
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe design and synthesis of nonlinear optical chromophores containing two short chromophores for an enhanced electro-optic activity†
Fenggang
Liu
 *a,
Gangzhi
Qin
a,
Ziheng
Li
a,
Ziwei
Wang
a,
Meishan
Peng
a,
Shuangke
Wu
a,
Chunlin
Li
a and
Yuhui
Yang
b
*a,
Gangzhi
Qin
a,
Ziheng
Li
a,
Ziwei
Wang
a,
Meishan
Peng
a,
Shuangke
Wu
a,
Chunlin
Li
a and
Yuhui
Yang
b
aSchool of Chemistry and Chemical Engineering, Guangzhou University, Guangzhou 510006, P. R. China. E-mail: liufg6@gzhu.edu.cn
bCollege of Materials Science and Engineering, Zhejiang Sci-Tech University, Hangzhou, 310018, P. R. China. E-mail: yhyang@zstu.edu.cn
First published on 10th December 2020
Abstract
A series of nonlinear optical chromophores A–D based on the diethylaminophenyl donor and tricyanofuran or phenyl-trifluoromethyl-tricyanofuran acceptors coupled through a tetraene bridge was synthesized and investigated. In particular, the donor and bridge sections of chromophores B and D were functionalized with a small chromophore alkylaniline cyanoacetate (B1) and dialkylaminobenzylidene malononitrile (D1) group, respectively, compared to the pentafluorobenzene group of chromophores A and C. Before poling, two small chromophores B1 and D1 with large dipole moment will greatly weaken the electrostatic interaction between chromophores by steric effect and electrostatic screening effect, thus increasing the poling efficiency. Although density functional theory calculations suggested that they have similar first-order hyperpolarizability, polymeric thin films doped with chromophores A–D exhibited the different r33 values of 115, 166, 213 and 276 pm V−1 at 1310 nm, respectively. The normalized r33 value of chromphores B and D was up to 7.72 × 10−19 and 17.25 × 10−19 pm cc per (V molecules), which is much higher than that of chromophores A and C. All these prove that introducing a small chromophore into the main chromophore can greatly improve the electro-optic coefficient of the chromophore by a rational molecular design.
1. Introduction
The information processes and transmission materials using photons as carriers have attracted extensive attention due to their advantages in transmission speed and bandwidth, as well as good parallelism, resistance to electromagnetic wave interference and high density. These materials have been widely used in optical communications and optical information processing, radar detection, optical switches and modulators.1–4 The key to the application of optoelectronic communication technology is whether electro-optical materials with excellent performance can be prepared.5 After years of development, the advantages of organic electro-optic materials have become increasingly apparent.6 Compared with inorganic materials (such as LiNbO3 and GaAs), organic electro-optic materials have high electro-optic coefficient, fast response speed, high bandwidth, low dielectric constant, good processability and integration.7 In addition, organic materials can be easily integrated with semiconductor microelectronic devices, which have broad application prospects.8The organic second-order nonlinear optical chromophore is the most important component of organic electro-optic materials. The chromophore is composed of donors, acceptors and bridges, usually with a large dipole moment.9 Many excellent chromophore molecules with large first-order hyperpolarizability were designed and synthesized.10 Among these, arylamine type donors,11,12 divinylthienyl (FTC),13 ring-locked tetraene (CLD) bridges,14 and tricyanofuran-based (TCF)15 or phenyl-trifluoromethyl-tricyanofuran (CF3-Ph-TCF) acceptors16 were the most common and effective structure. A chromophore with large first-order hyperpolarizability is a necessary condition to obtain a large electro-optic coefficient. However, chromophores with large first-order hyperpolarizability usually have large dipole moments. The electrostatic interaction between molecules will increase with an increase in the dipole moment. The dipole–dipole interaction between molecules will hinder the poling orientation of molecules in the electric field, thus reducing the polarization efficiency.
Hence, in the field of nonlinear optics, one major challenge is to effectively translate the hyperpolarizability (β) values of the chromophore into high bulk EO activities. Introducing sterically hindered groups was usually an effective way to weaken the dipole moment between molecules, thereby increasing the electro-optic coefficient. Various isolation groups, such as silane,17 various aromatic structures18,19 and alkane20,21 were introduced to the different parts of chromophores to increase the electro-optic coefficient by steric effect. At the same time, other properties of the chromophore, such as the solvability, thermal stability and poling orientation stability were enhanced accordingly.22 Based on the isolation principle, many novel structures derived from steric groups have been designed, such as hyperbranched chromophorse,23 star-shaped chromophores,24,25 H-shaped chromophores,26 multichromophore dendrimers27 and other dendritic chromophores28–30 in order to minimize unwanted electrostatic interactions.
Therefore, the following factors should be taken into consideration when selecting the steric groups: the first is the shape and size of the steric groups, which will affect the dipole–dipole interaction between the chromophores.31 The second is that the rigidity of the steric group will affect the mobility of the molecule in the polarization process, while the steric group with too large volume and rigidity will hinder the polarization process. The last one is the molecular weight of the steric group. If the molecular weight is too large, the content of the active component chromosphere in the electro-optic film will be reduced.32
However, the traditional steric groups usually only play the role of isolation, and may reduce the chromophore content in the electro-optic films to a certain extent due to the large molecular weight. The chromophore with electro-optic activity was rarely used as the isolation group.5,33,34 Therefore, we designed new functional steric groups, alkylaniline cyanoacetate group (B1) and dialkylaminobenzylidene malononitriles (D1) group. They are not just steric groups, but also small chromophores with donor and acceptor. Although these two small chromophores do not have great first-order hyperpolarizability, their dipole moments (μ) cannot be ignored. In addition to the steric effect, the B1 and D1 groups with considerable μ will greatly weaken the electrostatic interaction between the molecules by electrostatic screening effect before poling, thus increasing the electro-optic coefficient. Four nonlinear optical chromophores A–D based on the same diethylaminophenyl donor and TCF or CF3-Ph-TCF acceptor coupled through the CLD bridge was synthesized and systematically investigated, as shown in Fig. 1. In particular, the donor and bridge moiety of chromophores B and D have been modified with B1 and D1 functional groups, respectively. In order to prove the advantages of the newly introduced functional groups, we also synthesized chromophores A and C with classic pentafluorophenyl isolation groups for comparison.
