
Process condition-based tuneable selective catalysis of hydroxymethylfurfural (HMF) hydrogenation reactions to aromatic, saturated cyclic and linear poly-functional alcohols over Ni–Ce/Al2O3†
Brett
Pomeroy
,
M.
Grilc
and
B.
Likozar
 *
*
Department of Catalysis and Chemical Reaction Engineering, National Institute of Chemistry, Hajdrihova 19, 1000 Ljubljana, Slovenia
First published on 15th September 2021
Abstract
The related immense versatility of a ceria-promoted transition metal catalyst, utilized for the hydrogenation of 5-hydroxymethylfurfural (HMF), is demonstrated in this research study. We reveal a strategy to achieve considerable selective yields of three important high-value HMF-derived compounds by simply modifying the analysed reaction conditions and/or water-containing process medium.
Due to the rising concerns of climate change and pollution instigated by fossil fuel usage, lignocellulosic biomass as a renewable feedstock has been recognized as a sustainable replacement due to its availability and relatively low cost. Cellulose, the major component comprising approximately half of all lignocellulosic biomass, offers incredible potential as a source for chemicals and biofuels. In particular, 5-hydroxymethylfurfural (HMF) has received immense attention for bio-based chemical production with enormous potential, even being described as a “sleeping giant”.1 Obtained from the dehydration of glucose, HMF is a highly versatile intermediate which allows it to be converted into an assortment of value-added compounds. Notably, the diols 2,5-bis(hydroxymethyl)furan (BHMF) and 2,5-bis(hydroxymethyl)tetrahydrofuran (BHMTHF) have been considered extensively in the manufacturing of polyesters and heat insulating polymers which are presently made solely from fossil fuels.2,3 1,2,6-Hexanetriol (1,2,6-HT) can also be obtained from HMF and is a chemical that has tremendous value either directly as a moisturizing agent, solvent, and polymer cross-linker in certain resins and synthetic rubbers, or as an intermediate for the production of 1,6-hexandiol which is applied in the adhesive, coatings, and paint industry. 1,6-Hexanediol has an approximate global market value of 600 million USD, although it is currently produced in relatively low yields from adipic acid originating from non-renewable sources.4 Hydrogenation has been widely established as an efficient method to convert HMF into these aforementioned value-added chemicals. Typically, it entails moderate temperatures of up to 300 °C and high hydrogen pressures (up to 20 MPa) in the presence of a heterogeneous catalyst.5 In terms of selection of catalysts that are active for hydrogenation reactions, transition metals such as nickel are more desirable due to their low costs and availability despite their general inferior catalytic activity compared to noble metals such as Pt and Pd. Promoters in the form of a second metal (bimetallic) are necessary to improve their catalytic performance and tune their selectivity towards more favourable products. While a diverse collection of high-value chemicals can be obtained from HMF, most studies almost exclusively rely on a single catalyst to produce a single product, greatly limiting their economic feasibility and industrial utility. As shown in Table 1, several studies have independently achieved exceptional yields of BHMF, BHMTHF, or 1,2,6-HT, although they generally require the presence of noble metals and extremely high loadings in the case of using Ni, utilize toxic solvents like 1,4-dioxane, implement miniscule initial amounts of HMF, and/or require exceptionally long reaction times of 12+ hours.
| Catalyst | Cat. amount | HMF | Solvent | Temp. (°C) | Pres. (MPa) | Time (h) | Conv. (%) | Selectivity (%) (BHMF, BHMTHF, 1,2,6-HT) | Ref. | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Supported on Al2O3. b Supported on CeO2. c Supported on carbon. d Addition of CaO. | |||||||||||
| 20 mol% Cua | 0.016 g | 0.25 mmol | Methanol | 130 | 3 | 1 | 100 | 93 | 0 | 0 | 21 |
| 1 wt% Aua | 0.01 g | 0.1 M | Water | 60 | 6.5 | 2 | 100 | 96 | 0 | 0 | 22 |
| 1 wt% Rub | 0.2 g | 0.4 g | 1-BuOH/water | 130 | 2.8 | 12 | 100 | 0 | 91 | 3 | 13 |
| 74 wt% Nia | 0.2 g | 1.5 g | 1,4-Dioxane | 60 | 6 | 6 | 100 | 4 | 96 | 0 | 23 |
| RANEY® Ni (>93 wt%)a | 0.5 g | 1.5 g | 1,4-Dioxane | 100 | 1.5 | 30 | 100 | 0 | 96 | 0 | 24 |
| 1 wt% Ruc | 0.05 g | 0.4 g | 1-BuOH/water | 130 | 2.8 | 1 | 100 | 0 | 53 | 13 | 13 |
| 5 wt% Pt + 5 wt% Cob | 0.01 g | 0.2 mmol | Water | 135 | 3 | 24 | 100 | 0 | 41 | 42 | 25 |
| 10 wt% Ni + 15 wt% Cea | 0.1 g | 1 g | THF/water | 140 | 5 | 6 | 100 | 96 | 4 | 0 | This work |
| 1-BuOH | 190 | 5 | 3 | 100 | 0 | 88 | 9 | This work | |||
| THF/water | 190 | 5 | 3 | 100 | 0 | 73 | 26 | This work | |||
| THF/waterd | 190 | 5 | 1 | 100 | 2 | 39 | 59 | This work | |||
