
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polymeric near infrared emitters with bay-annulated indigo moieties†
Ana Clara B.
Rodrigues
 a,
Anika
Eckert
b,
João
Pina
a,
Ullrich
Scherf
b and
J. Sérgio
Seixas de Melo
*a
a,
Anika
Eckert
b,
João
Pina
a,
Ullrich
Scherf
b and
J. Sérgio
Seixas de Melo
*a
aUniversity of Coimbra, CQC, Department of Chemistry, P3004-535 Coimbra, Portugal. E-mail: sseixas@ci.uc.pt
bBergische Universität Wuppertal, Macromolecular Chemistry Group (buwmakro) and Wuppertal Center for Smart Materials and Systems (cm@s), Gauss-Str. 20, D-42119, Wuppertal, Germany
First published on 14th April 2021
Abstract
Three new copolymers based on bay-annulated indigo (BAI) building blocks were synthesized through Stille-type cross-coupling reactions, alternating the electron-deficient BAI units with thiophene-based donor moieties. These polymers showed absorption and fluorescence emission in the NIR region. Their spatial structures were found to prevent aggregation-caused quenching (ACQ) in the solid state. Photoluminescence at 77 K is found ca. four times more intense than at 293 K. Based on time-resolved photoluminescence experiments, the excited-state energy transfer hopping deactivation in the copolymers is discussed in terms of a Förster-type mechanism, with time constants ranging from 1010 to 1011 s−1.
Introduction
Natural dyes and pigments as indigo (Scheme 1) and its natural (indigoids) or synthetic derivatives are valued for their bright color and high photochemical stability and have been used since antiquity.1–5 A bay-annulated indigo derivative known as Cibalackrot (7,14-diphenyldiindolo[3,2,1-de;3′,2′,1′-ij][1,5]naphthyridine-6,13-dione), an industrial dye that has been known for over a century,6 has a highly rigid chemical structure, showing strong luminescence7 and is, as indigo, capable of singlet fission8,9 and can be used as an organic laser dye.10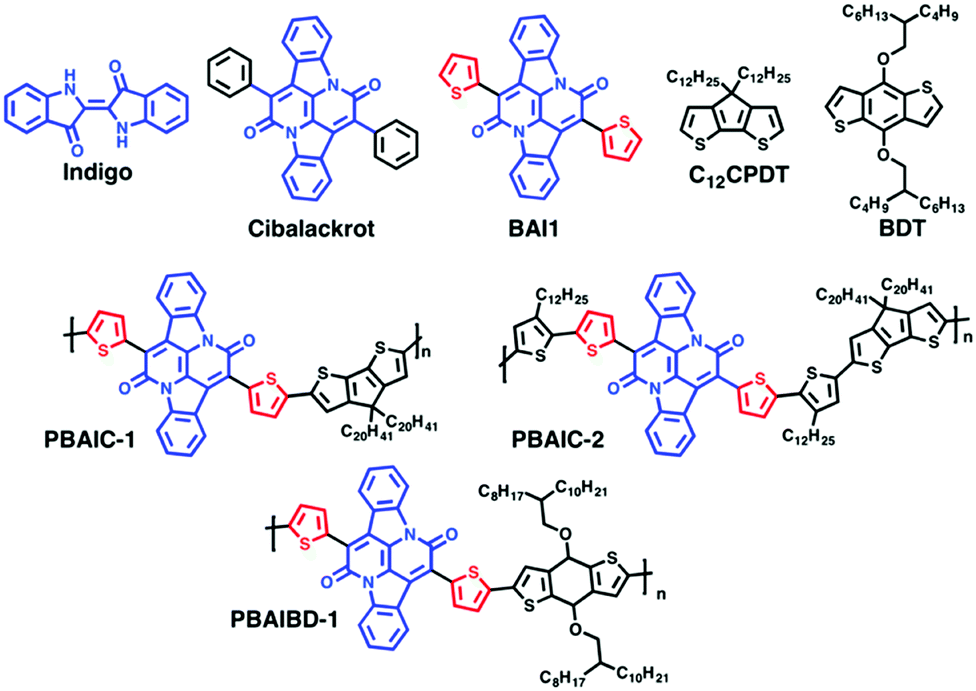 | ||
| Scheme 1 Structures and acronyms of indigo and its bay-annulated derivates, Cibalackrot and BAI1, together with the chemical structures of the thiophene-based building blocks (C12CPDT and BDT) used to prepare the NIR-emitting copolymers: PBAIC-1, PBAIC-2 and PBAIBD-1. | ||
The need for better performing and stable organic materials in organic electronics inspired the resurgence of historically long known molecules as indigo11,12 and their subsequent chemical transformation into new families of π-conjugated building blocks,12 also for construction of new macromolecular semiconductors.11 In this work, indigo was functionalized by reaction with 2-thienylacetyl chloride, as described previously,13,14 to produce the bay-annulated indigo derivative BAI1 (Scheme 1). The electron-deficient BAI1 moiety is a suitable building block for the generation of push–pull type conjugated polymers.15 In recent years copolymers comprising these building blocks have been studied for their suitability in OFETs and OPVs applications.13 Moreover, these donor–acceptor copolymers are also attractive in view of a potential application in organic solar cells16 and as NIR detectors. More specifically, near-infrared photodetection (NIR-OPD, @800–3000 nm)17 is valuable for numerous scientific as well as industrial (inspection, sorting, safety/security) and recreational applications. Nowadays, near-infrared photodetectors are commonly based on inorganic semiconductors such as InGaAs, InGaSb18 or HgCdTe,19 although organic-based NIR detectors18,20–24 have gained increasing interest. To date, a very limited amount of well performing NIR-OPDs have been reported.24 Recently, push–pull copolymers based on BAI indigo core were found to be suitable for photodetection beyond 1000 nm.19
Here, we investigate the ground- and excited-state characteristics of copolymers comprising BAI1 units, linked through thiophene-based donor units (cyclopentadithiophene CPDT- or benzodithiophene BDT-based) to verify the potential of such copolymers as NIR solid state emitters. Rational design considerations of the polymers indicate that the introduction of planarized and symmetrical thiophene donors (CPDT, 4H-cyclopenta[1,2-b:5,4-b′]dithiophene, and BDT, benzo[1,2-b:4,5-b′]dithiophene) should enable interchain π–π stacking and maximized on-chain electronic interaction, thus promoting the bulk charge transport. Furthermore, the donor blocks of the donor–acceptor copolymers have been structurally varied to evaluate their influence on its electronic properties. Additionally, and since the BAI acceptor units does not carry solubilizing side chains, the donor blocks have also been used to induce a sufficient solubility.
Experimental section
Materials
Reagents were purchased from Sigma-Aldrich, Fischer Scientific or TCI and used without further purification, unless otherwise indicated. For the photophysical studies, the solutions were prepared with solvents of spectroscopic grade or equivalent: 2-methyltetrahydrofuran (2-MeTHF, anhydrous/inhibitor free, Sigma-Aldrich), toluene (tol, 99+% for spectroscopy, Acros Organics) and analytical grade chloroform (Fischer Chemical). Silver nitrate (AgNO3, for analysis, Panreac), Ferrocene (for synthesis, Merck) and tetrabutylammonium hexafluorophosphate (NBu4PF6, ≥99.0% for electrochemical analysis, Sigma-Aldrich) were used in cyclic voltammetry assays without further purification.BAI1 was obtained from double annulation of indigo,13,19 as described in Scheme 1; 4,4-didodecyl-4H-cyclopenta[1,2-b:5,4-b′]dithiophene (C12CPDT) was synthesized as previously described.25 4,8-Bis((2-butyloctyl)oxy)benzo[1,2-b:4,5-b′]dithiophene (BDT) was obtained from TCI and used without further purification.
