
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEncapsulation of glycosylated porphyrins in silica nanoparticles to enhance the efficacy of cancer photodynamic therapy†
Wioleta
Borzęcka
abc,
Patrícia M. R.
Pereira
ad,
Rosa
Fernandes
 *de,
Tito
Trindade
*b,
Tomás
Torres
*cfg and
João P. C.
Tomé
*ah
*de,
Tito
Trindade
*b,
Tomás
Torres
*cfg and
João P. C.
Tomé
*ah
aQOPNA-LAQV-REQUINTE and Department of Chemistry, University of Aveiro, 3810-193 Aveiro, Portugal
bCICECO-Aveiro Institute of Materials and Department of Chemistry, University of Aveiro, 3810-193 Aveiro, Portugal
cDepartment of Organic Chemistry, Autonoma University of Madrid, 28049 Madrid, Spain
dCoimbra Institute for Clinical and Biomedical Research (iCBR), Faculty of Medicine, University of Coimbra, Azinhaga de Santa Comba, 3000-548 Coimbra, Portugal
eCNC.IBILI, University of Coimbra, 3000 Coimbra, Portugal
fIMDEA-Nanociencia, Campus de Cantoblanco, c/Faraday 9, 28049 Madrid, Spain
gInstitute for Advanced Research in Chemical Sciences (IAdChem), Universidad Autónoma de Madrid (UAM), 28049 Madrid, Spain
hCentro de Química Estrutural & Departamento de Engenharia Química, Instituto Superior Técnico, Universidade de Lisboa, 1049-001 Lisboa, Portugal. E-mail: jtome@tecnico.ulisboa.pt
First published on 14th December 2020
Abstract
In this study, we encapsulated S-galactosylated and S-glucosylated porphyrins (Pors) into amorphous silica nanoparticles (SNPs) to enhance the photodynamic therapy (PDT) activity. The resulting galacto- and gluco-nanoformulations were demonstrated to be spherical in shape with diameters of 197.3 ± 29.0 nm and 128.3 ± 22.2 nm. The galacto- and gluco-nanoparticles (NPs) were able to produce a high amount of singlet oxygen (1O2) and were stable under the conditions of the experiments. In vitro studies show that the nanoformulations were effectively taken up by the human bladder cancer cell lines HT-1376 and UM-UC-3. The PDT results show that these photoactive nanoformulations are 3 to 5 times more efficient than the non-encapsulated/free Pors. These Por–silica nanoformulations could be successfully used as novel nanocarriers for the delivery of photosensitizer materials for cancer PDT.
Introduction
PDT is a promising therapeutic procedure used in cancer treatment, which combines three components: a drug, visible or near-infrared light and molecular oxygen. On their own these elements do not have any cytotoxic effects; however, when combined they can produce cytotoxic reactive oxygen species (ROS). Increased levels of ROS can elicit oxidative stress and cancer destruction, via direct cancer cell damage and/or activation of an immune response against cancer cells.1–4 The advantage of using PDT over other cancer treatments (e.g. surgery, chemotherapy, and radiotherapy) is that it has lower toxic effects on biological systems.5 PDT was approved by the Food and Drug Administration (FDA) as a clinical protocol for cancer treatment more than 20 years ago, and since then many photosensitizers (PSs) have been developed for PDT, such as HpD, Photofrin, or Temoporfin.6 However, nowadays most of the available PSs are based on the tetrapyrrole structure such as porphyrins with larger hydrophobic groups attached and their solubility in aqueous solutions is often poor, restricting them in clinical applications. Moreover, PDT still has limitations for use as a general protocol to treat cancer. Thus, there is still a need to study alternatives to improve the efficiency of PDT and overcome its limitations, such as low effectiveness in treating large tumors or causing burns, swelling, pain, scarring in nearby healthy tissues or persistent skin photosensitization. For that, it is crucial to not only improve efficient irradiation systems to deliver the activating light, but also to develop new powerful and selective PSs. In the context of new PSs, NPs have recently emerged as promising vehicles for PDT agents, overcoming the limitations of PSs such as low water solubility and low targeting properties.7–11Using PSs in different nanoformulations can improve their biocompatibility, blood circulation, and selective accumulation in tumor tissues thanks to the enhanced permeability and retention effect (EPR).12–14 Among other nanomaterials, silica nanoparticles (SNPs) have emerged as promising vehicles for PDT owing to their biocompatibility, large surface area, controllable size formation, hydrophilic surface and ability of surface functionalization, hence the possibility for tumor targeting through surface modification.15–19 Moreover, amorphous silica shells can protect entrapped molecules against chemical and biochemical degradation, but at the same time control their release.20,21 There are already nanoparticle-based drug delivery platforms which were approved by the FDA and many others under clinical trials.22–26
Among other parameters, NPs’ interactions with cancer cells depend on the NPs’ size, which influences their active and passive cellular internalization, determining the therapeutic targeting.27 When compared with healthy cells, tumor cells have poor lymphatic drainage and leaky vasculature. Particles with a size ranging from 10 to 500 nm tend to accumulate inside tumor cells and their accumulation is facilitated by lymphatic filtration. On the contrary, very often much smaller ordinary drugs cannot remain in tumors because they return to circulation by a diffusion process.28,29
SNPs are platforms that allow covalent and non-covalent immobilization of PSs outside or inside their structures. Recent studies demonstrated that using SNPs combined with Pors could eliminate aggregation of Pors, the hydrophobic nature of PSs or reduced selectivity for targeted tissues and consequently increase the PDT efficiency.30,31 The pioneering work in this field was described in 2003 by Yan et al.,32 where meta-tetra(hydroxyphenyl)-chlorin embedded into silica nanoparticle platforms demonstrated the ability to generate 1O2. Moreover, the authors have shown that the 1O2 production by the SNPs is higher than the one from the free PS. From that time, scientists have put in a lot of effort to enhance PDT with PS–SNP. For instance, Gao et al.33 in a simple method enhanced the photodynamic selectivity of Pors adsorbed onto SNPs against breast cancer cells. In the same year, He et al.34 developed organically modified silica (ORMOSIL) nanoparticles encapsulated with protoporphyrin IX (PpIX) for direct two-photon PDT. In their research, ORMOSIL nanoparticles were able to successfully destroy HeLa cells. Ho et al.35 also used PpIX with mesoporous silica nanocarriers for selective cancer PDT. These highly efficient, non-cytotoxic drug delivery platforms designed for PDT were phospholipid-capped, PpIX-loaded and fluorescein isothiocyanate (FITC)-sensitized mesoporous silica nanocarriers derivatized with folate. These complex SNPs were effective to kill targeted HeLa human cervical epithelioid carcinoma cells and A549 human lung carcinoma cells in vitro and prevent further tumor growth. Miao X. et al.36 were able to overcome the hydrophobic nature of Photosan-II by loading it into hollow SNPs. By this, they eliminated the difficulties with delivery in the physiological environment and the low photophysical properties due to the aggregation of PSs, which decreased the production of 1O2 for PDT. These NPs enhance the photoactivity of the PS against QBC939 cholangiocarcinoma cells. Later, Wen et al.37 proved that these NPs in in vitro and in vivo experiments on liver cancer in nude mice were more efficient than the PS alone. Qian et al.38 developed Por (HPPH) doped colloidal mesoporous silica nanoparticles for three-photon PDT. The cytotoxic effect of HPPH doped SNP-mediated PDT against HeLa cells was proved.