We demonstrate for the first time the remarkable flexibility of a single, transition metal catalyst with relatively low metal loadings to form three major bio-based compounds from HMF in significantly high yields by only adjusting the reaction conditions and/or medium. Ceria as a catalyst dedicated for hydrogenation purposes is rather uncommon in the literature as it is typically applied for oxidation reactions due to its high redox properties and oxygen storage capacity.6,7 However, more unfamiliar is its capability to elicit both acidic-basic functionalities (amphoteric) depending on the catalyst preparation and reaction conditions.8,9 Additionally, its water tolerance, suppression of coke formation, and prevention of active metal sintering make ceria a favourable promoter.10–12 Based on the hypothesis of Dumesic et al. that metal oxides with high isoelectric points (IEP) are responsible for improved yields of ring saturated products by restricting acid-catalyzed degradation reactions, alumina was chosen as a support material due to its relatively high IEP/point zero charge (PZC), along with its high surface area and low cost.13 We acknowledge that there can be a discrepancy between IEP and PZC values, where the former is determined by electrophoresis and the latter is determined by potentiometric titration,14 but for simplicity purposes we report only PZC values in this study as these values were more frequently described in the literature. Two different alumina were investigated to validate this hypothesis; a pure γ-alumina with a reported PZC of 8.5, and another alumina with a lower PZC due to the presence of both α and γ phases.15,16 Furthermore, we have demonstrated the reaction to be effective in both monophasic water–THF and biphasic water–1-BuOH solvent systems, which together have been identified as solvent mixtures selective for HMF production from C6 sugars.17,18 Although presently THF is not classified as a green solvent like 1-BuOH, several studies have demonstrated the likely prospect of producing it from biomass-derived feedstocks such as maleic anhydride and furfural.18–20 Water as a co-solvent is obviously desired as a cheap and green solvent, while its presence also was determined to limit dehydration reactions and increase product selectivity towards the desired products.
The catalyst synthesis procedure, described in detail in the ESI,† is a rather simple, sequential incipient wetness impregnation of cerium initially, which was then oven dried and calcined at 400 °C under air, and then followed by nickel incorporation under the same conditions. SEM images with the corresponding EDXS elemental mappings (Fig. S1†) indicate that nickel and ceria are highly dispersed over the support, and the spent catalyst remains rather constant with no inclination of any major particle coarsening or sintering following reduction treatment or hydrogenation reaction. TEM images (Fig. S2†) identified Ce clusters on the catalyst surface made up of individual ∼9 nm Ce particles with a lattice fringe of 0.311 nm indicating a CeO2 (111) crystal plane. It was not possible to observe any distinct nickel particles in TEM. The diffraction lines corresponding with the CeO2 fluorite cubic cell (JCPDS# 00-004-0593) were clearly observed, although the peaks were generally quite broad indicating poor crystallinity (Fig. S3†). The ceria crystal size was estimated based on powder XRD spectra to be around 9 nm which grew slightly to 11–12 nm in the spent catalysts (Table S1†). No nickel peaks matching with NiO or Ni-containing alloys were detected in the XRD analysis of either Ni or Ni–Ce catalysts, as well as in either of the spent catalysts (Fig. S4†). This has been observed by others with similar Ni–Ce catalysts where Ni is proposed to be either in a highly dispersed amorphous state or in <4 nm NiO crystallites which are below the detection limit.26,27 XRD analysis of the mixed Al2O3 support clearly indicates the presence of an α-alumina phase which contributed to a lower PZC relative to pure γ-alumina.16 The specific surface area (SBET) and average pore volume (Vp) decreased marginally following Ni incorporation and for each sequential impregnation amongst the bimetallic Ni–Ce catalyst (Table S2†). This indicates that the Ni and Ce particles blocked some pores on the catalyst surface, and yet the catalyst retained a reasonably high specific surface area even amongst the spent catalysts between 147 and 172 m2 g−1. XPS analysis of the bimetallic Ni–Ce catalyst indicates the presence of both Ce3+ (v′ and u′) and Ce4+ (v, u, v′′, and u′′) oxidation states, although their precise quantification cannot be determined (Fig. S5†). The Ni2p3/2 and Ni2p1/2 spectra show bands with binding energies at 855.6 eV and 873.5 eV, respectively, correlating well with Ni2+ from Ni(OH)2.28,29 The Ce 3d spectra showed a slight increase in the peaks associated with Ce4+ and a decrease in the peaks associated with Ce3+ after Ni was incorporated into the 15Ce catalyst. This suggests that some of the ceria particles were oxidized from Ce3+ to Ce4+ in the presence of nickel species, possibly by weakening the interaction between Ce and the γ-Al2O3 support. The TPR profiles for different supported and doped catalysts along with pure oxides were obtained for comparative purposes, and are shown in Fig. S6.