
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect Diels–Alder reactions of furfural derivatives with maleimides†
Răzvan C.
Cioc
a,
Martin
Lutz
b,
Evgeny A.
Pidko
 c,
Marc
Crockatt
d,
Jan C.
van der Waal
d and
Pieter C. A.
Bruijnincx
*a
c,
Marc
Crockatt
d,
Jan C.
van der Waal
d and
Pieter C. A.
Bruijnincx
*a
aOrganic Chemistry and Catalysis, Debye Institute for Nanomaterials Science, Faculty of Science, Utrecht University, Universiteitsweg 99, 3584 CG, Utrecht, the Netherlands. E-mail: p.c.a.bruijnincx@uu.nl
bCrystal and Structural Chemistry, Bijvoet Centre for Biomolecular Research, Faculty of Science, Utrecht University, Padualaan 8, 3584 CH Utrecht, the Netherlands
cInorganic Systems Engineering, Department of Chemical Engineering, Faculty of Applied Sciences, Delft University of Technology, Van der Maasweg 9, 2629 HZ Delft, the Netherlands
dDepartment of Sustainable Process and Energy Systems, TNO, Leeghwaterstraat 44, 2628 CA Delft, the Netherlands
First published on 24th November 2020
Abstract
The Diels–Alder (DA) reaction of furans is a versatile tool in synthetic organic chemistry and in the production of sustainable building blocks and smart materials. Numerous experimental and theoretical investigations suggest that the diene scope is effectively limited to electron-rich furans, which excludes the most abundant and readily accessible renewable derivatives: furfural and its 5-hydroxymethyl homologue. Herein we show for the first time that electron-poor 2-formylfurans can also directly engage in Diels–Alder couplings. The key to success is the use of aqueous medium, which supplies an additional thermodynamic driving force by coupling the unfavorable DA equilibrium to the exergonic hydration of the carbonyl functionality in the adducts to form geminal diols. This finding enables the direct access to various novel DA adducts derived from renewable furfurals and maleimides, via a mild, simple and environmentally-friendly synthetic protocol.
Introduction
The Diels–Alder (DA) reaction of furan dienes towards 7-oxanorbornenes is an old yet powerful tool in organic chemistry, with applications ranging from natural product synthesis and renewable chemical commodities production to drug discovery and materials science.1 The reaction generally proceeds chemoselectively under mild conditions, often without the need for a catalyst. Depending on the furan/dienophile combination, yields may be low, however, as reactions may be kinetically slow or suffer from thermodynamic equilibrium limitations; moreover, the DA adduct is often quite labile, leading to facile cyclo-reversion back to the addends.As far as kinetics is concerned, it is well known that the substitution pattern of the furan diene strongly modulates reactivity. In line with the general Frontier Molecular Orbital (FMO) theory of [4 + 2] cycloadditions, numerous studies have shown that furans bearing electron-donating groups (e.g. H, Me, OMe, CH2OH, etc.) display good kinetics, while electron-poor furans (e.g. with CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O or COOR) are too sluggish, inactive substrates.2
O or COOR) are too sluggish, inactive substrates.2
Predicting the thermodynamics of such furan DA cycloadditions is much less straightforward. Being typically moderately exothermic and entropically disfavored, the Gibbs free energy of these reactions is generally in the order of only a few kJ mol−1. Thus, small changes in operating parameters (pressure, temperature, concentration, solvent) have a profound impact on the position of the DA equilibrium. Various strategies have been employed to overcome unfavorable thermodynamics (neat conditions, excess reactants, selective crystallization, elevated pressures, coupling with secondary reactions, etc.).
The solutions offered are generally quite case-specific, however, and small changes made in operating conditions to improve thermodynamics can adversely affect kinetics, and vice versa. For instance, in case of sluggish conversions, heating may improve kinetics but will negatively impact thermodynamics, since ΔS° < 0. In addition, small changes in the addends structure often impact the DA equilibrium in an intricate manner as well. For example, furan reacts faster with maleic anhydride than with maleimide, but the reaction with the latter is more exergonic.3 Similarly, methylated furans react more readily with itaconic anhydride than furan itself, but the equilibrium conversion is highest with the latter.4 Thus, the interplay between kinetics and thermodynamics in furan DA reactions is often rather subtle, making it challenging to understand, control and optimize them.