Thin films preparation
Polymer thin films were obtained with a desktop precision spin-coating system, model P6700 series from Speedline Technologies, as described elsewhere.26,27 Solutions for spin-coating were prepared by adding 2 mg of the polymers to 200 μL of chloroform solution, with stirring in the dark, at room temperature, overnight. Thin films from the samples were obtained by deposition of ca. 50 μL from a solution of the compounds into a circular sapphire substrate (10 mm diameter) followed by spin-coating (2500 rpm) in a nitrogen-saturated atmosphere (2 psi).Photophysical and electrochemical studies
Absorption and fluorescence spectra were recorded on Agilent Cary 5000 UV-Vis-NIR and Horiba-Jobin-Ivon SPEX Fluorog 3–22 spectrometers respectively. Absorption spectra of the transparent thin films were obtained in absorption mode using a clean sapphire substrate as the reference sample. The fluorescence spectra were corrected for the wavelength response of the system. For the steady state and time resolved emission experiments, the absorption at the excitation wavelength was kept below 0.1 values to avoid aggregation effects. Acquisition of the photoluminescence spectra of the polymers at NIR were performed with Hamamatsu R5509-42 photomultiplier, cooled to 193 K in a liquid nitrogen chamber (Products for Research model PC176TSCE-005), connected to the fluorimeter. The colour parameters were determined according to the CIE (Commission Internationale de l’Eclairage proceedings) 1931 scale diagram.28 The x and y colour parameters were determined with the acquisition of the transmittance spectra using Shimadzu UV-2600 equipped with Colour Analysis software.Fluorescence quantum yields (φF) of the polymers in solution were determined with three replicates, by William's method,29 using indocyanine green (IR-125 in ethanol, φF = 0.132)30 as the standard. The φF of the model monomers in solution were measured using the absolute method with a Hamamatsu Quantaurus QY absolute photoluminescence quantum yield spectrometer, model C11347 (integration sphere). The φF of the polymers in thin films were measured by the absolute method with an integrating sphere module, as described elsewhere.26,31,32 The following equation was used to determine the φF of the BAI polymers thin films:
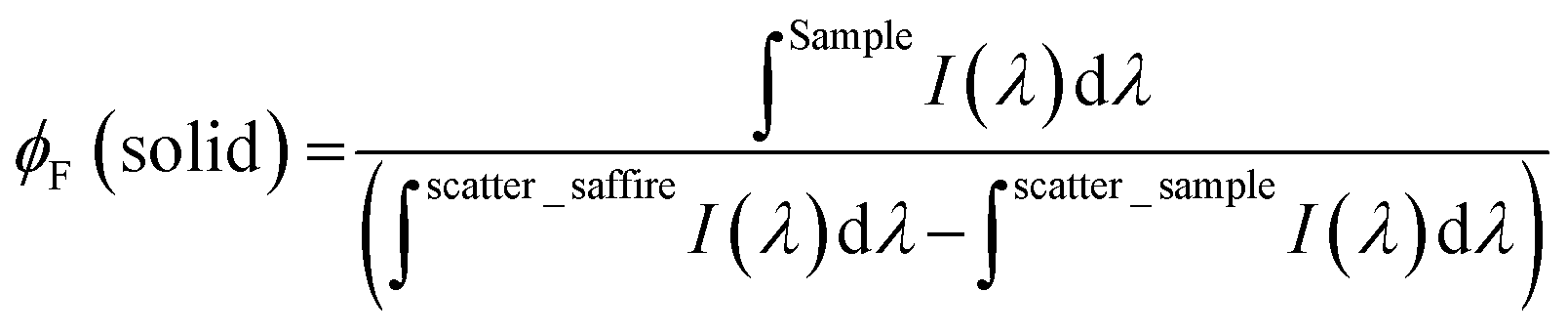 | (1) |
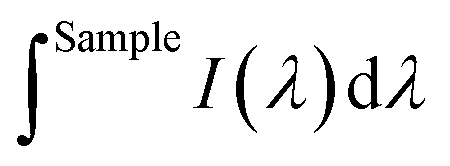 is the integrated area under the emission of the polymer thin film (which excludes the integration of the Rayleigh peak),
is the integrated area under the emission of the polymer thin film (which excludes the integration of the Rayleigh peak), 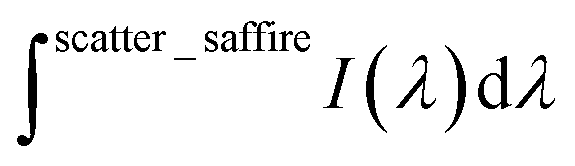 is the integrated area under the Rayleigh peak of a sample containing only a clean sapphire support, and
is the integrated area under the Rayleigh peak of a sample containing only a clean sapphire support, and 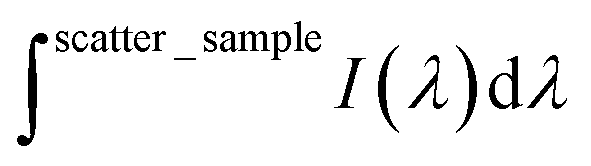 is the integrated area under the Rayleigh peak in the emission spectra of the polymer in the thin film.
is the integrated area under the Rayleigh peak in the emission spectra of the polymer in the thin film.
Fluorescence decays were measured using a home-built picosecond time-correlated single photon counting (ps-TCSPC) apparatus described elsewhere.33 Excitation was performed with the second harmonic, 372 nm or 402 nm (generated with a Spectra Physics GWU-23PS module) from a picosecond Spectra Physics mode-lock Tsunami laser (Ti:Sapphire) model 3950 (80 MHz repetition rate, tuning range 700–1000 nm), pumped by a Millennia Pro-10s, continuous wave, solid-state laser (532 nm). The fluorescence decays and the instrumental response function (IRF) were collected using 1024 channels in a time scale up to 24.4 ps per channel (using a Spectra Physics frequency divider, Pulse picker model 3980-2s, to reduce the fundamental laser repetition rate). Deconvolution of the fluorescence decay curves was performed using a modulation function method, as implemented by G. Striker in the SAND program, and previously reported in the literature.34
The time resolved ultrafast transient absorption measurements were collected in a broadband HELIOS spectrometer (350–1600 nm) from Ultrafast Systems as elsewhere described.35 The transient absorption data was obtained with excitation at 420 nm and 770 nm and probed in the 350–800 nm and 800–1600 nm range. The measurements in solution were obtained in a 2 mm quartz cuvette, with optical density ≈0.1–0.3 at the pump excitation wavelength. To avoid photodegradation low laser pump energies were used (≤100 nJ) at the excitation wavelengths 420 nm and 770 nm and the solutions were stirred during the experiments or kept in movement using a motorized translating sample holder. The spectral chirp of the data was corrected using Surface Xplorer PRO program from Ultrafast Systems. Global analysis of the data (using a sequential model) was performed using Glotaran software.36 Cyclic voltammetry experiments were carried out using an Autolab potentiostat/galvanostat PGSTAT204 running with NOVA 2.1 software and a three-electrode system in a one-compartment electrochemical cell of capacity 5 mL. A glassy carbon electrode (GCE) (d = 3 mm) was the working electrode, glassy carbon (GC) (d = 1.6 mm) wire was the counter electrode and Ag/Ag+ (0.01 M silver nitrate, AgNO3, in 0.1 M tetrabutylammonium hexafluorophosphate, NBu4PF6, in MeCN) was the reference electrode. Ferrocene/ferrocenium (Fc/FC+) redox couple were used as the internal reference. The reference and polymers were dissolved in 0.1 M NBu4PF6 in CHCl3 solution to acquire cyclic voltammograms with a 50 mV s−1 scan rate with a window potential between −1.6 V to 1.0 V (vs. Ag/Ag+).
