
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ vaccine application of inactivated CPMV nanoparticles for cancer immunotherapy†
Paul L.
Chariou
 a,
Veronique
Beiss
b,
Yifeng
Ma
b and
Nicole F.
Steinmetz
*abcdef
a,
Veronique
Beiss
b,
Yifeng
Ma
b and
Nicole F.
Steinmetz
*abcdef
aDepartment of Bioengineering, University of California-San Diego, La Jolla, CA 92039, USA. E-mail: nsteinmetz@ucsd.edu
bDepartment of NanoEngineering, University of California-San Diego, La Jolla, CA 92039, USA
cDepartment of Radiology, University of California-San Diego, La Jolla, CA 92039, USA
dMoores Cancer Center, University of California-San Diego, La Jolla, CA 92039, USA
eCenter for Nano-ImmunoEngineering, University of California-San Diego, La Jolla, CA 92039, USA
fInstitute for Materials Discovery and Design, University of California-San Diego, La Jolla, CA 92039, USA
First published on 3rd February 2021
Abstract
Cowpea mosaic virus (CPMV) is currently in the development pipeline for multiple biomedical applications, including cancer immunotherapy. In particular the application of CPMV as in situ vaccine has shown promise; here the plant viral nanoparticle is used as an adjuvant and is injected directly into a tumor to reverse immunosuppression and prime systemic anti-tumor immunity. Efficacy of this CPMV-based cancer immunotherapy has been demonstrated in multiple tumor mouse models and canine cancer patients. However, while CPMV is non-infectious to mammals, it is infectious to legumes and therefore, from a safety perspective, it is desired to develop non-infectious CPMV formulations. Non-infectious virus-like particles of CPMV devoid of nucleic acids have been produced; nevertheless, efficacy of such empty CPMV nanoparticles does not match efficacy of nucleic acid-laden CPMV. The multivalent capsid activates the innate immune system through pathogen pattern recognition receptors (PRRs) such as toll-like receptors (TLRs); the RNA cargo provides additional signaling through TLR-7/8, which boosts the efficacy of this adjuvant. Therefore, in this study, we set out to develop RNA-laden, but non-infectious CPMV. We report inactivation of CPMV using UV light and chemical inactivation using β-propiolactone (βPL) or formalin. 7.5 J cm−2 UV, 50 mM βPL or 1 mM formalin was determined to be sufficient to inactivate CPMV and prevented plant infection. We compared the immunogenicity of native CPMV and inactivated CPMV formulations in vitro and in vivo using RAW-Blue™ reporter cells and a murine syngeneic, orthotropic melanoma model (using B16F10 cells and C57BL6 mice). While the in vitro assay indicated activation of the RAW-Blue™ reporter cells by formaldehyde and UV-inactivated CPMV at levels comparable to native CPMV; βPL-inactivated CPMV appeared to have diminished activity. Tumor mouse model experiments indicate potent efficacy of the chemically-inactivated CPMV (UV-treated CPMV was not tested) leading to tumor regression and increased survival; efficacy was somewhat reduced when compared to CPMV, however these samples outperformed the empty CPMV nanoparticles. These results will facilitate the translational development of safe and potent CPMV-based cancer immunotherapies.
Introduction
In recent years, plant virus-based nanoparticles have been investigated for vaccine and immunotherapy applications to combat infectious diseases, cancers, and autoimmune diseases.1 Plant viruses are noninfectious to mammals and therefore are safer than their mammalian counterparts used in oncolytic therapies.2 They can be manufactured in a cost-effective manner and in large scales as viral nanoparticles (VNPs) as well as non-replicative virus-like particles (VLPs) devoid of their genomic payload.3,4Cowpea mosaic virus VNPs (CPMV) and VLPs thereof (termed empty CPMV or eCPMV5) have been developed for use as cancer immunotherapy. The native form of CPMV consists of a bipartite, positive-strand ssRNA virus forming a ∼30 nm icosahedron with pseudo T = 3 symmetry. The capsid consists of 60 identical copies of a large (L, 42 kDa) and small (S, 24 kDa) coat protein encapsidating RNA-1 (5.9 kb) and RNA-2 (3.5 kb) in separate but identical CPMV particles. eCPMV nanoparticles have identical protein composition but lack the viral genome; unlike other VLPs, eCPMV does not package host RNA.5,6 When applied as in situ vaccine, the CPMV or eCPMV formulation is administered directly into a tumor to reprogram the tumor microenvironment and launch systemic anti-tumor immunity. In previous work, we elucidated the mechanism of anti-tumor immune activation of the CPMV or eCPMV in situ vaccine.7,8 In brief, CPMV does not kill tumor cells directly, but rather acts on and activates the innate immune cells within the tumor microenvironment (TME). Intratumoral administration of CPMV results in upregulation of interleukin 6 (IL-6) and interferon γ (IFNγ) combined with the downregulation of interleukin 10 (IL-10) and transforming growth factor β (TGFβ); these changes in cytokine levels within the TME promotes the activation and repolarization of neutrophils and tumor associated macrophages (TAM) toward an anti-tumor phenotype.7,8 In addition, the in situ injection of CPMV induces the recruitment of natural killer cells as well as dendritic cells, which in turn convert myeloid cells into potent antigen presenting cells (APCs).7,8 CPMV acts as an adjuvant and the tumor serves as reservoir of antigens; processing of the antigens by the APCs in turn leads to activation of the adaptive immune system launching systemic anti-tumor immunity as evident by activation of effector memory CD4+ and cytotoxic CD8+ T cells to promote a systemic and tumor-specific immune response.7,8
Recent data indicate that CPMV may have a unique potency – as other VNPs such as Cowpea Chlorotic mottle virus (CCMV), Physalis mosaic virus (PhMV), and Sesbania mosaic virus (SeMV), when applied as in situ vaccine in mouse models of melanoma or ovarian cancer, do not match the efficacy observed with CPMV treatment.9 While both CPMV and eCPMV demonstrated potent antitumor response in mouse models7,8,10,11 and canine patients,12 the RNA containing CPMV formulations demonstrated higher efficacy than eCPMV through the activation of additional cytokines and immune cells, which ultimately led to an extended survival rate of tumor-bearing mice.13 The proteinaceous nanoparticle presents danger signals that activate the immune system through pathogen pattern recognition receptors (PRRs), and the presence of the RNA provides an additional danger signal.2 RNA activates TLR7/8, and induces type I interferon (IFN) secretion, which increases the potency of the CPMV-based vaccines.13,14 This phenomenon was also reported using papaya mosaic virus as an in situ vaccine.15
While CPMV is not infectious to mammals, it is infectious to legumes including Vigna unguiculata (i.e. cowpea) plants. From a translational point of view, it is thus important to develop RNA-laden but non-infectious CPMV that are potent but also safe to the environment and plants. CPMV nanoparticles are stable in a variety of environmental conditions. CPMV retains its structural integrity in a wide temperature and pH range (−80 °C to 60 °C, and pH 4.5 to 8.5); particles also withstand the presence of harsh chemicals, such as dimethyl sulfoxide at concentration of up to 50% by volume.16 CPMV particles are not sensitive to some methods of virus inactivation, including peptidase or hypochlorite treatment,17 but showed good response to ultra-violet (UV) light inactivation.18 To elaborate on the previous studies, here we investigated UV treatment vs. chemical treatment of CPMV to render it non-infectious while maintaining its potent efficacy as a cancer immunotherapy. We compared β-propiolactone (βPL) or formalin treatment with the previously reported UV inactivation method. These chemical treatment modalities are commonly used to produce non-virulent vaccines such as polio, hepatitis A, enterovirus 71, and influenza viruses vaccines.19,20 Of particular interest, these methods do not remove the RNA from the VNP, but rather create RNA damage preventing its translation and replication. UV light promotes dimerization of adjacent uracils as well as RNA–protein crosslinks.18 βPL promotes the alkylation or acylation of cytidine, deoxyadenosine, and deoxyguanosine. βPL treatment also leads to protein modifications; for example poliovirus proteins are more extensively modified by βPL than nucleic acid during viral inactivation.19 Specifically, βPL acylates and alkylates to a great extent cysteine, histidine, and methionine, and to a lesser extent aspartic acid, glutamic acid, lysine, serine, threonine, and tyrosine.19 Lastly, formalin causes RNA–protein and protein–protein covalent crosslinking.20 UV- or chemically-inactivated CPMV were produced; infectivity assays were performed using Vigna unguiculata. We then selected candidate formulations and compared the immunogenicity and anti-tumor efficacy of inactivated vs. native CPMV in RAW-Blue™ reporter cells and a dermal melanoma mouse model (using B16F10 cells and C57BL6 mice).
