
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanical, thermal, and microstructural analyses of thermoplastic poly(2-methoxyethyl acrylate)-based polyurethane by RAFT and polyaddition†
Shunsuke
Tazawa
a,
Tomoki
Maeda
 *ab and
Atsushi
Hotta
*a
*ab and
Atsushi
Hotta
*a
aDepartment of Mechanical Engineering, Keio University 3-14-1 Hiyoshi, Kohoku-ku, Yokohama 223-8522, Japan. E-mail: hotta@mech.keio.ac.jp
bFrontier Research Center for Applied Atomic Sciences, Ibaraki University 162-1 Shirakata, Tokai-mura, Naka-gun, Ibaraki 319-1106, Japan. E-mail: tomoki.maeda.polymer@vc.ibaraki.ac.jp
First published on 11th January 2021
Abstract
Mechanical and thermal properties of the newly synthesized solid poly(2-methoxyethyl acrylate) (PMEA)-based polyurethane (PU) were studied. The obtained PMEA-based PUs with various molecular weights were thermoplastic, synthesized by reversible addition–fragmentation chain transfer (RAFT) and polyaddition. Dynamic mechanical analysis (DMA) revealed that the storage modulus and the melting temperature of PMEA-based PU significantly increased from 1.5 × 104 Pa to 2.3 × 105 Pa and from 25 °C to 73 °C, respectively, as the molecular weight of PMEA-based PU increased. This enhancement in the properties could be due to the formation of an ordered structure in PMEA-based PU, studied using small-angle and wide angle X-ray scattering (SAXS, WAXS). Glass transition temperature, 5% weight-reduction temperature, and the amount of intermediate water of PMEA-based PU were also analyzed using differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA).
1. Introduction
Poly (2-methoxyethyl acrylate) (PMEA) is frequently used in biomedical coating due to its liquid-like properties. PMEA possessed excellent properties for medical applications, such as cell enrichment,1–4 low protein absorption,5 and antithrombogenicity.6,7 Among the properties, antithrombogenicity is particularly important for the devices that are in direct contact with blood.8–15 The glass transition temperature (Tg) of PMEA is found to be around −30 °C. Therefore, PMEA has been used for coating artificial organs,5,16,17 but it was found to be rather unstable in vivo, because PMEA turns to a viscous liquid at body temperature.To further extend its biomedical applications, various attempts to obtain solid PMEA at physiological temperature have been made for stable and long-lasting coating. Hirata et al. reported its blending with other polymers possessing a higher glass transition temperature (Tg). In their report, a solid PMEA-based material was obtained by mixing poly (methyl methacrylate) (PMMA) with a Tg of ∼110 °C into PMEA. The polymer blend could eventually achieve an antithrombogenic surface.18 Jankova et al. reported copolymerization with other polymers possessing higher Tg. In their report, solid PMEA was synthesized by copolymerizing MEA and 2-[3-(6-methyl-4-oxo-1,4-dihydropyrimidin-2-yl)ureido]ethyl methacrylate.19
Introducing hydrogen bonding into polymeric structures is known to be an efficient way to solidify materials. Polymers with hydrogen-bonding units can be solidified through intermolecular hydrogen bonding, which would make them thermoplastic, as hydrogen bonding is a type of reversible molecular bonding. Previously, we also reported a new type of thermoplastic solid polydimethylsiloxane (PDMS) by introducing L-phenylalanine-based hydrogen-bonding networks.20 In addition to the thermoplastic properties, self-healing properties could also be added to polymers by hydrogen bonding. For example, a self-healing supramolecular complex was reported by Cordier et al.21 A new self-healing copolymer synthesized by grafting amide-containing polyacrylate brushes on polystyrene backbones was also reported by Chen et al.22 A thermoplastic block copolymer containing hydrogen bonding based on N-acryloyl-L-phenylalanine and methyl acrylate was reported by Hendrich et al.23
Our group has been involved in the synthesis of thermoplastic polymers, including PMEA-based polyurethane (PU) by introducing hydrogen bonding via hydroxyl-terminated reversible addition–fragmentation chain transfer (RAFT) reagents and diisocyanate (Scheme 1).24,25 The new thermoplastic PMEA-based PU was found to be highly antithrombogenic. In this study, the mechanical and the thermal properties of PMEA-based PU were investigated by changing the molecular weight of PMEA-based PU. In more detail, the molecular weight of PMEA-based PU was controlled by the ratio of diisocyanate to PMEA with hydroxyl groups in both its ends (HO-PMEA-OH) synthesized by RAFT. Thermomechanical properties were analyzed by dynamic mechanical analysis (DMA). Structural analyses by small-angle and wide-angle X-ray scattering (SAXS and WAXS) were also conducted to study the enhancement of its mechanical properties. For the analysis of the thermal properties including thermal stability, the synthesized PMEA-based PU was analyzed using differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA). Finally, to discuss the excellent antithrombogenicity of PMEA-based PU, the intermediated water, which was effectively interacting with PMEA, was investigated using DSC.
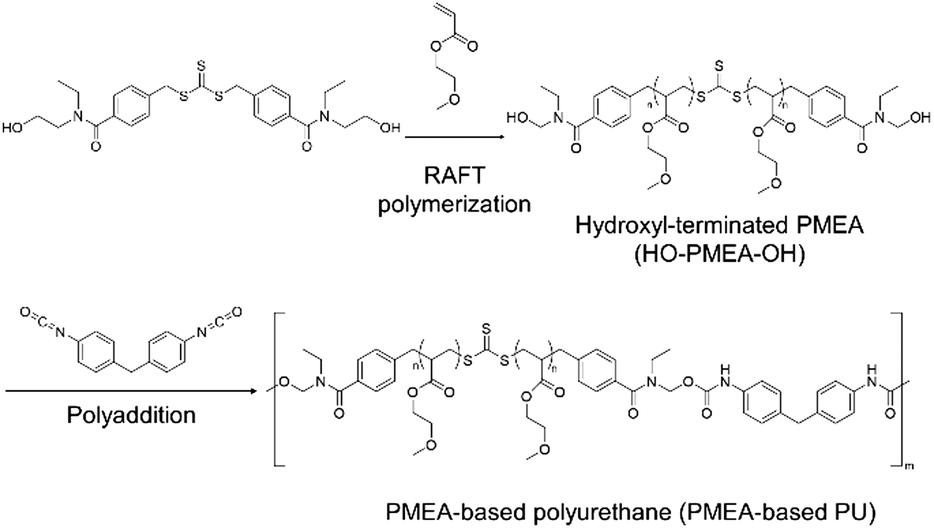 | ||
| Scheme 1 Synthetic procedure of PMEA-based PU. | ||
2. Experimental
2.1. Materials
2-Methoxyethyl acrylate (MEA) (Tokyo Chemical Industry, Tokyo, Japan) as a monomer, 2,2′-azobis[2-methyl-N-(2-hydroxyethyl) propionamide] (Wako Pure Chemical Industry, Tokyo, Japan) as an initiator, bis[4-[ethyl-(2-hydroxyethyl)carbamoyl]benzyl] trithiocarbonate (Wako Pure Chemical Industry, Tokyo, Japan) as a RAFT agent, and N,N-dimethylformamide (DMF, >99.9%) (Sigma-Aldrich, Tokyo, Japan) as a solvent were purchased and used for the polymerization of HO-PMEA-OH. The stabilizer in MEA was removed using an inhibitor remover (Sigma-Aldrich, Tokyo, Japan).4,4′-Methylenebis(phenyl isocyanate) (Tokyo Chemical Industry, Tokyo, Japan) as a polyaddition agent, dibutyltin dilaurate (Tokyo Chemical Industry, Tokyo, Japan) as a catalyst, and toluene (Wako Pure Chemical Industry, Tokyo, Japan), super-dehydrated toluene (Kanto Chemical Co., Inc., Tokyo, Japan), tetrahydrofuran (THF) (Wako Pure Chemical Industry, Tokyo, Japan) and diethyl ether (Wako Pure Chemical Industry, Tokyo, Japan) as solvents were purchased and used for the polymerization of PMEA-based PU.