Results and discussion
Synthesis and characterization
The general scheme for the synthesis of the monomeric BAI precursors BAI2 and BAI4 is depicted in Scheme 2. For the synthesis of 7,14-di(thiophen-2-yl)diindolo[3,2,1-de:3′,2′,1′-ij][1,5]naphthyridine-6,13-dione (BAI1) indigo is first subjected to a double annulation reaction to form the BAI core. Hereby, a twofold amidation followed by an intramolecular aldol condensation takes place.6,13,37 In the second step, the 2-positions of the thiophene rings are brominated to form 7,14-bis(5-bromothiophen-2-yl)diindolo[3,2,1-de:3′,2′,1′-ij][1,5]napht-hyridine-6,13-dione (BAI2). A subsequent Stille-type coupling reaction, catalyzed by tetrakis(triphenylphosphane)palladium(0) and tri-o-tolylphosphine as a ligand,13 of BAI2 and 2-(trimethylstannyl)-3-dodecylthiophene is used to extend the thiophene substituent into a bithiophene. 7,14-Bis(3′-dodecyl-[2,2′-bithiophene]-5-yl)diindolo[3,2,1-de:3′,2′,1′-ij][1,5]naphthyridine-6,13-dione (BAI3) is finally brominated in the absence of light into 7,14-bis(5′-bromo-3′-dodecyl-[2,2′-bithiophene]-5-yl)diindolo[3,2,1-de:3′,2′,1′-ij][1,5]naphthyridine-6,13-dione (BAI4). | ||
| Scheme 2 Synthesis of BAI monomers. Reaction conditions: (i) 2-thienylacetyl chloride, xylene, 145 °C, 48 h, 38.5%; (ii) NBS, chloroform, rt, 15 h, 69.7%; (iii) 2-(trimethylstannyl)-3-dodecylthiophene, Pd(PPh3)4, P(o-tol)3, toluene, 100 °C, 4 days, 1.5%; (iv) NBS, chloroform, rt, 12 h, 87.1%. | ||
The Stille-type cross-coupling reaction of the two different BAI monomers (BAI2 and BAI4) with distannylated CPDT (4,4-bis(2-octyldodecyl)-2,6-bis(trimethylstannyl)-4H-cyclopenta[1,2-b:5,4-b′]dithiophene-2,6-diyl, C9) was carried out as previously described for the synthesis of corresponding indigo-containing copolymers.38 Hereby, tetrakis(triphenylphosphane)palladium(0), Pd(PPh3)4, was used as a catalyst in a microwave-assisted coupling reaction. After irradiation at 125 °C for 25 minutes, the reaction mixture was stirred at 90 °C for a further 72 h (Scheme 3). The copolymers PBAIC-1 and PBAIC-2 were precipitated into cold methanol and fractionated by Soxhlet extraction. For the synthesis of the BAI-benzodithiophene copolymer PBAIBD-1, the reaction conditions were modified according to a report by He et al.13 Under inert conditions, tris(dibenzylideneacetone)dipalladium(0) as the catalyst and tri(o-tolylphosphane) as the ligand together with the monomers BD5 and BAI2 were dissolved in toluene and heated to 100 °C. After six days, the copolymer was precipitated and Soxhlet extracted (Scheme 3).
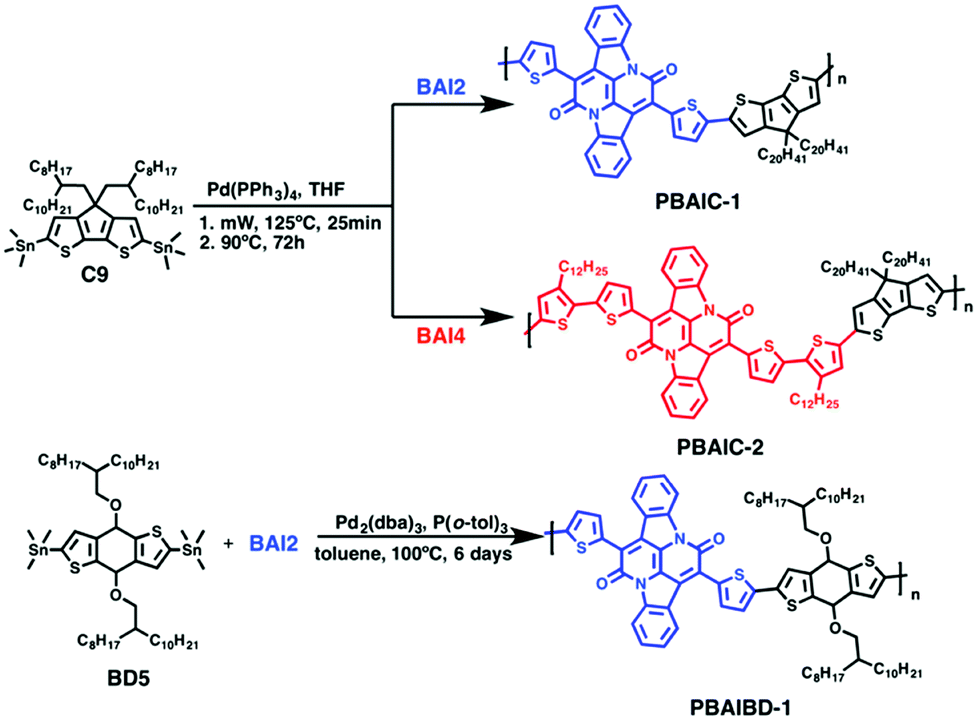 | ||
| Scheme 3 General synthetic scheme for the generation of the BAI-based copolymers: PBAIC-1, PBAIC-2 and PBAIBD-1. | ||
PBAIC-1 and PBAIBD-1 show moderate molecular weights 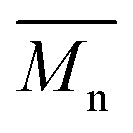 of 8–11 kg mol−1, with PD = 1.33 and 1.39, respectively, resulting possibly from a limited solubility of the products. The two additional dodecylthiophene moieties in copolymer PBAIC-2 increase the solubility of the polymer, thus probably causing its higher molecular weight and polydispersity (
of 8–11 kg mol−1, with PD = 1.33 and 1.39, respectively, resulting possibly from a limited solubility of the products. The two additional dodecylthiophene moieties in copolymer PBAIC-2 increase the solubility of the polymer, thus probably causing its higher molecular weight and polydispersity (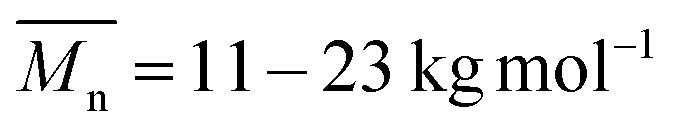 , PD = 1.94). Nevertheless, with a value below 2, the PDI index is still as expected for polycondensations. For further synthetical procedures and structural characterization, please consult the ESI.†
, PD = 1.94). Nevertheless, with a value below 2, the PDI index is still as expected for polycondensations. For further synthetical procedures and structural characterization, please consult the ESI.†
Optical and electrochemical properties
The photophysical properties of the three BAI copolymers were investigated in solution (2-MeTHF and toluene) and in thin films and compared to their monomeric units. In Fig. 1, the absorption and emission spectra of PBAIC-1 and PBAIC-2 in 2-MeTHF solution are compared with BAI1 and C12CPDT, PBAIBD-1 is compared to BAI1 and BDT. The change of the phenyl groups of Cibalackrot to the electron-richer thiophene groups in BAI1 lead to a bathochromic shift of the absorption maximum of ∼40 nm: 547 nm for Cibalackrot to 588 nm for BAI1 (Fig. SI4 and Table SI2, ESI†). Generally, incorporation of the BAI chromophores into the copolymers causes a significant red shift of the absorption and emission of these in comparison to BAI1.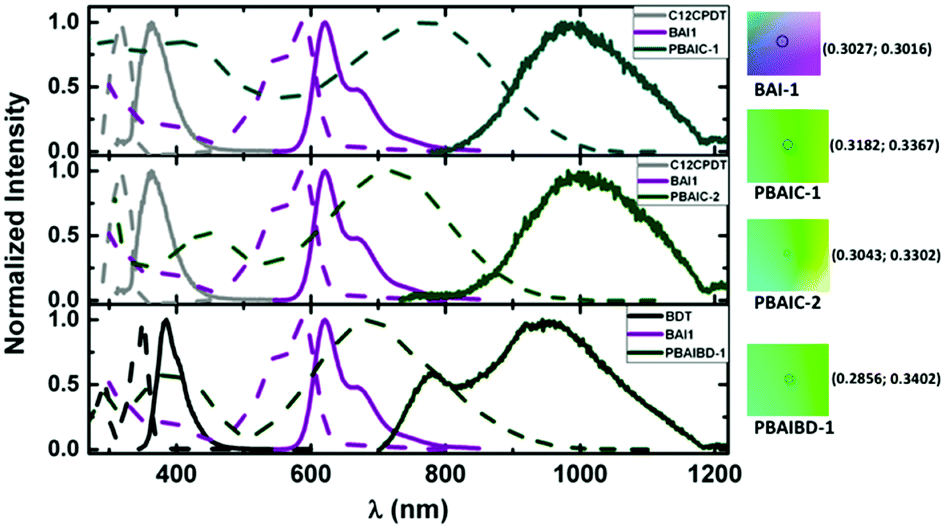 | ||
| Fig. 1 Normalized absorption (dashed lines) and emission spectra (solid lines) of BAI copolymers together with respective model chromophores in 2-MeTHF at 298 K. On the right side, the color parameters in the CIE 1931 chromaticity scale28 of the absorption spectra of BAI-1 and BAI copolymers in 2-MeTHF. | ||
