
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced toughness of hydroxyapatite–poly(ethylene terephthalate) composites by immersion in water†
Yui
Okuda
 a,
Ken
Hirota
a,
Tadashi
Mizutani
*a and
Yusuke
Numamoto
b
a,
Ken
Hirota
a,
Tadashi
Mizutani
*a and
Yusuke
Numamoto
b
aDepartment of Applied Chemistry, Graduate School of Science and Engineering, Doshisha University, Kyotanabe, Kyoto 610-0321, Japan. E-mail: tmizutan@mail.doshisha.ac.jp; Fax: +81-774-65-6794; Tel: +81-774-65-6623
bGoo Chemical Corporation, Limited, 58 Ijiri, Iseda-cho, Uji, Kyoto 611-0043, Japan
First published on 20th July 2021
Abstract
Water-soluble poly(ethylene terephthalate) bearing carboxylate groups on 14–22% of its phenylene moieties were coprecipitated with calcium ion and phosphate ion in water at room temperature, 70 °C, and 90 °C to obtain the composites of hydroxyapatite and the polymer. Composite powders with inorganic weight fractions of 30%, 50% and 70% were prepared to explore the effects of the inorganic weight fraction on the water resistance of the composites. The composite powder was compressed with hot pressing at 120 MPa and 120 °C. The compacts showed bending strengths of ∼42 MPa and elastic moduli of ∼4.2 GPa. After immersion in water for 1 week at 37 °C, the bending strengths of the compacts coprecipitated at 90 °C, containing 30 or 50 wt% hydroxyapatite, were ca. 70% of those of the pristine compacts. Moreover, the compacts with the inorganic weight fraction of 50% immersed in water did not break into two pieces in the bending test. The strain at break and the fracture energy were four times larger after immersion in water. Therefore, the immersion in water improved the toughness of the composite of hydroxyapatite and the carboxypolyester.
Introduction
Bones are formed by the precipitation of amorphous calcium phosphate in the gaps of collagen fibrils. Subsequent crystallization of the amorphous phase to hydroxyapatite occurs to afford the highly oriented composite of protein fibers and the platelet inorganic crystals.1 Bone is a lightweight, tough, ductile, and green material. Cortical bone contains ca. 70 wt% hydroxyapatite (HAP) or non-stoichiometric carbonated apatite.2–5 Non-collagenous proteins such as osteocalcin and osteopontin6–8 play important roles in biomineralization.8,9 Osteopontin contains serine and threonine, and some of their hydroxy groups are phosphorylated. It also contains the poly(aspartic acid) region.10 Osteocalcin contains γ-carboxyglutamic acid, and the –CH(COO−)2 group in the side chain can chelate Ca2+. These anionic groups bind to calcium ion and to the {100} surface of HAP.11 These functional groups serve as nucleation sites of hydroxyapatite and binding sites to the surface of hydroxyapatite crystals to regulate the crystal growth.12The interface between HAP and the organic phase in bone has been the subject of investigations. Citrate was shown to bind to the surface of HAP at a density of 1 molecule per 4 nm2 to endow the HAP surface with hydrophobicity.13 NMR studies indicated that there are polysaccharides on the surface of HAP.14 Adsorption of polar molecules such as citrate and polysaccharides indicates that the HAP surface is quite hydrophilic. These polar functional groups would stabilize the interface and possibly improve the mechanical properties, such as elastic moduli, a major function of hard tissues. Collagen also contains hydrophobic moieties such as proline, glycine, and alanine, and the total mol% of these hydrophobic amino acids is 63%.15 These hydrophobic moieties could confer water resistance to collagen and bone.
There have been many studies on hybridization of polymers and hydroxyapatite with a variety of protocols of hybridization, such as mechanical mixing of polymer with HAP crystals,16,17 polymerization of monomers in the presence of HAP,18 crystallization of HAP in polymer hydrogels,19–21 and coprecipitation of polymers and HAP.22–34 Bonfield et al.16,35 reported that mixing of high-density polyethylene with HAP realized composites whose elastic moduli can be controlled by the volume fraction of HAP in the range between 0.9 and 12.1 GPa, which covers a part of the range of the elastic moduli of natural bones (5–22 GPa).36 It should be noted, however, that the composite of hydrophobic polymer and HAP was not necessarily water-resistant. Ritchie and coworkers37 demonstrated that the bending strength of the composite of polylactic acid and HAP was reduced to 10–20% by exposure of the compact to humid air at 37 °C.
Among various strategies to prepare composites, coprecipitation of water-soluble polymer with calcium ion and phosphate ion is a facile route to obtain nanocrystals of hydroxyapatite hybridized with the polymer.38,39 The approach is also biomimetic and ecologically benign. Anionic functional groups in polymers can act as a heterogeneous nucleation site during coprecipitation, and are expected to lead to efficient hybridization at a molecular level.7,40–42 To prepare composite materials with excellent mechanical properties, the size and morphology of hydroxyapatite crystals should be controlled. The size and morphology of hydroxyapatite can be controlled by adjusting the functional groups of the polymer, the source of inorganic ions, concentrations of both the polymer and inorganic ions, temperature, pH and other coprecipitation parameters. For the importance of anionic functional groups in polymers, we previously reported that polyvinyl alcohol and starch bearing –OPO32− groups afforded a tough composite in which the phosphate groups lead to nucleation of HAP and possibly stabilize the organic/inorganic interface.43,44 Grohndahl et al.,45 Yao et al.,46 and we44 reported that anionic functional groups such as –COO− and –OPO32− effectively bind to HAP to regulate the crystal growth.
For practical use of these composites, a water absorption test is particularly important.47 Although polymer–hydroxyapatite composites were precipitated from water, these composites were not necessarily water-resistant, owing to the hydrophilic nature of the surface of HAP and the polymer. Previously, we reported that water-soluble polymers such as tapioca starch coprecipitated with HAP almost quantitatively, and the obtained composites exhibited bending strengths of 20–61 MPa.44 However, the HAP–starch composites absorbed water, and they became soft when soaked in water. Therefore, if we attempt to employ these composites as mechanical materials or bone substitutes, the effects of water on the mechanical properties of the composites should be clarified. The following two strategies are expected to be effective. (1) Introduction of hydrophobic moieties to the polymer will improve the water resistance of the composite. (2) Complementary polar functional groups on the polymer and those on the surface of the HAP crystals will cancel the polarity and render the interface hydrophobic, while free polar functional groups on the polymer and the HAP surface will be easily hydrated to lead to water-sensitive composites. Therefore, the ratio of polar groups and nonpolar groups in the polymer as well as the weight ratio of the polymer and hydroxyapatite are correlated and should be optimized to obtain water-resistant composites.
Koutsoukos and co-workers reported coprecipitation of HAP with copolymers having hydrophobic phenylene moieties.48 The composites of poly(ethylene terephthalate) have been studied for various applications.49–54 In this work, we employed poly(ethylene terephthalate) carrying carboxylate groups in some of the phenylene groups, as a water-soluble polymer containing hydrophobic moieties, to develop water-resistant composites. We focused on the effects of the hydrophilicity/hydrophobicity balance of the polymer as well as the polymer/HAP ratios on the water resistance of the composites in this study. We found that the composites containing 50 wt% HAP were water-resistant. The main chain of hydrophobic poly(ethylene terephthalate) played a significant role in the water resistance of the composites, and the composites after immersion in water retained ca. 70% of the bending strength of the pristine compact. The strain at break and the fracture energy of the composite were four times larger after it was immersed in water. Thus, the compact was tougher after immersion in water. We suggest that the carboxylate groups of the polymer confer water-solubility for coprecipitation to the polymer, interact with calcium ions to nucleate hydroxyapatite crystals, and contribute to a stable organic–inorganic interface, while the hydrophobic poly(ethylene terephthalate) main chains confer water-resistance to the composites.
Materials and methods
Materials
A 25 wt% aqueous solution (pH 6.5–8.5) of poly(ethylene terephthalate) bearing –COO−NH4+ groups in 14–22% of the phenylene groups, PLAS COAT Z-730, with a molecular weight of ca. 3000 was provided by Goo Chemical Corporation, Ltd. The polymer will be abbreviated as CPET. CaCl2·2H2O, Na2HPO4, NaOH, methanol and acetone were purchased from FUJIFILM Wako Pure Chemical Corporation.Instrumentation
X-Ray diffraction (XRD) spectra of the composite powder were recorded on a Rigaku SmartLab diffractometer with Cu-Kα radiation. The crystallite size of HAP, L, was calculated from the full-width of the diffraction peak at half of the maximum intensity (FWHM) of the (002) and (310) peaks using the Scherrer equation: L = 0.9λ/(B![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ), where λ is the wavelength of the X-ray, B measured in radians is the full-width at half maximum of the 2θ peak, and θ is the Bragg angle for the peak. Thermogravimetric and differential thermal analysis (TG/DTA) was performed with a Shimadzu DTA-60 thermal analysis instrument. The composite powder was heated in an alumina pan at a rate of 20 °C min−1 in air. Fourier transform infrared spectra (FT-IR) were recorded on a JASCO FT/IF-4600 spectrometer as KBr pellets. UV-visible spectra were obtained on an Agilent 8453 UV-visible spectrophotometer, using a 1 cm pathlength quartz cuvette. Scanning electron microscopy (SEM) images of the fracture surface were observed with a Hitachi High-Technology SU8020 scanning electron microscope.
cosθ), where λ is the wavelength of the X-ray, B measured in radians is the full-width at half maximum of the 2θ peak, and θ is the Bragg angle for the peak. Thermogravimetric and differential thermal analysis (TG/DTA) was performed with a Shimadzu DTA-60 thermal analysis instrument. The composite powder was heated in an alumina pan at a rate of 20 °C min−1 in air. Fourier transform infrared spectra (FT-IR) were recorded on a JASCO FT/IF-4600 spectrometer as KBr pellets. UV-visible spectra were obtained on an Agilent 8453 UV-visible spectrophotometer, using a 1 cm pathlength quartz cuvette. Scanning electron microscopy (SEM) images of the fracture surface were observed with a Hitachi High-Technology SU8020 scanning electron microscope.
