DOI:
10.1039/D0MA00613K
(Paper)
Mater. Adv., 2021,
2, 1621-1634
Anticorrosion allyl sulfonate graft chitosan/graphene oxide nanocomposite material
Received
18th August 2020
, Accepted 31st January 2021
First published on 1st February 2021
Abstract
Chitosan derivatives are easily synthesized since their amine-bound glucosamine molecular units allow for the introduction of reactive chemical groups within their polymer chains. These compounds also act as green corrosion inhibitor molecules for industrial metals with no adverse impact on the environment. In this stduy, we hereby present a synthesis route for introducing allyl sulfonate within the chitosan (CH) molecular chain via its amino groups by Michael addition reaction. Allyl sulfonate graft chitosan (CH(S)) was synthesized by a reaction involving CH and allyl sulfonate, catalyzed homogeneously by acetic acid in alkaline pH using sodium persulfate initiator. CH(S) was subsequently grafted onto graphene oxide (GO) nanosheets. The resultant organic/inorganic hybrid (CH(S)–GO) nanocomposite was then characterized using appropriate techniques and utilized as a corrosion inhibitor for an X70 pipeline steel substrate in a CO2 saturated NaCl electrolyte at 60 °C. The X70 pipeline steel grade is extensively used in various oilfield applications. This nanocomposite significantly inhibited corrosion, and this was attributed to the molecular adsorption and formation of protective polymeric chitosan-GO hybrid nanocomposite films on the metallic surface. The degree of corrosion inhibition between the CH(S)–GO nanocomposite and its CH(S) precursor was comparatively investigated; superior surface protection was revealed only in the presence of the hybrid nanocomposite. The physically adsorbed nanocomposite films on steel contributed to enhanced surface hydrophobicity due to the prevailing low surface energy GO nanosheets. The CH(S) copolymer analogue must have promoted corrosion inhibition but the hybrid (CH(S)–GO) polymer nanocomposite ensured a more compact coverage on steel surfaces, hence was effective as a corrosion inhibitor. Without this nanocomposite, anodic steel corrosion was significant within the test media due to irreversible actions of CO2 induced chloride dissolution episodes. This allyl sulfonate graft chitosan/graphene oxide nanocomposite material may have a future in oilfield chemistry as a corrosion-inhibitor additive in surface-treatment formulations.
1. Introduction
Most grades of steel are utilized in constructing auto parts in automotive industries as well as in fabricating transportation lines and storage structures for oil–gas fluids in petroleum industries.1 The applications of steel materials are enormous in both domestic and industrial spheres, however, their usage is significantly affected by corrosion. Protecting steel materials against corrosion has become vital for safeguarding the safety of steel structures and reducing the cost involved in overall repairs.2 To mitigate corrosion, there is a need for developing safe and sustainable strategies for protecting these fabricated structures. Of the numerous corrosion remediation techniques, the use of surface-active corrosion inhibitors has become one of the most applicable tools in controlling the scourge.1 Corrosion inhibitor formulations designed for metal surface protection are made of a combination of organic and inorganic components capable of forming barrier films needed to inhibit metallic dissolution by controlling the ingression of corrosion species within metal/solution interfaces.3 Most of these inhibitor components are film-forming polymeric compounds, and they are effective in protecting the internal walls of steel-based transport pipelines.1 As polymers, their effectiveness as corrosion inhibitor molecules may depend on factors related to their chemistry (e.g. solubilization) and molecular structures (e.g. molecular sizes). Both properties foster the adsorption of these molecules on metal surfaces.4 Metal/inhibitor interactions are also linked with the molecule's compatibility with metal–surface chemistry and surface charge, while its ability to adsorb on surfaces may also be linked with the type of corrosive medium used.2
Polymeric corrosion inhibitor molecules with heteroatoms (nitrogen, sulphur, oxygen, phosphorus, etc.) possess high electron density clouds that promote surface adsorption while those with multiple bonds also possess π bonds that act as adsorption sites.5 The electronic properties of organic molecules are fundamental in elucidating their surface activities against aqueous corrosion of metals,6–8 since the presence of unoccupied d-orbital spaces within corroded metals prompts the acceptance of free electrons from these organic compounds.5 This leads to the formation of metal–surface films in the liquid phase. Unlike the reduced surface coverage imposed by their monomer analogues, the molecular sizes of polymers ensure wider and more compact molecular coverages on these surfaces; hence are more effective corrosion inhibitors.1 If adsorbed monolayers of simple polymer films could significantly distort anodic or cathodic, those of polymer blends and composites would spontaneously sustain the degree of surface protective against corrosion. This is due to the synergistic effects of the individual polymer components. The higher inhibition effects by hybrid polymers provide multiple active sites of adsorption on charged metallic surfaces, in turn displacing preabsorbed water molecules. High molecular-weight polymer inhibitor blends and composites act as film-forming blankets that cover metallic surfaces, subsequently making their desorption almost irreversible compared to their monomers.1 Recently, our research group has investigated the use of chitosanic polymer derivatives for corrosion protection of pipeline steel in various corrosive media: glucosyloxyethyl acrylate graft chitosan against pipeline steel corrosion in 1 M HCl,9 carboxymethyl chitosan grafted poly(2-methyl-1-vinylimidazole) in CO2 saturated acidic oilfield formation water,10 poly(N-vinyl imidazole) grafted carboxymethyl chitosan in 1 M HCl,11 carboxymethyl chitosan grafted poly(2-methyl-1-vinylimidazole)/cerium molybdate nanocomposite against biocorrosion in Desulfovibrio ferrophilus culture.12 In summary, corrosion inhibition by chitosanic polymers proceeds upon molecular adsorption of metallic surfaces and this is followed by subsequent passive film formation as corrosion is inhibited.
There may be several limitations with the use of these polymers; one of the most outstanding could be linked with limitations of structural flexibility and metal–surface cohesive bonding.1 However, with the incorporation of inorganic metal nanoparticles, the resultant film-forming hybrid polymer nanocomposites possessing versatile and tunable physicomechanical and chemical properties act as bridges between macro and hybrid nanostructures.2 These new nanocomposites possess unique solution chemistries with aqueous dispersibility capable of metal adsorption. The presence of metal nanoparticles within these composites leads to enhanced corrosion resistance, unique solution stability and surface-active properties (e.g. hydrophobicity) that are not observed in normal polymers. The protective performance of several chitosanic nanocomposite systems incorporated with the following nanoparticles has been reported: Co/SnS2,13 Ag,14,15 graphene oxide (GO),16 ZnO/GO nano-hybrids,17 and Fe3O4,18 to mention but a few. In the present study, the allyl sulfonate graft chitosan/graphene oxide nanocomposite (CH(S)–GO) was used as a corrosion inhibitor for pipeline steel corrosion in CO2 saturated NaCl medium at 60 °C. This novel CH(S)–GO nanocomposite was synthesized by grafting allyl sulfonate graft chitosan (CH(S)) onto graphene oxide nanosheets. CH(S) was initially synthesized by an acetic acid catalyzed reaction involving chitosan (CHS) and allyl sulfonate precursors in alkaline pH. This is a coupling reaction between the amine functional chitosanic backbone and double-bond end-groups of reactive allyl sulfonate molecules. The innovation in the present study is the synthesis and corrosion-inhibition application of this new CH(S)–GO nanocomposite. Since molecular weights of adsorbing corrosion-inhibiting films directly contribute to enhanced inhibition,16 this allyl sulfonate graft chitosan copolymer was chosen for the present study to foster metal protection due to its size. High-molecular weight polymers of this nature are known for their enhanced metal–surface protection via physical adsorption, and so this is why CH(S)–GO in the present study is an advantage.15,16 The reason for the choice of this graft copolymer could also be linked with the presence of sulphur atoms on the graft allyl sulfonate moiety that would foster strong metal–surface binding, in turn promoting enhanced corrosion inhibition. This is one of the major advantages of using this graft copolymer cross-linked with allyl sulfonate, compared to straight-chain chitosan derivatives with no such side chains. The presence of functional oxygen heteroatoms from the chitosan ring also provides good adsorption affinity for steel-surface binding toward improved corrosion inhibition efficiency.16 Infusion of GO nanoparticles within the polymer matrices further enhances the anticorrosion performances of the nanocomposites due to their hydrophobicity and high surface area.16 The advancement in the present study is the GO modification of the chitosan polymer matrix for superior rigidity and hydrophobicity toward reinforced barrier properties. The resultant anticorrosion film from this nanocomposite would impede more diffusion paths of corrosive species as well as charge transfer between localized anodic and cathodic sites.15,16 With graphene oxide derivatives, corrosion inhibition is enhanced due to the factors related to some of their unique physical and chemical properties (e.g. significantly low surface energy, superhydrophobicity, high chemical resistance, thermal conductivity and mechanical strength).19–22 In aqueous phases, GO is able to form insoluble hydrophobic layers that further protect metal surfaces while also contributing to stable corrosion performance by virtue of its surface-bound chemical groups (e.g. hydroxyl, carboxyl, epoxides, and carbonyl groups).16,21,22 GO is a reliable precursor for most organically modified nanomaterials with potential for sustainable corrosion inhibition.
The allyl sulfonate graft chitosan/GO nanocomposite, like most chitosan derivatives, is an eco-friendly corrosion inhibitor compared to the toxic nitrate, dichromate and chromate-based anticorrosion formulations. The novel properties of this hybrid material span between those of the polymeric chitosan and the incorporated inorganic GO component.2 Generally, chitosan derivatives are benign, safe and effective corrosion inhibitor molecules that leave no adverse residual byproduct that impacts the environment. They are more effective and easier to synthesize since their amine-bound glucosamine molecular units allow for introduction of useful reactive chemical groups within these molecules.10
The reason for conducting the present investigation in CO2 saturated media could be linked with the present corrosion issues involving carbon dioxide gas (CO2), a causative corrosion agent for pipeline steel. CO2 is also a serious corrosive gas whose intermediate carbonic aicd product that significantly contributes to anodic degradation. CO2 is a byproduct of most chemical processes in the oil and gas industries. Corrosion of pipeline steel is severe in CO2 saturated brine media at acid pH, and this happens mostly when steel structures (e.g. pipeline materials) are exposed to chloride enriched oilfield formation media.23 These corrosion episodes increase during the transportation of fluids under heat-generating hydrodynamic conditions in product transportation pipeline assembly; these events also lead to the erosion of their internal walls. Left unchecked and untreated, corrosion leads to damaged transportation pipelines and loss of contents (by leakages). In order to avoid these economic losses, the production cost is heavily supported by service maintenance operations involving reliable corrosion protection programs in which the use of corrosion inhibitors is a major content.10 This carbon-based GO nanomaterial was introduced in this study in order to institute reduced wetness to the adsorbed protective nanocomposite films on steel during the corrosion inhibition process. The use of GO in surface science investigation like this one has attracted considerable attention due to its unique nanostructure as it comprises of segregated sp2 carbon domains as unique functionalities. It is also easily reduced by catalytic nitrile oxidation.24
2. Materials and methods
2.1. Reagents and chemicals
200 kDa molecular-mass chitosan (CH) was commercially purchased from Sigma Aldrich. Its degree of deacetylation stood at 88%. Sodium allyl sulfonate (C3H5NaO3S, 99%), sodium hydroxide (NaOH, ≥98%), acetone (≥99.9%), sodium persulfate (≥99%), hydrazine monohydrate (98%) and reduced graphene oxide (rGO) were also purchased from Sigma Aldrich. Acetic acid (≥99.7%) and sodium persulfate (Na2S2O8, ≥99%) were utilized as the catalyst and initiator, respectively; both were purchased from the same outlet. All reagents were used as purchased without purification.