Materials and methods
Preparation of native CPMV and eCPMV
Native CPMV was propagated in V. unguiculata (Burpee black-eyed pea no. 5) plants by mechanical inoculation; propagation and purification was as previously described.21 eCPMV was produced in Nicotiana benthamiana plants by agroinfiltration; propagation and purification was as previously described.5CPMV inactivation using UV light, β-propiolactone, and formalin
CPMV (1 mg mL−1) inactivation was conducted in 0.1 M potassium phosphate (KP) buffer (pH 7.0). Native CPMV was exposed to UV light at a wavelength of 254 nm (doses: 0.1, 0.2, 0.25, 0.5, 1, 5, 7.5, 10, and 15 J cm−2) using a UVP crosslinker (Analytik Jena AG). UV-inactivation was also performed using a UV light dose of 7.5 J cm−2 while changing the CPMV concentration (CPMV was diluted to 1, 2, 5 and 10 mg mL−1 in a total volume of 1 mL). Similarly, we induced the inactivation of native CPMV with β-propiolactone (βPL, Sigma-Aldrich) (doses: 1, 10, 50, 100, and 250 mM) for 24 h at 4 °C, followed by heat-inactivation of βPL for 2 h at 37 °C. CPMV was also exposed to formalin (Electron Microscopy Sciences) (doses: 0.1, 0.25, 0.5, 1, 10, 100, 250 mM) for 5 days at 37 °C. Chemically treated CPMV was centrifuged at 112![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 1 h to remove excess βPL or formalin.
000g for 1 h to remove excess βPL or formalin.
Black-eyed pea plants inoculation with native and inactivated CPMV
V. unguiculata (Burpee black-eyed pea no. 5 plants) were seeded in 3–1/4′′ square pots (Greenhouse Megastore) using Pro Mix BX soil (Greenhouse Megastore) and maintained in Conviron A1000 Reach-in growth chambers (Conviron) at 16/8 hour day/night cycles at 25 °C/22 °C and 60% relative humidity. When the plants were 10 days old, the primary leaves were mechanically inoculated with 40 μL (0.1 mg mL−1) of native or inactivated CPMV (see above); the leaves were lightly dusted with carborundum (Thermo Fisher Scientific) to promote mechanical lesions on the leaves enabling the virus infection. 10 plants were used per treatment arm; in addition, 10 plants were not treated. Leaves were imaged and harvested 10 days post-inoculation and stored at −80 °C until further processing.Viral RNA extraction
Inoculated leaves were exposed to liquid nitrogen for 30 s to 1 min in a mortar and subsequently pulverized into a thin powder using a pestle. The resulting leaf powder was suspended into 1 mL of UltraPure DNase/RNase free distilled water (Thermo Fisher Scientific) per gram of leaf material. The mixture was vortexed for 1 min and subsequently centrifuged at 13000g for 10 min. 500 μL of the supernatant was isolated and denatured with 1/4 vol. of 10% (w/v) SDS under heating for 10 min at 60 °C. Samples were treated with 2 volumes of UltraPure phenol:chloroform:isoamylalcohol (PCI 25:24:1, v/v/v) (Thermo Fisher Scientific), vortexed for 1 min, and centrifuged at 13000g for 10 min. The upper phase, which contains the RNA, was collected into a fresh tube and the PCI extraction was repeated an additional two times. 2 volumes of 100% (v/v) ethanol were added to the final RNA extract prior to purification using the Quick-RNA™ Miniprep kit (Zymo). The purified RNA product was suspended in 30 μL of UltraPure DNase/RNase free distilled water and stored at −20 °C until further analysis. The concentration was determined by UV-visible spectroscopy at 260 nm using the extinction coefficient for single-stranded RNA: 25 ng mL−1 cm−1.
Reverse transcription polymerase chain reaction (RT-PCR)
We used the SuperScript IV One-Step RT-PCR System kit (Thermo Fisher Scientific) according to the manufacturer's specifications. The RNA extracted from native and inactivated CPMV was reverse-transcribed and amplified into cDNA using the following reaction mixture: 2.5 μL forward primer (5′-GGTTCCCGCTTGCTTGGAGC-3′, 10 μM), 2.5 μL reverse primer (5′-GGAGGATTATAAATGTGCG-3′, 10 μM), 25 μL 2X Platinum SuperFi RT-PCR Master Mix, 0.5 μL SuperScript IV RT Mix, and 1 μg total RNA supplemented with nuclease-free water for a final volume of 50 μL. The RT-PCR conditions are summarized in Table 1.| Step | Temp. (°C) | Time (min:s) |
No. cycle |
|---|---|---|---|
| Reverse transcription | 50 | 10:00 |
1 |
| RT inactivation | 98 | 2:00 |
1 |
| Amplification | 98 | 0:10 |
30 |
| 50 | 0:10 |
||
| 72 | 0:30 |
||
| Final extension | 72 | 5:00 |
1 |
| Storage | 4 | ∞ | 1 |
Bioconjugation of cyanine 05 (Cy5) to CPMV
CPMV was labelled with sulfo-Cy5-NHS (Lumiprobe) targeting its solvent-exposed surface lysine residues (CPMV displays a total of 300 solvent-exposed surface lysine side chains per particle22). The reaction was carried out using 5 equivalents of sulfo-Cy5-NHS per coat protein in 0.1 M KP buffer (pH 7.0) at room temperature overnight, with agitation. The reaction mixture was then centrifuged at 112000g for 1 h to remove excess dyes, and resuspended in 0.1 M KP buffer (pH 7.0) overnight. Further purification to remove potential aggregates involved centrifugation at 13000g for 10 min. CPMV–Cy5 was eluted using PD Minitrap G-25 desalting columns (GE Healthcare) to remove excess free Cy5 dyes.
UV/vis spectroscopy
The UV/vis spectra of native, inactivated, and Cy5-labeled CPMV (CPMVCy5) were obtained using a NanoDrop Spectrophotometer (Thermo Fisher Scientific). Cy5 labelling was quantified based on the dye-to-CPMV ratio and the Beer–Lambert law. CPMV: ε(260 nm) = 8.1 mL mg−1 cm−1, molecular weight of CPMV = 5.6 × 106 g mol−1. Cy5: ε(647 nm) = 271000 M−1 cm−1, molecular weight of Cy5 = 777 g mol−1.
Dynamic light scattering (DLS)
A Zetasizer Nano ZSP/Zen5600 instrument (Malvern Panalytical) was operated to measure the hydrodynamic radii of native and inactivated CPMV (1 mg mL−1) nanoparticles. The particle diameter was calculated as the weighted mean of the intensity distribution.Transmission electron microscopy (TEM)
Formvar copper grids coated with carbon film (Electron Microscopy Sciences) were rendered more hydrophilic using the PELCO easiGlow operating system. Drops of CPMV (10 μL, 0.5 mg mL−1) were deposited onto the grids for 2 min at room temperature. The grids were then washed twice with deionized water for 30 s and subsequently stained twice with 2% (w/v) uranyl acetate for another 45 s. A Tecnai transmission electron microscope was used to capture images of the samples at 80 kV.Size exclusion chromatography (SEC)
200 μL of native and inactivated CPMV samples were eluted through a Superose 6 increase column using the ÄKTA Explorer chromatography system (GE Healthcare). The flow rate was set to 0.5 mL min−1 in 0.1 M KP buffer (pH 7.0) and the absorbance at 260 nm and 280 nm was recorded.Agarose gel electrophoresis
Agarose gels were run in 1× Tris–acetate–ETDA (TAE) running buffer in the presence of Nucleic Acid Gel Stain (GoldBio) diluted 1:20 000 (v/v). Native and inactivated CPMV formulations were analyzed using 3 μg of sample on 1.2% (w/v) agarose gels for 45 min at 110 V. Similarly, 1 μg of RNA extracted from native or inactivated CPMV was analyzed on 1.2% (w/v) agarose gels for 35 min at 110 V in the presence of a 1 kbp Plus DNA ladder (Invitrogen). Finally DNA amplicons were analyzed using 2.5 μL of RT-PCR product on 1.8% (w/v) agarose gels for 45 min at 110 V in the presence of a 100 bp Plus DNA ladder (Invitrogen). Gels were imaged before and after staining with Coomassie Brilliant Blue G-250 (0.25% w/v) using the FluorChem R imaging system under UV light or white light.