The shape of the absorption spectra is found similar for the three copolymers, with two broad absorption bands: one (I) in the 300–500 nm region and another (II), broader, at 550–1000 nm. The bathochromic shift of band I of the copolymers (PBAIC-1, PBAIC-2 and PBAIBD-1) that may be assigned to the donor units, in comparison to the absorption bands of the monomeric donor chromophores (BDT and CPDT), may indicate the conjugative interaction between the subunits. The interplay of first and second absorption band of the copolymers causes a green color of the copolymers, in contrast to the purple color characteristic of BAI1 (Fig. 1). The red shift of the second (II) absorption band in comparison with BAI1 can be associated to an extended π-conjugation and a strong donor–acceptor character of the copolymers. PBAIC-1 and PBAIC-2 (with the CPDT moiety as the electron donor unit) in 2-MeTHF solution display emission spectra with bands in the 800–1200 nm region, with peak maxima at 997 and 982 nm, respectively, while PBAIBD-1 shows an emission spectrum with two peaks: 778 nm (visible region) and 944 nm (NIR), see Table 1. The electronic spectra of the three copolymers in toluene (Fig. SI5, ESI†) are almost identical to the spectra in 2-MeTHF solutions (see Table SI3, ESI†). The introduction of the donor CPDT unit (from PBAIC-1), instead of BDT (PBAIBD-1), leads to a red-shift of the absorption either in solution, or in solid state. Nonetheless, solid state absorption bands are red-shifted relative to the solution spectra (Fig. SI5, ESI†). Fluorescence quantum yields (φF) of the BAI-containing copolymers obtained in 2-MeTHF, at 298 K and 77 K, and in thin films at 298 K (Table 1) are roughly similar to the solid state (thin films) values. The spatial structures of PBAIC-1, PBAIC-2 and PBAIBD-1 as donor–acceptor copolymers seem to preclude aggregation caused quenching (ACQ) effects. Moreover, the φF values of the BAI copolymers at 77 K are about four times higher than in solution at 293 K (see Fig. SI6 and Table SI4, ESI†), as a factor of interest for potential applications, e.g. as the photoactive layer of NIR organic photodiodes. The PL emission area of the three polymers presented a linear course with the increase of the absorption at the excitation wavelength, in solution (Fig. SI7, ESI†), ruling out aggregation effects of the polymer in 2-meTHF.
| Compound | Medium | λ abs (nm) | λ em (nm) | Δ SS (cm−1) | φ F |
|---|---|---|---|---|---|
| a φ F = 0.05 in methylcyclohexane in ref. 25. b sh = shoulder in the spectra; see the text for more details. | |||||
| C12CPDT | 2-meTHF | 316 | 363 | 4097 | 0.048a |
| BDT | 351 | 385 | 2516 | 0.089 | |
| BAI-1 | 550 (sh)b, 588 | 621, 669 (sh)b | 904 | 0.389 | |
| PBAIC-1 | 2-MeTHF | 404, 775 | 997 | 2873 | 0.015 ± 0.002 |
| Film | 392, 821 | 1074 | 2869 | 0.016 ± 0.001 | |
| PBAIC-2 | 2-MeTHF | 447, 716 | 982 | 3783 | 0.013 ± 0.002 |
| Film | 457, 753 | 1060 | 3846 | 0.011 ± 0.001 | |
| PBAIBD-1 | 2-MeTHF | 378, 688 | 944 | 3942 | 0.018 ± 0.002 |
| Film | 429, 749 | 1118 | 4407 | 0.0066 ± 0.0001 | |
The electrochemical properties and energy levels of BAI copolymers were determined in chloroform solution (Table 2). The characteristic, quasi-reversible double reduction of the amide groups, correlated to the BAI moiety, is prominent in the cyclic voltammogram of PBAIBD-1. Moreover, the oxidation and reduction potentials are similar to those found in the literature to D–A type polymers based on bay-annulated indigo and thiophene units.13,16,19
| Polymer | E onsetox (V) | E HOMO (eV) | E LUMO (eV) | E g (eV) | E g (eV) | E Ox1/2 (V) | E Red1/2 (V) |
|---|---|---|---|---|---|---|---|
| a E FOC = 0.064 V determined in 0.1 M NBu4PF6 in CHCl3. b Estimated from the onset of absorption. See eqn (3) and (4) and Table SI5 (ESI) for further details. | |||||||
| PBAIC-1 | 0.0929 | −4.96 | −3.57 | 1.39 | 1.48 | 0.27 | −1.15 |
| PBAIC-2 | 0.0909 | −4.95 | −3.54 | 1.42 | 1.29 | 0.23 | −1.18 |
| PBAIBD-1 | 0.514 | −5.38 | −3.85 | 1.52 | 1.54 | 0.25 | −1.12 |
The energies of the HOMO and LUMO levels were obtained according to eqn (2) and (3).39
| EHOMO = −(Eonsetox + 4.8) − EFOC | (2) |
| ELUMO = EHOMO + E0–0 | (3) |
The energy of the HOMO, EHOMO, was calculated from the onset oxidation potential, Eonsetox, plus 4.8, which is the difference between the reference energy level of ferrocene below the vacuum level and the oxidation potential of ferrocene, EFOC, given by the potential of FOC/FOC+vs. Ag/Ag+ measured by cyclic voltammetry in solution (Fig. 2). The LUMO energy, ELUMO, was obtained from the optical energy gap E0–0, calculated from the interception of the normalized UV-Vis absorption and fluorescence spectra, through eqn (3).