2.2. Pipeline steel material
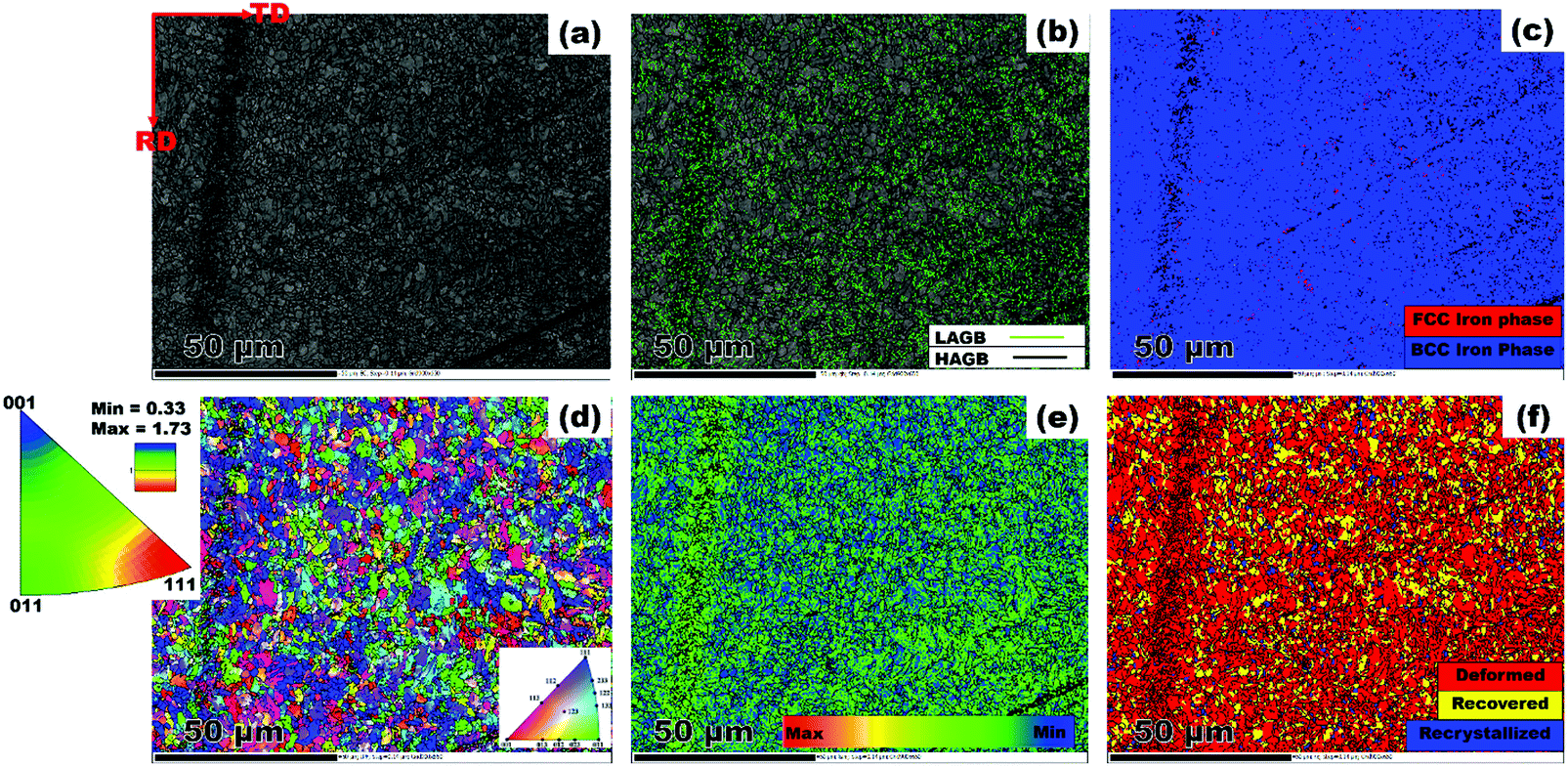
The metallic test substrate used in this study was a pipeline steel material belonging to a class of thermomechanically processed API (American Institute of Petroleum) 5L X70 grade donated by EVRAZ North America. The chemical composition of this steel is as follows: C: 0.047, Mn: 1.65, S: 0.0018, P: 0.009, Si: 0.18, Cu: 0.29, Ni: 0.07, Cr: 0.06, V: 0.001, Nb: 0.073, Mo: 0.247, Sn: 0.01, Al: 0.044, Ca: 0.0014, B: 0.0001, Ti: 0.022, N: 0.0099, O: 0.003, and Fe: balance. For the corrosion test, this metal was cut into small strips of 3 cm × 3 cm × 1 cm dimension before masking them to define a one centimeter squared geometric area. The microstructure of pipeline steel was established by the EBSD (electron backscatter diffraction) approach. A piece of steel specimen was machined out of the plate. Then, its surface RD – TD plane was mounted and polished with 500, 800, 1000, 1200, 2000 and 4000 μm abrasive papers. A mirror-like finish was achieved by further chemo-mechanical polishing in a colloidal silica suspension for 12 h. Electron diffraction patterns were collected using a field emission Hitachi SU6600 scanning electron microscope (SEM) that was fitted with an EBSD detector. An accelerating voltage of 30 kV and a step size of 0.14 were applied during scanning. The results were collected with the aid of Oxford Instrument's AZTEC 2.0 software, and later post-processed on the channel 5 software.
2.3. Preparation of allyl sulfonate graft chitosan
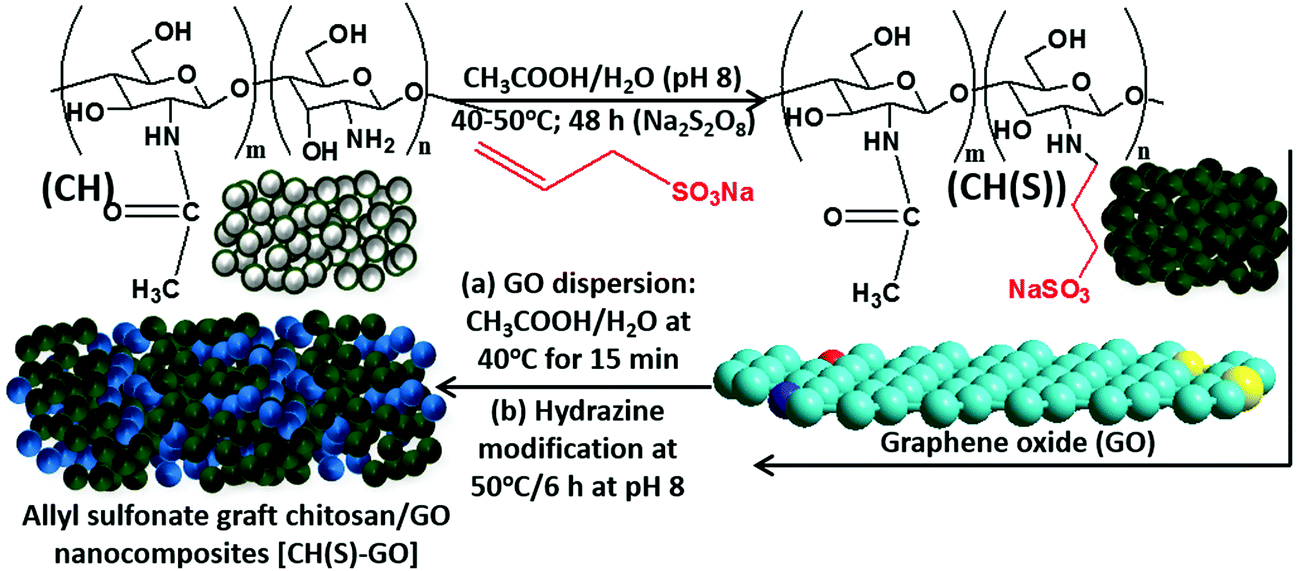
The synthesis of allyl sulfonate graft chitosan (CH(S)) was carried out by an acetic acid (CH3COOH) catalyzed reaction with a chitosan (CH) precursor in alkaline pH. About 2 g of CH was completely dissolved in 2 g of CH3COOH in 200 mL of water before adding premixed solution of 5 g of sodium allylsulfonate (C3H5NaO3S) and 1 g of persulfate (Na2S2O8). This suspension was continuously stirred for 1 h at 45 °C. The reaction was allowed to proceed at the same temperature for 48 h in a vacuum (as presented in Figure 1) after increasing the medium pH to 8 using 20% NaOH solution. Significant amounts of water and excess unreacted reagents were evaporated in a vacuum, leaving 80 mL of the product. It was then mixed with 500 mL of acetone to remove the unreacted C3H5NaO3S reactant. The resultant precipitate was repeatedly washed and filtered accordingly before finally drying overnight in a vacuum oven at 40 °C.
2.4. Preparation of the allyl sulfonate graft chitosan/graphene oxide nanocomposite
CH(S) was later grafted onto graphene oxide nanosheets via chemical grafting. This process was initiated after dispersing reduced graphene oxide (rGO) in water. The pre-washed CH(S) product was dissolved in 2 g of CH3COOH in 200 mL of water before dispersing graphene oxide pigments within the acidified CH hydrogel suspension. The medium was vigorously stirred (850 rpm) for 15 min at 40 °C (pH increased to 8 with ammonia solution). The medium was then sonicated for 30 min, filtered and then sparged with nitrogen gas. Before this, about 10 mL of hydrazine solution was slowly added during stirring, heated to 50 °C for 6 h and allowed to cool. The resultant nanocomposite was later weighed, stored for characterization using appropriate techniques and used as the corrosion inhibitor in this study. A similar procedure was reported by Haruna et al.25 for obtaining cyclodextrin–GO nanocomposites.
2.5. Characterization of the chitosan precursor and reaction products
Comparative analyses between the chitosan (CH) precursor, allyl sulfonate graft chitosan CH(S) and CH(S)–GO solid powdery products were performed using nuclear magnetic resonance (NMR) spectroscopy and attenuated total reflection Fourier transform infra-red spectroscopy (ATR-FTIR). NMR spectroscopy was conducted using an Avance III HD 600 MHz NMR spectrometer (Bruker) between both chitosan derivatives without rGO within the nanocomposites. The FTIR spectra were collected in transmittance mode after 32 scans from a Bio-RAD FTS-40 spectrophotometer (Bio-Rad). A comparative XRD analysis of CH, CH(S), rGO nanosheets and CH(S)–GO nanocomposites was performed using an X-ray diffractometer (Bruker D8 Discover XRD diffractometer). This experiment was conducted with CuKα characteristic radiation while the accompanying spectra present the 2θ angle ranging between 10 and 90 degrees with a 0.02-degree scanning step and 10° min−1 scan rate. Thermogravimetric analysis (TGA) experiments were conducted using 5 mg of samples of both materials using a TGA SDT Q600 analyzer after placing them in Pt crucibles. Weight loss-temperature curves of these samples were recorded and analysed using appropriate software from the instrument after analyses in a mixed N2/O2 atmosphere (both placed at 50 mL min−1 flow rate) at 5–20 °C min−1 heating rates. Inherent oxidative volatile products generated during the test were eliminated from the first N2 flow.9 The surface morphologies of both powders were also analyzed using a SEM at 1 kV acceleration voltage on precoated Au powder samples (10 nm; Quorum Q150T ES). Raman spectroscopy was carried out for rGO nanosheets using Renishaw Raman InVia reflex microscope (Renishaw) using a 514 nm edge laser. The Brunauer–Emmett–Teller (BET) surface area and nanopore volume of the rGO nanosheets were also measured using a BET instrument, Micromeritics ASAP 2020 and a porosimetry system, respectively. Prior to the BET tests, all test samples were completely degassed at 50 °C for 4 h in order to completely remove adsorbed moisture. Nitrogen gas was passed through the samples throughout the experiments.
2.6. Electrochemical corrosion tests
The electrochemical tests in this study were conducted by means of a potentiostat/galvanostat/ZRA (Interface 1000, Gamry Instruments) in a three-electrode set up. A Faraday cage was also utilized to avoid external interference. The Ag/AgCl (sat. KCl) was utilized as the reference electrode while platinum and steel strips were deployed as counter and working electrodes, respectively. The defined geometric area on each working electrode to the corrosive electrolyte (approximately 100 cm3) was 10 cm2. The corrosive media is this study was CO2 saturated brine (NaCl) spiked with 3 M acetic acid. This medium was purged with N2 for 10 min before saturating it with 99.99% CO2 for the duration of the test. The pH of the medium was 2. To avoid gas loss between each measurement, the CO2 gas was bubbled (10 L min−1) for 2 h each time and maintained at this pressure at 60 °C in a jacketed MultiPort Corrosion Cell Kit (Gamry). The frequency range for the electrochemical impedance spectroscopy (EIS) measurements was between 0.1 and 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Hz. EIS measurements were carried out at 10 mV peak-to-OCP amplitude sinusoidal perturbation; all measurements were conducted at open circuit potential (OCP) with reference to the Ag/AgCl (sat. KCl) electrode. The impedance results collected as EIS spectra were fitted with appropriate electrical equivalent circuit models in order to obtain different electrical parameters related to the properties of each tested system. This was accomplished by means of EChem Analyst software. Apart from having physical meanings, the chosen circuit model significantly minimized relative errors associated with each frequency point and also contributed to very low chi-squared error (less than 10−4).26 This direct-current test was also performed on each steel coupon after applying a corrosion potential between −0.25 and 0.25 V at 0.4 mV s−1 sweep rate. The corresponding magnitudes of corrosion current density (jcorr), also regarded as the corrosion rate, were obtained after Tafel fitting from both anodic and cathodic polarizations. Each test steel coupon was removed at the end of the tests and cleaned in accordance with the ASTM G3-89 (2010) standard.27
000 Hz. EIS measurements were carried out at 10 mV peak-to-OCP amplitude sinusoidal perturbation; all measurements were conducted at open circuit potential (OCP) with reference to the Ag/AgCl (sat. KCl) electrode. The impedance results collected as EIS spectra were fitted with appropriate electrical equivalent circuit models in order to obtain different electrical parameters related to the properties of each tested system. This was accomplished by means of EChem Analyst software. Apart from having physical meanings, the chosen circuit model significantly minimized relative errors associated with each frequency point and also contributed to very low chi-squared error (less than 10−4).26 This direct-current test was also performed on each steel coupon after applying a corrosion potential between −0.25 and 0.25 V at 0.4 mV s−1 sweep rate. The corresponding magnitudes of corrosion current density (jcorr), also regarded as the corrosion rate, were obtained after Tafel fitting from both anodic and cathodic polarizations. Each test steel coupon was removed at the end of the tests and cleaned in accordance with the ASTM G3-89 (2010) standard.27
2.7. Surface morphological and compositional analyses after corrosion tests
The corroded steel surfaces exposed to both acid media saturated with CO2 gas, with and without CH(S) and CH(S)–GO nanocomposites were investigated by means of scanning electron microscopy (SEM) and atomic force microscopy (AFM). SEM analyses were performed using a SU6600 SEM microscope (Hitachi High-Tech) after a 24 h exposure at 60 °C. All tests were confirmed by visual surface examinations. SEM analyses were also complemented by surface topological analyses of molecular adsorption on steel surfaces. AFM roughness quantification and image scans of surface areas of interest (10 × 10 μm) were performed in intermittent contact mode and at 1 Hz scan rate. All scans were probed with the aid of a silicon cantilever (whose properties are enlisted in Table 1), attached to a Model 4500 AFM instrument (Agilent Technologies). Gwyddion software was utilized for all post-imaging processing and analyses, including surface profiling and dimensional analyses of surface features. X-ray photoelectron spectroscopy (XPS) spectra of these adhering corrosion inhibitor/corrosion product aggregates were investigated for their chemical compositions. This surface analysis was conducted with the aid of a Kratos AXIS Supra X-ray Photoelectron Spectrometer with a monochromatized Al Kα X-ray radiation source. Spectrograms of these surfaces were collected from a C 1s internal reference with a binding energy of 285.0 eV. Their peak quantification and further simulations were conducted using a CasaXPS software, for both wide-scan and deconvoluted high-resolution XPS spectrograms. In order to determine the hydrophobicity of each surface, their aqueous contact angles (θow) were investigated using a DataPhysics Instrument. After measurements, the values of θow for each surface were extrapolated after surface analyses by the pendant drop method.
