
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aging effect on the co-crystallization behavior of the donor and acceptor crystals in aqueous dispersions†
Masaki
Takeda
 a,
Jun
Matsui
*b and
Akito
Masuhara
*ac
a,
Jun
Matsui
*b and
Akito
Masuhara
*ac
aGraduate School of Science and Engineering, Yamagata University, 4-3-16, Jonan, Yonezawa, Yamagata, Japan. E-mail: masuhara@yz.yamagata-u.ac.jp
bFaculty of Science, Yamagata University, 1-4-12, Kojirakawa-machi, Yamagata, Yamagata, Japan. E-mail: jun_m@sci.kj.yamagata-u.ac.jp
cResearch Center for Organic Electronics, Yamagata University, 4-3-16, Jonan, Yonezawa, Yamagata, Japan
First published on 4th March 2021
Abstract
Charge-transfer (CT) co-crystals composed of organic donor and acceptor molecules are attractive materials from both basic and applied perspectives. One of the most fascinating protocols for the formation of a CT crystal is the use of donor and acceptor crystals instead of their molecules as starting materials. However, the lack of studies has limited the universality of the method for future applications. In this study, we adapted the mixing of donor and acceptor crystal dispersions to fabricate CT co-crystals, and “phase-separated” crystals, which are a mixture of the donor and acceptor crystals. The aqueous dispersions of each crystal were prepared by the reprecipitation method, and mixed to form the targeted crystals. We controlled the co-crystallization behavior between the donor and acceptor crystals by aging of the dispersion, which was left standing in air for 24 h before mixing. CT co-crystals were formed in the non-aged mixture; in contrast, “phase-separated” crystals were obtained in the aged mixture. Current–voltage measurements using tetrathiafulvalene and tetracyanoquinodimethane revealed that the conventional CT co-crystals exhibited lower sheet resistance (103 to 104 Ω sq−1) than that of the “phase-separated” crystals, whereas the “phase-separated” crystals showed a photoconductive response.
Introduction
Charge-transfer (CT) co-crystals constructed from organic donor and accepter molecules have high potential for photoelectrical device applications such as organic field-effect transistors (OFETs) and light-emitting transistors (OLETs) owing to their superior properties and simple preparation process.1–6 Research on CT crystals has received considerable attention after the discovery of the molecular metal tetrathiafulvalene (TTF)–tetracyanoquinodimethane (TCNQ) co-crystal in 1973.7,8 So far, photoconductivity,9,10 ambipolar charge transport,11 and solid-state luminescence12–14 have been studied. Indeed, innovative properties such as stimuli-responsive behavior,15,16 Li-ion conduction,17,18 charge/discharge performance,19,20 and photothermal conversion21,22 have been found in recent years. A variety of properties give CT co-crystals practical applications, for instance, source/drain electrodes,23 rechargeable batteries,20 and seawater desalination22 using TTF–TCNQ co-crystals. The research history of CT co-crystals has covered over 40 years; however, they are still a central topic of organic crystals and electronics.There are numerous ways to prepare CT co-crystals. These methods can be divided into three categories: vapor-phase,24 liquid-phase,25 and solid-phase26 preparation. Particularly, liquid-phase preparation is a promising method because of its simple procedure and preparation of high-quality crystals. One of the unique directions of liquid-phase preparation is the mixing of nano/micro-sized donor and acceptor crystals in an aqueous dispersion.27–29 The conventional liquid-phase method mainly uses organic solvent, whereas mixing of dispersions employs water as a crystallization medium. The strategy was first reported in 2010 by Qichun Zhang et al.27 They initially prepared TTF and TCNQ microcrystal dispersions separately and then mixed them to form TTF–TCNQ nanowires. The same strategy was performed in 2011 using Ag nanoparticles and TCNQ microparticles.28 Inspired by this success, we have recently evaluated the micro/nanocrystallization process of TTF nanocrystals and TCNQ microcrystals via a charge-transfer induced reprecipitation method. In our process, an organic solution of TCNQ (or TTF) was injected into a TTF (or TCNQ) crystal dispersion. Using this process, we have succeeded in preparing nanorods as well as nanorod-coated-rhombic shape TTF–TCNQ crystals.29 As mentioned above, co-crystallization from donor and acceptor crystals in aqueous dispersions is a fascinating strategy for the formation of a CT co-crystal; however, there are very few reports on the subject, which restricts the applicability of the method.
The interfacial properties between the donor and acceptor crystal have also received considerable attention. This feature was first reported in 2008 on the TTF and TCNQ single-crystal interface, which exhibits metallic conduction.30 Based on this finding, the electric and photonic properties of organic crystal interfaces have been extensively studied in several materials.31–33 Recently, a high performance rechargeable battery has been reported using interfacial conduction.20 These reports indicate that organic donor and acceptor crystal interfaces have the potential for device applications. However, it is difficult to obtain a mixture of components without co-crystallization because of their high reactivity to form co-crystals, which densely stacks the molecules during preparation.
These reported studies suggested that a new strategy to separately fabricate CT co-crystals and a crystal mixture with CT at the interface is required. Here, we prepared two crystal structures, CT co-crystals and “phase-separated” crystals, which are a mixture of donor and acceptor crystals from the same donor acceptor pair using water as a poor medium. Initially, water dispersions of acceptor (TCNQ) and donor [TTF, phenothiazine (PTZ), pyrene, dibenzotetrathiafulvalene (DBTTF), or perylene] crystals were prepared using the reprecipitation method.34 Then, the acceptor and donor dispersions were mixed. We found that CT co-crystals or “phase-separated” crystals can be individually prepared by simply changing the aging time of the individual dispersion, especially the donor dispersion. Aging was performed by standing the donor and acceptor crystal dispersions for 24 h before mixing. We found that CT co-crystals were formed in the non-aged mixture. In contrast, the aged mixture prepared “phase-separated” crystals; the donor and acceptor crystals retained their shape without co-crystallization. This tendency was confirmed for most of the molecules that we used in this study, except the perylene/TCNQ mixture. Also, we studied the electric properties of the CT co-crystals and “phase-separated” crystals using TTF and TCNQ. The TTF–TCNQ co-crystals, which have CT states in the bulk, showed low sheet resistance owing to TTF/TCNQ segregated column formation. In contrast, the “phase-separated” crystals are unique in their crystal structure; CT states only occurred at the interface. Due to this unique structure, we found that the “phase separated” crystals showed photo-responsive conduction. Notably, we demonstrate that the CT states in the bulk and at an interface can be controlled by aging, which makes it possible to synthesize crystals with the desired properties using the same components. Indeed, the CT co-crystals would be useful in OFET and OLET device applications, while the “phase-separated” crystals can be applied to rechargeable batteries and organic photovoltaics, in which the interface plays an important role.
