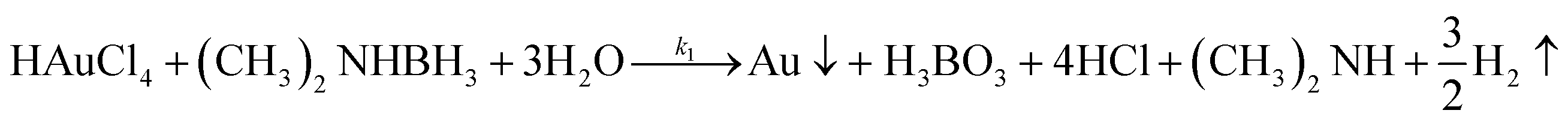DOI:
10.1039/D0MA00935K
(Paper)
Mater. Adv., 2021,
2, 488-496
M13 bacteriophage-templated gold nanowires as stretchable electrodes in perovskite solar cells†
Received
29th November 2020
, Accepted 13th December 2020
First published on 14th December 2020
Abstract
The synthesis and stretchable device application of bacteriophage-templated gold nanowires are demonstrated. Stretchable gold nanowires are synthesised by growing gold nanoparticles on a network of virus clones. Exploiting dimethylamine borane as a reducing agent, a spontaneous redox reaction between HAuCl4 and M13 bacteriophage wild-type virus—without the need for genetic engineering—produces a metal nanowire network with high transparency and conductivity. Virus, HAuCl4, and ascorbic acid additive concentrations are optimised for high optical conductivity. The optimal virus-templated gold nanowire electrodes from aqueous solutions exhibit a sheet resistance of 144.5 Ω sq−1 and transparency of 83% at 550 nm light wavelength. The virus-templated gold nanowire electrodes are transferred to polydimethylsiloxane for morphology roughness reduction as well as the application of its stretchability. Perovskite solar cells fabricated using virus-templated Au nanowires exhibit a power conversion efficiency of 9.28% and a stretchability of 8% owing to the intrinsic flexibility of the M13 bacteriophage template.
Introduction
The application of natural or nature-inspired synthetic biomaterials to electronics is a promising research field in the light of current plastic pollution and raw material shortage problems.1,2 The M13 bacteriophage is an environmentally friendly biomaterial, which has readily been used in electronics.3–10 In particular, the application of the M13 bacteriophage as a template for the assembly of conductive materials has attracted great attention.11–16 The M13 bacteriophage has a structure of single-stranded deoxyribonucleic acid (DNA) enclosed in a cylindrical capsid with 2700 copies of the pVIII protein and 5 copies of the pIII protein, resulting in 5-fold symmetry along the length of the virus. The pVIII proteins are oriented in such a way that the surface of the virus has both peptide termini, i.e., amino and carboxyl ends. Such characteristics make the M13 bacteriophage an excellent template for the synthesis of various functional nanowires as the carboxylate ends of the virus have an affinity for cationic metal particle binding and nucleation along the shaft of a virus.17,18 The flexible and filamentous M13 bacteriophage, with a high aspect ratio of approximately 6.5–880 nm is capable of forming electrodes with a large metal-coated network.3,4
In the wake of sustainable society, organometallic halide perovskite solar cells (PSCs) have emerged as a next-generation energy-harvester.19 As the paradigm of electronics is shifting to stretchable devices at the level of artificial skin and health-monitoring plasters,20 energy devices such as PSCs are following suit. Accordingly, research on low-cost and stretchable PSCs has progressed greatly in recent years, and it has been found that transparent conductors hold the key to the performance of stretchable and flexible PSCs.21–23 It is imperative that high-cost and inflexible metal oxide transparent conductors of PSCs are replaced by a low-cost and stretchable alternative. In this respect, carbon-based transparent conductors have shown promising potential.24–28 However, their conductivity has limitations and the use of chemical dopants has a stability issue.29,30 Moreover, most reported carbon electrodes are not solution-processable. Those that are solution-processable, such as reduced graphene oxide31 and double-walled carbon nanotubes,23 display limited performance. Metal nanowire electrodes, on the other hand, have the upper hand as they are solution-processable23,32–34 and highly conductive.35–37 However, their mechanical flexibility is low as metals are stiff and ductile. While nanowire entanglement may give rise to flexibility, a single string of metal wire can only stretch so far without being damaged or cut.38–40 Thus, it is essential that stretchable metal nanowire shafts are developed. The M13 bacteriophage, in this sense, can satisfy the need for a stretchable nanowire template. In fact, there have been a few M13 bacteriophage-templated electrodes reported to date.12–15,41–44 However, there are no reports on M13 bacteriophage-based stretchable transparent electrodes or their application to PSCs. Furthermore, the previous approach to M13 bacteriophage-templated electrodes required illumination45 or genetic engineering13,46,47 during synthesis. For example, an evolutionary screening process called bio-panning was used to selectively bind gold particles.17
In this study, we demonstrate the synthesis of environmentally friendly and scalable M13 bacteriophage-templated stretchable electrodes and their application as a transparent conductor in PSCs. Neither a diameter control48 nor a temperature control49 was necessary for the M13 bacteriophage-templated metal nanowire synthesis. Wild-type M13 bacteriophages were used directly to induce the reduction reaction of gold nanoparticles for the binding process using two commonly used reducing agents, sodium borohydride (NaBH4)50–52 and dimethylamine borane (DMAB).53–55 The concentrations of viruses, gold particles, and ascorbic acid additive were optimised for comparison. The optical conductivity of the virus-templated gold nanowire electrodes using NaBH4 was far superior to those using DMAB. The reason behind this performance difference was the formation of NaCl impurities and the rate of reaction. The resulting virus-templated Au nanowire electrode exhibited a sufficient optical conductivity of 144.5 Ω sq−1 with 83% transparency at 550 nm, which changed to 158.1 Ω sq−1 and 76% after the polydimethylsiloxane (PDMS) imprint transfer to alleviate the mal-effect of the rough morphology. The stretchable electrodes showed an excellent stretchability of 11% (change in resistance over original resistance of the film (ΔR/R)). PSCs were fabricated based on the stretchable virus-templated electrodes, which gave a power conversion efficiency (PCE) of 9.28% with negligible hysteresis and device stretchability of 8%. This performance is one of the highest among the reported stretchable metal nanowire-based PSCs.