Currently, synthetic applications of furan DA chemistry are dominated by electron-rich dienes such as furan itself, furfuryl alcohol and 2,5-dimethyl furan. To the best of our knowledge, no examples of direct DA reactions involving furfural (2-furan carboxaldehyde) or its homologue 5-hydroxymethylfurfural (5-HMF) have yet been reported; moreover, indirect strategies relying on redox-neutral chemical activation (acetalization,5 hydrazone formation6) are also scarce. That these substrates are not part of the current furan DA toolbox is unfortunate, as these are in fact the most readily accessible furans (and precursors to most other derivatives).7 Furthermore, having a reactive formyl handle on the DA product offers many opportunities for further synthetic upgrading. The current mismatch between furan diene availability, synthetic potential and reactivity in DA reactions is particularly pressing in the context of the production of biobased chemicals, e.g. high-value oxygenates such as phthalic anhydride and terephthalic acid.8 This field would greatly benefit from expansion of the furan diene scope beyond the ubiquitous methylated derivatives that give high yields for the DA reaction itself, but nevertheless show a low atom-, step- and redox-economy and thus poor sustainability for the overall process starting from the biomass resource (Scheme 1).9
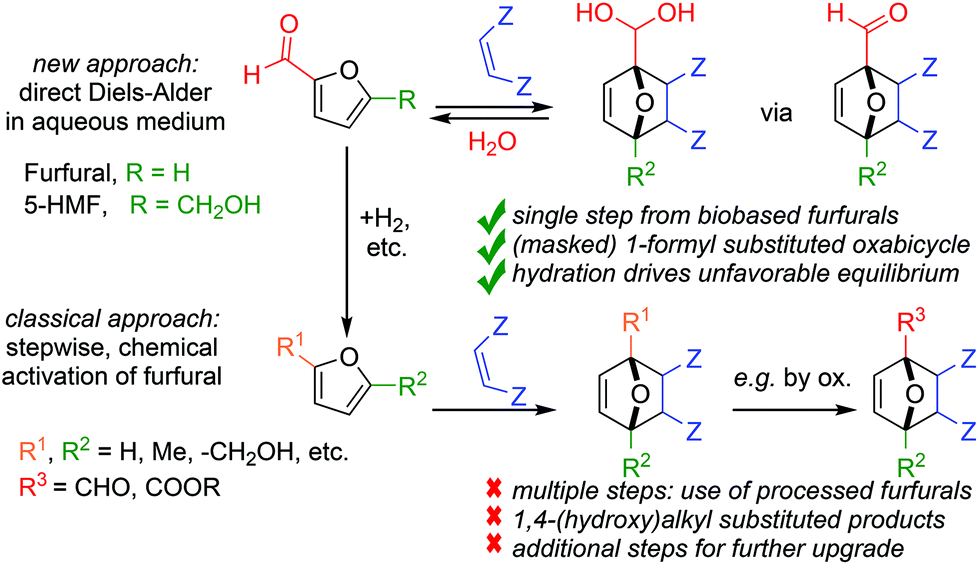 | ||
| Scheme 1 A new approach to direct Diels–Alder reaction of biobased formyl-functionalized furans. | ||
Acknowledging this major limitation in scope, we decided to take a closer look at the feasibility of employing furfural as diene. Using prototypical, reactive maleimide dienophiles, we chose to study the DA reaction in water as reaction medium, as water is well-known to improve both the kinetics and thermodynamics of other DA reactions10via the hydrophobic effect.11 Furfural has a fairly high solubility in water, which would not only make an ‘in-water’-type activation mechanism feasible,10c but also allows for concentrated solutions, anticipated to be favorable for both the reaction rate and the equilibrium.
Results and discussion
Reaction optimization
The first experiments between furfural 1a and N-methyl maleimide 2a in concentrated aqueous solution demonstrated the clean formation of DA adducts 3a (with a modest exo![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :endo selectivity of approx. 2:1). Intriguingly, the 1H-NMR spectrum of the crude reaction indicated that the adducts were exclusively present in the geminal diol form. This peculiar structural feature proved highly advantageous in downstream processing, as unreacted starting materials could be easily washed away with organic solvents leaving the DA adduct in the aqueous phase in essentially pure form (Fig. 1). Subsequent optimization experiments (see ESI for details†) showed that the highest yields (60–65%) could be obtained in concentrated aqueous solution (2 M), at 60 °C, and in the absence of catalysts or additives. 5-HMF (1b) could also be cleanly reacted to its DA adducts, again giving only the geminal diol product and allowing product purification by simple extraction.