Denaturing gel electrophoresis
Native and inactivated CPMV (12 μg) was supplemented with 4× LDS loading dye (Thermo Fisher Scientific) and denatured at 100 °C for 5 min. The L and S subunits were separated on 12% SDS-PAGE precast gels in 1× morpholinepropanesulfonic acid (MOPS) buffer (Thermo Fisher Scientific) for 40 min at 200 V and 120 mA in the presence of SeeBlue Plus2 ladder size markers (Thermo Fisher Scientific). Gels were stained with GelRed, (Biotium) and Coomassie Brilliant Blue G-250 (0.25% w/v) and subsequently imaged with the FluorChem R imaging system under UV light, white light, and MultiFluor red light using a 632 nm LED.Virus overlay binding protein assay (VOBPA)
Vimentin was diluted at a concentration of 1 mg mL−1 in 4 mM HCl and incubated at 100 °C for 10 min in the presence of 4× LDS loading dye. 5 μg of vimentin were loaded in each well of a 4–12% SDS-PAGE gel for 40 min at 200 V and 120 mA in the presence of SeeBlue Plus2 ladder size markers. The gel was then transferred to a membrane at 160 mA for 1 h. The resulted blotted vimentin was denatured and gently rocked for 10 min twice at room temperature using denaturing buffer (6 M guanidine–HCl, 2 mM EDTA, 50 mM DTT, 50 mM Tris/HCl, final pH of 8.3). Then, the membrane was moved to 4 °C and gently rocked for 10 min in 20 mL of renaturing buffer (10 mM Tris/HCl, 150 mM NaCl, 2 mM EDTA, 2 mM DTT, 0.1% Triton X-100, final pH 7.3) supplemented with 7.64 g of guanidine–HCl (4 M). After 10 min, 6.5 mL of renaturing buffer was added, followed by another 13.5 mL, and 40 mL in intervals of 10 min, gradually diluting the guanidine–HCl to 1 M. All solution was removed and another 20 mL of fresh renaturing buffer was added for 10 min (0 M guanidine–HCl). The membrane was then moved to room temperature and gently rocked overnight in 5% (w/v) milk solution in renaturing buffer. 100 μg of CPMV, UV-CPMV, βPL-CPMV, Form-CPMV were incubated individually with the membrane in renaturing buffer supplemented with 1% (w/v) milk and 5% (v/v) glycerol. Membranes were washed 4 times for 5 min in TBST. A 1:5000 dilution of primary rabbit anti-CPMV antibody were added in 5% (w/v) milk/TBST and gently rocked for 1 h. Membranes were once again washed 4 times for 5 min in TBST. A 1:5000 dilution of secondary HRP-labeled goat anti-rabbit antibody was added in 5% (w/v) milk/TBST for 1 h. Membranes were washed 4 times for 5 min in TBST. The DAB/Ni Peroxidase Substrate Kit (Vector Biolabs) was used to stain the membranes prior to imaging with the FluorChem R imaging system under white light.
Cell culture
B16F10 (ATCC CRL-6475, mouse skin melanoma) were cultured in Dulbecco's modified Eagle medium (DMEM, ATCC) with L-glutamine supplemented with 10% (v/v) fetal bovine serum (FBS) and 1% (v/v) penicillin–streptomycin. RAW-Blue™ cells (Invivogen) were maintained according to manufacturer's recommendation in DMEM with L-glutamine supplemented with 10% (v/v) heat inactivated FBS, 1% (v/v) penicillin–streptomycin, 100 μg mL−1 Normocin, 200 μg mL−1 Zeocin. Cells were grown at 37 °C and 5% CO2.Flow cytometry
RAW-Blue™ cells were grown to 70% confluency, washed twice with PBS, and collected with a cell scraper. Cells were pelleted at 500 g for 5 min and resuspended in 1 mL of test medium (DMEM with L-glutamine supplemented with 10% (v/v) heat inactivated FBS and 1% (v/v) penicillin–streptomycin). A Countess II FL Automated Cell Counter (Thermo Scientific) was used to prepare a 1500000 cells mL−1 stock solution in test medium. Cells (300000 cells/200 μL media per well) were seeded on 96-well v-bottom plates, and triplicates of CPMVCy5, UV-CPMVCy5, βPL-CPMVCy5, Form-CPMVCy5 were added at a concentration 100000 particles per cell and incubated for 1 h and 8 h at 37 °C and 5% CO2. Cells were then washed twice in FACS buffer (0.1 mL 0.5 M EDTA, 0.5 mL FBS, and 1.25 mL 1 M HEPES pH 7.0 in Ca2+ and Mg2+ free PBS (50 mL total volume)) and fixed with 2% (v/v) paraformaldehyde in FACS buffer for 10 min at room temperature and subsequently washed twice more. Samples were analyzed using a BD Accuri™ C6 (BD Biosciences) and processed with the FlowJo software (https://www.flowjo.com/).
RAW-Blue™ cell activation assay
The assay was performed as per manufacturer's recommendation. RAW-Blue™ cells were grown to 70% confluency, washed twice with PBS, and collected with a cell scraper. Cells were pelleted at 500 g for 5 min and resuspended in 1 mL of test medium (DMEM with L-glutamine supplemented with 10% (v/v) heat inactivated FBS and 1% (v/v) penicillin–streptomycin). A Countess II FL Automated Cell Counter (Thermo Scientific) was used to prepare a 550000 cells per mL stock solution in test medium. Cells (100000 cells/200 μL media per well) were seeded on 96-well flat-bottom plates, and triplicates of CPMV, UV-CPMV, βPL-CPMV, Form-CPMV were added at a concentration of 5 μg per well and incubated for 18 h at 37 °C and 5% CO2. Triplicate negative controls of cells only and media only, as well as a triplicate control of 1× lipopolysaccharide (LPS, Thermo Fisher Scientific) were also tested. Subsequently, 20 μL of media per well was incubated with 180 μL of QUANTI-Blue solution (Invivogen; serves as substrate for secreted embryonic alkaline phosphatase, SEAP, which serves as the reporter gene in RAW-Blue™ cells) for 2 h at 37 °C and 5% CO2. The SEAP level was quantified using an Infinite M200 plate reader (Tecan) at 655 nm. Statistical significance was quantified using a 1 way ANOVA test; P value < 0.0001 = ****, P value < 0.001 = ***, P value < 0.01 = **, P value < 0.05 = *, ns = non-significant data.
Tumor mouse model and in situ vaccination
Female C57BL/6J mice were purchased from The Jackson Laboratory. Mice used in experiments were 8 week old and mouse studies were approved by the Institutional Animal Care and Use Committee of the University of California, San Diego. The murine B16F10 melanoma cell line (ATCC) was implanted (200000 cells in 30 μL) by intradermal injection in the mice's right flank. Tumors were apparent by day 6 and animals; animals that did not bear tumors by day 6 were excluded from the study. 10 μL PBS (n = 9), 100 μg of CPMV (n = 10), βPL-CPMV (n = 10), or Form-CPMV (n = 10) in 10 μL of 0.1 M KP buffer (pH 7.0) were injected into the base of the tumors on days 7, 12, and 17, and 22. The mice were monitored every second or third day for signs of tumor progression (weight, tumor volume, and signs of visual pain). Tumor volume was calculated based on the following equation: tumor volume = (tumor length2 × tumor width)/2. Animals were sacrificed when their tumor reached 1000 mm3, their weight decreased by 15%, or when they showed visible signs of pain. Surviving animals were monitored until day 60. On day 80, surviving animals were re-challenged with B16F10 (200000 cells in 30 μL), and monitored for an additional 40 days. No animals enrolled in the experiments were excluded during analysis. Statistical significance was quantified using a one-way ANOVA test; P value < 0.0001 = ****, P value < 0.001 = ***, P value < 0.01 = **, P value < 0.05 = *, ns = non-significant data.
Results and discussion
Nanoparticle characterization
We assessed the effect of UV light, βPL, and formalin treatments on CPMV's structural integrity and genome stability (Scheme 1). The potential sites of UV light mediated dimerization of adjacent uracils are highlighted in Fig. S1 (ESI†); RNA-1 and RNA-2 of CPMV contain a total of 2905 uracil nucleotides, including more than 700 sites of adjacent uracils. Amino acid prone to βPL mediated alkylation or acylation or formalin crosslinking have also been highlighted on the interior and exterior surface of CPMV in Fig. S2 (ESI†). Native and treated CPMV particles were characterized by a combination of dynamic light scattering (DLS), transmission electron microscopy (TEM), and size exclusion chromatography (SEC) to assess their physical state (Fig. 1 and Fig. S3–S7, ESI†). Data were consistent and indicated that for most conditions tested, CPMV particles exposed to UV, βPL or formalin remained structurally sound; 30 nm-sized particles were imaged in TEM and measured by DLS; also, SEC elution profiles were in agreement showing the typical elution profiles (at ∼12 mL from the Superose6 10/300 column) with a 260:280 nm ratio of 1.7 indicative of intact CPMV particles.