Results and discussion
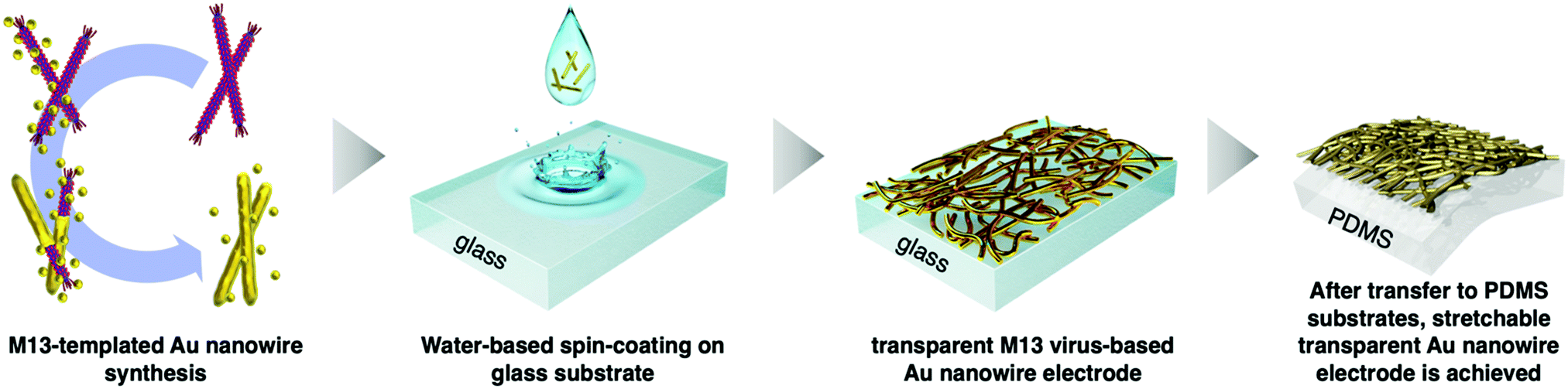
Virus-templated nanowires were fabricated by growing metal particles on wild-type M13 bacteriophage in water and drop-casting the solution onto glass substrates, which were later transferred onto polydimethylsiloxane (PDMS) for device application as stretchable electrodes (Fig. 1). The wild-type M13 bacteriophage possesses amino acids with carboxylate ends, namely, aspartate and glutamate. These functional groups are known to bind to positive metal ions via ion exchange.56 Initially, we tried to bind silver metal particles to the M13 bacteriophages to produce virus-templated Ag nanowires. Varying amounts of AgNO3 were added to the aqueous M13 bacteriophage solution at different concentrations. This was followed by the addition of ascorbic acid, which has been reported to help metal growth,57–65 and reducing agents,66–76 which promote the reduction of the added metal ions. However, the virus-templated Ag nanowire electrodes did not exhibit any conductivity (Fig. S1, ESI†). Bio-transmission electron microscopy (bio-TEM) images confirm that the virus network forms a nice nanowire (Fig. S2, ESI†). This indicates that the reason for the lack of conductivity is that the wild-type M13 bacteriophages do not have sufficient ionic interaction strength to bind to Ag nanoparticles as strongly as to Au nanoparticles.77 This explains why previous studies report genetically modified 4E virus—not the wild-type—when producing virus-templated Au nanowire electrodes.45
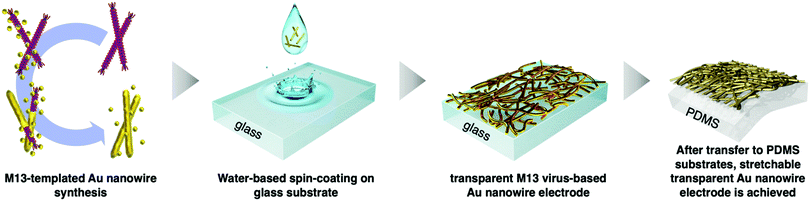 |
| | Fig. 1 Schematic illustration of the growth, thin-film fabrication, and transfer of the M13 bacteriophage-templated gold nanowire for stretchable electrode applications. | |
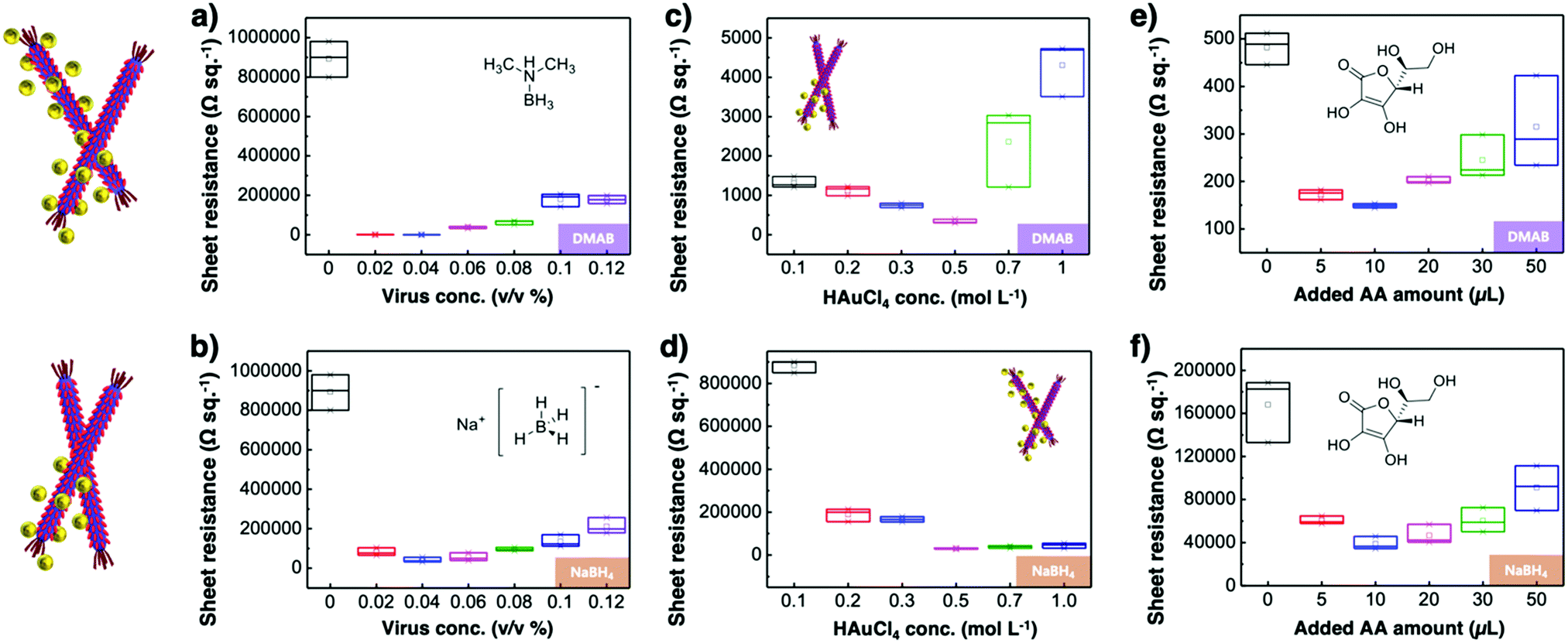
Virus-templated Au nanowire electrodes had drastically higher conductivity and transparency than their Ag nanowire electrode counterparts. Fig. 2 and Fig. S3 (ESI†) show the sheet resistance of virus-templated Au nanowire electrodes on glass measured using the 4 probe method over the virus, HAuCl4, and ascorbic acid (AA) concentrations under two different reducing agents, namely, 4-(dimethylamino)benzaldehyde (DMAB) (eqn (1)) and (eqn (2)) and NaBH4 (eqn (3)). Fig. 2a and b show the resistance of the electrodes when 0.5 mol L−1 HAuCl4 was used without AA. The results reveal that the amount of virus affects the sheet resistance greatly. It is interesting that the sheet resistance values of the virus-based electrodes were significantly lower when DMAB rather than NaBH4 was used as a reducing agent. A relatively small amount of the M13 virus (0.04% v/v) was optimal with both DMAB and NaBH4. This is because the M13 bacteriophages themselves are insulators and serve only as a growth template for metal ions. Changing the amount of HAuCl4 influenced the overall sheet resistance as well, though at a relatively lesser degree. While increasing concentration decreased sheet resistance, 0.5 mol L−1 was found to be the optimal concentration for both DMAB and NaBH4 (Fig. 2c and d). Adding AA decreased sheet resistance even further. The optimal AA concentration was 10 μL, which resulted in sheet resistance of approximately 144.5 and 36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 Ω sq−1 for the DMAB-added and NABH4-added virus-templated Au nanowires, respectively.
500 Ω sq−1 for the DMAB-added and NABH4-added virus-templated Au nanowires, respectively.