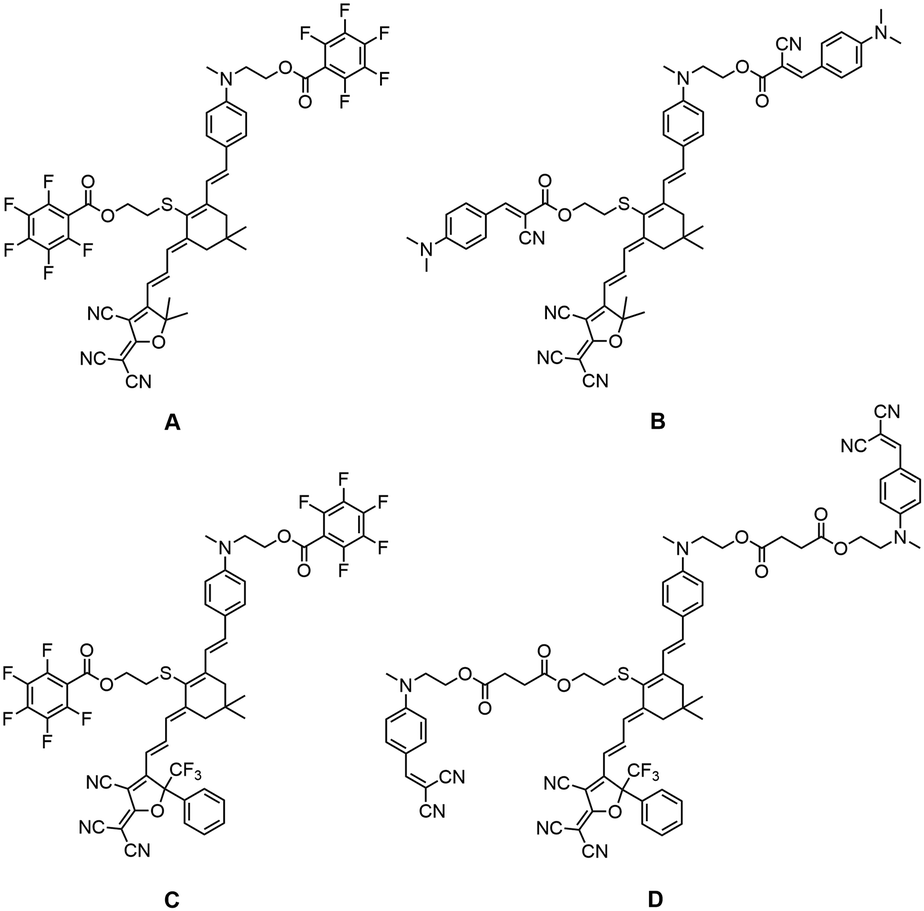 | ||
| Fig. 1 Chemical structure for chromophores A–D. | ||
2. Results and discussion
2.1 Synthesis and characterization of chromophores
The synthetic route and details of chromophores A–D are presented in Fig. 2. The synthesis of chromophores A–D requires the following seven-step reaction, starting from the diethylaminophenyl donor compounds 2: epoxy isophorone 1 was connected to donor 2 through Knoevenagel condensation, using sodium ethoxide as the base to yield compound 3. Then, the tert-butyldimethylsilyl group was used to protect the hydroxyl group of compound 3 to afford compound 4. Then, compound 4 was reacted with diethyl(cyanomethyl)phosphonate to give trienenitrile 5 by the Wittig–Horner reaction. Aldehyde 6 was obtained by reduction of the nitrile group of compound 5. The TBDMS group on compound 6 was deprotected by the action of hydrochloric acid, and the hydroxyl group was yielded to facilitate the next step of compound 7. The aldehyde 8a–c was obtained by connecting the isolated group R1–R3 with the hydroxyl groups of compound 7 using the Steglich esterification. The final condensations with the tricyanovinyldihydrofuran (TCF) acceptor or phenyl-trifluoromethyl-tricyanofuran acceptors (CF3-Ph-TCF) gave all four chromophores A–D as green solids. The four chromophores were fully characterized by 1H NMR and 13C NMR and HRMS. The four chromophores possess good solubility in most common organic solvents, such as acetone, ethyl acetate and methylene chloride. | ||
| Fig. 2 Synthetic routes for chromophores A–D. | ||
2.2. Thermal stability
The thermo gravimetric analysis (TGA) data were measured at a heating rate of 10 °C min−1 under a nitrogen atmosphere to compare the thermal stability of the four chromophores. As shown in Fig. 3 and Table 1, the decomposition temperatures (Td) of the four chromophores were higher than 220 °C (226–273 °C). Chromophore B has the highest decomposition temperature, followed by chromophore D, chromophore A and chromophore C. All four chromophores were thermally stable to withstand the poling process and EO device applications.35 | ||
| Fig. 3 TGA curves of chromophores A–D with a heating rate of 10 °C min−1 under a nitrogen atmosphere. | ||
| Cmpd | T d (°C) | λ max | λ max | Δλc | λ max |
|---|---|---|---|---|---|
| a λ max (nm) values were measured in chloroform. b λ max (nm) values were measured in dioxane. c Δλ (nm) was the difference between aλmax and bλmax. d λ max (nm) values were measured in film. | |||||
| A | 235 | 640 | 601 | 39 | 624 |
| B | 273 | 660 | 627 | 33 | 665 |
| C | 226 | 723 | 663 | 60 | 732 |
| D | 253 | 731 | 673 | 58 | 792 |
2.3. Optical properties
The UV-visible absorption spectra of the four chromophores A–D were measured in six kinds of solvents with different dielectric constants and in PMMA films in order to study the optical properties of the four chromophores. As shown in Fig. 4, 5, Fig. S1 (ESI†) and Table 1, chromophores A, B, C and D show the maximum absorption (λmax) values of 640 nm, 660 nm, 723 nm and 731 nm in chloroform, respectively. The λmax values of chromophores B and D were both redshifted (ca. 9–20 nm) compared to that of the unmodified chromophores A and C due to the electron withdrawing effect of the pentafluorobenzene group in chromophores A and C.35 Because of the stronger acceptor CF3-Ph-TCF, chromophores C and D exhibited more red-shifted maximum absorption wavelengths in comparison with chromophores A and B. For chromophores B and D, another strong absorption band with a λmax in chloroform at 427 nm and 429 nm was from the two alkylaniline cyanoacetate and dialkylaminobenzylidene malononitriles groups, respectively.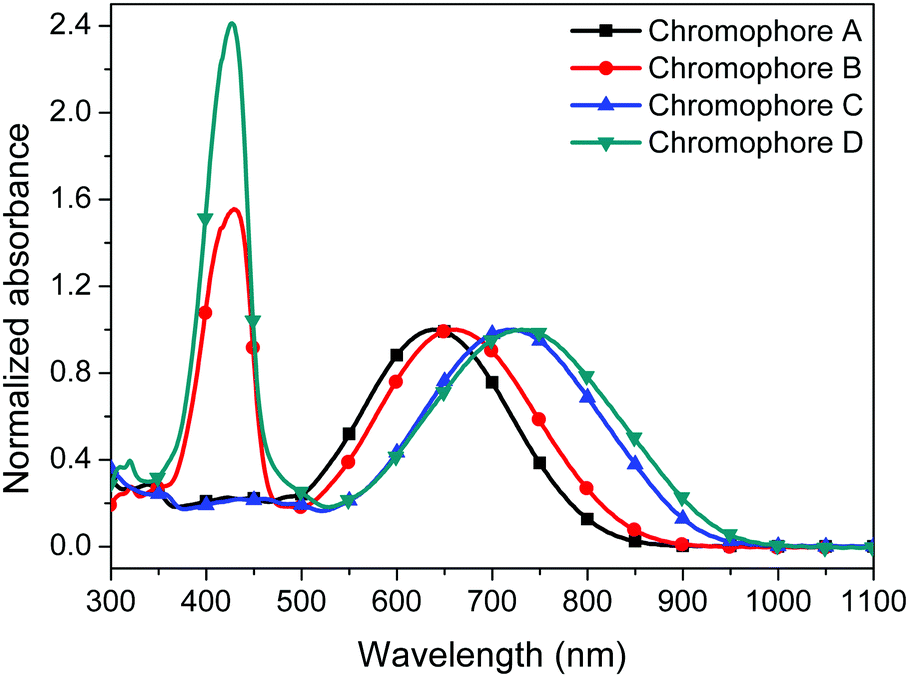 | ||
| Fig. 4 UV-Vis absorption spectra of chromophores A–D in chloroform. | ||
 | ||
| Fig. 5 UV-Vis absorption spectra of chromophores A–D in films. | ||
We also tested the UV absorption of these chromophores in film. The absorption maxima (λmax) of chromophores A–D in PMMA are 624 nm, 665 nm, 732 nm and 792 nm, respectively. The absorption in the film was different from the absorption in chloroform. This was probably due to the different interactions between the chromophores and the polymers within different structures. It is also interesting to note that chromophores B and D differ much more strongly from those of chromophores A and C, respectively, in the solid than in solution.