:endo selectivity of approx. 2:1). Intriguingly, the 1H-NMR spectrum of the crude reaction indicated that the adducts were exclusively present in the geminal diol form. This peculiar structural feature proved highly advantageous in downstream processing, as unreacted starting materials could be easily washed away with organic solvents leaving the DA adduct in the aqueous phase in essentially pure form (Fig. 1). Subsequent optimization experiments (see ESI for details†) showed that the highest yields (60–65%) could be obtained in concentrated aqueous solution (2 M), at 60 °C, and in the absence of catalysts or additives. 5-HMF (1b) could also be cleanly reacted to its DA adducts, again giving only the geminal diol product and allowing product purification by simple extraction.
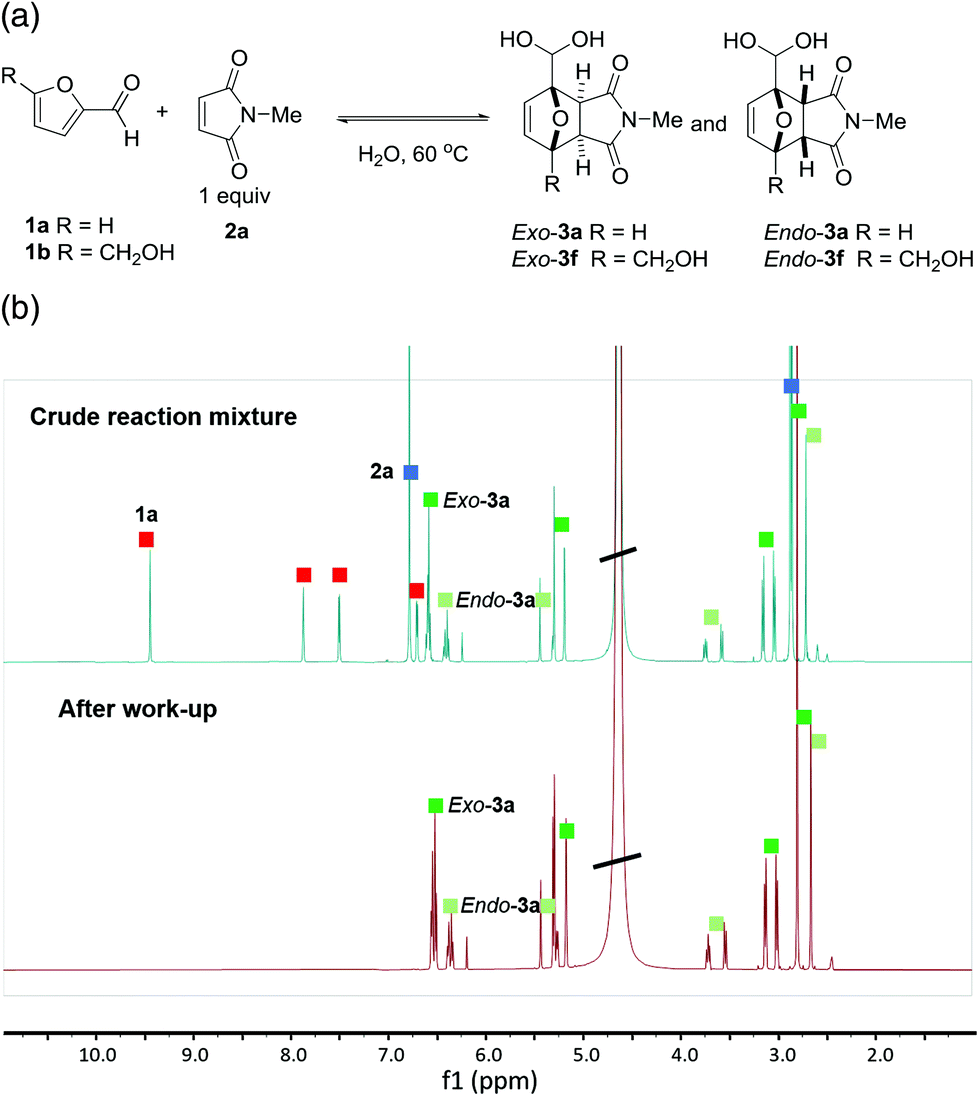 | ||
| Fig. 1 (a) First examples of the direct DA cycloaddition of formyl-substituted furans furfural 1a and 5-HMF 1b (with maleimide 2a). (b) The DA adducts can be readily purified by extractive work-up, as exemplified by 3a. 1H-NMR spectrum: crude reaction mixture (above); purified adducts 3a (below). | ||
The typical kinetic profiles of a reversible DA reaction were obtained, with stereoselectivity for the thermodynamically-preferred exo-isomer gradually increasing in time (Fig. 2).12 Conversion of 1a reached equilibrium within 60 h, at which point the exo-3a:endo-3a ratio was approx. 5:1. The nearly identical profiles of total adduct yield and furfural conversion highlight the reaction's high chemoselectivity. For dienophile 2a, hydrolysis to maleic acid occurred to a very limited extent (typically 1–2% in 16 h). The chemoselectivity for 5-HMF 1b was similarly high, but the endo isomer was still the major product after 60 h.13,14
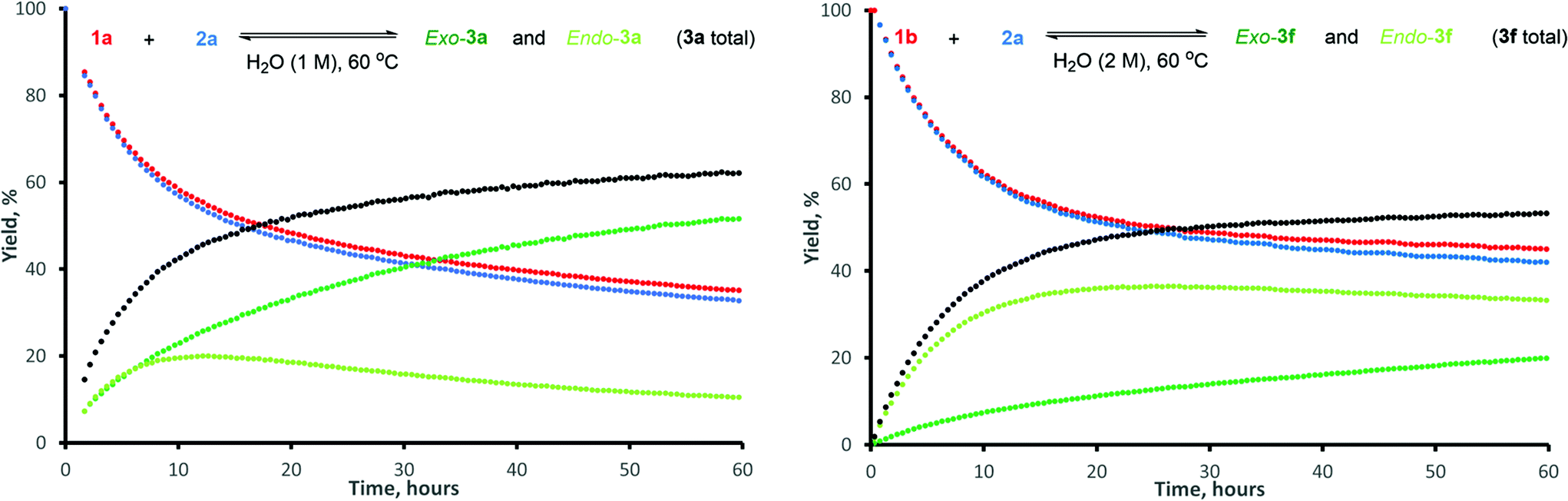 | ||
| Fig. 2 Kinetic traces for the reaction between: furfural (1a) and N-methyl maleimide (2a), left; 5-HMF (1b) and N-methyl maleimide (2a), right. | ||
Reaction scope
With the optimal conditions in hand, the substrate scope was further explored (Table 1). The title reaction between furfural 1a and N-methyl maleimide 2a afforded 58% total yield of adducts on a 4 mmol scale; promisingly, scaling-up to 70 mmol proved successful (entry 2) as well as the recycling of recovered unreacted starting materials (entry 3). Free maleimide 2b also worked, giving only a slightly lower conversion and chemoselectivity (entry 4). The reaction still performed well with N-ethyl maleimide, but higher homologues (N-Pr, N-Ph) showed reduced conversion and yields, as a result of their lower aqueous solubility. Noteworthy, these more lipophilic adducts showed an increased preference for dehydration of the geminal diol back to the aldehyde 3′ and partitioning to the organic phase (entry 4 vs. 1, 5 and 6).|