Table 1 Properties of the silicon cantilever used in this study
| Force constant |
Resonant frequency |
Curvature radius |
Tip shape |
| 50 N m−1 |
170 kHz |
< 10 nm |
Standard/conical |
3. Results and discussion
3.1. Microstructure analysis of the test steel substrate
The results from the microstructural examination are presented in Fig. 2. First, it is obvious that the steel structure is made up of refined grains. The band contrast image shown in Fig. 2(a) confirms the dominant presence of bainitic-ferrite grains across the scanned area. Such a decrease in grain size is required to achieve the necessary strength required in high strength pipeline steel. The deployment of thermomechanically controlled processing (TMCP) in the production of steel plate warrants intense deformation followed by accelerated cooling, which gives rise to the end microstructure.28 These authors identified TMCP as a means of improving the mechanical properties of steel through grain refinement. This implies that the processing technique adopted in producing the current pipeline steel favoured the development of fine grains. Similarly, low angle grain boundaries (LAGBs) marked in lime colour are seen dispersed amongst prominent manifestation of high angle grain boundaries (HAGBs) represented as black lines in Fig. 2(b). Grain boundaries are categorised with respect to the misorientation angle (θ). The LAGBs are classified within 5° < θ < 15°, while the HAGBs include grains with θ value ranging from 15° < θ < 62.5°. Also, the phase distribution map presented in Fig. 2(c) indicates a strong presence of body centered cubic (BCC) iron in a ferrite phase. Insignificant manifestations of face centered cubic (FCC) iron are red spots, which correspond to the austenite phase. One can simply say that the steel is primarily made up of BCC iron. The matching IPF (inverse pole figure) orientation map is presented in Fig. 2d. Although the grains are randomly oriented, the [111]||ND grains (blue coloured) are in relatively larger proportion with slight deviation towards [011]||ND (green coloured). Similar grain orientation patterns have been established elsewhere in a previous study on X70 pipeline steel.29 In addition, the IPF attached to Fig. 2d clearly shows higher intensity at the [111] pole compared to the [011] pole. This corroborates the earlier claim that [111] grains featured more in the scanned area. However, increased rolling deformations impart a higher degree of local average misorientation (LAM). According to Fig. 2(e) the steel comprises of more misoriented grains. The LAM analysis was performed on grains having θ values that are less than 5°. In a similar manner, highly deformed grain structures (red coloured region) are noticeable in Fig. 2(f). The fraction of the deformed region amounted to 85.4%; whereas the recovered and recrystallized regions accounted for only 8.3 and 6.4%, respectively. The microstructure observed in the test material consists of features that are quite typical of hot rolled steel.
3.2. NMR and FTIR spectroscopy of synthesized allyl sulfonate graft chitosan copolymers
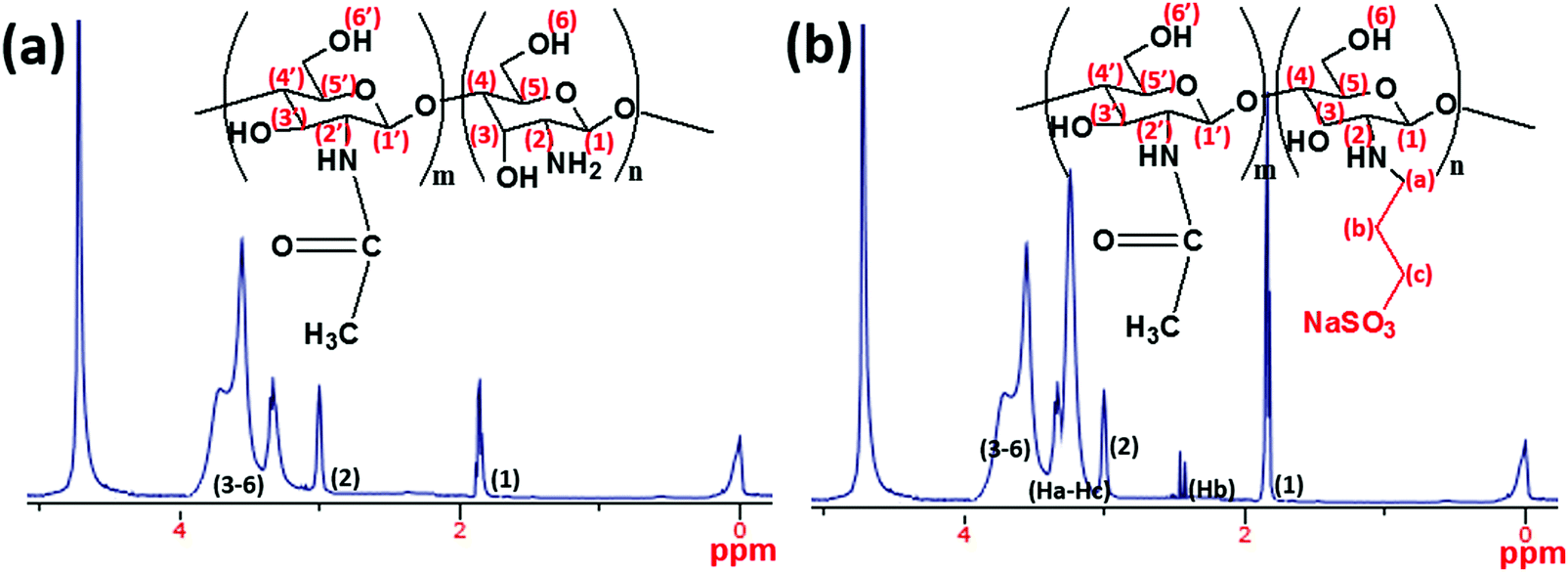
In the present study, a synthesis route for introducing allylsulfonate within chitosan (CH) molecular chain via its amino groups was presented via Michael addition reaction. Most importantly, this reaction was chosen in order to exploit the susceptibility of the sulfonate group to metal–surface interactions during corrosion inhibition. The final product is an allyl sulfonate graft chitosan (CH(S)) copolymer with a 90% computed product yield. It was formed after reacting both precursors via their chitosanic amino chemical group and the allyl double bonds.30 The elaborate synthetic formation scheme is presented in Fig. 1. Before modification with rGO, the resultant amorphous and fibrous chitosanic powder was characterized using appropriate spectroscopic techniques. The comparative proton 1H NMR spectra of this chitosan derivative CH(S) and its CH precursor are presented in Fig. 3(a and b). Fig. 3(b) depicts the NMR spectrum of the new allylsulfonate graft chitosan copolymer derivative. When compared to proton signals of the CH precursor, the new peaks between 2 and 3.5 ppm could be attributed to Ha–Hc protons after the substitution of the N-alkylated group to the amino group by the allyl double bonds. The proton signal of the –CH2 group for CH(S) is very intense at 1.89 ppm relative to CH. A closer view at this spectrum also reveals that a singlet peak could be consistent with the presence of acetyl proton of the N-acetylglucosamine (GlcNAc) at 1.85 ppm.30,31 Apart from the survival of this acetyl proton (H1), there is also another singlet at 2.98 ppm due to H2 of GlcNAc and N-alkylated GlcNAc groups. The proton peaks between 3.52 and 3.72 ppm are consistent with protons H3 to H6.
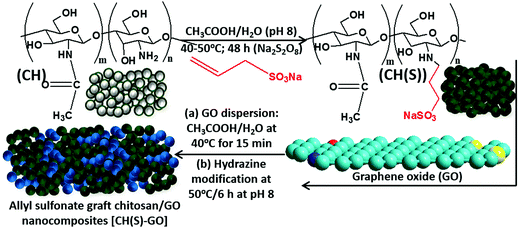 |
| | Fig. 1 Formation scheme for the allyl sulfonate graft chitosan/graphene oxide (CH(S)–GO) nanocomposite from chitosan: allyl sulfonate graft chitosan CH(S) was synthesized by an acetic-acid catalyzed reaction involving chitosan (CHS) and allyl sulfonate in alkaline pH in the presence of a sodium persulfate initiator. | |
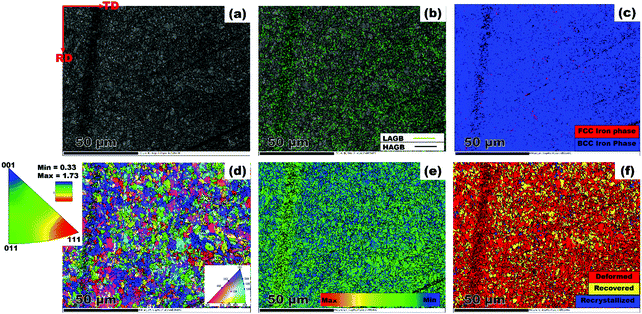 |
| | Fig. 2 EBSD maps showing (a) band contrast (b) grain boundary character (c) phase map (d) IPF orientation distribution (e) LAM (f) area fraction of recrystallized, recovered and deformed regions. | |
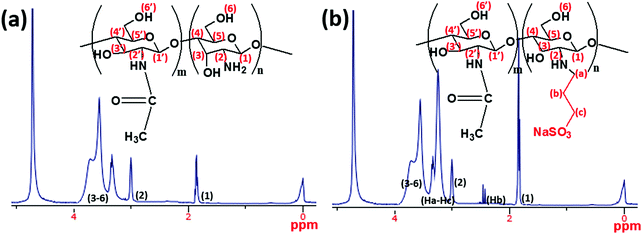 |
| | Fig. 3
1H NMR spectra of (a) chitosan (CH) and (b) the synthesized allyl sulfonate graft chitosan (CH(S)) copolymer utilized as a corrosion inhibitor within the present study (solvent: premixed CD3COOD and D2O). Broadened peaks are due to inherent interactions between some chitosanic protons. | |
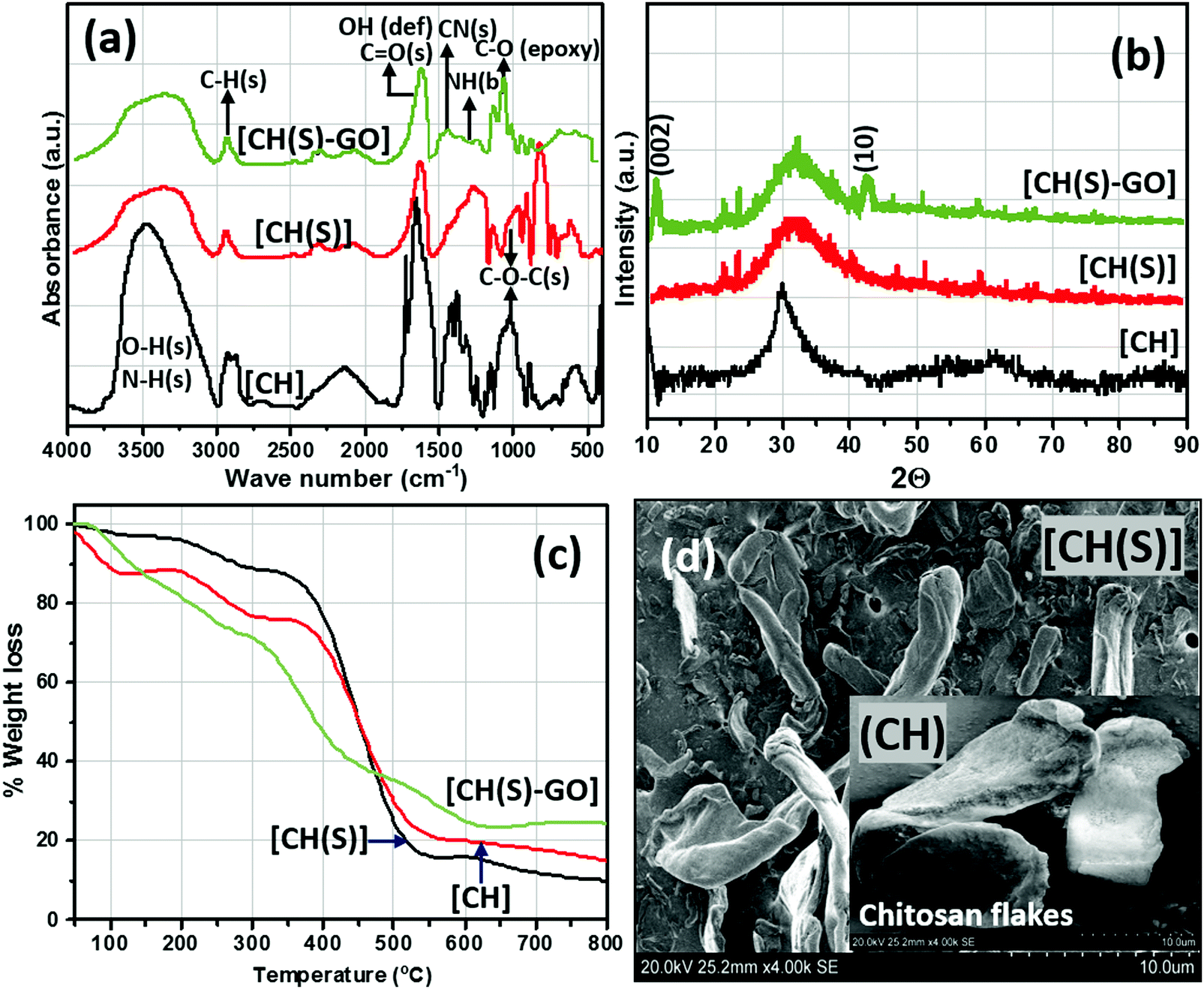
After grafting CH(S) onto the graphene oxide nanosheets via chemical grafting, the resultant nanocomposite was characterized using appropriate techniques. Fig. 4(a) depicts the FTIR spectra of the as-synthesized allyl sulfonate graft chitosan/graphene oxide (CH(S)–GO) nanocomposite and the CH(S) product as well as its CHS precursor. Common among these spectra are stretching vibration broad envelops of –OH, –NH at 3450 cm−1 and C–H at 2880 cm−1 as well as a glucosamine carbonyl IR peak at 1733 cm−1 from the ester bond. Peaks consistent with glycosidic C–O–C linkage bonds are located around 1040–1150 cm−1 while those due to symmetric and asymmetric stretching vibrations of the glucosamine carbonyl group and OH deformation could be seen around 1500–1750 cm−1, respectively.32,33 There are also N–H and C–N bending vibration peaks at 1560 and 1500 cm−1 while those at 1380 and 1420 cm−1 are consistent with C–N axial stretching and N–H deformation, respectively.34 In comparison with the spectrum of CH, the IR absorption signal at 2920 cm−1 for the CH(S) spectrum is consistent with the –CH2− bending vibration. This is suggestive of complete chemical grafting of the allylsulfonate chemical group within the chitosanic chain. The observed IR peaks at 1050 and 1195 cm−1 are also due to the contributions of the sulfonate group.30 Since the CH(S)–GO nanocomposite was incorporated with rGO nanosheets, its FTIR spectrum shows characteristic peaks consistent with rGO. Peaks linked with peroxide/epoxy chemical groups and O–H deformation are found at 957 and 1404 cm−1, respectively, while those of alkoxy and epoxy carbonyl stretching vibrations are located at 1049 and 1227 cm−1. The presence of these peaks is due to inherent hydroxyl, carboxyl and epoxide chemical groups on the rGO nanosheets.35 The results of both spectroscopic techniques reveal a successful grafting of the allylsulfonate group within the chitosanic molecular chain via its amino groups via the Michael addition reaction. Secondly, CH(S) grafted graphene oxide (GO) particles reveal absorption peaks consistent with both primary components, CH(S) copolymers and GO nanosheets.