2.4 Theoretical calculations
The density functional theory (DFT) calculations were carried out using the Gaussian 09 program package at the R-B3LYP level by employing the split valence 6-31g(d) basis set to compare the dipole moment (μ), HOMO–LUMO energy gaps ΔE (DFT) and first hyperpolarizability (β) of the four chromophores.36,37 All chromophores were assumed to be in trans-configuration. All of the calculated data are summarized in Table 2. Fig. S2 (ESI†) describes the electron density distribution of the HOMO and LUMO structures of the chromophores. The density of the ground and excited state electron is asymmetric along the dipolar axis of the molecules.| Cmpd | ΔE(DFT)a (eV) | β tot (10−30 esu) | μ (D) | r 33 (pm V−1) |
|---|---|---|---|---|
| a Calculated from DFT calculations. b The first-order hyperpolarizability calculated from DFT calculations. c The total dipole moment. d At 30 wt%. | ||||
| A | 2.293 | 904.5 | 20.82 | 115 |
| B | 2.202 | 1058.1 | 23.58 | 166 |
| C | 2.029 | 1085.2 | 22.69 | 213 |
| D | 2.009 | 1183.9 | 23.26 | 276 |
| B1 | 3.567 | 42.9 | 9.06 | — |
| D1 | 3.509 | 39.0 | 10.77 | — |
The HOMO–LUMO energy gaps ΔE (DFT) of the four chromophores was calculated, as shown in Table 2. The trend of this data is consistent with the absorption of the UV absorption wavelength. In general, smaller energy differences between the molecules correlated with the redshift of the UV absorption wavelength.
The β value of the four chromophores was also calculated using DFT calculations. The first hyperpolarizability of chromophore C with CF3-Ph-TCF is larger than that of chromophore A due to the smaller energy gap caused by the stronger donor.38 In the same way, the first hyperpolarizability of compound D is greater than that of compound B. The first hyperpolarizability of chromophore A is smaller than that of compound B with the same TCF acceptor due to the larger energy gap caused by the electron withdrawing effect of the pentafluorophenyl group.
Because the alkylaniline cyanoacetate group (B1) and dialkylaminobenzylidene malononitrile group (D1) can also be regarded as small chromophores, we calculated the first-order hyperpolarizability of these two functional groups. The calculated β value of the B1 and D1 groups was 42.9 × 10−30 esu and 39.0 × 10−30 esu, respectively. These values were negligible relative to the first-order hyperpolarizability of the large chromophore (∼1000 × 10−30 esu). Therefore, the introduction of these two functionalized groups does not have a large impact on the first-order hyperpolarizability of the chromophore. However, in terms of the dipole moment, the values of these small chromophores were worth mentioning. The dipole moments of B1 and D1 were 9.06 Debye and 10.77 Debye, respectively. This value cannot be ignored relative to the dipole moment of the large chromophore (∼20 Debye). It should be noted that the dipole moments of the complex chromophores will depend on the conformation of the side chains.
2.5 Electro-optic performance
To test the effect of different functional groups on the electro-optic coefficient of the chromophores, electro-optic films with different doping concentrations were prepared. Different contents of chromophores were physically doped with polymer PMMA, and then dibromoethane was added. To spin cast a chromophore film requires a solution of approximately 8–10 wt%. After a night of stirring, the mixed solution was filtered through a 0.2 μm PTFE filter and then spin-coated onto selected ITO glass. The prepared electro-optic film was dried in a vacuum drying oven overnight. The corona polarization process was carried out at 5–10 °C higher than the glass transition temperature (Tg) of the Electro-optic (EO) film. The electro-optic coefficients of the EO films were measured by Teng-Man simple reflection method, at the wavelength of 1310 nm using a carefully selected thin ITO electrode (resistivity ≤100 ohm sq−1).5,39The electro-optic coefficient of the electro-optic film is usually related to the following parameters: the hyperpolarizability (β) is the micro electro-optic coefficient of the chromophore molecule; the chromophore number density (N) is the content of effective chromophore components in the electro-optic film.40 The poling efficiency means how many chromophore molecules are polarized and oriented. If the intermolecular dipole–dipole interaction can be ignored, the electro-optic coefficient will increase with N, β of the chromophore and the poling efficiency. At low doping concentration, the electrostatic interaction between the chromophores is weak, so the r33 value of the chromophore is mainly related to the chromophore number density and first-order hyperpolarizability of the chromophores.41,42 As the content of the chromophores in the electro-optic film gradually increases, the electrostatic interaction between the molecules cannot be ignored. Strong dipole–dipole interactions between the molecules will hinder the polarization orientation of the molecules under the action of an electric field. So, although the chromophores have similar first-order hyperpolarizability, their macro electro-optic coefficients still differ greatly because of the difference in the steric hindrance groups.
In order to study the influence of the functionalized groups on the electro-optic coefficients of the chromophores, the doping of PMMA films with 10–40 wt% chromophores was prepared. We first tested the electro-optic coefficients of different chromophores at 10 wt% doping concentration. For the four chromophores, the poled films of A/PMMA, B/PMMA, C/PMMA and D/PMMA gave r33 values of 46, 58, 84 and 95 pm V−1, respectively. The electro-optic coefficients of C/PMMA were larger than that of A/PMMA, and the electro-optic coefficients of D/PMMA were larger than that of B/PMMA, illustrating that the increased acceptor strength of the chromophores can increase their r33 value due to the larger hyperpolarizability and better isolation effect. The electro-optic coefficients at higher doping concentrations were also measured, as shown in Fig. 6.