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No | Product | Furan | R1 | Maleimide | R2 | Recovery 1a, % | exo-3, % | endo-3, % | Total aqueous, % | exo-3′, % | endo-3′, % | Total organic, % |
| a Recovery of 1 and yields of 3 and 3′ were determined by 1H-NMR analysis of the crude reaction mixture with external standard, after extractive work-up. b 70 mmol scale. c 10 mmol scale with recycled 1 and 2. d Minor amount of unidentified side products observed. e Biphasic reaction mixture. f DMSO used as cosolvent; reaction mixtures were biphasic in the absence of cosolvent. g Mainly present in the hydrated form 3i. h Products were bis-geminal diols. i 2-Acetylfuran was used as diene. j Isolated yield (silicagel chromatography) and crude yield were identical. | ||||||||||||
| 1 | 3a | 1a | H | 2a | Me | 37 | 40 | 18 | 58 | 3 | 1 | 4 |
| 2b | 3a | 1a | H | 2a | Me | 38 | 35 | 16 | 51 | 5 | 1 | 6 |
| 3c | 3a | 1a | H | 2a | Me | 38 | 34 | 18 | 52 | 4 | 2 | 6 |
| 4d | 3b | 1a | H | 2b | H | 53 | 30 | 8 | 38 | 0 | 0 | 0 |
| 5 | 3c | 1a | H | 2c | Et | 48 | 28 | 8 | 36 | 6 | 1 | 7 |
| 6e | 3d | 1a | H | 2d | nPr | 70 | 7 | 1 | 8 | 11 | 1 | 12 |
| 7e,f | 3e | 1a | H | 2e | Ph | 81 | 1 | 0 | 1 | 5 | 1 | 6 |
| 8 | 3f | 1b | CH2OH | 2a | Me | 51 | 13 | 37 | 50 | 0 | 0 | 0 |
| 9f | 3g | 1c | CH2OMe | 2a | Me | 67 | 5 | 7 | 12 | 3 | 3 | 6 |
| 10f | 3h | 1d | Me | 2a | Me | 80 | 8 | 3 | 11 | 3 | 0 | 3 |
| 11e | 3i | 1e | Br | 2a | Me | 70 | 5 | 12 | 17 | 1 | 9g | 10 |
| 12f | 3j | 1f | CHO | 2a | Me | 97 | 2h | 5h | 7 | 0 | 0 | 0 |
| 13f | 3k | 1g | COOH | 2a | Me | 94 | 4 | 1 | 5 | 0 | 0 | 0 |
| 14e | 3l | 1h | Hi | 2a | Me | 61 | 0 | 0 | 0 | 32j | Trace | 0 |
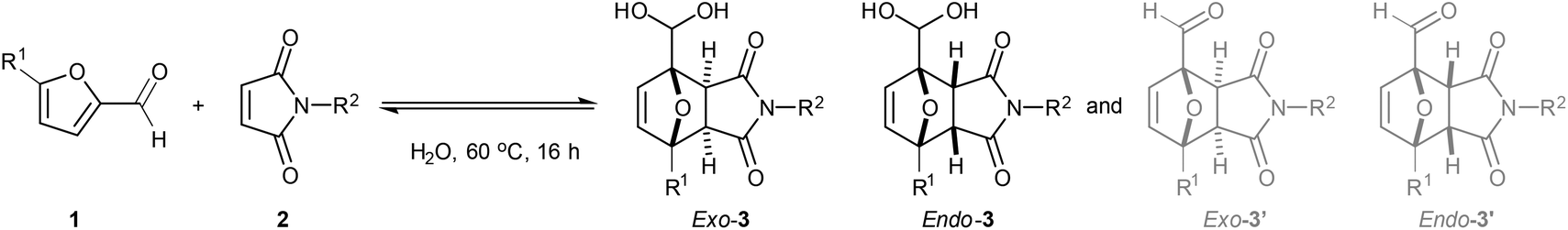
Next, we studied the performance of 5-substituted furfurals as dienes. 5-HMF 1b afforded a 50% yield of cycloadducts 3f (mainly endo, entry 8), considerably higher than the methylated derivative 1c (18% total yield). 5-Methylfurfural 1d showed a lower conversion, possibly due to less favorable thermodynamics4 or lower aqueous miscibility (entry 10), as did the bromo derivative 1e (entry 11). Remarkably, the presence of a second electron withdrawing-substituent (CHO, entry 12 and COOH, entry 13) still allowed for DA adduct formation, albeit in a very small amount; notably, the adducts of 2,5-diformylfuran 1f were found to be symmetric bis-geminal diols. Finally, 2-acetylfuran 1h also proved a suitable diene, giving 32% of exo-3′l. Interestingly, the geminal diol was not formed in this case.
Illustrative of the sensitivity of the DA reaction to small structural changes, no DA adducts could be detected upon variation of the dienophile, as neither acrylonitrile, maleic acid, fumaric acid, acetylenedicarboxylic acid or 4-cyclopentene-1,3-dione were found to react with furfural under these conditions. Presumably, in the case of the first three dienophiles, the HOMO–LUMO gap is too large for the reaction to occur with a noticeable rate at 60 °C.15 For the latter two, dienophile stability was an issue, given the tarry, insoluble product observed after reaction.