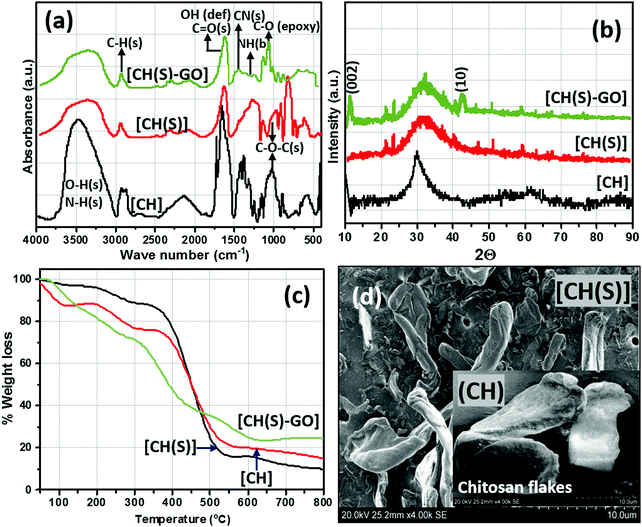 |
| | Fig. 4 FTIR (a) XRD (b) and TGA (c) curves and SEM micrographs (d) of CH(S) and CHS(S)–GO nanocomposites; inset: SEM of CHS powder of unmodified chitosan (CH) compared to the as-synthesized allyl sulfonate graft chitosan (CH(S)) and allyl sulfonate graft chitosan/graphene oxide (CH(S)–GO) nanocomposite. | |
3.3. Characterization of allyl sulfonate graft chitosan/GO nanocomposites and rGO nanosheets
The XRD patterns of the as-synthesized allyl sulfonate graft chitosan CH(S), its nanocomposite (CH(S)–GO) and the CH precursor are depicted in Fig. 4(b). Between these three materials, their X-ray diffractograms show strong reflection between 2θ = 20 and 35° and a few at 21.3° and 22.4°, except for pure chitosan (CH). There are wide peaks between 2θ = 25 and 40° that could be ascribed to highly crystalline chitosan (due to the influences of inter-molecular and extra-molecular hydrogen bonding). There are resolved diffraction peaks linked with 002 and 10 reduced GO planes for the CH(S)–GO nanocomposite between 2θ = 11 and 2θ = 42.26°, respectively, and this may be indicative of the prevalence of short-range order within stacked graphene layers.36 The observed differences could be attributed to inherent degrees of crystallinity as a result of reduced GO content and extra-molecular hydrogen bonding.30 Apart from the inorganic content, between CH and its graft copolymer derivatives, the differences in fingerprint peaks (2θ) at 21.1° and after 40° could be due to varying degrees of crystallinity between these chitosanic materials. This is consistent with the disruption of the molecular structure with the presence of sodium allyl sulfonate moieties grafted to the chitosan polymer chain.30 These differences are also reflected in their thermogravimetric (TG) curves depicted in Fig. 4(c). These curves were recorded under an Ar atmosphere, and regions of higher weight loss are contributions linked with the compactness of their organic polymer structures and inorganic GO content.9,30 Compared to unmodified chitosan (CH), the SEM of the as-synthesized CH(S) copolymer shows differences in fibrous morphology after allyl sulfonate grafting (Fig. 4d). The surface morphology of the CH flakes shows larger and agglomerated particles and less amorphous compared to that of the CH(S) copolymer.9 Prior to SEM imaging, these vacuum-dried powdery CH and CH(S) grains were simply placed on conducting tapes on SEM sample holders with no further treatments, except for thin film Au coating (10 nm; Quorum Q150T ES). The presented morphologies are consistent for dried samples. Solubilization and drying of synthesized CH(S) powder could have affected its morphology compared to that of as-received CH.
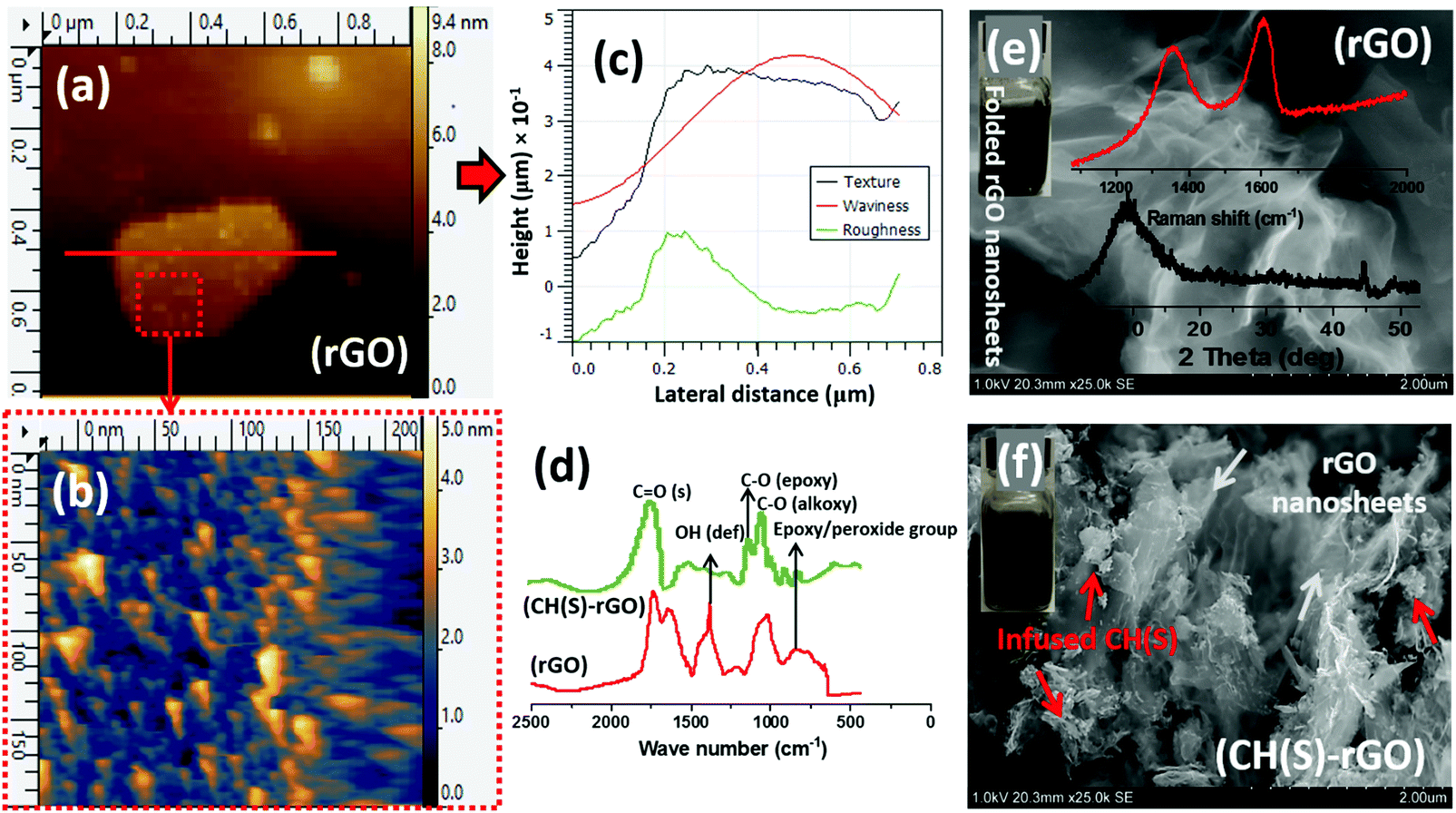
The surface morphologies and chemical makeup of the reduced graphene oxide in this study were also investigated using AFM and SEM techniques as well as ATR-FTIR spectroscopy, respectively. After the modification step presented in section 2.4, the reduced GO nanosheets and CH(S)–GO nanocomposites were evenly dispersed in water at 60 °C. Their suspension remained stable for several days without the need for capping reagents. The AFM micrographs (Fig. 5a and b) show distinct monolayers and still preserved individual nanosheet motifs37 while the adjacent height profile shows variations of some surface parameters mapped from the surface AFM topology (Fig. 5c). The measured thickness in these nanosheets is between 1 and 1.5 nm and few nm of lateral dimension. AFM scans were collected in tapping mode while Gwyddion software was utilized for post-imaging processing, surface profiling and dimensional analyses of the rGO nanosheet. Between the ATR-FTIR spectra of both rGO nanosheets and CH(S)–GO nanocomposites, there are distinct peaks consistent with C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching and O–H deformation vibration at 1733 and 1404 cm−1, respectively. There are also peaks at 1227 and 1049 cm−1, attributed to C–O from epoxy and alkoxy stretching vibrations, respectively, and these chemical groups are due to inherent residual oxygen, hydroxyl, periphery carboxyl and epoxide groups on the rGO nanosheet surfaces. There is also a peak at 957 cm−1 consistent with epoxy (or peroxide) groups.38 He et al.38 have opined that inherent interactions between hydrogen bonds within these functionalities further disrupt the π–π stacking between nanosheets in turn, averting particulate agglomeration. The glucosamine carbonyl IR peak at 1733 cm−1 and the glycosidic C–O–C linkage bonds are in the range around 1040–1150 cm−1.32,33 The SEM micrographs of rGO nanosheet powder (Fig. 5e) show rGO nanosheets with folded and corrugated morphology while allyl sulfonate graft chitosan copolymer powder could be observed infused within rGO sheets within the nanocomposite (Fig. 5f). Raman and XRD patterns of rGO nanosheets are also shown in the inset. The first spectrum shows D mode due to DGO vacancies while the D mode to G mode ratio (ID/IG) is more than unity due to the contributions of hydrazine.37 On the Raman spectrum, the D band is lined with the symmetry A1g mode and G band with the E2g mode of sp2 carbon atoms.37 The BET-derived surface area and pore volume of these rGO nanosheets were measured to be 3.5581 m2 g−1 and 0.0244 cm3 g−1.