† Pure NiO demonstrated a distinct reduction peak between approximately 240 °C and 400 °C, assigned to the reduction of NiO to Ni0. Thus, all catalysts were reduced prior to activity tests at 400 °C as they were expected to fully reduce all unalloyed Ni species. Pure CeO2 resulted in moderate hydrogen consumption at elevated temperatures, although when impregnated onto the γ-Al2O3 support, the reducibility was essentially eliminated. This suggests that Ce is interacting strongly with the alumina support, potentially forming a CeAlO3 alloy.30,31 As a result of this, the Ni–Ce bimetallic catalyst demonstrated superior reducibility compared to the Ni monometallic catalyst. The presence of CeAlO3 has been reported to lessen the interaction between Ni and Al2O3, restricting NiO crystalline growth, improving dispersion, and enhancing reducibility.32,33 The reducibility of Ni supported on either alumina support proved to be practically equal below 400 °C. NH3/CO2/CO temperature-programmed desorption mass spectrometry (TPD-MS) was performed to quantify the acidic, basic, and metallic sites, respectively, and the calculated values are presented in Table S3.† The principal difference between Ni and Ni–Ce catalysts was the number of acidic sites, where ceria incorporation reduced the acidity to almost half compared to the monometallic Ni catalyst (289 vs. 502 μmol g−1), and yet the acid strength remained consistent as shown in Fig. S7.† The concentration of basic sites was found to be identical in the Ni and Ni–Ce catalysts at 70 μmol g−1, and the concentration of metallic sites obtained from Ni was slightly higher at 71 μmol g−1 compared to that with ceria promotion at 61 μmol g−1. Thus, the differences in the concentration of sites between the catalysts suggest that the ratio between the acidic and basic sites could be an important aspect in the distinct formation of BHMTHF and 1,2,6-HT.
Based on all detected intermediates and products during catalytic activity tests, a detailed reaction scheme is proposed in Scheme 1. The colours for each compound presented in the reaction network correspond with the colours displayed in all forthcoming figures depicting product selectivity and concentrations.
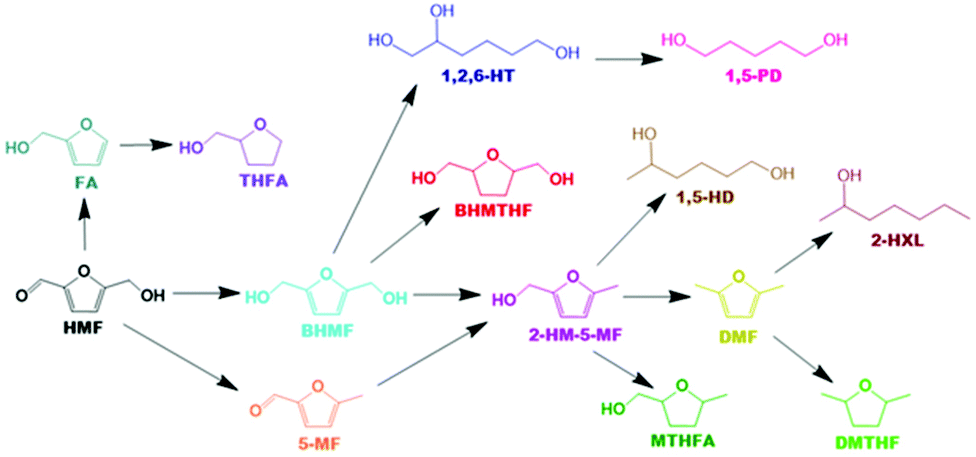 | ||
| Scheme 1 Proposed reaction network of HMF hydrogenation. | ||
It was not possible under our reaction conditions to cleave the C–O bond in the saturated furan ring to convert BHMTHF directly to 1,2,6-HT, which has been reported to be possible by several other groups.4,34,35 As shown in Fig. 1a, the product selectivity between the different γ-Al2O3 supports and dopants varied significantly. In the absence of nickel, the monometallic Ce catalyst demonstrated negligible activity with only 2% conversion. When Ni was supported on the mixed γ and α-Al2O3, complete conversion was achieved with an approximate product selectivity of 50% BHMF, 30% BHMTHF, and 3% 1,2,6-HT, with the remaining products being mostly 2-HM-5-MF and THFA. When Ni was supported on pure γ-alumina, ring saturation reactions increased considerably yielding mostly BHMTHF along with moderate amounts of MTHFA. Ring opened products also emerged which was mostly comprised of 1,5-HD. As the reducibility was almost identical, the presence of α-Al2O3 in the support likely contributed to the poor performance compared to the support which was determined to be exclusively γ-Al2O3, possibly as a result of the lower PZC. The incorporation of Ce prior to Ni resulted in dehydration reactions being significantly impeded and in their place, BHMTHF production increased up to 70%. The yield from the ring opening reaction forming 1,2,6-HT also increased to 9%. Subsequently, an activity test was performed with lanthanum in place of ceria to validate the beneficial effect of ceria. When ceria was replaced with lanthanum, complete conversion was still achieved but ring saturation and ring opening reactions were substantially hampered, resulting in primarily only BHMF. As observed in TPR, regardless of the substantial improvement in Ni reducibility with La impregnation instead of Ce, the Ni–La catalyst gave the lowest amount of BHMTHF and 1,2,6-HT (Fig. S6†). In conclusion, these results suggest that nickel reducibility was enhanced with the addition of ceria, although it does not appear to be the leading cause for the higher yields of BHMTHF and 1,2,6-HT. These results suggest that the higher PZC associated with the pure γ-alumina support in addition to ceria promotion could play a crucial role in the optimal formation of BHMTHF and 1,2,6-HT. Fig. 1b presents the selectivity of the detected intermediates and products as a function of time from the activity test performed with the 5Ni10Ce catalyst. In an attempt to optimize the yields of BHMTHF and 1,2,6-HT as well as reduce the reaction time, various Ni and Ce loadings were investigated with shorter activity tests of only 3 hours (Fig. S8†). From the results it can be concluded that the catalyst composition containing 10 wt% Ni and 15 wt% Ce performed the best with the highest selectivity of BHMTHF and 1,2,6-HT. Thus, this catalyst composition was subjected to further catalytic activity tests in order to optimize BHMTHF and 1,2,6-HT production.