Follow-up chemistry
The geminal diol in adduct 3 is an unusual,16 but versatile functional group for further conversion towards value-added molecules (Scheme 2). Additions of N-based nucleophiles to the (masked) carbonyl proceeded readily and the insoluble products were conveniently isolated by filtration. Notably, with N,N-dimethylhydrazine, spontaneous dehydration resulted in aromatization, as reported in related chemistry.6 Varying the hydrazine allowed for the tosyl hydrazone to be prepared in excellent yield and a single crystal X-ray diffraction analysis of (racemic) exo-5 provided unambiguous diastereochemistry assignment. The oxime of adduct 3a was isolated in 69% yield. Next, the geminal diol 3a could be subjected to a Pinnick-type oxidation,17 leading to the selective precipitation of the major exo-7 isomer. Finally, careful hydrogenation of the alkene moiety in 3, a strategy previously developed to stabilize DA adducts,18 gave 8 with minimal overreduction of the diol to the primary alcohol.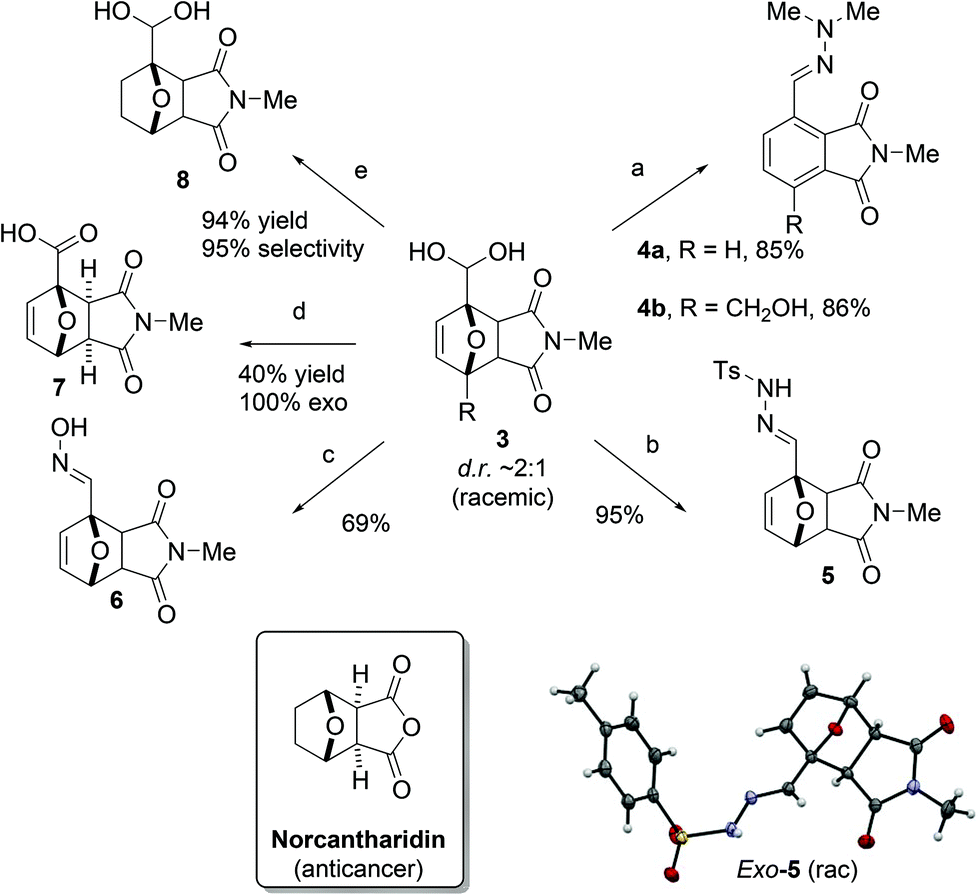 | ||
| Scheme 2 Follow-up chemistry starting with adduct 3a. Reagents and conditions: a. Me2N-NH2, rt; b. TsNH-NH2, rt; c. (NH3OH)Cl, NaOH, rt; d. NaClO2, H2O2, KH2PO4, rt. e. H2, Pd/C, rt. | ||
These results demonstrate some of the many downstream chemical diversification options offered by the now more highly substituted 7-oxabicyclo[2.2.1]heptane motif. This scaffold is encountered in natural products1f,i and bioactive molecules (e.g. norcantharidin and its amide/imide derivatives)19 and can be readily upgraded into complex synthetic targets.20 Thus, our novel DA coupling between furfurals and maleimides provides a valuable expansion of the organic chemist's toolbox. In addition, the approach may inspire the development of novel DA-based routes towards renewable building blocks, e.g. high-value biobased aromatics, directly from the most readily accessible oxygenated furanics.
Noteworthy, all furfural-derived DA adducts synthesized herein are novel compounds. Our two-stage aqueous protocol is simple, environmentally-friendly and allows for the facile recycling of unreacted starting materials; moreover, high-purity products can be easily obtained without the need for chromatography. Importantly, the aqueous cycloaddition route is superior to alternative reaction sequences: control experiments showed that other furfural derivatives such as the tosyl hydrazone and the oxime are poor dienes in coupling with 2a.
Mechanistic studies
In these unexpectedly efficient DA reactions, water plays a critical role both as a solvent and as a reactant in geminal diol formation. Two different pathways may account for the formation of the geminal diol adducts 3, depending on whether aldehyde hydration occurs prior or subsequent to DA coupling. Density functional theory (DFT) calculations at the PBE0-D3(SMD(water))/6-311+G(d) level of theory suggests that the two pathways have similar overall activation barriers and are thus kinetically indistinguishable (see Fig. 3 and ESI for details†). Hydration of furfural significantly increases its reactivity as diene (>20 kJ mol−1 reduction in the ΔG‡ for cycloaddition), as the strongly electron-withdrawing