2.2. Polymerization of HO-PMEA-OH
HO-PMEA-OH was synthesized by RAFT polymerization in the same manner as described in our previous paper.24 In brief, for the synthesis of HO-PMEA-OH with a molecular weight of 8000 (HO-PMEA-OH 8 k), 200 mmol (26 g) of MEA, 80 mL of DMF, and 4.0 mmol (2.08 g) of bis[4-[ethyl-(2-hydroxyethyl) carbamoyl] benzyl] trithiocarbonate were added into a dried reaction flask. Then, the freeze–pump–thaw cycle was repeated three times. After the removal of oxygen, 2.0 mmol (0.58 g) of 2′-azobis [2-methyl-N-(2-hydroxyethyl) propionamide] was added into the flask, and the RAFT polymerization was started at 70 °C under an argon atmosphere for 24 h. Finally, the reaction solution was precipitated in distilled water and the precipitate was dried under vacuum. In this study, in addition to HO-PMEA-OH 8 k, HO-PMEA-OH with a molecular weight of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (HO-PMEA-OH 80 k) was also synthesized.
000 (HO-PMEA-OH 80 k) was also synthesized.
2.3. Polymerization of PMEA-based polyurethane (PU)
PMEA-based PU was synthesized by the polyaddition in the same manner as described in our previous paper.24 In brief, for the synthesis of PMEA-based PU with a molecular weight of 80000 (PMEA-based PU 80 k), HO-PMEA-OH 8 k (3.0 g, 0.38 mmol) dissolved in toluene was poured into a dried reaction flask. After the evaporation of toluene at 50 °C under vacuum to remove water in HO-PMEA-OH, HO-PMEA-OH was again dissolved in 20 mL of super-dehydrated toluene. Then, a 5.0 mL solution of 4,4′-methylenebis(phenyl isocyanate) (0.19 g, 0.76 mmol) in super-dehydrated toluene and dibutyltin dilaurate (0.06 g, 0.10 mmol) were added into the reaction flask, and the reaction was conducted at 50 °C under an argon atmosphere for 48 h. Finally, the reaction solution was precipitated in diethyl ether. In this study, in addition to PMEA-based PU 80 k, PMEA-based PU with a molecular weight of 50000 (PMEA-based PU 50 k) was also synthesized.
Furthermore, for the mechanical, thermal, and structural analyses, solid films were prepared. In detail, PMEA-based PU was dissolved in 5.0 mL of THF, and the solution was poured into a polytetrafluoroethylene mold, before it was dried at room temperature for 40 h, which was followed by the drying process at room temperature under vacuum for 24 h.
2.4. Chemical structure analysis by NMR
The chemical structures of HO-PMEA-OH and PMEA-based PU were analyzed by 1H-NMR using an ECA500 instrument (JEOL Ltd, Tokyo, Japan). DMSO-d6 was used as the solvent for HO-PMEA-OH and PMEA-based PU 50 k, and CDCl3 was used for PMEA-based PU 80 k.![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) – + CH2–Ph), δ 1.32–1.83 (brm; −C2CH–), δ 4.08 (s; –OC2CH2OCH3), δ 3.46 (s; –OCH2C2OCH3), δ 3.23 (s; –OCH2CH2OC3), δ 7.18 (d; H–Ph) and 7.23 (d; H–Ph).26
– + CH2–Ph), δ 1.32–1.83 (brm; –C2CH–), δ 4.08 (s; –OC2CH2OCH3), δ 3.46 (s; –OCH2C2OCH3), δ 3.23 (s; –OCH2CH2OC3), δ 3.77 (s; –Ph–
– + CH2–Ph), δ 1.32–1.83 (brm; −C2CH–), δ 4.08 (s; –OC2CH2OCH3), δ 3.46 (s; –OCH2C2OCH3), δ 3.23 (s; –OCH2CH2OC3), δ 7.18 (d; H–Ph) and 7.23 (d; H–Ph).26
– + CH2–Ph), δ 1.32–1.83 (brm; –C2CH–), δ 4.08 (s; –OC2CH2OCH3), δ 3.46 (s; –OCH2C2OCH3), δ 3.23 (s; –OCH2CH2OC3), δ 3.77 (s; –Ph–![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) 2–Ph–), δ 9.54 (s; –CON–), δ 7.04–7.36 (brm; H–Ph).27,28
– + CH2–Ph), δ 1.42–2.06 (brm; –C2CH–), δ 4.19 (s; –OC2CH2OCH3), δ 3.56 (s; –OCH2C2OCH3), δ 3.35 (s; –OCH2CH2OC3), δ 3.89 (s; –Ph–2–Ph–), δ 6.76–6.90 (s; –CON–), δ 6.90–7.34 (brm; H–Ph).29
2–Ph–), δ 9.54 (s; –CON–), δ 7.04–7.36 (brm; H–Ph).27,28
– + CH2–Ph), δ 1.42–2.06 (brm; –C2CH–), δ 4.19 (s; –OC2CH2OCH3), δ 3.56 (s; –OCH2C2OCH3), δ 3.35 (s; –OCH2CH2OC3), δ 3.89 (s; –Ph–2–Ph–), δ 6.76–6.90 (s; –CON–), δ 6.90–7.34 (brm; H–Ph).29
2.5. Molecular weight analysis by SEC