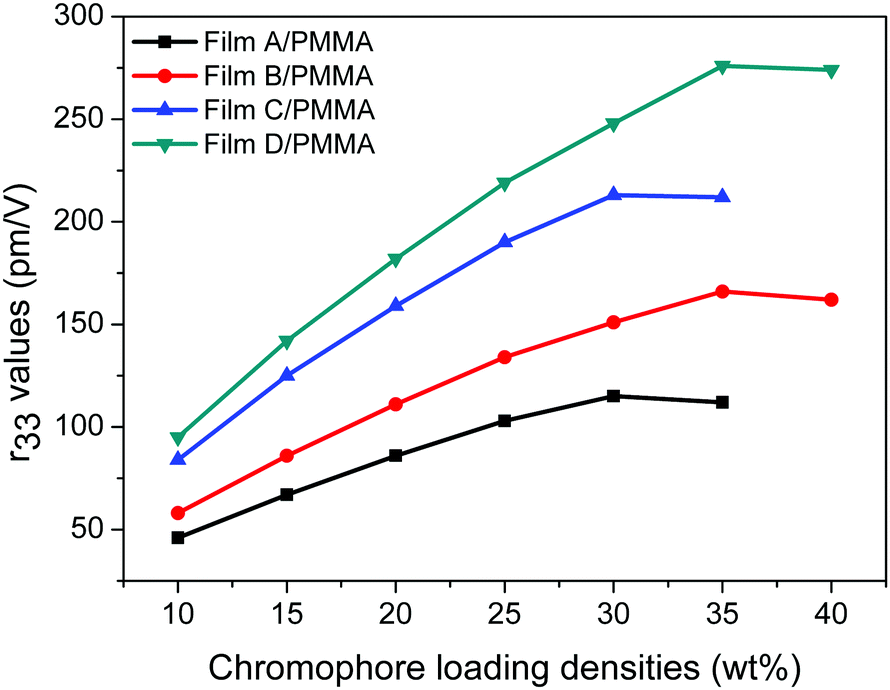 | ||
| Fig. 6 The r33 values of the NLO thin films as a function of the chromophore loading densities. | ||
The electro-optic coefficients of film-A/PMMA gradually increased from 46 pm V−1 (at 10 wt%) to 115 pm V−1 (at 30 wt%) and dropped to 112 pm V−1 (at 35 wt%). A similar trend was also observed for film-C/PMMA, whose r33 values gradually increased from 84 pm V−1 (at 10 wt%) to 213 pm V−1 (at 30 wt%), and dropped to 212 pm V−1 (at 35 wt%). However, the maximum r33 value of chromophores B and D was obtained at higher doping concentration. The electro-optic coefficients of film-B/PMMA were gradually improved from 58 pm V−1 (10 wt%) to 166 pm V−1 (35 wt%), while the r33 value dropped to 162 pm V−1 (40 wt%). The electro-optic coefficients of film-B/PMMA were gradually improved from 95 pm V−1 (10 wt%) to 276 pm V−1 (35 wt%), while the r33 value dropped to 274 pm V−1 (40 wt%).
The β value of chromophores B and D are 48% and 9% higher than that of chromophores A and C, respectively. Their r33 values vary greatly, especially at high doping concentrations. At 10 wt% doping concentration, the electro-optic coefficients of chromophores B and D are 26% and 13% higher than that of chromophores A and C, respectively. This is mainly due to the large first-order hyperpolarizabilities of chromophores B and D. However, at 35 wt% doping concentration, the electro-optic coefficients of chromophores B and D are 48% and 30% higher than that of chromophores A and C, respectively.
The number density of the chromophore will affect the r33 value of the chromophore, so the number density of each chromophore at 30 wt% doping concentration was also calculated, as shown in Table 3. The chromophore number density of film A/PMMA, film B/PMMA, film C/PMMA and film D/PMMA is 2.17 × 1020, 2.15 × 1020, 1.94 × 1020 and 1.60 × 1020 molecules per cm3, respectively. By normalizing the electro-optic coefficients for the chromophore number density (at 35 wt%): dividing the observed r33 value by chromophore number density (N), values of 5.16 × 10−19 pm cc per (V molecules), 7.72 × 10−19 pm cc per (V molecules), 10.93 × 10−19 pm cc per (V molecules) and 17.25 × 10−19 pm cc per (V molecules) were obtained for films A–D/PMMA, respectively.
With the increase of the doping concentration, the electrostatic interaction between the molecules becomes stronger and stronger. The dipole–dipole interaction between the molecules will hinder the polarization orientation of the chromophores, so the introduction of steric groups can effectively improve the polarization efficiency and electro-optic coefficient. Pentafluorobenzene is a very effective isolating group. The r33 value of the chromophore with a pentafluorobenzene isolating group was much larger than that for the chromophores without the isolating group.43
The dispersion enhancement, larger β value and larger poling efficiency may be responsible for the greater r33 values of chromophores B and D compared to chromophores A and C. The isolation effect of the alkylaniline cyanoacetate group (B1) and dialkylaminobenzylidene malononitrile (D1) group should be similar to pentafluorobenzene, or even slightly weaker, because of the similar rigidity and size. However, the poling efficiency and electro-optic coefficient of chromophores B and D are much greater than those of chromophores A and C due to the functions of the B1 and D1 groups. Before poling, the chromophore molecules tend to be aligned in antiparallel orientation due to the electrostatic interactions between the molecules. Dipole–dipole interactions between the molecules will hinder the poling orientation of the molecules under an electric field. The overall dipole moment of chromophores B and D was lowered by the screening effect of the two B1 and D1 groups around the primary chromophores B and D. The small chromophores B1 and D1 on the bridge and donor part of chromophores B and D will greatly weaken the electrostatic interaction between the molecules before the poling process. Then, the small chromophores B1 and D1 will be orientated under the action of an external electric field; thus, the dipole moment of chromophores B and D is enhanced. The process described above will greatly improve the polarization efficiency and electro-optic coefficient of chromophores B and D. It should be mentioned that the r33 value of film B and D/PMMA are very high compared to the EO films, which were doped with chromophores with the diethylaminophenyl donor and the TCF or CF3-Ph-TCF acceptor.11,35,44–46
3. Conclusions