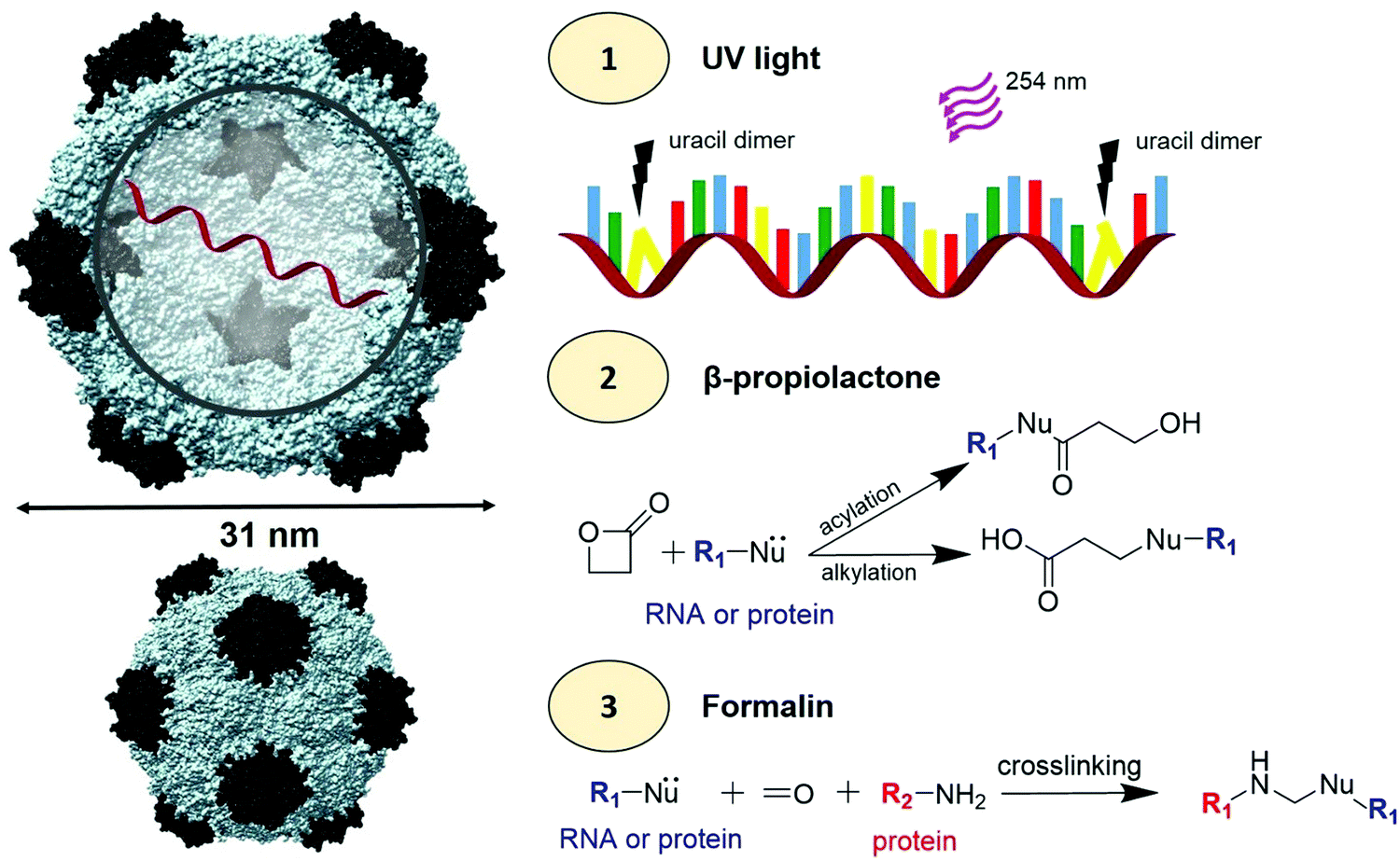 | ||
| Scheme 1 CPMV viral inactivation. Left: Structure of CPMV using the UCSF Chimera software (PDB ID: 1NY7); the small coat protein is shown in black and the large coat protein is shown in grey. RNA-1 and RNA-2 are encapsidated in different but identical CPMV particles (shown is one RNA strand). To the right, (1) inactivation of RNA using a 254 nm wavelength UV light to promote uracil dimers. In the RNA schematic, blue = adenine, green = cytosine, yellow = uracil, and red = guanine (2) βPL induced acylation and alkylation of RNA and proteins. (3) Formalin induced cross-linking of RNA and proteins. | ||
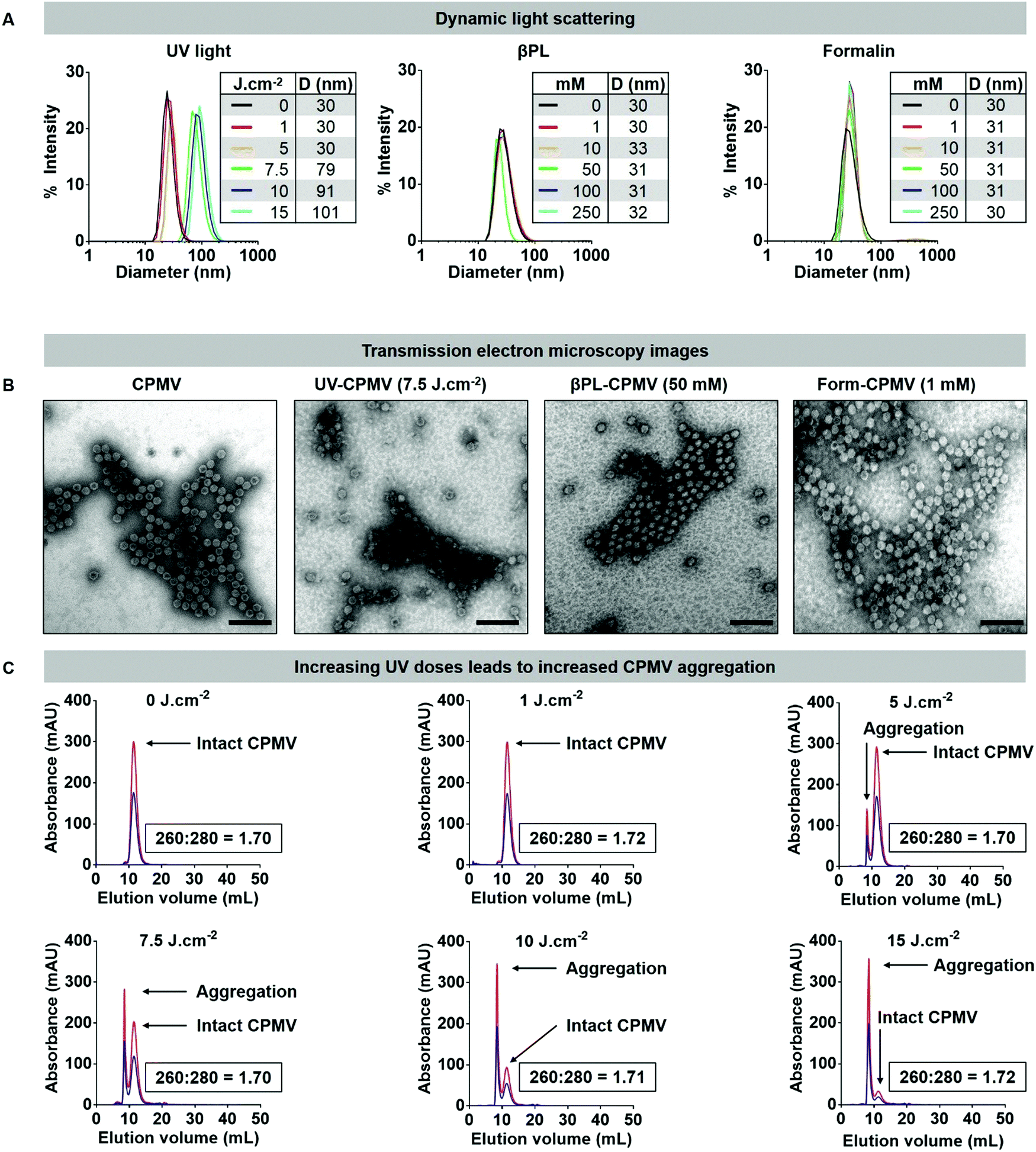 | ||
| Fig. 1 Structural integrity of UV, βPL, and formalin inactivated CPMV particles. (A) Dynamic light scattering of CPMV treated with (left) UV light (0, 1, 5, 7.5, 10, and 15 J cm−2), (middle) βPL (0, 1, 10, 50, 100, 250 mM), and (right) formalin (0, 0.5, 1, 10, 100, 250 mM). Table summarizes the average hydrodynamic diameter (denoted as D in nm) of each formulations. (B) TEM images of the native and inactivated CPMV formulations (negatively stained with UAc). Scale bars correspond to 100 nm. (C) Size exclusion chromatography (SEC) of CPMV treated with UV light. Blue = 260 nm, red = 280 nm. The ratio of RNA to coat protein (260:280) is included in each panel; additional SEC panels are presented in the ESI.† | ||
However, treatment of CPMV with UV using dosage of 7.5 J cm−2 and higher resulted in particle aggregation. DLS measurement indicated radii of 71, 91, and 101 nm for CPMV treated with 7.5, 10, and 15 J cm−2, respectively (Fig. 1A). This was also consistent with apparent particle aggregation in the TEM images and SEC elution profiles (Fig. 1B, C and Fig. S3–S5, ESI†). While not apparent in DLS, SEC data indicate some degree of aggregation of CPMV treated with a UV dosage of 5 J cm−2. The data attests to the robustness of the CPMV nanoparticle and also highlights the potency of UV irradiation to induce protein crosslinking and particle aggregation.
UV/vis spectroscopy gave insight into the RNA-to-protein ratio; for intact CPMV, the absorbance ratio at 260:280 nm is 1.7. Data revealed that the RNA to protein ratio remained close to 1.7 for most treatment conditions (Fig. 2A). There was however a decrease in the 260:280 ratio of Form-CPMV treated with 100 mM (260:280 = 1.58) and 250 mM (260:280 = 1.53) formalin. UV/vis spectroscopy and SEC were in good agreement (Fig. S4 and S5, ESI†). While a lower 260:280 nm ratio may indicate loss of RNA, data indicate that these changes in absorbance may be attributed to extensive RNA and RNA–protein crosslinking rather than RNA loss. Indeed, extensive RNA crosslinking was observed when RNA was extracted from formalin treated CPMV and analyzed by UV/vis spectroscopy (Fig. 2B) or agarose gel electrophoresis (Fig. 2C). Treatment with 1 J cm−2 UV light or 1 mM βPL or 1 mM formalin had little effect on the RNA, and RNA-1 and RNA-2 were detectable as sharp bands in the gels. At higher dosage or concentration – independent of the treatment modality, RNA crosslinking was apparent: for UV and βPL-treated samples, extracted RNA had higher mobility and individual RNAs could not be resolved, which would indicate inter- and/or intramolecular RNA crosslinking resulting in a more compact form of the RNA enhancing its mobility. For the formalin-treated samples, extensive RNA–RNA crosslinking and likely more extensive RNA–protein crosslinking occurred resulting in loss of mobility of the RNA (Fig. 2C).
