
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSurface-initiated polymerization of mussel-inspired dopamine for hydrophilic coatings†
Tzu-Yu
Kuo
a and
Yi-Chang
Chung
 *ab
*ab
aDepartment of Chemical and Materials Engineering, National University of Kaohsiung, Kaohsiung, Taiwan, Republic of China. E-mail: ycchung@nuk.edu.tw
bResearch Center of Biomimetics and Medicare Technology, National University of Kaohsiung, Kaohsiung, Taiwan, Republic of China
First published on 19th July 2021
Abstract
Inspired by mussels, dopamine with catechol and amino groups in the molecular structure has been broadly studied for its universal attachment, biocompatibility and ease of use. Dopamine is known to undergo autoxidation in the presence of oxygen and transform polydopamine particles. This method utilizes a rapid redox reaction to polymerize dopamine; however, there are some disadvantages, such as difficulty in controlling the reaction, appearance of homopolymers in solution, unstoppable polymerization and so on, resulting in little industrial or commercial development of dopamine-based surface coatings. In this study, we carried out free-radical polymerization of dopamine. Via this unorthodox method, a very thin polydopamine-modified coating can be fabricated. It can produce a reaction localized on the surface to functionalize or modify it, making it a promising tool for surface modification.
Introduction
Surface modification for functionalization of a material has always been a major strategy to manipulate interfacial interaction between material surfaces and the objects to which they are applied without changing the bulk properties of the interior. Due to the differences of surface energy and chemical intermolecular forces on material surfaces, finding a universal surface treatment should be a difficult and persisting research topic for scientists. In recent years, Messersmith and his colleagues have announced a broadly-applicable surface treatment on many material surfaces,1,2 including an omniphobic material, polytetrafluoroethylene.3 Inspired by the adhesive byssus secreted by mussels, dopamine has received much attention on account of its universal attachment,1,2,4–9 biocompatibility,10–12 and ease of use.5,11,13Since the advent of polydopamine (PDA) coatings, dopamine research has sprung up for surface modifications of materials, producing special thin films on surfaces. The well-known principle is that dopamine can be oxidized in alkaline environments and spontaneously polymerizes to form polydopamine.7,14–16 Nevertheless, the polymerization mechanism of dopamine and how dopamine aggregates into PDA coatings are still unsolved mysteries. Bielawski and co-workers employed a broad range of solid state spectroscopic and crystallographic techniques to analyze the structure of PDA,17 announcing that dopamine is aggregated via hydrogen bonding and π–π stacking. They propose that PDA is not a covalent polymer but a supermolecule instead. Some other research also supports that dopamine can aggregate through noncovalent bonding.18,19 On the other hand, Liebscher and co-workers investigated PDA structure using solid-state nuclear magnetic resonance, electrospray ionization high-resolution mass spectrometry, and X-ray photoelectron spectroscopy,20 demonstrating that PDA will not consist of single indoline units if just held together by hydrogen bonding, implying that carbon–carbon connections between the monomer units exist. Furthermore, some other researchers changed the polymerization conditions from alkaline to acidic aqueous solutions to obtain a dense surface of fine PDA aggregates. However, once one applies the PDA coatings for surface modifications, the most prevalent problem will be that much more dopamine self-polymerizes in the aqueous/alkaline solutions and gradually turns the color of solutions black.21 This leads to some practical problems for real productive applications due to unstoppable and uncontrollable polymerization of dopamine as well as formation of PDA particles in the bulk solution rather than modification on surfaces only. Moreover, PDA and its derivatives are considered excellent “primers” for electroless plating of metal coatings, and they are also helpful for successive grafting of functional polymers on the outmost layers, which has gained much attention in studies and applications,4,5,11,13,22–27 especially using the combination of other conventional polymer coatings via preparation of “one pot” reactions.4,13,28
According to the literature review,29 during the formation of byssus threads, the mechanism is assumed to be lack of carbon–carbon formation (like PDA) in the process of attachment on surfaces. Therefore, we designed a simple test to detect the generation of free radicals around the byssus of mussels using DPPH (1,1-diphenyl-2-picrylhydrazyl radical 2,2-diphenyl-1-(2,4,6-trinitrophenyl)hydrazyl) bleaching test (in Fig. S1, ESI†). The test result was positive, implying that absorbance decreases as byssi form, and so the DPPH underwent a reaction with the generated free radicals. It inspired us to generate new ideas about the growth of PDA coatings, which probably relies on some other effects.