In the present work 5,10,15,20-tetrakis(4-1′-thio-glucosyl-2,3,5,6-tetrafluorophenyl)porphyrin (SGlc-Por, PS 1) and 5,10,15,20-tetrakis(4-1′-thio-galactosyl-2,3,5,6-tetrafluorophenyl)porphyrin (SGal-Por, PS 2) were chosen as PS (Scheme 1) platforms because there are a number of reports on the potential of glycosylated porphyrins as PDT agents.39–43 In our research we used Pors with S-glycoside bonds rather than O-glycoside because drugs bearing saccharides with O-glycoside linkages are readily hydrolyzed by a variety of enzymatic and non-enzymatic acid/base reactions resulting in short half-lives.44 It is known that aggregation of the PS decreases the efficiency of 1O2 generation. Thus, we decided to deliver PSs inside cancer cells by the use of nanovehicles which could also enhance the stability of these PSs in aqueous media. Hence, PSs were non-covalently encapsulated into an amorphous silica matrix, and then these nanomaterials were studied in cancer PDT.
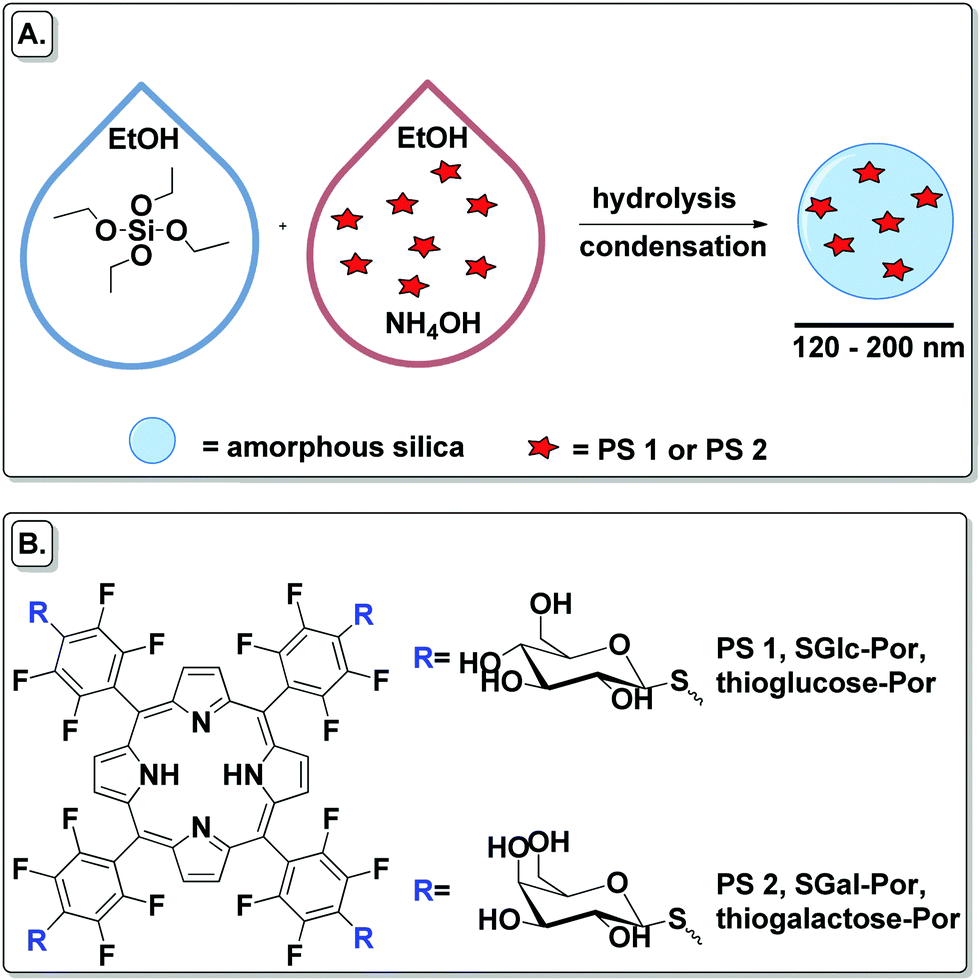 | ||
| Scheme 1 (A) Schematic preparation of PSs encapsulated into SNPs. (B) Structures of SGlc-Por and SGal-Por used as free and encapsulated PSs. | ||
Results and discussion
Synthesis of the PSs
S-Glycoside porphyrins PS 1 and PS 2 were synthesized according to literature procedures presented elsewhere.39–43 Briefly, protected carbohydrate porphyrins were obtained by nucleophilic substitution of the p-fluorine of the corresponding free-base porphyrin 5,10,15,20-tetrakis(pentafluorophenyl)porphyrin (TPPF20) with acetyl 2,3,4,6-tetra-O-acetyl-1-thio-D-glucopyranoside or acetyl 2,3,4,6-tetra-O-acetyl-1-thio-D-galactopyranoside. Applying a simple synthesis, these Pors bearing four carbohydrate moieties conjugated via S-glycoside bonds were obtained in high yields. Final deprotection of the carbohydrate moieties using alkaline hydrolysis afforded the final unprotected S-glycoside porphyrins PS 1 and PS 2.Preparation of SNPs and their PS–SNP hybrids
SNPs and their Por–silica nanoformulations were prepared by the Stöber method in which the hydrolysis and condensation of tetraethyl orthosilicate (TEOS) is facilitated by a base in ethanol and water (Scheme 1).45To obtain a Por–silica nanoformulation with photodynamic properties it is necessary that the amount of PS inside the NP generates reactive oxygen species and/or that the Por photo-properties do not change much during the nanoformulation steps. The conditions of the NPs’ preparation were optimized by varying the amount of base NH4OH, TEOS and PS (Tables 1–3). Compound PS 1 was chosen to optimize the reaction conditions. Generally, in a 15 mL falcon tube, each PS was dissolved in EtOH and then NH4OH was added (the total volume of this mixture was 6.25 mL). The mixture was sonicated for 5 min and TEOS dissolved in EtOH was added (the total volume of this mixture was 1.25 mL). The reaction was incubated for 24 h at 25 °C under continuous agitation (250 rpm) in an incubator shaker (IKA KS 4000 i control) in a horizontal position. After that time, the NPs were isolated by centrifugation (15 mL falcon tubes, 6000 rpm, 30 min) and washed with EtOH. The final NPs were air-dried.