formyl substituent is converted into a weakly electron-donating bis-hydroxyalkyl group. This effect can be compared to acetalization, which is known to enhance furfural reactivity towards dienophiles.5 On the other hand, hydrated furfural (1′a) is a high-energy intermediate and is thus formed only to a very small extent in aqueous solution (indeed, we could not detect this species by 1H-NMR). Most likely, less than 1% of furfural is hydrated at equilibrium; for comparison, the pKhydration of benzaldehyde is 2.21 The enhanced reactivity of 1′a is thus canceled out by its low concentration. The overall kinetic barrier for this pathway (approx. 100 kJ mol−1) is very similar to the ΔG‡ of the direct Diels–Alder cycloaddition (1a + 2a to form 3′a) and in good accordance with prior DFT data for related systems.2d,14 While the calculations did not show a discernable kinetic effect, they do showcase the impact of hydration on the thermodynamics of the overall process. The reaction 3′a + H2O to form 3a provides a substantial thermodynamic drive (−12 to −15 kJ mol−1) to an otherwise barely exergonic cycloaddition (ΔG° is −2 to −7 kJ mol−1). These results suggest that it is not, as typically assumed, kinetics that hampers the desired DA reaction, but thermodynamics. This outcome is rather unusual, as reactivity (and the lack thereof) in furan cycloadditions is generally explained in terms of FMO theory-derived kinetic arguments.22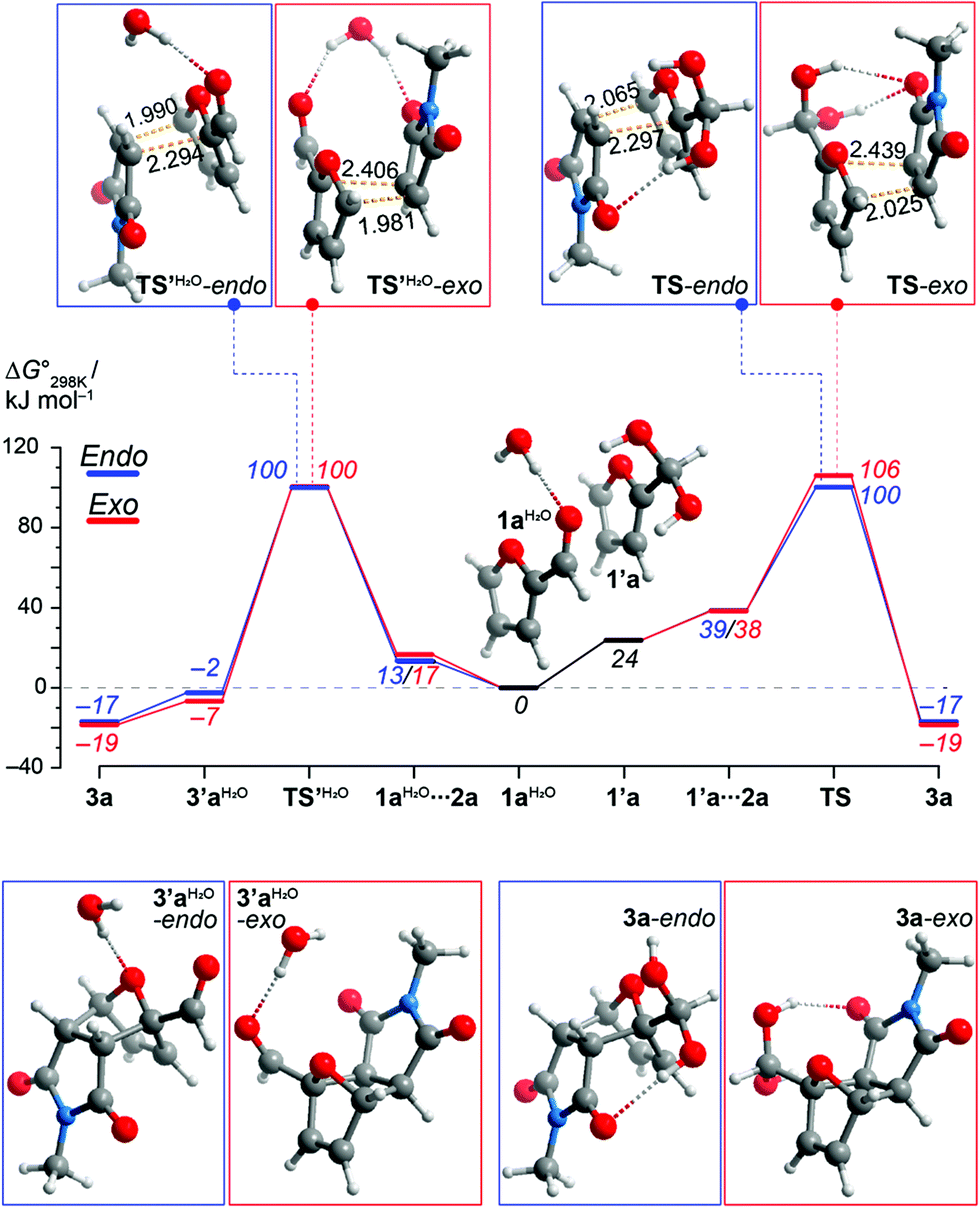 | ||
| Fig. 3 DFT-computed Gibbs free energy diagram for the reaction between furfural 1a and maleimide 2a in water, with hydration prior to (right hand side) or after (left) DA addition. | ||
To the best of our knowledge, in nearly a century of DA chemistry, we here present the first direct observation and spectroscopic characterization of cycloadducts of furfural.22 The crucial role of water is highlighted by the experimentally determined endergonic nature (K = 0.02 M−1 for exo-3′a formation at 60 °C) of the reaction between 1a and 2a when run neat or in organic solvents (see ESI†). Intriguingly, attempts to isolate the free aldehyde 3′a by column chromatography afforded the hydrated form 3a instead. In aqueous solution, the extent of hydration of 3a is >99% (by 1H-NMR). Obviously, the adjacent electron-withdrawing ether group greatly enhances the electrophilicity of the carbonyl (cf., degrees of hydration for 2-chloro-isobutanal and isobutanal are 83% and 28%).21,23 Thus, the fairly exergonic hydration of the primary Diels–Alder adducts 3′ in aqueous media provides the critical thermodynamic drive to shift the equilibrium to the product side. On the other hand, hydration seems not to be relevant in the case of 2-acetylfuran 1h as substrate (for comparison, acetone is <1% hydrated in aqueous solution21): both in aqueous media and under neat conditions, adduct 3′l is formed exclusively in the carbonyl form (at comparable conversion). This observation indicates that physical interactions involving water (hydrogen bonding, hydrophobic effect) are of secondary importance in this system and the major role of water is to pull the equilibrium to the product side by chemically trapping the adducts 3′.