Experimental
Materials
Tetrathiafulvalene (TTF, >98.0%), tetracyanoquinodimethane (TCNQ, >98.0%), phenothiazine (PTZ, >98.0%), pyrene (>97.0%), and perylene (>98.0%) were purchased from Tokyo Chemical Industry. Dibenzothetrathiafulvelene (DBTTF, 97%) was purchased from Sigma-Aldrich. Tetrahydrofuran (THF, special grade) was purchased from Kanto Chemical Co., Inc. Ultra-pure water (Milli-Q Q-POD1, 18.2 MΩ cm) was used in the laboratory setup. All reagents were used without purification.Preparation
The aqueous dispersion of the crystals was prepared by the conventional reprecipitation method.34 The donor or TCNQ solution [200 μL dissolved in THF (20 mM)] was injected into 10 mL ultra-pure water with vigorous stirring. The color of the water changed from transparent to cloudy immediately after injecting the solutions. After preparation of the donor and acceptor dispersions, the obtained aqueous dispersions were mixed to observe the co-crystallization behavior. For the non-aged mixture, the aqueous dispersions were mixed within several minutes after preparation. Additionally, for the aged mixture, the aqueous dispersion was left for 24 h and then mixed. The mixed dispersions were kept in air for 2 h before characterization.Characterization
The absorption spectra were measured using a JASCO V-670 spectrophotometer with an integrating sphere. The aqueous dispersions were filled in a 1 mm quartz cell without dilution. Powder X-ray diffraction (XRD) patterns of the crystals were measured using a RIGAKU RINT Ultima-III X-ray diffractometer on a membrane filter (pore size: 0.1 μm) with Cu Kα radiation (λ = 0.15418 nm) at 45 kV and 200 mA. The aqueous dispersion (30 mL for the component dispersion and 60 mL for the mixed dispersion) was filtered through a membrane filter via vacuum filtration. Field emission scanning electron microscopy (FESEM) and energy-dispersive X-ray spectrometry (EDS) were performed using a Hitachi SU8000 scanning electron microscope on the membrane filter (pore size: 0.05 μm or 0.1 μm) at 5 kV and 5 mA for the FESEM observation and 10 kV and 20 mA for the EDS mapping. The contrast of the FESEM and EDS images was changed after the measurement to observe the crystal clearly. The merged EDS mapping images were made using ImageJ. Dynamic light scattering (DLS) was performed using a Malvern Zetasizer Nano ZS with a red laser (633 nm). The aqueous dispersion with aging was ultra-sonicated to disperse the aggregated crystals before the measurement. The current–voltage (I–V) characteristics were measured using a Tektronix 4200A-SCS with two tungsten terminals with APOLLOWAVE α100. The crystals for the I–V measurement were collected by vacuum filtration on a membrane filter (pore size: 0.01 μm). The photoconductivity was measured using the same setup under white light illumination (9.9 mW cm−2).Results and discussion
Aging effect on the co-crystallization behavior between TTF and TCNQ crystals
First, we investigated the aging effect on the co-crystallization behavior by employing donor TTF and acceptor TCNQ (Fig. 1a). The aqueous dispersion of the crystals was prepared by the conventional reprecipitation method.34 TTF (or TCNQ) dissolved in THF was injected into ultra-pure water under vigorous stirring to obtain a TTF (or TCNQ) crystal aqueous dispersion. These aqueous dispersions were mixed within a few minutes after preparation (non-aged mixture). Additionally, the aqueous dispersion was kept at room temperature (approximately 20–25 °C) for 24 h, and then the obtained dispersions were mixed (aged mixture). The mixed dispersions were kept in air for 2 h without stirring before characterization.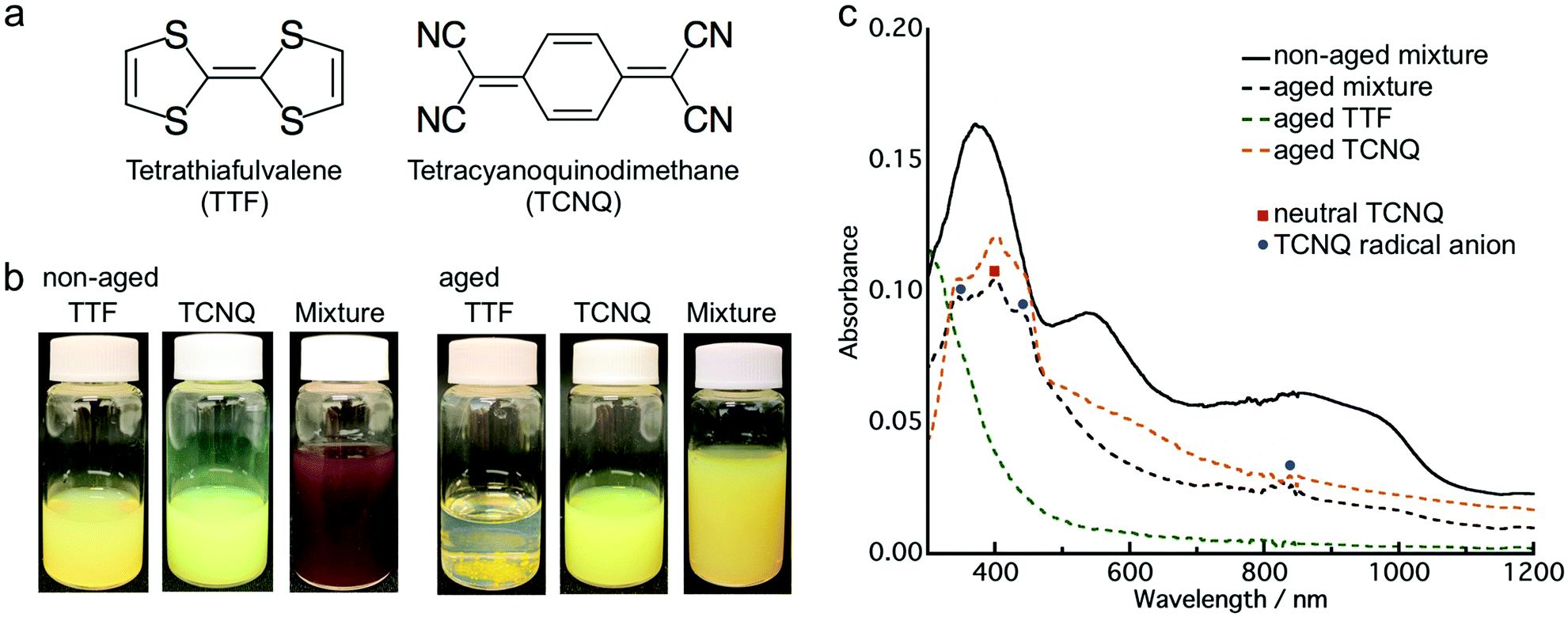 | ||
| Fig. 1 (a) Molecular structure of TTF and TCNQ. (b) Photographs of the TTF dispersion, TCNQ dispersion, non-aged mixture, and aged mixture. (c) Absorption spectra of the non-aged mixture (black solid line), aged mixture (black dashed line), aged TTF (green dashed line) and aged TCNQ (orange dashed line). | ||