The molecular weights of HO-PMEA-OH and PMEA-based PU were analyzed by size exclusion chromatography (SEC) using a Prominence HPLC system (Shimadzu Co. Ltd., Tokyo, Japan) with a column of Shodex GPC K804L (Showa Denko K.K., Tokyo, Japan). Chloroform was used as the eluent, and polystyrene (Showa Denko K.K., Tokyo, Japan) was used for the calibration.2.6. Hydrogen-bond unit analysis by FTIR
The formation of hydrogen-bond units as crosslinking points was analyzed by Fourier transform infrared spectroscopy (FTIR) in ATR mode using an ALPHA-E instrument (Bruker Optics K.K., Kanagawa, Japan). The resolution was set at 4 cm−1, and the number of scans was set at 64.2.7. Thermomechanical analysis
The thermomechanical properties of HO-PMEA-OH and PMEA-based PU were analyzed by dynamic mechanical analysis (DMA) using an ARES-G2 instrument (TA Instruments Inc., Tokyo, Japan). A circular plate with a diameter of 8 mm was used, and the frequency was set at 1 Hz. Temperature was changed from −60 °C to 130 °C, and the heating rate was set at 5 °C min−1. The state of the sample was defined as solid, when the storage modulus G′ was higher than the loss modulus G′′. The crossover point of G′ and G′′ was defined as the melting point.2.8. Structural analysis
The structural analysis was conducted using a NANO-Viewer (Rigaku Co., Tokyo, Japan). SAXS and WAXS measurements were conducted by changing the sample–detector distance as 97 mm, 290 mm, and 875 mm. An X-ray (λ = 1.54 Å) passing through three slits (1st slit: 0.2 mm, 2nd slit: 0.1 mm, and 3rd slit: 0.25 mm) was used. The scattered X-ray was detected using a PILATUS 100 K detector and the obtained 2D profiles were circumferentially averaged to get the 1D profiles.2.9. DSC analysis
DSC analysis was conducted using a DSC25 instrument (TA Instruments Inc., Tokyo, Japan). 10–15 mg of PMEA-based PU was used for the analysis. The hydrated PMEA-based PU sample was prepared by dipping the sample in 1 mL of distilled water for 24 h and by removing the distilled water from the surface. As for the measurement sequence, the sample was heated to 40 °C, cooled from 40 °C to −100 °C, and heated again from −100 °C to 50 °C. The cooling and the heating rates were set at 5 °C min−1.The ratio of intermediate water to all water was calculated using eqn (1).
| Ratio of intermediate water (%) = ΔHCrystal/334 Jg−1 × 100 | (1) |
2.10. Thermogravimetric analysis (TGA)
The thermal stability of PMEA-based PU was analyzed by TGA using a DTG-60 instrument (Shimadzu, Tokyo, Japan). The sample (20 mg) in a pan was heated from room temperature to 600 °C under a N2 atmosphere. The heating rate was set at 10 °C min−1, and the flow rate of N2 was set at 100 mL min−1. α-Alumina (Shimadzu, Tokyo, Japan) was used as the reference.3. Results and discussion
3.1. Chemical structures of HO-PMEA-OH and PMEA-based PU
1H-NMR measurements were carried out to confirm the synthesis of HO-PMEA-OH and PMEA-based PU. Fig. 1 shows the typical NMR spectrum of HO-PMEA-OH with a molecular weight of 8000 g mol−1 (HO-PMEA-OH 8 k). It was confirmed that HO-PMEA-OH (the prepolymer for the synthesis of PMEA-based PU) was successfully polymerized via RAFT polymerization. Fig. 2 shows the NMR spectrum of PMEA-based PU with a molecular weight of 47000 g mol−1 (PMEA-based PU 50 k). After the chemical reaction of HO-PMEA-OH with diisocyanate, further peaks due to the reaction appeared in addition to the original peaks observed in the NMR spectrum of HO-PMEA-OH. Therefore, it was confirmed that PMEA-based PU was successfully polymerized via polyaddition.
 | ||
| Fig. 1 NMR spectrum of HO-PMEA-OH 8 k. | ||
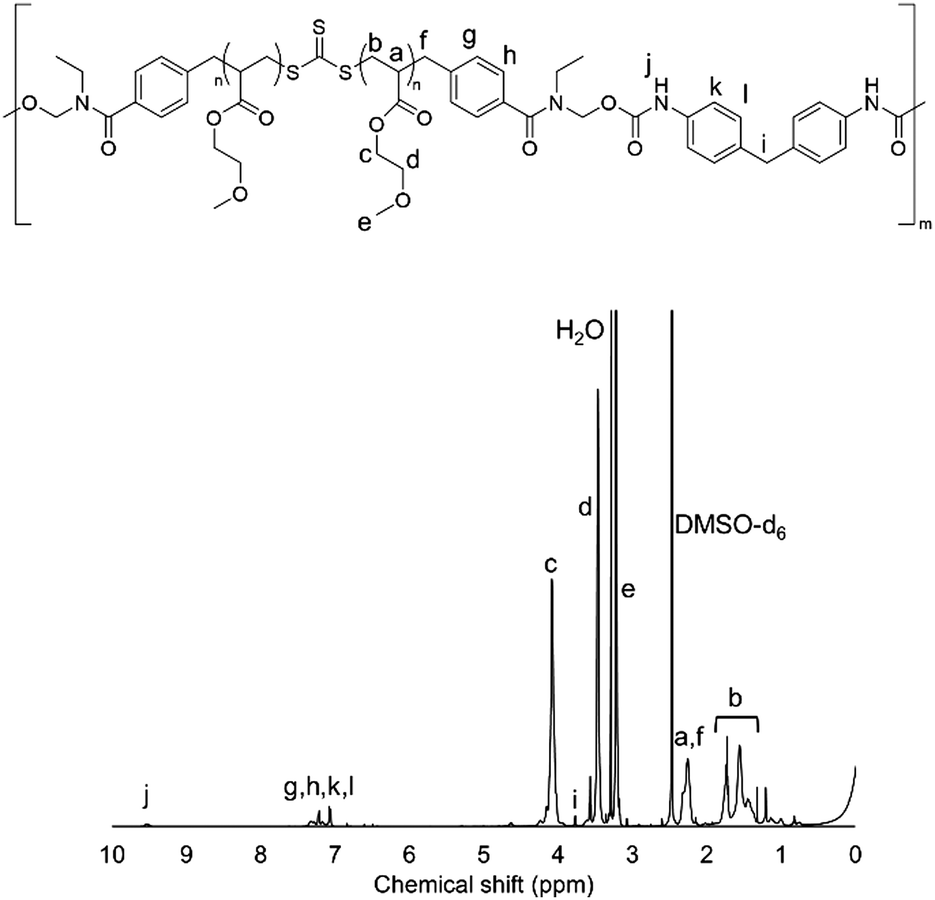 | ||
| Fig. 2 NMR spectrum of PMEA-based PU 50 k. | ||
HO-PMEA-OH and PMEA-based PU were synthesized by changing the molecular weights, which were analyzed through SEC measurements (the SEC curves were presented in Fig. S1, ESI†), and the results are all listed in Table 1.