Nonetheless, in view of the observations of predecessors, we believe that free-radical polymerization of dopamine is a feasible method. We decided to use an initiator to polymerize dopamine. Our research provides convincing evidence that PDA is polymerized via covalent bonding. In order to offer further proof that dopamine can be polymerized by free-radical polymerization, we copolymerized dopamine with the zwitterionic material SBMA. The results were analyzed with some instruments on surfaces, confirming that dopamine can be polymerized by free-radical polymerization. This research brings new insight into the controllable polymerization of dopamine and functional monomers.
Experimental
Materials
Dopamine hydrochloride (98%) and initiator potassium persulfate (KPS) were purchased from Acros Organics, part of Thermo Fisher Scientific Inc. (USA). N,N-Dimethyl-N-(2-methacryloyloxyethyl)-N-(3-sulfopropyl)ammonium betaine (SBMA) was supplied by Taiwan Hopax Chemicals. Mfg. Co., Ltd. The surface modifying reagents including caffeic acid (CA) (99%) and 2,2′-azobis(2-methylpropionamidine) dihydrochloride (AIBA) (97%) were obtained from Alfa Aesar (USA) and MilliporeSigma (USA) respectively. All other organic solvents were bought from Echo Chem. Co., Ltd. (Taiwan). DPPH was also obtained from MilliporeSigma (USA).Preparation of polySBMA (PSBMA) homopolymer
Firstly, the PSBMA was prepared using SBMA of 6 g and 5% KPS (initiator) in water. The mixture was purged with N2 gas for 10 min and then polymerized at 60 °C for 5 h, after which the crude polymer was rinsed with EtOH several times. After drying, the PSBMA was kept in a desiccator.Immobilization of initiator on substrate surface
A cleaned microscope slide was put into caffeic acid alcoholic solution (2 μg ml−1) for use as a substrate. Subsequently, hydrogen peroxide (0.05 ml ml−1) was added as an oxidant and kept at 60 °C for 4 h, after which the substrate was rinsed for 10 min. Then, AIBA (4 μg ml−1) was prepared in sodium carbonate aqueous solution (1 μg ml−1) as an initiator. The substrate coated with CA was then immersed into the solution. AIBA was grafted on CA via an amidation reaction at 37 °C for 4 h and rinsed for 10 min. In order to confirm whether the initiator was grafted and kept releasing free radicals on the substrate, a frequently-used DPPH method was employed for the examination.Surface-initiated polymerization and IPN on surfaces
