.25 |
| 12 | 40 | 20 | 60 | 4:2 |
5.83 |
| 13 | 30 | 30 | 60 | 3:3 |
5.42 |
| 14 | 20 | 40 | 60 | 2:4 |
4.16 |
| 15 | 10 | 50 | 60 | 1:5 |
3.33 |
| 16 | 60 | 0 | 60 | — | 2.99 |
| 17 | 0 | 60 | 60 | — | 1.02 |
The rate of formation was linearly increased (Table 4, entries 1–9) with an increase in the reaction volume. With 12 mL of the solvent, the rate was 1.22 × 10−4 M h−1, which was appreciably increased to 6.25 × 10−4 M h−1 with 60 mL of solvent. The larger volume of solvent was found to be very effective to dissolve the gases, which allows for higher concentrations of CO2 and H2 in the reaction system. Thus more solubility in the larger reaction volume enhances the available concentrations of these gases, resulting in an increased rate.
To observe the effect of the ratio of the used solvent on the rate, the ratio (v/v) of methanol and water was suitably varied from 7:1 to 1:5 by maintaining the total reaction volume 60 mL under identical conditions of the other parameters, and the results are listed in Table 4. A decreasing trend in the rate (Table 4, entries 10–15) was observed with a decreasing volume of methanol from 5:1 to 1:5. The maximum rate (6.25 × 10−4 M h−1) was with the ratio of 5:1, and on further increasing the volume of methanol the rates were almost constant at the ratios of 6:1 and 7:1 (Table 4, entries 9 and 10) indicating that the ratio of 5:1 is effective to maintain the best performance of the catalyst to produce formic acid. The rate using only methanol as a solvent without water was 2.99 × 10−4 M h−1 (Table 4, entry 16), and with only water (Table 4, entry 17) it was 1.02 × 10−4 M h−1. The higher rate using only methanol as the solvent is due to the higher solubilities of both the gases in it. An effective synergy of the mixed methane–water solvent was observed to enhance the rate. These rates obtained from the individual solvents methanol and water were effectively increased with the mixed solvent to 6.25 × 10−4 M h−1 at the ratio of 5:1.
Reaction mechanism
Based on the kinetic first-order dependence in terms of the amounts of the catalyst and partial pressures of CO2 and H2 and the experimental observations, a reaction mechanistic route of CO2 hydrogenation to formic acid over Ru-HT is proposed in Scheme 1via two plausible reaction pathways. | ||
| Scheme 1 Proposed reaction mechanisms of CO2 hydrogenation to formic acid over Ru-HT. | ||
The first one is a stepwise mechanism in which the inserted metal ion of Ru-HT (1) triggers the H2 molecule to form hydride bonds with the ruthenium ion for the formation of the intermediate (2). This is in line with the experimentally observed first-order kinetic dependence with respect to the catalyst and H2. The reactivity of H2 facilitates its interaction to react with the catalyst first. Based on the first-order dependence with CO2 and comparatively lower reactivity of it, in the next step, the reaction proceeds with the absorption of CO2 on the surface of the catalyst and the bond between carbon and the hydride of the metal ion for the formation of formate ion with the metal.
The second one is based on the contribution of water to the reaction, which is more precise concerning an effectually active solvent molecule in the mechanistic path. Consequently, the dissociation of the coordinated water increases the scope for the successive insertion and coordination of CO2. The reaction proceeds quickly and easily since H2 gets activated first by the catalyst (1) to form Ru–H2 (Ru-dihydride) (2) under the applied reaction conditions because the reactivity of CO2 is less than that of H2.48–51 The reaction of Ru-hydride (2) with CO2 through the insertion of CO2 into one of the hydride bonds forms the intermediate formate (3). In the rate-determining step, the reductive elimination of formate (3) yields formic acid and the coordinatively unsaturated ruthenium species.36,48–50 To complete the catalytic cycle, the rapid oxidative addition of dihydrogen regenerates the catalyst (1).