O stretching and O–H deformation vibration at 1733 and 1404 cm−1, respectively. There are also peaks at 1227 and 1049 cm−1, attributed to C–O from epoxy and alkoxy stretching vibrations, respectively, and these chemical groups are due to inherent residual oxygen, hydroxyl, periphery carboxyl and epoxide groups on the rGO nanosheet surfaces. There is also a peak at 957 cm−1 consistent with epoxy (or peroxide) groups.38 He et al.38 have opined that inherent interactions between hydrogen bonds within these functionalities further disrupt the π–π stacking between nanosheets in turn, averting particulate agglomeration. The glucosamine carbonyl IR peak at 1733 cm−1 and the glycosidic C–O–C linkage bonds are in the range around 1040–1150 cm−1.32,33 The SEM micrographs of rGO nanosheet powder (Fig. 5e) show rGO nanosheets with folded and corrugated morphology while allyl sulfonate graft chitosan copolymer powder could be observed infused within rGO sheets within the nanocomposite (Fig. 5f). Raman and XRD patterns of rGO nanosheets are also shown in the inset. The first spectrum shows D mode due to DGO vacancies while the D mode to G mode ratio (ID/IG) is more than unity due to the contributions of hydrazine.37 On the Raman spectrum, the D band is lined with the symmetry A1g mode and G band with the E2g mode of sp2 carbon atoms.37 The BET-derived surface area and pore volume of these rGO nanosheets were measured to be 3.5581 m2 g−1 and 0.0244 cm3 g−1.
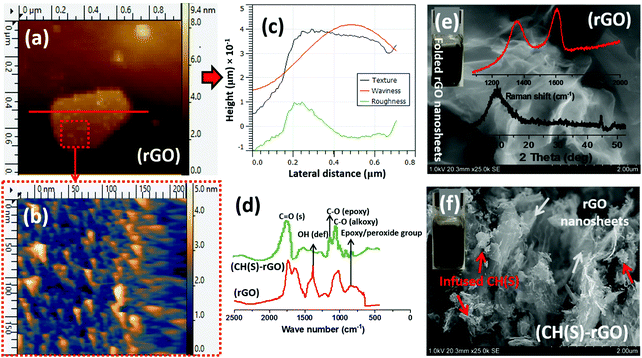 |
| | Fig. 5 AFM micrographs showing rGO nanosheets (a and b); (c) height profile showing variations of some surface parameters mapped from the surface AFM topology. Comparative (d) ATR-FTIR spectra and SEM micrographs of reduced GO nanosheet powder (e) and allyl sulfonate graft chitosan/graphene oxide (CH(S)–GO) nanocomposite powder (f). Inset: Raman and XRD of reduced GO nanosheets; aqueous suspensions (0.1 mg mL−1 rGO and CH(S)–GO nanocomposites). | |
3.4. Examination of inhibition by electrochemical techniques
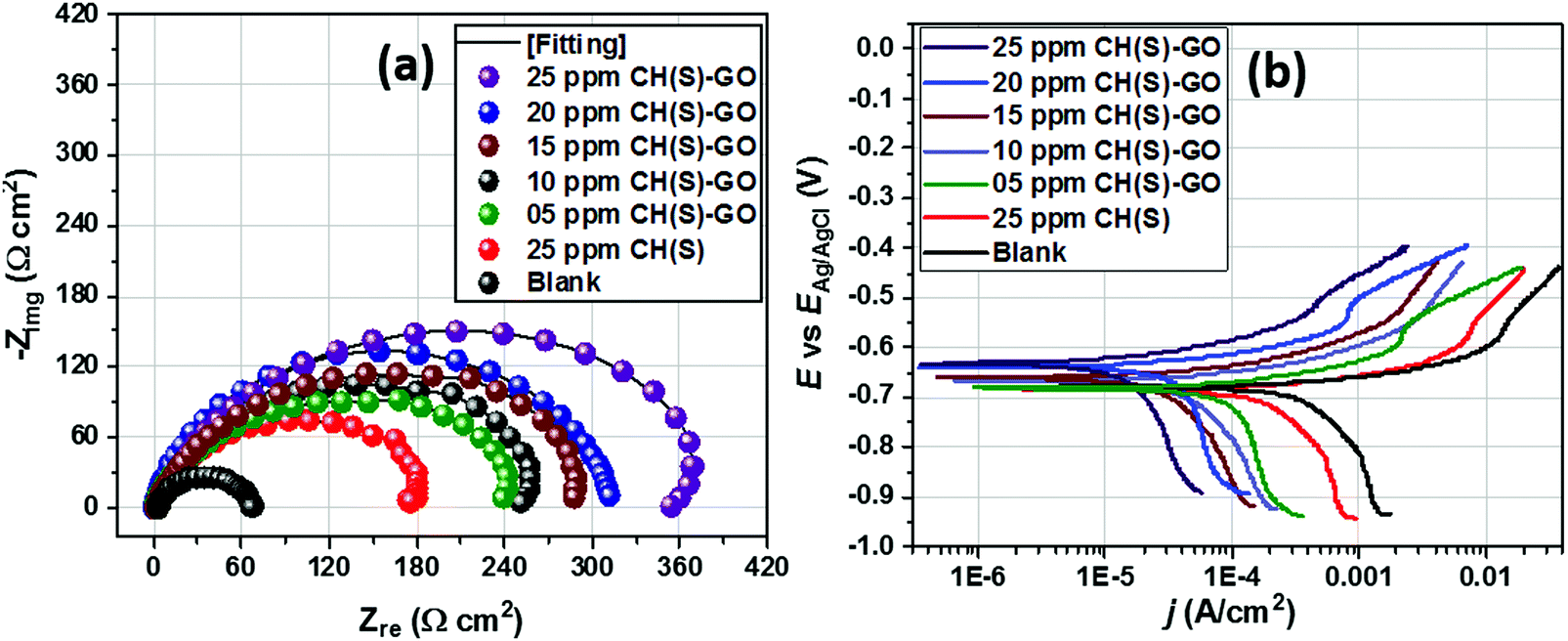
The EIS technique is a suitable tool for the characterization of adhering inhibitor layered formations on metal surfaces due to its ability to probe kinetic controls and transport processes associated with corrosion.26 In the present study, EIS was utilized in studying the electrochemical processes connected with the dissolution of the steel substrate in the presence of a hybrid film-forming CH(S)–GO polymer nanocomposite. The impedance curves for steel substrates in CO2 saturated NaCl electrolytes containing different concentrations of nanocomposites are depicted in Fig. 7(a). These curves are single-loop capacitive semicircles, and this could be linked with reactions associated with charge transfers.9 Wider curves are consistent with concentrations of nanocomposites that offered the most inhibition against steel corrosion, and this could be attributed to the formation of passive inhibitor films on these metallic surfaces.9–11 This trend is also consistent with an increase in charge transfer resistance (Rct) with increasing nanocomposite concentrations; magnitudes of Rct up to 61.4, 214.7, 290.0 and 361.4 Ω cm2 were recorded for 0, 5, 15 and 25 ppm. These impedance curves were fitted into an appropriate circuit model (Rsoln(Qdl(Rct))) and the aforementioned values of Rct as well as other electrochemical parameters were obtained as presented within the supporting information (Table 2). Rct and Rsoln denote the values of resistance associated with the charge transfer and the acidic solution, respectively. Qdl is the constant phase element (CPE, Yo), and it is the capacitance components introduced to make up for defects within the double layer and metallic surface.10 Its impedance is defined as 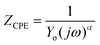 . Here, the magnitude of j, an imaginary factor, is equal to
. Here, the magnitude of j, an imaginary factor, is equal to 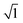 ; ω is equivalent to 2πf, where ω (rad s−1) and f (Hz) are angular and real frequencies. α and Yo are associated with the nature of electroactive species and phase shift, respectively. The magnitudes of Qdl were observed to decrease with the increasing nanocomposite concentration, and this was consistent with the gathering of passive films that gradually displaced adsorbed water molecules at the metallic steel surfaces. These physical surface events isolated the metallic surfaces from further corrosion. Also, the corrosion inhibition of metallic substrates is controlled by transient responses of associated reactions at metal–electrolyte interfaces as well as the presence of adhering corrosion products/passive inhibitor films.25
; ω is equivalent to 2πf, where ω (rad s−1) and f (Hz) are angular and real frequencies. α and Yo are associated with the nature of electroactive species and phase shift, respectively. The magnitudes of Qdl were observed to decrease with the increasing nanocomposite concentration, and this was consistent with the gathering of passive films that gradually displaced adsorbed water molecules at the metallic steel surfaces. These physical surface events isolated the metallic surfaces from further corrosion. Also, the corrosion inhibition of metallic substrates is controlled by transient responses of associated reactions at metal–electrolyte interfaces as well as the presence of adhering corrosion products/passive inhibitor films.25
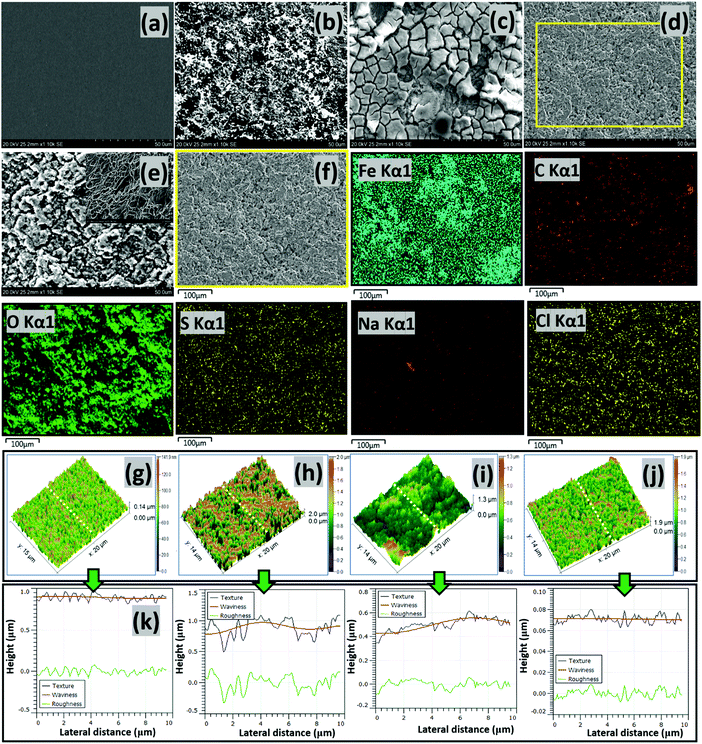 |
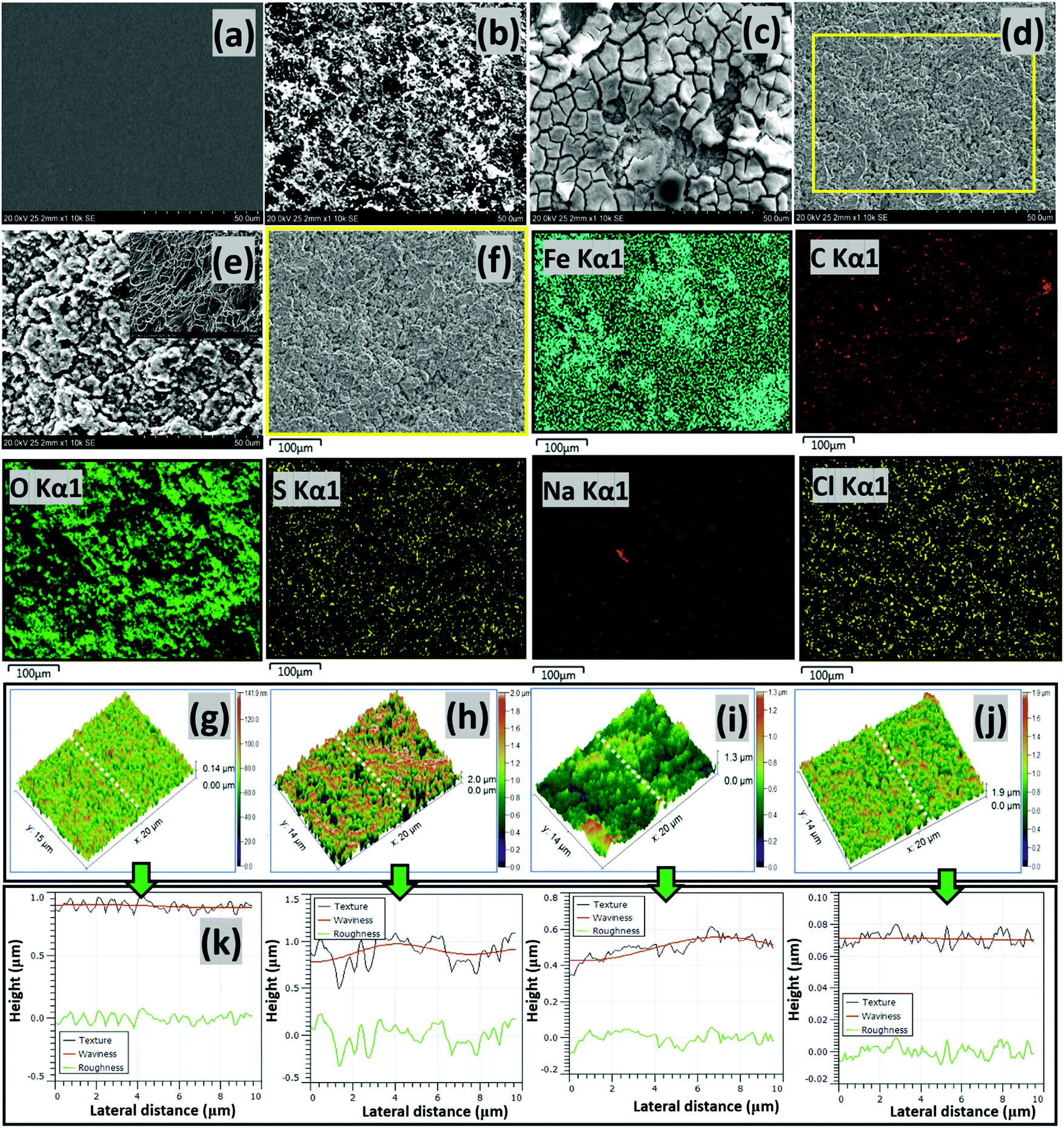
| | Fig. 6 SEM micrographs of precleaned untested (a) and tested (b) steel surfaces exposed to the CO2 saturated NaCl electrolyte without corrosion inhibitors, as well as tested steel strips within electrolytes containing the CH(S) (c) and CH(S)–GO nanocomposite (d) at 60 °C. Surface morphologies of tested steel strips showing adsorbed corrosion product/carbonate scales (e) and adsorbed CH(S)/GO films (f) on steel substrates as well as corresponding EDS micrographs showing selected elements (Fe, C, O, S, Na and Cl (Kα1)) from adsorbed CH(S)–GO nanocomposite films. Inset: surface morphology showing the extent of surface pits (e) on steel at high magnification; Fe, Na and Cl originate from the base metal and electrolyte, respectively. The AFM micrographs of precleaned untested (g) and tested (h) steel surfaces exposed to the CO2 saturated NaCl electrolyte without corrosion inhibitors, as well as tested steel strips within electrolytes containing CH(S) (i) and CH(S)–GO nanocomposite (j); the third column depicts surface line profiles (k) showing the roughness variation for steel substrates exposed to these media. | |
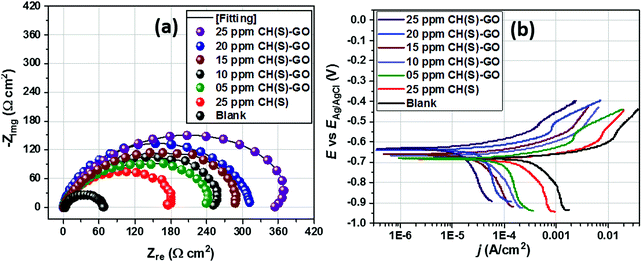 |
| | Fig. 7 Nyquist (a) and polarization (b) curves for steel substrates exposed to CO2 saturated NaCl media modified with different concentrations of hybrid CH(S)–GO nanocomposites compared to the unmodified allyl sulphonate graft chitosan polymer (CH(S)). | |
Table 2 EIS and polarization parameters for metallic steel substrates exposed to CO2 saturated NaCl solutions with CH(S)–GO nanocomposites and CH(S) contents
| System under study |
EIS |
Potentiodynamic polarization |
|
R
soln (Ω cm2) |
R
ct (Ω cm2) |
CPE, Yo (μF cm−2 s−(1−αc)) |
η% |
E
corr (V vs. Ref.) |
j
corr (μA cm−2) |
β
a (mV dec−1) |
β
c (mV dec−1) |
η% |
| Values of goodness of fit (χ2) are less than 10−4; values of α within this study are unity or close to unity. The magnitudes of corrosion inhibition efficiency (η%) were also computed from values of Rct and jcorr obtained from the EIS and polarization techniques, respectively. For each technique, these computed parameters were obtained in relative terms between steel substrates within the blank and in the presence of inhibitors. |
| Blank (0 ppm) |
2.3 |
61.4 |
658.8 |
— |
−0.68 |
855.0 |
97.3 |
147.5 |
— |
| 25 ppm CH(S) |
4.5 |
185.1 |