3.2. Analysis of the chemical structures and hydrogen bonding in PMEA-based PU
To investigate the chemical structures in the synthesized PMEA-based PU, FTIR analysis was conducted. Fig. 3 and 4 both present the FTIR spectra of the synthesized PMEA at different wavenumbers. As shown in Fig. 3, the peaks at 1610 cm−1 and ∼1630 cm−1 were observed in HO-PMEA-OH 8 k in addition to the peaks derived from C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O at 1728 cm−1, and OCH3 and OCH2 at 2800–3000 cm−1 of PMEA.30,31 The peak at 1610 cm−1 derives from the CC ring stretching of the benzene rings of the RAFT agent, and the peak at ∼1630 cm−1 comes from the CO stretching of the amide bonds combined with the benzene rings of the RAFT agent. In a previous study, Tang et al. reported that the peak of the CC ring stretching in aromatic polyamide composed of trimesoyl chloride and 1,3-benzenediamine was found at 1609 cm−1.32 Fukumaru et al. also reported that the peak of the CO stretching was at 1640 cm−1 in the tert-butyldimethylsilyl (TBS)-functionalized poly(p-phenylene benzobisoxazole) (PPBO) precursor.33
O at 1728 cm−1, and OCH3 and OCH2 at 2800–3000 cm−1 of PMEA.30,31 The peak at 1610 cm−1 derives from the CC ring stretching of the benzene rings of the RAFT agent, and the peak at ∼1630 cm−1 comes from the CO stretching of the amide bonds combined with the benzene rings of the RAFT agent. In a previous study, Tang et al. reported that the peak of the CC ring stretching in aromatic polyamide composed of trimesoyl chloride and 1,3-benzenediamine was found at 1609 cm−1.32 Fukumaru et al. also reported that the peak of the CO stretching was at 1640 cm−1 in the tert-butyldimethylsilyl (TBS)-functionalized poly(p-phenylene benzobisoxazole) (PPBO) precursor.33
 | ||
| Fig. 3 FTIR spectra of PMEA-based PU (1450 cm−1–1800 cm−1). | ||
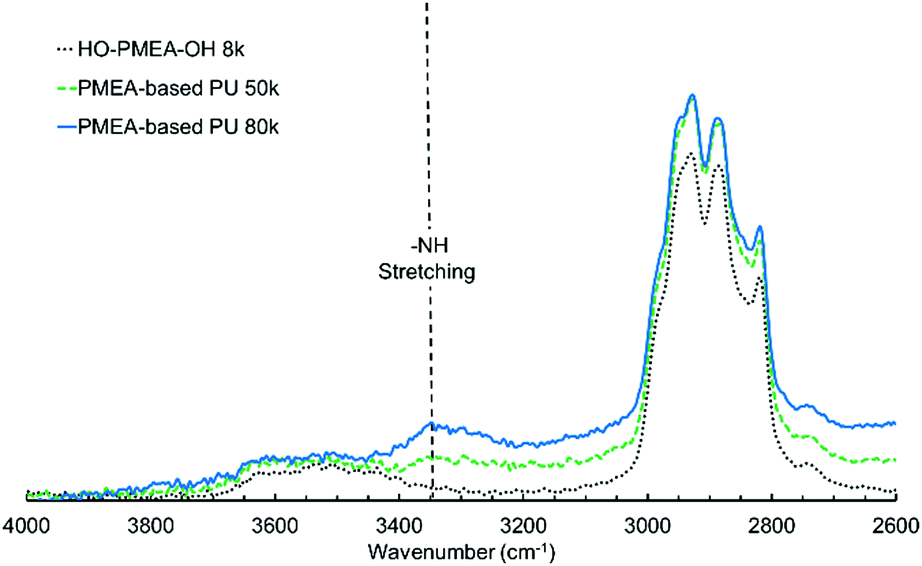 | ||
| Fig. 4 FTIR spectra of PMEA-based PU (2600 cm−1–4000 cm−1). | ||
In addition to the peaks observed in HO-PMEA-OH 8 k, the peaks at 1535 cm−1 and 1599 cm−1 were newly detected after the polyaddition of HO-PMEA-OH, which could only be observed for PMEA-based PU (i.e. PMEA-based PU of 80 k and 50 k). The peaks between 1500 cm−1 and 1560 cm−1 derive from the combination of the N–H in plane bending and the C–N stretching (amide II), and the peak at 1599 cm−1 derives from the CC ring stretching of the benzene rings of diisocyanate. In fact, Xu et al. previously reported that the peak of amide II could be observed between 1500 cm−1 and 1600 cm−1 in a urethane elastomer composed of polycarbonate diols, isophorone diamine, and isophorone diisocyanate.34 Xiang et al. also reported that the peaks of amide II were found between 1480 cm−1 and 1560 cm−1 in polyurethane composed of hydroxyl-terminated butadiene–acrylonitrile copolymer, hexamethylene diisocyanate, and 3,3′-dimethyl-4,4′-diamino dicyclohexyl methane.35 Moreover, Stefanović et al. reported that the peak of the CC ring stretching of diisocyanate (4,4′-methylenebis(phenyl isocyanate)) was detected at 1600 cm−1 in polyurethane synthesized from hydroxyl-terminated poly(propylene oxide)-b-PDMS-b-poly(propylene oxide) using 4,4′-methylenebis(phenyl isocyanate).28 Therefore, it was indicated that the polyaddition using diisocyanate was successfully conducted.
The existence of hydrogen bonding was also confirmed by the FTIR spectra. It was known that the peaks originating from free CO and hydrogen-bonded CO stretching in urethane could be observed at 1719 cm−1 and 1704 cm−1, respectively.36 As presented in Fig. 3, however, the peak between 1700 cm−1 and 1750 cm−1 originating from the MEA monomers of PMEA-based PU 80 k and PMEA-based PU 50 k overlapped with the peak of HO-PMEA-OH 8 k. Since the molar ratio of CO in MEA to that of CO in urethane was approximately 25:1, a significantly large CO peak of the MEA could easily involve the urethane-derived CO peak. As shown in Fig. 4, the broad peak centered at ∼3340 cm−1 derives from the hydrogen-bonded N–H stretching. Jena et al. reported that the peak of the stretching vibration of the amide bonds could be observed at 3340 cm−1 in the hyper-branched polyurethane-urea composed of glycerol, 2,2-bis(methylol)propionic acid, and isophorone diisocyanate.37 Aoki et al. also reported that the peak of the stretching vibration of the amide bonds was detected at ∼3320 cm−1 in polyurethane composed of propanediol and diisocyanate.38 It was, therefore, considered that hydrogen bonding was successfully introduced into PMEA molecules, which should greatly contribute to the solidification of PMEA.