Conclusions
This work demonstrates the successful use of unfunctionalized furfural derivatives as dienes in Diels–Alder cycloadditions. Thus, a variety of novel DA adducts could be readily obtained following an aqueous protocol in line with the principles of green chemistry. In addition, our mechanistic investigation implies that the well-known limitations of Diels–Alder chemistry with furfural and other related electron-poor furans are of thermodynamic rather than kinetic nature and can be circumvented. The new insights provided can guide further efforts in the development of new chemistry for these oxygenated, electron-poor furans, e.g. to allow the more sustainable valorization of the key biobased platform molecules furfural and 5-HMF. Studies on the expansion of the scope of this method in terms of substrates, nucleophilic solvent use (see ESI for preliminary results†) and downstream conversion are currently underway.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by The Netherlands Organization for Scientific Research (NWO LIFT grant 741.018.408). NWO is acknowledged for providing access to SurfSARA supercomputing facilities. E.A. Pidko acknowledges funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement no. 725686). The X-ray diffractometer has been financed by NWO. Dr T.N. Ran and Dr J. T. B. H. Jastrzebski (Utrecht University) and Mr I. Chernyshov (ITMO University, St. Petersburg) are acknowledged for technical assistance.Notes and references
- Selection of recent reviews and monographs: (a) H. Sun, C. P. Kabb, M. B. Sims and B. S. Sumerlin, Prog. Polym. Sci., 2019, 89, 61 CrossRef CAS; (b) M. Vauthier, L. Jierry, J. C. Oliveira, L. Hassouna, V. Roucoules and F. Bally-Le Gall, Adv. Funct. Mater., 2019, 29, 1806765 CrossRef; (c) H. Takikawa, A. Nishii, T. Sakaib and K. Suzuki, Chem. Soc. Rev., 2018, 47, 8030 RSC; (d) T. Elschner, F. Obst and T. Heinze, Macromol. Biosci., 2018, 18, 1800258 CrossRef; (e) C. D. Spicer, E. T. Pashuck and M. M. Stevens, Chem. Rev., 2018, 118, 7702 CrossRef CAS; (f) S. Roscales and J. Plumet, Heterocycles, 2015, 90, 741 CrossRef CAS; (g) G. Delaittre, N. K. Guimard and C. Barner-Kowollik, Acc. Chem. Res., 2015, 48, 1296 CrossRef CAS; (h) M. Gregoritza and F. P. Brandl, Eur. J. Pharm. Biopharm., 2015, 97, 438 CrossRef CAS; (i) A. J. Moreno-Vargas and P. Vogel, in Synthesis of Saturated Oxygenated Heterocycles I: 5- and 6-Membered Rings, ed. J. Cossy, Springer-Verlag, Berlin Heidelberg, 2014, 141–188 Search PubMed; (j) C. L. Hugelshofer and T. Magauer, Synthesis, 2014, 46, 1279 CrossRef; (k) C. E. Puerto Galvis, L. Y. Vargas Méndez and V. V. Kouznetsov, Chem. Biol. Drug Des., 2013, 82, 477 CrossRef CAS; (l) Y.-L. Liu and T.-W. Chuo, Polym. Chem., 2013, 4, 2194 RSC; (m) A. Gandini, Prog. Polym. Sci., 2013, 38, 1 CrossRef CAS.
- (a) Y. Qiu, J. Phys. Org. Chem., 2015, 28, 370 CrossRef CAS; (b) R. W. Foster, L. Benhamou, M. J. Porter, D.-K. Bučar, H. C. Hailes, C. J. Tame and T. D. Sheppard, Chem. – Eur. J., 2015, 21, 6107 CrossRef CAS; (c) V. Froidevaux, M. Borne, E. Laborbe, R. Auvergne, A. Gandini and B. Boutevin, RSC Adv., 2015, 5, 37742 RSC; (d) R. C. Boutelle and B. H. Northrop, J. Org. Chem., 2011, 76, 7994 CrossRef CAS; (e) W. G. Dauben and H. O. Krabbenhoft, J. Am. Chem. Soc., 1976, 98, 1992 CrossRef CAS A curious exception to this rule would be intramolecular Diels–Alder reactions with furans bearing halogen or nitro substituents at the 2-position, see for instance: (f) T. Y. Cowie, M. Veguillas, R. L. Rae, M. Rougé, J. M. Żurek, A. W. Prentice, M. J. Paterson and M. W. P. Bebbington, J. Org. Chem., 2017, 82, 6656 CrossRef CAS; (g) S. N. Pieniazek and K. N. Houk, J. Org. Chem., 2006, 71, 5432 CrossRef; (h) K. R. Crawford, S. K. Bur, C. S. Straub and A. Padwa, Org. Lett., 2003, 5, 3337 CrossRef CAS; (i) Z. Klepo and K. Jakopcic, J. Heterocycl. Chem., 1987, 24, 1787 CrossRef CAS.
- L. Rulíšek, P. Šebek, Z. Havlas, R. Hrabal, P. Čapek and A. Svatoš, J. Org. Chem., 2005, 70, 6295 CrossRef.
- A. D. Pehere, S. Xu, S. K. Thompson, M. A. Hillmyer and T. R. Hoye, Org. Lett., 2016, 18, 2584 CrossRef CAS.
- (a) H. Chang, G. W. Huber and J. Dumesic, ChemSusChem, 2020, 13, 5213 CrossRef CAS; (b) I. Scodeller, S. Mansouri, D. Morvan, E. Muller, K. de Oliveira Vigier, R. Wischert and F. Jérôme, Angew. Chem., Int. Ed., 2018, 130, 10670 CrossRef; (c) A. Guidi, V. Theurillat-Moritz and P. Vogel, Tetrahedron: Asymmetry, 1996, 7, 3153 CrossRef CAS; (d) J.-L. Métral, J. Lauterwein and P. Vogel, Helv. Chim. Acta, 1986, 69, 1287 CrossRef; (e) T. Kametani, T. Suzuki, A. Tomino and K. Unno, Chem. Pharm. Bull., 1982, 30, 167 CrossRef CAS; (f) J. Maślińska-Solich, J. Chem. Soc., Perkin Trans. 1, 1975, 606 RSC.
- (a) M. Crockatt and J. H. Urbanus, (TNO), WO2017/146581, 2017; (b) S. Higson, F. Subrizi, T. D. Sheppard and H. C. Hailes, Green Chem., 2016, 18, 1855 RSC; (c) M. V. Gil, V. Luque-Agudo, E. Román and J. A. Serrano, Synlett, 2014, 25, 2179 CrossRef CAS; (d) K. T. Potts and E. B. Walsh, J. Org. Chem., 1984, 49, 4099 CrossRef CAS.