| NP | NH4OH [M] | Total amount of final NPs [mg] | % of PS in final NPs [%] | Size of NPs [nm] |
|---|---|---|---|---|
| NP 1 | 0.16 | — | — | — |
| NP 2 | 0.32 | 20.0 | 7.95 | 58.4 ± 6.5 |
| NP 3 | 0.64 | 24.0 | 5.67 | 266.6 ± 24.4 |
| NP | TEOS [mM] | Total amount of final NPs [mg] | % of PS in final NPs [%] | Size of NPs [nm] |
|---|---|---|---|---|
| NP 2 | 50 | 20 | 7.95 | 58.4 ± 6.5 |
| NP 4 | 300 | 134.6 | 1.27 | 57.5 ± 10.7 |
| NP | PS [mM] | Total amount of final NPs [mg] | % of PS in final NPs [%] | Size of NPs [nm] |
|---|---|---|---|---|
| NP 5 | 0.2 | 19.7 | 3.41 | 48.1 ± 4.6 |
| NP 2 | 0.4 | 20 | 7.95 | 58.4 ± 6.5 |
| NP 6 | 1.6 | 25.5 | 26.27 | 104.1 ± 6.2 |
| NP 7 | 4.0 | 23.0 | 48.7 | 197.3 ± 29.0 |
PSs encapsulation optimization studies
Keeping the amount of PS and TEOS constant, three reactions with different amounts of base were performed to determine the amount of encapsulated PS versus the amount of base (Table 1). In this case, PS 1 (2.977 μmol, 0.4 mM) was dissolved in EtOH and then NH4OH was added. The mixture was sonicated for 5 min and TEOS (0.375 mmol, 50 mM) dissolved in EtOH was added. After 24 h reaction, the NPs were washed and dried at room temperature.A high concentration of NH4OH resulted in a small amount of encapsulated PS 1 in the NPs. A concentration of 0.16 M NH4OH in the reaction mixture did not allow the formation of nanoformulations (NP 1). The concentration of base also influences the size of the NPs. After increasing the concentration of NH4OH by two times, the size of the NPs increased by almost 5 times (NP 2versusNP 3). The highest concentration of PS 1 in the final NPs was observed for NP 2 where a 0.32 M concentration of NH4OH was used; thus, for further experiments, these conditions were used for the synthesis of the nanoformulations.
Keeping the amounts of PS and base constant, the dependence of the amount of encapsulated PS on the amount of TEOS was determined. For this, PS 1 (2.977 μmol, 0.4 mM) was dissolved in EtOH and then NH4OH (2.4 mmol, 0.32 M) was added. The mixture was sonicated for 5 min and TEOS dissolved in EtOH was added (Table 2). After 24 h reaction, the NPs were washed and air-dried.
The decrease of TEOS in the reaction medium by six times increases the amount of PS in the final NPs by 6.3 times. Thus, the concentration of TEOS was kept as 50 mM for the next studies.
Then, we determined how the amount of encapsulated PS depends on the amount of PS in the reaction mixture (keeping the amounts of TEOS and base constant). For this, PS 1 was dissolved in EtOH and then NH4OH (2.4 mmol, 0.32 M) was added. The mixture was sonicated for 5 min and TEOS (0.375 mmol, 50 mM) dissolved in EtOH was added (Table 3). After 24 h, the NPs were washed and air-dried.
A high concentration of PS in the reaction mixture resulted in more effective encapsulation of the PS into the NPs (Table 3). Moreover, the size of the NPs is changing with the same tendency. This is probably because when the amount of PS was increased in the reaction medium, silica forms bigger pores and the NPs become bigger.
Optimized PS–SNP formulation protocol
The most successful results were obtained with the preparation of NP 7 and, consequently, NPs encapsulating compound PS 2 (NP 8, Table 4) were prepared according to the same procedure. In both cases, the amount of encapsulated PS was almost the same. The same protocol was used to synthesize silica nanoparticles in the absence of a PS (SNP) for further comparison and characterization (for details, please see the ESI†).| NP | PS 1 [mM] | PS 2 [mM] | Total amount of final NPs [mg] | % of PS in final NPs [%] | Size of NPs [nm] |
|---|---|---|---|---|---|
| NP 7 | 4.0 | – | 23.0 | 48.7 | 197.3 ± 29.0 |
| NP 8 | – | 4.0 | 28.2 | 36.0 | 128.3 ± 22.2 |
SNP and PS–NP characterization
The selected NPs were morphologically and chemically characterized by imaging and spectroscopic techniques. For details, please see the ESI.†The average size of all NPs was measured by transmission electron microscopy (TEM, Fig S5–S7, ESI†). For the final materials, NP 7 and NP 8, dynamic light scattering (DLS) was also used to determine the size distribution (Fig. S8 and S9, ESI†). The particle size distribution was measured by TEM after drying the sample, and in water dispersion by DLS. The difference in the particle sizes measured by TEM (NP 7, 197.3 ± 29.0 nm; NP 8, 128.3 ± 22.2 nm) and DLS (NP 7, 233.5 nm; NP 8, 133.2 nm) showed the common difference between the mean hydrodynamic diameter (measured by DLS) and the size (measured by TEM). Typically the hydrodynamic diameter obtained by DLS is larger than the size gathered by TEM. These NPs have a uniform size distribution and are regular in terms of size and shape. As previously mentioned, particles with a size ranging from 10 to 500 nm accumulate inside tumor cells and therefore the developed NPs have an appropriate size for passive targeting to tumor tissues.
The UV-vis absorption spectra of NP 7 and NP 8 were collected after dispersing, respectively, 0.535 and 0.510 mg nanoformulations in 3 mL of distilled water (Fig. S12, ESI†). The nanoformulations show the typical spectra of a free base Por, with the Soret band at 403 nm (NP 7) and 407 nm (NP 8).
The EDS spectra show the chemical composition of NP 7 and NP 8, where a small percentage of sulphur must be related to the thio-carbohydrate moieties of the Pors (Fig. S10 and S11, ESI†). PS 1, PS 2, SNP and the corresponding NP 7 and NP 8 were also analysed by FT-IR (Fig. S13, ESI†). The spectra of both NP formulations show some features of the porphyrins (PS 1 and PS 2) and the sole silica nanoparticles (SNP).