3.3. Thermomechanical properties of PMEA-based PU
DMA was carried out to investigate the thermoplasticity of the synthesized PMEA-based PU. Fig. 5 shows the DMA results of the synthesized PMEA-based PU. As shown in Fig. 5, at the lower temperature region (from −60 °C to ∼−30 °C), G′ of PMEA-based PU was constant. In detail, G′ values of PMEA-based PU 50 k and PMEA-based PU 80 k were 1.9 × 106 Pa and 1.1 × 107 Pa, respectively. After the glass transition temperature, G′ decreased drastically. Tg values of PMEA-based 50 k and PMEA-based PU 80 k obtained from the tanδ results (Fig. S2, ESI†) were −30.7 °C and −24.8 °C, respectively. At around the ambient temperature (from 0 °C to 30 °C), the solid-state region (i.e. the region where the storage modulus G′ was higher than the loss modulus G′′) appeared, and accordingly, the melting point emerged by introducing hydrogen bonding. In detail, G′ values of PMEA-based PU 50 k and PMEA-based PU 80 k were 1.5 × 104 Pa and 2.3 × 105 Pa, respectively, and G′ of PMEA-based PU 80 k was higher than G′′ around 25 °C.
 | ||
| Fig. 5 DMA curves of PMEA-based PU with different molecular weights. | ||
As a reference, the DMA results of HO-PMEA-OH are shown in Fig. S3 (ESI†). HO-PMEA-OH 8 k is the prepolymer for the synthesis of PMEA-based PU and HO-PMEA-OH 80 k is the PMEA of the same molecular weight as PMEA-based PU 80 k but without hydrogen bonding. In detail, as presented in Fig. S3(a) (ESI†), G′ of HO-PMEA-OH 8 k was 19 Pa, which was lower than the G′′ at around 25 °C. As shown in Fig. S3(b) (ESI†), the G′ of HO-PMEA-OH 80 k around 25 °C was found to be 2.5 × 103 Pa, which was lower than the G′′ around 25 °C and was 92 times lower than that of PMEA-based PU 80 k.
In the region after the glass transition, from the results presented in Fig. 5 and Fig. S3 (ESI†), PMEA-based PU 80 k was in the solid state around 25 °C, while the prepolymer of PMEA-based PU (i.e. HO-PMEA-OH 8 k) and the PMEA of the same molecular weight as PMEA-based PU 80 k but without hydrogen bonding (i.e. HO-PMEA-OH 80 k) were in the liquid state around 25 °C. Additionally, G′ of PMEA-based PU increased, as the molecular weight of PMEA-based PU increased, and thus the amount of hydrogen bonding per molecular chain increased. Therefore, it was confirmed that the thermomechanical properties of PMEA-based PU was significantly improved by introducing diisocyanate-based hydrogen bonding. In previous studies, the enhancement of the mechanical properties by introducing hydrogen-bonding was reported, where the importance of the control of the amount of hydrogen bonding was carefully discussed. For example, Hayashi et al. reported that the plateau modulus of their triblock copolymer-based elastomer with acrylamide-based hydrogen bonding was ∼3 times higher than that of the triblock copolymer-based elastomer without hydrogen bonding.39 In addition, Stefanovic et al. also reported that, by increasing the amount of hydrogen bonding, the storage modulus has increased from 2.3 × 108 Pa to 4.9 × 108 Pa at 25 °C for polyurethane synthesized from hydroxyl-terminated poly(propylene oxide)-b-PDMS-b-poly(propylene oxide) using 4,4′-methylenebis(phenyl isocyanate).28
The melting points of PMEA-based PU 50 k and 80 k were also determined to be at 25 °C and 73 °C, respectively, from the crossover points of the G′ and the G′′ curves. It was, therefore, confirmed that the melting temperature was increased by increasing the amount of hydrogen bonding per molecular chain. In the previous study, Stefanovic et al. reported that the melting temperature was increased from 140 °C to 158 °C for their polyurethane synthesized from hydroxyl-terminated poly(propylene oxide)-b-PDMS-b-poly(propylene oxide) using 4,4′-methylenebis(phenyl isocyanate) by increasing the amount of hydrogen bonding.28
In order to discuss the enhancement of the mechanical and the thermal properties of PMEA-based PU by increasing the molecular weight, the structural analyses were performed by SAXS and WAXS. Fig. 6 shows the SAXS and WAXS profiles of PMEA-based PU. As shown in Fig. 6, I (q) of PMEA-based PU 80 k exhibited deviations from the decrease according to the power law of q−4 in the q range below 0.2 Å−1, and a sharp peak was observed at 0.51 Å−1 in addition to the broad peaks of PMEA-based PU 80 k. In contrast, I (q) of PMEA-based PU 50 k decreased according to the power law of q−4 below 0.03 Å−1, and no sharp peak was observed other than the broad peaks of PMEA-based PU 50 k. Therefore, it was confirmed that microphase-separated disorder structures with diffused phase-boundary and crystalline lattice planes (d ∼1.2 nm) were formed, which was observed