| | 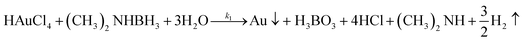 | (1) |
| | 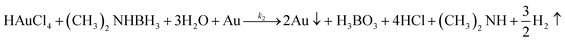 | (2) |
| | | HAuCl4 + NaBH4 → Au0 + B(OH)3 + NaCl + H3O+ | (3) |
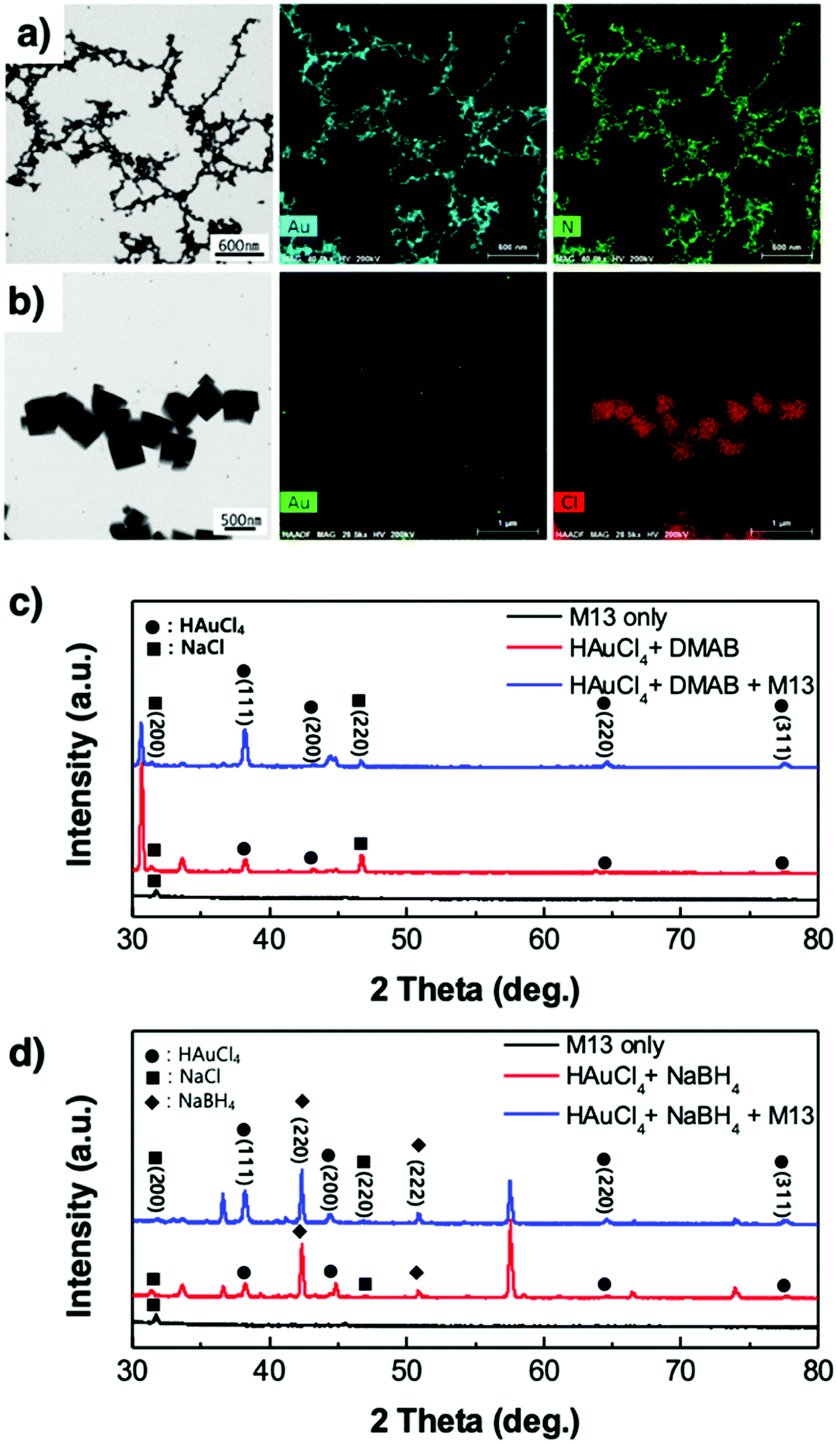
Bio-TEM images show the virus-templated Au nanowire network, the effect of the type of reducing agent, M13 bacteriophage concentration, and HAuCl
4 concentration. Both the virus-templated Au nanowires made by DMAB and NaBH
4 show a well-connected nanowire network, but those made by DMAB show particles around the network (Fig. S4, ESI
†). Delving further into the study, bio-TEM images with different virus concentrations show that Au particles have a round shape, and an increase in the virus concentration decreases the number of particles (Fig. S5, ESI
†). Increasing the HAuCl
4 concentration increases the round-shaped particles around the network (Fig. S6, ESI
†). Energy-dispersive X-ray spectroscopy (EDS) analysis confirms that the virus nanowires were fully covered by Au (
Fig. 3a). On the other hand, the virus-templated Au nanowire made by NaBH
4 contains cubic particles, which are mixed with circular Au particles (Fig. S7, ESI
†). From the EDS analysis, we suspect that these particles are NaCl, which is the reason for the poor optical conductivity of the virus-templated Au nanowires made by NaBH
4 (
Fig. 3b) (
eqn (3)).
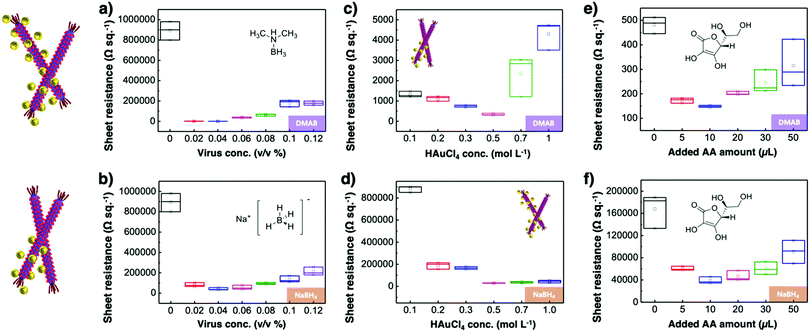 |
| | Fig. 2 Sheet resistance according to (a) and (b) virus concentrations, (c) and (d) HAuCl4 concentrations, and (e) and (f) AA concentrations under DMAB and NaBH4 reducing agents. | |
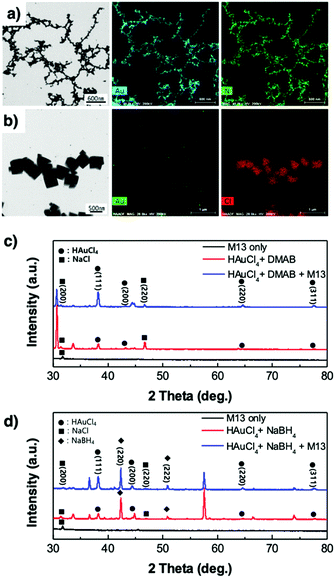 |
| | Fig. 3 STEM images and EDS analysis of the virus-templated Au nanowires formed under (a) DMAB and (b) NaBH4. XRD spectra of the virus-templated Au nanowires formed under (c) DMAB and (d) NaBH4. | |
X-Ray diffraction spectroscopy (XRD) was used to analyse the virus-templated Au nanowires. The XRD spectrum of HAuCl4 mixed with DMAB shows peaks corresponding to Au (Fig. 3c and Fig. S8, ESI†), which intensify upon the addition of the M13 bacteriophage, proving that the presence of the M13 virus enhances Au formation. In the case of the virus-templated Au nanowires made by NaBH4, a mixture of HAuCl3 and NaBH4 formed not only Au but also NaCl, as evidenced by both XRD and EDS (Fig. 3b, d, and Fig. S9, ESI†) (eqn (3)). The presence of the M13 virus enhanced the intensity of the Au peaks as expected, and the suppression of the NaBH4 peaks upon the addition of the M13 viruses corroborates this (Fig. S10, ESI†). This reveals that the nano-particle growth of the M13 viruses is not as good when NaBH4 is used as a reducing agent.
