
Surface-functionalized mesoporous gallosilicate catalysts for the efficient and sustainable upgrading of glycerol to solketal†
Alvise
Vivian‡
 a,
Loraine
Soumoy‡
a,
Luca
Fusaro
a,
Sonia
Fiorilli
b,
Damien P.
Debecker
*c and
Carmela
Aprile
*a
a,
Loraine
Soumoy‡
a,
Luca
Fusaro
a,
Sonia
Fiorilli
b,
Damien P.
Debecker
*c and
Carmela
Aprile
*a
aUniversity of Namur, Department of Chemistry, Unit of Nanomaterials Chemistry, 5000 Namur, Belgium. E-mail: carmela.aprile@unamur.be
bDepartment of Applied Science and Technology, Politecnico di Torino, Institute of Chemistry, 10129 Torino, Italy
cUniversité catholique de Louvain (UCLouvain), Institute of Condensed Matter and Nanosciences (IMCN), 1348 Louvain-la-Neuve, Belgium. E-mail: damien.debecker@uclouvain.be
First published on 9th October 2020
Abstract
Two series of functionalized mesoporous Ga silicates were prepared in a straightforward and sustainable one-pot procedure using different alkyl silanes. The efficacy of the adopted co-synthetic approach based on aerosol processing has been proved by 29Si solid-state NMR experiments revealing a degree of functionalization close to the theoretical value. The successful incorporation of gallium as single sites within the silica framework was confirmed via71Ga solid-state magic-angle-spinning NMR measurements. These materials were tested as catalysts for the synthesis of solketal from glycerol at low temperature and under solventless conditions. A systematic study evidenced the importance of a careful tuning of surface polarity, achievable with surface functionalization as well as with different thermal treatments. The solids functionalized with a low degree of methyl groups (5%) displayed enhanced performances compared to the non-functionalized analogues, highlighting the highly beneficial role of surface hydrophobicity as well as the importance of the careful tuning of the hydrophilic/hydrophobic balance. The best functionalized catalysts proved to be easily reusable for multiple catalytic runs. With such a high-performance catalyst in hand, we propose a process which shows a favorable E-factor, indicating that the production of solketal can be envisaged in a sustainable way.
Introduction
In 2015, the United Nations Member States adopted their “2030 Agenda for Sustainable Development”, accelerating the transition toward, inter alia, more sustainable mobility and industry.1,2 In this context, the conversion of biomass into biofuels plays a pivotal role. Increasing the amount of biofuel in traditional fuels is now needed in order to fulfill the requirements of the European Commission: a minimum of 14% of renewable energy will be required in road and rail transport by 2030.3 On the biofuel market, biodiesel is gaining a growing importance due to its good compatibility with commercial diesel engines.4 Nevertheless, its production via transesterification of triglycerides coming from natural lipid sources yields 10 wt% of glycerol as a by-product.5 The more intense production of biodiesel over the last few years caused an oversupply of glycerol on the market.6 Its price consequently suffered a drastic decrease and nowadays it is even considered as a waste product. However, in the context of green chemistry, glycerol represents a very interesting building block as it directly comes from renewable feedstocks and can be produced via low energy consumption compared to petro-chemical based reagents.7Glycerol offers tremendous potential to produce value-added chemicals and the synthesis of solketal, through an acetalization reaction with acetone, is a particularly relevant pathway.8 On the one hand, solketal has various applications as a solvent in large-scale production (paint, ink formulations),9 in the pharmaceutical industry (as an additive for injectables, a vehicle for drugs)9 and even as a fuel additive.10,11 The latter provides opportunities for biorefineries to develop their circular economy. On the other hand, the valorization pathway fulfills some of the 12 Principles of Green Chemistry established by Anastas and Warner:7 glycerol and even acetone can be produced from renewable feedstocks (from triglycerides and hemicellulose,13 respectively), the reaction can be classified as a highly atom economical process (88%) and with a potentially low E-factor.12,13
Acetalization of glycerol is usually performed in the presence of homogeneous Brønsted or Lewis acid catalysts such as H2SO4, HCl, SnCl4 or Sn(OAc)2.10,14 Despite the high intrinsic activity of these homogeneous catalysts, scientists are working hard to develop heterogeneous catalysts, which can be recovered, reused or even implemented in continuous flow processes, thereby improving the sustainability of the process.15 The use of zeolites, sulfonic acid functionalized silicates, K 10 montmorillonites, zirconia-based catalysts or gallo-silicates for the acetalization of glycerol has already shown promising results.16–21 More recently, we proposed a new class of gallo-silicate catalysts synthesized via the aerosol-assisted sol–gel process that show outstanding performances in the conversion of glycerol into solketal.22