- (a) C. Xu, E. Paone, D. Rodríguez-Padrón, R. Luque and F. Mauriello, Chem. Soc. Rev., 2020, 49, 4273 RSC; (b) H. Xia, S. Xu, H. Hu, J. An and C. Li, RSC Adv., 2018, 8, 30875 RSC; (c) F. A. Kucherov, L. V. Romashov, K. I. Galkin and V. P. Ananikov, ACS Sustainable Chem. Eng., 2018, 6, 8064 CrossRef CAS; (d) L. Hua, L. Lin, Z. Wu, S. Zhou and S. Liu, Renewable Sustainable Energy Rev., 2017, 74, 230 CrossRef; (e) R. Mariscal, P. Maireles-Torres, M. Ojeda, I. Sádaba and M. López Granados, Energy Environ. Sci., 2016, 9, 1144 RSC.
- (a) E. Mahmoud, D. A. Watson and R. F. Lobo, Green Chem., 2014, 16, 167 RSC; (b) C. L. Williams, C.-C. Chang, P. Do, N. Nikbin, S. Caratzoulas, D. G. Vlachos, R. F. Lobo, W. Fan and P. J. Dauenhauer, ACS Catal., 2012, 2, 935 CrossRef CAS.
- (a) J. J. Pacheco and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 8363 CrossRef CAS; (b) J. K. Ogunjobi, T. J. Farmer, C. R. McElroy, S. W. Breeden, D. J. Macquarrie, D. Thornthwaite and J. H. Clark, ACS Sustainable Chem. Eng., 2019, 7, 8183 CrossRef CAS.
- For a selection of reviews, see. (a) T. Kitanosono and S. Kobayashi, Chem. – Eur. J., 2020, 26, 9408 CrossRef CAS; (b) M. B. Gawande, V. D. B. Bonifácio, R. Luque, P. S. Branco and R. S. Varma, Chem. Soc. Rev., 2013, 42, 5522 RSC; (c) R. N. Butler and A. G. Coyne, Chem. Rev., 2010, 110, 6302 CrossRef CAS; (d) A. Chanda and V. V. Fokin, Chem. Rev., 2009, 109, 725 CrossRef CAS; (e) S. Otto and J. B. F. N. Engberts, Pure Appl. Chem., 2000, 72, 1365 CAS; (f) U. Pindur,, G. Lutz and C. Otto, Chem. Rev., 1993, 93, 741 CrossRef.
- (a) W. Blokzijl and J. B. F. N. Engberts, Angew. Chem., Int. Ed., 1993, 32, 1545 CrossRef; (b) D. C. Rideout and R. Breslow, J. Am. Chem. Soc., 1980, 102, 7816 CrossRef CAS.
- A similar kinetic profile, in which the exo/endo ratio continues to change at equilibrium conversion, was also observed in: J. C. de Oliveira, M.-P. Laborie and V. Roucoules, Molecules, 2020, 25, 243 CrossRef.
- Note: At 40 °C, the endo/exo ratio was found to be approx. 7:1. Ananikov et al. have determined computationally that the exo-adduct of 1b with 2b is 2 kJ mol−1 more stable than the endo stereoisomer; the latter is expected to form faster, see 14.
- F. A. Kucherov, K. I. Galkin, E. G. Gordeev and V. P. Ananikov, Green Chem., 2017, 19, 4858 RSC.
- R. Lu, F. Lu, X. Si, H. Jiang, Q. Huang, W. Yu, X. Kong and J. Xu, ChemSusChem, 2018, 11, 1621 CrossRef CAS.
- (a) A. F. Crespi, A. J. Byrne, D. Vega, A. K. Chattah, G. A. Monti and J. M. Lázaro-Martínez, J. Phys. Chem. A, 2018, 122, 601 CrossRef CAS; (b) A. F. Crespi, D. Vega, A. K. Chattah, G. A. Monti, G. Y. Buldain and J. M. Lázaro-Martínez, J. Phys. Chem. A, 2016, 120, 7778 CrossRef CAS.
- E. Dalcanale and F. Montanari, J. Org. Chem., 1986, 51, 567 CrossRef CAS.
- (a) S. Thiyagarajan, H. C. Genuino, M. Śliwa, J. C. van der Waal, E. de Jong, J. van Haveren, B. M. Weckhuysen, P. C. A. Bruijnincx and D. S. van Es, ChemSusChem, 2015, 8, 3052 CrossRef CAS; (b) S. Thiyagarajan, H. C. Genuino, J. C. van der Waal, E. de Jong, B. M. Weckhuysen, J. van Haveren, P. C. A. Bruijnincx and D. S. van Es, Angew. Chem., Int. Ed., 2015, 55, 1368 CrossRef; (c) H. C. Genuino, S. Thiyagarajan, J. C. van der Waal, E. de Jong, J. van Haveren, D. S. van Es, B. M. Weckhuysen and P. C. A. Bruijnincx, ChemSusChem, 2016, 10, 277 CrossRef.
- For reviews, see ref. 1k and. (a) L. Seabrooks and L. Hu, Acta Pharm. Sin. B, 2017, 7, 409 CrossRef; (b) G. Wang, J. Dong and L. Deng, Curr. Med. Chem., 2018, 25, 2034 CrossRef CAS For a recent study on norcantharidin derivative synthesis via furan DA addition, see. K. I. Galkin, F. A. Kucherov, O. N. Markov, K. S. Egorova, A. V. Posvyatenko and V. P. Ananikov, Molecules, 2017, 22, 2210 CrossRef.
- C. S. Schindler and E. M. Carreira, Chem. Soc. Rev., 2009, 38, 3222 RSC.
- S. H. Hilal, L. L. Bornander and L. A. Carreira, QSAR Comb. Sci., 2005, 24, 631 CrossRef CAS.
- The endergonic nature of Diels–Alder cycloadditions involving furfural has also been suggested in ref. 2d and 5b, based on computational data; attempts to experimentally evidence the formation of the targeted cycloadducts failed in these studies.
- Note: This property is additionally evidenced by the fact concentrating an aqueous solution of 8 yields a sticky solid consisting of acetal oligomers rather than a well-defined aldehyde.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2023757. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0gc03558k |
| This journal is © The Royal Society of Chemistry 2021 |