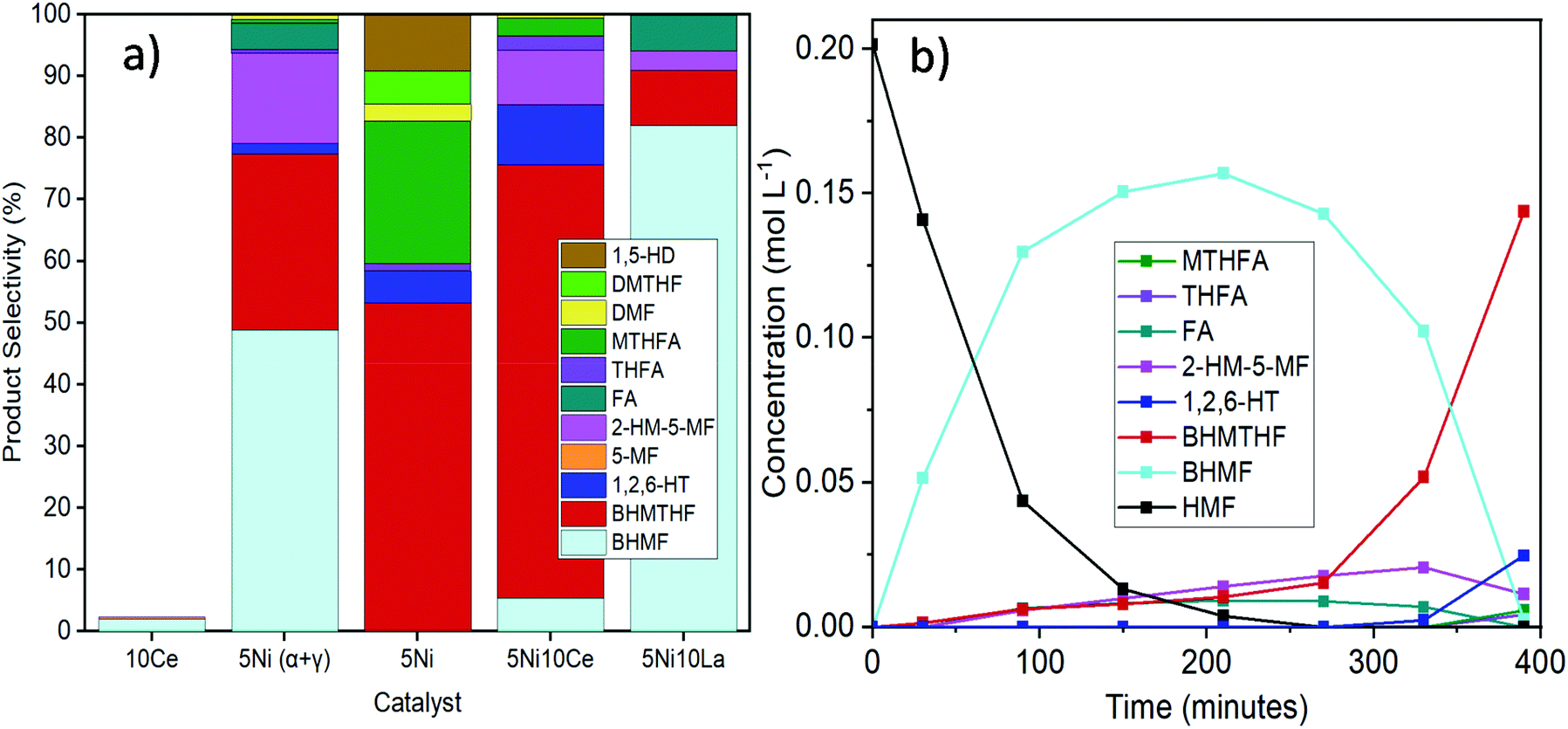 | ||
| Fig. 1 (a) Comparison of product selectivity between activity tests using different supports and dopants (reaction conditions: 200 °C, 50 bar of H2, 6 hours, THF as the solvent). (b) Demonstration of the product selectivity as a function of time with the 5Ni10Ce catalyst. | ||
Activity tests were carried out with reaction temperatures ranging from 140–200 °C to investigate the influence of temperature, as shown in Fig. 2. When using only THF as the solvent, temperatures below 170 °C were inadequate in converting past the initial reduction of the aldehyde reaction to produce BHMF. The highest BHMTHF selectivity of 85% was achieved at 170 °C. As the reaction temperatures were increased above 170 °C, successive dehydration reactions occurred forming MTHFA and the ring opened products increased including the formation of 1,5-HD and 1,5-PD. When water was incorporated into the reaction solvent system, dehydration reactions were virtually terminated causing BHMTHF and 1,2,6-HT to be practically the only final products. As the reaction temperature increased from 170 °C to 200 °C in both THF and H2O/THF solvent systems, the ratio between cis:trans isomers of BHMTHF decreased from 5.4/5.6 to around 4.7 (according to GC analysis and was verified by 13C NMR as shown in Fig. S9 and S10†). This is one of the highest trans-BHMTHF isomer formations ever achieved from HMF as most of the literature exclusively report >95% cis isomer.36,37 This is likely a result of the lower Ni loadings and higher reaction temperatures compared to most other studies, allowing the intermediate to more readily desorb following syn addition of the initial C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, flip over, and readsorb to subsequently hydrogenate the second CC from the opposite side to produce the trans-isomer. The highest 1,2,6-HT selectivity of 26% was achieved at 190 °C. The maximum BHMF selectivity of 96% in this study was also achieved at 140 °C when the reaction time was extended to 6 hours. A similar behaviour was observed when water was added to the reaction using the 10Ni catalyst, and yet the 1,2,6-HT selectivity was lower relative to the 10Ni15Ce catalyst, reaching only 10% (Fig. S11a†). An additional reaction was performed using 100 mg each of 10Ni/Al2O3 and 15Ce/Al2O3 which resulted in a minor increase of 1,2,6-HT selectivity to 15%, although it was still inferior to that with the bimetallic 10Ni15Ce catalyst (Fig. S11b†). This implies that the close proximity and/or interface between Ni and Ce particles is necessary for the enhanced ring opening yielding 1,2,6-HT.