In recent years, aerosol processes have emerged as efficient methods to synthesize heterogeneous catalysts displaying controlled properties for their application in a wide range of reactions.23 For example, transition metal-doped silica and titania for oxidation of volatile organic compounds,24 metal oxides for electrocatalysis,25 titanosilicate catalysts for epoxidation,26 or even supported metal nanoparticles for hydrogenation reactions27 have already been prepared by aerosol processes and shown excellent catalytic performances. In particular, the “aerosol-assisted sol–gel process” (AASG) emerged as an efficient tool to synthesize mesoporous silica-based solids featuring metal atoms inserted as single sites in their framework.28 In this synthesis procedure, a sol containing a templating agent (e.g. a surfactant), a solvent, silica and a metal precursor is nebulized into droplets that are gas-transported towards a drying chamber or tube, leading to the evaporation of the solvent. This fast evaporation triggers the self-assembly of the template used as a sacrificial agent to generate pores and the rapid polycondensation of the inorganic precursors, yielding homogeneous materials with a spherical shape. Compared to the traditional sol–gel route, the aerosol process presents several advantages including reduction of waste production, a limited number of steps and low environmental impact. Moreover, the possibility to produce the solid in a continuous way makes it industrially attractive.23,29–31
Regarding the conversion of glycerol into solketal, the development of highly performing heterogeneous catalysts has not yet been achieved. In order to design more efficient heterogeneous catalysts, it is well known that multiple characteristics can be optimized such as the nature and number of active sites, mass transport of reactants and products to and from the active sites, surface area, pore size distribution, etc. However, an important and less studied parameter is the role played by the hydrophilic and hydrophobic properties of the catalyst surface. The latter influences strongly the adsorption of reagents and the desorption of products or by-products to/from the catalytic surface, thus determining the performances of the solid. Considering the mechanism of the acetalization of glycerol, the key role played by the hydrophilic and hydrophobic properties of the catalyst surface can be easily understood. On the one hand, the hydrophilicity of the catalytic surface will favor glycerol adsorption in proximity to the active sites. On the other hand, more hydrophobic surfaces would help the removal of water produced as a by-product of the reaction, locally shifting the reaction towards the formation of solketal. In fact, the elimination of water using a Dean–Stark apparatus has already proved to have a positive effect on the solketal yield.10 As the surface of silicates is decorated by silanol groups, its hydrophilic–hydrophobic balance can be tuned by alkylation procedures using a co-synthetic or a post-synthetic approach.28,32 In 2006, Ji et al. reported the synthesis of functionalized mesoporous silicates via an aerosol process but their use in catalysis was not reported.33 Recently, we reported on aerosol-made methyl-functionalized stanno-silicate catalysts and their application in the production of ethyl lactate.28 To the best of our knowledge, the role played by the hydrophobic/hydrophilic properties of the catalytic surface in the acetalization of glycerol to solketal was never systematically investigated.
Here, we take advantage of the aerosol-assisted sol–gel process to develop a co-synthesis approach and obtain, in a one-pot and green procedure, a novel class of alkyl-functionalized gallosilicates. The specific functionalization of the catalyst surface is presented as a way to further improve catalytic performances while taking into account the sustainability of the process.
Experimental section
Materials
Pluronic® P-123 (Merck), Pluronic® F127 (Merck), hydrochloric acid 2 M (Roth), tetraethyl orthosilicate (TEOS 97%, TCI), methyltriethoxysilane (MTES 98%, TCI), propyltriethoxysilane (PrTES 98%, TCI), phenyltriethoxysilane (PhTES 99%, TCI) and (Ga(NO3)3·xH2O, ROTH). Absolute ethanol (analytical grade) was supplied from Fisher Scientific.Catalyst preparation
To synthesize functionalized Ga-silicates (Ga-P-x and Ga-F-x), a mixture of two silica sources, tetraethyl orthosilicate (TEOS) and methyltriethoxysilane (MTES) for methyl functionalized solids, propyltriethoxysilane (PrTES) for propyl functionalized solids or phenyltriethoxysilane (PhTES) for phenyl functionalized solids was prepared. In the synthesis of methyl functionalized solids, the quantity of MTES was chosen to reach three degrees of functionalization: 5, 10 and 15 mol%, corresponding to the molar MTES/(MTES + TEOS) × 100% ratio. For propyl and phenyl functionalized solids, the amount of PrTES or PhTES was adjusted to achieve a degree of functionalization of 5 mol%. Pluronic F127 and P123 were selected as structure directing agents. The detailed amounts of TEOS and alkylsilane used for each material are reported in Table S1.†TEOS and MTES or PrTES or PhTES were hydrolyzed in an aqueous solution of HCl 0.02 M (20 g) to yield solution 1. Solution 2 was prepared by dissolving Pluronic® P123 or F127 (3.9 g) in absolute ethanol (45 g). Solutions 1 and 2 were respectively stirred for 15 hours and 40 minutes at room temperature. Then, the gallium precursor (Ga(NO3)3·xH2O) was added to solution 1 in order to have a Si/Ga molar ratio equal to 74. Solutions 1 and 2 were mixed together and then stirred for 30 minutes. The resulting solution was atomized with a 6-Jet 9306A atomizer from TSI. Then, the aerosol was passed through a tubular quartz tube heated at 350 °C. A cellulose nitrate filter was used to collect the powders, further dried for one night at 80 °C and calcined under static air at 250 °C for 8 h (heating rate of 1 °C min−1).