After initiator immobilization, the glass substrate was put into DA (6 μg ml−1)/SBMA (4 μg ml−1) and DA (6 μg ml−1)/PSBMA (1.5 μg ml−1) aqueous solutions at 80 °C in nitrogen for 8 h, respectively. The pH values for those solutions were around neural: 7.55 for DA solution, 7.64 for DA/SBMA, and 7.56 for DA/PSBMA. After being rinsed for 10 min and dried, the samples were characterized by scanning electron microscope (SEM), atomic force microscopy (AFM), X-ray photoelectron spectroscopy (XPS), UV-vis spectrophotometry, and water contact angle measurement Fig. 1.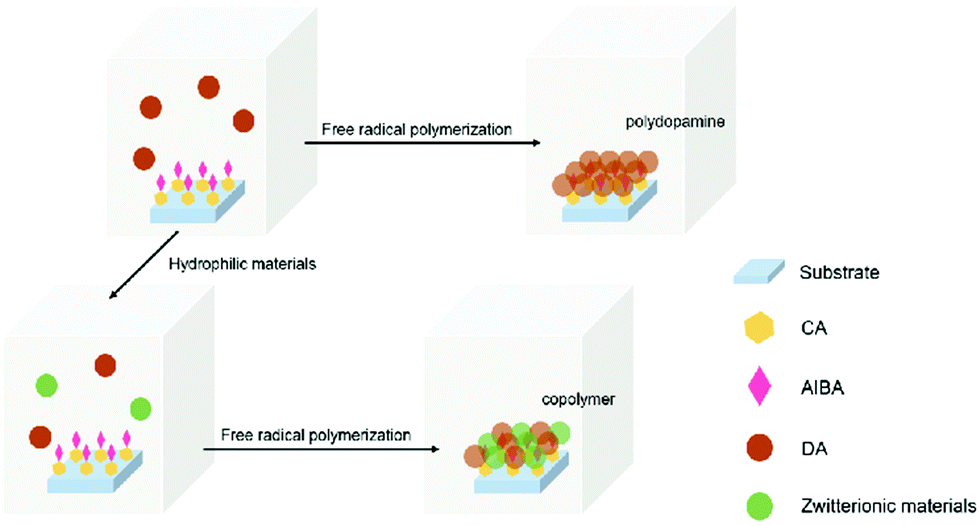 | ||
| Fig. 1 Surface-initiated free-radical polymerization process. | ||
Results and discussion
The surface grafting on the glass was performed using a concept of surficial interpenetration network (IPN) via surface-initiated graft polymerization of DA, DA/PSBMA and DA/SBMA. We focused on limited polymerization on the substrate surface in order to avoid formation of bulk polymers. Therefore, multiple surface reactions on the substrate were carried out and then characterized to verify the achievement of reactions. XPS analysis was carried out on the glass substrates before and after they were coated by the caffeic acid in EtOH. After the glass was coated with caffeic acid and then rinsed off, the Na 1s, Zn 2p1 and Zn 2p3 signals on the prime glass surface disappeared in the surveyed spectra (in Fig. 2), demonstrating that the coverage of caffeic acid thin layer formation on the glass surface. XPS analysis of immobilization of AIBA on the surface showed that the characterized peaks were enhanced at approximately 285.0 eV for C–C group of C 1s, and 400.0 eV for amine of N 1s, indicating that AIBA was successfully immobilized on the glass surface by CA, as shown in Fig. 3.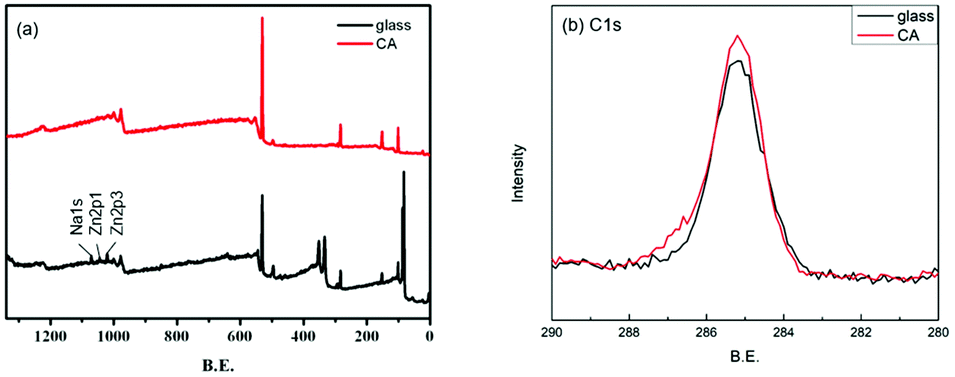 | ||
| Fig. 2 XPS spectra of the glass substrate before and after being coated by CA. (a) survey and (b) C 1s spectra of samples. | ||
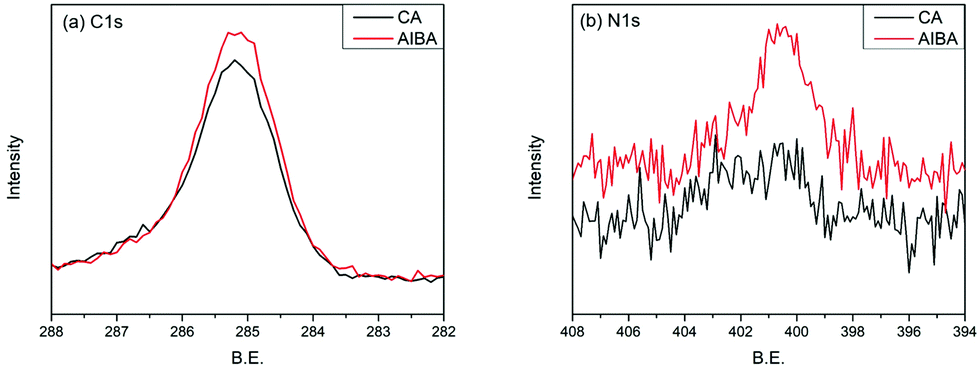 | ||
| Fig. 3 XPS spectra of CA coating immobilized with AIBA. (a) C 1s spectra, (b) N 1s spectra. | ||
DPPH is widely used to examine the ability of compounds to release free radicals or hydrogen donors. We prepared a 0.1 mM DPPH EtOH solution. The glass substrate grafted with initiators was put into DPPH solution at 60 °C for 2 h. When DPPH was reduced, the color of the DPPH solution changed from purple to yellow (shown in Fig. S2, ESI†), making the UV-vis absorption at 517 nm decrease from 1.07 to 0.78, which can be used to quantize the free radicals on the surface (in Fig. S3, ESI†). This result also proved that the initiator was grafted on the substrate and successfully released free radicals.