Preparation of CPET–HAP composites by coprecipitation of CPET, Ca2+, and PO43−
| Sample | ICfeed (wt%) | 25 wt% CPET aq. (g) | 0.2 M Na2HPO4 aq. (mL) | 1 M NaOH aq. (mL) | 0.2 M CaCl2 aq. (mL) | Coprecip. temp. (°C) | Aging temp. (°C) |
|---|---|---|---|---|---|---|---|
| 1 | 30 | 21.76 | 69.7 | 18.6 | 116 | r.t. | r.t. |
| 2 | 50 | 9.32 | 69.7 | 18.6 | 116 | r.t. | r.t. |
| 3 | 70 | 4.0 | 69.7 | 18.6 | 116 | r.t. | r.t. |
| 4 | 30 | 21.76 | 69.7 | 18.6 | 116 | r.t. | 70 |
| 5 | 50 | 9.32 | 69.7 | 18.6 | 116 | r.t. | 70 |
| 6 | 70 | 4.0 | 69.7 | 18.6 | 116 | r.t. | 70 |
| 7 | 30 | 21.76 | 69.7 | 18.6 | 116 | r.t. | 90 |
| 8 | 50 | 9.32 | 69.7 | 18.6 | 116 | r.t. | 90 |
| 9 | 70 | 4.0 | 69.7 | 18.6 | 116 | r.t. | 90 |
| 10 | 30 | 21.76 | 69.7 | 18.6 | 116 | 70 | 70 |
| 11 | 50 | 9.32 | 69.7 | 18.6 | 116 | 70 | 70 |
| 12 | 70 | 4.0 | 69.7 | 18.6 | 116 | 70 | 70 |
| 13 | 30 | 21.76 | 69.7 | 18.6 | 116 | 90 | 90 |
| 14 | 50 | 9.32 | 69.7 | 18.6 | 116 | 90 | 90 |
| 15 | 70 | 4.0 | 69.7 | 18.6 | 116 | 90 | 90 |
:1) was added to precipitate CPET–Na+. The supernatant was removed by decantation. To the turbid suspension was added distilled water to obtain white precipitates. The precipitates were filtered and washed with water, followed by washing with a small amount of methanol. The white precipitates were dried in vacuo at 80 °C for 1 h.
Densification of the composite powder
The composite powder was placed in a mold with a rectangular slot with dimensions of 4 mm × 13 mm, and it was uniaxially pressed at 120 MPa at 120 °C for 5 min to obtain a white rectangular parallelepiped with dimensions of 4 mm × 13 mm × 1.5–2.0 mm.Three-point bending test of the composite compacts
A three-point bending test was carried out using a bending testing machine (MZ-603, Maruto Instrument Co., Ltd, Japan) with a cross-head speed of 0.5 mm min−1. Details of the three-point bending test were described in a previous paper.44 Fracture energy was calculated from the area between the stress–strain curve and the horizontal axis. Standard errors of the mean of the mechanical properties were determined based on 3–5 independent determinations. The fracture surface was observed using SEM.Water resistance test of the composite compacts
The compacts were immersed in water (20 mL) either at room temperature for 24 h or at 37 °C for 1 week. After immersion in water, the compact was wiped with paper to remove the water on the surface, and the increase in weight divided by the weight of the dry compact, Δw/w, was determined: Δw/w = weight increase/dry weight × 100 (%). The compacts were then immediately subjected to a three-point bending test in order to evaluate the effects of water immersion on the mechanical properties. The UV-visible spectra of the aqueous phase after immersion of the compacts were recorded to evaluate the amounts of dissolved CPET. The CPET–NH4+ solution at a concentration of 8 mg L−1 showed absorbance of 0.44 at 240 nm. The concentrations of CPET in the aqueous phase were determined based on the absorbance at 240 nm.Results and discussion
Hybridization of hydroxyapatite with CPET by coprecipitation
Water-soluble poly(ethylene terephthalate), CPET, has carboxy groups on 14–22% of the phenylene groups in the main chain of poly(ethylene terephthalate) (Scheme 1). The carboxy groups render the polymer water-soluble, and coprecipitation can be carried out in water; meanwhile, the hydrophobic main chain would confer water-resistance to the composite. Hybridization was performed by coprecipitation of the polymer and the inorganic ions in water. To an alkaline aqueous solution containing CPET and phosphate ions was added aqueous calcium chloride to precipitate CPET–HAP composites at three temperatures: room temperature, 70 °C, and 90 °C. The suspensions were aged at room temperature, 70 °C, or 90 °C (Scheme 2). The coprecipitation conditions are summarized in Table 1. Table 2 shows that the yields of the coprecipitated powder were 82–95%.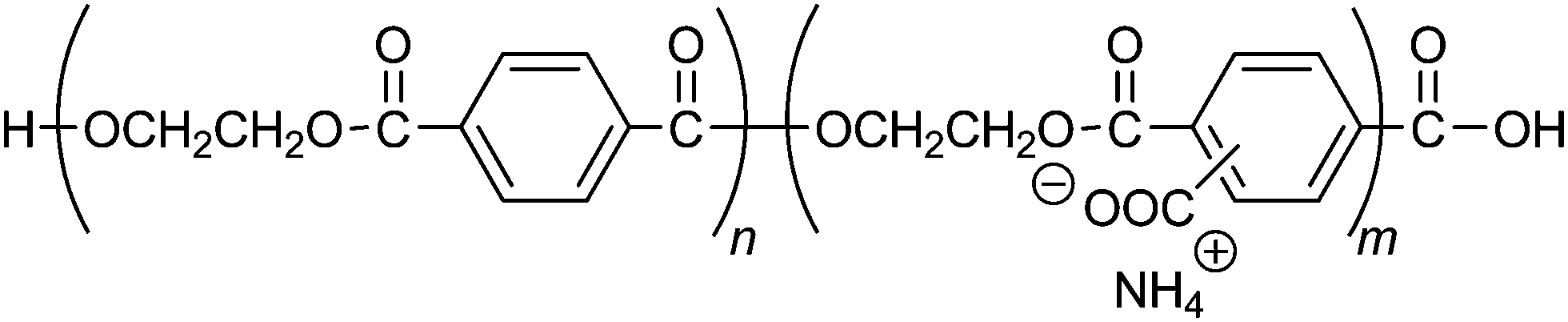 | ||
| Scheme 1 Structure of CPET. | ||
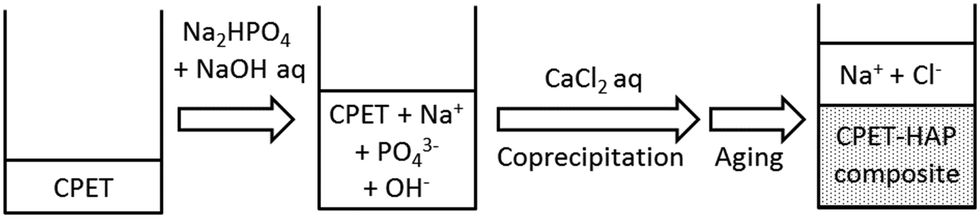 | ||
| Scheme 2 Coprecipitation of CPET and hydroxyapatite. | ||
| Sample | Y obs, g (Yobs/Ycalcd%)b | IC (wt%) | Crystallite size along c-axis (nm) | Crystallite size along a-axis (nm) | Aspect ratio c/a |
|---|---|---|---|---|---|
| a XRD showed that only an amorphous inorganic phase formed. b Calculated yield, Ycalc, was obtained by assuming that all calcium ions and phosphate ions were precipitated as HAP, Ca10(PO4)6(OH)2, and all CPET was incorporated in the precipitate. | |||||
| 1 | 7.1 (91) | 25 | |||
| 2 | 4.4 (95) | 45 | 16 | 5 | 3.3 |
| 3 | 3.0 (89) | 63 | 14 | 6 | 2.6 |
| 4 | 7.1 (91) | 25 | 22 | 6 | 3.9 |
| 5 | 4.4 (94) | 46 | 21 | 6 | 3.7 |
| 6 | 3.0 (89) | 65 | 20 | 7 | 3.0 |
| 7 | 7.0 (91) | 26 | 23 | 6 | 3.8 |
| 8 | 4.4 (94) | 46 | 26 | 7 | 3.8 |
| 9 | 2.9 (87) | 65 | 25 | 7 | 3.3 |
| 10 | 6.9 (89) | 26 | 26 | 6 | 4.1 |
| 11 | 3.8 (82) | 47 | 33 | 9 | 3.7 |
| 12 | 2.8 (83) | 68 | 35 | 9 | 3.8 |
| 13 | 6.9 (89) | 26 | 31 | 8 | 4.1 |
| 14 | 3.7 (79) | 49 | 36 | 8 | 4.7 |
| 15 | 2.8 (84) | 68 | 33 | 9 | 3.8 |
The weight fraction of the inorganic phase in the composites was determined by thermogravimetry. The composite powder was heated from room temperature to 100 °C in air at a rate of 20 °C min−1, maintained at 100 °C for 10 min, heated to 1000 °C at a rate of 20 °C min−1, and maintained at 1000 °C for 10 min. The weight% of the inorganic components in the composites (IC) was calculated as a ratio of the weight at 1000 °C to that at 100 °C. We used the feed ratios of CPET to Ca2+/PO43− ions to prepare composites with inorganic weight fractions of 30%, 50%, and 70%, assuming that all the inorganic ions were precipitated as HAP and all CPET was incorporated in the composite powder. As shown in Table 2, the ratios of HAP to CPET in the composites determined by TG agreed well with those of the feed ratios, indicating that inorganic/organic weight ratios in the composites can be easily controlled by adjusting the feed ratios.25
The XRD patterns of the composite powder of samples 1–15 are shown in Fig. 1 and Fig. S1 (ESI†). Sample 1, coprecipitated at room temperature in the presence of a high concentration of CPET, exhibited only a broad diffraction pattern, showing that amorphous calcium phosphate formed. Except for sample 1, the characteristic diffraction peaks due to HAP were observed. The peaks are indexed based on the hexagonal HAP in Fig. 1. The diffraction peaks of samples 2–3, coprecipitated and aged at room temperature, were broad, showing that crystallization of HAP at room temperature proceeded slowly. The crystallite sizes were calculated using the Scherrer formula and are listed in Table 2. The crystallite size of HAP increased as the coprecipitation temperature and aging temperature were increased. Aizawa et al.55 reported that HAP crystals obtained by homogeneous precipitation grew to the c-axis to form fiber-like crystals. We also observed that the HAP crystal grew to the c-axis, and the length of the fiber-like crystals was 26–36 nm when coprecipitation was carried out at 70–90 °C.