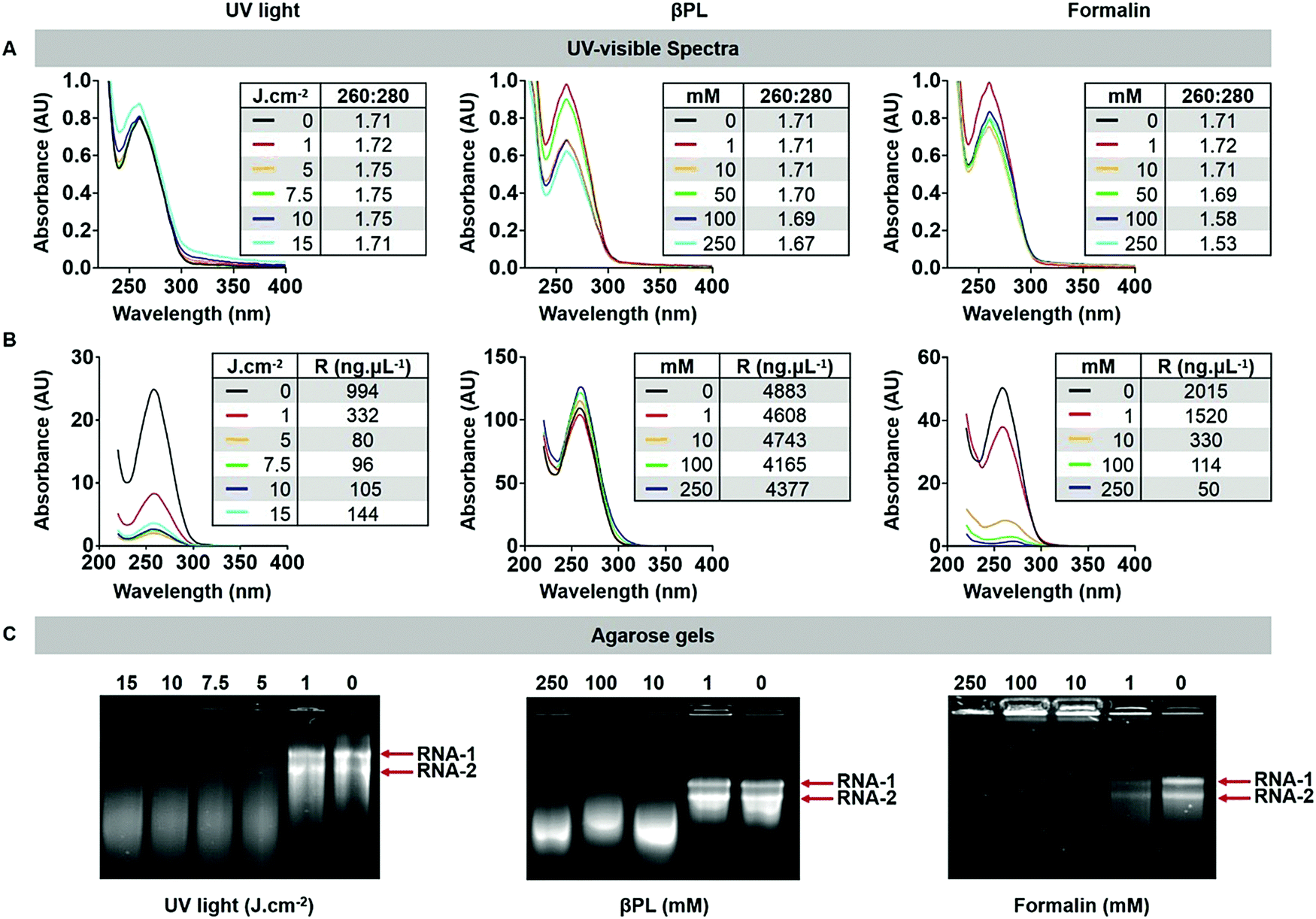 | ||
| Fig. 2 CPMV RNA integrity following UV, βPL, and formalin treatment. (A) UV-visible light spectra of inactivated CPMV and corresponding 260:280 nm ratio. (B) UV-visible light spectra of RNA extracted from treated CPMV and corresponding yields. (C) Agarose electrophoretic gel of the RNA extracted from UV-CPMV, βPL-CPMV, and Form-CPMV under UV light. | ||
To gain further insights into whether and to what degree RNA–protein crosslinking occurred, intact and denatured CPMV particles pre- and post-inactivation were analyzed by native gel electrophoresis (agarose GE) and denatured samples were analyzed by SDS-PAGE. All gels were imaged after staining for proteins (Coomassie) and nucleic acid (Gel Red) under white light and UV light, respectively. In the native agarose GE, two distinctive protein bands were observed due to the presence of two electrophoretic (fast and slow) forms of CPMV particles (Fig. 3A); the fast form of CPMV is the result of a 24 amino acid loss at the C-terminus of the small coat protein due to proteolysis in plant cells.5 In all samples, nucleic acids and proteins traveled through the agarose gel together, confirming the entrapment of the RNA within the CPMV particles. CPMV treated with doses of 5, 7.5, 10 and 15 J cm−2 of UV showed signs of particle aggregation, as indicated by the presence of a smear instead of two distinct bands (this is consistent with DLS and SEC data). Compared to CPMV, βPL-CPMV and Form-CPMV traveled farther through the gel proportionally to the concentration of βPL and formalin used to inactivate CPMV. These results could reflect a change in overall particle charge due to the action of βPL (alkylation and acylation) and formalin (crosslinking) on proteins and RNA.
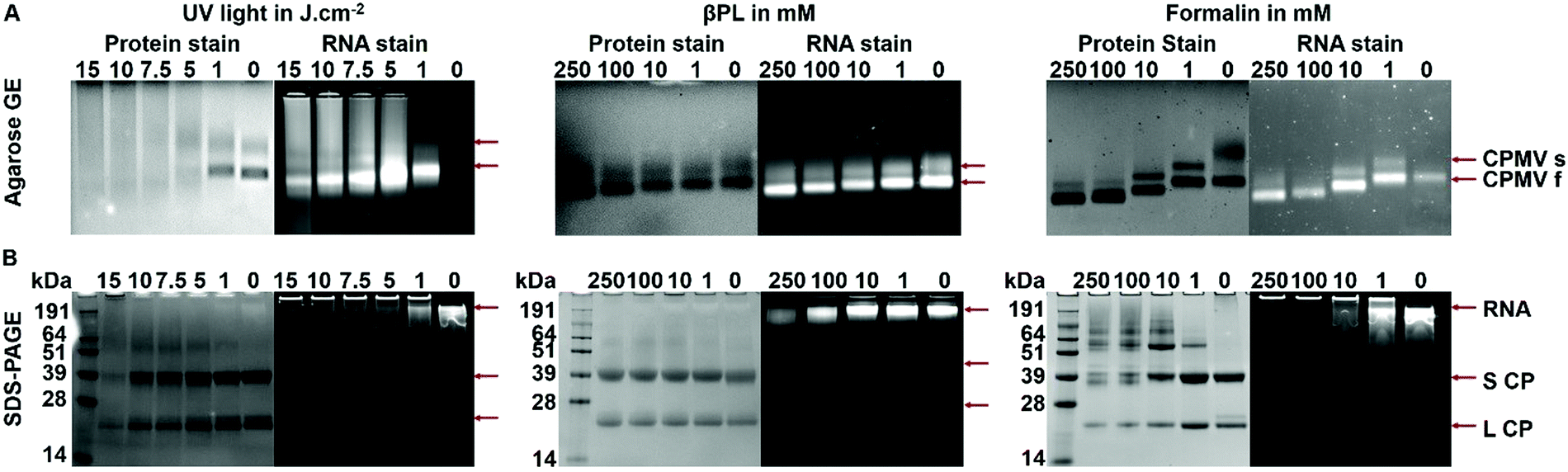 | ||
| Fig. 3 Gel electrophoretic mobility of native and inactivated CPMV. (A) Non-denaturing agarose gel electrophoresis images of CPMV treated with UV light, βPL, and formalin under white light after Coomassie staining (protein detection), and UV light after GelRed staining (RNA detection). Note that the RNA stain corresponding to the native CPMV in the upper left panel appears very faint due to the increased signal intensity of crosslinked, UV treated RNA. (B) Corresponding denaturing SDS-PAGE under white light after Coomassie staining, and under UV light after GelRed staining. | ||
Next, samples were denatured and the L and S coat protein subunits were separated and analyzed by SDS-PAGE; L and S were visualized as single bands at 42 kDa and 24 kDa, respectively (Fig. 3B). The band intensity of UV-CPMV coat proteins (CP) decreased with increasing dose of UV-radiation, indicative of the presence of particle aggregates resulting in reduced yields of free L and S protein. In particular, aggregation resulted in smeared proteins when CPMV was treated with 5 J cm−2 or higher doses of UV light. βPL-CPMV showed no sign of aggregation or protein regardless of the dose of βPL used during treatment. In contrast, the higher the dose of formalin, the more inter-CP crosslinking was observed; and this effect was apparent at doses as low as 0.5 mM (Fig. S7, ESI†). GelRed staining was added to SDS-PAGE gels to assess the RNA content of particles. RNA from UV-CPMV and Form-CPMV particles did not travel through the gel (but rather were detectable in the wells), most likely due to intra-RNA and RNA-coat protein crosslinking. RNA from βPL-CPMV matched the profile of that released from CPMV; however, at high βPL concentrations RNA breakage was observed (as evident by increased electrophoretic mobility) – this is also consistent with UV-visible spectroscopy and SEC data as described above.