In conclusion, we have developed and systematically investigated a series of nonlinear optical chromophores A–C based on the diethylaminophenyl donor and tricyanofuran or phenyl-trifluoromethyl-tricyanofuran acceptors coupled through a tetraene bridge. The donor and bridge moiety of the chromophores B and D were functionalized with small chromophore alkylaniline cyanoacetate (B1) and dialkylaminobenzylidene malononitrile (D1) group, respectively. While the chromophores A and C were functionalized with the pentafluorobenzene group. In particular, the alkylaniline cyanoacetate and dialkylaminobenzylidene malononitrile groups were small chromophores with donor and acceptor. Before poling, two B1 and D1 groups with considerable dipole moment will greatly weaken the electrostatic interaction between the chromophores by electrostatic screening and steric effect. Then, the poling efficiency was increased with weakened dipole–dipole interaction. Although the DFT calculation suggests that the β values of the four chromophores A–D were similar owing to their similar D–π–A structure, the electro-optic coefficients of the four chromophores were quite different. As a result, the electro-optic films doped with chromophores A–D exhibited different r33 values of 115, 166, 213 and 276 pm V−1 at 1310 nm, respectively. The normalized r33 values of chromphores B and D were up to 7.72 × 10−19 and 17.25 × 10−19 pm cc per (V molecules), respectively, which is much higher than that of chromophores A and C without the small chromophore. This indicates that the small chromophores, alkylaniline cyanoacetate and dialkylaminobenzylidene malononitriles, introduced in the donor and bridge of the chromophores can improve the poling efficiency more effectively than the pentafluorobenzene group, thus greatly improving the r33 value of the chromophores. In the meantime, the thermal decomposition temperature of the small chromophore-containing chromophores B and D was high than 250 °C. The high thermal stability, together with the large electro-optic coefficient, suggests the potential use of these new chromophores in advanced electro-optic devices.4. Experimental
4.1 Materials and instrument
All chemicals used were bought from chemical companies, and used directly without special purification. Methylene chloride (DCM), DMF and tetrahydrofuran (THF) were all commercial ultra-dry reagents (water < 50 ppm L−1). Compounds 2–7, chromophore A and chromophore C were synthesized according to literature.34 The NMR data (1H NMR and 13C NMR) were measured on an Advance Bruker 500 M (500 MHz) NMR spectrometer. The mass spectrometry data were tested on a MALDI-TOF (Matrix Assisted Laser Desorption/Ionization of Flight) instrument on a BIFLEXIII (Bruker, Inc.,) spectrometer. The TGA data were measured on a PerkinElmer TGA4000 with a heating rate of 10 °C min−1 under nitrogen atmosphere. The UV-Vis spectra were collected on a Shimadzu UV1800 photospectrometer.4.2 Syntheses
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8 to 1:5) as the eluent. The compound 8b was obtained as a red oil in 65% yield (0.52 g, 0.65 mmol). MS (MALDI) (M+, C47H51N5O5S): calcd: 798.02; found: 798.14. 1H NMR (500 MHz, CDCl3) δ 10.15 (d, J = 8.0 Hz, 1H, CHO), 7.93 (dd, J = 14.5, 7.9 Hz, 3H, CH), 7.85 (dd, J = 14.7, 9.1 Hz, 4H, ArH, CH), 7.40 (d, J = 8.9 Hz, 2H, ArH), 6.97 (d, J = 8.0 Hz, 1H, ArH), 6.83 (d, J = 16.2 Hz, 1H, CH), 6.67–6.66 (m, 3H, ArH), 6.65–6.64 (m, 3H, ArH), 4.38 (t, J = 5.7 Hz, 2H, NCH2), 4.31 (t, J = 6.5 Hz, 2H, OCH2), 3.67 (t, J = 5.7 Hz, 2H, OCH2), 3.09 (s, 6H, NCH3), 3.09 (s, 6H, NCH3), 3.02 (s, 3H, NCH3), 2.90 (t, J = 6.5 Hz, 2H, SCH2), 2.78 (s, 2H, CH2), 2.51 (s, 2H, CH2), 1.04 (s, 6H, CH3). 13C NMR (126 MHz, CDCl3) δ 191.51, 164.27, 163.88, 156.21, 154.85, 154.66, 153.63, 151.16, 149.31, 135.23, 134.15, 129.03, 127.02, 126.83, 125.19, 124.64, 119.27, 117.38, 112.09, 111.43, 93.42, 93.03, 63.97, 62.93, 60.34, 50.70, 41.63, 39.97, 38.85, 30.08, 28.31.
:6 to 1:4) as the eluent to afford chromophore B as a deep green solid in 68% yield (0.67 g, 0.68 mmol). HRMS (ESI) (M+, C59H60N8O5S): calcd: 993.4486; found: 993.4478. 1H NMR (500 MHz, CDCl3) δ 8.17–8.12 (m, 1H, ArH), 7.98 (d, J = 8.7 Hz, 2H, ArH), 7.93–7.87 (m, 4H, CH, ArH), 7.57 (d, J = 14.8 Hz, 1H, CH), 7.45 (d, J = 8.9 Hz, 2H, ArH), 6.94 (d, J = 16.0 Hz, 1H, CH), 6.75–6.65 (m, 6H, ArH), 6.46 (d, J = 14.8 Hz, 1H, CH), 5.30 (s, 1H,CH), 4.43 (t, J = 5.7 Hz, 2H, NCH2), 4.30 (t, J = 5.9 Hz, 2H, OCH2), 3.74 (t, J = 5.7 Hz, 2H, OCH2), 3.14 (s, 6H, NCH3), 3.10 (s, 6H, NCH3), 3.09 (s, 3H, NCH3), 2.96 (t, J = 5.9 Hz, 2H, SCH2), 2.55 (s, 2H, CH2), 2.52 (s, 2H, CH2), 1.65 (s, 3H, CH3), 1.60 (s, 3H, CH3), 1.03 (s, 6H, CH3). 13C NMR (126 MHz, CDCl3) δ 176.27, 173.55, 164.32, 164.02, 154.91, 154.45, 153.76, 151.92, 149.72, 144.81, 136.49, 134.26, 129.42, 128.75, 128.12, 125.29, 124.83, 119.20, 117.48, 117.21, 112.55, 112.21, 111.92, 111.57, 97.18, 94.22, 93.02, 63.42, 62.89, 55.17, 53.39, 50.80, 41.78, 41.11, 40.01, 39.03, 34.26, 30.34, 28.36, 26.17.
:8 to 1:5) as the eluent. The compound 8c was obtained as a red oil in 68% yield (0.69 g, 0.68 mmol). MS (MALDI) (M+, C57H61N7O9S): calcd: 1020.22; found: 1020.31. 1H NMR (500 MHz, CDCl3) δ 10.05 (d, J = 8.1 Hz, 1H, CHO), 7.84 (d, J = 16.1 Hz, 1H, CH), 7.70–7.67 (m, 4H, CH, ArH), 7.36 (d, J = 8.9 Hz, 2H, ArH), 7.34 (d, J = 2.6 Hz, 2H, ArH), 6.89–6.77 (m, 2H, CH), 6.69–6.56 (m, 6H, ArH), 4.23–4.18 (m, 6H, NCH2), 4.03 (t, J = 7.1 Hz, 4H, OCH2), 3.66–3.62 (m, 4H, OCH2), 3.55 (t, J = 5.8 Hz, 2H, OCH2), 3.04 (s, 3H, NCH3), 3.03 (s, 3H, NCH3), 2.94 (s, 3H, NCH3), 2.76–2.69 (m, 2H, OCH2), 2.67–2.65 (m, 2H, SCH2), 2.50–2.47 (m, 4H, SCH2, OCH2), 2.10 (s, 2H, CH2), 1.95 (s, 2H, CH2), 0.97 (s, 6H, CH3). 13C NMR (126 MHz, CDCl3) δ 190.99, 171.59, 171.38, 170.55, 157.63, 155.97, 153.15, 150.63, 149.16, 134.86, 133.41, 128.55, 126.78, 126.39, 124.73, 124.28, 119.28, 115.47, 114.45, 111.68, 111.40, 71.68, 62.54, 61.31, 60.93, 59.89, 50.23, 41.21, 38.50, 38.27, 33.10, 29.57, 28.69, 27.83.