C, flip over, and readsorb to subsequently hydrogenate the second CC from the opposite side to produce the trans-isomer. The highest 1,2,6-HT selectivity of 26% was achieved at 190 °C. The maximum BHMF selectivity of 96% in this study was also achieved at 140 °C when the reaction time was extended to 6 hours. A similar behaviour was observed when water was added to the reaction using the 10Ni catalyst, and yet the 1,2,6-HT selectivity was lower relative to the 10Ni15Ce catalyst, reaching only 10% (Fig. S11a†). An additional reaction was performed using 100 mg each of 10Ni/Al2O3 and 15Ce/Al2O3 which resulted in a minor increase of 1,2,6-HT selectivity to 15%, although it was still inferior to that with the bimetallic 10Ni15Ce catalyst (Fig. S11b†). This implies that the close proximity and/or interface between Ni and Ce particles is necessary for the enhanced ring opening yielding 1,2,6-HT.
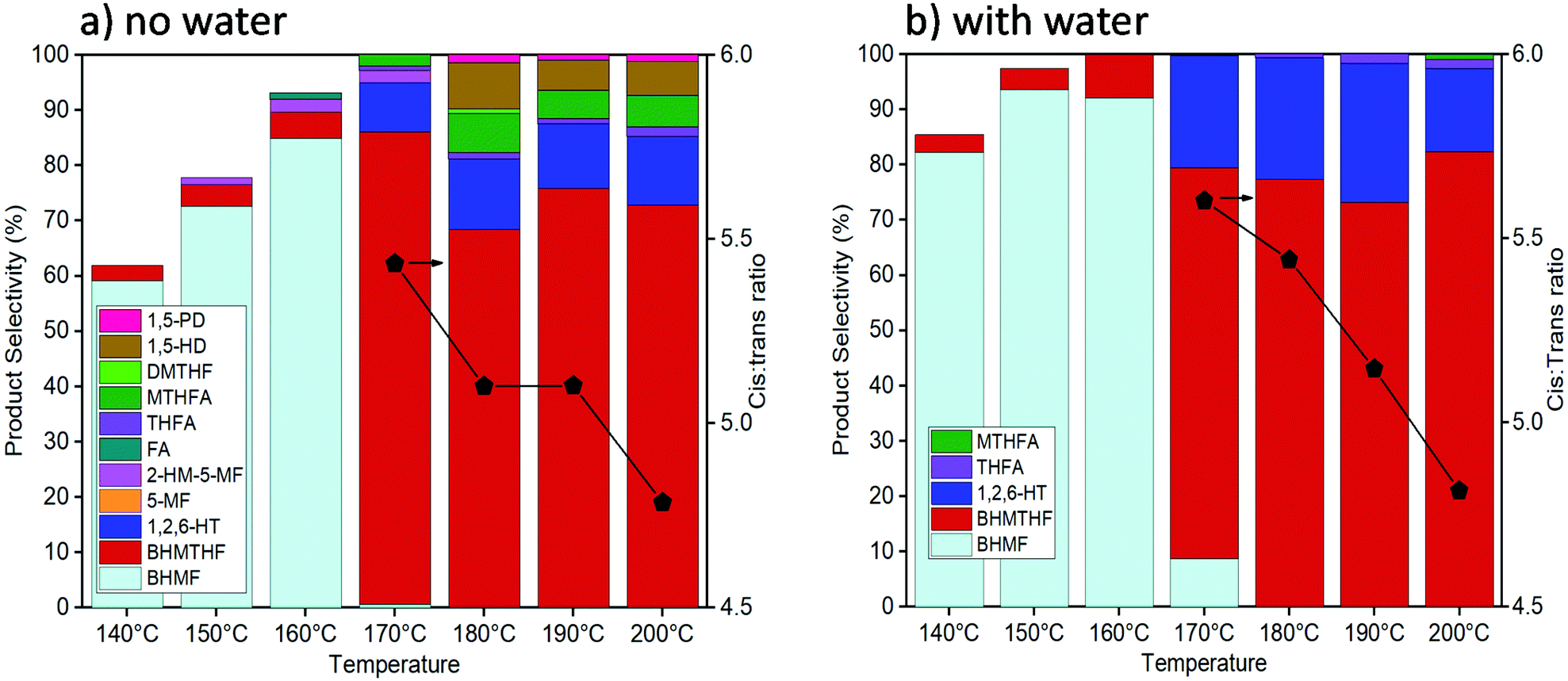 | ||
| Fig. 2 Activity tests with the reaction temperature from 140–200 °C with pure THF (a) and 5 vol% H2O in THF (b) as the solvent (reaction conditions: 10Ni15Ce catalyst, 50 bar of H2, 3 hours). | ||
As shown in Table 2, comparable results were also obtained on applying monophasic 1-BuOH and biphasic water-1-BuOH solvent systems, and even the highest BHMTHF selectivity of 88% was achieved in this study. To supplement the assumption that the point zero charge is a crucial aspect in the hydrogenation of HMF as previously cited, a selection of metal oxides with different point zero charges were added for activity tests utilizing water–THF as the solvent. Each activity test included 0.1 g of the 10Ni15Ce/γ-Al2O3 catalyst along with 0.5 g of metal oxide, and the results are presented in Table 2. Specifically, metal oxides were chosen considering their intrinsically high PZC (ZnO (∼9), CaO (∼11), and MgO (∼12)) and that they are all relatively insoluble in water, permitting straightforward filtration prior to the analysis of liquid samples.38–40
Interestingly, ZnO addition influenced the product selectivity trivially compared to its absence. The incorporation of MgO substantially increased the ring opening formation yielding 1,2,6-HT in up to 42%, although its performance was inferior to that of CaO despite its higher PZC. The addition of CaO drastically aided in the production of 1,2,6-HT, achieving a selectivity of 59% in just 1 hour after achieving the desired reaction temperature, which is the maximum accomplished in this study and also one of the highest reported in the literature. These conclusions were contrary to the results achieved by Dumesic et al., previously stated above, who reported a distinct