SEM images in Fig. 4 show that there were no particles deposited after allocating the glass substrates without immobilized initiator, even in a free-radical-polymerized PDA aqueous solution. Via the free-radical polymerization of DA on the initiator-immobilized glass substrate, we could find homogeneous deposition of PDA nanoparticles about 30 nm in size. The copolymer of DA/SBMA deposited on the surface tended to form a thin composite layer including aggregated particles where the average size was increased to approximately 50–100 nm. The IPN polymer entanglement of PDA/PSBMA was deposited and formed heterogeneous aggregates on the surface. SEM images can verify that the initiation of free radicals has generated grafted nanoparticle/PSBMA composite films, a wide variety of which can be produced via this method as compared to the commonly-used self-polymerization of dopamine.
 | ||
| Fig. 4 SEM images of glass substrate before and after surface-initiated free-radical polymerization in monomer solutions. (a) Glass substrate in DA solution without initiator immobilized, and initiator-immobilized substrate (b) in DA solution, (c) in DA/SBMA solution, and (d) in DA/PSBMA solution. | ||
In AFM analysis, it was found that there were no particles on the glass substrate without immobilized initiator, even when immersed in a DA aqueous solution, as shown in Fig. 5(a). PDA nanoparticles appeared due to the initiation via immobilized initiators on the glass substrate in DA aqueous solution, as Fig. 5(b) shows. Regardless of whether we copolymerized DA with SBMA or blended it with PSBMA, the complex was deposited and formed greater particles as shown in Fig. 5(c and d), corresponding to the results of SEM analysis.
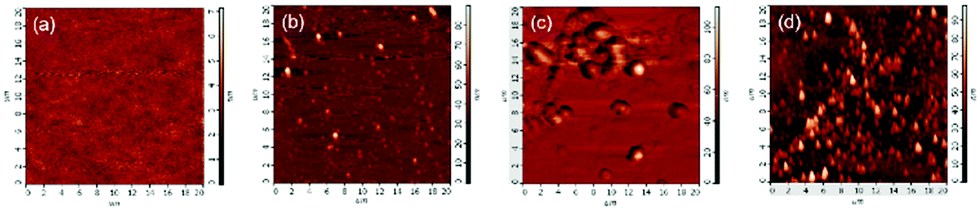 | ||
| Fig. 5 AFM images of glass substrate before and after surface-initiated free-radical polymerization in monomer solutions. (a) Glass substrate in DA solution without initiator immobilized, and initiator-immobilized substrate (b) in DA solution, (c) in DA/SBMA solution, and (d) in DA/PSBMA solution. | ||
It has been confirmed that PSBMA can be entangled with PDA in some literature.4,13 With the successful free-radical polymerization of PDA in this work, the copolymerization of DA and SBMA has become an attemptable strategy. XPS analysis illustrates that the characteristic peaks appear at approximately 285.0 eV, 286.5 eV, and 289.0 eV for C–C, C–O, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups (in Fig. 6(a)); at 400.0 eV and 402.0 eV for amine and ammonium groups (in Fig. 6(b)); and at 168.0 eV for sulfite groups (in Fig. 6(c)), respectively. The results indicate the existence of PDA/PSBMA formation on the surface. The intensity of C–O, CO, ammonium and sulfonate groups of DA/SBMA copolymer and IPN of PDA/PSBMA are quite similar, showing entanglement of polymer chains deposited on the surfaces. The appearance of characteristic N+–R4 and SO3− peaks demonstrates that PSBMA was entangled with PDA.13 It means both addition of SBMA and PSBMA complexed with DA can be formed as hydrophilic top layers due to free-radical initiation. The deconvolution of N 1s and C 1s peaks is shown in Fig. S4 (ESI†) and its ratios of functional groups on the surface can be seen in Table. 1. The list shows that more quaternary ammonium (N+) groups of SBMA are present after polymerization of DA in DA/PSBMA solution than those in DA/SBMA solution (copolymerization of DA and SBMA).