Characterization
Transmission electron microscopy (TEM) images were obtained using a Philips Tecnai 10 microscope operating at 80 kV. Nitrogen adsorption–desorption analyses were carried out at 77 K with a volumetric adsorption analyzer (Micromeritics ASAP 2420). Prior to the analysis, the samples were pre-treated for 8 hours at 150 °C under reduced pressure (0.1 mbar). The Brunauer–Emmett–Teller (BET) method was applied in the 0.05–0.30P/P0 range to calculate the specific surface area, while the pore size distributions were calculated from the adsorption isotherm using the Barrett–Joyner–Halenda (BJH) method. The micropore volume and micropore surface area were calculated by the t-plot method that was applied to the adsorption branch of the isotherms in the thickness range of 3.5–5.0 Å. The micropore diameter was evaluated by the DFT method using cylinder geometry, N2 – cylindrical pores – oxide surface model. Powder X-ray diffraction (XRD) patterns were recorded on a PANalytical X′pert diffractometer with Cu Kα radiation (k = 1.54178 Å). Inductively coupled plasma optical emission spectroscopy was employed to determine the chemical composition of the materials using an Optima 8000 ICP-OES spectrometer. The silicon environment and the coordination of the gallium atoms were studied by 29Si Magic Angle Spinning (MAS) and 71Ga MAS Nuclear Magnetic Resonance (NMR), respectively.29Si NMR spectra were recorded at room temperature on a Bruker Avance-500 spectrometer operating at 11.7 T (99.3 MHz for 29Si) using a 4 mm CP-MAS Bruker probe. The sample was packed in a 4 mm zirconia rotor and measured with a spinning frequency of 8000 Hz. Quantitative direct excitation (DE-MAS) 29Si spectra were recorded using the following acquisition parameters: 300 s relaxation delay, 3 μs (90°) excitation pulse, 52 ms acquisition time. Cross polarization (CP-MAS) spectra were recorded using a 5 s relaxation delay and 5 ms contact time. The processing comprised exponential multiplication of the FID with a line broadening factor of 30 Hz, zero-filling, Fourier transform, phase and baseline corrections. The chemical shift scale was calibrated at room temperature with respect to a sample of solid 3-(trimethylsilyl)-1-propanesulfonic acid sodium salt (DSS) (0.0 ppm).
71Ga NMR spectra were recorded at room temperature on a Bruker Avance-500 spectrometer operating at 11.7 T using a 2.5 mm CP-MAS Bruker probe. The sample was packed in a 2.5 mm zirconia rotor and measured with a spinning frequency of 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Hz. The spectra were recorded using a Hahn echo sequence and the following acquisition parameters: a relaxation time of 0.3 s, an excitation of 2.6 μs (90°) and 16 ms acquisition time. Data processing includes the multiplication of the FID (Free Induction Decay) by a line broadening factor of 1000 Hz, zero-filling, Fourier transformation and phase and baseline corrections. Fourier transformation from the echo maximum ensured a flat baseline with only zero-order phase correction. The chemical shift scale was calibrated at room temperature with respect to a sample of solid gallium nitrate (0.0 ppm).
000 Hz. The spectra were recorded using a Hahn echo sequence and the following acquisition parameters: a relaxation time of 0.3 s, an excitation of 2.6 μs (90°) and 16 ms acquisition time. Data processing includes the multiplication of the FID (Free Induction Decay) by a line broadening factor of 1000 Hz, zero-filling, Fourier transformation and phase and baseline corrections. Fourier transformation from the echo maximum ensured a flat baseline with only zero-order phase correction. The chemical shift scale was calibrated at room temperature with respect to a sample of solid gallium nitrate (0.0 ppm).
Vapor-phase water adsorption isotherms were acquired at 295 K, using a surface characterization analyzer (Micromeritics 3Flex). Prior to the analysis, the samples were pre-treated for 8 hours at 180 °C under reduced pressure (0.07 mbar). Water was purified using freeze–pump–thaw cycles.
Thermogravimetric analysis (TGA) was performed on a Mettler Toledo TGA/DSC 3+ STARe System. The samples were analysed using a temperature ramp from 30 °C to 800 °C at 10 °C min−1 under an air flow (60 mL min−1).
FT-IR measurements were performed in transmission mode by using a Bruker Tensor 27 spectrometer equipped with a liquid nitrogen-cooled mercury–cadmium–telluride (MCT) detector, operating at a 2 cm−1 resolution. A pre-treatment was carried out using a standard vacuum frame, in an IR cell equipped with KBr windows. The sample was pressed into self-standing wafers and outgassed for 1 h at 400 °C before adsorption of ammonia at room temperature.
Adsorption of ammonia was studied in the pressure range of 0.01–20.0 mbar: the reversible fraction of the adsorbed ammonia was then removed by prolonged outgassing at room temperature.