The presence of water with methanol enhanced the formation of formic acid. However, the solubility of both gases in water is significantly less than that of methanol.47 On the one hand, the solubility of CO2 is more than twice, and that of H2 is more than 5 times greater in methanol than it is in water. On the other hand, five times excess volume of methanol with respect to water is taken during the catalytic reaction. Thus, in the volume of both solvents taken, in terms of the solubility, the contribution of methanol as a solvent is greater to enhance the reaction rate. Here the role of water to enhance the rate of formation is small concerning its solvent properties due to the higher solubility of both gases in methanol compared with water. The contribution of water appeared to be more specific regarding the molecular effect functioning in the mechanistic path. During the reaction, the molecular effect in terms of the operative mechanism was contributed by molecular water, which increases the dissociation of the coordinated water and eases the consequential insertion and coordination of CO2 to a high amount.
The aquated Ru intermediate (4) reacted with one of the oxygens of CO2 for its coordination via the H-bonding interaction of the water, which caused the formation of the next intermediate (5). The insertion of CO2 into the Ru–H bond during formate formation (6) caused stabilization of the transition state between intermediates (5) and (6). This stabilization polarizes the fragment of CO2 by enhancing the electrophilicity of CO2 on the carbon and the susceptibility of hydride transfer from Ru to yield formic acid along with the intermediate (7). This intermediate (7), undergoing rapid oxidative addition of dihydrogen, formed the catalyst (1).
Conclusions
Kinetic, thermodynamic and mechanistic aspects of the ruthenium-inserted hydrotalcite (Ru-HT) heterogeneous catalyst were investigated for hydrogenation of CO2 for the selective formation of formic acid. The rates were determined in terms of the formation of formic acid. Kinetic investigations were performed as a function of the amount of catalyst, the partial pressures of the individual gases CO2 and H2, total pressure, agitation speed, temperature, reaction volume and the volumetric ratio (v/v) of the used mixed solvent mixture of methanol and water. The rate of formation of formic acid depended significantly on all these studied parameters. The rate of formation followed first-order kinetics with respect to the amount of catalyst and the partial pressures of the reacting gases. All the parameters were varied over a wide range of values to observe their effects on the rate and for their kinetic parametric optimisation. The maximum rate of 6.25 × 10−4 M h−1 was observed under the following optimised kinetic reaction conditions: 100 mg of catalyst, pCO2 = 30 bar, pH2 = 30 bar, temperature = 60 °C, agitation speed = 800 rpm and a methanol–water (v:v = 5:1) solvent. The activation enthalpy and entropy determined from the temperature dependence were very favourable under the employed reaction conditions. Based on the kinetic findings and the experimental results, two reaction mechanisms for the Ru-HT-catalyzed synthesis of formic acid were proposed, wherein the molecular effect of water was observed to be efficiently operative.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
CSMCRI communication No. IMC 03, CSIR-CSMCRI – 155/2018. The authors thank Council of Scientific and Industrial Research (CSIR), New Delhi, India for the financial support through Network Project on Clean Coal Technology (Tap Coal, CSC 0102) and Analytical Division and Central Instrumentation Facility of CSIR Central Salt and Marine Chemicals Research Institute, Bhavnagar, for providing instrumental analysis.Notes and references
- (a) W.-H. Wang, Y. Himeda, J. T. Muckerman, G. F. Manbeck and E. Fujita, Chem. Rev., 2015, 115, 12936 CrossRef CAS PubMed; (b) M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS PubMed; (c) G. Centi, E. A. Quadrelli and S. Perathoner, Energy Environ. Sci., 2013, 6, 1711 RSC; (d) M. Grasemann and G. Laurenczy, Energy Environ. Sci., 2012, 5, 8171 RSC; (e) C. Federsel, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 6254 ( Angew. Chem. , 2010 , 122 , 6392 ) CrossRef CAS PubMed; (f) Y. Inoue, H. Izumida, Y. Sasaki and H. Hashimoto, Chem. Lett., 1976, 863 CrossRef CAS.