by WAXS and SAXS, when the molecular weight and the amount of hydrogen bonding increased. In the previous study, Bonart and Muller reported the state of segregation in the urethane elastomers evaluated by SAXS.40 Koberstein also reported the SAXS results of the microphase-separated structures with the diffused phase-boundary in the segmented polyurethane elastomers.41,42 Nozaki et al. also reported that the PU elastomers synthesized with poly(oxytetramethylene) glycol, trans-1,4-bis(isocyanatomethyl)cyclohexane, and 1,4-butanediol exhibited a microphase-separated structure with diffused phase-boundary.43 Moreover, Xiang et al. reported that polyurethanes composed of ethylene glycol, hexamethylene diisocyanate, and hydroxyl-terminated butadiene-acrylonitrile copolymer had a crystalline structure with an interplane distance of 1.33 nm.44
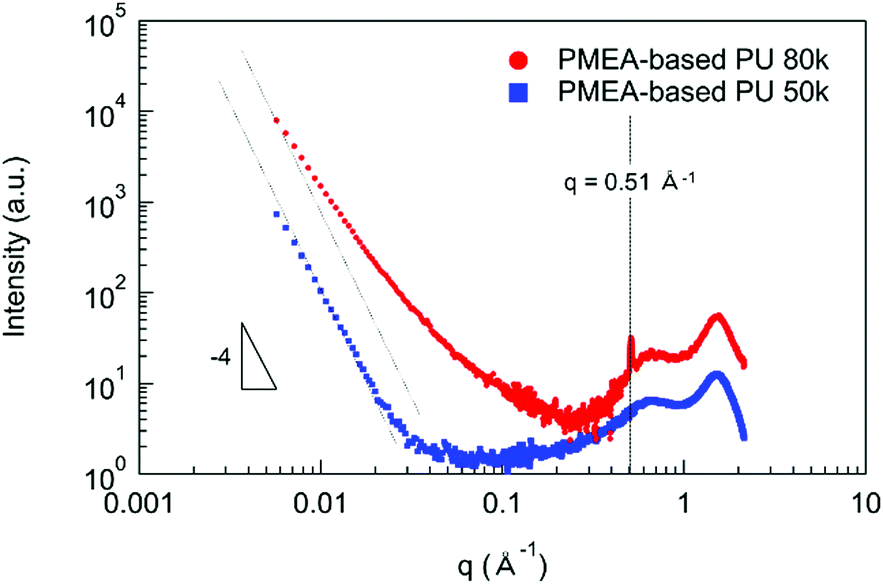 | ||
| Fig. 6 SAXS and WAXS profiles of PMEA-based PU. | ||
3.4. Thermal properties of PMEA-based PU
The thermal properties of PMEA-based PU were evaluated by DSC (Fig. 7). As shown in Fig. 7, the glass transition was clearly observed for all samples and the glass transition temperature Tg of PMEA-based PU was slightly higher than the Tg of HO-PMEA-OH. Moreover, Tg of PMEA-based PU was slightly increased as the molecular weight of PMEA-based PU increased. The thermal properties of the synthesized PMEA are summarized in Table 2. In detail, Tg values of PMEA-based PU 50 k and PMEA-based PU 80 k determined by the DSC measurements were −35.2 °C and −29.3 °C, respectively, while those of HO-PMEA-OH 8 k and HO-PMEA-OH 80 k were –41.5 °C and −38.0 °C, respectively. The increase of Tg could be largely attributed to the formation of hydrogen bonding. In fact, Hayashi et al. also reported that the Tg of poly(4-vinylpyridine)-b-[poly(butyl acrylate)-co-polyacrylamide]-b-poly(4-vinylpyridine) with acrylamide-based hydrogen bonding was 22 °C higher than that of poly(4-vinylpyridine)-b-[poly(butyl acrylate)]-b-poly(4-vinylpyridine) without hydrogen bonding.39 | ||
| Fig. 7 DSC curves of the synthesized PMEAs. | ||
| Sample | T g [°C] | 5% weight-reduction temperature [°C] |
|---|---|---|
| HO-PMEA-OH 8 k | −41.5 | 331 |
| HO-PMEA-OH 80 k | −38.0 | 334 |
| PMEA-based PU 50 k | −35.2 | 276 |
| PMEA-based PU 80 k | −29.3 | 278 |
Furthermore, thermogravimetric analysis (TGA) was conducted to investigate the thermal stability of PMEA-based PU with different molecular weights (Fig. 8). As shown in Fig. 8, the initial degradation of PMEA-based PUs started at a lower temperature than HO-PMEA-OH, and then the drastic degradation happened almost at the same temperature as HO-PMEA-OH. PMEA-based PUs gradually started losing their weight from ∼200 °C before the drastic decrease at ∼350 °C, while HO-PMEA-OH started losing their weight from ∼300 °C. The initial slow degradation of PMEA-based PU around 200 °C could be due to the degradation of the urethane bond, and the drastic degradation around 350 °C after the initial degradation could result from the degradation of the main polymeric chains of PMEA. In addition, as shown in Fig. 8 and Fig. S4 (ESI†), both PMEA-based PU and HO-PMEA-OH with different molecular weights also exhibited similar TGA curves, respectively, indicating that the molecular weight did not largely affect the thermal stability of the synthesized PMEA. In detail, 5% weight-reduction temperatures of PMEA-based PU 50 k and 80 k were 276 °C and 278 °C, respectively, while those of HO-PMEA-OH 8 k and 80 k were 331 °C and 334 °C, respectively (Table 2).
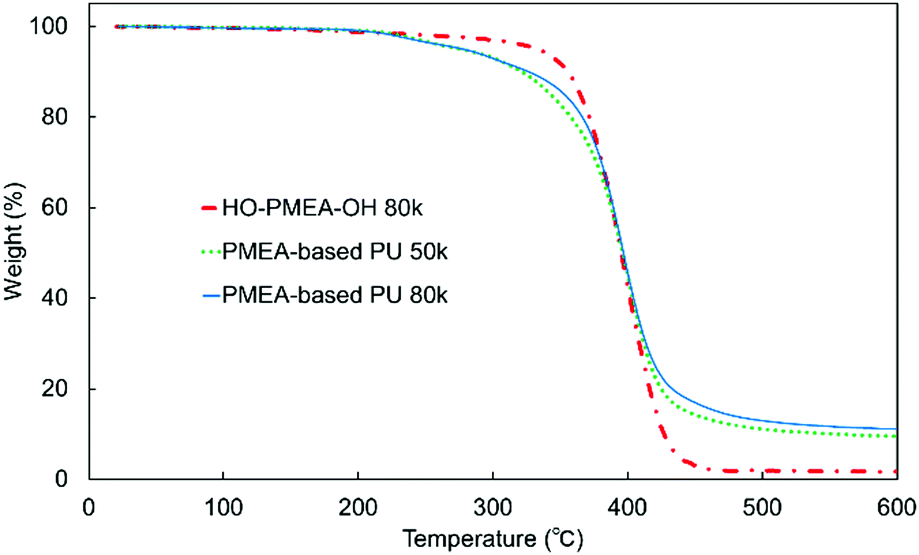 | ||
| Fig. 8 TGA curves of the synthesized PMEAs. | ||
According to previous papers on the degradation of urethane bonds, the bond was found to start degrading around 200 °C.45,46 For example, Gaina et al. reported that the synthesized polyurethane composed of 2-[N,N-bis(2-methyl-2-hydroxyethyl)amino]furfuryl, poly(tetramethylene ether) glycol and 4,4′-dibenzyldiisocyanate degraded from 200 °C due to the breakup of the urethane linkage.46 In contrast, according to previous papers on the degradation of PMEA, PMEA was found to degrade between ∼300 °C and ∼450 °C, a much higher temperature than the urethane bond. For example, Jankova et al. reported that PMEA synthesized by free radical polymerization drastically degraded between ∼300 °C and ∼450 °C.19
3.5. Intermediate water in PMEA-based PU