245.5 |
66.8 |
−0.68 |
250.0 |
90.4 |
162.1 |
70.7 |
| 5 ppm CH(S)–GO |
5.4 |
214.7 |
196.6 |
70.4 |
−0.67 |
225.0 |
97.8 |
150.4 |
73.7 |
| 10 ppm CH(S)–GO |
4.5 |
244.5 |
178.7 |
74.9 |
−0.67 |
200.0 |
112.7 |
182.7 |
76.6 |
| 15 ppm CH(S)–GO |
2.1 |
290.0 |
149.5 |
78.8 |
−0.66 |
180.0 |
99.1 |
143.2 |
78.9 |
| 20 ppm CH(S)–GO |
5.2 |
309.9 |
111.2 |
80.2 |
−0.64 |
149.0 |
85.2 |
154.4 |
82.6 |
| 25 ppm CH(S)–GO |
5.2 |
361.4 |
66.8 |
83.0 |
−0.63 |
88.0 |
99.5 |
172.7 |
89.7 |
The kinetics of electron transfer during steel corrosion within this CO2 saturated NaCl medium after application of active dissolution potentials was also investigated using the potentiodynamic polarization technique.11,21 The polarization curves for this metal in nanocomposite doped NaCl media are presented in Fig. 7(b); these results complement those obtained from the EIS technique. After appropriate theoretical fitting, the associated Tafel parameters were collected and are presented in Table 2. The magnitudes of corrosion current density (jcorr) decreased for steel substrates exposed to the CO2 saturated NaCl electrolyte with increasing nanocomposite concentrations, relative to the untreated corrosive chloride medium. This process was initiated by steel surface coverage by protective nanocomposite films and subsequent impedance of the corrosive currents of chloride ions. This trend of jcorr values is consistent with corrosion inhibition in the presence of adsorbed nanocomposites on steel.11 Magnitudes of jcorr up to 855, 225, 180 and 88 μA cm−2 were recorded for 0, 5, 15 and 25 ppm nanocomposites. Corrosion inhibition could also have been fostered by molecular adsorption due to the electron transfer and covalent bond formation between partially occupied Fe+2 orbitals.9–11 Interfacial adsorption of protective nanocomposite films on steel by chemisorption could have been possible via donor–acceptor interactions. Values of Ecorr are also shifted to noble or positive values with increasing nanocomposite concentrations, and this is suggestive of the predominantly anodic dissolution mechanism for steel substrates in the presence of the nanocomposite.10 Enhanced corrosion inhibition was realized for the 25 ppm CH(S)–GO nanocomposite (η% = 89.7) compared to the same concentration of its CH(S) graft polymer analogue (η% = 70.7).
3.5. Analyses of metal surface morphologies after corrosion tests
The results obtained from the two electrochemical techniques have revealed inhibition in steel corrosion at inceased nanocomposite concentration. This led to in-depth analyses of metallic surfaces after complete exposure to nanocomposite doped CO2 saturated saline medium at 60 °C for 24 h using SEM and AFM. Fig. 6 depicts the SEM micrographs of untested and abraded substrate (a) as well as treated steel substrates within saline without (b) and with 25 ppm allyl sulfonate graft chitosan CH(S) (c) and CH(S)–GO nanocomposites (d). Compared to the unimpacted metallic surface, chloride-induced and CO2 corrosion attacks had led to unimpeded surface damage, observed as surface protrusions and grooves.10 There were also evidence of significant pitting and formation of corrosion products and crystalline carbonate scales due to CO2 led steel dissolution within the blank corrosive medium.2,10 The presented surface morphology also shows the extent of surface pits on steel at high magnification after removing the adsorbed corrosion product/carbonate scales (e). However, a significant improvement in corrosion inhibition is observed in the presence of nanocomposites due to the formation of protective inhibiting organic CH(S) and CH(S)–GO organic/inorganic hybrid nanocomposite films on the steel substrate (f). Film formation is confirmed in the presented EDS micrographs; C, O and S are the elemental composition of the adsorbed nanocomposite; Fe, Na and Cl originate from the base metal and electrolyte, respectively. This also led to the reduction in the surface roughness for steel in NaCl due to the reduced corrosion attack and impedance of corrosive ion currents as passive nanocomposite films formed on steel by molecular adsorption.2,10 The scales on the steel in CH(S) doped medium are broken due to the excessive dehydration effect during surface drying prior to imaging while the protective films in the presence of the hybrid nanocomposite are more orderly formed and morphologically compact.
The results from SEM morphology were also complemented by AFM surface analysis; similar trends in surface morphology were recorded from both techniques as depicted in Fig. 6(g–j). The roughness of the steel surface reduced upon addition of the nanocomposite. The mean surface roughness (RRMS) of the steel substrate within the undoped CO2 saturated saline electrolyte was 667 nm while 456 and 301 nm were recorded for steel substrates within CH(S) and CH(S)–GO nanocomposite doped medium. This trend shows evidence of corrosion inhibition in the presence of CH(S) and CH(S)–GO. A lower value of surface roughness (114 nm) was recorded for the untested surface, since it was carefully polished and has an even film-free surface structure. The nanocomposite films are usually multilayered and add extra roughness to corrosion impacted surfaces; this trend could be attributed to the corrosion inhibition in line with steel surface roughness lowering. This corrosion inhibiting property of nanocomposite is consistent with its adsorption at the Fe substrate/chloride electrolyte interface. Corrosion inhibition is a consequence of mass transport processes involving nanocomposite adsorption and its film formation on steel.2,10 Molecular adsorption could be a physical process or chemisorption as a result of electrostatic interactions between charged surfaces and inhibitor molecules. The variation of different surface parameters (texture, waviness and roughness) could be seen on the last column of Fig. 6(k). These parameters were profiled from the same AFM micrographs mapped in three-dimensions. The surface of the steel substrate exposed to the blank CO2 saturated chloride electrolyte was significantly rough and textured with uneven microstructural morphology due to unhindered CO2-induced saline corrosion. This was not observed in the presence of nanocomposites. The prevalence of iron carbonate scale formations on the surface of the substrates is due to the reaction between both ferrous and carbonate ions as depicted in eqn (1).10
3.6. XPS analyses of adhering protective films on tested steel surfaces
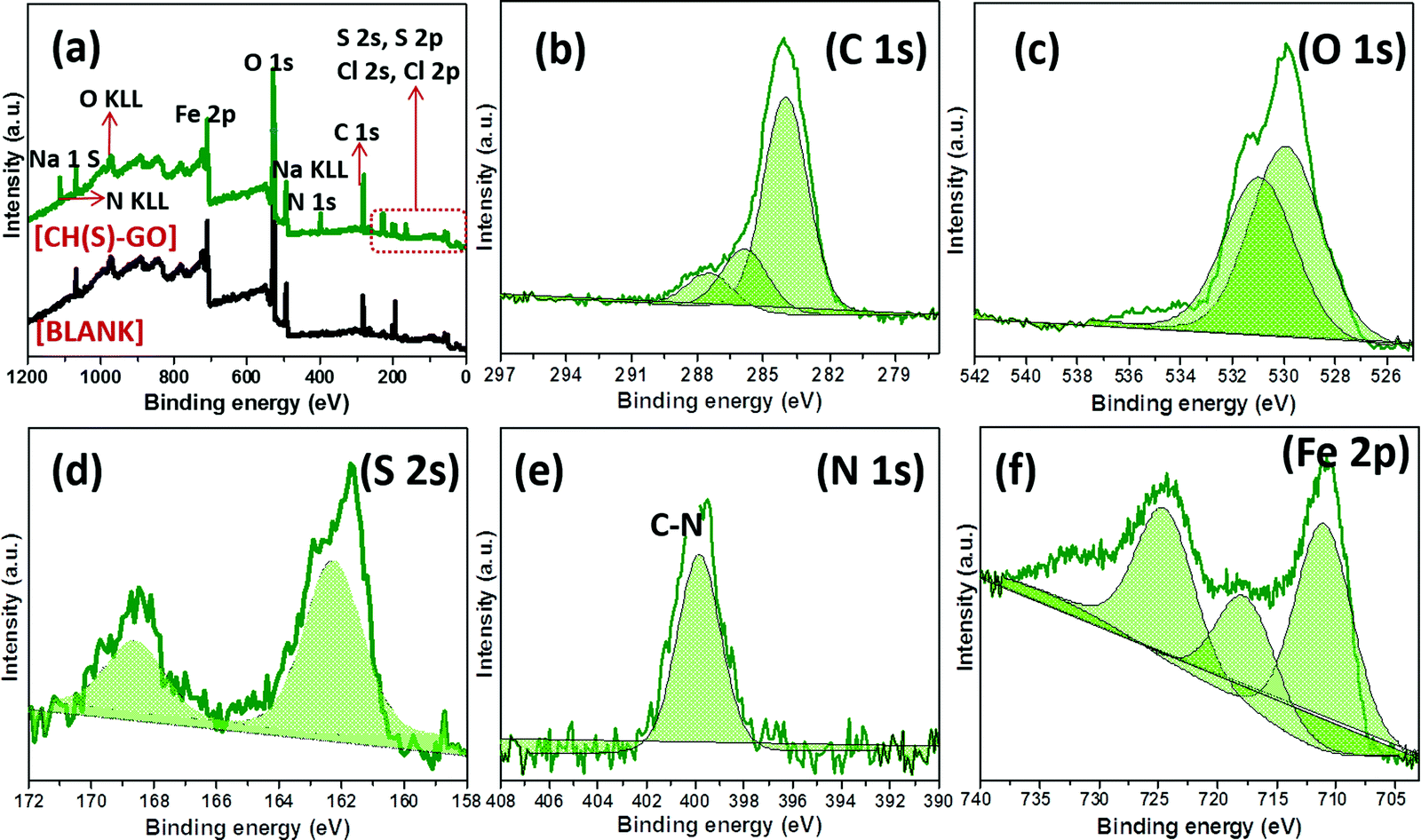
Evidence of metal–surface molecular adsorption by the hybrid CH(S)–GO nanocomposite was probed by XPS technique. Wide-scan XPS spectrograms of these adhering corrosion inhibitor/corrosion product aggregates are presented in Fig. 8, without (blank) and with the CH(S)–GO inhibitor in the CO2 saturated NaCl media. Varying chemical compositions are revealed on both spectrograms within the range of binding energy under study (0 to 1200 eV). Elemental components consistent with hybrid nanocomposites are depicted in Fig. 8(a); this spectrogram shows core level peaks N 1s, S 2s, O 1s, C 1s, etc.9–11 This confirmed the adsorption of the inhibitor aggregates as protective inhibitor films on the charged metallic steel surface, and these films contain surface functional groups containing oxygen (absorbed water and hydroxyl/carboxyl groups) from the reduced GO nanosheets. This carbon-based material is bound by the sp2 network bearing within its electrically conductive reduced GO network.36 Without this inhibitor nanocomposite, corrosion of steel also led to the formation of corrosion product scales on the metallic surface within the CO2 saturated NaCl acidified medium due to the accumulation of FeCO3 scales at 60 °C.9,35 Peaks consistent with these carbonate scales could also be found at Fe 2p (at 709.5 eV), C 1s (at 