O groups (in Fig. 6(a)); at 400.0 eV and 402.0 eV for amine and ammonium groups (in Fig. 6(b)); and at 168.0 eV for sulfite groups (in Fig. 6(c)), respectively. The results indicate the existence of PDA/PSBMA formation on the surface. The intensity of C–O, CO, ammonium and sulfonate groups of DA/SBMA copolymer and IPN of PDA/PSBMA are quite similar, showing entanglement of polymer chains deposited on the surfaces. The appearance of characteristic N+–R4 and SO3− peaks demonstrates that PSBMA was entangled with PDA.13 It means both addition of SBMA and PSBMA complexed with DA can be formed as hydrophilic top layers due to free-radical initiation. The deconvolution of N 1s and C 1s peaks is shown in Fig. S4 (ESI†) and its ratios of functional groups on the surface can be seen in Table. 1. The list shows that more quaternary ammonium (N+) groups of SBMA are present after polymerization of DA in DA/PSBMA solution than those in DA/SBMA solution (copolymerization of DA and SBMA).
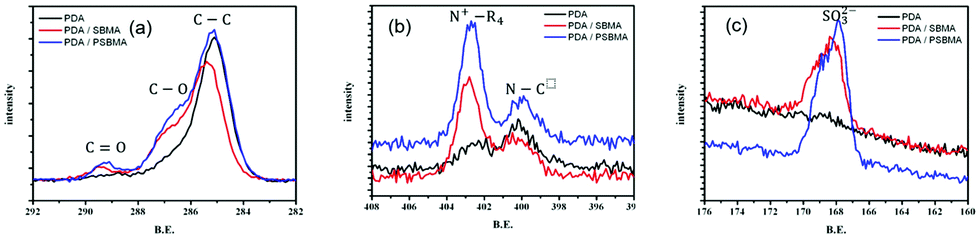 | ||
| Fig. 6 (a) C 1s, (b) N 1s, (c) S 2p XPS spectra of PDA, and the co-deposited modified layers, PDA/SBMA and PDA/PSBMA. | ||
| Ratios of surface groups | in DA | in DA/SBMA | in DA/PSBMA |
|---|---|---|---|
| C–O/C–C | 0.27 | 0.55 | 0.60 |
| COO/C–C | 0.04 | 0.09 | 0.08 |
| N(+)/N(0) | 0.46 | 1.52 | 2.04 |
| N/C | 0.13 | 0.13 | 0.11 |
In addition to observing the microstructure through SEM and AFM analysis, we also used water contact angle (WCA) analysis to inspect the surface modification, as can be seen in Fig. 7. The WCA of the unmodified glass substrate was about 58.7 ± 7.3°. After the substrate was coated with caffeic acid, the WCA decreased to 35.7 ± 4.0°. When the AIBA initiator was immobilized, the WCA increased to 48.1 ± 7.7°. Following free-radical polymerization in DA solution, DA/SBMA solution and DA/PSBMA solution, respectively, the WCAs became 36.4° ± 3.7°, 25.1° ± 8.5° and 16.4° ± 5.6° after rinsing once to get rid of monomers or small molecule impurities. Predictably, the presence of SBMA with zwitterionic groups attached resulted in a significant decrease in WCAs. In order to completely remove weakly-attached homopolymers, the modified substrates were subjected to ultrasonic shock for 1 min, after which the WCAs slightly increased to 44.5° ± 7.5°, 40.6° ± 4.8° and 20.2° ± 3.0°, respectively. The results demonstrate that PSBMA could be firmly attached via PDA formation (like semi-IPN on the surface), compared to surface copolymerization of DA and SBMA monomers. The WCA result is between that of the pure PSBMA and PDA modifications on the surface of materials,4,30,31 showing the zwitterionic groups fully exposed in water. Besides, our use of alcoholic solution to fabricate the coating step-by-step might be crucial, compared to the literature using alkaline or aqueous solution. Water, with its high surface energy, might reject wetting on some hydrophobic polymer surfaces, leading to low efficiency of deposition. The water contact angle data on the deposition of caffeic acid on various polymer surfaces can be seen, showing the wettability of the hydrophobic surface and lowered contact angles. Once the WCA on the substrates could be lowered down, the PDA and its co-deposition of hydrophilic polymers in aqueous solution can be easily attached on the substrate surface.