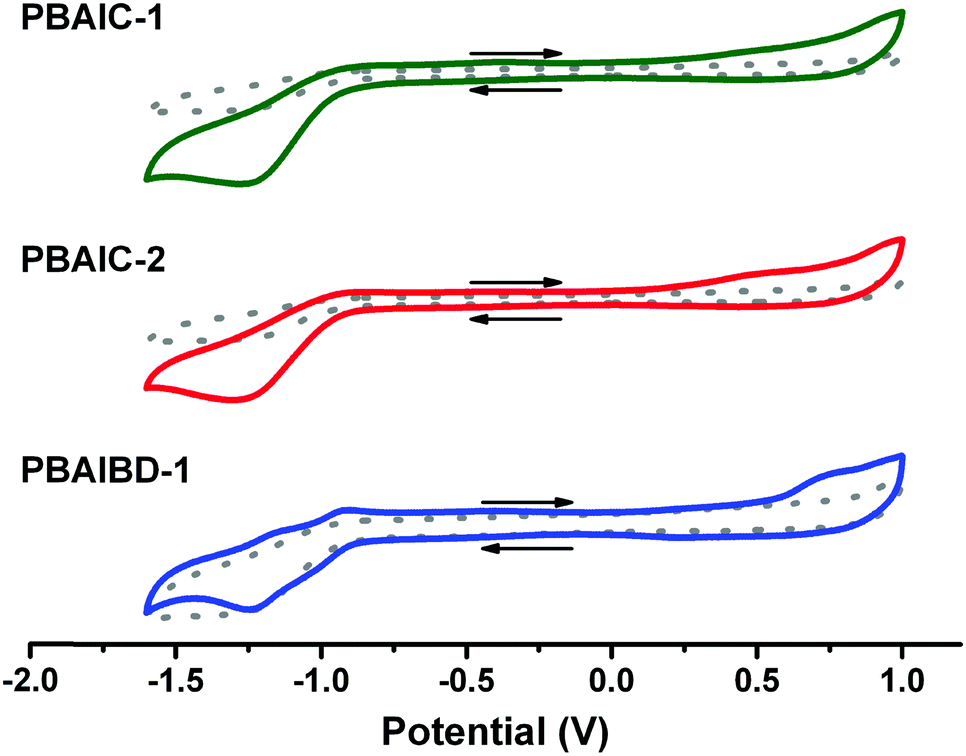 | ||
| Fig. 2 Cyclic voltammograms of the BAI copolymers in CHCl3 solution obtained with a scan rate of 100 mV s−1. Dotted lines represent the blank voltammogram, acquired with the electrolyte solution (0.1 M NBu4PF6 in CHCl3). | ||
The HOMO-levels of the CPDT-based polymers are similar with values of −4.96 eV (PBAIC-1) and −4.95 eV (PBAIC-2), respectively, and correlate well with values determined from the onset of absorption (Table SI5, ESI†). The LUMO-levels, about −3.5 to −3.9 eV, is closely correlated to the ELUMO = −3.6 eV, previously attributed to the model compound BAI1.40CPDT-based polymers also presented smaller band gaps than PBAIBD-1 (built with BDT as donor unit), thus, CPDT may be a stronger electron donor unit than BDT and, therefore, lead to higher HOMO energy levels (Table 2).
The band gap energies of BAI copolymers vary between 1.3–1.5 eV, when determined either by CV, or UV-spectroscopy (Table 2). Narrow band gap (Eg) conjugated polymers, 1.1 ≤ Eg ≤ 2.1 eV, have especially low lying LUMO energies making them excellent candidates for n-type OFET materials.41 Moreover, these BAI copolymers also display optical absorption in the near-IR region of the electromagnetic spectrum, ideal for OPV devices.21 Also, because these polymers contain both donor and acceptor moieties on their backbones, they are promising candidates for single-component polymer solar cells by replacing polymer blends used as active layers in manufacturing all-polymer solar cells, and thus, overcome drawbacks such as miscibility between the donor and acceptor polymer components and gradual phase separation of polymer blend films.42–44
Time-resolved spectroscopy studies
Femtosecond transient absorption (fs-TA) was further used to characterize the excited state dynamics of the copolymers and two model monomers (BAI1 and BDT) in solution and in thin films (Fig. SI8–SI11 and Table SI6, ESI†). From Fig. 3, it is seen that the fs-TA spectra of the copolymers are composed of a broad positive excited state absorption (ESA) in the 880–1600 nm range, and negative TA bands between 350 nm and 955 nm matching with those in Fig. 1 attributed to ground-state absorption (GSA).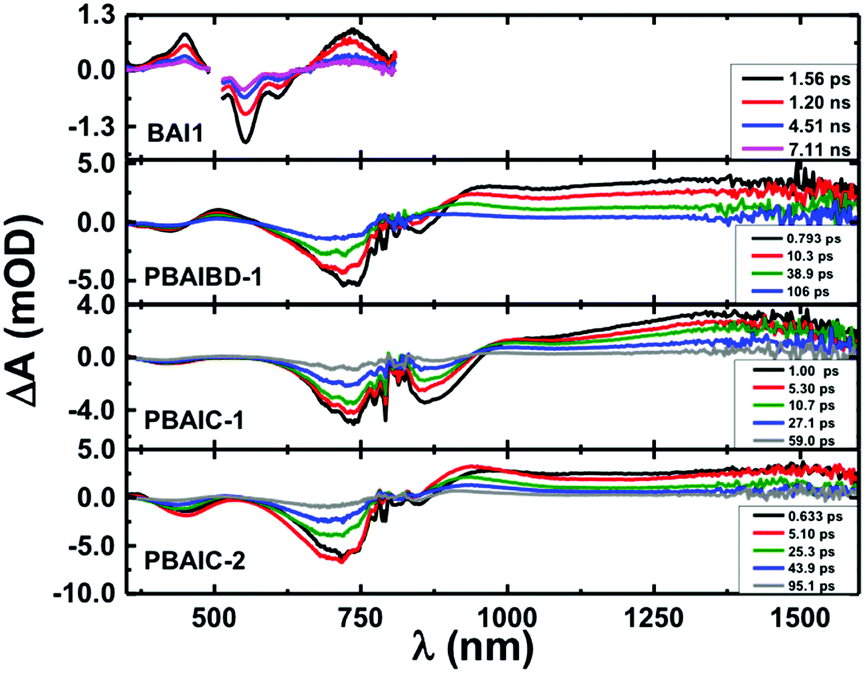 | ||
| Fig. 3 Room temperature time resolved transient absorption spectra (fs-TA) for the BAI copolymers and monomeric BAI1 in aerated 2Me-THF solution collected with excitation at 770 nm and 500 nm, respectively. The data was collected in the 350–825 nm and 800–1600 nm ranges and merged together using Surface Xplorer software with normalization of the two surfaces in the 800–825 nm range. This explains why the spectral data in the 770–830 nm region shows some noise. | ||
In solution after about ∼5 ps (PBAIC-2) and ∼10 ps (PBAIBD-1) the initially formed ESA band blue-shifts by ∼20 nm and thereon the band maxima remain unchanged. Since there are no additional bands at longer decay times (that could be attributed, e.g., to triplet excited state formation in the polymers) the positive TA bands are attributed to the S1 → Sn ESA. This behavior contrasts with that of the monomeric BAI1 chromophore where in addition to the S1 → Sn ESA (in the 350–480 nm and 665–800 nm), GSA, and stimulated emission in the (510–630 nm) bands, a triplet–triplet absorption band is also observed with a triplet lifetime of τT = 54 μs (presented in the ns-TA absorption spectra, Fig. SI9, ESI†). For PBAIBD-1 a characteristic ESA band is seen in the 460–560 range, as also found for the monomeric BDT unit. However, contrary to monomeric BDT, in PBAIBD-1 this band disappears after 106 ps (see Fig. SI10, SI11, ESI,† and Fig. 2). For monomeric BDT the S1 → Sn ESA band was found to be strongly overlapped with the T1 → Tn ESA band, as shown by comparison with the triplet absorption spectra obtained by ns-TA, (τT = 99 μs), Fig. SI10 (ESI†). The data support (i) the pronounced conjugation between BAI1 and BDT moieties in the PBAIBD-1 copolymer, which promotes electron-delocalization, as shown by the significant red-shift of the absorption band when compared with the monomeric BAI1 (Fig. 1), and (ii) the absence of triplet state formation in the polymers, due to the fast deactivation of the excited state. The mechanism behind this will be discussed below.