In this work, the intermediate water in PMEA-based PU was analyzed by DSC (Fig. 9). The intermediate water is known to be composed of water molecules strongly interacting with polymers (PMEA molecular chains). It is known that the intermediate water contributes largely to the enhancement of the blood compatibility of PMEA.47Fig. 9 shows the DSC curves measured for unhydrated and hydrated PMEA-based PU. The hydrated PMEA-based PU showed the crystallization peak of intermediate water at −35 °C and the melting peak of water at ∼0 °C through the DSC measurements, while the original PMEA-based PU (i.e. the unhydrated PMEA-based PU) showed only Tg at −29 °C. In general, such crystallization peak and the melting peak should derive from the crystallization of intermediate water and the melting of frozen water, respectively.48 Actually, in the previous study, it was reported that PMEA and PMEA-based copolymers exhibited the peak of the cold crystallization at ∼−40 °C in DSC curves.49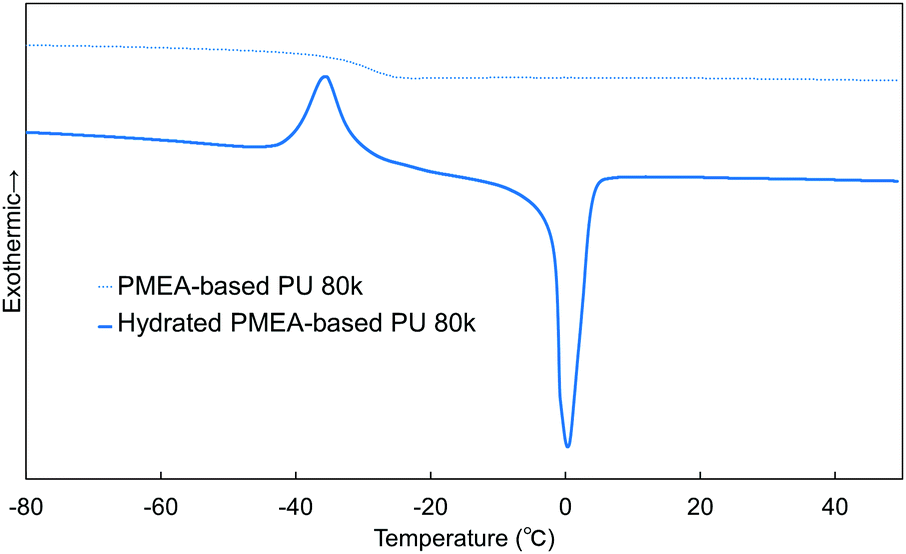 | ||
| Fig. 9 DSC curves of the unhydrated and hydrated PMEA-based PUs. | ||
In addition, the weight ratio of intermediate water to all absorbed water was calculated from the enthalpy of cold crystallization. The ratio of intermediate water in PMEA-based PU 80 k was found to be 3.6 ± 0.4 wt%, high enough to show excellent antithrombogenicity. In the previous study, Kobayashi et al. reported that the threshold of the ratio of the intermediate water for high antithrombogenicity was 1.0 wt% in poly(ω-methoxyalkyl acrylate)s.48 Therefore, it was concluded that the intermediate water was effectively formed in PMEA-based PU even after introducing hydrogen bonding.
4. Conclusions
Mechanical and thermal properties of thermoplastic PMEA-based PU synthesized by RAFT polymerization using hydroxyl-terminated RAFT reagents and polyaddition using diisocyanate were investigated by changing the molecular weight of PMEA-based PU. It was found from DMA analyses that PMEA-based PU 50 k and PMEA-based PU 80 k were in the solid state below 25 °C and 73 °C, respectively, while hydroxyl-terminated PMEA (HO-PMEA-OH 80 k) was in the liquid state after the glass transition region. In contrast, below the glass transition temperature, all PMEAs were in the solid state, where Tg values were detected from −20 °C to −45 °C by DMA and DSC. In addition, the storage modulus G′ was also increased from 1.5 × 104 Pa to 2.3 × 105 Pa by increasing the molecular weight of PMEA-based PU from 50 k to 80 k. The enhancement of the mechanical and thermal properties could be due to the formation of microphase-separated disorder structures with the diffused phase-boundary in PMEA-based PU, revealed by small-angle and wide-angle X-ray scattering (SAXS and WAXS). Furthermore, the Tg of PMEA-based PU was also increased from −35.2 °C to −29.3 °C with the increase in molecular weight, whereas the thermal stability of PMEA-based PU was not much affected by the increase in the molecular weight. Finally, PMEA-based PU was found to possess a sufficient amount of intermediate water compared to the conventional antithrombogenic materials.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported in part by Grant-in-Aid for Scientific Research (A) (No. 19H00831 to A. H.), in part by Grant-in-Aid for Challenging Exploratory Research (No. 19K22067 to A. H.), in part by Grant-in-Aid for Scientific Research (B) (No. 20H02023 to T. M.), and in part by Grant-in-Aid for JSPS Fellows (No. 19J12154 to S. T.) from the Japan Society for the Promotion of Science (JSPS: “KAKENHI”).Notes and references
- T. Hoshiba, E. Nemoto, K. Sato, T. Orui, T. Otaki, A. Yoshihiro and M. Tanaka, PLoS One, 2015, e0136066 CrossRef.
- M. Miyazaki, T. Maeda, K. Hirashima, N. Kurokawa, K. Nagahama and A. Hotta, Polymer, 2017, 115, 246–254 CrossRef CAS.
- Y. Okayama, K. Nakahara, X. Arouette, T. Ninomiya, Y. Matsumoto, Y. Orimo, A. Hotta, M. Omiya and N. Miki, J. Micromech. Microeng., 2010, 095018 CrossRef.
- Y. Oishi, M. Nakaya, E. Matsui and A. Hotta, Composites, Part A, 2015, 73, 72–79 CrossRef CAS.
- A. Melle, A. Balaceanu, M. Kather, Y. Wu, E. Gau, W. Sun, X. Huang, X. Shi, M. Karperien and A. Pich, J. Mater. Chem. B, 2016, 4, 5127–5137 RSC.
- C. Sato, M. Aoki and M. Tanaka, Colloids Surf., B, 2016, 145, 586–596 CrossRef CAS.
- K. Akamatsu, T. Furue, F. Han and S. Nakao, Sep. Purif. Technol., 2013, 102, 157–162 CrossRef CAS.
- T. Maeda, K. Hagiwara, S. Yoshida, T. Hasebe and A. Hotta, J. Appl. Polym. Sci., 2014, 131, 40606 CrossRef.
- K. Bito, T. Hasebe, S. Maegawa, T. Maeda, T. Matsumoto, T. Suzuki and A. Hotta, J. Biomed. Mater. Res., Part A, 2017, 105, 3384–3391 CrossRef CAS.
- C. Xu, A. E. Kuriakose, D. Truong, P. Punnakitikashem, K. T. Nguyen and Y. Hong, J. Mater. Chem. B, 2018, 6, 7288–7297 RSC.
- X. Liu, L. Yuan, D. Li, Z. Tang, Y. Wang, G. Chen, H. Chen and J. L. Brash, J. Mater. Chem. B, 2014, 2, 5718–5738 RSC.