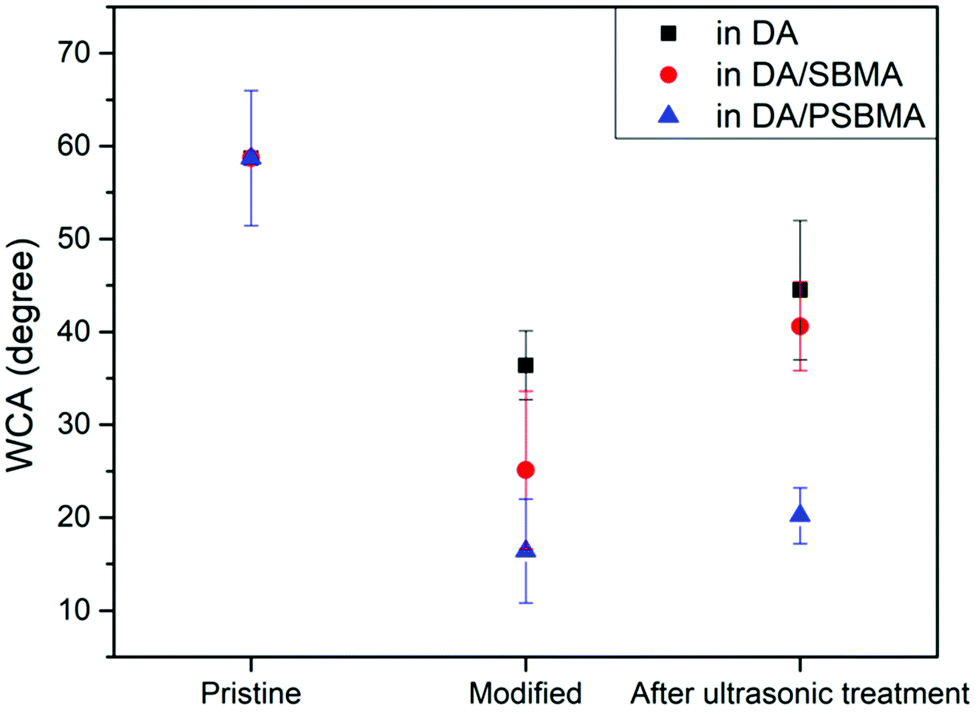 | ||
| Fig. 7 WCA measurements on substrate, and after surface polymerization in DA solution, DA/SBMA solution, and DA/PSBMA solution, respectively. | ||
It is well-known that DA can spontaneously undergo autoxidation in the presence of oxygen, forming a PDA polymer. The rate of polymerization is relatively fast compared to some other polymerization reactions, especially in alkaline environments, and the color of the solution turns dark brown within several minutes. As the polymerization time increases, the color changes to deep black and some particles precipitate on the bottom of the container. Nevertheless, the self-polymerization of DA can be slowed down in the presence of oxygen in an acidic solution, so that the DA aqueous solution can be kept transparent for a certain time.21,32 To support the hypothesis of radical-driven polymerization of DA, the PDA aqueous solution was tested via adding initiators in the bulk solution and could be seen to gradually turn orange-brown, as can be seen in Fig. 8. It might show that only insignificant amounts of bulky fused rings of PDA particles formed in this condition (i.e., no precipitation). Some mechanisms have been suggested to explain the free-radical polymerization of DA, corresponding to the light-brown color formation, instead of dark brown.33,34 Furthermore, we employed DA polymerization via immobilization of the initiator on the surface and found that the aqueous solution could remain transparent and colorless, due to the limited reaction occurring only on the surface and absence of PDA formation in the bulk solution (in Fig. 8(d–f)). A homopolymer, analogue of initiator-bound polydopamine, was synthesized in a similar process, thus proving that the surface initiation of dopamine polymerization can be used to control the thin polydopamine layer coating on a polymer surface. The homopolymer could be found as a small precipitation at the bottom of the test tube, evidencing that the polymerization of dopamine monomer can happen under the free-radical initiation in a mild pH and O2-free condition. The polymer precipitation was collected, dispersed in D2O after rinsing, and characterized by NMR. The NMR spectra (Fig. S5, ESI†) shows the chemically-equivalent protons in the structures of the analogue, compared to the polydopamine that was synthesized with the conventional autoxidation reaction in an alkaline solution. Because we could immobilize initiators onto the substrate surfaces to carry out surficial PDA polymerization via radical-driven reaction, the aqueous solution could be reused for surface modification several times.