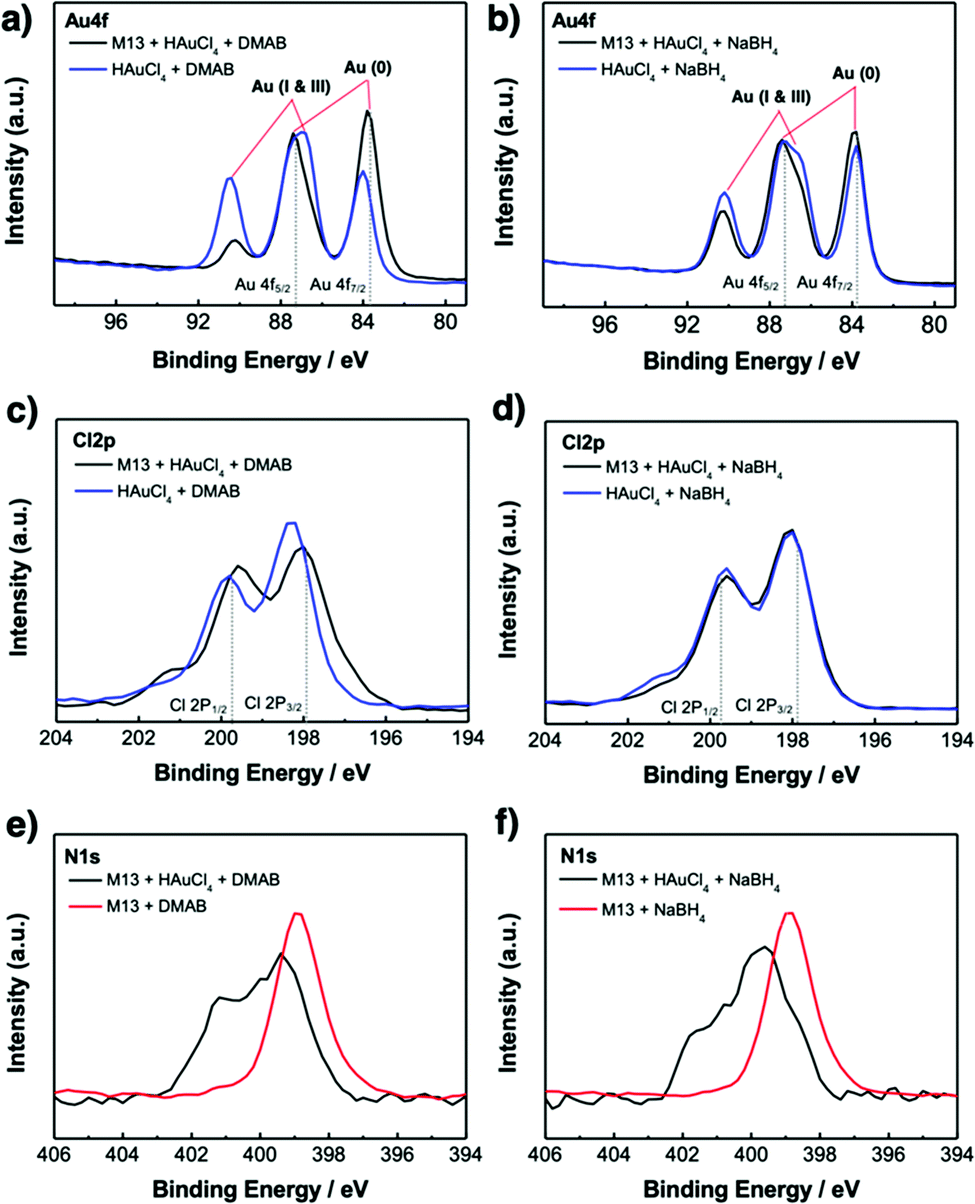
X-Ray photoelectron spectroscopy (XPS) was used to investigate the chemistry occurring within the virus-templated Au nanowires. The Au 4f peaks of the HAuCl4 + DMAB mixture and the M13 virus + HAuCl4 + DMAB mixture show that the intensity of the Au 4f7/2 peak, which corresponds to Au(0) metal particles, increases upon the addition of the M13 virus (Fig. 4a and Fig. S11, ESI†).78–80 At the same time, the Au 4f5/2 peak convolution between 86 eV (Au(I)) and 87 eV (Au(III)) as well as the peak at 91 eV, which corresponds to Au(I & III), decreased upon the addition of the M13 virus, indicating the transition of HAuCl4 to Au particles. On the other hand, the Au 4f peaks of the HAuCl4 + NaBH4 mixture and the M13 virus + HAuCl4 + NaBH4 mixture show that the intensity of the Au 4f7/2 peak increases slightly, which indicates that Au particles are generated in a relatively small quantity (Fig. 4b and Fig. S11, ESI†). A decrease in the peaks responsible for Au(I) and Au(II) was observed, but to a smaller degree compared to that in the case of DMAB. This confirms our finding from TEM and XRD that a greater amount of Au particles was generated when DMAB was used as a reductant as opposed to NaBH4. The Cl 2p peaks are composed of two species, namely Cl 2p3/2 at 197.6 eV, which represents adsorbed chloride ions on metals,81,82 and Cl 2p1/2 at 199.3 eV, which represents reduced Cl ions, such as HCl.83,84 The Cl 2p3/2 peak decreases while the Cl 2p1/2 peak increases upon the addition of the M13 virus to a mixture of HAuCl4 and DMAB, indicating that the reduction of Cl in HAuCl4 has taken place (Fig. 4c and Fig. S12, ESI†). However, there is no change in the HAuCl4 and NaBH4 mixture in the presence of the M13 virus (Fig. 4d and Fig. S12, ESI†). The position of the N 1s peaks reveals the M13 viruses functioning as a template for the Au particles as the peak shifts to a higher binding energy when there are elements attached to the virus surface. Both the virus-templated Au nanowires with DMAB and NaBH4 manifest upshifts of N 1s peaks, yet a greater peak shift is observed for the DMAB-added system over the NaBH4-added system, confirming a stronger interaction at the virus surface (Fig. 4e and f).85 Both DMAB and NaBH4 contain boron. XPS profiles show that the boron species disappeared upon the addition of HAuCl4 (Fig. S13, ESI†). This evidences that a reaction took place, but we are unsure as to why boron cannot be detected after the reaction (eqn (1)), (eqn (2)), and (eqn (3)). As evidenced by various analyses thus far, the Au-binding M13 bio-template under DMAB enabled a higher yield synthesis of Au nanowires than under NaBH4. We conjecture that one of the reasons for this can be found in the chemical reaction mechanism. Barring the formation of NaCl, the reaction between HAuCl4 and NaBH4 is generally a one-step quick reaction with a relatively weak reducing power of NaBH4 (eqn (3)).50–52 However, being a much stronger reducing agent than NaBH4, DMAB induces faster reaction kinetics and forms Au at a higher growth rate (eqn (1)) and (eqn (2)).53–55 The greater growth rate than etching rate due to Cl ions indicates that when DMAB is used as a reducing agent, the Au nanowire quality is bound to be high.86 In addition to this, reduction with NaBH4 favours alkaline conditions, whereas reduction with DMAB favours acidic conditions. Since the solution turns acidic when we add HAuCl4 and the reduction reaction by DMAB produces H3BO3, the conditions stay acidic throughout the reaction which favours DMAB over NaBH4.87
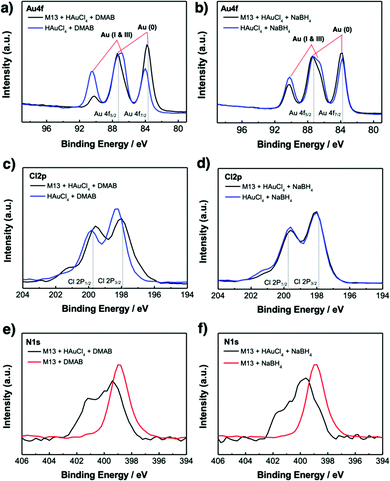 |
| | Fig. 4 XPS measurements of Au 4f peaks of the virus-templated Au nanowires made under (a) DMAB or (b) NaBH4 (blue). (c) Cl 2p peaks of the virus-templated Au nanowires made under (c) DMAB or (d) NaBH4. N 1s peaks of the virus-templated Au nanowires made under (e) DMAB and (f) NaBH4. | |
Photoelectron yield spectroscopy (PYS) was used to analyse the work function of the DMAB-added virus-templated Au nanowires and bare M13 viruses (Fig. S14a, ESI†). The bare M13 viruses have a work function of 6.0 eV. However, the DMAB-added virus-templated Au nanowires have a work function of approximately 5.3 eV.23,88 We suspect the mixture of Au(0) + Au(I) + Au(III) in the M13 virus-templated Au nanowires leads to 5.3 eV instead of 5.0 eV. This confirms the presence of Au nanoparticles on the viruses as well as the fact that the virus-templated Au nanowires have a good work function value for use as a transparent electrode in PSCs.