- J. Z. Zhang, Z. Li, H. Wang and C. Y. Wang, J. Mol. Catal. A: Chem., 1996, 112, 9 CrossRef CAS.
- R. S. Shukla, S. D. Bhatt, R. B. Thorat and R. V. Jasra, Appl. Catal., A, 2005, 294, 111 CrossRef CAS.
- C. Fellay, P. J. Dyson and G. A. Laurenczy, Angew. Chem., Int. Ed., 2008, 47, 3966 CrossRef CAS PubMed.
- (a) Y. Kiso and K. Saeki, Jpn Pat., 5236617, 1977 Search PubMed; (b) J. Z. Zhang, Z. Li, H. Wang and C. Y. Wang, J. Mol. Catal. A: Chem., 1996, 112, 9 CrossRef CAS.
- W. Gan, P. J. Dyson and G. Laurenczy, ChemCatChem, 2013, 5, 3124 CrossRef CAS.
- T. Schaub and R. A. Paciello, Angew. Chem., Int. Ed., 2011, 50, 7278 CrossRef CAS PubMed.
- G. A. Olah, A. Goeppert and G. K. Surya Prakash, Beyond Oil and Gas: The Methanol Economy, Weinheim, Wiley-YCH, 2006 Search PubMed.
- S. Saeidi, N. A. S. Amin and M. R. Rahimpour, J. CO2 Util., 2014, 5, 66 CrossRef CAS.
- R. W. Dorner, D. R. Hardy, F. W. Williams and H. D. Willauer, Energy Environ. Sci., 2010, 3, 884 RSC.
- (a) J. Klankermayer, S. Wesselbaum, K. Beydoun and W. Leitner, Angew. Chem., Int. Ed., 2016, 55, 7296 ( Angew. Chem. , 2016 , 128 , 7416 ) CrossRef CAS PubMed; (b) J. Klankermayer and W. Leitner, Philos. Trans. R. Soc., A, 2016, 374, 20150315 CrossRef PubMed; (c) W. Leitner, Angew. Chem., Int. Ed. Engl., 1995, 34, 2207 ( Angew. Chem. , 1995 , 107 , 2391 ) CrossRef CAS; (d) H. Hayashi, S. Ogo and S. Fukuzumi, Chem. Commun., 2004, 2714 RSC; (e) W. Leitner, E. Dinjus and F. Gaßner, in CO2 Chemistry-Aqueous-Phase Organometallic Catalysis, ed. B. Cornils and W. A. Hermann, Wiley-VCH, Weinheim, 1998, p. 488 Search PubMed.
- M. S. Maru, P. Patel, N. H. Khan and R. S. Shukla, Curr. Catal., 2020, 9, 59 CrossRef CAS.
- (a) P. Patel, S. Nandi, M. S. Maru, R. I. Kureshy and N. H. Khan, J. CO2 Util., 2018, 25, 310 CrossRef CAS; (b) M. S. Maru, S. Ram, R. S. Shukla and N. H. Khan, J. Mol. Catal., 2018, 446, 23 CrossRef CAS.
- M. S. Maru, S. Ram, J. H. Advani and R. S. Shukla, ChemistrySelect, 2017, 2, 3823 CrossRef CAS.
- M. D. Khokhar, R. S. Shukla and R. V. Jasra, J. Mol. Catal., 2015, 400, 1 CrossRef CAS.
- M. D. Khokhar, R. S. Shukla and R. V. Jasra, React. Kinet., Mech. Catal., 2015, 114, 265 CrossRef CAS.
- (a) N. Sudheesh and R. S. Shukla, Appl. Catal., A, 2014, 473, 116 CrossRef CAS; (b) N. Sudheesh and R. S. Shukla, Appl. Catal., A, 2013, 453, 159 CrossRef CAS.
- (a) N. Sudheesh, J. N. Parmar and R. S. Shukla, Appl. Catal., A, 2012, 415–416, 124 CrossRef CAS; (b) N. Sudheesh, A. K. Chaturvedi and R. S. Shukla, Appl. Catal., A, 2011, 409–410, 99 CrossRef CAS.