Fig. 1b presents photographs of the aqueous dispersions, which clearly depict the aging effect. After the mixing of yellow TTF and yellow-green TCNQ dispersions, the non-aged mixture appeared dark brown, which indicates co-crystallization of TTF and TCNQ. The color change of the non-aged mixture was completed within 1 min after mixing. In contrast, the aged mixture remained yellow, which implied that co-crystallization between TTF and TCNQ crystals did not occur. Fig. 1c shows the absorption spectra of the non-aged and aged mixture. Note that the absorption spectra involved an absorption background of light scattering from the crystals. Distinct from the non-aged TTF and TCNQ crystals (Fig. S1, ESI†), the non-aged mixture shows broad absorption in the whole range due to the formation of TTF–TCNQ crystals. The absorption spectrum of the aged mixture is different to the non-aged mixture, and the absorption of neutral TCNQ (399 nm) and TCNQ radical anions (346 nm, 433 nm and 825 nm) was confirmed.35,36 The absorption spectrum of the aged mixture is similar to the sum of the spectra of the aged TTF and TCNQ dispersions. The above results demonstrate that the aging of the TTF and TCNQ aqueous dispersions before mixing affects the CT progression between the crystals.
The FESEM observation shows the morphological differences of the crystals in the dispersion. TTF formed into a spherical shape (Fig. 2a and d), while TCNQ crystallized into a rhombic shape (Fig. 2b and e). The average size of the TTF crystals was 340 nm before aging, and smaller (120 nm) and larger TTF (340 nm) crystals co-existed after aging (Fig. S2a, ESI†). In contrast, the TCNQ crystals showed a similar average size (340 nm) regardless of aging (Fig. S2b, ESI†). As shown in Fig. 2c and f, the crystal morphology in the mixture is completely different. The non-aged mixture exhibits an urchin-like morphology, whereas the aged mixture displays a spherical/rhombic morphology. EDS mapping of the urchin-like morphology in the non-aged mixture showed sulfur and nitrogen atoms through the whole crystal, indicating the formation of TTF–TCNQ co-crystals (Fig. S3a, ESI†). We have reported that this crystal was formed via co-crystallization between the TTF and TCNQ crystals.29 The spherical TTF crystals react with rhombic TCNQ crystals, and TTF–TCNQ co-crystals can be grown on the TCNQ surface. In sharp contrast, EDS mapping of the spherical/rhombic morphology in the aged mixture displayed sulfur and nitrogen identified on the individual crystals (Fig. S3b, ESI†), indicating that “phase-separated” crystals of spherical TTF and rhombic TCNQ were formed.
 | ||
| Fig. 2 FESEM images of the (a) non-aged TTF crystal, (b) non-aged TCNQ crystal, (c) non-aged mixture, (d) aged TTF crystal, (e) aged TCNQ crystal, and (f) aged mixture. | ||
To reveal the structure of the crystals, a powder XRD measurement was conducted. Fig. S4 (ESI†) shows the XRD patterns of the TTF and TCNQ crystals. The XRD patterns were not changed with aging, revealing that the aging did not affect the crystal structure of the individual crystals. The spherical TTF crystals belong to the space group of P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , with cell parameters of a = 8.379 Å, b = 12.906 Å, c = 8.145 Å, α = 98.91°, β = 96.62°, and γ = 100.44°, which is regarded as the β-phase of TTF.37 The rhombic TCNQ crystals belong to the space group of C2/c, with cell parameters of a = 8.906 Å, b = 7.06 Å, c = 16.395 Å, α = γ = 90°, and β = 98.53°.38Fig. 3 shows the XRD patterns of the non-aged and aged mixture. The diffraction peaks observed at 5.3 nm−1, 6.9 nm−1, and 10.6 nm−1 in the TTF–TCNQ co-crystals were attributed to the (100), (002) and (200) planes, whose positions are similar to those of the TTF–TCNQ bulk crystals; thus, the TTF–TCNQ co-crystals formed by dispersion mixing belong to the space group of P21/c, with cell parameters of a = 12.298 Å, b = 3.819 Å, c = 18.468 Å, α = γ = 90°, and β = 104.46°.39 Note that diffraction peaks from the TTF and TCNQ crystals were observed in the aged mixture [e.g., the (020) plane of TTF at 10.0 nm−1, and the (022) plane of TCNQ at 19.4 nm−1]. These peaks were not found in the non-aged mixture. The XRD results clearly showed that the non-aged mixture formed only TTF–TCNQ co-crystals; in contrast, the aged mixture contained TTF–TCNQ co-crystal as well as TTF and TCNQ crystals.