The amount of encapsulated PS inside the NPs was calculated by UV-vis spectrophotometry. The final NPs were washed with EtOH until no typical Soret and Q bands were observed in the rinse solvent. The final concentration of PS in the NPs [%] was calculated by subtracting the non-encapsulated PS, determined by absorption measurements, in the rinse solvent.46NP 7 (0.289 μmol mg−1) has a slightly higher concentration of PS per mg of final material than NP 8 (0.215 μmol mg−1).
Singlet oxygen generation study
Singlet oxygen (1O2) was determined by an indirect chemical method using 1,3-diphenylisobenzofuran (DPBF) as a 1O2 quencher (for details, please refer to the ESI†). 1O2 was determined for both nanoformulations (NP 7 and NP 8) and their corresponding PSs (PS 1 and PS 2) (Fig. 1) in at least three independent experiments. The free PS 1 and PS 2 were tested at concentrations of 0.5 μM. NPs composed of PS 1 and PS 2 were tested at concentrations of PS: 12.6 μM (NP 7) and 1.6 μM (NP 8). Both free PSs oxidized DPBF in a similar manner. In spite of the different amount of PS inside the NPs and the different size of these nanoformulations, the DPBF kinetic decay was similar in the two nanoformulations. From this study, we can observe that 0.131 mg of NP 7 could produce the same amount of 1O2 as 0.022 mg of NP 8, suggesting that NP 8 is 6 times more efficient in terms of 1O2 generation. Although NP 8 (128.3 ± 22.2 nm) is smaller than NP 7 (197.3 ± 29.0 nm) and has a slightly lower amount of PS per mg of NPs (Table 4), these NPs produce a higher amount of 1O2. This could be due to the fact that in the case of smaller NPs oxygen can penetrate better their pores and 1O2 has a shorter way to go out from the nanoformulation than in bigger NPs. Thus, NP 8 is more effective in 1O2 production than NP 7 while the corresponding free Pors (PS 1 and PS 2) produce the same amount of 1O2 under equal experimental conditions.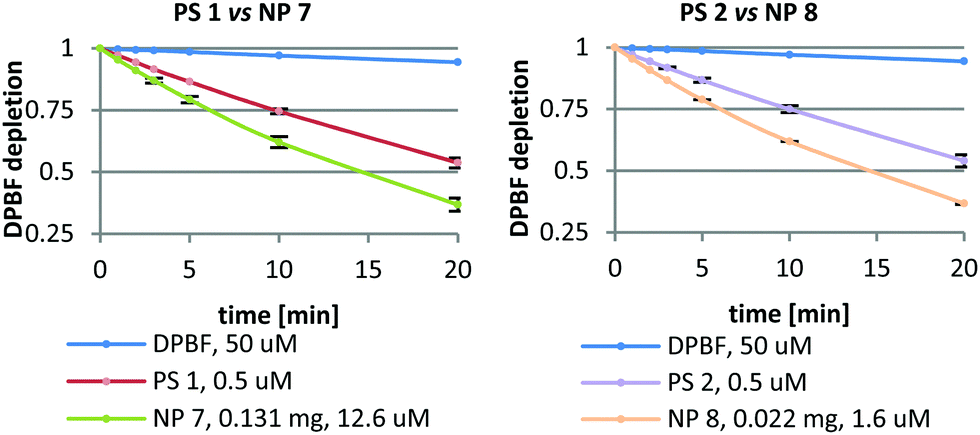 | ||
| Fig. 1 1O2 generation by PS 1, PS 2 and their corresponding NPs (NP 7 and NP 8) where each point represents the mean of at least three independent experiments, and has a standard deviation lower than 3%. | ||
In vitro studies
In vitro studies were carried out on two human bladder cancer cell lines, HT-1376 and UM-UC-3. These cell lines are suitable as in vitro models for the testing of new glyco-porphyrinoids, since the glyco-binding proteins – glucose transporter (GLUT1) and galactose-binding protein (galectin-1) – are abundant but differentially expressed between these two cell lines.47 Previous studies have demonstrated that these two proteins have a key role in the uptake and further phototoxicity of galactodendritic PSs.47–51Cellular uptake of the PSs and their nanoformulations
Preliminary uptake studies were performed for glucosylated PS 1 and galactosylated PS 2 (Fig. 2). Bladder cancer cells were incubated with increasing concentrations of PS (0, 2.5, 5 and 10 μM prepared in PBS, maximum 0.5% DMSO v/v) for 4 h in darkness. Fluorescence spectroscopy studies demonstrated that PS 1 accumulation was higher in HT-1376 than in UM-UC-3 cancer cells. On the other hand, the uptake of PS 2 was higher in UM-UC-3 than in HT-1376 cancer cells.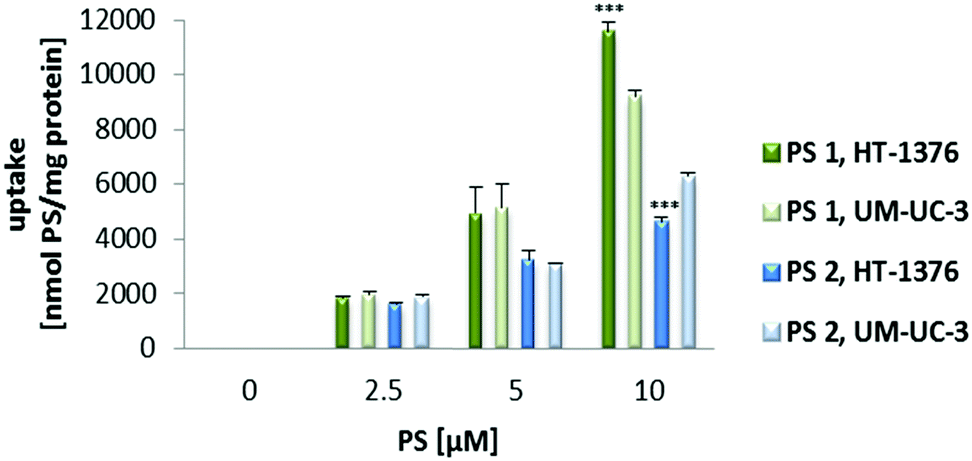 | ||
| Fig. 2 Cellular uptake of PS 1 and PS 2 (0–10 μM in PBS) by UM-UC-3 and HT-1376 bladder cancer cells. Cells were incubated with the PS for 4 h and the uptake was determined by fluorescence spectroscopy. Data are means ± s.e.m. of at least three independent experiments performed in triplicate. ***P < 0.001 compared to the PS uptake by UM-UC-3 cancer cells, using Student's t test. | ||
Next, the uptake of NP 7 and NP 8 was evaluated by fluorescence spectroscopy and fluorescence microscopy (Fig. 3 and 4) after incubating UM-UC-3 and HT-1376 bladder cancer cells in the dark with different concentrations of the new NPs.