improvement in BHMTHF yields and a decrease in ring opening reactions when similarly adding 500 mg of MgO into the same reaction mixture together with 1 wt% Ru supported on CeO2 as the catalyst.13 This suggests that transient intermediate(s) are likely present in very low coverages and/or concentrations (undetectable by GC in the liquid phase) that are formed on the catalyst surface, desorb, and then react further on a different site located either on the alumina-supported catalyst, or more primarily on the separate metal oxide particles with high IEP/PZC. Furthermore, these results propose that the ring-opening reaction yielding 1,2,6-HT does not occur exclusively on the metal NP, but predominantly on separate active sites that do not require close proximity between the metal NP and/or support. The results obtained during recyclability tests also complement our hypothesis on the key role that surface charge likely has in this reaction. As demonstrated in Fig. S12, the 10Ni15Ce catalyst maintained consistently high conversions above 90% for 5 continuous cycles of reactions in both THF and water![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :THF solvent systems. However, the product selectivity changed significantly following the initial activity test where ring saturation and ring opening reactions were considerably impeded, leaving BHMF as the main product. NH3/CO2/CO-TPD analysis of the spent catalysts revealed considerable changes to the concentration of sites following reactions (Table S3†). Specifically, the concentration of basic sites was ∼200% higher in the spent 10Ni15Ce catalysts compared to that before the reaction, while the concentration of metallic sites slightly increased. As this was independent of the inclusion of water, we believe this was a consequence of the presence of ceria which has been reported to permit extensive oxygen mobility from the bulk phase towards the catalyst surface via oxygen vacancies through (reverse) oxygen spillover effects.41,42 This increase in O2− moieties (and decrease of acidic sites) on the surface of the catalyst is expected to lower its PZC relative to before the reaction, similar to what has been described elsewhere.43,44 These results suggest that the strong negative charge of the catalyst is possibly inducing a repulsive effect on the furan ring, preventing it from effectively adsorbing, although still allowing adsorption via the carbonyl group. Overall, it would appear that the surface charge of the catalyst is undeniably an important factor of adsorption configurations during HMF hydrogenation, although additional aspects need to be carefully considered.