, with cell parameters of a = 8.379 Å, b = 12.906 Å, c = 8.145 Å, α = 98.91°, β = 96.62°, and γ = 100.44°, which is regarded as the β-phase of TTF.37 The rhombic TCNQ crystals belong to the space group of C2/c, with cell parameters of a = 8.906 Å, b = 7.06 Å, c = 16.395 Å, α = γ = 90°, and β = 98.53°.38Fig. 3 shows the XRD patterns of the non-aged and aged mixture. The diffraction peaks observed at 5.3 nm−1, 6.9 nm−1, and 10.6 nm−1 in the TTF–TCNQ co-crystals were attributed to the (100), (002) and (200) planes, whose positions are similar to those of the TTF–TCNQ bulk crystals; thus, the TTF–TCNQ co-crystals formed by dispersion mixing belong to the space group of P21/c, with cell parameters of a = 12.298 Å, b = 3.819 Å, c = 18.468 Å, α = γ = 90°, and β = 104.46°.39 Note that diffraction peaks from the TTF and TCNQ crystals were observed in the aged mixture [e.g., the (020) plane of TTF at 10.0 nm−1, and the (022) plane of TCNQ at 19.4 nm−1]. These peaks were not found in the non-aged mixture. The XRD results clearly showed that the non-aged mixture formed only TTF–TCNQ co-crystals; in contrast, the aged mixture contained TTF–TCNQ co-crystal as well as TTF and TCNQ crystals.
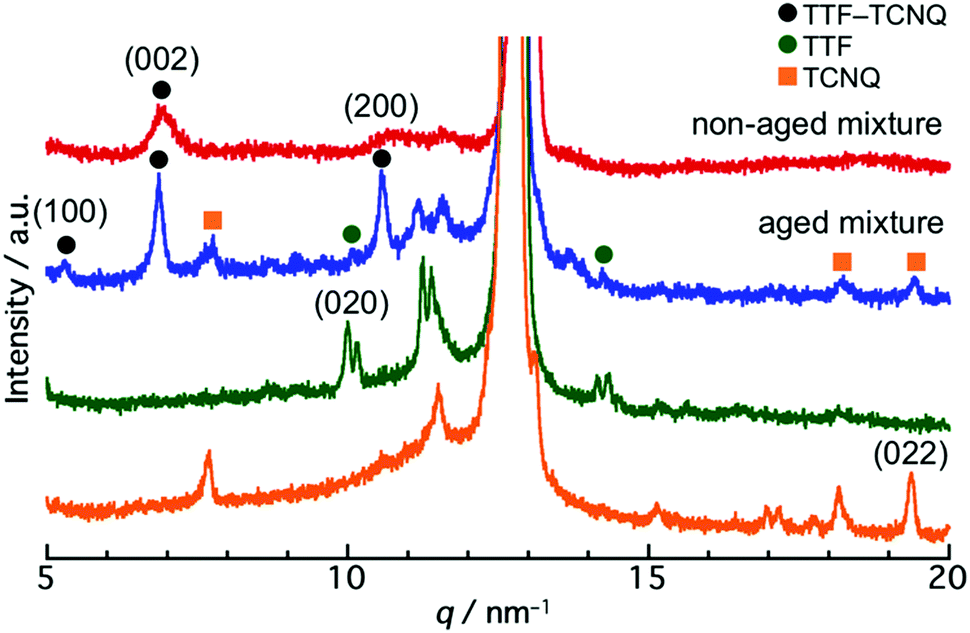 | ||
| Fig. 3 XRD patterns of the non-aged mixture (red line), aged mixture (blue line), aged TTF crystal (green line), and aged TCNQ crystal (orange line). The diffraction peaks at 11.5 nm−1 and 12.8 nm−1 indicate the diffraction from the filter. | ||
It is difficult to define the precise factors that determine the final product morphologies; however, the potential reason could be the state of the crystals. Crystals prepared by the reprecipitation method often form a metastable state because of their fast crystallization, and the crystals change from metastable to stable states over time.40–42 The transition can be observed in the size change of the crystals. Fig. 4 shows the normalized crystal size [V(t)/V(0)] vs. time (t) measurement by DLS.43V(t) is the crystal size at time t, and V(0) is the crystal size at 0 min. The normalized TTF crystal size gradually changed from 1.0 to 0.6 after 600 min, and the size was not changed above 600 min, indicating that the TTF crystals formed a stable state over 600 min. In contrast, the TCNQ crystals do not exhibit significant size changes over time (1.0 to 1.1 after 1400 min), which shows that the TCNQ crystals form a stable state immediately after reprecipitation. In the non-aged mixture, the TTF crystals still exist in a metastable state and the reaction between TTF and TCNQ crystals proceeded spontaneously to form a stable state as a TTF–TCNQ co-crystal. Conversely, in the aged mixture, the TTF crystals already exist in the stable state and it would be difficult for co-crystallization between the crystals to proceed because of their high stability. Consequently, the TTF and TCNQ crystals retained their shape without co-crystallization.