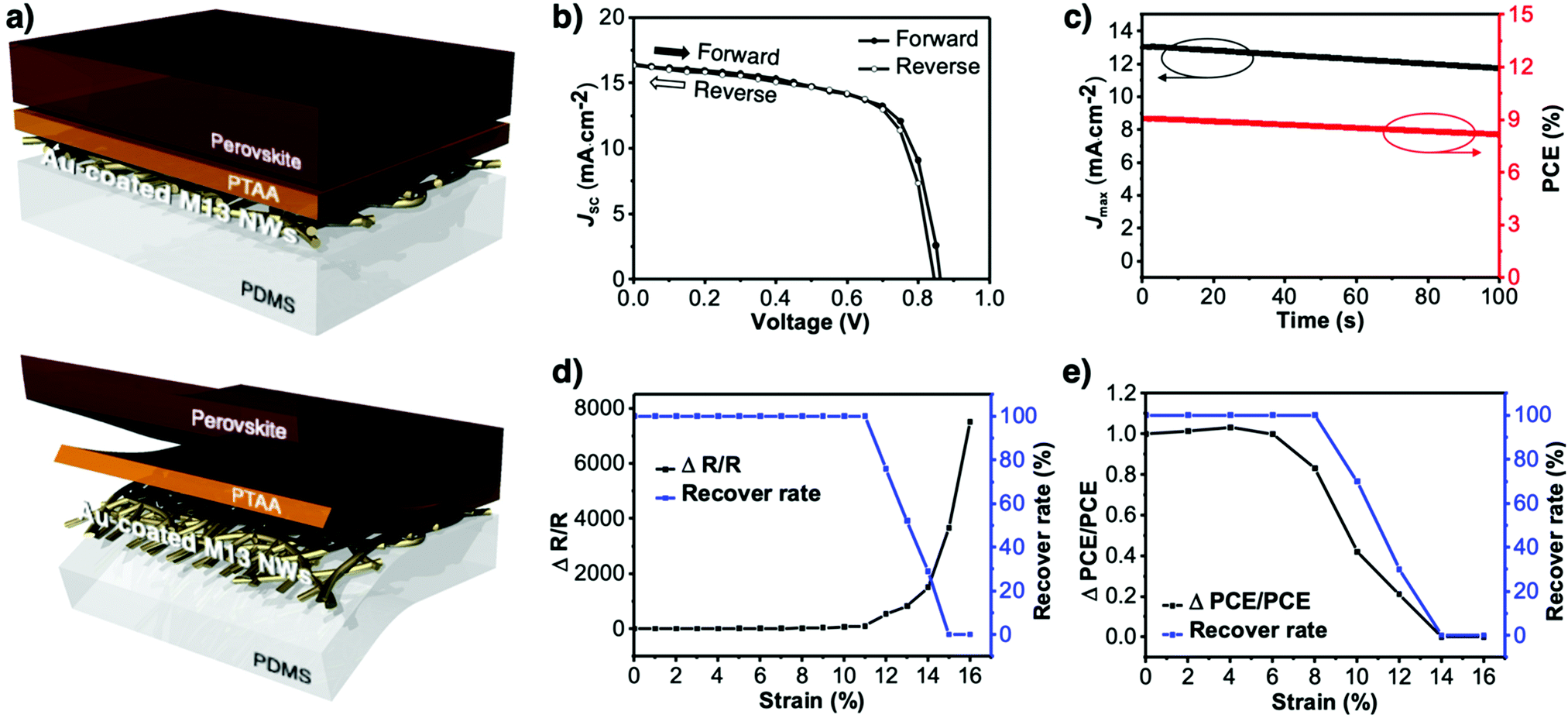
The morphology of the virus-templated Au nanowires was analysed using atomic force microscopy (AFM). The virus-templated Au nanowires on glass substrates showed micrometre-scale roughness due to the random stacking of the nanowires (Fig. 5a). To reduce the roughness and enable stretchable device application, the nanowires were transferred to PDMS films by a stamping method. The embedded nanowires exhibited an average roughness (Ra) of 43 nm. The roughness was further reduced to 1.2 nm by applying poly(triaryl amine) (PTAA) on top (Fig. S15, ESI†). Such roughness values are sufficiently low for the fabrication of PSCs. The conductivity and transmittance were retained after transferring to the stretchable PDMS substrate (Fig. S16, ESI†). The optical conductivity of the virus-templated Au nanowires was slightly lower than the previously reported values of metal nanowires (Table S1 and Fig. S17, ESI†). PSCs were fabricated in a structural configuration of PDMS/virus-templated Au nanowires/PTAA/MA0.6FA0.4PbI2.9Br0.1/C60/bathocuproine (BCP)/Au (Fig. 6a). The devices exhibited a PCE of 9.28% with negligible hysteresis (Fig. 6b and Fig. S18, S19, ESI;† and Table 1). Moreover, maximum power point tracking (MPPT) data showed a power output of 9.1%, which was retained rather well over 100 s in air without encapsulation (Fig. 6c). The MPPT power output dropped rather fast. We attribute this to the humid atmosphere (RH ∼ 60%) at the time and place of the measurement. According to our time-evolution of UV-vis absorption spectra shows that both the perovskite films on ITO and Au nanowires showed the same degradation rate (Fig. S20, ESI†). Furthermore, a stretchability test was performed on both the virus-templated Au nanowire electrode and PSC (Fig. 6d and e). Fig. 6d shows that the electrode can stretch up to 10% strain without a decrease in conductivity, and it loses the ability to recover conductivity when stretched to more than 11% strain. Fig. 6e shows that the virus-templated Au nanowire-based PSC can be stretched axially up to 8% of its original dimension or width. The obtained PCE and stretchability is one of the best among the reported transparent nanowire electrode-based PSCs (Table 2 and Fig. S21, ESI†).89–94
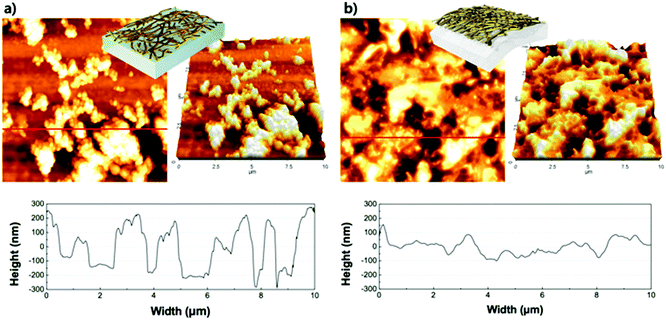 |
| | Fig. 5 AFM images (10 × 10 μm2) of M13 bacteriophage-templated Au nanowires on (a) glass, and (b) the surface after transfer to PDMS. | |
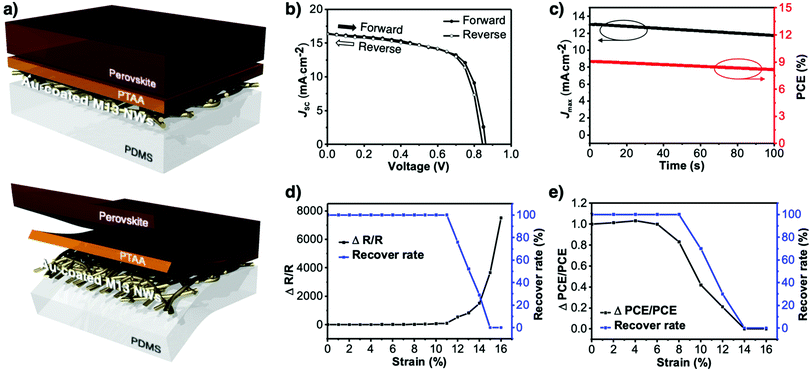 |
| | Fig. 6 (a) Illustrations of the virus-templated Au nanowires on PDMS with PTAA and perovskite layers coated on top. (b) J–V curves of the virus-templated Au nanowire-based stretchable PSC under forward and reverse bias. (c) MPPT data of the of the virus-templated Au nanowire-based stretchable PSC. (d) Stretchability data of the virus-templated Au nanowire electrode. (e) Stretchability data of the virus-templated Au nanowire-based PSC. | |
Table 1 Photovoltaic parameters of the virus-templated Au nanowire-based stretchable PSCs under 1 sun (AM1.5G, 100 mW cm−2)
|
|
Forward |
Reverse |
|
V
OC (V) |
0.86 |
0.85 |
|
J
SC (mA cm−2) |
13.1 |
13.3 |
| FF |
0.66 |
0.66 |
| PCE (%) |
9.28 |
9.11 |
| Hysteresis index |
0.021 |
Table 2 Reported PCEs of metal nanowire electrode-based PSCs
| Year |
Material type |
PCE (%) |
Ref. |
| 2016 |
Ag |
8.44 |
89
|
| 2017 |
Cu |
8.65 |
90
|
| 2018 |
Cu@Ni |
12.2 |
91
|
| 2018 |
Ag |
7.88 |
92
|
| 2016 |
Ag/graphene |
10.4 |
93
|
| 2019 |
Ag/GO |
9.62 |
94
|
| 2020 |
Au |
9.28 |
This work |
Conclusions
Although the photovoltaic performance of the virus-templated gold nanowire-based PSCs is less than ideal at this stage, the versatility and potential of the biologically synthesised metal nanowires are clear. Virus-templated nanowires function well with Au nanoparticles under DMAB reductant. The virus concentration, HAuCl4 concentration, and AA additive concentration were optimised to give a sheet resistance of 144.5 Ω sq−1 and 83% at 550 nm on glass. The nanowires were transferred to PDMS substrates to test their stretchable application, which resulted in a PCE of 9.28% and a stretchability of 8%. This performance is one of the highest reported for stretchable metal nanowire-based PSCs and demonstrates the first stretchable biomaterial-templated electrode.