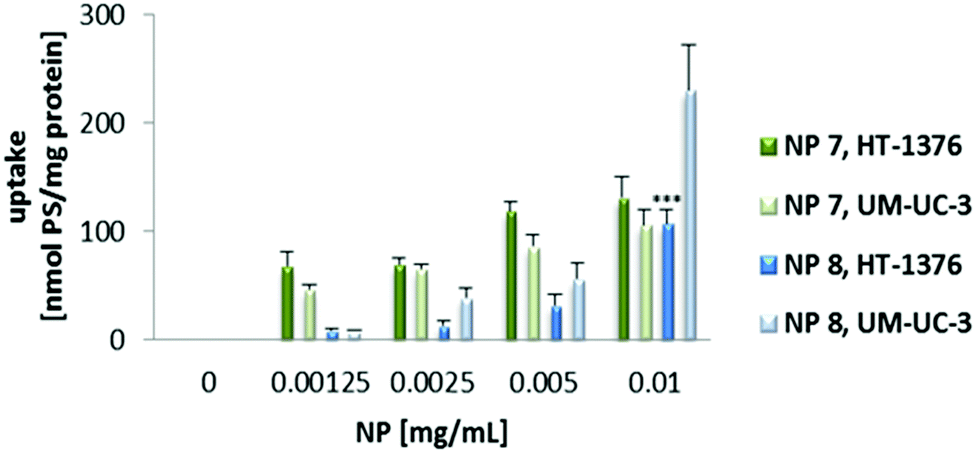 | ||
| Fig. 3 Cellular uptake of NP 7 and NP 8. UM-UC-3 and HT-1376 bladder cancer cells were incubated with NP 7 (0–0.010 mg mL−1, 0–2.89 μM of PS 1 in RPMI medium) and NP 8 (0–0.010 mg mL−1, 0–2.15 μM of PS 2 in RPMI medium) overnight. Data are means ± s.e.m. of at least three independent experiments performed in triplicate. ***P < 0.001 compared to the NP uptake by UM-UC-3 cancer cells, using Student's t test. | ||
 | ||
| Fig. 4 Representative fluorescence images of UM-UC-3 and HT-1376 bladder cancer cells after overnight incubation with NP 7 (red, 0.010 mg mL−1, 2.89 μM of PS 1) or NP 8 (red, 0.010 mg mL−1, 2.15 μM of PS 2) in darkness and the cell nucleus stained with DAPI (blue). Scale bars, 20 μm. | ||
Uptake experiments of both NP 7 and NP 8 (for concentrations ranging from 0 to 0.010 mg mL−1) demonstrated that intracellular accumulation was negligible when the cells were incubated with the NPs for 4 h (data not shown). Further studies were performed by incubating cancer cells with the PS nanoformulations overnight. When the cells were incubated overnight with RPMI medium containing NP solutions, the intracellular accumulation was dependent on the concentration of the NPs and cell line. Interestingly, the uptake of PS 1 and PS 2 performed with overnight incubation (PS solutions prepared in cell culture medium) was lower (data not shown) when compared with the uptake of 4 h (PS solutions prepared in PBS buffer). Since we had different incubation conditions, a limitation of this study was that the medium was not present for incubation of the PSs for 4 h. Because of this limitation, the PS uptake may not be comparable for 4 h and 24 h. In addition, the presence of the medium may induce some aggregation effect, both when cells are incubated with the free PSs and after their release from nanoparticles. The uptake of NP 7 was higher in HT-1376 cells (which contain high levels of GLUT1 protein) when compared with UM-UC-3 cancer cells (Fig. 3). On the other hand, the NP 8 uptake was higher in UM-UC-3 cells (which contain high levels of galectin-1 protein) than in HT-1376 cancer cells. Considering the levels of galectin-1 and GLUT1 proteins, the uptake of NP 7 is higher in HT-1376 cells because these NPs contain Por bearing glucose moieties. On the contrary, the higher uptake of NP 8 in UM-UC-3 cancer cells was observed because of Por bearing galactose moieties in the pores of these nanoformulations.
Further studies performed by fluorescence microscopy (Fig. 4) showed preferential intracellular accumulation of NP 7 and NP 8 in HT-1376 and UM-UC-3 bladder cancer cells, respectively, when the cells were incubated overnight with 0.010 mg mL−1 of the NP formulations (2.89 μM of PS 1 in the case of NP 7 and 2.15 μM of PS 2 for NP 8).
Dark toxicity and phototoxicity
The toxicity of PS 1, PS 2, NP 7 and NP 8 in UM-UC-3 and HT-1376 bladder cancer cells in the dark was evaluated using the well-known MTT assay (Fig. S15 and S16, ESI†). This colorimetric assay determines the cell metabolic activity, by assessing the ability of living bladder cancer cells to reduce yellow 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide (MTT) to purple formazan. After overnight incubation of cancer cells (in the dark) with the NPs (0–0.010 mg mL−1 in RPMI medium) or 4 h incubation with the PSs (0–10 μM in PBS), none of the PSs or new NPs induced dark toxicity in the cancer cells (Fig. S15 and S16, ESI†). Moreover, during the 1O2 generation studies we observed that the NPs are very much stable in the solution of DMF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (9:1, by volume) in which all experiments were performed (Fig. S3 and S5, ESI†). However, during the biological experiments, slow release of the PS from the silica matrix was detected after 4 h and 18 h incubation in PBS buffer or RPMI medium (Fig. S14, ESI†). Higher release in the case of NP 8 than in NP 7 was noted. It could be explained by the fact that NP 7 (197.3 ± 29.0 nm) is bigger than NP 8 (128.3 ± 22.2 nm) and thus the PS is more concentrated in NP 8. As a result the photosensitizer could be released more easily from NP 8.
:H2O (9:1, by volume) in which all experiments were performed (Fig. S3 and S5, ESI†). However, during the biological experiments, slow release of the PS from the silica matrix was detected after 4 h and 18 h incubation in PBS buffer or RPMI medium (Fig. S14, ESI†). Higher release in the case of NP 8 than in NP 7 was noted. It could be explained by the fact that NP 7 (197.3 ± 29.0 nm) is bigger than NP 8 (128.3 ± 22.2 nm) and thus the PS is more concentrated in NP 8. As a result the photosensitizer could be released more easily from NP 8.