Production of solketal from glycerol and acetone
Results and discussion
Two series of functionalized Ga-silicates (Si/Ga atomic ratio of 74) were prepared via a co-synthetic approach exploiting the aerosol-assisted sol–gel process and using three different alkyl-silanes: methyl, propyl and phenyltriethoxysilane. In addition, two structure-directing agents from the Pluronic family (F127 and P123) were selected in order to study their impact on the morphological properties and on the catalytic activity of the materials.Initially, Ga-based solids were synthesized by selecting three degrees of methyl-functionalization: 5, 10 and 15 mol% (see the Experimental section for more information). These materials were denoted as Ga-P-x-Me and Ga-F-x-Me, where P or F indicates the employed templating agent (P123 or F127 respectively) and x indicates the nominal degree of functionalization. In order to investigate the effect of different alkyl moieties on the textural and surface properties of the solids, four additional functionalized materials were synthesized using only 5 mol% of propyl or phenyltriethoxysilane. The materials were denoted as Ga-P(or F)-5-Pr and Ga-P(or F)-5-Ph (where 5 indicates the degree of functionalization, Pr or Ph indicates the presence of propyl or phenyl moieties respectively). All the above-mentioned functionalized Ga-silicates were then compared with their non-functionalized analogues synthesized under the same conditions, respectively, named Ga-P-0 and Ga-F-0.
After the synthesis, particular attention should be paid to retain the alkyl-functionalization while the surfactant is being removed from the pores. In the literature, several methods including solvent extraction34 or calcination35 have been reported to remove Pluronics from silica-based porous solids. However, taking inspiration from one of our previous work, a mild calcination treatment was here selected.28 We found that the best compromise between the total removal of the templating agent and the almost full preservation of the functionalization can be achieved with calcination at 250 °C for 8 hours.
Thus, all the solids were calcined at the selected temperature and analyzed by performing quantitative 29Si solid-state magic angle spinning (MAS) NMR experiments. Fig. 1 shows an example of quantitative 29Si direct excitation (DE) MAS spectra of the Ga-P-x-Me series. The spectra of the other materials can be found in the ESI (Fig. S1–S4†). The characteristic contributions typical of an amorphous silica matrix can be observed clearly: Q4 [(SiO)4Si] (around −111 ppm), Q3 [(SiO)3SiOH] (around −102 ppm) and Q2 [(SiO)2Si(OH)2] (around −92 ppm) signals can be found in all samples. Two additional signals at −66 and −59 ppm can be seen in the 29Si NMR spectra of methyl-functionalized solids and can be assigned to MeSi(OSi)3 (T3) and Me(HO)Si(OSi)2 (T2) species.28 These contributions can be found as well in the whole series of functionalized solids (see Fig. S1–S4†) and they attest of the successful functionalization of the surface. 29Si DE-MAS NMR spectra highlight also that the theoretical percentage of organic moieties is largely preserved for all materials (see Table S2†). These results corroborate the efficiency of the synthetic procedure including the mild calcination treatment.
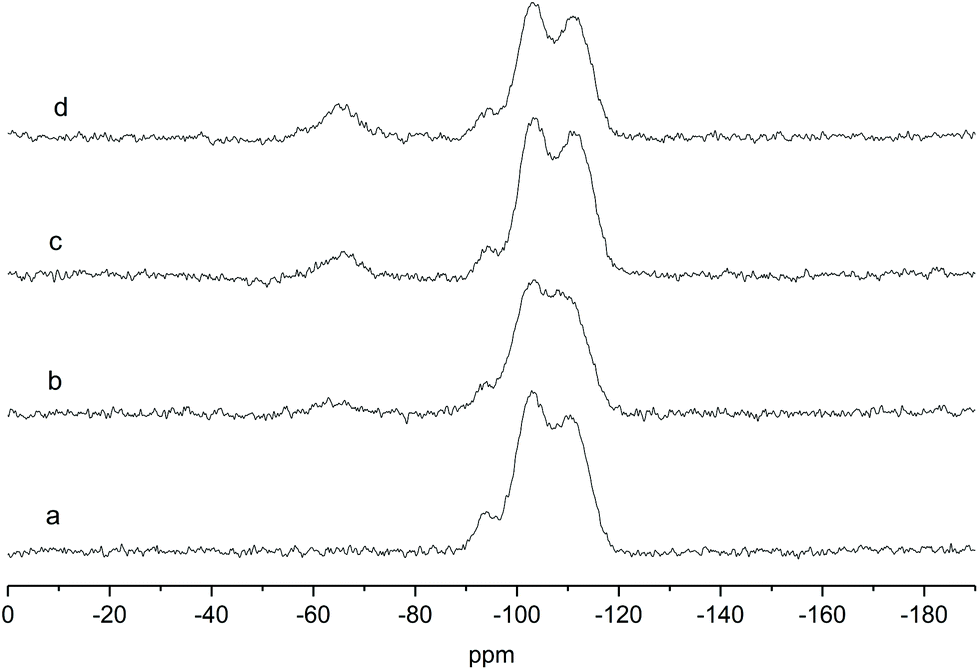 | ||
| Fig. 1 Solid-state direct excitation 29Si MAS NMR spectra of (a) Ga-P-0 (b) Ga-P-5-Me (c) Ga-P-10-Me and (d) Ga-P-15-Me after calcination at 250 °C for 8h. | ||
The gallium loading was evaluated by inductively coupled plasma optical emission spectroscopy (ICP-OES). The experimental composition – expressed in mmol of gallium per gram of catalyst – was very close to the nominal composition of 2.2 mmol g−1 (Table S2†) for all the solids. Nevertheless, the formation of catalytically active acid sites, fundamental for the application of these solids as catalysts, is strictly related to the insertion of Ga as single sites. The isomorphous substitution of silicon atoms by trivalent elements, such as gallium, is known to induce a combination of Brønsted and Lewis acid sites within the silica framework.21 The coordination environment of gallium was therefore elucidated by solid-state 71Ga NMR spectroscopy. In addition to the intrinsic difficulties of 71Ga and due to the low gallium loading replacing silicon in an amorphous environment, investigations were performed under magic angle spinning (MAS) at 25 kHz and at a high magnetic field (11.7 T).22
Fig. 2 shows the MAS NMR spectra of Ga-P-0, Ga-P-5-Me, Ga-P-10-Me and Ga-P-15-Me. The sharp peak centered at +140 ppm, observed for all the materials, is ascribed to the tetrahedral framework of gallium and it constitutes proof of the incorporation of mainly single-site Ga in the silica framework.36,37 This contribution has already been observed for other gallo-silicates reported in the literature. The presence of an additional weak contribution at 0 ppm can be assigned to the presence of minor amounts of extra-framework Ga species (see Fig. S5† for a comparison with a pure Ga2O3 spectrum). These species are supposed to have an octahedral environment and have already been reported not to be catalytically active.21 Similar results were obtained on analyzing the environment of Ga in the case of Ga-F-0 (Fig. S5†). 71Ga NMR measurements indicate that neither the presence of methyl-functional groups nor their amount affects the incorporation of gallium in the silica framework of the materials. It appears also that the chemical environment of gallium is not influenced by the nature of the templating agent.