Together, data indicate that at doses higher than 1 J cm−2 UV light or 1 mM βPL or 1 mM formalin, RNA and protein modifications and crosslinking occur. At high concentration of UV light (≥5 J cm−2) CPMV particle aggregation is observed, and at high βPL concentrations (≥10 mM), RNA breakage is indicated.
UV light- and chemically-treated CPMV exhibit diminished infectivity
To determine the dose of UV, βPL, or formalin required to inactivate CPMV, we mechanically inoculated 40 μg of UV light- and chemically-treated CPMV formulations onto primary leaves of cowpea plants. Plants were monitored for the appearance of symptoms at 10 days post inoculation. Ten cowpea plants were inoculated per treatment arm, and a negative (not infected) and positive (infected with untreated CPMV) group were included. Necrotic or chlorotic lesions were observed in leaves infected with less than 7.5 J cm−2 treated UV-CPMV, 10 mM treated βPL-CPMV, and 1 mM treated Form-CPMV (Fig. 4). Reverse transcription (RT) polymerase chain reaction (PCR) was carried out on the total RNA content extracted from individual leaves to further attest for the presence of CPMV infection (Fig. S8–S11, ESI†). One leaf from five different plants was randomly selected in each treatment group. The forward primer (5′-GGTTCCCGCTTGCTTGGAGC-3′, RNA-2 nucleotide position 2630–2649) and reverse primer (5′-GGAGGATTATAAATGTGCG-3′, RNA-2 nucleotide position 2823–2805) were previously reported to yield high amounts of amplicons, 177 bps in size.23 RT-PCR products were analyzed by agarose gel electrophoresis and confirmed that leaves mechanically inoculated with CPMV inactivated through UV treatment at doses of 7.5 J cm−2 and higher were indeed effectively inactivated: CPMV infection was not detectable by symptoms or RT-PCR (Fig. 4). Similarly, CPMV formulations treated with 50 mM βPL-CPMV and 1 mM Form-CPMV were confirmed to be fully inactivated. It is worth mentioning that leaves inoculated with 10 mM βPL-CPMV showed no visual symptoms of infection, yet these leaves tested positive by RT-PCR – the latter is a more sensitive assay. | ||
| Fig. 4 Analysis of the infectivity of UV-CPMV, βPL-CPMV, and Form-CPMV using V. unguiculata (black eyed pea no. 5) plants. Depiction of individual leaves infected with CPMV, UV-CPMV, βPL-CPMV, or Form-CPMV at various doses; positive red symbols represent leaves showing visual symptoms of infection, while green negative symbols depicts leaves that show no symptoms of infection. Total RNA was extracted from the leaves and RT-PCR amplicons were obtained proportionally to the CPMV infectivity level. Green box indicates minimal dose required to inactivate CPMV. For the leaf inoculated with 7.5 J cm−2 treated UV-CPMV, the burnt lesion observed at the tip of the leaf was caused by residual carborundum during the inoculation process and is not a sign of infection. | ||
Based on the inactivation studies and infection assays in plants, for all subsequent experiments, we used the inactivated CPMV formulation obtained from 7.5 J cm−2 UV, 50 mM βPL, and 1 mM formalin. The UV-CPMV (7.5 J cm−2) sample was structurally intact but to some degree aggregated, and there was evidence of RNA–RNA and RNA–protein crosslinking. The βPL-CPMV (50 mM) sample remained structurally sound and monodisperse but its RNA was severely damaged. And the Form-CPMV (1 mM) sample also retained its size and monodispersity even though protein–protein, protein–RNA, and RNA–RNA crosslinking occurred.
CPMV inactivation by UV light was previously reported and only required a dose of 2.5 J cm−2 to prevent infection.18 This difference highlights one of the hurdles to standardizing UV inactivation across systems; we employed a UVP crosslinker (Analytik Jena AG) delivering 7 mW cm−2, and samples were prepared using a 1 mg mL−1 solution placed 20 cm away from the UV source. Rae et al. relied on a Stratalinker 1800 UV Crosslinker (Stratagene) delivering 3 mW cm−2, and their samples were irradiated 15 cm from the UV source at a concentration of 2 mg mL−1. In addition, the volume to surface area ratio of the prepared samples could have also influenced the results. Single-stranded RNA containing mammalian viruses have also been inactivated to produce vaccines. Among them hepatitis A,24 HIV,25 and influenza26 reported inactivation doses of 0.3, 1, and 1 J cm−2, respectively. The need for use of higher dosage to inactivate the plant virus may reflect the higher stability of the plant viral nanoparticle and its requirement to remain stable in the environment when exposed to UV light. As aforementioned, βPL and formalin are commonly used to produce inactivated mammalian virus vaccines.19,20 The dose required to inactivate CPMV using βPL (50 mM) and formalin (1 mM) is very similar to those reported in the literature for mammalian viruses. For example, the eastern equine encephalitis and poliomyelititis type II,27 equine herpesvirus type I,28 HIV,29 and the influenza virus30 have been successfully inactivated with 5–60 mM βPL; similarly, hepatitis A,31 HIV,29 influenza A virus,32 Japanese encephalitis virus,33 and rabies34 were successfully inactivated using 5–120 mM of formalin.
Effect of inactivation on the chemical reactivity of CPMV
CPMV viral capsids contain 300 solvent-exposed lysine residues that can be chemically modified to impart new functionalities through isothiocyanate and N-hydroxysuccinimide (NHS)–ester coupling. Examples include the conjugation of targeting ligands (e.g. folic acid35), therapeutics (e.g. doxorubicin36), and fluorescent dyes (e.g. Alexa Fluor dyes21). To verify that CPMV retained its chemical reactivity, CPMV, UV-CPMV, βPL-CPMV, and Form-CPMV were incubated with 5 molar excess of sulfo-cyanine 5-NHS per coat protein overnight, followed by centrifugation and desalting column techniques to remove the excess dyes. The level of Cy5 labeling of the CPMVCy5, UV-CPMVCy5, βPL-CPMVCy5, and Form-CPMVCy5 formulation was quantified using UV-visible spectroscopy; all samples produced dyes per particle values similar to CPMV (30 to 35 dyes per particle) (Fig. 5A). SDS-PAGE analysis further confirmed the chemical conjugation of Cy5 to both the L and S coat protein subunits (Fig. 5B). Thus, UV, βPL and formalin inactivated CPMV particles retain similar surface chemical reactivity. | ||
| Fig. 5 Chemical reactivity and biological activity of inactivated CPMV. (A) UV-visible spectra of native and inactivated CPMV conjugated to Cy5, and table reporting the number of Cy5 dye conjugated per CPMV particles. (B) Corresponding denaturing SDS-PAGE electrophoresis gels under white light after Coomassie staining, and under red fluorescent light. (C) Schematic of a VOPBA assay against vimentin (on the left), SDS-PAGE gel of vimentin under with light after Coomassie staining (middle, red box), and corresponding white light image of CPMV binding to vimentin during VOBPA assay (on the right). (D) Activation of RAW-Blue™ cells by CPMV, UV-CPMV, βPL-CPMV, and Form-CPMV and eCPMV. (E) Corresponding rate of cellular internalization measured by flow cytometry after 1 h and 8 h of incubation with RAW-Blue™ cells. Statistical significance was obtained using a 1 way ANOVA test; P value < 0.0001 = ****, P value < 0.001 = ***, P value < 0.01 = **, P value < 0.05 = *, ns = non-significant data. | ||
Effect of inactivation on the biological activity of CPMV
Although CPMV is a plant virus which does not infect mammalian cells, CPMV is known to bind to the mammalian protein vimentin; while a cytoskeleton protein, vimentin is also surface displayed and secreted by immune cells, epithelial cells, as well as cancer cells (e.g. breast cancer and colon adenocarcinoma) and plays a role in inflammation and cell migration.21,37,38 These natural CPMV–vimentin interactions can be harnessed for example to target CPMV to tumor neovasculature.39 Here, we tested whether the UV or chemical inactivation of CPMV would alter the CPMV–vimentin binding. Using a Virus Overlay Protein Binding Assay (VOPBA; for details see Methods Section), we observed no difference in vimentin binding comparing CPMV, UV-CPMV, βPL-CPMV and Form-CPMV, indicating that bio-specificity of the plant viral nanoparticles is retained (Fig. 5C).To determine whether inactivated CPMV retained its potent immunogenicity, we first assayed immunogenicity using the RAW-Blue™ assay (Fig. 5D). RAW-Blue™ cells are derived from murine RAW264.7 macrophages and express numerous pattern-recognition receptors such as toll-like receptors, nucleotide-binding oligomerization domain like receptors, retinoic acid-inducible gene-I like receptors, and C-type lectin receptors. Upon activation of the receptors, RAW-Blue™ cells secrete alkaline phosphatase (secreted embryonic alkaline phosphatase, SEAP), which serves as the reporter (enzymatic activity is the quantitative read out). While this assay does not give any information about which innate pathways the CPMV particle activate, the assay provides a quantitative read out and allows to compare the relative immunogenicity of the various CPMV formulations. 5 μg of CPMV, UV-CPMV, βPL-CPMV, Form-CPMV and eCPMV were incubated with RAW-Blue™ cells for 18 h before the cell's SEAP levels were quantified (Fig. 5D). A positive (1× LPS) and a negative control (no particles) were included. CPMV, UV-CPMV, and Form-CPMV showed significant and comparable levels of activation in the RAW-Blue™ assay: CPMV resulted in a 1.9-fold increase of SEAP level, and UV-CPMV and Form-CPMV resulted in 2.2-fold increase of SEAP levels (there was no statistical significance difference comparing these groups). Of note is that βPL-CPMV appeared to be less effective, matching eCPMV, in activating RAW-Blue™ and SEAP levels were increased only by 1.3-fold – therefore CPMV, UV-CPMV and Form-CPMV were almost 2-fold more effective (P value < 0.001).