- (a) N. Sudheesh, S. K. Sharma, R. S. Shukla and R. V. Jasra, J. Mol. Catal. A: Chem., 2010, 316, 23 CrossRef CAS; (b) N. Sudheesh, S. K. Sharma, R. S. Shukla and R. V. Jasra, J. Mol. Catal. A: Chem., 2008, 296, 61 CrossRef CAS.
- S. K. Sharma, R. S. Shukla, P. A. Parikh and R. V. Jasra, J. Mol. Catal., 2009, 304, 33 CrossRef CAS.
- V. K. Srivastava, S. K. Sharma, R. S. Shukla and R. V. Jasra, Ind. Eng. Chem. Res., 2008, 47, 3795 CrossRef CAS.
- (a) S. K. Sharma, V. K. Srivastava, R. S. Shukla, P. A. Parikh and R. V. Jasra, New J. Chem., 2007, 31, 277 RSC; (b) R. V. Jasra, V. K. Srivastava, R. S. Shukla, H. C. Bajaj and S. D. Bhatt, US Pat., 7294745 B2, 2007 Search PubMed; (c) V. K. Srivastava, S. K. Sharma, R. S. Shukla and R. V. Jasra, Catal. Commun., 2006, 7, 879 CrossRef CAS.
- F. Basile, L. Basini, G. Fornasari, M. Gazzano, F. Trifiro and A. Vaccari, Chem. Commun., 1996, 2435 RSC.
- K. Kaneda, T. Yamashita, T. Matsushita and K. Ebitani, J. Org. Chem., 1998, 63, 1750 CrossRef CAS.
- T. Matsushita, K. Ebitani and K. Kaneda, Chem. Commun., 1999, 265 RSC.
- (a) K. Motokura, D. Nishimura, K. Mori, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2004, 126, 5662 CrossRef CAS PubMed; (b) K. Motokura, T. Mizugaki, K. Ebitani and K. Kaneda, Tetrahedron Lett., 2004, 45, 6029 CrossRef CAS.
- F. Basile, G. Fornasari, M. Gazzano and A. Vaccari, J. Mater. Chem., 2002, 12, 3296 RSC.
- B. B. İsmail, Y. Mehmet, S. K. Gülşah and B. Ahmet, Turk. J. Chem., 2020, 44, 364 CrossRef PubMed.
- J. A. Martins, A. C. Faria, M. A. Soria, C. V. Miguel, A. E. Rodrigues and L. M. Madeira, Catalysts, 2019, 9, 1008 CrossRef CAS.
- W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703 RSC.
- S. Moret, P. J. Dyson and G. Laurenczy, Nat. Commun., 2014, 5, 1 Search PubMed.
- G. Laurenczy, F. Joó and L. Nádasdi, Inorg. Chem., 2000, 39, 5083 CrossRef CAS PubMed.
- A. Xin, Z. Yizan, Z. Qiang and W. Jinfu, Chin. J. Chem. Eng., 2009, 17, 88 CrossRef.
- J. Ye, C.-J. Liu, D. Mei and Q. Ge, J. Catal., 2014, 317, 44 CrossRef CAS.
- R. J. Berger, E. H. Stitt, G. B. Martin, F. Kapteijn and J. A. Moulijn, CATTECH, 2001, 5, 30 CrossRef.
- (a) R. Salomão, L. M. Milena, M. H. Wakamatsu and V. C. Pandolfelli, Ceram. Int., 2011, 37, 3063 CrossRef; (b) S. K. Sharma, P. A. Parikh and R. V. Jasra, J. Mol. Catal. A: Chem., 2010, 317, 27 CrossRef CAS.
- F. Cavani, F. Trifiro and A. Vaccari, Catal. Today, 1991, 11, 173 CrossRef CAS.