Global analysis of representative kinetic traces of the copolymers shows that transient decays are well fitted with the sum of three exponentials. The best-fit results and pre-exponential values are presented in Table 3 (also in Table SI6 and Fig. SI11 in the ESI†). Either in solution, or in thin films, the copolymers show a first decay transient with values in the 0.71–8.9 ps range, a second one with values in the 14.9–58.4 ps range and a longer decay transient with values ranging from 55.6 ps to 1948 ps. BAI1 transient lifetimes of 66 ps, 5201 ps and 54 μs were determined. The latter was fixed in the analysis to the triplet lifetime obtained by ns-TA (Fig. SI9, ESI†). The two shortest decay times are in agreement with the double-exponential decay behavior obtained from the fluorescence decays of BAI1 (fluorescence lifetimes, τF = 140 ps and 4680 ps, Fig. SI12, ESI†). BDT transient decays are well fitted with a double exponential decay law, with transient lifetimes of 313 ps (also in agreement with τF = 560 ps obtained by TCSPC, Fig. SI13, ESI†) and 99 μs, that was fixed in the analysis to the triplet lifetime obtained by ns-TA (Fig. SI14 in ESI†).
| Polymer | λ (nm) | a i | k ET(1) (s−1) | k ET(2) (s−1) | ||
|---|---|---|---|---|---|---|
| a NA = not applicable. Shorter τ1 (<10 ps) component attributed to solvation dynamics. | ||||||
| PBAIC-1 | τ i (ps) | 0.71 | 19.4 | 114.7 | ||
| 911 | −0.494 | −0.506 | 0.059 | NAa | 4.55 × 1010 | |
| 1200 | 0.194 | 0.645 | 0.160 | |||
| PBAIC-2 | τ i (ps) | 2.4 | 36.7 | 230.8 | ||
| 911 | −0.560 | 0.895 | 0.105 | 4.11 × 1011 | 2.12 × 1010 | |
| 1200 | 0.232 | 0.746 | 0.022 | |||
| PBAIBD-1 | τ i (ps) | 8.9 | 49.7 | 163.1 | ||
| 505 | 0.506 | 0.196 | 0.298 | |||
| 911 | 0.440 | −0.271 | 0.560 | 1.11 × 1011 | 1.83 × 1010 | |
| 1200 | 0.292 | 0.848 | 0.642 | |||
The shortest decay time component (τ1) which, at shorter emission wavelengths is associated to a negative pre-exponential for the PBAIC-1 copolymer (Table 3), should be considered to be associated to solvent dynamics (in THF with characteristic times ranging from 0.43 ps to 0.94 ps),45 or attributed to vibrational relaxation due to the depopulation of a hot vibrational state generated through the 770 nm excitation (since excitation was not performed in the lowest energy 0–0 electronic transition).38 Yet, this is not the case with PBAIC-2 and PBAIBD-1, where a longer component is observed with 2.4 ps and 8.9 ps (Table 3), respectively, in addition to a longer τ3 component in the order of hundreds of ps. These τ1 and τ2 decay components are associated to excitation energy migration within different segments of the polymer chain (higher to lower energy conjugated segments) ending up in the τ3 decay component which mirrors the decay of the more relaxed segment to the ground state.
From the donor–acceptor (D–A) character of the copolymers, one can determine the energy transfer (kET) rate constant from:38,46–48
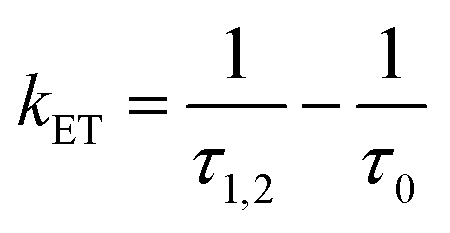 | (4) |
ID(t) = exp(−t/τ0)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp[−(4/3)π3/2R03CA(t/τ0)1/2] exp[−(4/3)π3/2R03CA(t/τ0)1/2] | (5) |
Although energy transfer leads to nonexponential decays, the ID(t) for PBAIC-2 and PBAIBD-1 can be fitted with a sum of two exponentials terms (Fig. 4), which corresponded to the two shortest decay times in these copolymers (Table 3).