:THF solvent systems. However, the product selectivity changed significantly following the initial activity test where ring saturation and ring opening reactions were considerably impeded, leaving BHMF as the main product. NH3/CO2/CO-TPD analysis of the spent catalysts revealed considerable changes to the concentration of sites following reactions (Table S3†). Specifically, the concentration of basic sites was ∼200% higher in the spent 10Ni15Ce catalysts compared to that before the reaction, while the concentration of metallic sites slightly increased. As this was independent of the inclusion of water, we believe this was a consequence of the presence of ceria which has been reported to permit extensive oxygen mobility from the bulk phase towards the catalyst surface via oxygen vacancies through (reverse) oxygen spillover effects.41,42 This increase in O2− moieties (and decrease of acidic sites) on the surface of the catalyst is expected to lower its PZC relative to before the reaction, similar to what has been described elsewhere.43,44 These results suggest that the strong negative charge of the catalyst is possibly inducing a repulsive effect on the furan ring, preventing it from effectively adsorbing, although still allowing adsorption via the carbonyl group. Overall, it would appear that the surface charge of the catalyst is undeniably an important factor of adsorption configurations during HMF hydrogenation, although additional aspects need to be carefully considered.
Conclusions
We demonstrate for the first time the capability of a single, alumina-supported, transition metal catalyst to generate high yields of three, industrially relevant, high-value products from the hydrogenation of HMF by only changing the reaction conditions and/or medium. Furthermore, we demonstrate in this study the beneficial influence water has on limiting dehydration reactions and facilitating ring opening reactions in a biphasic solvent system in which one of the phases is water, or in monophasic systems containing water. The addition of solid metal oxides with high point zero charges further improved ring opening to form 1,2,6-HT, attaining one of the highest selectivities reported in the literature. Future work is underway to investigate the deciding factor(s) for regulating furan ring hydrogenation and ring opening reactions.Author contributions
All authors made equal contributions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to appreciatively acknowledge the financial support from the EU Framework Program for Research and Innovation Horizon 2020 under Grant agreement no. 814416 (Reaxpro) and the ARRS (Program P2-0152 and postdoctoral research project Z2-9200).References
- K. I. Galkin and V. P. Ananikov, ChemSusChem, 2019, 1–8 Search PubMed.
- R. J. Van Putten, J. C. Van Der Waal, E. De Jong, C. B. Rasrendra, H. J. Heeres and J. G. De Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed.
- C. Moreau, M. N. Belgacem and A. Gandini, Top. Catal., 2004, 27, 11–30 CrossRef CAS.
- J. He, S. P. Burt, M. Ball, D. Zhao, I. Hermans, J. A. Dumesic and G. W. Huber, ACS Catal., 2018, 8, 1427–1439 CrossRef CAS.
- A. V. Bridgwater, Biomass Bioenergy, 2012, 38, 68–94 CrossRef CAS.
- G. M. Mullen, E. J. Evans, B. C. Siegert, N. R. Miller, B. K. Rosselet, I. Sabzevari, A. Brush, Z. Duan and C. Buddie Mullins, React. Chem. Eng., 2018, 3, 75–85 RSC.
- C. Papadopoulos, K. Kappis, J. Papavasiliou, J. Vakros, M. Kuśmierz, W. Gac, Y. Georgiou, Y. Deligiannakis and G. Avgouropoulos, Catal. Today, 2019, 355, 647–653 CrossRef.
- Z. Wu, A. K. P. Mann, M. Li and S. H. Overbury, J. Phys. Chem. C, 2015, 119, 7340–7350 CrossRef CAS.
- L. Vivier and D. Duprez, ChemSusChem, 2010, 3, 654–678 CrossRef CAS PubMed.
- Z. Zhang, Y. Wang, J. Lu, M. Wang, J. Zhang, X. Liu and F. Wang, Inorganics, 2017, 5, 1–11 Search PubMed.
- N. Miletić, U. Izquierdo, I. Obregón, K. Bizkarra, I. Agirrezabal-Telleria, L. V. Bario and P. L. Arias, Catal. Sci. Technol., 2015, 5, 1704–1715 RSC.
- L. Yang, L. Pastor-Pérez, S. Gu, A. Sepúlveda-Escribano and T. R. Reina, Appl. Catal., B, 2018, 232, 464–471 CrossRef CAS.
- R. Alamillo, M. Tucker, M. Chia, Y. Pagán-Torres and J. Dumesic, Green Chem., 2012, 14, 1413–1419 RSC.
- M. Kosmulski, Adv. Colloid Interface Sci., 2016, 238, 1–61 CrossRef CAS PubMed.
- A. Pintar, J. Batista and I. Muševič, Appl. Catal., B, 2004, 52, 49–60 CrossRef CAS.
- J. Sung, L. Zhang, C. Tian, Y. R. Shen and G. A. Waychunas, J. Phys. Chem. C, 2011, 115, 13887–13893 CrossRef CAS.
- M. Sayed, N. Warlin, C. Hulteberg, I. Munslow, S. Lundmark, O. Pajalic, P. Tunå, B. Zhang, S. H. Pyo and R. Hatti-Kaul, Green Chem., 2020, 22, 5402–5413 RSC.