Experimental
M13 bacteriophage synthesis
The M13 bacteriophage was purchased from New England Biolabs (Ipswich, MA, USA). The product (M13KE, NEB # N0316) was mass-produced according to the following protocol: the M13 bacteriophage was amplified in an Escherichia coli (E. coli, ER2738) culture and purified by precipitation using polyethylene glycol followed by dispersion. Then it was refined through a membrane with 0.45 μm pores (Minisart® NML, Syringe Filter 16555), and centrifuged repeatedly. The accuracy of the cultured product was confirmed by DNA sequencing analysis (BIONICS, Seoul, Republic of Korea). The M13 bacteriophage concentration was measured using Nano-drop 2000 (Evolution 300, Thermo Fisher Scientific, Waltham, MA, USA). Previously reported formulae and absorbance spectral values were used for the concentration calculation.10
Metal nanowire growth
The M13 bacteriophage-templated stretchable and transparent electrodes were prepared by dispersing different concentrations of wild-type M13 bacteriophage in 1 mL of DI water (0, 0.02, 0.04, 0.06, 0.08, 0.1, and 0.12% v/v). Then, 0.5 M targeting metal ion solution (gold(III) chloride hydrate, HAuCl4 or silver nitrate, AgNO3, Sigma Aldrich) was added to the prepared M13 bacteriophage solution. At this time, the concentrations of the metal ions in the M13 bacteriophage solution were 0.1, 0.2, 0.3, 0.5, 0.7, and 1.0 mol L−1. Subsequently, 0, 5, 10, 20, 30, and 50 μL of 100 mM ascorbic acid (AA, Sigma Aldrich) were added to the reaction mixture. Finally, after adding 10 μL of 1 M 4-(dimethylamino)benzaldehyde (DMAB, Sigma Aldrich) or NaBH4 (Sigma Aldrich) as a reducing agent, the mixture was reacted at 25 °C for 3 h. The methods of synthesising silver and gold are the same.
Substrate transfer
After 50 μL drop-casting of metal nanowires on a glass substrate, it was completely filmed in a vacuum dry oven (60 °C for 12 h). Then, PDMS was poured into the nanowire film and hardened. When the glass substrate and PDMS were slowly separated, the metal nanowire film was fixed to the PDMS and peeled off the glass substrate.
Device fabrication
Pre-patterned ITO/glass substrates (12 Ω sq−1, 20 × 20 mm2) (AMG Tech) were cleaned through sonication in acetone, isopropyl alcohol, and deionised water for 15 min in that order and stored in a drying oven at 120 °C for 20 min. PTAA solution was prepared by dissolving 8 mg of PTAA (Sigma Aldrich) in 1 mL of 2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane (F4 TCNQ) solution, which was prepared by dissolving F4 TCNQ in chlorobenzene (Sigma Aldrich) at a concentration of 1 wt%. Then, the PTAA layer was formed by spin-coating the solution on the DWNT (or ITO) substrates at 4000 rpm for 30 s followed by annealing on a hotplate at a temperature of 100 °C for 10 min. To obtain a reproducible coating of the perovskite film on the hydrophobic PTAA layer, poly[(9,9-bis(30-((N,N-dimethyl)-N-ethylammonium)-propyl)-2,7-fluorene)-alt-2,7-(9,9-dioctylfluorene)]dibromide (PFN-P2) (1-Material) was introduced as an interfacial compatibilizer. Its precursor solution was prepared by dissolving PFN-P2 in methanol with a concentration of 0.4 mg mL−1 and spin-coated on the PTAA layer at 4000 rpm for 20 s. Perovskite layer with a composition of MA0.6FA0.4PbI2.9Br0.1 precursor solution was prepared by dissolving 461 mg of PbI2 (Tokyo Chemical Industry), 79.5 mg of methylammonium iodide (Tokyo Chemical Industry), 68.8 mg of formamidinium iodide (Tokyo Chemical Industry), 11.2 mg of methylammonium bromide (Sigma Aldrich), and 75 μL of urea-added dimethylsulfoxide (DMSO) (Sigma Aldrich) in 0.55 mL of N-dimethylformamide (Sigma Aldrich). The urea solution was prepared by dissolving urea (Sigma Aldrich) in DMSO at a concentration of 44.4 mg mL−1 to induce large grain crystallisation of the perovskite film. The perovskite precursor solution was stirred for 1 h at room temperature. The solution was spin-coated at 4000 rpm for 20 s. Diethyl ether antisolvent (3–500 μL) was applied 7 s after the beginning of the spin-coating. A transparent film of perovskite intermediate phase was formed and changed into a black perovskite film after annealing at a temperature of 130 °C for 20 min. The whole spin-coating process was conducted at a controlled temperature (25 °C) and relative humidity (<10%). Then C60 (20 nm) and BCP (6 nm) layers were deposited through a square metal mask of 1.5 × 1.5 cm2. Furthermore, a Cu electrode was deposited to complete devices with an area of 0.2 × 0.2 cm2 (0.04 cm2) under a pressure of <5.0 × 10−6 torr inside a thermal evaporator.
Material and device characterization
J–V curves of perovskite solar cells under light were measured using a source metre (Keithley 2400, Tektronix) at a step voltage of 20 mV and a delay time of 50 ms in both the forward and reverse scan directions. AM1.5G illumination was simulated using a solar simulator (Sol3A Class, Oriel) with a KG-5-filtered silicon standard cell. J–V curves under dark conditions were measured using a probe station built inside a dark-shield box. Grazing-incidence XRD 2θ scans were performed on a Jordan Valley D1 diffractometer using copper Kα1 radiation and a parallel beam source. In the 2θ scans, the scattering angle 2θ between the incident beam and the diffracted beam changed, whereas the incident angle ω between the incident beam and the sample surface was fixed at 1°. Core-level photoemission measurements were performed by XPS (PerkinElmer, 5400MC) using monochromatic Al Kα radiation. XPS resolution was estimated to be 1 eV, and the energy offset was calibrated using the surface C 1s peak. TEM measurements were conducted using high-resolution scanning/transmission electron microscopy (Talos F200X, FEI and H-7600, Hitachi). Optical transmittance and absorption spectra were obtained using a UV-vis-NIR spectrophotometer (V-570, JASCO). Surface topography measurements were conducted using an atomic force microscope (NX10, Park Systems) in non-contact mode. The van der Pauw method of four-probe measurement was conducted using a probe station and indium metal pieces.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Creative Materials Discovery Program of the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (NRF-2017M3D1A1039287), the Ministry of Education (NRF-2020R1A6A3A13075717), and the Korean government (NRF-2020R1G1A1101578). Part of this work was supported by JSPS KAKENHI Grant Numbers JP18H05329, JP20H00220, and by JST, CREST Grant Number JPMJCR20B5, Japan.
Notes and references
- M. Irimia-Vladu, P. A. Troshin, M. Reisinger, L. Shmygleva, Y. Kanbur, G. Schwabegger, M. Bodea, R. Schwödiauer, A. Mumyatov, J. W. Fergus, V. F. Razumov, H. Sitter, N. S. Sariciftci and S. Bauer, Adv. Funct. Mater., 2010, 20, 4069 CrossRef CAS.
- M. Muskovich and C. J. Bettinger, Adv. Healthcare Mater., 2012, 1, 248 CrossRef CAS.
- C. Mao, Science, 2004, 303, 213 CrossRef CAS.
- B. Y. Lee, J. Zhang, C. Zueger, W.-J. Chung, S. Y. Yoo, E. Wang, J. Meyer, R. Ramesh and S.-W. Lee, Nat. Nanotechnol., 2012, 7, 351 CrossRef CAS.