After confirming the uptake and non-dark toxicity of the PSs and their new NP formulations in UM-UC-3 and HT-1376 bladder cancer cells, their toxicity after light irradiation was equally evaluated using the MTT assay (Fig. 5 and 6). UM-UC-3 and HT-1376 bladder cancer cells were incubated for 4 h with the PSs (0–10 μM in PBS) or overnight with the NPs (NP 7: 0–0.010 mg mL−1, 0–2.89 μM of PS 1 and NP 8: 0–0.010 mg mL−1, 0–2.15 μM of PS 2) and then irradiated with an optical fiber emitting white light for 40 min (12 mW cm−2). No cytotoxicity was observed in the untreated (cells incubated in the absence of PSs or NPs) sham irradiated cells.
 | ||
| Fig. 5 Phototoxicity of PS 1 and PS 2 (0–10 μM in PBS) determined 24 h after PDT treatment using the MTT assay. The percentage of cytotoxicity was calculated relative to control cells (cells incubated with PBS and then irradiated). Data are means ± s.e.m. of at least three independent experiments performed in triplicate. **P < 0.01, ***P < 0.001 compared to MTT reduction in control cells, using Student's t test. $$P < 0.01, $$$P < 0.001 compared to MTT reduction by UM-UC-3 cancer cells, using Student's t test. | ||
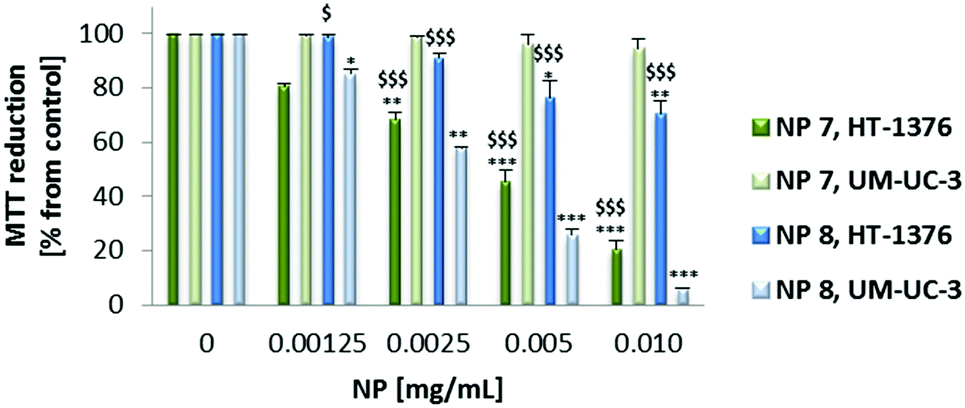 | ||
| Fig. 6 Phototoxicity of NP 7 (0–0.010 mg mL−1, 0–2.89 μM of PS 1) and NP 8 (0–0.010 mg mL−1, 0–2.15 μM of PS 2) determined 24 h after PDT treatment using the MTT assay. The percentage of cytotoxicity was calculated relative to control cells (cells incubated with RPMI medium and then irradiated). Data are means ± s.e.m. of at least three independent experiments performed in triplicate. *P < 0.05, **P < 0.01, ***P < 0.001 compared to MTT reduction in control cells, using Student's t test. $$P < 0.01, $$$P < 0.001 compared to MTT reduction by UM-UC-3 cancer cells, using Student's t test. | ||
Both the PSs and NPs induced phototoxicity in UM-UC-3 and HT-1376 bladder cancer cells in a concentration- and cell line-dependent manner. However, in the case of the NPs the phototoxicity is much more dependent on the cell line type than on the PSs (Fig. 5 and 6). NP 7 led to significantly higher phototoxicity on HT-1376 cells compared to UM-UC-3 cells (Fig. 6). The phototoxicity of NP 8 was higher in UM-UC-3 than in HT-1376 bladder cancer cells (Fig. 8). Taking into account the levels of galectin-1 and GLUT1 proteins, the phototoxicity of NP 7 is higher in HT-1376 cells compared to UM-UC-3 cells because the uptake of these NPs was higher due to the presence of Por bearing glucose moieties. All the same, the higher phototoxicity of NP 8 in UM-UC-3 cancer cells was due to the higher uptake by these cells because of Por bearing galactose moieties. The different size of the two nanoformulations could also have some influence on these data; however, under PDT it is expected that the main biological behavior arises from the PS concentration and less from the nanoparticle size. SNPs without PSs did not induce phototoxicity in UM-UC-3 and HT-1376 bladder cancer cells (data not shown).
Taking into account the concentration of PSs inside the NPs, the phototoxicity was shown to be higher with the NP formulations than with the free PSs (Fig. 5–7). From the below figure it is clear that these new nanomaterials are more efficient in PDT than the corresponding free Pors (Fig. 7). NP 7 with 2.89 μM of PS 1 was able to induce a pronounced decrease in the HT-1376 cell viability, which was similar to 10 μM of free PS 1. Likewise, NP 8 with 2.15 μM of PS 2 was able to induce a similar decrease in the UM-UC-3 cell viability to 10 μM of free PS 2. Thus, these NPs are around 3–5 times more effective in photodynamic therapy activity than the respective free Pors.
 | ||
| Fig. 7 Phototoxicity of PS 1 (2.5 μM, 10 μM), PS 2 (2.5 μM, 10 μM), NP 7 (2.89 μM of PS 1) and NP 8 (2.15 μM of PS 2) determined 24 h after PDT treatment using the MTT assay. The percentage of cytotoxicity was calculated relative to control cells (cells incubated with RPMI medium and then irradiated). Data are means ± s.e.m. of at least three independent experiments performed in triplicate. **P < 0.01, ***P < 0.001 compared to MTT reduction in control cells, using Student's t test. $$P < 0.01, $$$P < 0.001 compared to MTT reduction by UM-UC-3 cancer cells, using Student's t test. | ||
The role of cytotoxic ROS generated after PDT with 0.010 mg mL−1 of NP 7 (2.89 μM of PS 1) and NP 8 (2.15 μM of PS 2) was evaluated using sodium azide, histidine52 and cysteine53 as ROS quenchers (Fig. 8). When PDT experiments were performed with cells in the presence of non-toxic concentrations of ROS quenchers, there was a reduction in the phototoxicity induced by the new NPs. The data show that 1O2 should have a high effect on the phototoxicity induced by NP 7 or NP 8, since the phototoxicity was highly reduced when PDT experiments were performed with 1O2 quenchers (sodium azide and histidine).