- G. H. Gunasekar, K. Park, K.-D. Jung and S. Yoon, Inorg. Chem. Front., 2016, 3, 882 RSC.
- B. Wiyantoko, P. Kurniawati, T. E. Purbaningtias and I. Fatimah, Procedia Chem., 2015, 17, 21 CrossRef CAS.
- (a) S. Pichaikaran and P. Arumugam, Green Chem., 2016, 18, 2888 RSC; (b) Md. T. Uddin, Y. Nicolas, C. Olivier, L. Servant, T. Toupance, S. Li, A. Klein and W. Jaegermann, Phys. Chem. Chem. Phys., 2015, 17, 5090 RSC; (c) N. Ullah and S. Omanovic, Mater. Chem. Phys., 2015, 159, 119 CrossRef CAS; (d) A. Ballarini, P. Benito, G. Fornasari, O. Scelza and A. Vaccari, Int. J. Hydrogen Energy, 2013, 38, 15128 CrossRef CAS.
- (a) J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-ray Photoelectron Spectroscopy: A Reference Book of Standard Spectra for Identification and Interpretation of XPS Data, PerkinElmer, Boca Raton, FL, 1992 Search PubMed; (b) R. E. Shepherd, A. Proctor, W. W. Henderson and T. K. Myser, Inorg. Chem., 1987, 26, 2440 CrossRef CAS.
- (a) A. Jana, J. Mondal, P. Borah, S. Mondal, A. Bhaumik and Y. Zhao, Chem. Commun., 2015, 5151, 10746 RSC; (b) D. J. Morgan, Surf. Interface Anal., 2015, 47, 1072 CrossRef CAS.
- (a) A. Dibenedetto, P. Stufano, F. Nocito and M. Aresta, ChemSusChem, 2011, 4, 1311 CrossRef CAS PubMed; (b) S. Ogo, R. Kabe, H. Hayashi, R. Harada and S. Fukuzumi, Dalton Trans., 2006, 4657 RSC; (c) Y. Ohnishi, T. Matsunaga, Y. Nakao, H. Sato and S. Sakaki, Organometallics, 2006, 25, 3352 CrossRef CAS; (d) Y. Ohnishi, T. Matsunaga, Y. Nakao, H. Sato and S. Sakaki, J. Am. Chem. Soc., 2005, 127, 4021 CrossRef CAS PubMed.
- M. M. Taquikhan, S. B. Halligudi and S. Shukla, J. Mol. Catal., 1989, 57, 47 CrossRef.
- J. A. Martins, A. C. Faria, M. A. Soria, C. V. Migual, A. E. Rodrigues and L. M. Maderia, Catalysts, 2019, 9, 1008 CrossRef CAS.
- P. Kumar, R. C. Korosec and U. L. Stanger, J. Colloid Interface Sci., 2021, 585, 549 CrossRef CAS PubMed.
- H. Stephen and T. Stephen, Solubilities of Inorganic and Organic Compounds part I and II, Pergamon Press, New York, 1963, vol. I Search PubMed.
- (a) P. G. Jessop, F. Joý and C.-C. Tai, Coord. Chem. Rev., 2004, 248, 2425 CrossRef CAS; (b) P. G. Jessop, T. Ikariya and R. Noyori, Chem. Rev., 1995, 95, 259 CrossRef CAS.
- F. Joo, Aqueous Organometallic Catalysis, Kluwer Academic Publishers, Dordrecht, 2001 Search PubMed.
- (a) W. Leitner, E. Dinjus and F. GaBtier, J. Organomet. Chem., 1994, 475, 257 CrossRef CAS; (b) F. Gassner and W. Leitner, J. Chem. Soc., Chem. Commun., 1993, 1465 RSC; (c) P. G. Jessop, T. Ikariya and R. Noyori, Nature, 1994, 368, 231 CrossRef CAS.
- J. C. Tsai and K. M. Nicholas, J. Am. Chem. Soc., 1992, 114, 5117 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00431j |
| This journal is © The Royal Society of Chemistry 2021 |