- D. P. Pires, S. Cleto, S. Sillankorva, J. Azeredo and T. K. Lu, Microbiol. Mol. Biol. Rev., 2016, 80, 523 CrossRef CAS.
- J. M. L. Bernard and M. B. Francis, Front. Microbiol., 2014, 5, 1 Search PubMed.
- D.-M. Shin, H. J. Han, W.-G. Kim, E. Kim, C. Kim, S. W. Hong, H. K. Kim, J.-W. Oh and Y.-H. Hwang, Energy Environ. Sci., 2015, 8, 3198 RSC.
- J.-W. Oh, W.-J. Chung, K. Heo, H.-E. Jin, B. Y. Lee, E. Wang, C. Zueger, W. Wong, J. Meyer, C. Kim, S.-Y. Lee, W.-G. Kim, M. Zemla, M. Auer, A. Hexemer and S.-W. Lee, Nat. Commun., 2014, 5, 3043 CrossRef.
- J.-S. Moon, W.-G. Kim, D.-M. Shin, S.-Y. Lee, C. Kim, Y. Lee, J. Han, K. Kim, S. Y. Yoo and J.-W. Oh, Chem. Sci., 2017, 8, 921 RSC.
- W.-J. Chung, J.-W. Oh, K. Kwak, B. Y. Lee, J. Meyer, E. Wang, A. Hexemer and S.-W. Lee, Nature, 2011, 478, 364 CrossRef CAS.
- C. Mao, C. E. Flynn, A. Hayhurst, R. Sweeney, J. Qi, G. Georgiou, B. Iverson and A. M. Belcher, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 6946 CrossRef CAS.
- K. T. Nam, Science, 2006, 312, 885 CrossRef CAS.
- Y. J. Lee, H. Yi, W.-J. Kim, K. Kang, D. S. Yun, M. S. Strano, G. Ceder and A. M. Belcher, Science, 2009, 324, 1051 CAS.
- F. Li and Q. Wang, Small, 2014, 10, 230 CrossRef CAS.
- M. Janczuk-Richter, I. Marinović, J. Niedziółka-Jönsson and K. Szot-Karpińska, Electrochem. Commun., 2019, 99, 11 CrossRef CAS.
- N. K. Petty, T. J. Evans, P. C. Fineran and G. P. C. Salmond, Trends Biotechnol., 2007, 25, 7 CrossRef CAS.
- Y. Huang, C.-Y. Chiang, S. K. Lee, Y. Gao, E. L. Hu, J. De Yoreo and A. M. Belcher, Nano Lett., 2005, 5, 1429 CrossRef CAS.
- S.-K. Lee, D. S. Yun and A. M. Belcher, Biomacromolecules, 2006, 7, 14 CrossRef CAS.
- N.-G. Park, M. Grätzel, T. Miyasaka, K. Zhu and K. Emery, Nat. Energy, 2016, 1, 16152 CrossRef CAS.
- J. Rogers, Z. Bao and T.-W. Lee, Acc. Chem. Res., 2019, 52, 521 CrossRef CAS.
- M. Park, H. J. Kim, I. Jeong, J. Lee, H. Lee, H. J. Son, D. Kim and M. J. Ko, Adv. Energy Mater., 2015, 5, 1501406 CrossRef.
- B. J. Kim, D. H. Kim, Y.-Y. Lee, H.-W. Shin, G. S. Han, J. S. Hong, K. Mahmood, T. K. Ahn, Y.-C. Joo, K. S. Hong, N.-G. Park, S. Lee and H. S. Jung, Energy Environ. Sci., 2015, 8, 916 RSC.
- I. Jeon, J. Yoon, U. Kim, C. Lee, R. Xiang, A. Shawky, J. Xi, J. Byeon, H. M. Lee, M. Choi, S. Maruyama and Y. Matsuo, Adv. Energy Mater., 2019, 9, 1901204 CrossRef.
- I. Jeon, R. Xiang, A. Shawky, Y. Matsuo and S. Maruyama, Adv. Energy Mater., 2019, 9, 1801312 CrossRef.
- I. Jeon, Y. Matsuo and S. Maruyama, Top. Curr. Chem., 2018, 376, 4 CrossRef.
- J. Yoon, H. Sung, G. Lee, W. Cho, N. Ahn, H. S. Jung and M. Choi, Energy Environ. Sci., 2017, 10, 337 RSC.
- H. Sung, N. Ahn, M. S. Jang, J. Lee, H. Yoon, N. Park and M. Choi, Adv. Energy Mater., 2016, 6, 1501873 CrossRef.
- I. Jeon, J. Yoon, N. Ahn, M. Atwa, C. Delacou, A. Anisimov, E. I. Kauppinen, M. Choi, S. Maruyama and Y. Matsuo, J. Phys. Chem. Lett., 2017, 8, 5395 CrossRef CAS.
- B. Kumanek and D. Janas, J. Mater. Sci., 2019, 54, 7397–7427 CrossRef CAS.
- I. Jeon, C. Delacou, H. Okada, G. E. Morse, T.-H. Han, Y. Sato, A. Anisimov, K. Suenaga, E. I. Kauppinen, S. Maruyama and Y. Matsuo, J. Mater. Chem. A, 2018, 6, 14553 RSC.
- M. Savchak, N. Borodinov, R. Burtovyy, M. Anayee, K. Hu, R. Ma, A. Grant, H. Li, D. B. Cutshall, Y. Wen, G. Koley, W. R. Harrell, G. Chumanov, V. Tsukruk and I. Luzinov, ACS Appl. Mater. Interfaces, 2018, 10, 3975 CrossRef CAS.
- E. Lee, J. Ahn, H.-C. Kwon, S. Ma, K. Kim, S. Yun and J. Moon, Adv. Energy Mater., 2018, 8, 1702182 CrossRef.
- B. Li, S. Ye, I. E. Stewart, S. Alvarez and B. J. Wiley, Nano Lett., 2015, 15, 6722 CrossRef CAS.
- Y. Zhang, J. Guo, D. Xu, Y. Sun and F. Yan, ACS Appl. Mater. Interfaces, 2017, 9, 25465 CrossRef CAS.
- L. Lan, T. Yin, C. Jiang, X. Li, Y. Yao, Z. Wang, S. Qu, Z. Ye, J. Ping and Y. Ying, Nano Energy, 2019, 62, 319 CrossRef CAS.
- S. Lin, H. Wang, X. Zhang, D. Wang, D. Zu, J. Song, Z. Liu, Y. Huang, K. Huang, N. Tao, Z. Li, X. Bai, B. Li, M. Lei, Z. Yu and H. Wu, Nano Energy, 2019, 62, 111 CrossRef CAS.
- S. Kang, J. Jeong, S. Cho, Y. J. Yoon, S. Park, S. Lim, J. Y. Kim and H. Ko, J. Mater. Chem. A, 2019, 7, 1107 RSC.
- S. Hong, H. Lee, J. Lee, J. Kwon, S. Han, Y. D. Suh, H. Cho, J. Shin, J. Yeo and S. H. Ko, Adv. Mater., 2015, 27, 4744 CrossRef CAS.
- J. H. Cho, D. J. Kang, N.-S. Jang, K.-H. Kim, P. Won, S. H. Ko and J.-M. Kim, ACS Appl. Mater. Interfaces, 2017, 9, 40905 CrossRef CAS.
- H. Lee, I. Kim, M. Kim and H. Lee, Nanoscale, 2016, 8, 1789 RSC.
- Y. J. Lee, Y. Lee, D. Oh, T. Chen, G. Ceder and A. M. Belcher, Nano Lett., 2010, 10, 2433 CrossRef CAS.
- R. A. Blaik, E. Lan, Y. Huang and B. Dunn, ACS Nano, 2016, 10, 324 CrossRef CAS.
- Y. Lee, J. Kim, D. S. Yun, Y. S. Nam, Y. Shao-Horn and A. M. Belcher, Energy Environ. Sci., 2012, 5, 8328 RSC.
- X. Dang, H. Yi, M.-H. Ham, J. Qi, D. S. Yun, R. Ladewski, M. S. Strano, P. T. Hammond and A. M. Belcher, Nat. Nanotechnol., 2011, 6, 377 CrossRef CAS.