284.4 eV) and O 1 s (531.4 eV) while those related to the corrosive electrolyte are located at 1071.2 eV (Na 1 s), 507.3 eV (Na KLL), 270.1 eV (Cl 1s) and 198.8 eV (Cl 2p). The deconvoluted high-resolution XPS spectra for selected elements were also recorded in the presence of the CH(S)–GO nanocomposite; these spectra are presented in Fig. 8(b–f). The C 1s XPS spectrogram (b) shows triplet peaks and could be centers for metal adsorption. The peak at 285.0 eV could be ascribed to the C–C bond from the adsorbed hybrid protective nanocomposite film39,40 while peaks at 286.5 eV (C–N or C–O–C bond) and 287.9 eV (O–CO bond from carbonyl) are also observed.41 Peaks at 287.3 and 289.2 eV could also be ascribed to FeCO3 scales on steel. On the O 1s XPS spectrogram (c), there are two sub-peaks at 530.5 and 531.5 eV consistent with C–O and HO–CO/or C–O–C, respectively.9–11 Peaks at 530 eV could be assigned to metal–oxygen bonds from metal hydrated oxide and carbonate scales.10 Lu et al.39 have opined that the peak at 529.8 eV could be attributed to O2− from adhering carbonate scales. As presented in the S 2p spectrogram (d), the S peak is deconvoluted into S2p3/2 and S2p1/2 doublets at binding energy zoned between 160 and 171 eV. These peaks could be due to the non-oxidized S sulphonate group, specifically highlighting the C–S bond.42,43 The single peak at 400 eV on the N 1s spectrogram (e) is consistent with C–N bonds from the carbon attached to the amine group abound deacetylated units of D-glucosamine moieties or that of the acetylated unit of the N-acetyl-D-glucosamine. These chemical groups were attached to the adsorbed protective nanocomposite films on steel.44 The Fe 2p spectrogram (f) shows low and high distinct spins from p-orbital electrons. However, Fe 2p XPS spectrum (f) shows a satellite peak at 721 eV while the other peaks with prominent heights could be linked with metallic iron, Fe2+ and/or Fe3+.40
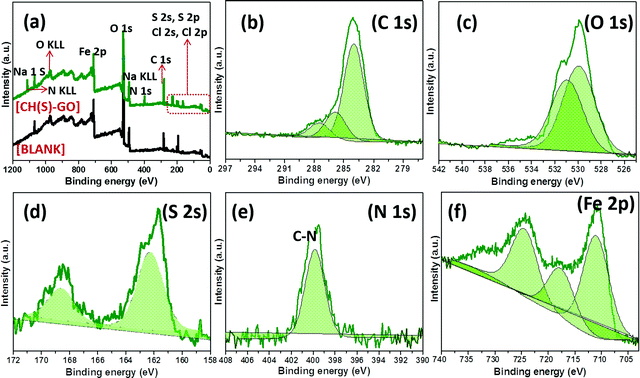 |
| | Fig. 8 XPS spectra of adhering protective nanocomposite films/carbonate scales on steel substrates after exposure to CO2 saturated NaCl at 60 °C modified with allyl sulfonate graft chitosan/graphene oxide nanocomposites (CH(S)–GO); (a) wide scans with (green) and without (black) the CH(S)–GO nanocomposite. High-resolution spectra (b–f) were collected for steel dispersed within the test corrosive medium modified only with the CH(S)–GO nanocomposite. | |
3.7. Surface contact angles of impacted steel surfaces
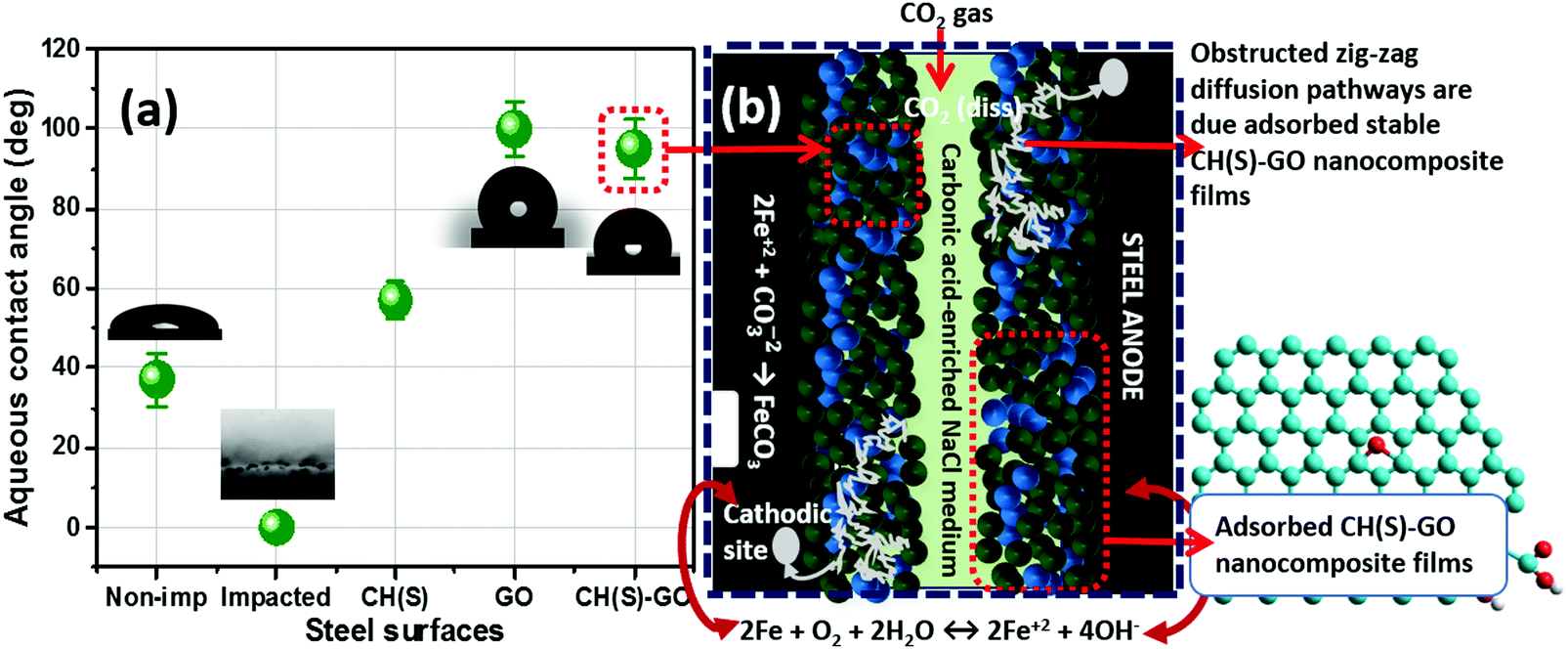
We have established from experimental evidence obtained from surface analytical techniques that corrosion inhibition by the CH(S)–GO nanocomposite was due to the metal–surface molecular adsorption. To further confirm this, the surface contact angle (θow) of each impacted steel surface was measured in a view to assessing surface hydrophobicity using the sessile drop method. Fig. 9(a) depicts the magnitudes of θow collected at room temperature after the corrosion test, relative to untreated metallic surfaces. While the unimpacted (untreated) surface recorded a value of θow up to 39°, the magnitude of this parameter for treated metallic substrate within the blank CO2 saturated NaCl stood at zero degree (0°) due to surface evenness in the presence of adsorbed FeCO3 scales.10 Without the organic–inorganic hybrid inhibitor molecules within the corrodent, steel significantly corroded. As observed in Fig. 6, this surface is roughened with scale-rich and amorphous corrosion product aggregates due to the impact of CO2 corrosion and brine-induced anodic dissolution. Higher values of θow (98°) for CH(S)–GO nanocomposite (100° for reduced GO) treated steel relative to CH(S) (58°) could be attributed to the formation of hydrophobic graphene oxide films. These physically adsorbed protective inhibitor films on steel reveal unsettled globular shaped water droplets due to prevailing low surface energy reduced GO nanosheet grafted hybrid nanocomposites.45–47 The trend of θow values in Fig. 9 shows a correlation between surface wettability and molecular adsorption in the presence of nanocomposites with significantly reduced solid–liquid contact area.
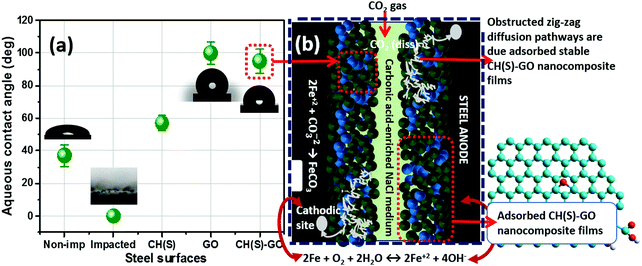 |
| | Fig. 9 (a) Variation of values of aqueous contact-angles of non-impacted and impacted steel surfaces; (b) schematics showing the proposed mechanism of corrosion inhibition by allyl sulfonate graft chitosan/graphene oxide (CH(S)–GO) nanocomposites adsorbed on the steel surface after exposure to CO2 saturated NaCl at 60 °C. At this temperature, the adsorption of the hybrid nanocomposite is also accompanied by FeCO3 scaling and the cathodic formation of HCO3− (2H2CO3+ 2e− → H2+ 2HCO3−) and CO32− (2HCO3− + 2e− → H2+ 2CO32−) at 4 < pH < 6.10 The adsorption of the CH(S)–GO nanocomposite significantly reduced anodic steel dissolution within the media, in turn forming chemical stable and hydrophobic protective films. These compact films further obstruct the diffusion pathways of corrosive ions toward the metal surfaces.8–10 | |
3.8. Proposed mechanism of corrosion inhibition by hybrid nanocomposites
Experimental evidence has revealed severe metal surface pits as consequences of synergistic chloride and CO2 corrosion attacks as presented in the SEM and AFM micrographs (Fig. 6). These anodic dissolution episodes are isolated and localized without the CH(S)–GO nanocomposite within the corrosion media. However, these surface events differ significantly in the presence of corrosion inhibitor nanocomposites after their adsorption on steel.10–12 This complex adsorption mechanism is depicted in the schematics shown in Fig. 9(b). It shows the proposed monolayer adsorption of the allyl sulfonate graft chitosan/graphene oxide (CH(S)–GO) nanocomposite on the steel surface, leading to its corrosion inhibition in CO2 saturated NaCl at 60 °C. At this temperature, the adsorption of the nanocomposites is also accompanied by FeCO3 (eqn (1)) scaling and two cathodic reactions involving the formation of carbonate and bicarbonates.10,11 Weak carbonic acid was also formed when the CO2 gas was dissolved within the aqueous medium (eqn (2)), leading to lower pH values (eqn (3)). This acid further initiated steel corrosion, leading to the anodic reaction that involved Fe2+ ions going into the medium (eqn (4)).10
The adsorption of the CH(S)–GO nanocomposites significantly reduced anodic steel dissolution, in turn forming stable and hydrophobic protective films. The presence of these compact films further obstructed the diffusion pathways of corrosive ions toward the metal surfaces. These CH(S)–GO nanocomposites covered the metal surfaces by physisorption and could also have formed protective surface films due to donor–acceptor electron exchanges. These electron exchanges originate from metal-inhibitor type reactions leading to the electron pair interactions between the chitosanic moiety and vacant 3d-orbitals of the metallic steel substrate.10–13 Here, the chemical structure of the nanocomposite comes into play. The grafted chitosanic copolymer comprises of long polymeric chain bound lone pair electrons that can act as metal–surface bonding sites for strong coordination bond formation. As presented in the results obtained from the XPS technique, these protective (CH(S)–GO) nanocomposite films on steel contain hybrid polymeric graphene oxide and chitosanic functionalities required for stable corrosion-resistance, relative to unmodified allyl sulphonate graft chitosan (CH(S)). If adsorbed monolayers of polymer CH(S) films could significantly distorted anodic or cathodic reaction, those of polymeric nanocomposites would sustain an enhanced degree of surface protection against corrosion due to inherent rGO nanosheets within the composite. Corrosion inhibition was due to the synergistic effects of the individual polymeric components.