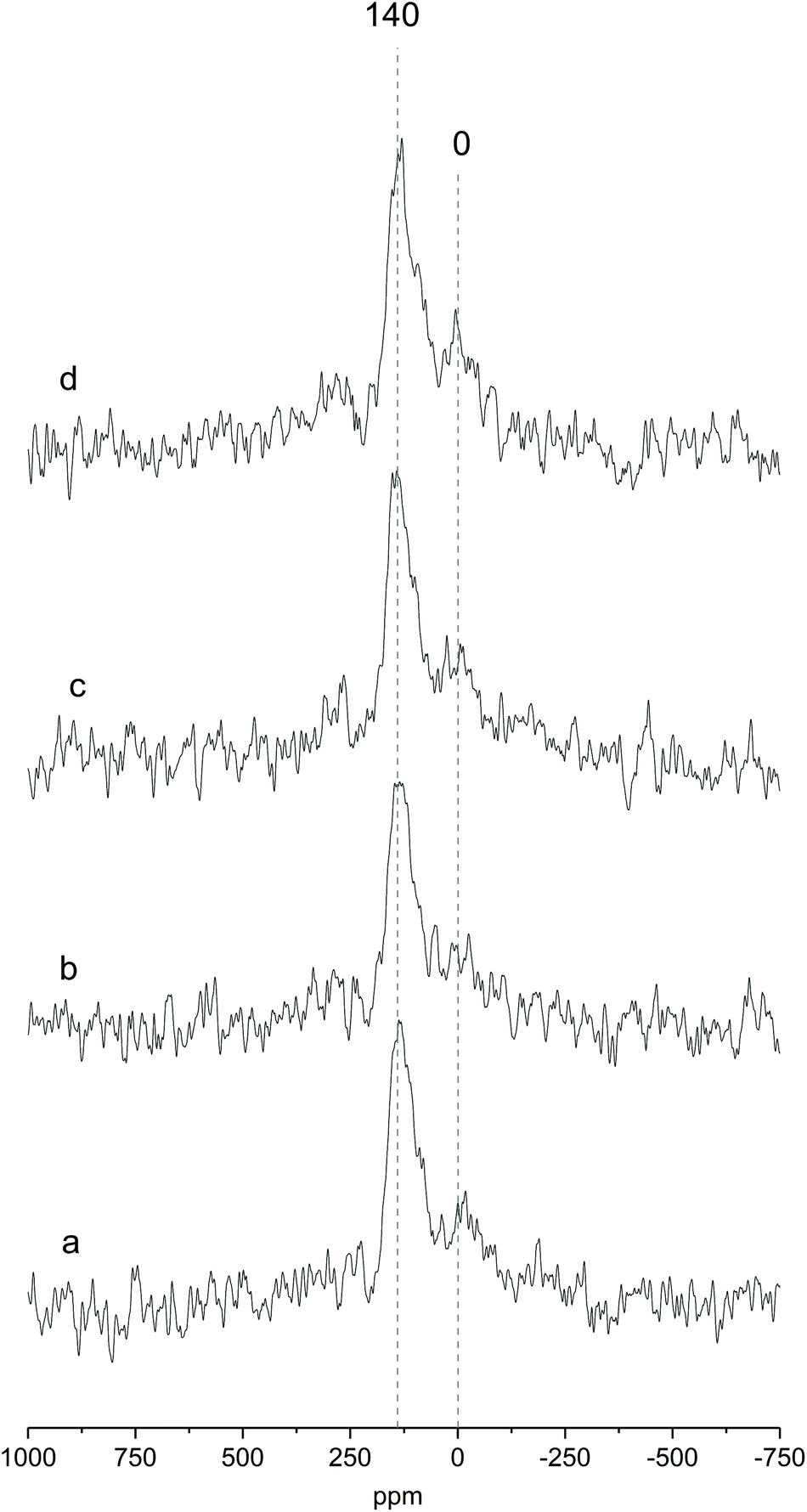 | ||
| Fig. 2 Solid-state 71Ga MAS NMR spectra of (a) Ga-P-0, (b) Ga-P-5-Me, (c) Ga-P-10-Me and (d) Ga-P-15-Me after calcination at 250 °C for 8 h. The spectra were recorded using a Hahn echo sequence (see the Experimental section). | ||
The sample morphology was investigated by transmission electron microscopy (TEM). All solids are mesoporous and display a spherical shape. The average pore diameter lies between 6 and 7 nm, typical of aerosol solids templated with Pluronic P123 or F127.28,38 When F127 was used as a surfactant, a well-organized structure was obtained even when the methyl-functionalization was increased to 15% (Fig. 3e–h). Interestingly, a slight deformation of the structure in the external part of the particles was visible for the materials synthesized in the presence of Pluronic P123 (Fig. 3a–d). This deformation was more evident with the increasing amount of methyl-functionalization. The major modification of the structure with the appearance of some “onion like” organizations is clearly visible on increasing the functionalization to 15%. This behavior was previously observed and has been ascribed to the different balance among the hydrophilic part (poly(ethylene oxide)) and the hydrophobic part (poly(propylene oxide)) of the two triblock polymers.28 As indicated by their formula, the templating agent Pluronic P123 ((PEO)20-(PPO)70-(PEO)20) has a lower number of hydrophilic units than Pluronic F127 ((PEO)100-(PPO)65-(PEO)100).
 | ||
| Fig. 3 TEM micrographs of (a) Ga-P-0, (b) Ga-P-5-Me, (c) Ga-P-10-Me, and (d) Ga-P-15-Me and of (e) Ga-F-0, (f) Ga-F-5-Me, (g) Ga-F-10-Me, and (h) Ga-F-15-Me. | ||