 | ||
| Fig. 8 Optical appearances of PDA generated (a) in acidic environments, (b) by free-radical polymerization and (c) in alkaline environments. Also, the appearances of DA aqueous solution (d) before free-radical polymerization, (e) after polymerization on the surface of substrate via immobilized initiators, and (f) after addition of initiators into solution for polymerization. | ||
Conclusions
We employed an initiator-immobilization technique to restrict the reaction to the intended surface in order to reduce PDA polymerization in the bulk solution. Meanwhile, from the incorporation of a free-radical initiator, we observed that the controlled and restrained radicals could attack the dopamine and initiate the formation of PDA. That might be a better alternative (compared with the frequently-used alkaline stimulation method) for surface modification under mild pH conditions. Furthermore, the surface reaction avoided producing homopolymers and precipitative contamination, allowing the expensive dopamine solution to be reused. The formation of nanoparticles of complex deposited on the surface require further detailed exploration to find optimal reaction conditions (for instance, reaction time, monomer concentration, and so on) for practical surface modification applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the projects funded by the Ministry of Science and Technology of Taiwan R.O.C. under MOST 105-2221-E-390 -028 -MY3 and 107-2218-E-390 -004.Notes and references
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed.
- H. Lee, Y. Lee, A. R. Statz, J. Rho, T. G. Park and P. B. Messersmith, Adv. Mater., 2008, 20, 1619–1623 CrossRef CAS PubMed.
- B. Shen, B. Xiong and H. Wu, Biomicrofluidics, 2015, 9, 044111 CrossRef PubMed.
- J. Kuang and P. B. Messersmith, Langmuir, 2012, 28, 7258–7266 CrossRef CAS PubMed.
- W. Sheng, B. Li, X. Wang, B. Dai, B. Yu, X. Jia and F. Zhou, Chem. Sci., 2015, 6, 2068–2073 RSC.
- J. H. L. Beal, A. Bubendorfer, T. Kemmitt, I. Hoek and W. Mike Arnold, Biomicrofluidics, 2012, 6, 036503 CrossRef PubMed.
- Y. Liu, K. Ai and L. Lu, Chem. Rev., 2014, 114, 5057–5115 CrossRef CAS PubMed.
- E. Faure, C. Falentin-Daudré, C. Jérôme, J. Lyskawa, D. Fournier, P. Woisel and C. Detrembleur, Prog. Polym. Sci., 2013, 38, 236–270 CrossRef CAS.
- J. Ryu, S. H. Ku, H. Lee and C. B. Park, Adv. Funct. Mater., 2010, 20, 2132–2139 CrossRef CAS.
- I. S. Kwon and C. J. Bettinger, J. Mater. Chem. B, 2018, 6, 6895–6903 RSC.
- Y. Yang, P. Qi, Y. Ding, M. F. Maitz, Z. Yang, Q. Tu, K. Xiong, Y. Leng and N. Huang, J. Mater. Chem. B, 2014, 3, 72–81 RSC.
- X. Zhang, S. Wang, L. Xu, L. Feng, Y. Ji, L. Tao, S. Li and Y. Wei, Nanoscale, 2012, 4, 5581 RSC.