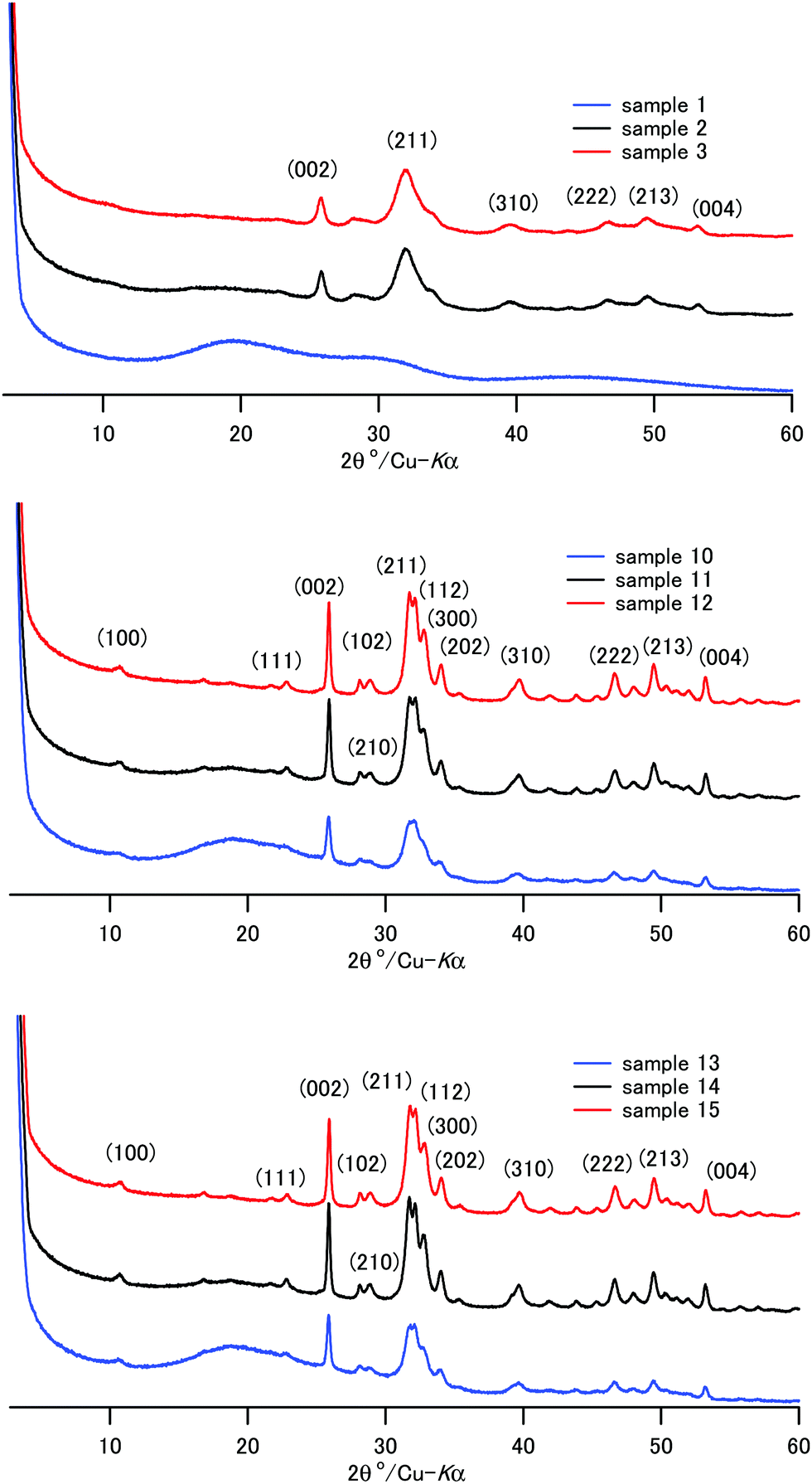 | ||
| Fig. 1 The XRD patterns of samples 1–3 and 10–15. | ||
Crystals of apatite of fish bone are platelet-shaped and are 2 nm in thickness, 20 nm in width, and 30 nm in height.56 Samples 12 and 15 coprecipitated at 70 °C/90 °C and IC of 70 wt% showed a similar crystallite size and aspect ratio to those of the apatite of bone. Li and coworkers reported that the nanometer size of HAP crystal makes significant contributions to the mechanical properties of HAP–polyamide composites.57 Uniform dispersion of nanometer-sized particles with a high aspect ratio leads to a wide interface area, and this would result in superior mechanical properties of bone and the composites. Table 2 shows that the aspect ratios, c/a, were 2.6–4.7, and they increased as the coprecipitation temperature was increased. For the present composites, coprecipitation at 70–90 °C helped the crystallization of HAP to create a “brick and mortar” structure58 in the composite.
An energy dispersion X-ray spectroscopy study showed that the atomic ratio Ca/P/C/O = 12:9:37:40 for sample 14 (Fig. S6, ESI†). The Ca/P ratio was 1.3, and the value is close to the chemical formula of HAP (Ca10(PO4)3(OH)2, Ca/P = 1.6).
The thermogravimetry (TG) and differential thermal analysis (DTA) traces of the composites containing 30 wt% of HAP are shown in Fig. 2. Other TG/DTA traces are shown in Fig. S2 (ESI†). All the composite samples exhibited a weight loss started at 310 °C with an exothermic peak in DTA, and the weight loss ended at 530 °C. This weight loss was ascribed to the thermal decomposition of CPET.
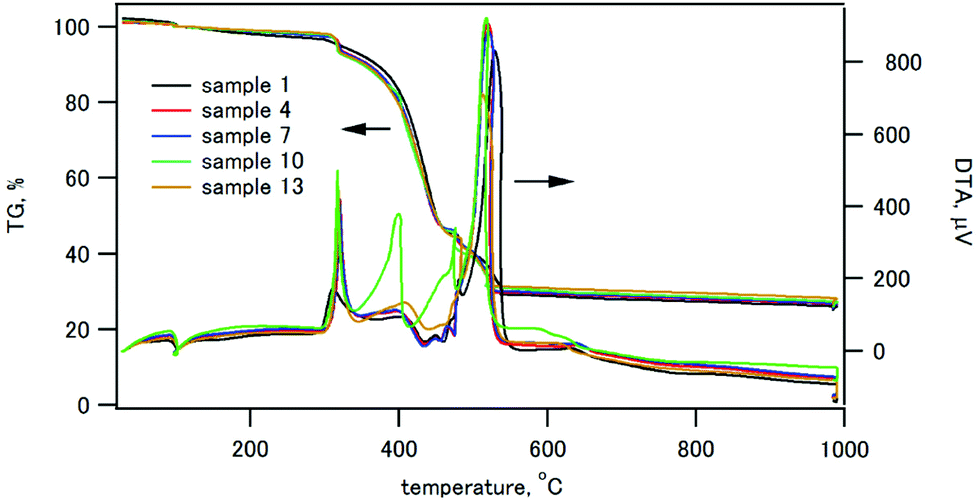 | ||
| Fig. 2 TG/DTA traces of the composites containing 30 wt% HAP. | ||
Infrared spectra of the composite powder
The infrared spectra of sample 11, CPET–Na+, and HAP are shown in Fig. 3. The infrared spectrum of sample 11 showed peaks at 561 and 605 cm−1 (O–P–O bending), 730 cm−1 (C–H out-of-plane bending vibration of the p-substituted aromatic ring), 1029 cm−1 (P–O stretching), 1102 cm−1 (ester C–O–C symmetric stretching and P–O stretching), 1270 cm−1 (ester O–C–O antisymmetric stretching), 1722 cm−1 (ester C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching), 2965 cm−1 (C–H stretching of ethylene), and 3432 cm−1 (O–H stretching). Fig. 4 shows the expanded spectra between 1350 and 1800 cm−1 of the composite with the inorganic weight fraction of 70%. The symmetric and antisymmetric stretching of the carboxylate group appeared at 1410 cm−1 and in a region between 1550 and 1610 cm−1, respectively. The antisymmetric stretching of the carboxylate group may overlap with the CC stretching of the benzene ring, and the signal assignment is not straightforward.
O stretching), 2965 cm−1 (C–H stretching of ethylene), and 3432 cm−1 (O–H stretching). Fig. 4 shows the expanded spectra between 1350 and 1800 cm−1 of the composite with the inorganic weight fraction of 70%. The symmetric and antisymmetric stretching of the carboxylate group appeared at 1410 cm−1 and in a region between 1550 and 1610 cm−1, respectively. The antisymmetric stretching of the carboxylate group may overlap with the CC stretching of the benzene ring, and the signal assignment is not straightforward.
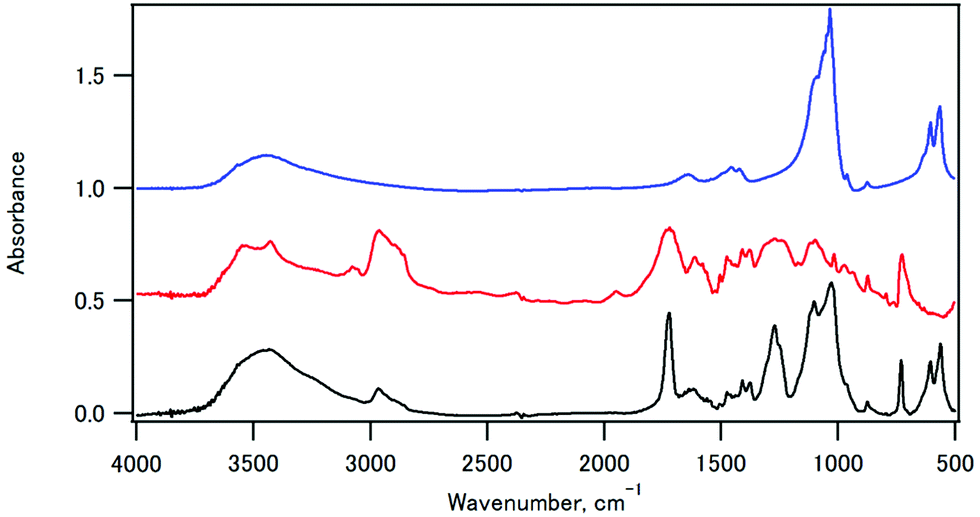 | ||
| Fig. 3 The infrared spectra of sample 11 (black), CPET–Na+ (red), and HAP (blue). | ||
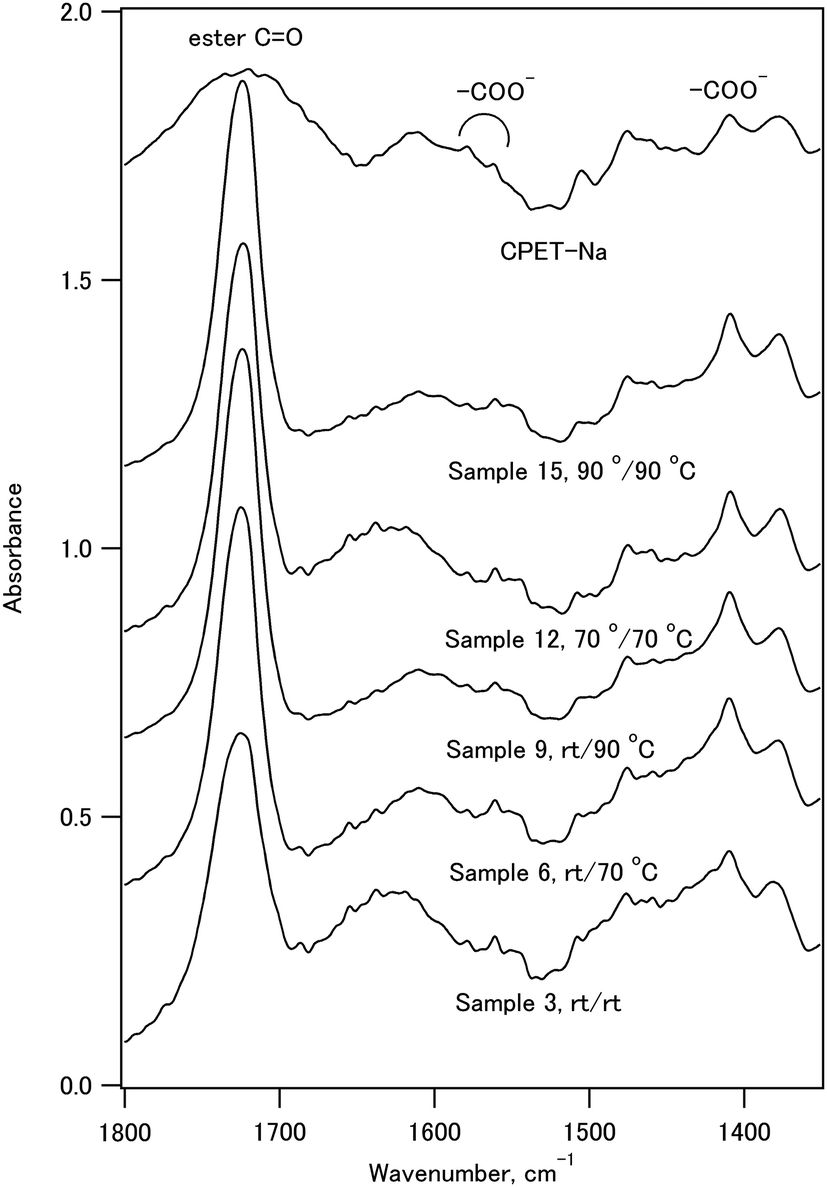 | ||
| Fig. 4 The infrared spectra of the composite powder with IC of 70% and CPET–Na+. | ||
The full width at half maximum (FWHM) values of the ester carbonyl stretching peak of the composites and CPET–Na+ are shown in Fig. 5. It is interesting to note that the FWHM values of the composites were smaller than that of CPET–Na+. The FWHM values decreased with increasing HAP content from 30% to 70%. The increase of FWHM reveals the increasing diversity of the main chain conformations of CPET, where there are many conformers with respect to the co-planarity of the ester group and the benzene ring. Since the fraction of CPET on the surface of the HAP crystals increased as the HAP content increased, the observation suggests that the CPET on the HAP crystal surface has a narrower ester peak than the bulk CPET. The small FWHM values can be ascribed to a well-defined conformation of CPET on the HAP crystal surface. The FWHM values coprecipitated and aged at room temperature were large, and heating above 70 °C resulted in smaller FWHM. Therefore, annealing above 70 °C lead to a stable conformation of the polymer to reduce the FWHM values.