| | | CO2(aq) + H2O(l) ↔ H2CO3(aq) | (3) |
4. Conclusions
In this study, allyl sulfonate graft chitosan (CH(S)) was synthesized by an acetic-acid catalyzed reaction involving chitosan (CH) and allyl sulfonate in an alkaline reaction bath. CH(S) was subsequently grafted onto graphene oxide nanosheets. The resultant organic/inorganic hybrid (CH(S)–GO) nanocomposite was then characterized using appropriate techniques and utilized as a corrosion inhibitor for an X70 pipeline steel substrate in a CO2 saturated NaCl electrolyte. Significant corrosion reduction was realized in the presence of the hybrid (CH(S)–GO) nanocomposite. Corrosion inhibition was attributed to the molecular adsorption and formation of protective nanocomposite films on the steel surface. However, dissolution of steel in the nanocomposite doped corrosive electrolyte exhibited a predominantly anodic kinetics. Evidence of molecular adsorption was elucidated by means of surface analytical techniques. Morphological and compositional results from SEM (including AFM) and XPS techniques revealed the presence of adsorbed nanocomposite/carbonate scale aggregates on less corroded steel surfaces. The degree of corrosion inhibition between the (CH(S)–GO) nanocomposite and its CH(S) precursor has been experimentally established, and superior surface protection is revealed in the presence of the nanocomposite. These physically adsorbed protective nanocomposite films on steel contributed to enhanced surface hydrophobicity due to the prevailing low surface energy instigated by the reduced GO nanosheet contents. While the CH(S) copolymer analogue contributed to significant surface coverage, the (CH(S)–GO) polymer nanocomposite adsorption accorded a broad metal–surface protection, hence, was a more effective corrosion inhibitor. The CH(S)–GO polymer nanocomposite offered sustained surface protection against corrosion due to the synergistic effects of its individual polymer components. Without the hybrid nanocomposite, anodic steel corrosion was significant within the undoped electrolyte, and corrosion was attributed to the irreversible action of the CO2 saturated chloride solutions. Before corrosion tests, the pipeline steel substrates utilized within this study was characterized by means of SEM and EBSD techniques. The microstructure of pipeline steel substrates utilized within this study was characterized by using the EBSD technique. Mostly refined bainitic-ferrite grains with orientation deviation towards [111]||ND and [011]||ND directions were observed across the steel structure. Also, the steel displayed significant proportion of deformed regions and increased intensity of grain misorientation.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
The University of Saskatchewan is acknowledged for providing the facilities for this study. This experimental work was initiated and completed while UE was still at the Department of Mechanical Engineering, University of Saskatchewan, Saskatchewan, Canada.
References
- U. Eduok, E. Jossou, A. Tiamiyu, J. Omale and J. Szpunar, Ind. Eng. Chem. Res., 2017, 56, 5586–5597 CrossRef CAS.
- U. Eduok, E. Jossou and J. Szpunar, J. Mol. Liq., 2017, 241, 684–693 CrossRef CAS.
- M. E. Olvera-Martínez, J. Mendoza-Flores and J. Genesca, J. Loss Prev. Process Ind., 2015, 35, 19–28 CrossRef.
- E. Stupnisek- Lisac, S. Podbrscek and T. Soric, J. Appl. Electrochem., 1994, 24, 779–784 CrossRef CAS.
- N. O. Obi-Egbedi, I. B. Obot and M. I. El-Khaiary, J. Mol. Struct., 2011, 1002, 86–96 CrossRef CAS.
- J. Wang, D. Liu, S. Cao, S. Pan, H. Luo, T. Wang, H. Ding, B. B. Mamba and J. Gui, J. Mol. Liq., 2020, 312, 113436 CrossRef CAS.
- I. B. Obot and U. M. Eduok, J. Mol. Liq., 2017, 246, 66–90 CrossRef CAS.
- Y. El Aoufir, R. Aslam, F. Lazrak, R. Marzouki, S. Kaya, S. Skal, A. Ghanimi, I. H. Ali, A. Guenbour, H. Lgaz and I. M. Chung, J. Mol. Liq., 2020, 303, 112631 CrossRef CAS.
- U. Eduok, E. Ohaeri, J. Szpunar and I. Akpan, J. Mol. Liq., 2020, 315, 113772 CrossRef CAS.
- U. Eduok, E. Ohaeri and J. Szpunar, Ind. Eng. Chem. Res., 2019, 58, 7179–7192 CrossRef CAS.
- U. Eduok, E. Ohaeri and J. Szpunar, Electrochim. Acta, 2018, 278, 302–312 CrossRef CAS.
- U. Eduok and J. Szpunar, ECS Meeting Abstracts, 2019, 8, 853 Search PubMed.
- M. Srivastava, S. K. Srivastava, Nikhil, G. Ji and R. Prakash, Int. J. Biol. Macromol., 2019, 140, 177–187 CrossRef CAS.
- D. D. Divakar, N. T. Jastaniyah, H. G. Altamimi, Y. O. Alnakhli, Muzaheed, A. A. Alkheraif and S. Haleem, Int. J. Biol. Macromol., 2018, 108, 790–797 CrossRef CAS.
- E. Badr, H. H. H. Hefni, S. H. Shafek and S. M. Shaban, Int. J. Biol. Macromol., 2020, 157, 187–201 CrossRef CAS.
- R. K. Gupta, M. Malviya, K. R. Ansari, H. Lgaz, D. S. Chauhan and M. A. Quraishi, Mater. Chem. Phys., 2019, 236, 121727 CrossRef CAS.
- H. J. Majidi, A. Mirzaee, S. M. Jafari, M. Amiri, M. Shahrousvand and A. Babaei, Int. J. Biol. Macromol., 2020, 148, 1190–1200 CrossRef.
- S. Singh, G. Singh, N. Bala and K. Aggarwa, Int. J. Biol. Macromol., 2020, 151, 519–528 CrossRef CAS.
- A. B. Ikhe, A. B. Kale, J. Jeong, M. J. Reece, S.-H. Choi and M. Pyo, Corros. Sci., 2016, 109, 238–245 CrossRef CAS.
- E. M. Fayyad, K. K. Sadasivuni, D. Ponnamma and M. A. A. Al-Maadeed, Carbohydr. Polym., 2016, 151, 871–878 CrossRef CAS.
- N. Kirkland, T. Schiller, N. Medhekar and N. Birbilis, Corros. Sci., 2012, 56, 1–4 CrossRef CAS.
- D. Prasai, J. C. Tuberquia, R. R. Harl, G.
K. Jennings and K. I. Bolotin, ACS Nano, 2012, 6, 1102–1108 CrossRef CAS.
- H. Tian, W. Li, B. Hou and D. Wang, Corros. Sci., 2017, 117, 43–48 CrossRef CAS.
- W. Bai, Q. Sheng and J. Zheng, Talanta, 2016, 150, 302–309 CrossRef CAS.
- K. Haruna, T. A. Saleh, I. B. Obot and S. A. Umoren, Prog. Org. Coat., 2019, 128, 157–167 CrossRef CAS.
- S. J. Garcia, T. A. Markley, J. M. C. Mol and A. E. Hughes, Corros. Sci., 2013, 69, 346–358 CrossRef CAS.
- ASTM G3 - 89 (2010) Standard Practice for Conventions Applicable to Electrochemical Measurements in Corrosion Testing. (https://www.astm.org/DATABASE.CART/HISTORICAL/G3-89R10.htm).
- S. Vervynckt, K. Verbeken, B. Lopez and J. J. Jonas, Int. Mater. Rev., 2012, 57, 187–207 CrossRef CAS.
- E. Ohaeri, J. Omale, A. Tiamiyu, K. M. M. Rahaman and J. Szpunar, J. Mater. Eng. Perform., 2018, 27, 4533–4547 CrossRef CAS.
- M. Jiang, K. Wang, J. F. Kennedy, J. Nie, Q. Yu and G. Ma, Int. J. Biol. Macromol., 2010, 47, 696–699 CrossRef CAS.
- Y. Wu, Y. L. Zheng, C. C. Wang, J. H. Hu and S. K. Fu, Carbohydr. Polym., 2005, 59, 165–171 CrossRef CAS.
- R. A. Ahmed, R. A. Farghali and A. M. Fekry, Int. J. Electrochem. Sci., 2012, 7, 7270–7282 CAS.
- C. J. E. De Souza, M. M. Pereira and H. S. Mansur, J. Mater. Sci. Mater. Med., 2009, 20, 553–561 CrossRef.
- E. M. Fayyad, K. K. Sadasivuni, D. Ponnamma and M. A. A. Al-Maadeed, Carbohydr. Polym., 2016, 151, 871–878 CrossRef CAS.
- D. He, Z. Peng, W. Gong, Y. Luo, P. Zhao and L. Kong, RSC Adv., 2015, 5, 11966–11972 RSC.
- L. Stobinski, B. Lesiak, A. Malolepszy, M. Mazurkiewicz, B. Mierzwa, J. Zemek, P. Jiricek and I. Bieloshapka, J. Electron Spectrosc., 2014, 195, 145–154 CrossRef CAS.
- J. Zhang, H. Yang, G. Shen, P. Cheng, J. Zhang and S. Guo, Chem. Commun., 2010, 46, 1112–1114 RSC.
- D. He, Z. Peng, W. Gong, Y. Luo, P. Zhao and L. Kong, RSC Adv., 2015, 5, 11966–11972 RSC.
- Y. Lu, H. Jing, Y. Han, Z. Feng and L. Xu, Appl. Surf. Sci., 2016, 389, 609–611 CrossRef CAS.
- N. Z. N. Hashima, E. H. Anouar, K. Kassim, H. M. Zaki, A. I. Alharthib and Z. Embong, Appl. Surf. Sci., 2019, 476, 861–877 CrossRef.
- D. H. Wang, Y. Hu, J. J. Zhao, L. L. Zeng, X. M. Tao, W. Chen and H. Holey, J. Mater. Chem. A, 2014, 2, 17415–17420 RSC.
- M. Ahangar, M. Izadi, T. Shahrabi and I. Mohammadi, J. Mol. Liq., 2020, 314, 113617 CrossRef CAS.
- A. Abdul Razzaq, Y. Yao, R. Shah, P. Qi, L. Miao, M. Chen, X. Zhao, Y. Peng and Z. Deng, Energy Storage Mater, 2019, 16, 194–202 CrossRef.
- S. B. Jiang, L. H. Jiang, Z. Y. Wang, M. Jin, S. Bai, S. Song and X. Yan, Construct. Build. Mater., 2017, 150, 238–247 CrossRef CAS.
- T. Ghosh, P. Bardhan, M. Mandal and N. Karak, Mater. Sci. Eng., C, 2019, 105, 110055 CrossRef.
- E. Udabe, M. Forsyth, A. Somers and D. Mecerreyes, Mater. Adv., 2020, 1, 584–589 RSC.
- A. Ul-Hamid, Mater. Adv., 2020, 1, 1012–1037 RSC.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *,
Enyinnaya
Ohaeri
and
Jerzy
Szpunar
*,
Enyinnaya
Ohaeri
and
Jerzy
Szpunar
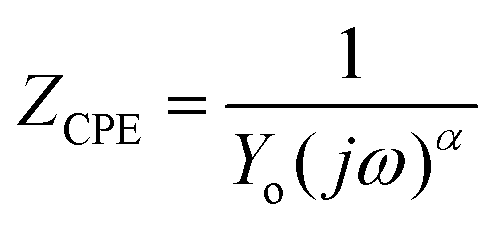 . Here, the magnitude of j, an imaginary factor, is equal to
. Here, the magnitude of j, an imaginary factor, is equal to 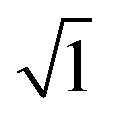 ; ω is equivalent to 2πf, where ω (rad s−1) and f (Hz) are angular and real frequencies. α and Yo are associated with the nature of electroactive species and phase shift, respectively. The magnitudes of Qdl were observed to decrease with the increasing nanocomposite concentration, and this was consistent with the gathering of passive films that gradually displaced adsorbed water molecules at the metallic steel surfaces. These physical surface events isolated the metallic surfaces from further corrosion. Also, the corrosion inhibition of metallic substrates is controlled by transient responses of associated reactions at metal–electrolyte interfaces as well as the presence of adhering corrosion products/passive inhibitor films.25
; ω is equivalent to 2πf, where ω (rad s−1) and f (Hz) are angular and real frequencies. α and Yo are associated with the nature of electroactive species and phase shift, respectively. The magnitudes of Qdl were observed to decrease with the increasing nanocomposite concentration, and this was consistent with the gathering of passive films that gradually displaced adsorbed water molecules at the metallic steel surfaces. These physical surface events isolated the metallic surfaces from further corrosion. Also, the corrosion inhibition of metallic substrates is controlled by transient responses of associated reactions at metal–electrolyte interfaces as well as the presence of adhering corrosion products/passive inhibitor films.25