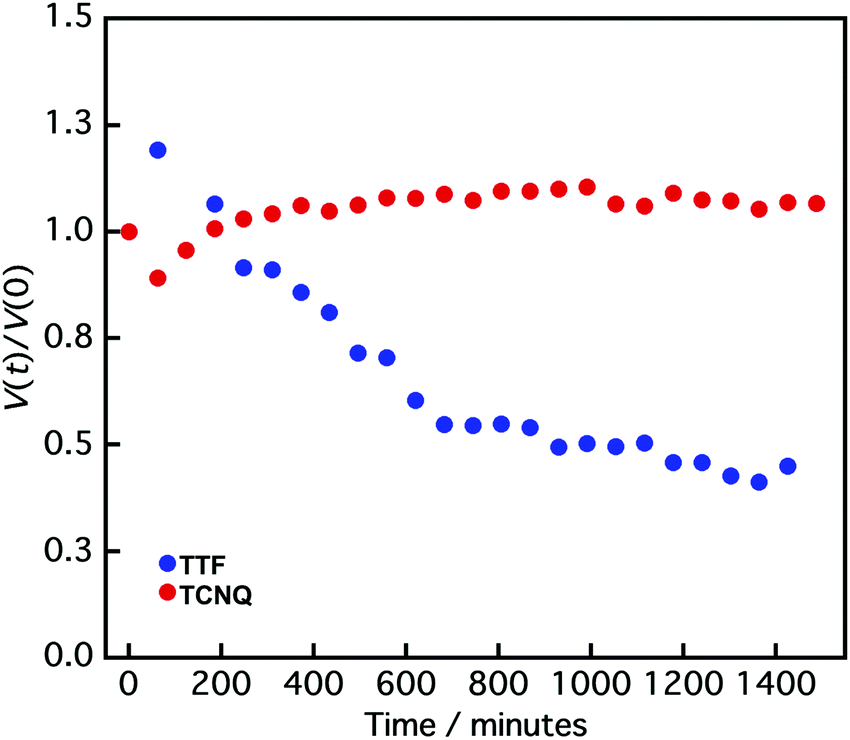 | ||
| Fig. 4 Normalized crystal size [V(t)/V(0)] vs. time (t) of TTF (blue circle) and TCNQ crystals (red circle) measured by DLS. V(t) is the crystal size at time t, and V(0) is the crystal size at 0 min. | ||
As mentioned above, the state of the TTF crystals plays a more important role to determine the co-crystallization behavior in the TTF/TCNQ mixture. In fact, the non-aged TTF/aged TCNQ mixture forms a brown dispersion, indicating the formation of TTF–TCNQ co-crystals, whereas the aged TTF/non-aged TCNQ mixture remains yellow, showing that co-crystallization has not proceeded between the crystals (Fig. S5, ESI†). The FESEM observation of the non-aged TTF/aged TCNQ mixture showed nanodots on the rhombic crystal surface. Conversely, the aged TTF/non-aged TCNQ mixture demonstrated a spherical/rhombic morphology.
To summarize the above results, we considered that the aging of the component crystal dispersion improved the crystallinity of the crystals, which reduced the reactivity of the crystals to form the CT co-crystals. In fact, the powder XRD patterns of TCNQ crystals showed a narrowing of the Bragg diffraction peak width (Fig. S4b, ESI†) and that for DBTTF (Fig. S7c, ESI†) exhibited a clear peak of (110) at 6.9 nm−1 by aging.
Aging effect on the co-crystallization behavior between typical donor crystals and TCNQ crystals
To gain a deeper understanding of the aging effect on the co-crystallization behaviour between the donor and TCNQ crystals, we employed typical donors (PTZ, pyrene, DBTTF, and perylene, Fig. 5; upper part) and mixed them with the TCNQ aqueous dispersion. The aqueous dispersions were prepared by the reprecipitation method,34 and underwent the same procedure as the TTF and TCNQ cases. | ||
| Fig. 5 Upper part: molecular structure of PTZ, pyrene, DBTTF, and perylene. Middle part: absorption spectra of the non-aged mixture (solid line) and aged mixture (dashed line). Lower part: XRD patterns of the non-aged mixture (red line), aged mixture (blue line), aged donor crystals (green line), and aged TCNQ crystal (orange line). The diffraction peaks at 11.5 nm−1 and 12.8 nm−1 indicate the diffraction from the filter. | ||
We find that aging affected the co-crystallization behavior in the PTZ/TCNQ, pyrene/TCNQ, and DBTTF/TCNQ mixture, which has a similar tendency to that of the TTF/TCNQ systems. Fig. 5 (middle part) shows the absorption spectra of the non-aged and aged mixtures. In the non-aged mixture, new absorption which was not confirmed in the donor and TCNQ dispersion (Fig. S6a–c, ESI†) was clearly confirmed over 700 nm, at 580–830 nm, and over 770 nm in the PTZ/TCNQ, pyrene/TCNQ, and DBTTF/TCNQ mixtures, respectively. These absorption bands correspond to CT absorption from the donor to TCNQ, indicating the formation of PTZ–TCNQ,44 pyrene–TCNQ,45 and DBTTF–TCNQ46 co-crystals. On the contrary, the spectra of the aged mixtures were the sum of the donor and TCNQ absorption spectra, which indicates that CT formation was negligible in the aged mixtures (Fig. S6e–g, ESI†).
The XRD results also supported the difference in co-crystallization behavior between the non-aged and aged mixtures. The powder XRD patterns for the donor crystals were almost the same as that of the reported bulk crystal (Fig. S7 and Table S1, ESI†). Moreover, the peak positions were not changed with aging, which supported that the aging did not affect the crystal structure. Fig. 5 (lower part) shows the XRD pattern of the non-aged and aged mixtures. The non-aged mixtures of PTZ/TCNQ, pyrene/TCNQ, and DBTTF/TCNQ contained diffraction originating from the donor crystals and co-crystals (the crystal structure of the CT co-crystals belongs to the literature47–50). On the other hand, in the aged mixture, diffraction from the donors and TCNQ crystals was clearly confirmed [e.g., the (022) plane of TCNQ at 19.4 nm−1 was confirmed in all combinations], and small diffraction from the co-crystals was also observed in PTZ/TCNQ and pyrene/TCNQ. These results indicate that the non-aged mixture dominantly formed CT co-crystals, whereas the aged mixture produced “phase-separated” crystals.
The formation of CT co-crystals or “phase-separated” crystals was also confirmed by their crystal morphology. As shown in Fig. S8 (ESI†), cubic-like PTZ (550 nm), sphere-like pyrene (250 nm), and cubic-like DBTTF (420 nm) were confirmed in the non-aged donor dispersion, while cubic/rhombic-like PTZ (640 nm), cubic-like pyrene (2520 nm), and cubic-like DBTTF (510 nm) were observed in the aged donor dispersion. The crystal size is larger after aging, suggesting the transition of the crystal from a metastable to a stable state, which causes a difference in the co-crystallization behavior. As shown in Fig. S9 (ESI†), the crystal morphology is completely different between the non-aged and aged mixtures. The CT co-crystal shapes were rod-like in the PTZ/TCNQ and pyrene/TCNQ mixtures, and cubic-like in the DBTTF/TCNQ mixture. On the other hand, the aged mixture showed “phase-separated” crystals; crystal shapes of both the donor and acceptor were clearly observed.