As a consequence, the methyl groups of MTES could have more affinity with the hydrophobic unit of P123 than with F127. Since the methyl group of MTES is hydrophobic enough to present cosurfactant properties, it will be preferentially located nearby the hydrophobic shell of the micelle, causing a structural deformation in the final material. This behaviour may induce a variation of the interfacial curvature favouring the transition through a lamellar phase.
In the case of Ga-P-5-Pr and Ga-P-5-Ph, well-organized spherical particles were observed (Fig. S6†). Thus, longer organic chains such as in the case of propyl moieties or bulkier phenyl groups did not cause substantial deformation of the structure.
The low angle powder XRD patterns of Ga-silicates templated with P123 or F127 (Fig. S7†) featured mainly first order d100 diffraction peaks, indicating ordered materials. The d-spacing associated with the P123 and F127 series is respectively 9 nm and 11 nm, that is typical of ordered mesostructured silicates assembled using a non-ionic surfactant. For both the series, N2-physisorption measurements showed type-IV isotherms characterized by considerable hysteresis loops. P123 templated solids present H1 hysteresis loops with steeper branches, typical of materials with a narrow distribution of relatively uniform mesopores (Fig. 4 and Fig. S8†). F127 templated materials exhibit a H2 hysteresis loop characteristic of cage-type mesopores with a mean pore diameter of 7 nm (Fig. 4 and Fig. S8†). All solids display a high specific surface area and narrow pore size distribution. An overview of the textural properties of the two series of functionalized Ga-silicates is reported in Table 1.
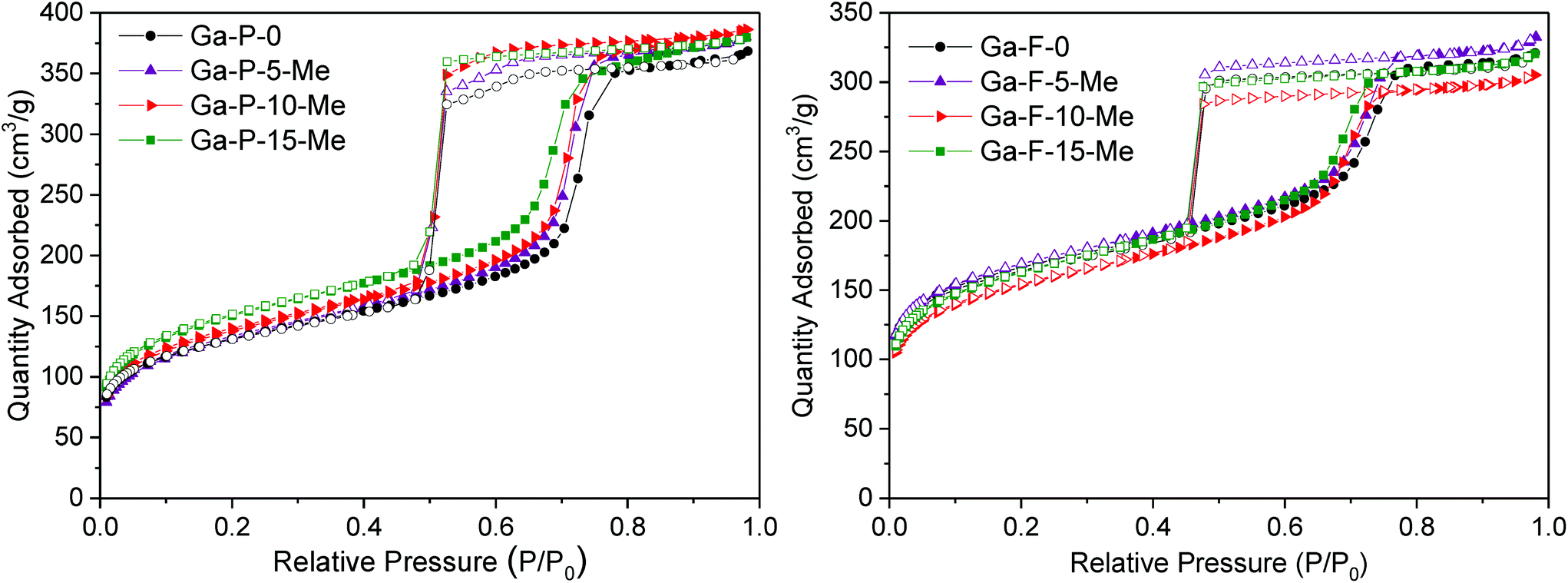 | ||
| Fig. 4 Nitrogen adsorption/desorption isotherms of Ga-P-0, Ga-P-5-Me, Ga-P-10-Me and Ga-P-15-Me (left) and Ga-F-0, Ga-F-5-Me, Ga-F-10-Me and Ga-F-15-Me (right) after calcination at 250 °C for 8 h. | ||
| Material | BET SSA (m2 g−1) | BJH pore width (nm) | Pore volume (cm3 g−1) | Gaa (mmol g−1) | % funct.b |
|---|---|---|---|---|---|