:6 to 1:4) as the eluent to afford chromophore D as a deep green solid in 71% yield (0.93 g, 0.71 mmol). HRMS (ESI) (M+, C73H67F3N10O9S): calcd: 1317.4844; found: 1317.4813. 1H NMR (500 MHz, CDCl3) δ 7.95 (d, J = 15.9 Hz, 1H, CH), 7.81–7.72 (m, 4H, ArH), 7.59–7.48 (m, 4H, CH), 7.46–7.38 (m, 5H, ArH), 7.00 (d, J = 15.9 Hz, 1H, CH), 6.75–6.66 (m, 8H, ArH), 6.56 (d, J = 14.6 Hz, 1H, CH), 4.30–4.25 (m, 8H, NCH2, OCH2), 4.14–4.06 (m, 4H, OCH2), 3.70 (q, J = 5.9 Hz, 4H, OCH2), 3.64 (t, J = 5.9 Hz, 2H, OCH2), 3.11 (s, 3H, NCH3), 3.09 (s, 3H, NCH3), 3.04 (s, 3H, NCH3), 2.77 (t, J = 6.5 Hz, 2H, SCH2), 2.58–2.54 (m, 4H, OCH2), 2.28 (s, 2H,CH2), 2.03 (s, 2H, CH2), 0.97 (s, 3H, CH3), 0.87 (s, 3H, CH3). 13C NMR (126 MHz, CDCl3) δ 174.51, 171.05, 170.76, 170.09, 161.91, 157.05, 156.39, 153.30, 152.53, 149.25, 146.40, 137.02, 132.81, 130.43, 129.10, 128.43, 127.94, 127.44, 125.83, 124.16, 123.79, 118.80, 116.39, 114.76, 113.73, 111.18, 110.82, 110.22, 109.73, 95.58, 95.20, 94.95, 71.68, 61.98, 60.67, 60.32, 59.36, 57.10, 49.66, 40.75, 40.11, 38.87, 37.85, 33.19, 32.51, 29.35, 29.05, 27.79, 27.58, 27.29, 26.78, 20.03, 13.20.
:8 to 1:5) as the eluent. The compound 8b was obtained as a red oil in 65% yield (0.52 g, 0.65 mmol). MS (MALDI) (M+, C47H51N5O5S): calcd: 798.02; found: 798.14. 1H NMR (500 MHz, CDCl3) δ 10.15 (d, J = 8.0 Hz, 1H, CHO), 7.93 (dd, J = 14.5, 7.9 Hz, 3H, CH), 7.85 (dd, J = 14.7, 9.1 Hz, 4H, ArH, CH), 7.40 (d, J = 8.9 Hz, 2H, ArH), 6.97 (d, J = 8.0 Hz, 1H, ArH), 6.83 (d, J = 16.2 Hz, 1H, CH), 6.67–6.66 (m, 3H, ArH), 6.65–6.64 (m, 3H, ArH), 4.38 (t, J = 5.7 Hz, 2H, NCH2), 4.31 (t, J = 6.5 Hz, 2H, OCH2), 3.67 (t, J = 5.7 Hz, 2H, OCH2), 3.09 (s, 6H, NCH3), 3.09 (s, 6H, NCH3), 3.02 (s, 3H, NCH3), 2.90 (t, J = 6.5 Hz, 2H, SCH2), 2.78 (s, 2H, CH2), 2.51 (s, 2H, CH2), 1.04 (s, 6H, CH3). 13C NMR (126 MHz, CDCl3) δ 191.51, 164.27, 163.88, 156.21, 154.85, 154.66, 153.63, 151.16, 149.31, 135.23, 134.15, 129.03, 127.02, 126.83, 125.19, 124.64, 119.27, 117.38, 112.09, 111.43, 93.42, 93.03, 63.97, 62.93, 60.34, 50.70, 41.63, 39.97, 38.85, 30.08, 28.31.
:6 to 1:4) as the eluent to afford chromophore B as a deep green solid in 68% yield (0.67 g, 0.68 mmol). HRMS (ESI) (M+, C59H60N8O5S): calcd: 993.4486; found: 993.4478. 1H NMR (500 MHz, CDCl3) δ 8.17–8.12 (m, 1H, ArH), 7.98 (d, J = 8.7 Hz, 2H, ArH), 7.93–7.87 (m, 4H, CH, ArH), 7.57 (d, J = 14.8 Hz, 1H, CH), 7.45 (d, J = 8.9 Hz, 2H, ArH), 6.94 (d, J = 16.0 Hz, 1H, CH), 6.75–6.65 (m, 6H, ArH), 6.46 (d, J = 14.8 Hz, 1H, CH), 5.30 (s, 1H,CH), 4.43 (t, J = 5.7 Hz, 2H, NCH2), 4.30 (t, J = 5.9 Hz, 2H, OCH2), 3.74 (t, J = 5.7 Hz, 2H, OCH2), 3.14 (s, 6H, NCH3), 3.10 (s, 6H, NCH3), 3.09 (s, 3H, NCH3), 2.96 (t, J = 5.9 Hz, 2H, SCH2), 2.55 (s, 2H, CH2), 2.52 (s, 2H, CH2), 1.65 (s, 3H, CH3), 1.60 (s, 3H, CH3), 1.03 (s, 6H, CH3). 13C NMR (126 MHz, CDCl3) δ 176.27, 173.55, 164.32, 164.02, 154.91, 154.45, 153.76, 151.92, 149.72, 144.81, 136.49, 134.26, 129.42, 128.75, 128.12, 125.29, 124.83, 119.20, 117.48, 117.21, 112.55, 112.21, 111.92, 111.57, 97.18, 94.22, 93.02, 63.42, 62.89, 55.17, 53.39, 50.80, 41.78, 41.11, 40.01, 39.03, 34.26, 30.34, 28.36, 26.17.
:8 to 1:5) as the eluent. The compound 8c was obtained as a red oil in 68% yield (0.69 g, 0.68 mmol). MS (MALDI) (M+, C57H61N7O9S): calcd: 1020.22; found: 1020.31. 1H NMR (500 MHz, CDCl3) δ 10.05 (d, J = 8.1 Hz, 1H, CHO), 7.84 (d, J = 16.1 Hz, 1H, CH), 7.70–7.67 (m, 4H, CH, ArH), 7.36 (d, J = 8.9 Hz, 2H, ArH), 7.34 (d, J = 2.6 Hz, 2H, ArH), 6.89–6.77 (m, 2H, CH), 6.69–6.56 (m, 6H, ArH), 4.23–4.18 (m, 6H, NCH2), 4.03 (t, J = 7.1 Hz, 4H, OCH2), 3.66–3.62 (m, 4H, OCH2), 3.55 (t, J = 5.8 Hz, 2H, OCH2), 3.04 (s, 3H, NCH3), 3.03 (s, 3H, NCH3), 2.94 (s, 3H, NCH3), 2.76–2.69 (m, 2H, OCH2), 2.67–2.65 (m, 2H, SCH2), 2.50–2.47 (m, 4H, SCH2, OCH2), 2.10 (s, 2H, CH2), 1.95 (s, 2H, CH2), 0.97 (s, 6H, CH3). 13C NMR (126 MHz, CDCl3) δ 190.99, 171.59, 171.38, 170.55, 157.63, 155.97, 153.15, 150.63, 149.16, 134.86, 133.41, 128.55, 126.78, 126.39, 124.73, 124.28, 119.28, 115.47, 114.45, 111.68, 111.40, 71.68, 62.54, 61.31, 60.93, 59.89, 50.23, 41.21, 38.50, 38.27, 33.10, 29.57, 28.69, 27.83.