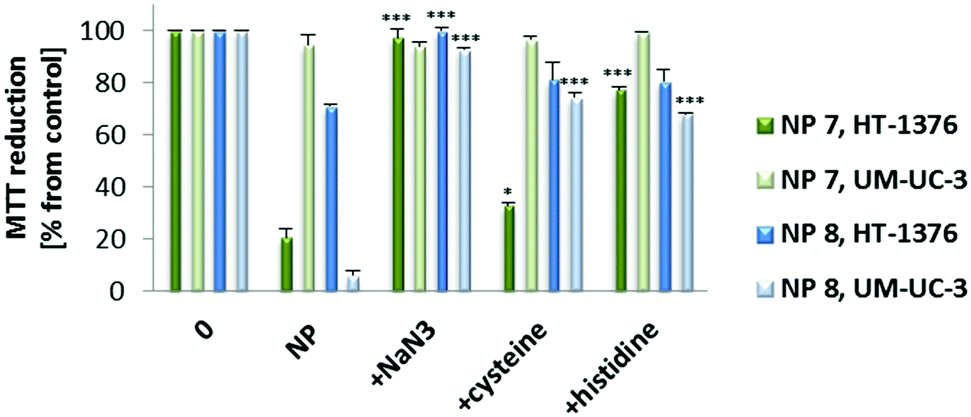 | ||
| Fig. 8 Phototoxicity after PDT with NP 7 (0.010 mg mL−1, 2.89 μM of PS 1) and NP 8 (0.010 mg mL−1, 2.15 μM of PS 2) in the presence of 50 nM of ROS quenchers (sodium azide, histidine and cysteine) in HT-1376 and UM-UC-3 cells. Cytotoxicity was assessed 24 h after treatment using the MTT assay. The percentage of cytotoxicity was calculated relative to control cells (untreated cells). Data are means ± s.e.m. of at least three independent experiments performed in triplicate. *P < 0.05, ***P < 0.001 compared to MTT reduction (%) 24 h after PDT with NPs using Student's t test. | ||
Conclusions
In summary, two types of new glyco-PS loaded SNPs were successfully prepared after modification and optimization of the well-known Stöber method. Both novel materials were able to produce 1O2 and hence in vitro studies with two human bladder cancer cell lines, HT-1376 and UM-UC-3, were performed. Despite slightly lower efficiency in terms of singlet oxygen generation compared to non-immobilized PSs, the nanocarriers offer an alternative route to develop new platforms for PDT. Overall, these NPs demonstrated 3–5 times higher therapeutic efficacy in vitro compared with the corresponding free PSs. In the presented work, we demonstrated that S-glycoside Pors encapsulated into a silica matrix by a straightforward Stöber method were more efficient in in vitro PDT against two human bladder cancer cell lines, HT-1376 and UM-UC-3, than non-encapsulated PSs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Thanks are due to FCT/MEC for the financial support to QOPNA (FCT UID/QUI/00062/2019), CICECO-Aveiro Institute of Materials (UIDB/50011/2020), CIBB (UIDB/04539/2020) and CQE (UIDB/00100/2020) research units, through national funds and where applicable cofinanced by the FEDER, within the PT2020 Partnership Agreement. The authors also acknowledge FCT for the doctoral research fellowship SFRH/BD/85941/2012 (to PMRP). The work was also supported by the Spanish MINECO (CTQ2017-85393-P) and ERA-NET/MINECO (EuroNanoMed2017-191/PCIN-2017-042) and Portuguese COMPETE (POCI-01-0145-FEDER-007440). We further wish to thank the Seventh Framework Programme (FP7-People-2012-ITN) for funding the SO2S project (grant agreement number: 316975).Notes and references
- R. Bonnett, Chem. Soc. Rev., 1995, 24, 19–33 RSC.
- P.-C. Lo, M. S. Rodríguez-Morgade, R. K. Pandey, D. K. P. Ng, T. Torres and F. Dumoulin, Chem. Soc. Rev., 2020, 49, 1041–1056 RSC.
- D. E. J. G. J. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380–387 CrossRef CAS.
- I. J. Macdonald and T. J. Dougherty, J. Porphyrins Phthalocyanines, 2001, 05, 105–129 CrossRef CAS.
- C. Liang, X. Zhang, Z. Wang, W. Wang, M. Yang and X. Dong, J. Mater. Chem. B, 2020, 8, 4748–4763 RSC.
- H. Abrahamse and M. R. Hamblin, Biochem. J., 2016, 473, 347–364 CrossRef CAS.
- D. K. Chatterjee, L. S. Fong and Y. Zhang, Adv. Drug Delivery Rev., 2008, 60, 1627–1637 CrossRef CAS.
- N. M. Idris, M. K. Gnanasammandhan, J. Zhang, P. C. Ho, R. Mahendran and Y. Zhang, Nat. Med., 2012, 18, 1580–1586 CrossRef CAS.
- H. S. Qian, H. C. Guo, P. C.-L. Ho, R. Mahendran and Y. Zhang, Small, 2009, 5, 2285–2290 CrossRef CAS.
- Y. Zhou, X. Liang and Z. Dai, Nanoscale, 2016, 8, 12394–12405 RSC.
- V. Almeida-Marrero, E. van de Winckel, E. Anaya-Plaza, T. Torres and A. de la Escosura, Chem. Soc. Rev., 2018, 47, 7369–7400 RSC.
- P. Couleaud, V. Morosini, C. Frochot, S. Richeter, L. Raehma and J.-O. Durand, Nanoscale, 2010, 2, 1083–1095 RSC.
- H. Kobayashi, R. Watanabe and P. L. Choyke, Theranostics, 2014, 4, 81–89 CrossRef CAS.
- H. Maeda, Bioconjugate Chem., 2010, 21, 797–802 CrossRef CAS.
- L. Tang and J. Cheng, Nano Today, 2013, 8, 290–312 CrossRef CAS.
- S. Heidegger, Go, A. Schmidt, S. Niedermayer, C. Argyo, S. Endres, T. Bein and C. Bourquin, Nanoscale, 2016, 8, 938–948 RSC.
- W. Q. Lim, S. Z. F. Phua, H. V. Xu, S. Sreejith and Y. Zhao, Nanoscale, 2016, 8, 12510–12519 RSC.
- X. Yao, X. Chen, C. He, L. Chen and X. Chen, J. Mater. Chem. B, 2015, 3, 4707–4714 RSC.
- J. G. Croissant, S. Picard, D. Aggad, M. Klausen, C. Mauriello Jimenez, M. Maynadier, O. Mongin, G. Clermont, E. Genin, X. Cattoën, M. Wong Chi Man, L. Raehm, M. Garcia, M. Gary-Bobo, M. Blanchard-Desce and J.-O. Durand, J. Mater. Chem. B, 2016, 4, 5567–5574 RSC.
- V. Biju, Chem. Soc. Rev., 2014, 43, 744–764 RSC.