- K. Enomoto, T. Hasebe, R. Asakawa, A. Kamijo, Y. Yoshimoto, T. Suzuki, K. Takahashi and A. Hotta, Diamond Relat. Mater., 2010, 19, 806–813 CrossRef CAS.
- T. Hoshida, D. Tsubone, K. Takada, H. Kodama, T. Hasebe, A. Kamijo, T. Suzuki and A. Hotta, Surf. Coat. Technol., 2007, 202, 1089–1093 CrossRef CAS.
- H. Ezura, K. Ichijo, H. Hasegawa, K. Yamamoto, A. Hotta and T. Suzuki, Vacuum, 2008, 82, 476–481 CrossRef CAS.
- H. Kodama, A. Shirakura, A. Hotta and T. Suzuki, Surf. Coat. Technol., 2006, 201, 913–917 CrossRef CAS.
- K. Sato, S. Kobayashi, M. Kusakari, S. Watahiki, M. Oikawa, T. Hoshiba and M. Tanaka, Macromol. Biosci., 2015, 15, 1296–1303 CrossRef CAS.
- T. Hirata, H. Matsuno, M. Tanaka and K. Tanaka, Phys. Chem. Chem. Phys., 2011, 13, 4928–4934 RSC.
- T. Hirata, H. Matsuno, D. Kawaguchi, N. L. Yamada, M. Tanaka and K. Tanaka, Phys. Chem. Chem. Phys., 2015, 17, 17399–17405 RSC.
- K. Jankova, I. Javakhishvili, S. Kobayashi, R. Koguchi, D. Murakami, T. Sonoda and M. Tanaka, ACS Appl. Bio Mater., 2019, 2, 4154–4161 CrossRef CAS.
- S. Tazawa, A. Shimojima, T. Maeda and A. Hotta, J. Appl. Polym. Sci., 2018, 135, 45419 CrossRef.
- P. Cordier, F. Tournilhac, C. Soulie-Ziakovic and L. Leibler, Nature, 2008, 451, 977–980 CrossRef CAS.
- Y. L. Chen, A. M. Kushner, G. A. Williams and Z. B. Guan, Nat. Chem., 2012, 4, 467–472 CrossRef CAS.
- M. Hendrich, L. Lewerdomski and P. Vana, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 2809–2819 CrossRef CAS.
- S. Tazawa, T. Maeda, M. Nakayama and A. Hotta, Macromol. Rapid Commun., 2020, 2000346 CrossRef CAS.
- R. C. Coffin, S. J. Diamanti, A. Hotta, V. Khanna, E. J. Kramer, G. H. Fredrickson and G. C. Bazan, Chem. Commun., 2007, 3550–3552, 10.1039/b705808j.
- A. Sudo, T. Hamaguchi, N. Aoyagi and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 318–326 CrossRef CAS.
- C. G. Gong and H. W. Gibson, J. Am. Chem. Soc., 1997, 119, 8585–8591 CrossRef CAS.
- I. S. Stefanovic, M. Spirkova, R. Poreba, M. Steinhart, S. Ostojic, V. Tesevic and M. V. Pergal, Ind. Eng. Chem. Res., 2016, 55, 3960–3973 CrossRef CAS.
- X. Liu, B. Yang, Z. Hou, N. Zhang and Y. Gao, Mater. Sci. Eng., C, 2019, 104, 109952 CrossRef CAS.
- S. Ye, S. Morita, G. Li, H. Noda, M. Tanaka, K. Uosaki and M. Osawa, Macromolecules, 2003, 36, 5694–5703 CrossRef CAS.
- S. Morita, M. Tanaka and Y. Ozaki, Langmuir, 2007, 23, 3750–3761 CrossRef CAS.
- C. Y. Y. Tang, Y. N. Kwon and J. O. Leckie, Desalination, 2009, 242, 149–167 CrossRef CAS.
- T. Fukumaru, T. Fujigaya and N. Nakashima, Macromolecules, 2012, 45, 4247–4253 CrossRef CAS.
- Z. Xu, X. Wang and H. Huang, J. Appl. Polym. Sci., 2020, 49575 CrossRef CAS.
- D. Xiang, M. Liu, G. Chen, T. Zhang, L. Liu and Y. Liang, RSC Adv., 2017, 7, 55610–55619 RSC.
- K. Kojio, T. Fukumaru and M. Furukawa, Macromolecules, 2004, 37, 3287–3291 CrossRef CAS.
- K. K. Jena, D. K. Chattopadhyay and K. Raju, Eur. Polym. J., 2007, 43, 1825–1837 CrossRef CAS.
- D. Aoki and H. Ajiro, Macromolecules, 2017, 50, 6529–6538 CrossRef CAS.
- M. Hayashi, S. Matsushima, A. Noro and Y. Matsushita, Macromolecules, 2015, 48, 421–431 CrossRef CAS.
- R. Bonart and E. H. Muller, J. Macromol. Sci., Part B: Phys., 1974, 10, 345–357 CrossRef CAS.
- J. T. Koberstein and R. S. Stein, J. Macromol. Sci., Part B: Phys., 1983, 21, 1439–1472 CAS.
- J. T. Koberstein and R. S. Stein, J. Macromol. Sci., Part B: Phys., 1983, 21, 2181–2200 CAS.
- S. Nozaki, S. Masuda, K. Kamitani, K. Kojio, A. Takahara, G. Kuwarnura, D. Hasegawa, K. Moorthi, K. Mita and S. Yamasaki, Macromolecules, 2017, 50, 1008–1015 CrossRef CAS.
- D. Xiang, J. He, T. Cui, L. Liu, Q. S. Shi, L. C. Ma and Y. Liang, Macromolecules, 2018, 51, 6369–6379 CrossRef CAS.
- C. Carré, L. Bonnet and L. Avérous, RSC Adv., 2014, 4, 54018–54025 RSC.
- C. Gaina, O. Ursache, V. Gaina and C. D. Varganici, eXPRESS Polym. Lett., 2013, 7, 636–650 CrossRef CAS.
- D. Murakami, N. Mawatari, T. Sonoda, A. Kashiwazaki and M. Tanaka, Langmuir, 2019, 35, 2808–2813 CrossRef CAS.
- S. Kobayashi, M. Wakui, Y. Iwata and M. Tanaka, Biomacromolecules, 2017, 18, 4214–4223 CrossRef CAS.
- R. Koguchi, K. Jankova, N. Tanabe, Y. Amino, Y. Hayasaka, D. Kobayashi, T. Miyajima, K. Yamamoto and M. Tanaka, Biomacromolecules, 2019, 20, 2265–2275 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00816h |
| This journal is © The Royal Society of Chemistry 2021 |