- R. Zhou, P.-F. Ren, H.-C. Yang and Z.-K. Xu, J. Membr. Sci., 2014, 466, 18–25 CrossRef CAS.
- M. E. Lynge, R. van der Westen, A. Postma and B. Städler, Nanoscale, 2011, 3, 4916 RSC.
- Q. Ye, F. Zhou and W. Liu, Chem. Soc. Rev., 2011, 40, 4244–4258 RSC.
- R. A. Zangmeister, T. A. Morris and M. J. Tarlov, Langmuir, 2013, 29, 8619–8628 CrossRef CAS PubMed.
- D. R. Dreyer, D. J. Miller, B. D. Freeman, D. R. Paul and C. W. Bielawski, Langmuir, 2012, 28, 6428–6435 CrossRef CAS PubMed.
- S. Hong, Y. S. Na, S. Choi, I. T. Song, W. Y. Kim and H. Lee, Adv. Funct. Mater., 2012, 22, 4711–4717 CrossRef CAS.
- R. Mrówczyński, R. Turcu, C. Leostean, H. A. Scheidt and J. Liebscher, Mater. Chem. Phys., 2013, 138, 295–302 CrossRef.
- J. Liebscher, R. Mrówczyński, H. A. Scheidt, C. Filip, N. D. Hădade, R. Turcu, A. Bende and S. Beck, Langmuir, 2013, 29, 10539–10548 CrossRef CAS PubMed.
- T.-P. Chen, T. Liu, T.-L. Su and J. Liang, Langmuir, 2017, 33, 5863–5871 CrossRef CAS.
- L. J. Duan, Y. Liu, J. Kim and D. J. Chung, J. Appl. Polym. Sci., 2013, 130, 131–137 CrossRef CAS.
- E. Shin, S. W. Ju, L. An, E. Ahn, J.-S. Ahn, B.-S. Kim and B. K. Ahn, ACS Appl. Mater. Interfaces, 2018, 10, 1520–1527 CrossRef CAS PubMed.
- X. Fan, L. Lin, J. L. Dalsin and P. B. Messersmith, J. Am. Chem. Soc., 2005, 127, 15843–15847 CrossRef CAS PubMed.
- C. Zhang, M.-Q. Ma, T.-T. Chen, H. Zhang, D.-F. Hu, B.-H. Wu, J. Ji and Z.-K. Xu, ACS Appl. Mater. Interfaces, 2017, 9, 34356–34366 CrossRef CAS PubMed.
- C. Zobrist, J. Sobocinski, J. Lyskawa, D. Fournier, V. Miri, M. Traisnel, M. Jimenez and P. Woisel, Macromolecules, 2011, 44, 5883–5892 CrossRef CAS.
- B. P. Lee, P. B. Messersmith, J. N. Israelachvili and J. H. Waite, Annu. Rev. Mater. Res., 2011, 41, 99–132 CrossRef CAS PubMed.
- C. Zhao, F. Zuo, Z. Liao, Z. Qin, S. Du and Z. Zhao, Macromol. Rapid Commun., 2015, 36, 909–915 CrossRef CAS PubMed.
- J. H. Waite, J. Exp. Biol., 2017, 220, 517–530 CrossRef.
- M.-C. Sin, P.-T. Lou, C.-H. Cho, A. Chinnathambi, S. A. Alharbi and Y. Chang, Colloids Surf., B, 2015, 127, 54–64 CrossRef CAS PubMed.
- L. Chen, L. Tan, S. Liu, L. Bai and Y. Wang, J. Biomater. Sci., Polym. Ed., 2014, 25, 766–785 CrossRef CAS.
- H. Wei, J. Ren, B. Han, L. Xu, L. Han and L. Jia, Colloids Surf., B, 2013, 110, 22–28 CrossRef CAS PubMed.
- X. Kang, W. Cai, S. Zhang and S. Cui, Polym. Chem., 2017, 8, 860–864 RSC.
- V. Ball, D. D. Frari, V. Toniazzo and D. Ruch, J. Colloid Interface Sci., 2012, 386, 366–372 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00908c |
| This journal is © The Royal Society of Chemistry 2021 |