- C. R. Steven, G. A. Busby, C. Mather, B. Tariq, M. L. Briuglia, D. A. Lamprou, A. J. Urquhart, M. H. Grant and S. V. Patwardhan, J. Mater. Chem. B, 2014, 2, 5028–5042 RSC.
- C. M. Dawidczyk, C. Kim, J. H. Park, L. M. Russell, K. H. Lee, M. G. Pomper and P. C. Searson, J. Controlled Release, 2014, 187, 133–144 CrossRef CAS.
- G. Pillai, SOJ Pharm. Pharm. Sci, 2014, 1, 1–13 Search PubMed.
- G. Pillai and M. L. Ceballos-Coronel, SAGE Open Med., 2013, 1, 1 Search PubMed.
- G. Obaid, M. Broekgaarden, A.-L. Bulin, H.-C. Huang, J. Kuriakose, J. Liu and T. Hasan, Nanoscale, 2016, 8, 12471–12503 RSC.
- D. Lombardo, M. A. Kiselev and M. T. Caccamo, J. Nanomater., 2019, 2019, 26 Search PubMed.
- L. Shang, K. Nienhaus and G. U. Nienhaus, J. Nanobiotechnol., 2014, 12, 5 CrossRef.
- V. Torchilin, Adv. Drug Delivery Rev., 2011, 63, 131–135 CrossRef CAS.
- S. Biswas and V. P. Torchilin, Adv. Drug Delivery Rev., 2014, 66, 26–41 CrossRef CAS.
- F. Figueira, J. A. S. Cavaleiro and J. P. C. Tomé, J. Porphyrins Phthalocyanines, 2011, 15, 517–533 CrossRef CAS.
- W. Borzęcka, T. Trindade, T. Torres and J. Tomé, Curr. Pharm. Des., 2016, 22, 6021–6038 CrossRef.
- R. K. Fei Yan, Photochem. Photobiol., 2003, 78, 587–591 CrossRef.
- W. Li, W. Lu, Z. Fan, X. Zhu, A. Reed, B. Newton, Y. Zhang, S. Courtney, P. T. Tiyyagura, S. Li, E. Butler, H. Yu, P. C. Ray and R. Gao, J. Mater. Chem., 2012, 22, 12701–12708 RSC.
- J. Qian, D. Wang, F. Cai, Q. Zhan, Y. Wang and S. He, Biomaterials, 2012, 33, 4851–4860 CrossRef CAS.
- I. T. Teng, Y.-J. Chang, L.-S. Wang, H.-Y. Lu, L.-C. Wu, C.-M. Yang, C.-C. Chiu, C.-H. Yang, S.-L. Hsu and J.-a. A. Ho, Biomaterials, 2013, 34, 7462–7470 CrossRef CAS.
- X. Deng, L. Xiong, L. Lin, G. Xiong and X. Miao, Photodiagn. Photodyn. Ther., 2013, 10, 460–469 CrossRef CAS.
- Z.-T. Liu, L. Xiong, Z.-P. Liu, X.-Y. Miao, L.-W. Lin and Y. Wen, Nanoscale Res. Lett., 2014, 9, 319–327 CrossRef.
- L. Dongyu, Z. Hequn, C. Liliang, Z. Xinyuan and Q. Jun, Opt. Quantum Electron., 2015, 47, 3081–3090 CrossRef.
- X. Chen, L. Hui, D. A. Foster and C. M. Drain, Biochemistry, 2004, 43, 10918–10929 CrossRef CAS.
- S. Shiho HirohHirohara, M. Obata, H. Alitomo, K. Sharyo, T. Ando, S. Yano and M. Tanihara, Bioconjugate Chem., 2009, 20, 944–952 CrossRef.
- S. Singh, A. Aggarwal, S. Thompson, J. P. C. Tomé, X. Zhu, D. Samaroo, M. Vinodu, R. Gao and C. M. Drain, Bioconjugate Chem., 2010, 21, 2136–2146 CrossRef CAS.
- S. Hirohara, M. Nishida, K. Sharyo, M. Obata, T. Ando and M. Tanihara, Bioorg. Med. Chem., 2010, 18, 1526–1535 CrossRef CAS.
- S. Hirohara, M. Obata, H. Alitomo, K. Sharyo, T. Ando, M. Tanihara and S. Yano, J. Photochem. Photobiol., B, 2009, 97, 22–33 CrossRef CAS.
- X. Chen, L. Hui, D. A. Foster and C. M. Drain, Biochemistry, 2004, 43, 10918–10929 CrossRef CAS.
- W. Stober and A. Fink, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- C. M. B. Carvalho, E. Alves, L. Costa, J. P. C. Tomé, M. A. F. Faustino, M. G. P. M. S. Neves, A. C. Tomé, J. A. S. Cavaleiro, A. Almeida, Â. Cunha, Z. Lin and J. Rocha, ACS Nano, 2010, 4, 7133–7140 CrossRef CAS.
- P. M. R. Pereira, S. Silva, J. A. S. Cavaleiro, C. A. F. Ribeiro, J. P. C. Tomé and R. Fernandes, PLoS One, 2014, 9, e95529 CrossRef.
- S. Silva, P. M. R. Pereira, P. Silva, F. A. A. Paz, M. A. F. Faustino, J. A. S. Cavaleiro and J. P. C. Tome, Chem. Commun., 2012, 48, 3608–3610 RSC.
- P. M. R. Pereira, S. Silva, J. S. Ramalho, C. M. Gomes, H. Girão, J. A. S. Cavaleiro, C. A. F. Ribeiro, J. P. C. Tomé and R. Fernandes, Eur. J. Cancer, 2016, 68, 60–69 CrossRef CAS.
- P. M. R. Pereira, S. Silva, M. Bispo, M. Zuzarte, C. Gomes, H. Girão, J. A. S. Cavaleiro, C. A. F. Ribeiro, J. P. C. Tomé and R. Fernandes, Bioconjugate Chem., 2016, 27, 2762–2769 CrossRef CAS.
- P. M. R. Pereira, W. Rizvi, N. V. S. D. K. Bhupathiraju, N. Berisha, R. Fernandes, J. P. C. Tomé and C. M. Drain, Bioconjugate Chem., 2018, 29, 306–315 CrossRef CAS.
- M. Bancirova, Luminescence, 2011, 26, 685–688 CrossRef CAS.
- O. I. Aruoma, B. Halliwell, B. M. Hoey and J. Butler, Free Radical Biol. Med., 1989, 6, 593–597 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NPs’ preparation and characterization, singlet oxygen studies and dark toxicity studies. See DOI: 10.1039/d0ma00830c |
| This journal is © The Royal Society of Chemistry 2021 |