Interestingly, there were no differences in the absorption spectra and XRD patterns in the non-aged and aged mixtures of perylene/TCNQ. The absorption was the sum of the component dispersions (Fig. S6d and h, ESI†), and diffractions from perylene and TCNQ were obtained in both the non-aged and aged mixtures (Fig. 5; middle part). The DLS measurements (Fig. S8i, ESI†) before and after aging showed perylene crystals in similar sizes (280 nm and 240 nm), indicating that perylene forms a stable state immediately after preparation. Therefore, we inferred that a reaction did not occur between the stable perylene and TCNQ crystals. In fact, cubic perylene and rhombic TCNQ co-existed in the non-aged and aged mixtures (Fig. S9d and h, ESI†).
Comparison of the co-crystallization behavior of the donor/TCNQ mixtures shows that the water solubility affects the CT progression between the crystals. The water solubilities of PTZ, pyrene, and perylene are 1.594 × 10−3, 1.350 × 10−4, and 4.037 × 10−7 mg mL−1, respectively, at room temperature (25 °C).51 Unfortunately, there is no information on the water solubility of DBTTF. According to the XRD results (Fig. 5; middle part), the PTZ and pyrene crystals, which have relatively high solubility (10−3 to 10−4 mg mL−1), appeared to form CT co-crystals with TCNQ, even after aging. For instance, the (040) plane at 9.9 nm−1 from PTZ–TCNQ co-crystals was confirmed in the aged PTZ/TCNQ mixture, and the (010) plane at 6.5 nm−1 from pyrene–TCNQ co-crystals was observed in the aged pyrene/TCNQ mixture. In the mixing of aqueous dispersions, the driving force of co-crystallization would be the dissolution of the molecules from the crystal surface; thus, higher solubility leads to the dissolution of the crystals, which accelerates the co-crystallization. In other words, higher solubility in water is favourable to form CT co-crystals, while lower solubility is beneficial to form “phase-separated” crystals.
Conductivity and photoconductivity study
To explore the applicability of the CT co-crystals and “phase-separated” crystals prepared by mixing of aqueous dispersions, we evaluated the conductivity and photoconductivity properties using TTF and TCNQ. The TTF–TCNQ co-crystals and “phase-separated” crystals were collected on a membrane filter by vacuum filtration and used for the measurement (Fig. S10a, ESI†). FESEM images of the TTF–TCNQ co-crystals and “phase-separated” crystals on a filter are shown in Fig. S10b (ESI†). I–V measurements were performed at five points in the same sample. Fig. 6a and b show the I–V curves of the TTF–TCNQ co-crystals and “phase-separated” crystals in the dark and under white light illumination. Nonlinear and linear curves of the TTF–TCNQ co-crystals and “phase-separated” crystals were observed, respectively. The sheet resistance calculated from the resistance of the sample and measurement area was approximately 103 to 104 Ω sq−1 in the TTF–TCNQ co-crystals. The value appears similar to that of TTF–TCNQ thin films on a CT crystal surface,52 indicating the potential for conductive material applications. We also expected that low resistance would be observed in the “phase-separated” crystals owing to the interfacial conductivity between the TTF and TCNQ crystals; however, the sheet resistance was on the order of 1011 Ω sq−1, which was approximately 107 to 108 Ω sq−1 higher than the reported value at the crystal interface (1 × 103 to 3 × 104 Ω sq−1).30 Interestingly, the TTF–TCNQ co-crystals did not present any differences between the dark and illuminated conditions, while a photoresponse was observed in the “phase-separated” crystals, which demonstrates their potential application in photoelectrical devices.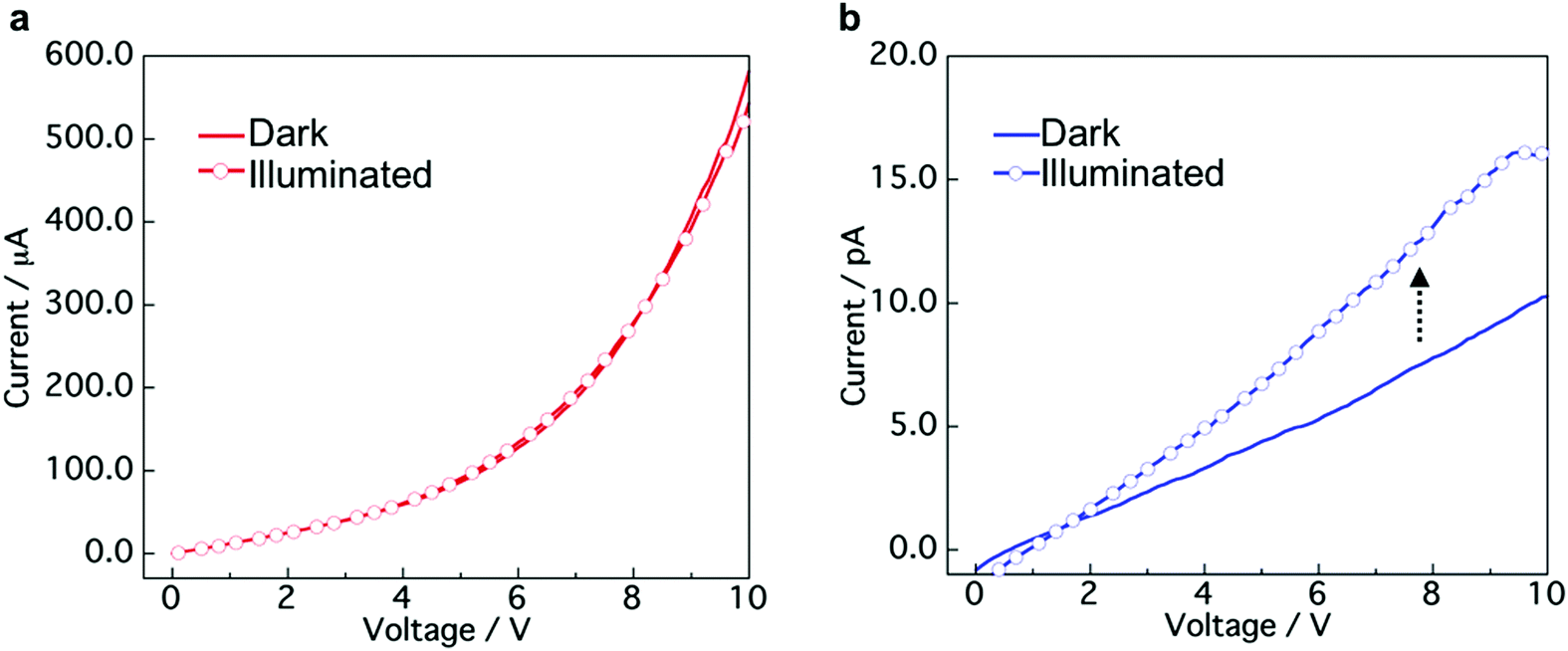 | ||
| Fig. 6 I–V curves of (a) TTF–TCNQ co-crystals and (b) “phase-separated” crystals in the dark and under white light illumination conditions (9.9 mW cm−2). | ||
The TTF–TCNQ co-crystals formed a conventional segregated stacked crystal structure, which is known to exhibit high conductivity. Therefore, the co-crystals showed a low sheet resistance. In contrast, the TTF/TCNQ “phase-separated” crystals only form a CT state at the crystal interface, which indicates that most of the crystal is an insulator. Light irradiation of the “phase-separated” crystals excites TTF and TCNQ to produce photocarriers.53 The photocarriers migrated to the crystal interface and transferred to the electrode using the CT state at the interface.