These differences in SEAP secretion levels may either be attributed to the chemistry of the CPMV formulation or this could be attributed to differences in cellular uptake rates. To probe for the latter, we used flow cytometry to measure CPMV cell uptake. Data indicate that all particle formulations showed similar internalization rates; and indeed βPL-CPMV were internalized to a lesser degree (Fig. 5E). We hypothesize that the diminished immunogenicity of βPL treated CPMV can be explained by a combination of reduced cell uptake and more severe RNA damage including RNA breakage (as indicated by shortening of the RNA and lower A260:280 nm ratios).
Lastly, we used a dermal mouse tumor model to validate the efficacy of the chemically-inactivated CPMV probes vs. the native CPMV; we also considered an eCPMV nanoparticle, as we have previously shown that eCPMV does not match the efficacy of CPMV when used as in situ vaccine.13 UV-inactivation was not tested and these experiments will be carried out in future work. Here, we thought to test the chemically-inactivated CPMV nanoparticles, because these showed distinct behavior in vitro. Our goal was not to establish the mechanism of action of the individual CPMV formulation, but to determine if they retain their anti-tumoral properties. Dermal tumors were induced by transdermal injection of murine B16F10 melanoma cells using C57BL/6J mice (for details, see Methods sections). Seven days post-tumor challenge, tumor-bearing mice (n = 10, tumor sizes ∼30 mm3) were treated by intratumoral administration of 100 μg CPMV/10 μL; the treatment arms were CPMV, eCPMV, βPL-CPMV, Form-CPMV, or PBS. Three additional boosts were administered on day 12, 17, and 22. Mice were sacrificed once the tumor volume reached 1000 mm3, their weight decreased by 15%, or when visible signs of pain were observed (Fig. 6 and Fig. S11, ESI†). The PBS control group was characterized by a median overall survival (mOS) of 23 days and 100% of the mice were sacrificed by day 25 due to tumor progression beyond the ethical limit. In stark contrast, 7/10 mice treated with CPMV experienced complete disease regression and were tumor free by day 32 (n = 1), 37 (n = 5), and 39 (n = 1). The remaining 3/10 mice did not experience tumor growth until day 30 but had to be sacrificed on day 35, 38 and 40 due to visible symptoms of pain and weight loss. Previous data indicate that CPMV is well tolerated at doses of up to 100 mg kg−1 of mouse body weight; therefore we do not attribute the weight loss observed in some animals as toxicity from the CPMV nanoparticle.8,40 However the intratumoral treatment and resulting local immune response causes some tumors to collapse on themselves, and an ulcer is formed. In some animals, this ulcer can cause pain and loss in appetite. Compared to PBS, eCPMV treated mice exhibited a slower disease progression with a mOS of 28 days but ultimately all mice were sacrificed by day 33 due to their tumor volume. We have previously observed efficacy of eCPMV8 – however the efficacy of eCPMV vs. CPMV is reduced.13 We hypothesize that the RNA provides additional signaling through TLR-7/8 which enhances the potency of RNA-laden CPMV.
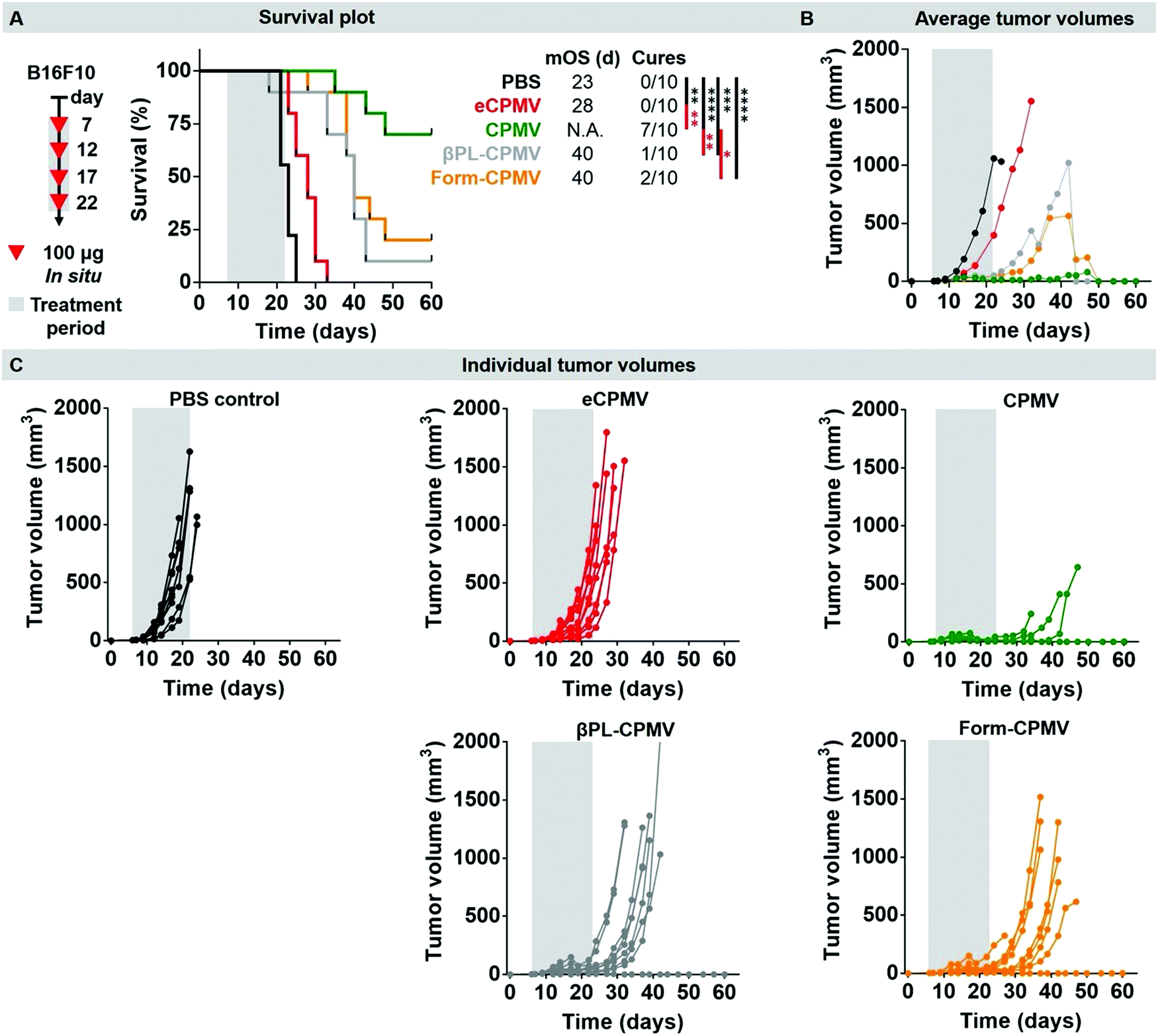 | ||
| Fig. 6 In situ vaccine efficacy of inactivated CPMV nanoparticles in a dermal mouse tumor model. (A) Survival plot of C57BL/6J mice (n = 10) transdermally injected with B16F10 melanoma cells and subsequently treated with PBS (black), eCPMV (red), CPMV (green), βPL-CPMV (grey), or Form-CPMV (orange). (B) Temporal average tumor volume of the corresponding treatment groups. (C) Individual tumor volume progression in each treatment group. Statistical significance was obtained using a 1 way ANOVA test; P value < 0.0001 = ****, P value < 0.001 = ***, P value < 0.01 = **, P value < 0.05 = *, ns = non-significant data. | ||
The chemically-inactivated CPMV nanoparticles demonstrated efficacy against the dermal melanoma as evident by reduced tumor burden and increased survival compared to PBS and eCPMV. βPL-CPMV and Form-CPMV treatment exhibited somewhat diminished anti-tumoral efficacy characterized by a mOS of 40 days and complete tumor regression in 1 βPL-CPMV and 2 Form-CPMV treated mice. This data highlights that the RAW-Blue™ and macrophage uptake studies only give limited insights into the efficacy of CPMV nanoparticles; this is expected as the situation in vivo is not realistically mimicked by nanoparticle–cell studies using a single cell type in 2D culture. Nevertheless, the in vitro assays can provide clues. There was no apparent difference comparing the efficacy of βPL-CPMV vs. Form-CPMV, which may indicate that either formulation would be a suitable candidate for future translational development. The somewhat reduced efficacy compared to CPMV may be explained by cross-linking of the RNA to itself and to protein, which reduced recognition by TLRs.