:6 to 1:4) as the eluent to afford chromophore D as a deep green solid in 71% yield (0.93 g, 0.71 mmol). HRMS (ESI) (M+, C73H67F3N10O9S): calcd: 1317.4844; found: 1317.4813. 1H NMR (500 MHz, CDCl3) δ 7.95 (d, J = 15.9 Hz, 1H, CH), 7.81–7.72 (m, 4H, ArH), 7.59–7.48 (m, 4H, CH), 7.46–7.38 (m, 5H, ArH), 7.00 (d, J = 15.9 Hz, 1H, CH), 6.75–6.66 (m, 8H, ArH), 6.56 (d, J = 14.6 Hz, 1H, CH), 4.30–4.25 (m, 8H, NCH2, OCH2), 4.14–4.06 (m, 4H, OCH2), 3.70 (q, J = 5.9 Hz, 4H, OCH2), 3.64 (t, J = 5.9 Hz, 2H, OCH2), 3.11 (s, 3H, NCH3), 3.09 (s, 3H, NCH3), 3.04 (s, 3H, NCH3), 2.77 (t, J = 6.5 Hz, 2H, SCH2), 2.58–2.54 (m, 4H, OCH2), 2.28 (s, 2H,CH2), 2.03 (s, 2H, CH2), 0.97 (s, 3H, CH3), 0.87 (s, 3H, CH3). 13C NMR (126 MHz, CDCl3) δ 174.51, 171.05, 170.76, 170.09, 161.91, 157.05, 156.39, 153.30, 152.53, 149.25, 146.40, 137.02, 132.81, 130.43, 129.10, 128.43, 127.94, 127.44, 125.83, 124.16, 123.79, 118.80, 116.39, 114.76, 113.73, 111.18, 110.82, 110.22, 109.73, 95.58, 95.20, 94.95, 71.68, 61.98, 60.67, 60.32, 59.36, 57.10, 49.66, 40.75, 40.11, 38.87, 37.85, 33.19, 32.51, 29.35, 29.05, 27.79, 27.58, 27.29, 26.78, 20.03, 13.20.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the National Natural Science Foundation of China (No. 21805049) and Guangzhou Municipal Science and Technology Project (No. 201904010176) for the financial support.Notes and references
- Y. A. Getmanenko, T. G. Allen, H. Kim, J. M. Hales, B. Sandhu, M. S. Fonari, K. Y. Suponitsky, Y. Zhang, V. N. Khrustalev, J. D. Matichak, T. V. Timofeeva, S. Barlow, S.-H. Chi, J. W. Perry and S. R. Marder, Adv. Funct. Mater., 2018, 28, 1804073 CrossRef.
- J. Luo, X.-H. Zhou and A. K. Y. Jen, J. Mater. Chem., 2009, 19, 7410–7424 RSC.
- W. Heni, Y. Kutuvantavida, C. Haffner, H. Zwickel, C. Kieninger, S. Wolf, M. Lauermann, Y. Fedoryshyn, A. F. Tillack, L. E. Johnson, D. L. Elder, B. H. Robinson, W. Freude, C. Koos, J. Leuthold and L. R. Dalton, ACS Photonics, 2017, 4, 1576–1590 CrossRef CAS.
- M. Ayata, Y. Fedoryshyn, W. Heni, B. Baeuerle, A. Josten, M. Zahner, U. Koch, Y. Salamin, C. Hoessbacher, C. Haffner, D. L. Elder, L. R. Dalton and J. Leuthold, Science, 2017, 358, 630–632 CrossRef CAS.
- D. L. Elder, C. Haffner, W. Heni, Y. Fedoryshyn, K. E. Garrett, L. E. Johnson, R. A. Campbell, J. D. Avila, B. H. Robinson, J. Leuthold and L. R. Dalton, Chem. Mater., 2017, 29, 6457–6471 CrossRef CAS.
- H. Xu, F. Liu, D. L. Elder, L. E. Johnson, Y. de Coene, K. Clays, B. H. Robinson and L. R. Dalton, Chem. Mater., 2020, 32, 1408–1421 CrossRef CAS.
- P. A. Sullivan and L. R. Dalton, Acc. Chem. Res., 2010, 43, 10–18 CrossRef CAS.
- L. R. Dalton, P. A. Sullivan and D. H. Bale, Chem. Rev., 2010, 110, 25–55 CrossRef CAS.
- Z. Li, Q. Li and J. Qin, Polym. Chem., 2011, 2, 2723–2740 RSC.
- S. Pascal, Y. A. Getmanenko, Y. Zhang, I. Davydenko, N. Minh Hoang, G. Pilet, S. Redon, Y. Bretonniere, O. Maury, I. Ledoux-Rak, S. Barlow, S. R. Marder and C. Andraud, Chem. Mater., 2018, 30, 3410–3418 CrossRef CAS.
- Y.-J. Cheng, J. Luo, S. Huang, X. Zhou, Z. Shi, T.-D. Kim, D. H. Bale, S. Takahashi, A. Yick, B. M. Polishak, S.-H. Jang, L. R. Dalton, P. J. Reid, W. H. Steier and A. K. Y. Jen, Chem. Mater., 2008, 20, 5047–5054 CrossRef CAS.
- O. Kwon, S. Barlow, S. A. Odom, L. Beverina, N. J. Thompson, E. Zojer, J. L. Bredas and S. R. Marder, J. Phys. Chem. A, 2005, 109, 9346–9352 CrossRef CAS.
- Y. Yang, H. Xu, F. Liu, H. Wang, G. Deng, P. Si, H. Huang, S. Bo, J. Liu, L. Qiu, Z. Zhen and X. Liu, J. Mater. Chem. C, 2014, 2, 5124–5132 RSC.
- D. Briers, L. De Cremer, G. Koeckelberghs, S. Foerier, T. Verbiest and C. Samyn, Macromol. Rapid Commun., 2007, 28, 942–947 CrossRef CAS.
- M. Q. He, T. M. Leslie and J. A. Sinicropi, Chem. Mater., 2002, 14, 2393–2400 CrossRef CAS.
- S. Liu, M. A. Haller, H. Ma, L. R. Dalton, S. H. Jang and A. K. Y. Jen, Adv. Mater., 2003, 15, 603–607 CrossRef CAS.