a mmol of gallium per gram of catalyst determined by ICP-OES analysis.
b
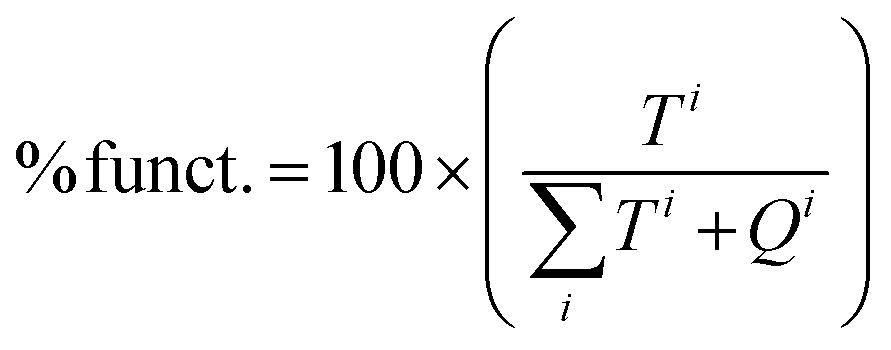 . .
|
|||||
| Ga-P-0 | 440 | 7.8 | 0.6 | 2.0 | — |
| Ga-P-5-Me | 410 | 7.1 | 0.6 | 1.9 | 4 |
| Ga-P-10-Me | 430 | 7.3 | 0.5 | 1.8 | 8 |
| Ga-P-15-Me | 480 | 6.6 | 0.5 | 1.8 | 13 |
| Ga-P-5-Pr | 500 | 7.0 | 0.6 | 1.8 | <3 |
| Ga-P-5-Ph | 460 | 6.6 | 0.5 | 2.0 | <3 |
| Ga-F-0 | 550 | 7.8 | 0.5 | 2.1 | — |
| Ga-F-5-Me | 500 | 7.5 | 0.4 | 1.8 | 3 |
| Ga-F-10-Me | 460 | 6.9 | 0.4 | 1.9 | 8 |
| Ga-F-15-Me | 500 | 6.8 | 0.4 | 1.9 | 14 |
| Ga-F-5-Pr | 550 | 7.1 | 0.5 | 2.0 | <3 |
| Ga-F-5-Ph | 450 | 6.3 | 0.4 | 2.0 | <3 |
The reaction selected in this work is represented by the acetalization of glycerol with acetone to produce solketal. Based on literature data, a proposed mechanism for the target reaction catalyzed by a Brønsted or Lewis acid catalyst is shown in Fig. S9.†19,39 The effective insertion of single-site tetrahedral gallium, proved by solid-state 71Ga NMR should give rise to the desired acidic properties essential for promoting the upgrading of glycerol into solketal.
In Table 2 the catalytic performances in the conversion of glycerol into solketal for the whole set of solids are presented. Considering sustainability as a key aspect of the process, the tests were carried out under solvent free conditions at 50 °C. A kinetic study using Ga-P-0 or Ga-F-0 as the catalyst was performed (Fig. S10†). The formation of solketal was followed from 1 hour to 16 hours in order to select the adequate reaction time to compare the activity of our catalysts in the kinetic regime (initial activity). It appears that a short reaction time of two hours represents the best compromise between an adequate solketal yield allowing an accurate quantification and a meaningful comparison among the catalysts far enough from the equilibrium conversion.