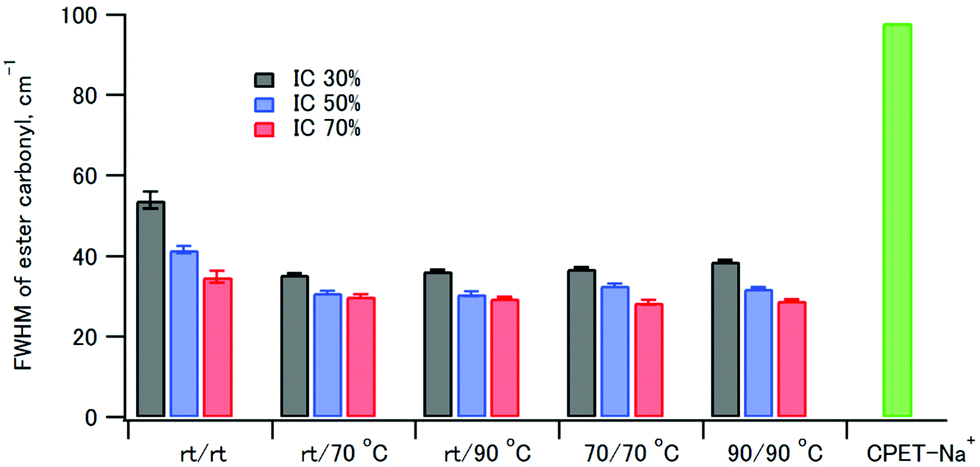 | ||
| Fig. 5 FWHM of the ester carbonyl stretching at 1725 cm−1 of samples 1–15 and CPET–Na+. | ||
Fig. 6 compares the P–O stretching peaks of the composites with IC of 50%. The 1020 cm−1 component is characteristic of poorly crystalline apatites, and the 1030 cm−1 component is characteristic of highly crystalline apatite.59,60 The absorbance at 1015 nm−1 decreased as the coprecipitation temperature was increased from room temperature to 90 °C, indicating that the crystallinity of HAP increased. These observations are fully consistent with the XRD studies.
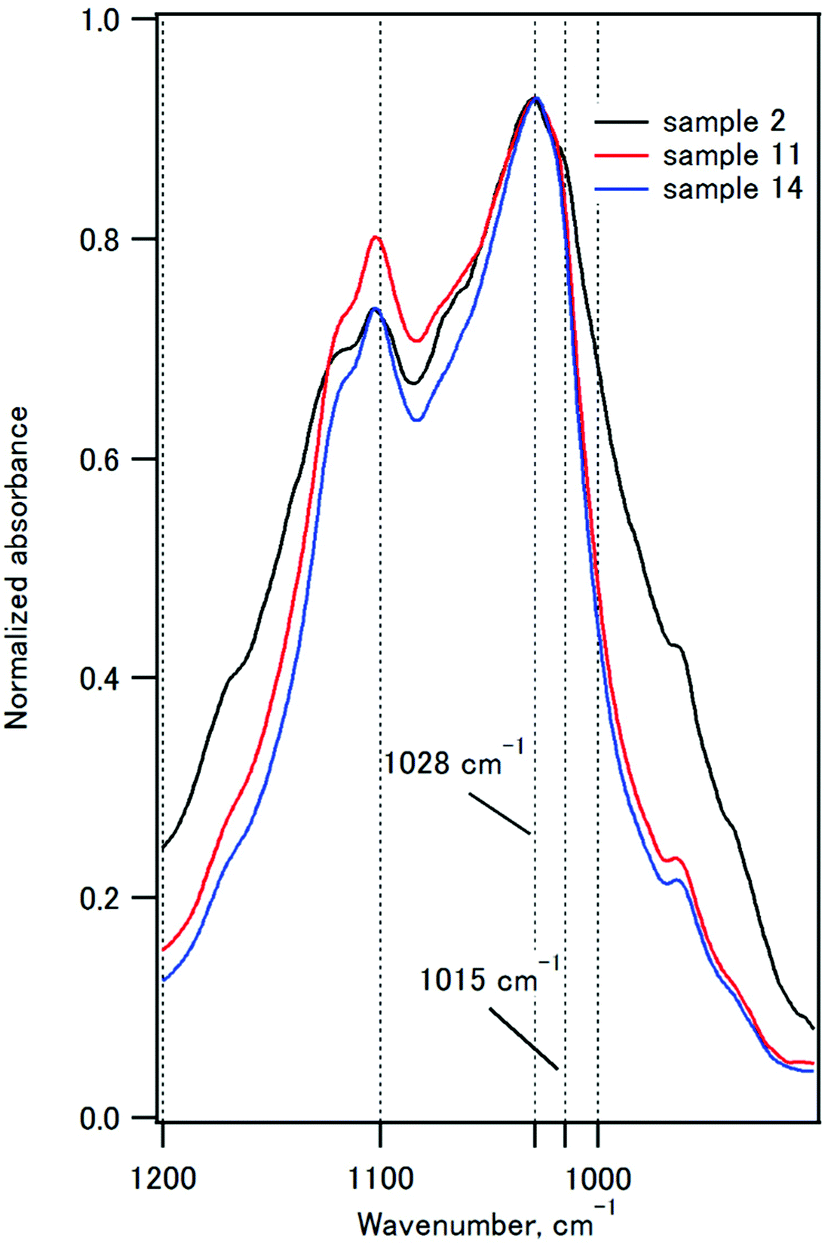 | ||
| Fig. 6 The P–O stretching region in the infrared spectra of samples 2, 11, and 14. | ||
Densification of the composite powder
The composite powder was uniaxially pressed at 120 °C and at 120 MPa for 5 min to obtain compacts with dimensions of 4 × 13 × 1.5–2 mm. The densities of the compacts, dobs, were determined from the weights and the volumes of the compacts and are listed in Table 3. Fig. 7 illustrates the plot of the relative density, dobs/dcalc, against IC. The calculated density, dcalc, was obtained from the organic/inorganic weight ratios, the density of CPET (1.24 g cm−3), and the density of HAP (3.10 g cm−3).43 The relative density decreased with increasing IC for all samples 1–15, showing that high inorganic contents decreased the efficiency of the densification process. CPET is a thermoplastic with a softening point of 80–85 °C. Heating at 120 °C during hot pressing softened the polymer phase to help densification.| Sample | σ b (MPa) | ε b (%) | E b1 (GPa) | E b2 (GPa) | G f (MJ m−3) | d obs (g cm−3) |
|---|---|---|---|---|---|---|
| a Averages of at least three independent determinations are shown with standard error of the means. | ||||||
| 1 | 24 ± 1 | 2.0 ± 0.1 | 0.7 ± 0.2 | 1.8 ± 0.2 | 0.18 ± 0.01 | 1.337 ± 0.006 |
| 2 | 30 ± 1 | 2.2 ± 0.1 | 0.7 ± 0.1 | 2.1 ± 0.2 | 0.26 ± 0.01 | 1.51 ± 0.01 |
| 3 | 19 ± 0.5 | 1.1 ± 0.1 | 1.0 ± 0.2 | 2.3 ± 0.1 | 0.09 ± 0.01 | 1.74 ± 0.03 |
| 4 | 25 ± 2 | 1.7 ± 0.1 | 1.03 ± 0.03 | 1.9 ± 0.1 | 0.19 ± 0.02 | 1.365 ± 0.004 |
| 5 | 31 ± 1 | 2.2 ± 0.2 | 1.1 ± 0.3 | 2.2 ± 0.2 | 0.29 ± 0.01 | 1.532 ± 0.005 |
| 6 | 35 ± 2 | 1.7 ± 0.2 | 1.2 ± 0.1 | 3.8 ± 0.7 | 0.22 ± 0.04 | 1.71 ± 0.03 |
| 7 | 28 ± 4 | 1.7 ± 0.1 | 0.9 ± 0.1 | 2.0 ± 0.1 | 0.20 ± 0.04 | 1.39 ± 0.01 |
| 8 | 31 ± 1 | 2.4 ± 0.1 | 0.8 ± 0.1 | 2.1 ± 0.1 | 0.30 ± 0.01 | 1.533 ± 0.009 |
| 9 | 38 ± 1 | 1.7 ± 0.1 | 1.2 ± 0.3 | 3.4 ± 0.1 | 0.25 ± 0.01 | 1.73 ± 0.01 |
| 10 | 42 ± 2 | 2.3 ± 0.2 | 1.1 ± 0.1 | 2.5 ± 0.1 | 0.41 ± 0.06 | 1.406 ± 0.004 |
| 11 | 34 ± 2 | 1.8 ± 0.1 | 1.2 ± 0.2 | 2.8 ± 0.1 | 0.25 ± 0.02 | 1.61 ± 0.01 |
| 12 | 39 ± 2 | 1.5 ± 0.1 | 1.9 ± 0.2 | 3.6 ± 0.2 | 0.25 ± 0.02 | 1.81 ± 0.02 |
| 13 | 32 ± 1 | 1.7 ± 0.1 | 0.5 ± 0.1 | 1.9 ± 0.1 | 0.33 ± 0.02 | 1.077 ± 0.002 |
| 14 | 32 ± 2 | 1.8 ± 0.2 | 1.2 ± 0.2 | 2.55 ± 0.01 | 0.24 ± 0.03 | 1.59 ± 0.01 |
| 15 | 42 ± 1 | 1.59 ± 0.05 | 1.5 ± 0.1 | 4.2 ± 0.1 | 0.26 ± 0.01 | 1.86 ± 0.02 |
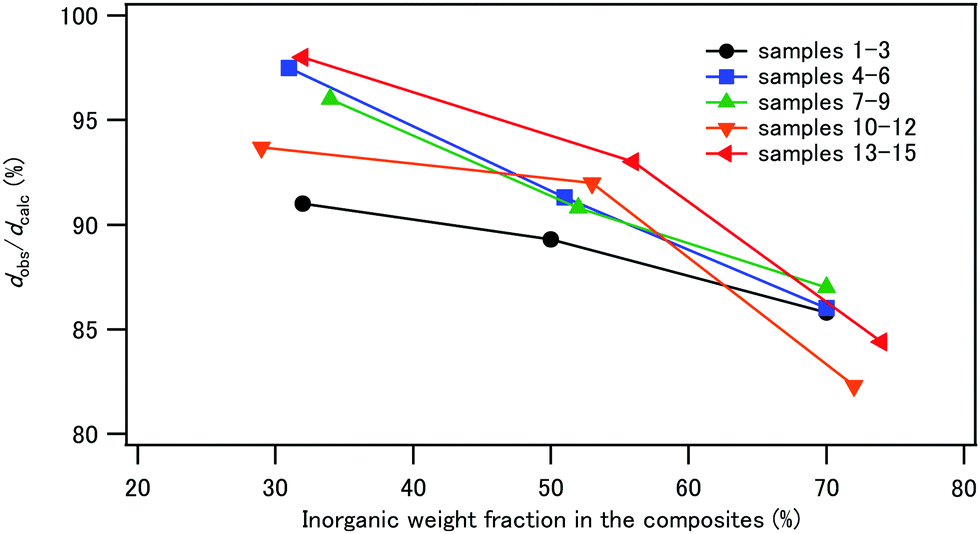 | ||
| Fig. 7 Plot of the relative density of the compacts against the inorganic weight fraction, IC. | ||
The photographs of the compacts are shown in Fig. 8. The compacts with IC of 30% were relatively transparent, and the compacts became less transparent with increasing IC in the composites. With respect to the coprecipitation/aging temperatures, sample 8 with IC 50 wt%, coprecipitated at room temperature and aged at 90 °C, was transparent. This is in contrast to the observations that the compacts of the composites of hydroxyapatite and starch or cellulose nanofibers were opaque.44 The refractive indices of HAP and PET are 1.641 and 1.57, respectively.61,62 The similar refractive indices of the filler and the matrix may be one of the reasons for the transparency, i.e., weak light scattering.
 | ||