- K. T. Nam, Y. J. Lee, E. M. Krauland, S. T. Kottmann and A. M. Belcher, ACS Nano, 2008, 2, 1480 CrossRef CAS.
- B. R. Peelle, E. M. Krauland, K. D. Wittrup and A. M. Belcher, Langmuir, 2005, 21, 6929 CrossRef CAS.
- P. Selvakannan, A. Swami, D. Srisathiyanarayanan, P. S. Shirude, R. Pasricha, A. B. Mandale and M. Sastry, Langmuir, 2004, 20, 7825 CrossRef CAS.
- Y. Liu, Y. Chen, R. Shi, L. Cao, Z. Wang, T. Sun, J. Lin, J. Liu and W. Huang, RSC Adv., 2017, 7, 4891 RSC.
- H. Ding, Y. Zhang, G. Yang, S. Zhang, L. Yu and P. Zhang, RSC Adv., 2016, 6, 8096 RSC.
- H. C. Brown and B. C. S. Rao, J. Am. Chem. Soc., 1956, 78, 2582 CrossRef CAS.
- D. Todd, J. Chem. Educ., 1979, 56, 540 CrossRef CAS.
- A. Gharatape, S. Davaran, R. Salehi and H. Hamishehkar, RSC Adv., 2016, 6, 111482 RSC.
- J. H. Billman and J. W. McDowell, J. Org. Chem., 1961, 26, 1437 CrossRef CAS.
- M. Wojnicki, E. Rudnik, M. Luty-Błocho, K. Pacławski and K. Fitzner, Hydrometallurgy, 2012, 43, 127 Search PubMed.
- U. Sanyal and B. R. Jagirdar, Inorg. Chem., 2012, 51, 13023 CrossRef CAS.
- S. Joly, R. Kane, L. Radzilowski, T. Wang, A. Wu, R. E. Cohen, E. L. Thomas and M. F. Rubner, Langmuir, 2000, 16, 1354 CrossRef CAS.
- F. Kim, K. Sohn, J. Wu and J. Huang, J. Am. Chem. Soc., 2008, 130, 14442 CrossRef CAS.
- B. P. Khanal and E. R. Zubarev, ACS Nano, 2019, 13, 2370 CAS.
- N. R. Jana, L. Gearheart and C. J. Murphy, J. Phys. Chem. B, 2001, 105, 4065 CrossRef CAS.
- J. Rodríguez-Fernández, J. Pérez-Juste, F. J. García de Abajo and L. M. Liz-Marzán, Langmuir, 2006, 22, 7007 CrossRef.
- C. J. Ward, R. Tronndorf, A. S. Eustes, M. L. Auad and E. W. Davis, J. Nanomater., 2014, 1 Search PubMed.
- C. L. John, S. L. Strating, K. A. Shephard and J. X. Zhao, RSC Adv., 2013, 3, 10909 RSC.
- B. Zümreoglu-Karan, J. Nanopart. Res., 2009, 11, 1099 CrossRef.
- M. M. T. Khan and A. E. Martell, J. Am. Chem. Soc., 1967, 89, 4176 CrossRef CAS.
- L. Malassis, R. Dreyfus, R. J. Murphy, L. A. Hough, B. Donnio and C. B. Murray, RSC Adv., 2016, 6, 33092 RSC.
- H. Kang, J. T. Buchman, R. S. Rodriguez, H. L. Ring, J. He, K. C. Bantz and C. L. Haynes, Chem. Rev., 2019, 119, 664 CrossRef CAS.
- N. E. Larm, J. A. Thon, Y. Vazmitsel, J. L. Atwood and G. A. Baker, Nanoscale Adv., 2019, 1, 4665 RSC.
- E. Alegria, A. Ribeiro, M. Mendes, A. Ferraria, A. do Rego and A. Pombeiro, Nanomaterials, 2018, 8, 320 CrossRef.
- J. Haber and K. Sokolov, Langmuir, 2017, 33, 10525 CrossRef CAS.
- E. G. Wrigglesworth and J. H. Johnston, RSC Adv., 2017, 7, 45757 RSC.
- M. N. Martin, J. I. Basham, P. Chando and S.-K. Eah, Langmuir, 2010, 26, 7410 CrossRef CAS.
- P. Suchomel, L. Kvitek, R. Prucek, A. Panacek, A. Halder, S. Vajda and R. Zboril, Sci. Rep., 2018, 8, 4589 CrossRef.
- C. Lin, K. Tao, D. Hua, Z. Ma and S. Zhou, Molecules, 2013, 18, 12609 CrossRef CAS.
- M. Dasog, W. Hou and R. W. J. Scott, Chem. Commun., 2011, 47, 8569 RSC.
- N. S. K. Gowthaman, B. Sinduja and S. A. John, RSC Adv., 2016, 6, 63433 RSC.
- P. J. Rivero, J. Goicoechea, A. Urrutia and F. J. Arregui, Nanoscale Res. Lett., 2013, 8, 101 CrossRef.
- K. T. Nam, R. Wartena, P. J. Yoo, F. W. Liau, Y. J. Lee, Y.-M. Chiang, P. T. Hammond and A. M. Belcher, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17227 CrossRef CAS.
- Au XPS Reference from Cardiff University, can be found under http://sites.cardiff.ac.uk/xpsaccess/reference/gold/.
- J. Radnik, C. Mohr and P. Claus, Phys. Chem. Chem. Phys., 2003, 5, 172 RSC.
- M. Sankar, Q. He, M. Morad, J. Pritchard, S. J. Freakley, J. K. Edwards, S. H. Taylor, D. J. Morgan, A. F. Carley, D. W. Knight, C. J. Kiely and G. J. Hutchings, ACS Nano, 2012, 6, 6600 CrossRef CAS.
- C. Han, L. Ge, C. Chen, Y. Li, Z. Zhao, X. Xiao, Z. Li and J. Zhang, J. Mater. Chem. A, 2014, 2, 12594 RSC.
- C. A. Strydom, J. F. van Staden and H. J. Strydom, J. Electroanal. Chem. Interfacial Electrochem., 1990, 277, 165 CrossRef CAS.
- P. Parent, J. Lasne, G. Marcotte and C. Laffon, Phys. Chem. Chem. Phys., 2011, 13, 7142 RSC.
- J. Yang, Y. Zhao, J. Zhang and C. Zheng, Fuel, 2016, 167, 366 CrossRef CAS.
- D. Dong, Y. Zhang, S. Sutaria, A. Konarov and P. Chen, PLoS One, 2013, 8, e82332 CrossRef.
- J. Ma and M. Zhan, RSC Adv., 2014, 4, 21060 RSC.
- J. Huang, C. Ding, Y. Yang, G. Liu and W.-B. Cai, Chin. J. Catal., 2019, 40, 1895 CrossRef.
- I. Jeon, S. Zeljkovic, K. Kondo, M. Yoshizawa and Y. Matsuo, ACS Appl. Mater. Interfaces, 2016, 8, 29866 CrossRef CAS.
- A. Kim, H. Lee, H. C. Kwon, H. S. Jung, N. G. Park, S. Jeong and J. Moon, Nanoscale, 2016, 8, 6308–6316 RSC.
- H. Hwang, J. Ahn, E. Lee, K. Kim, H. C. Kwon and J. Moon, Nanoscale, 2017, 9, 17207–17211 RSC.
- K. Kim, H. C. Kwon, S. Ma, E. Lee, S. C. Yoon, G. Jang, H. Yang and J. Moon, ACS Appl. Mater. Interfaces, 2018, 10, 30337 CrossRef CAS.
- Y. Jin, Y. Sun, K. Wang, Y. Chen, Z. Liang, Y. Xu and F. Xiao, Nano Res., 2018, 11, 1998 CrossRef CAS.
- H. Dong, Z. Wu, Y. Jiang, W. Liu, X. Li, B. Jiao and X. Hou, ACS Appl. Mater. Interfaces, 2016, 8, 31212–31221 CrossRef CAS.
- H. Chen, M. Li, X. Wen, Y. Yang, D. He, W. C. Choy and H. Lu, Nanomaterials, 2019, 9, 193 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00935k |
| ‡ J. H. and J.-S. N. contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  b,
Kyusun
Kim
b,
Eun Jung
Choi
c,
Jong-Min
Lee
b,
Kyusun
Kim
b,
Eun Jung
Choi
c,
Jong-Min
Lee