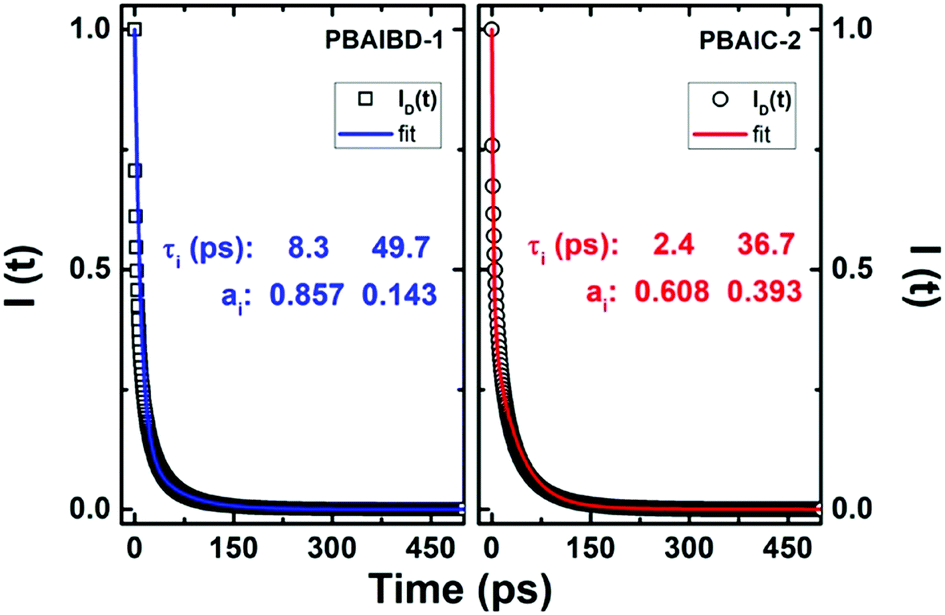 | ||
| Fig. 4 Plot of the fluorescence decay function of the donor, ID(t), in the presence of acceptors (open circles and squares) according to eqn (5) and respective double exponential fit, I(t) = a1exp(−t/τ1) + a2exp(−t/τ2), blue and red lines for the PBAIBD-1 and PBAIC-2 copolymers. | ||
For PBAIC-2, τ2 is dominant particularly at longer wavelengths (representing ∼91% of the total decay) and should be therefore seen as governing the ET process (i.e., the ET process involving more distant D–A pairs); for PBAIBD-1 similar contributions were found for the two decay times, 50%, indicating that short and distant D–A pairs contribute equally to the on-chain hopping decay mechanism. Additionally, the rate constants for the ET process varies from 1.83 × 1010 to 4.11 × 1011 s−1 (Table 3), characteristic of conjugated organic polymers.38,49
Conclusions
In this work, three new BAI-based donor–acceptor copolymers were synthesized, and their photoexcitation decay mechanisms investigated in solution and thin films. Even though the φF of these copolymers is found from 1 up to 2% in solution, the film values are basically identical to solution, thus showing that ACQ is not taking place. While in solution, the BAI-containing copolymers presented broad long wavelength absorption bands, with maxima varying between 688–775 nm, in thin films, the red-shifted emission bands reach 900–1300 nm, indicating that these copolymers are a promising class of low-bandgap, circa 1.4–1.5 eV, copolymers with solid state absorption and emission widely shifted into the NIR region. The decays in the PBAIBD-1 and PBAIC-2 copolymers were further associated to an excitation energy transfer hopping that could be rationalized with a Förster-type mechanism with differing donor–acceptor contributions.Our work uncovers not only the need for the development of new NIR-absorber polymers to featured applications e.g. in sensing and photovoltaics, but also reinforces the use of bay-annulated indigo as a building block for the generation of push–pull type conjugated polymers.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Project “Hylight” (no. 031625) 02/SAICT/2017, PTDC/QUI-QFI/31625/2017, which is funded by the Portuguese Science Foundation and Compete Centro 2020. We acknowledge funding by Fundo Europeu de Desenvolvimento Regional (FEDER) through Programa Operacional Factores de Competitividade (COMPETE) and project ROTEIRO/0152/2013. The Coimbra Chemistry Centre is supported by the Fundação para a Ciência e a Tecnologia (FCT, UID/QUI/00313/2019–2020). The research leading to these results has received funding from Laserlab-Europe (grant agreement no. 284464, EC's Seventh Framework Programme). We also acknowledge Dr Daniela Pinheiro, for the assistance in the electrochemical studies, and Dr Sybille Allard, for GPC spectral data.Notes and references
- M. J. Melo, J. L. Ferreira, A. J. Parola and J. S. S. de Melo, Appl. Photochem., 2016, 499–530, DOI:10.1007/978-3-319-31671-0_13
.
- J. Pina, D. Sarmento, M. Accoto, P. L. Gentili, L. Vaccaro, A. Galvao and J. S. Seixas de Melo, J. Phys. Chem. B, 2017, 121, 2308–2318 CrossRef CAS PubMed
- J. Sérgio Seixas de Melo, Photochemistry, 2018, 45, 68–100 Search PubMed
- J. Sérgio Seixas de Melo, Photochemistry, 2020, 47, 196–216, 10.1039/9781788016520-00196
- D. Pinheiro, M. Pineiro, A. M. Galvão and J. S. Seixas de Melo, Chem. Sci., 2021, 12, 303–313 RSC
- G. Engi, Zeitschrift für Angew. Chem., 1914, 27, 144–148 CrossRef CAS
- J. Seixas de Melo, R. Rondao, H. D. Burrows, M. J. Melo, S. Navaratnam, R. Edge and G. Voss, J. Phys. Chem. A, 2006, 110, 13653–13661 CrossRef CAS PubMed
- M. B. Smith and J. Michl, Annu. Rev. Phys. Chem., 2013, 64, 361–386 CrossRef CAS PubMed
- J. L. Ryerson, A. Zaykov, L. E. Aguilar Suarez, R. W. A. Havenith, B. R. Stepp, P. I. Dron, J. Kaleta, A. Akdag, S. J. Teat, T. F. Magnera, J. R. Miller, Z. Havlas, R. Broer, S. Faraji, J. Michl and J. C. Johnson, J. Chem. Phys., 2019, 151, 184903 CrossRef PubMed
- A. Shukla, N. R. Wallwork, X. Li, J. Sobus, V. T. N. Mai, S. K. M. McGregor, K. Chen, R. J. Lepage, E. H. Krenske, E. G. Moore, E. B. Namdas and S. C. Lo, Adv. Opt. Mater., 2019, 1901350, DOI:10.1002/adom.201901350
- E. D. Glowacki, G. Voss and N. S. Sariciftci, Adv. Mater., 2013, 25, 6783–6800 CrossRef CAS PubMed
- T. Furuyama, D. Tamura, H. Maeda and M. Segi, Tetrahedron Lett., 2018, 59, 2913–2916 CrossRef CAS
- B. He, A. B. Pun, D. Zherebetskyy, Y. Liu, F. Liu, L. M. Klivansky, A. M. McGough, B. A. Zhang, K. Lo, T. P. Russell, L. Wang and Y. Liu, J. Am. Chem. Soc., 2014, 136, 15093–15101 CrossRef CAS
- M. A. Kolaczkowski, B. He and Y. Liu, Org. Lett., 2016, 18, 5224–5227 CrossRef CAS PubMed
- B. He, W. T. Neo, T. L. Chen, L. M. Klivansky, H. Wang, T. Tan, S. J. Teat, J. Xu and Y. Liu, ACS Sustainable Chem. Eng., 2016, 4, 2797–2805 CrossRef CAS