| Fig. 8 Photographs showing the transparency of the compacts of samples 1–15 with 1.6 mm thickness on a paper printed with “CPET–HAP”. | ||
Mechanical properties of the composite compacts
A three-point bending test of the compacts was performed. Representative stress–strain curves are shown in Fig. S3 (ESI†). The curves showed that the initial slope was smaller than the slope at break. The bending elastic moduli were thus calculated both from the initial slope, Eb1, and the slope at break, Eb2. Table 3 summarizes the bending strengths, the strains at break, the elastic moduli, the fracture energies, and the densities of the composite compacts.Fig. 9 shows the bending strengths of the compacts: as the coprecipitation/aging temperatures were increased, the bending strengths increased. This can be ascribed to the formation of highly crystalline HAP interacting with well-orientated CPET at higher temperatures. For the composites with the inorganic weight fractions of 30% and 50%, the bending strength of the compact coprecipitated at 70 °C was larger than that at 90 °C. The composite of phosphorylated starch and HAP (IC 70%) prepared in a similar fashion showed a strain at break of 2–3%.43 In comparison, the CPET–HAP composite (IC 70%) showed smaller strains at break of 1.1–2.4%. The elastic modulus of sample 12 (IC 72%) was 3.6 GPa, while that of phosphorylated tapioca starch–HAP (IC 67%) was 2.7 GPa.44 These observations indicate that the CPET–HAP composite with the inorganic weight fraction of ca. 70% was stiffer than the corresponding starch–HAP composite.
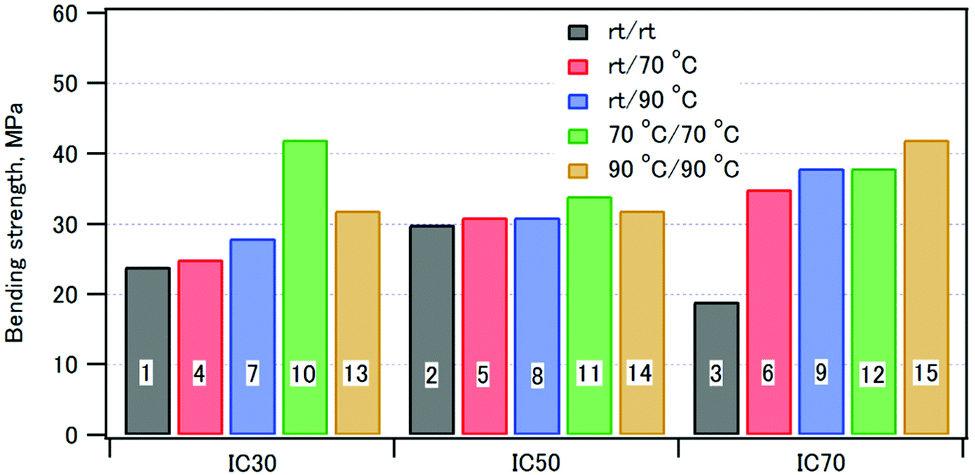 | ||
| Fig. 9 The bending strengths of samples 1–15. | ||
Fig. 10 shows that the elastic moduli of the composites linearly increased with increasing volume fraction of HAP. The rule of mixture for elastic moduli,63Eb2 = EfVf + EmVm, is applicable for these composites. Ef and Em are the elastic moduli of HAP and CPET, respectively, and Vf and Vm are the volume fractions of HAP and CPET, respectively. The least-squares analysis for the plots of samples 10–12 indicates that the elastic modulus of HAP (Ef) was 5.4 GPa and that of CPET (Em) was 1.9 GPa. Similarly, the elastic modulus of HAP (Ef) was 7.4 GPa and that of CPET (Em) was 0.9 GPa for samples 13–15.
 | ||
| Fig. 10 Plot of the elastic moduli (Eb2) against the volume fraction (Vf) of HAP for the compressed samples prepared from the composite powder coprecipitated/aged at various temperatures. Least-squares line fit according to the rule of mixtures are shown. Samples 10–12, coprecipitated and aged at 70 °C, gave Eb2 = 3.4Vf + 1.9, Ef = 5.4 GPa, and Em = 1.9 GPa. Samples 13–15, coprecipitated and aged at 90 °C, gave Eb2 = 6.6Vf + 0.9, Ef = 7.4 GPa, and Em = 0.9 GPa. | ||
If the composite is used as synthetic implant materials, matching of the elastic modulus of the composite with that of bone (4.7–19.3 GPa)64 is important.16 The elastic moduli of the CPET–HAP composite compacts were 1.8–4.2 GPa, smaller than those of bone. Bonfield et al. reported that the composite of high-density polyethylene and HAP followed the rule of mixture, and Ef = 18.9 GPa and Em = 0.8 GPa.16 Trau and coworkers65 reported that the tensile elastic moduli of the polyester–HAP composites ranged from 3.0 to 7.0 GPa, and they increased linearly with increasing HAP weight% in the composites. Both of these composites were prepared by mixing HAP and polyethylene or polyester. Their larger Ef values than that of the CPET–HAP composites indicate that the reinforcement by HAP is very efficient, which can be ascribed to the different protocols of hybridization of the compacts.
The compacts were fragile: the compacts broke in half in the bending test. The fracture surface of sample 14 is shown in Fig. 11(a). The surface is slightly rough, and the roughness increased for the fracture surface of the compacts immersed in water (vide infra).