Conclusions
In conclusion, CT co-crystals or “phase-separated” crystals, which are a mixture of donor and acceptor crystals, were prepared by mixing of aqueous dispersions with different aging time. Evidently, aging of the aqueous dispersion affects the co-crystallization behavior between the donor and TCNQ crystals. The non-aged mixing facilitates the formation of the CT co-crystals, while aged mixing is favourable to form the “phase-separated” crystals. This tendency was confirmed in TTF/TCNQ, PTZ/TCNQ, pyrene/TCNQ, and DBTTF/TCNQ mixtures. Perylene crystals were not co-crystallized with TCNQ even before aging owing to their high stability and low solubility in water. The conductivity and photoconductivity measurements of the CT co-crystals and “phase-separated” crystals using TTF and TCNQ showed their potential for photoelectrical device applications. Furthermore, the “phase-separated” crystal dispersion can be used to prepare a thin film with multiple crystal interfaces between donor and acceptor crystals. We believe that the insights obtained in this study will aid in the better understanding of the co-crystallization behavior of the crystals in aqueous dispersions and realize advanced functions due to the unique micro/nanocrystal morphologies.Author contributions
M. T. performed all experiments and prepared the manuscript. J. M. and A. M. designed the concept of the research and revised the manuscript. All authors have approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Prof. Junji Kido, Prof. Takayuki Chiba, and Ms Hinako Ebe from Yamagata University for their support in performing the digital microscope observation and I–V measurements. This work was partially supported by the Grant-in-Aid for JSPS Fellows (JP 20J10289) and a research grant from the Japan Power Academy.References
- W. Zhu, H. Dong, Y. Zhen and W. Hu, Sci. China Mater., 2015, 58, 854–859 CrossRef.
- J. Zhang, W. Xu, P. Sheng, G. Zhao and D. Zhu, Acc. Chem. Res., 2017, 50, 1654–1662 CrossRef PubMed.
- H. Jiang, P. Hu, J. Ye, K. K. Zhang, Y. Long, W. Hu and C. Kloc, J. Mater. Chem. C, 2018, 6, 1884–1902 RSC.
- L. Sun, W. Zhu, F. Yang, B. Li, X. Ren, X. Zhang and W. Hu, Phys. Chem. Chem. Phys., 2018, 20, 6009–6023 RSC.
- L. Sun, Y. Wang, F. Yang, X. Zhang and W. Hu, Adv. Mater., 2019, 31, 1902328 CrossRef PubMed.
- Y. Huang, Z. Wang, Z. Chen and Q. Zhang, Angew. Chem., Int. Ed., 2019, 58, 9696–9711 CrossRef PubMed.
- J. Ferraris, D. O. Cowan, V. Walatka and J. H. Perlstein, J. Am. Chem. Soc., 1973, 95, 948–949 CrossRef.
- L. B. Coleman, M. J. Cohen, D. J. Sandman, F. G. Yamagishi, A. F. Garito and A. J. Heeger, Solid State Commun., 1973, 12, 1125–1132 CrossRef.
- J. Tsutsumi, H. Matsui, T. Yamada, R. Kumai and T. Hasegawa, J. Phys. Chem. C, 2012, 116, 23957–23964 CrossRef.
- W. Zhu, Y. Yi, Y. Zhen and W. Hu, Small, 2015, 11, 2150–2156 CrossRef PubMed.
- T. Salzillo, A. Campos and M. M. Torrent, J. Mater. Chem. C, 2019, 7, 10257–10263 RSC.
- Y.-L. Lei, Y. Jin, D.-Y. Zhou, W. Gu, X.-B. Shi, L.-S. Liao and S.-T. Lee, Adv. Mater., 2012, 24, 5345–5351 CrossRef PubMed.
- Y.-Q. Sun, Y.-L. Lei, X.-H. Sun, S.-T. Lee and L.-S. Liao, Chem. Mater., 2015, 27, 1157–1163 CrossRef.
- K. Liu, Y. Lei and H. Fu, Chem. Mater., 2020, 32, 5162–5172 CrossRef.
- Y. Sun, Y. Lei, H. Dong, Y. Zhen and W. Hu, J. Am. Chem. Soc., 2018, 140, 6186–6189 CrossRef PubMed.
- L. Sun, F. Yang, X. Zhang and W. Fu, Mater. Chem. Front, 2020, 4, 715–728 RSC.
- Y.-S. Guan, Y. Hu, H. Zhang, G. Wu, H. Yan and S. Ren, Chem. Commun., 2019, 55, 7179–7182 RSC.
- K. H. Sato, M. Umeki, T. Tezuka and K. Oyaizu, ACS Appl. Electron. Mater., 2020, 2, 2211–2217 CrossRef.
- S. Lee, J. Hong, S.-K. Jung, K. Ku, G. Kwon, W. M. Seong, H. Kim, G. Yoon, I. Kang, K. Hong, H. W. Jang and K. Kang, Energy Storage Mater., 2019, 20, 462–469 CrossRef.