- Y. Yang, J. Liu, M. Zhang, F. Liu, H. Wang, S. Bo, Z. Zhen, L. Qiu and X. Liu, J. Mater. Chem. C, 2015, 3, 3913–3921 RSC.
- J. D. Luo, Y. J. Cheng, T. D. Kim, S. Hau, S. H. Jang, Z. W. Shi, X. H. Zhou and A. K. Y. Jen, Org. Lett., 2006, 8, 1387–1390 CrossRef CAS.
- S. R. Hammond, O. Clot, K. A. Firestone, D. H. Bale, D. Lao, M. Haller, G. D. Phelan, B. Carlson, A. K. Y. Jen, P. J. Reid and L. R. Dalton, Chem. Mater., 2008, 20, 3425–3434 CrossRef CAS.
- X. Zhang, I. Aoki, X. Piao, S. Inoue, H. Tazawa, S. Yokoyama and A. Otomo, Tetrahedron Lett., 2010, 51, 5873–5876 CrossRef CAS.
- Y. Yang, S. Bo, H. Wang, F. Liu, J. Liu, L. Qiu, Z. Zhen and X. Liu, Dyes Pigm., 2015, 122, 139–146 CrossRef CAS.
- F. Liu, M. Zhang, H. Xiao, Y. Yang, H. Wang, J. Liu, S. Bo, Z. Zhen, X. Liu and L. Qiu, J. Mater. Chem. C, 2015, 3, 9283–9291 RSC.
- Y. W. Bai, N. H. Song, J. P. Gao, X. Sun, X. M. Wang, G. M. Yu and Z. Y. Wang, J. Am. Chem. Soc., 2005, 127, 2060–2061 CrossRef CAS.
- P. Gopalan, H. E. Katz, D. J. McGee, C. Erben, T. Zielinski, D. Bousquet, D. Muller, J. Grazul and Y. Olsson, J. Am. Chem. Soc., 2004, 126, 1741–1747 CrossRef CAS.
- Z. a. Li, P. Chen, Y. Xie, Z. Li and J. Qin, Adv. Electron. Mater., 2017, 3, 1700138 CrossRef.
- H. Xu, F. Liu, S. Chen, W. Shi, Z. Xie, J. Wang, Z. Zhai, J. Liu, H. Li and J. Wang, Dyes Pigm., 2019, 165, 144–150 CrossRef CAS.
- P. A. Sullivan, H. Rommel, Y. Liao, B. C. Olbricht, A. J. P. Akelaitis, K. A. Firestone, J.-W. Kang, J. Luo, J. A. Davies, D. H. Choi, B. E. Eichinger, P. J. Reid, A. Chen, A. K. Y. Jen, B. H. Robinson and L. R. Dalton, J. Am. Chem. Soc., 2007, 129, 7523–7530 CrossRef CAS.
- G. Liu, Q. Liao, H. Deng, W. Zhao, P. Chen, R. Tang, Q. Li and Z. Li, J. Mater. Chem. C, 2019, 7, 7344–7351 RSC.
- X. Zang, G. Liu, Q. Li, Z. A. Li and Z. Li, Macromolecules, 2020, 53, 4012–4021 CrossRef CAS.
- H. Xu, J. Liu, J. Liu, C. Yu, Z. Zhai, G. Qina and F. Liu, Mater. Chem. Front., 2020, 4, 168–175 RSC.
- J. Wu, C. Peng, H. Xiao, S. Bo, L. Qiu, Z. Zhen and X. Liu, Dyes Pigm., 2014, 104, 15–23 CrossRef CAS.
- H. Ma, S. Liu, J. D. Luo, S. Suresh, L. Liu, S. H. Kang, M. Haller, T. Sassa, L. R. Dalton and A. K. Y. Jen, Adv. Funct. Mater., 2002, 12, 565–574 CrossRef CAS.
- W. Jin, P. V. Johnston, D. L. Elder, K. T. Manner, K. E. Garrett, W. Kaminsky, R. Xu, B. H. Robinson and L. R. Dalton, J. Mater. Chem. C, 2016, 4, 3119–3124 RSC.
- J. Lei, C. Guo, F. Liu, S. Chen, W.-J. Shi, Z. Wang, Z. Zhai, S. Mo and J. Wang, Dyes Pigm., 2019, 170, 107607 CrossRef CAS.
- F. Liu, S. Chen, S. Mo, G. Qin, C. Yu, W. Zhang, W.-J. Shi, P. Chen, H. Xu and M. Fu, J. Mater. Chem. C, 2019, 7, 8019–8028 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, 2009 Search PubMed.
- I. Davydenko, S. Benis, S. B. Shiring, J. Simon, R. Sharma, T. G. Allen, S.-H. Chi, Q. Zhang, Y. A. Getmanenko, T. C. Parker, J. W. Perry, J.-L. Bredas, D. J. Hagan, E. W. Van Stryland, S. Barlow and S. R. Marder, J. Mater. Chem. C, 2018, 6, 3613–3620 RSC.
- F. Liu, H. Zhang, H. Xiao, H. Xu, S. Bo, L. Qiu, Z. Zhen, L. Lai, S. Chen and J. Wang, Dyes Pigm., 2018, 157, 55–63 CrossRef CAS.
- L. R. Dalton, P. A. Sullivan, D. H. Bale and B. C. Bricht, Solid-State Electron., 2007, 51, 1263–1277 CrossRef CAS.
- L. R. Dalton, D. Lao, B. C. Olbricht, S. Benight, D. H. Bale, J. A. Davies, T. Ewy, S. R. Hammond and P. A. Sullivan, Opt. Mater., 2010, 32, 658–668 CrossRef CAS.
- J. Wu, Q. Li, W. Wang, G. Deng, X. Zhang, Z. Li, H. Xiao and J. Liu, Mater. Lett., 2017, 199, 72–74 CrossRef CAS.
- H. Zhang, H. Xiao, F. Liu, F. Huo, Y. He, Z. Chen, X. Liu, S. Bo, L. Qiu and Z. Zhen, J. Mater. Chem. C, 2017, 5, 1675–1684 RSC.
- J. D. Luo, H. Ma, M. Haller, A. K. Y. Jen and R. R. Barto, Chem. Commun., 2002, 888–889 RSC.
- C. Zhang, C. G. Wang, J. L. Yang, L. R. Dalton, G. L. Sun, H. Zhang and W. H. Steier, Macromolecules, 2001, 34, 235–243 CrossRef CAS.
- X.-H. Zhou, J. Davies, S. Huang, J. Luo, Z. Shi, B. Polishak, Y.-J. Cheng, T.-D. Kim, L. Johnson and A. Jen, J. Mater. Chem., 2011, 21, 4437–4444 RSC.
- X.-H. Zhou, J. Luo, J. A. Davies, S. Huang and A. K. Y. Jen, J. Mater. Chem., 2012, 22, 16390–16398 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00677g |
| This journal is © The Royal Society of Chemistry 2021 |