 | ||
| Fig. 11 SEM images of the fracture surface of sample 14; (a) the compact fragment after the three-point bending test when the compact was not immersed in water, (b) the compact fragment after the three-point bending test when the compact had been immersed in water at room temperature for 24 h prior to the bending test, and (c) the compact fragment after the three-point bending test when the compact had been immersed in water at 37 °C for 1 week prior to the bending test. | ||
Water-resistance of the composite compacts
The compacts were immersed in distilled water either at room temperature for 24 h or at 37 °C for 1 week. The compacts retained their shape after immersion in water. Up to a 5% weight increase was observed, as shown in Fig. 12. There is a trend that Δw/w increased with increasing the inorganic fraction in the composite. As shown in Fig. 7, the compacts with IC 70% had a large pore volume. One of the reasons of the larger weight increase of the compacts with IC 70% is their larger pore volume to absorb water. In contrast, the compacts of phosphorylated tapioca starch–HAP composites coprecipitated at room temperature reported previously44 showed Δw/w of 210% (IC 30%), 132% (IC 50%) and 100% (IC 70%) after immersion in water at room temperature for 24 h. The tapioca starch–HAP compacts became too soft to perform a three-point bending test. Therefore, HAP–CPET composites with the ethylene terephthalate chain displayed much better water-resistance than the HAP–starch composites.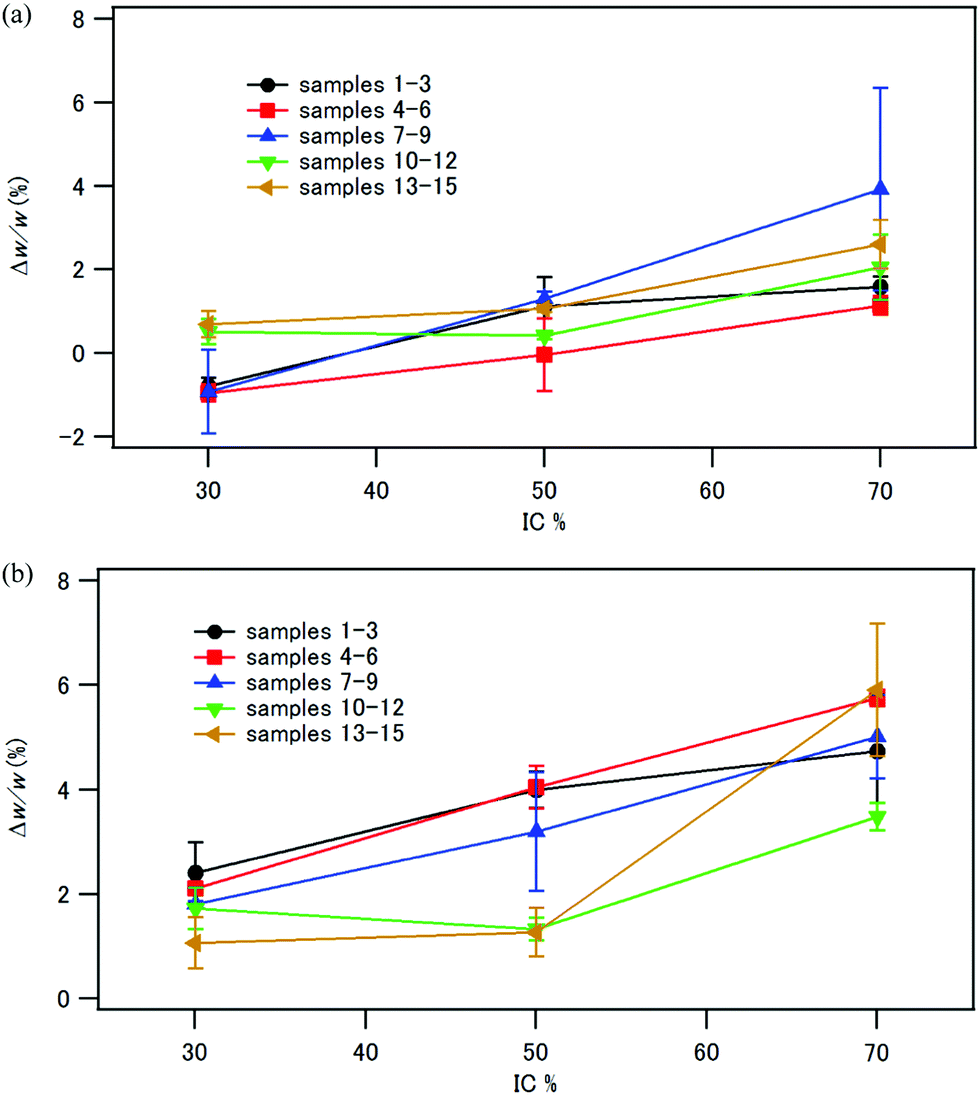 | ||
| Fig. 12 Weight increase Δw/w (%) of samples 1–15 after immersion in water at room temperature for 24 h (a), and at 37 °C for 1 week (b). Error bars (standard error of the means) are shown based on 3–5 independent determinations. | ||
The aqueous phase after immersion of the composite compacts exhibited an absorption peak at 220–270 nm in the UV-visible spectra owing to the terephthalate chromophore (see Fig. S4, ESI†), and the absorbance at 240 nm was used to estimate the concentration of the dissolved CPET in water. The concentrations of CPET in the aqueous phase were very low (Table S1, ESI†). Up to 8.4 mg L−1 of CPET was dissolved in the aqueous phase. The solubilities of CPET–Na+ and CPET–Ca2+ at 37 °C determined by UV-visible spectroscopy were 1.1 g L−1 and 4.0 mg L−1, respectively. Therefore, the concentration of CPET in the aqueous phase is much lower than the solubility of CPET–Na+. In Fig. S5 (ESI†), the amounts of CPET dissolved in water after immersion are shown as the ratios of the weight of dissolved CPET in water to the weight of the CPET in the composite compact. Up to 0.27% CPET was dissolved in water after immersion in water for 1 week. These observations indicated that most of the CPET in the composites was bound to HAP or Ca2+ and not soluble in water. The increase in CPET in the aqueous phase with increasing IC can be attributed to the larger water absorption of the composites with IC 70 wt%. There was little difference in the dissolved amount of CPET among the composites coprecipitated under various conditions.
Fig. 13 illustrates the mechanical properties of the compacts immersed in water relative to those of the pristine compacts (see also Tables S2 and S3, ESI†). Fig. 13 (a)–(d) shows the results after immersion in water at room temperature for 24 h, and Fig. 13 (e)–(h) shows those after immersion in water at 37 °C for 1 week. The compacts with the inorganic weight fraction of 70%, samples 3, 6, 9, and 15, all collapsed quite easily in the bending test. It is worth noting that sample 14, which has an inorganic weight fraction of 50% and was coprecipitated/aged at 90 °C, exhibited a larger bending strength (120%), larger strain at break (160%), and larger fracture energy (180%) after immersion in water at room temperature for 24 h (Fig. 13a, c and d). After immersion in water at 37 °C for 1 week, the strain at break and the fracture energy were four times larger than those of the pristine sample (Fig. 13g and h). The bending strength of sample 14 was 69% of that of the pristine sample (Fig. 13e).
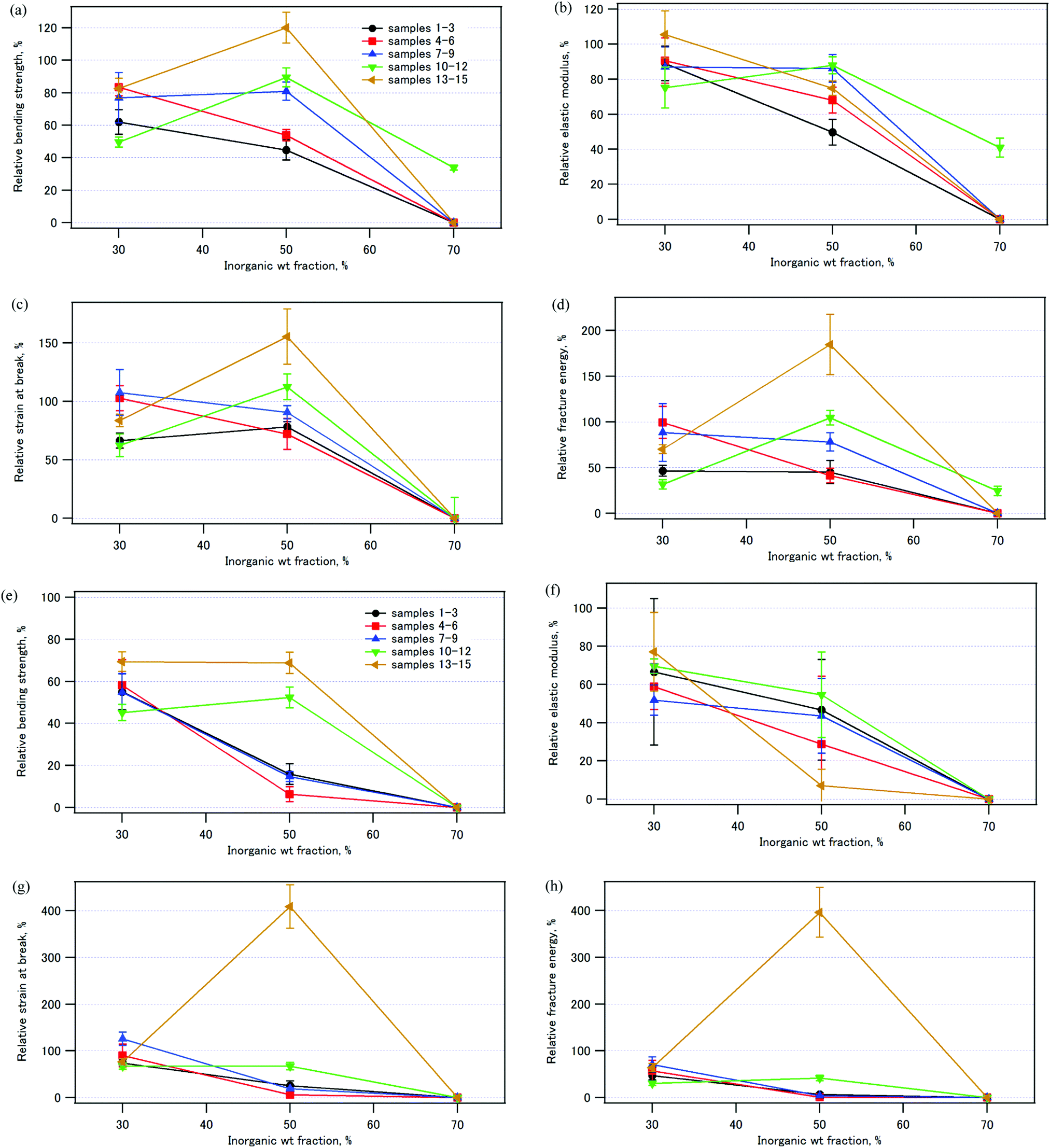 | ||
| Fig. 13 The ratios of the mechanical properties of the samples soaked in water to those of the pristine samples. (a) and (e) Bending strength, (b) and (f) elastic moduli, (c) and (g) strain at break, (d) and (h) fracture energies. (a)–(d) Samples were soaked in water at room temperature for 24 h. (e)–(h) Samples were soaked in water at 37 °C for 1 week. Averages of 3–4 determinations. | ||
Fig. 14 compares the stress–strain curves of sample 14, which has 50% IC and was coprecipitated and aged at 90 °C before and after immersion in water. After immersion in water at 37 °C for 1 week, the compact became softer, and the strain at break was as large as 7–8%. After the three-point bending test, the compact did not break in half. The fracture surface of sample 14 after water immersion is shown in Fig. 11(b and c). The fracture surface of sample 14 after immersion in water seems to be rougher, and the observation suggests that the matrix or the interface was softer by immersion in water. The role of water molecules on the mechanical properties can be attributed to the following two factors: (1) the polymer phase absorbed water to be more flexible, and (2) the interface between HAP and the polymer became a gel-like structure.
 | ||
| Fig. 14 Representative stress–strain curves of sample 14 (IC 50%, coprecipitation and aging at 90 °C). Pristine sample (black), sample soaked at room temperature for 24 h (blue), and sample soaked at 37 °C for 1 week (red). | ||
It should be noted that the effects of water on the mechanical properties of the composites depended on the inorganic weight fraction of the composites. The compacts with IC 50% were water resistant, while those with IC 70% were not. For the composites coprecipitated at 70 °C or 90 °C, the relative bending strength, the strain at break, and the fracture energy decreased in the order: IC 50% > IC 30% ≫ IC 70%. Therefore, there seems to be an optimum organic/inorganic ratio for water resistance of the composites. For the samples with IC 70%, there could be free HAP surface not covered by CPET, and the HAP surface is so hydrophilic that the composites were easily broken after immersion in water. For sample 14 with IC 50%, we speculate that most of the HAP crystal surface was covered with the carboxylate groups of CPET, and hydration of the HAP surface was suppressed. There may be a critical value of IC, IC*, depending on the hydrophilicity of the polymer, and the composites are water-resistant for IC < IC*.