Conclusions
We developed protocols yielding non-infectious CPMV particles using 7.5 J cm−2 UV, 50 mM βPL and 1 mM formalin. While UV treatment led to some degree of nanoparticle aggregation at the required inactivation dose of 7.5 J cm−2, the resulting UV-CPMV remained its structure, chemical reactivity, and most importantly retained their biological activity; chemically-inactivated CPMV was tested in vivo and data demonstrate efficacy of the nanoparticle formulations against dermal melanoma; these formulations were less effective when compared to CPMV yet more effective when compared to eCPMV. Together, these findings presented here pave the way for the translational development of (environmentally) safe yet potent CPMV-based nanoparticles for use as in situ vaccine or immunotherapy and vaccine application.Conflicts of interest
Dr. Steinmetz is a co-founder of and has a financial interest with Mosaic ImmunoEngineering Inc. The other authors declare no potential conflict of interest.Acknowledgements
This work was funded in part by a grant from the National Institute of Health, U01-CA218292, to NFS.References
- I. Balke and A. Zeltins, Adv. Drug Delivery Rev., 2019, 119 CrossRef CAS.
- M. F. Bachmann and G. T. Jennings, Nat. Rev. Immunol., 2010, 10, 787 CrossRef CAS.
- M.-A. D’Aoust, M. M.-J. Couture, N. Charland, S. Trépanier, N. Landry, F. Ors and L.-P. Vézina, Plant Biotechnol. J., 2010, 8, 607 CrossRef.
- M. Tschofen, D. Knopp, E. Hood and E. Stöger, Annu. Rev. Anal. Chem., 2016, 9, 271 CrossRef.
- K. Saunders, F. Sainsbury and G. P. Lomonossoff, Virology, 2009, 393, 329 CrossRef CAS.
- E. L. Hesketh, Y. Meshcheriakova, R. F. Thompson, G. P. Lomonossoff and N. A. Ranson, Sci. Rep., 2017, 7, 539 CrossRef.
- C. Wang, S. N. Fiering and N. F. Steinmetz, Adv. Ther., 2019, 2, 1900003 CrossRef.
- P. H. Lizotte, A. M. Wen, M. R. Sheen, J. Fields, P. Rojanasopondist, N. F. Steinmetz and S. Fiering, Nat. Nanotechnol., 2016, 11, 295 CrossRef CAS.
- S. Shukla, C. Wang, V. Beiss, H. Cai, T. Washington, A. A. Murray, X. Gong, Z. Zhao, H. Masarapu and A. Zlotnick, et al. , Biomater. Sci., 2020, 8, 5489–5503 RSC.
- A. Kerstetter-Fogle, S. Shukla, C. Wang, V. Beiss, P. L. R. Harris, A. E. Sloan and N. F. Steinmetz, Cancers, 2019, 11, 515 CrossRef CAS.
- R. Patel, A. E. Czapar, S. Fiering, N. L. Oleinick and N. F. Steinmetz, ACS Omega, 2018, 3, 3702 CrossRef CAS.
- P. J. Hoopes, R. J. Wagner, K. Duval, K. Kang, D. J. Gladstone, K. L. Moodie, M. Crary-Burney, H. Ariaspulido, F. A. Veliz and N. F. Steinmetz, et al. , Mol. Pharmaceutics, 2018, 15, 3717 CrossRef CAS.
- C. Wang, V. Beiss and N. F. Steinmetz, J. Virol., 2019, 93, e00129 CAS.
- M. M. Albakri, F. A. Veliz, S. N. Fiering, N. F. Steinmetz and S. F. Sieg, Immunology, 2020, 159, 183 CrossRef CAS.
- M.-È. Lebel, K. Chartrand, E. Tarrab, P. Savard, D. Leclerc and A. Lamarre, Nano Lett., 2016, 16, 1826 CrossRef CAS.
- Q. Wang, E. Kaltgrad, T. Lin, J. E. Johnson and M. G. Finn, Chem. Biol., 2002, 9, 805–811 CrossRef CAS.
- C. S. Rae, I. Wei Khor, Q. Wang, G. Destito, M. J. Gonzalez, P. Singh, D. M. Thomas, M. N. Estrada, E. Powell and M. G. Finn, et al. , Virology, 2005, 343, 224 CrossRef CAS.
- C. Rae, K. J. Koudelka, G. Destito, M. N. Estrada, M. J. Gonzalez and M. Manchester, PLoS One, 2008, 3, e3315 CrossRef.
- J. P. Uittenbogaard, B. Zomer, P. Hoogerhout and B. Metz, J. Biol. Chem., 2011, 286, 36198 CrossRef CAS.
- T. Wilton, G. Dunn, D. Eastwood, P. D. Minor and J. Martin, J. Virol., 2014, 88, 11955 CrossRef.
- H. S. Leong, N. F. Steinmetz, A. Ablack, G. Destito, A. Zijlstra, H. Stuhlmann, M. Manchester and J. D. Lewis, Nat. Protoc., 2010, 5, 1406 CrossRef CAS.
- A. Chatterji, W. F. Ochoa, M. Paine, B. R. Ratna, J. E. Johnson and T. Lin, Chem. Biol., 2004, 11, 855 CrossRef CAS.
- N. F. Steinmetz, Viral Capsids as Programmable Nanobuilding Blocks, John Innes Centre, Norwich, UK, 2007 Search PubMed.
- J. Jean, R. Morales-Rayas, M.-N. Anoman and S. Lamhoujeb, Food Microbiol., 2011, 28, 568 CrossRef CAS.
- E. E. Henderson, G. Tudor and J.-Y. Yang, Radiat. Res., 1992, 131, 169 CrossRef CAS.
- Y. Furuya, M. Regner, M. Lobigs, A. Koskinen, A. Mullbacher and M. Alsharifi, J. Gen. Virol., 2010, 91, 1450 CrossRef CAS.
- G. A. Logrippo, Ann. N. Y. Acad. Sci., 1960, 83, 578 CrossRef CAS.
- T. M. Campbell, M. J. Studdert and M. H. Blackney, Vet. Microbiol., 1982, 7, 535 CrossRef CAS.
- E. Race, C. A. Stein, M. D. Wigg, A. Baksh, M. Addawe, P. Frezza and J. S. Oxford, Vaccine, 1995, 13, 1567 CrossRef CAS.
- E. I. Budowsky, E. A. Friedman, N. V. Zheleznova and F. S. Noskov, Vaccine, 1991, 9, 398 CrossRef CAS.
- G. G. Frösner, W. Stephan and H. Dichtelmüller, Eur. J. Clin. Microbiol., 1983, 2, 355 Search PubMed.
- A. Takada, S. Matsushita, A. Ninomiya, Y. Kawaoka and H. Kida, Vaccine, 2003, 21, 3212 CrossRef CAS.
- Y.-C. Fan, H.-C. Chiu, L.-K. Chen, G.-J. J. Chang and S.-S. Chiou, PLoS Neglected Trop. Dis., 2015, 9, e0004167 CrossRef.
- R. E. Kissling and D. R. Reese, J. Immunol., 1963, 91, 362 CAS.
- Y. Lu and P. S. Low, Adv. Drug Delivery Rev., 2012, 64, 342 CrossRef.
- A. A. A. Aljabali, S. Shukla, G. P. Lomonossoff, N. F. Steinmetz and D. J. Evans, Mol. Pharmaceutics, 2013, 10, 3 CrossRef CAS.
- P. H. Beatty and J. D. Lewis, Biomater. Based Phages Viruses, 2019, 145, 130 CAS.
- K. J. Koudelka, G. Destito, E. M. Plummer, S. A. Trauger, G. Siuzdak and M. Manchester, PLoS Pathog., 2009, 5, e1000417 CrossRef.
- N. F. Steinmetz, A. L. Ablack, J. L. Hickey, J. Ablack, B. Manocha, J. S. Mymryk, L. G. Luyt and J. D. Lewis, Small, 2011, 7, 1664 CrossRef CAS.
- P. Singh, D. Prasuhn, R. M. Yeh, G. Destito, C. S. Rae, K. Osborn, M. G. Finn and M. Manchester, J. Controlled Release, 2007, 120, 41 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00752h |
| This journal is © The Royal Society of Chemistry 2021 |