- J. Li, W. Zhang, S. Xu and C. Hu, Front. Chem., 2020, 8 DOI:10.3389/fchem.2020.00070.
- J. P. Lange and S. H. Wadman, ChemSusChem, 2020, 13, 5329–5337 CrossRef CAS PubMed.
- J. Kanetaka, T. Asano and S. Masamune, Ind. Eng. Chem., 1970, 62, 24–32 CrossRef CAS.
- K. T. V. Rao, Y. Hu, Z. Yuan, Y. Zhang and C. C. Xu, Appl. Catal., A, 2021 DOI:10.1016/j.apcata.2020.117892.
- J. Ohyama, A. Esaki, Y. Yamamoto, S. Arai and A. Satsuma, RSC Adv., 2013, 3, 1033–1036 RSC.
- X. Kong, R. Zheng, Y. Zhu, G. Ding, Y. Zhu and Y. W. Li, Green Chem., 2015, 17, 2504–2514 RSC.
- X. Kong, Y. Zhu, H. Zheng, F. Dong, Y. Zhu and Y. W. Li, RSC Adv., 2014, 4, 60467–60472 RSC.
- H. Kataoka, D. Kosuge, K. Ogura, J. Ohyama and A. Satsuma, Catal. Today, 2020, 352, 60–65 CrossRef.
- P. V. R. Rao, V. P. Kumar, G. S. Rao and K. V. R. Chary, Catal. Sci. Technol., 2012, 2, 1665–1673 RSC.
- R. Guo, Y. Zhou, W. Pan, J. Hong, W. Zhen, Q. Jin, C. Ding and S. Guo, J. Ind. Eng. Chem., 2013, 19, 2022–2025 CrossRef CAS.
- K. Han, T. Kreuger, B. Mei and G. Mul, ACS Catal., 2017, 7, 1610–1614 CrossRef CAS PubMed.
- X. H. Liu, W. Liu, X. K. Lv, F. Yang, X. Wei, Z. D. Zhang and D. J. Sellmyer, J. Appl. Phys., 2010 DOI:10.1063/1.3374468.
- S. Damyanova, C. A. Perez, M. Schmal and J. M. C. Bueno, Appl. Catal., A, 2002, 234, 271–282 CrossRef CAS.
- A. Iriondo, V. L. Barrio, J. F. Cambra, P. L. Arias, M. B. Guemez, M. C. Sanchez-Sanchez, R. M. Navarro and J. L. G. Fierro, Int. J. Hydrogen Energy, 2010, 35, 11622–11633 CrossRef CAS.
- K. Kamonsuangkasem, S. Therdthianwong, A. Therdthianwong and N. Thammajak, Appl. Catal., B, 2017, 218, 650–663 CrossRef CAS.
- X. Yang, J. Da, H. Yu and H. Wang, Fuel, 2016, 179, 353–361 CrossRef CAS.
- J. He, S. P. Burt, M. R. Ball, I. Hermans, J. A. Dumesic and G. W. Huber, Appl. Catal., B, 2019, 258, 117945 CrossRef.
- T. Buntara, S. Noel, P. H. Phua, I. Melián-Cabrera, J. G. De Vries and H. J. Heeres, Angew. Chem., Int. Ed., 2011, 50, 7083–7087 CrossRef CAS PubMed.
- X. Hu, R. J. M. Westerhof, L. Wu, D. Dong and C. Z. Li, Green Chem., 2015, 17, 219–224 RSC.
- S. H. Krishna, D. J. McClelland, Q. A. Rashke, J. A. Dumesic and G. W. Huber, Green Chem., 2017, 19, 1278–1285 RSC.
- P. Tengvall, Protein interactions with biomaterials, Elsevier Ltd., 2011, vol. 4 Search PubMed.
- N. A. Oladoja, I. A. Ololade, V. O. Olatujoye and T. A. Akinnifesi, Water, Air, Soil Pollut., 2012, 223, 1861–1876 CrossRef CAS.
- M. Farooq and A. Ramli, Natl. Postgrad. Conf., 2011, 2011 DOI:10.1109/NatPC.2011.6136447.
- A. Parastaev, V. Muravev, E. Osta, A. J. F. van Hoof, T. F. Kimpel, N. Kosinov and E. J. M. Hensen, Natl. Catal., 2020, 3, 526–533 CrossRef CAS.
- G. Balducci, M. S. Islam, J. Kaspar, P. Fornasiero and M. Graziani, Chem. Mater., 2000, 12, 677–681 CrossRef CAS.
- X. Hao, L. Quach, J. Korah, W. A. Spieker and J. R. Regalbuto, J. Mol. Catal. A: Chem., 2004, 219, 97–107 CrossRef CAS.
- M. H. Derkani, A. J. Fletcher, M. Fedorov, W. Abdallah, B. Sauerer, J. Anderson and Z. J. Zhang, Colloids Interfaces, 2019 DOI:10.3390/colloids3040062.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc02086b |
| This journal is © The Royal Society of Chemistry 2021 |