Molecular modeling studies indicated that if CPET is adsorbed on HAP in a face-on fashion, there is one COOH group per 3.3–5.2 nm2. Schmidt-Rohr et al. reported that the density of citrate on the {100} surface of HAP of natural bone is one citrate molecule per 4 nm2.13 Therefore, the density of carboxylate groups of CPET is close to the density of binding sites of carboxylate groups on the {100} plane of HAP. Water-resistance of the composites can be attributed to matching of the density of the carboxylate groups of CPET and that of the binding sites on the surface of HAP crystals.
Conclusions
A water-soluble polymer with the hydrophobic PET main chain substituted with carboxylate groups was coprecipitated with calcium and phosphate ions to obtain the polymer–hydroxyapatite composite powder. The organic–inorganic weight ratios and the crystallite sizes of the hydroxyapatite were easily controlled by the coprecipitation conditions, such as the polymer/inorganic source feed ratios and the reaction temperatures. Composite powders of the polymer–hydroxyapatite in weight ratios of 30/70, 50/50, and 70/30 were prepared. The crystallite sizes of HAP in the composites grew as the coprecipitation temperature was increased, and they were 26–36 nm when the coprecipitation was performed at 70 °C or 90 °C. The composite powder was compressed with hot pressing at 120 MPa and at 120 °C. The compacts showed bending strengths of ∼42 MPa and elastic moduli of ∼4.2 GPa. The elastic moduli of the compacts increased linearly with increasing volume fraction of HAP in the composites. The compressed compacts prepared from the composites coprecipitated at 90 °C showed water-resistance, and the composite with 50 wt% HAP showed 69% of the bending strength of the pristine composite after being soaked in water at 37 °C for 1 week. The strain at break was four times larger than that of the pristine composite. Therefore, the toughness of the compact was increased by immersion in water for 1 week. Given an appropriate organic to inorganic weight ratio, hybridization of a polymer with both carboxylate groups and hydrophobic moieties with HAP is expected to be a promising strategy for the preparation of ecologically friendly materials.Author contributions
Yui Okuda: investigation, methodology, formal analysis, validation, writing – original draft; Ken Hirota: supervision, methodology; Tadashi Mizutani: conceptualization, supervision, writing – original draft, writing – review and editing, funding acquisition; Yusuke Numamoto: supervision, investigation.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research (C) (grant number 17K05982) from the Japan Society for the Promotion of Science.References
- L. B. Gower, Biomimetic Model Systems for Investigating the Amorphous Precursor Pathway and Its Role in Biomineralization, Chem. Rev., 2008, 108, 4551–4627 CrossRef CAS PubMed.
- Y. Suetsugu, Y. Takahashi, F. P. Okamura and J. Tanaka, Structure Analysis of A-Type Carbonate Apatite by a Single-Crystal X-Ray Diffraction Method, J. Solid State Chem., 2000, 155, 292–297 CrossRef CAS.
- T. I. Ivanova, O. V. Frank-Kamenetskaya, A. B. Kol'tsov and V. L. Ugolkov, Crystal Structure of Calcium-Deficient Carbonated Hydroxyapatite. Thermal Decomposition, J. Solid State Chem., 2001, 160, 340–349 CrossRef CAS.
- F. Ren, X. Lu and Y. Leng, Ab initio simulation on the crystal structure and elastic properties of carbonated apatite, J. Mech. Behav. Biomed. Mater., 2013, 26, 59–67 CrossRef CAS.
- L. C. Palmer, C. J. Newcomb, S. R. Kaltz, E. D. Spoerke and S. I. Stupp, Biomimetic Systems for Hydroxyapatite Mineralization Inspired By Bone and Enamel, Chem. Rev., 2008, 108, 4754–4783 CrossRef CAS PubMed.
- E. Duman, E. S. Kehribar, R. E. Ahan, E. Yuca and U. O. S. Seker, Biomineralization of Calcium Phosphate Crystals Controlled by Protein. Protein Interactions, ACS Biomater. Sci. Eng., 2019, 5, 4750–4763 CrossRef CAS PubMed.
- J. Kirkham, S. J. Brookes, R. C. Shore, S. R. Wood, D. A. Smith, J. Zhang, H. Chen and C. Robinson, Physico-chemical properties of crystal surfaces in matrix-mineral interactions during mammalian biomineralisation, Curr. Opin. Colloid Interface Sci., 2002, 7, 124–132 CrossRef CAS.
- A. L. Boskey, Phosphoproteins and Biomineralization, Phosphoproteins and Biomineralization, Phosphorus Sulfur Relat. Elem., 1999, 144, 189–192 CrossRef.
- C. J. S. Ibsen, D. Gebauer and H. Birkedal, Osteopontin Stabilizes Metastable States Prior to Nucleation during Apatite Formation, Chem. Mater., 2016, 28, 8550–8555 CrossRef CAS.
- A. George and A. Veis, Phosphorylated Proteins and Control over Apatite Nucleation, Crystal Growth, and Inhibition, Chem. Rev., 2008, 108, 4670–4693 CrossRef CAS.
- M. L. Zoch, T. L. Clemens and R. C. Riddle, New insights into the biology of osteocalcin, Bone, 2016, 82, 42–49 CrossRef CAS PubMed.
- J. Li, B. A. Baker, X. Mou, N. Ren, J. Qiu, R. I. Boughton and H. Liu, Biopolymer/Calcium Phosphate Scaffolds for Bone Tissue Engineering, Adv. Healthcare Mater., 2014, 3, 469–484 CrossRef CAS.
- Y.-Y. Hu, A. Rawal and K. Schmidt-Rohr, Strongly bound citrate stabilizes the apatite nanocrystals in bone, Proc. Natl. Acad. Sci. U. S. A., 2010, 52, 22425–22429 CrossRef.
- E. R. Wise, S. Maltsev, M. E. Davies, M. J. Duer, C. Jaeger, N. Loveridge, R. C. Murray and D. G. Reid, The Organic-Mineral Interface in Bone Is Predominantly Polysaccharide, Chem. Mater., 2007, 19, 5055–5057 CrossRef CAS.
- M. M. Maestro, J. Turnay, N. Olmo, P. Fernandez, D. Suarez, J. M. G. Paez, S. Urillo, M. A. Lizarbe and E. Jorge-Herrero, Biochemical and mechanical behavior of ostrich pericardium as a new biomaterial, Acta Biomater., 2006, 2, 213–219 CrossRef PubMed.
- W. Bonfield, M. D. Grynpas, A. E. Tully, J. Bowman and J. Abram, Hydroxyapatite reinforced polyethylene – a mechanically compatible implant material for bone replacement, Biomaterials, 1981, 2, 185–186 CrossRef CAS.
- J. L. Moreau, M. D. Weir and H. H. K. Xu, Self-setting collagen-calcium phosphate bone cement: Mechanical and cellular properties, J. Biomed. Mater. Res., Part A, 2009, 91A, 605–613 CrossRef CAS PubMed.
- C. C. P. M. Verheyen, J. R. d. Wijn, C. A. v. Blitterswijk and K. d. Groot, Evaluation of hydroxylapatite/poly(L-lactide) composites: Mechanical behavior, J. Biomed. Mater. Res., 1992, 26, 1277–1296 CrossRef CAS PubMed.
- T. Taguchi, A. Kishida and M. Akashi, Hydroxyapatite Formation on/in Poly(vinyl alcohol) Hydrogen Matrices Using a Novel Alternate Soaking Process, Chem. Lett., 1998, 711–712 CrossRef CAS.
- J. Song, E. Saiz and C. R. Bertozzi, A New Approach to Mineralization of Biocompatible Hydrogel Scaffolds: An Efficient Process toward 3-Dimensional Bonelike Composites, J. Am. Chem. Soc., 2003, 125, 1236–1243 CrossRef CAS PubMed.
- T. Iwatsubo, R. Kishi, T. Miura, T. Ohzono and T. Yamaguchi, Formation of Hydroxyapatite Skeletal Materials from Hydrogel Matrices via Artificial Biomineralization, J. Phys. Chem. B, 2015, 119, 8793–8799 CrossRef CAS PubMed.
- M. Kikuchi, S. Itoh, S. Ichinose, K. Shinomiya and J. Tanaka, Self-organization mechanism in a bone-like hydroxyapatite/collagen nanocomposite synthesized in vitro and its biological reaction in vivo, Biomaterials, 2001, 22, 1705–1711 CrossRef CAS.
- I. Yamaguchi, K. Tokuchi, H. Fukuzaki, Y. Koyama, K. Takakuda, H. Monma and J. Tanaka, Preparation and microstructure analysis of chitosan/hydroxyapatite nanocomposites, J. Biomed. Mater. Res., 2001, 55, 20–27 CrossRef CAS PubMed.
- I. Yamaguchi, S. Iizuka, A. Osaka, H. Monmad and J. Tanaka, The effect of citric acid addition on chitosan/hydroxyapatite composites, Colloids Surf., A, 2003, 214, 111–118 CrossRef CAS.
- Z. Li, L. Yubao, Y. Aiping, P. Xuelin, W. Xuejiang and Z. Xiang, Preparation and in vitro investigation of chitosan/nano-hydroxyapatite composite used as bone substitute materials, J. Mater. Sci.: Mater. Med., 2005, 16, 213–219 CrossRef CAS PubMed.
- B. Li, X. Wang, J. Ma and L. Huang, Preparation of phosphorylated chitosan/chitosan/hydroxyapatite composites by co-precipitation method, Adv. Mater. Res., 2009, 79–82, 401–404 CAS.
- M. S. Sadjadi, M. Meskinfam, B. Sadeghi, H. Jazdarreh and K. Zare, In situ biomimetic synthesis, characterization and in vitro investigation of bone-like nanohydroxyapatite in starch matrix, Mater. Chem. Phys., 2010, 124, 217–222 CrossRef CAS.
- C. Y. Zhang, J. Chen, Z. Zhuang, T. Zhang, X. P. Wang and Q. F. Fang, In Situ Hybridization and Characterization of Fibrous Hydroxyapatite/Chitosan Nanocomposite, J. Appl. Polym. Sci., 2012, 124, 397–402 CrossRef CAS.