- J. Zhu, T. Li, K. Shi, J. Wang, Y. Lin, G. Yu and X. Zhan, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 213–220 CrossRef CAS
- J. Qi, W. Qiao and Z. Y. Wang, Chem. Rec., 2016, 16, 1531–1548 CrossRef CAS PubMed
- D. Li, S. Yip, F. Li, H. Zhang, Y. Meng, X. Bu, X. Kang, C. Lan, C. Liu and J. C. Ho, Adv. Opt. Mater., 2020, 8, 2001201 CrossRef CAS
- F. Verstraeten, S. Gielen, P. Verstappen, J. Kesters, E. Georgitzikis, J. Raymakers, D. Cheyns, P. Malinowski, M. Daenen, L. Lutsen, K. Vandewal and W. Maes, J. Mater. Chem. C, 2018, 6, 11645–11650 RSC
- X. Liu, Y. Lin, Y. Liao, J. Wu and Y. Zheng, J. Mater. Chem. C, 2018, 6, 3499–3513 RSC
- T. J. Wen, D. Wang, L. Tao, Y. Xiao, Y. D. Tao, Y. Li, X. Lu, Y. Fang, C. Z. Li, H. Chen and D. Yang, ACS Appl. Mater. Interfaces, 2020, 12, 39515–39523 CrossRef CAS PubMed
- D. S. Leem, K. H. Lee, N. Li, B. W. Park, T. Choi, T. Ro, O. K. Kwon, Y. N. Kwon, T. N. Ng and S. Kim, Adv. Opt. Mater., 2020, 2001682, DOI:10.1002/adom.202001682
- G. Yuan, H. Lv, H. Liu, H. He, Q. Sun, X. Zhang and S. wang, Dyes Pigm., 2020, 183, 108674 CrossRef CAS
- F. Verstraeten, S. Gielen, P. Verstappen, J. Raymakers, H. Penxten, L. Lutsen, K. Vandewal and W. Maes, J. Mater. Chem. C, 2020, 8, 10098–10103 RSC
- J. Pina, A. Eckert, U. Scherf, A. M. Galvão and J. S. Seixas de Melo, Mater. Chem. Front., 2018, 2, 149–156 RSC
- J. Pina, J. Seixas de Melo, H. D. Burrows, A. Bilge, T. Farrell, M. Forster and U. Scherf, J. Phys. Chem. B, 2006, 110, 15100–15106 CrossRef CAS PubMed
- A. C. B. Rodrigues, I. S. Geisler, P. Klein, J. Pina, F. J. H. Neuhaus, E. Dreher, C. W. Lehmann, U. Scherf and J. S. Seixas de Melo, J. Mater. Chem. C, 2020, 8, 2248–2257 RSC
- T. Smith and J. Guild, Trans. Opt. Soc., London, 1931, 33, 73–134 CrossRef
- A. T. R. Williams, S. A. Winfield and J. N. Miller, Analyst, 1983, 108, 1067–1071 RSC
- K. Rurack and M. Spieles, Anal. Chem., 2011, 83, 1232–1242 CrossRef CAS PubMed
- J. C. de Mello, H. F. Wittmann and R. H. Friend, Adv. Mater., 1997, 9, 230–232 CrossRef CAS
- L. O. Pålsson and A. P. Monkman, Adv. Mater., 2002, 14, 757–758 CrossRef
- J. Pina, J. Seixas de Melo, H. D. Burrows, A. L. Maçanita, F. Galbrecht, T. Bünnagel and U. Scherf, Macromolecules, 2009, 42, 1710–1719 CrossRef CAS
- G. Striker, V. Subramaniam, C. A. M. Seidel and A. Volkmer, J. Phys. Chem. B, 1999, 103, 8612–8617 CrossRef CAS
- J. Pina, M. J. R. P. Queiroz and J. S. Seixas de Melo, Photochem. Photobiol. Sci., 2016, 15, 1029–1038 CrossRef CAS PubMed
- J. J. Snellenburg, S. P. Laptenok, R. Seger, K. M. Mullen and I. H. M. V. Stokkum, J. Statistical Software, 2012, 49, 1–22 Search PubMed
- R. Tobler, Helv. Chim. Acta, 1945, 28, 901–911 CrossRef CAS
- J. Pina, M. Alnady, A. Eckert, U. Scherf and J. S. Seixas de Melo, Mater. Chem. Front., 2018, 2, 281–290 RSC
- R. C. Pereira, A. D. R. Pontinha, M. Pineiro and J. S. Seixas de Melo, Dyes Pigm., 2019, 166, 203–210 CrossRef CAS
- M. A. Kolaczkowski and Y. Liu, Chem. Rec., 2019, 19, 1062–1077 CrossRef CAS PubMed
- K. J. Fallon, N. Wijeyasinghe, E. F. Manley, S. D. Dimitrov, S. A. Yousaf, R. S. Ashraf, W. Duffy, A. A. Y. Guilbert, D. M. E. Freeman, M. Al-Hashimi, J. Nelson, J. R. Durrant, L. X. Chen, I. McCulloch, T. J. Marks, T. M. Clarke, T. D. Anthopoulos and H. Bronstein, Chem. Mater., 2016, 28, 8366–8378 CrossRef CAS
- Y. U. Kim, B. S. Ma, Y. Kim, S. H. Park, H. Kang, H. J. Yoon, M. J. Cho, T.-S. Kim, J. H. Kim and D. H. Choi, Chem. Eng. J., 2021, 415, 128952, DOI:10.1016/j.cej.2021.128952
- C. G. Park, S. H. Park, Y. Kim, T. L. Nguyen, H. Y. Woo, H. Kang, H. J. Yoon, S. Park, M. J. Cho and D. H. Choi, J. Mater. Chem. A, 2019, 7, 21280–21289 RSC
- N. Y. Kwon, S. H. Park, H. Kang, A. V. Takaloo, A. K. Harit, H. Y. Woo, T. G. Kim, H. J. Yoon, M. J. Cho and D. H. Choi, J. Mater. Chem. A, 2020, 8, 20091–20100 RSC
- M. L. Horng, J. A. Gardecki, A. Papazyan and M. Maroncelli, J. Phys. Chem., 1995, 99, 17311–17337 CrossRef CAS
-
J. S. Seixas de Melo, J. Pina, F. B. Dias and A. L. Maçanita, Appl. Photochem., 2013, ch. 15, pp. 533–585 DOI:10.1007/978-90-481-3830-2_15
- J. Pina, J. S. Seixas de Melo, J.-M. Koenen, S. Jung and U. Scherf, J. Phys. Chem. C, 2013, 117, 3718–3728 CrossRef CAS
- J. Pina, J. S. Seixas de Melo, N. Koenen and U. Scherf, J. Phys. Chem. B, 2013, 117, 7370–7380 CrossRef CAS PubMed
- B. Ferreira, P. F. da Silva, J. S. Seixas de Melo, J. Pina and A. Macanita, J. Phys. Chem. B, 2012, 116, 2347–2355 CrossRef CAS PubMed
- A. Yekta, M. A. Winnik, J. P. S. Farinha and J. M. G. Martinho, J. Phys. Chem. A, 1997, 101, 1787–1792 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: For detailed synthesis, structure characterization and further photophysical data. See DOI: 10.1039/d1ma00260k |
| This journal is © The Royal Society of Chemistry 2021 |