- Y. Fujihara, H. Kobayashi, S. Takaishi, T. Tomai, M. Yamashita and I. Honma, ACS Appl. Mater. Interfaces, 2020, 12, 25748–25755 CrossRef PubMed.
- Y. Wang, W. Zhu, W. Du, X. Liu, X. Zhang, H. Dong and W. Hu, Angew. Chem., Int. Ed., 2018, 57, 3963–3967 CrossRef PubMed.
- S. Tian, Z. Huang, J. Tan, X. Cui, Y. Xiao, Y. Wan, X. Li, Q. Zhao, S. Li and C.-S. Lee, ACS Energy Lett., 2020, 5, 2698–2705 CrossRef.
- M. Hiraoka, T. Hasegawa, T. Yamada, Y. Takahashi, S. Horiuchi and Y. Tokura, Adv. Mater., 2007, 19, 3248–3251 CrossRef.
- M. Sakai, M. Iizuka, M. Nakamura and K. Kudo, Jpn J. Appl. Phys., 2003, 42, 2488–2491 CrossRef.
- M. Oyama, Q. Wei, M. Mukaida, Y. Kawanishi and T. Ishida, Synth. Met., 2017, 230, 12–17 CrossRef.
- D. Braga, L. Maini and F. Grepioni, Chem. Soc. Rev., 2013, 42, 7638–7648 RSC.
- J. Xiao, Z. Yin, H. Li, Q. Zhang, F. Boey, H. Zhang and Q. Zhang, J. Am. Chem. Soc., 2010, 132, 6926–6928 CrossRef PubMed.
- J. Xiao, Z. Yin, Y. Wu, J. Guo, Y. Cheng, H. Li, Y. Huang, Q. Zhang, J. Ma, F. Boey, H. Zhang and Q. Zhang, Small, 2011, 7, 1242–1246 CrossRef PubMed.
- M. Takeda, K. Umemoto, T. Nohara, K. Tozawa, J. Matsui and A. Masuhara, J. Electrochem. Soc., 2019, 166, B3131–B3135 CrossRef.
- H. Alves, A. S. Molinari, H. Xie and A. F. Morpurgo, Nat. Mater., 2008, 7, 574–580 CrossRef PubMed.
- Y. Takahashi, K. Hayakawa, T. Naito and T. Inabe, J. Phys. Chem. C, 2012, 116, 700–703 CrossRef.
- Y. Takahashi, K. Hayakawa, K. Takayama, S. Yokokura, J. Harada, H. Hasegawa and T. Inabe, Chem. Mater., 2014, 26, 993–998 CrossRef.
- Y. Krupskaya, I. G. Lezama and A. F. Morpurgo, Adv. Funct. Mater., 2016, 26, 2334–2340 CrossRef.
- H. Nakanishi and H. Oikawa, in Single Organic Nanoparticles, ed. H. Masuhara, H. Nakanishi and K. Sasaki, Springer, Berlin, 1st edn, 2003, ch. 2, pp. 17–31 Search PubMed.
- D. de Caro, M. Souque, C. Faulmann, Y. Coppel, L. Valade, J. Fraxedas, O. Vendier and F. Courtade, Langmuir, 2013, 29, 8983–8988 CrossRef PubMed.
- H. Tamiya, H. Nakano and T. Iimori, J. Lumin., 2017, 192, 203–207 CrossRef.
- A. Ellern, J. Bernstein, J. Y. Becker, S. Zamir, L. Shahal and S. Cohen, Chem. Mater., 1994, 6, 1378–1385 CrossRef.
- R. E. Long, R. A. Sparks and K. N. Trueblood, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1965, 18, 932–939 CrossRef.
- T. J. Kistenmacher, T. E. Phillips and D. O. Cowan, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1974, B30, 763–768 CrossRef.
- J. Mori, Y. Miyashita, D. Oliveira, H. Kasai, H. Oikawa and H. Nakanishi, J. Cryst. Growth, 2009, 311, 553–555 CrossRef.
- L. Huang, Q. Liao, Q. Shi, H. Fu, J. Ma and J. Yao, J. Mater. Chem., 2010, 20, 159–166 RSC.
- M. Nishida and E. R. V. Keuren, MRS Commun., 2011, 1, 7–11 CrossRef.
- F. Wang, V. N. Richards, S. P. Shields and W. E. Buhro, Chem. Mater., 2014, 26, 5–21 CrossRef.
- J.-J. Li, S.-H. Zhang, F.-X. Wang, H.-D. Wu, L.-Y. Shi and G.-B. Pan, Mater. Lett., 2018, 210, 161–164 CrossRef.
- R. J. Dillon and C. J. Bardeen, J. Phys. Chem. A, 2012, 116, 5145–5150 CrossRef PubMed.
- H.-D. Wu, F.-X. Wang, Y. Xiao and G.-B. Pan, J. Mater. Chem. C, 2013, 1, 2286–2289 RSC.
- H. Kobatashi, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1974, B30, 1010–1017 CrossRef.
- C. K. Prout, I. J. Tickle and J. D. Wright, J. Chem. Soc., Perkin Trans. 2, 1973, 528–530 RSC.
- M. A. Dobrowolski, G. Garbarino, M. Mezouar, A. Ciesielski and M. K. Cyrański, CrystEngComm, 2014, 16, 415–429 RSC.
- H. Kobayashi and J. Nakayama, Bull. Chem. Soc. Jpn., 1981, 54, 2408–2411 CrossRef.
- S. H. Yalkowsky, Y. He and P. Jain, Handbook of Aqueous Solubility Data, CRC Press, Boca Raton, 2016 Search PubMed.
- Y. Takahashi, T. Mikasa, K. Hayakawa, S. Yokokura, H. Hasegawa, J. Harada and T. Inabe, J. Phys. Chem. C, 2016, 120, 17537–17545 CrossRef.
- H. Jiang, X. Yang, Z. Cui, Y. Liu, H. Li and W. Hu, Appl. Phys. Lett., 2009, 94, 123308 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Absorption spectra, XRD patterns, FESEM images, DLS measurements, photographs, EDS mapping, digital microscope images, I–V curves (PDF). See DOI: 10.1039/d0ma01001d |
| This journal is © The Royal Society of Chemistry 2021 |