- T. Onoki, A. Nakahira, T. Tago, Y. Hasegawa and T. Kuno, Novel low temperature processing techniques for apatite ceramics and chitosan polymer composite bulk materials and its mechanical properties, Appl. Surf. Sci., 2012, 262, 263–266 CrossRef CAS.
- X.-M. Pu, Z.-Z. Sun, Z.-Q. Hou, Y. Yang, Q.-Q. Yao and Q.-Q. Zhang, Fabrication of chitosan/hydroxylapatite composite rods with a layer-by-layer structure for fracture fixation, J. Biomed. Mater. Res., Part B, 2012, 100B, 1179–1189 CrossRef CAS.
- J.-E. Won, A. El-Fiqi, S.-H. Jegal, C.-M. Han, E.-J. Lee, J. C. Knowles and H.-W. Kim, Gelatin-apatite bone mimetic co-precipitates incorporated within biopolymer matrix to improve mechanical and biological properties useful for hard tissue repair, J. Biomater. Appl., 2014, 28, 1213–1225 CrossRef PubMed.
- V. O. Kollath, S. Mullens, J. Luyten, K. Traina and R. Cloots, Protein-calcium phosphate nanocomposites: benchmarking protein loading via physical and chemical modifications against co-precipitation, RSC Adv., 2015, 5, 55625–55632 RSC.
- N. A. Zakharov, L. I. Demina, A. D. Aliev, M. R. Kiselev, V. V. Matveev, M. A. Orlov, T. V. Zakharova and N. T. Kuznetsov, Synthesis and Properties of Calcium Hydroxyapatite/Silk Fibroin Organomineral Composites, Inorg. Mater., 2017, 53, 333–342 CrossRef CAS.
- M. Shakir, I. Zia, A. Rehman and R. Ullah, Fabrication and characterization of nanoengineered biocompatible n-HA/chitosan-tamarind seed polysaccharide: Bio-inspired nanocomposites for bone tissue engineering, Int. J. Biol. Macromol., 2018, 111, 903–916 CrossRef CAS PubMed.
- J. Huang, L. D. Silvio, M. Wang, K. E. Tanner and W. Bonfield, In vitro mechanical and biological assessment of hydroxyapatite-reinforced polyethylene composite, J. Mater. Sci.: Mater. Med., 1997, 8, 775–779 CrossRef CAS PubMed.
- J.-Y. Rho, L. Kuhn-Spearing and P. Zioupos, Mechanical properties and the hierarchical structure of bone, Med. Eng. Phys., 1998, 20, 92–102 CrossRef CAS PubMed.
- R. E. Neuendorf, E. Saiz, A. P. Tomsia and R. O. Ritchie, Adhesion between biodegradable polymers and hydroxyapatite: Relevance to synthetic bone-like materials and tissue engineering scaffolds, Acta Biomater., 2008, 4, 1288–1296 CrossRef CAS PubMed.
- S. I. Stupp and G. W. Ciegler, Organoapatite: Materials for artificial bone. I. Synthesis and microstructure, J. Biomed. Mater. Res., 1992, 26, 169–183 CrossRef CAS.
- M. Kikuchi, J. Tanaka, S. ltoh, S. Lchinose, Y. Koyama, K. Takakuda, K. Nagaoka and S. Tanaka, Preparation of Hydroxyapatite/Collagen Composites Using Biomimetic Process and their Biocompatibility, Mater. Res. Soc. Symp. Proc., 2000, 599, 51–53 CrossRef CAS.
- E. Bertoni, A. Bigi, G. Falini, S. Panzavolta and N. Roveri, Hydroxyapatite/polyacrylic acid nanocrystals, J. Mater. Chem., 1999, 9, 779–782 RSC.
- N. Pramanik, S. K. Biswas and P. Pramanik, Synthesis and Characterization of Hydroxyapatite/Poly(Vinyl Alcohol Phosphate) Nanocomposite Biomaterials, Int. J. Appl. Ceram. Technol., 2008, 5, 20–28 CrossRef CAS.
- Y. Zhou, Q. Liu, Z. Zhao, W. Wang, L. Zheng and Z. Li, Preparation and Characterization of Phosphorylated Collagen and Hydroxyapatite Composite as a Potential Bone Substitute, Chem. Lett., 2013, 42, 83–85 CrossRef CAS.
- A. Kusakabe, K. Hirota and T. Mizutani, Crystallisation of hydroxyapatite in phosphorylated poly(vinyl alcohol) as a synthetic route to tough mechanical hybrid materials, Mater. Sci. Eng., C, 2017, 70, 487–493 CrossRef CAS.
- Y. Okuda, K. Hirota, T. Mizutani and Y. Aoyama, Co-precipitation of tapioca starch and hydroxyapatite. Effects of phosphorylation of starch on mechanical properties of the composites, Results Mater., 2019, 3, 100035 CrossRef.
- R. J. Coleman, K. S. Jack, S. Perrier and L. Grondahl, Hydroxyapatite Mineralization in the Presence of Anionic Polymers, Cryst. Growth Des., 2013, 13, 4252–4259 CrossRef CAS.
- W. Fang, H. Zhang, J. Yin, B. Yang, Y. Zhang, J. Li and F. Yao, Hydroxyapatite Crystal Formation in the Presence of Polysaccharide, Cryst. Growth Des., 2016, 16, 1247–1255 CrossRef CAS.
- P. S. Theocaris, G. C. Papanicolaou and E. A. Kontou, Interrelation between Moisture Absorption, Mechanical Behavior, and Extent of the Boundary Interphase in Particulate Composites, J. Appl. Polym. Sci., 1983, 28, 3145–3153 CrossRef CAS.
- E. Dalas, J. K. Kallitsis and P. G. Koutsoukos, Crystallization of Hydroxyapatite on Polymers, Langmuir, 1991, 7, 1822–1826 CrossRef CAS.
- P. Siriphannon and P. Monvisade, Poly(ethylene terephthalate)/hydroxyapatite biomaterials: Preparation, characterization, and in vitro bioactivity, J. Biomed. Mater. Res., Part A, 2009, 88A, 464–469 CrossRef CAS PubMed.
- S. T. Mosavi-Mirkolaei, S. K. Najafi and M. Tajvidi, Physical and Mechanical Properties of Wood-Plastic Composites Made with Microfibrillar Blends of LDPE, HDPE and PET, Fibers Polym., 2019, 20, 2156–2165 CrossRef CAS.
- Y. Hibino, A. Oyanea, K. Shitomi and H. Miyaji, Technique for simple apatite coating on a dental resin composite with lightcuring through a micro-rough apatite layer, Mater. Sci. Eng., C, 2020, 116, 111146 CrossRef CAS PubMed.
- S. A. P. Sughanthy, M. N. M. Ansari and A. Atiqah, Dynamic mechanical analysis of polyethylene terephthalate/hydroxyapatite biocomposites for tissue engineering applications, J. Mater. Res. Technol., 2020, 9, 2350–2356 CrossRef CAS.
- X. Zhang, S. Zhao, M. G. Mohamed, S.-W. Kuo and Z. Xin, Crystallization behaviors of poly(ethylene terephthalate) (PET) with monosilane isobutyl-polyhedral oligomeric silsesquioxanes (POSS), J. Mater. Sci., 2020, 55, 14642–14655 CrossRef CAS.
- Y. Tian, Y. Yang, J. Tan, D. Ding, Z. Song, Q. Tao, X. Zheng, T. Hu, X. Gong and C. Wu, Ionization.liquid-crystalline polymer synergistically reinforced poly(ethylene terephthalate) due to interfacial compatibilization by ion-dipole interactions, J. Appl. Polym. Sci., 2021, 138, e50127 CrossRef.
- M. Aizawa, T. Matsuura and Z. Zhuang, Syntheses of Single-Crystal Apatite Particles with Preferred Orientation to the a- and c-Axes as Models of Hard Tissue and Their Applications, Biol. Pharm. Bull., 2013, 36, 1654–1661 CrossRef CAS PubMed.
- C. Burger, H.-w. Zhou, H. Wang, I. Sics, B. S. Hsiao, B. Chu, L. Graham and M. J. Glimcher, Lateral Packing of Mineral Crystals in Bone Collagen Fibrils, Biophys. J., 2008, 95, 1985–1992 CrossRef CAS PubMed.
- X. Wang, Y. Li, J. Wei and K. D. Groot, Development of biomimetic nano-hydroxyapatite/poly(hexamethylene adipamide) composites, Biomaterials, 2002, 23, 4787–4791 CrossRef CAS.
- F. Barthelat, Z. Yin and M. J. Buehler, Structure and mechanics of interfaces in biological materials, Nat. Rev. Mater., 2016, 1, 16007 CrossRef CAS.
- C. Rey, K. Besharh, R. Griffin and M. J. Glimcher, Structural Studies of the Mineral Phase of Calcifying Cartilage, J. Bone Miner. Res., 1991, 6, 515–525 CrossRef CAS PubMed.
- D. Magne, P. Pilet, P. Weiss and G. Daculsi, Fourier Transform Infrared Microspectroscopic Investigation of the Maturation of Nonstoichiometric Apatites in Mineralized Tissues: A Horse Dentin Study, Bone, 2001, 29, 547–552 CrossRef CAS.
- A. A. Ivanova, M. A. Surmeneva, R. A. Surmenev and D. Depla, Influence of deposition conditions on the composition, texture and microstructure of RF-magnetron sputter-deposited hydroxyapatite thin films, Thin Solid Films, 2015, 591, 368–374 CrossRef CAS.
- H. Nakamura, Y. Shirakawa, H. Kitamura, T. Yamada, Z. Shidara, T. Yokozuka, P. Nguyen, T. Takahashi and S. Takahashi, Blended polyethylene terephthalate and polyethylene naphthalate polymers for scintillation base substrates, Radiat. Meas., 2013, 59, 172–175 CrossRef CAS.
- D. S. Schnur and D. Lee, Stiffness and inelastic deformation in acrylic-titanium composite implant materials under compression, J. Biomed. Mater. Res., 1983, 17, 973–991 CrossRef CAS PubMed.
- C. I. Albert, J. Jameson and G. Harris, Design and validation of bending test method for characterization of miniature pediatric cortical bone specimens, Proc. Inst. Mech. Eng., Part H, 2013, 227, 105–113 CrossRef PubMed.
- B. Rai, W. Noohom, P. H. Kithva, L. Grondahl and M. Trau, Bionanohydroxyapatite/Poly(3-hydroxybutyrate-co-3-hydroxyvalerate) Composites with Improved Particle Dispersion and Superior Mechanical Properties, Chem. Mater., 2008, 20, 2802–2808 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00512j |
| This journal is © The Royal Society of Chemistry 2021 |