:4
|
|
|||||
|---|---|---|---|---|---|
| Entry | Material | Y SK (%) | Sel.SKa (%) | TOFb | TONc |
| a Sel.SK = mol solketal/(mol solketal + mol 2,2-dimethyl-1,3-dioxan-5-ol). The presence of the by-product (2,2-dimethyl-1,3-dioxan-5-ol) was identified by 1H liquid state NMR (Fig. S13†). b TOF (TurnOver Frequency) here defined as mol of solketal per mol of Ga × time. c TON (TurnOver Number) here defined as mol of solketal per mol of Ga after 2 hours of reaction. | |||||
| 1 | Ga-P-0 | 21 | 84 | 532 | 1014 |
| 2 | Ga-P-5-Me | 25 | 85 | 663 | 1326 |
| 3 | Ga-P-10-Me | 18 | 82 | 506 | 1012 |
| 4 | Ga-P-15-Me | 18 | 82 | 500 | 1000 |
| 5 | Ga-P-5-Pr | 17 | 83 | 462 | 923 |
| 6 | Ga-P-5-Ph | 13 | 79 | 328 | 656 |
| 7 | Ga-F-0 | 10 | 81 | 242 | 484 |
| 8 | Ga-F-5-Me | 16 | 83 | 434 | 867 |
| 9 | Ga-F-10-Me | 12 | 82 | 311 | 622 |
| 10 | Ga-F-15-Me | 9 | 79 | 240 | 479 |
| 11 | Ga-F-5-Pr | 10 | 80 | 243 | 485 |
| 12 | Ga-F-5-Ph | 9 | 77 | 224 | 447 |
To verify the reproducibility of the catalytic results, the materials were tested three times under the same reaction conditions. The associated error calculated was around 2% in absolute value, proving the high reproducibility of the tests.
The comparison between functionalized and non-functionalized catalysts, synthesized under the same conditions (compare entries 1 and 7 with 2 and 8 in Table 2), revealed that the 5% methylated solids Ga-P-5-Me and Ga-F-5-Me present improved catalytic activity in terms of TON (defined as moles of solketal produced per mole of Ga), even when the activity is normalized to the BET surface area (Fig. S11†). From this comparison emerged a positive influence of the methylation on the performances of the catalysts. It is important to underline that only a low degree of methylation allows the enhancement of the catalytic activity of the solids. This phenomenon was already reported by our group, in the case of functionalized Sn-silicates used in the production of ethyl lactate.28 The observed enhancement in terms of TON could be attributed to the surface functionalization rather than to the presence of active sites (gallium single sites) in a different coordination environment. Moreover, the presence of bulkier functional groups in the formulation (see entries 5, 6 and 11, 12) leads to a lowering of the activity. To deeply investigate this point, water vapor sorption experiments were performed at 295 K on Ga-P-0, Ga-P-5-Me, Ga-P-5-Pr and Ga-P-5-Ph (Fig. S12,† left). In order to meaningfully compare our solids, the amount of water adsorbed was normalized by the BET specific surface area. The quantitative evaluation of the water affinity of the catalyst surface was featured at low relative pressure of the isotherm (in our case, 0.15), as indicated in the literature.40 As illustrated in Fig. S12 and Table S3† (compare entries 1 and 2), a reduced affinity for water at low relative pressure is observed for Ga-P-5-Me compared to Ga-P-0. These data suggest that the addition of methyl groups allows tuning the hydrophilic–hydrophobic balance to more hydrophobic surfaces. The less hydrophilic surface of Ga-P-5-Me may have a role in glycerol acetalization by locally helping the removal of water from the active sites and therefore favoring the formation of solketal. The quicker removal of water from the hydrophobic surface may help preserving the integrity of the acid sites of the catalyst. In comparison with Ga-P-5-Me, the water uptake at low relative pressure is lower for Ga-P-5-Pr and Ga-P-5-Ph (compare entries 2 with 3 and 4 in Table S3†). This result highlights that the presence of bulkier groups in the catalysts induces more hydrophobic surfaces. Water sorption measurements evidence that the presence of too bulky hydrophobic moieties has a detrimental effect on the catalytic activity, hindering probably the adsorption of glycerol in proximity to the active sites.
No further improvement in the catalyst performance was observed when the methylation degree is brought to 10% (see entries 3 and 9) or to 15% (see entries 4 and 10). To go further, a series of Ga-P-x methylated materials were analyzed by thermogravimetric analysis (TGA). In order to evaluate the hydrophobic/hydrophilic nature of our solids, the weight loss was quantified at 140 °C and normalized by the BET specific surface area. As illustrated in Fig. S12† (right), when the percentage of methyl moieties increases, the desorption of physisorbed water decreases. These results indicate that an enhancement of the percentage of methylation allows achieving a more hydrophobic surface. All these results confirm that the fine tuning of the hydrophilic–hydrophobic balance of the catalytic surface plays a crucial role in the design of more active catalysts.
Surprisingly the solids synthesized employing P123 as the surfactant display predominantly better catalytic performances than the corresponding materials of the F127 series (compare entries 1 to 6 with 7 to 12). Considering the 71Ga MAS NMR analysis, the insertion of Ga as a single site is very similar on Ga-P-0 and Ga-F-0 (Fig. S5†). Their textural properties deserve therefore in-depth characterization to better understand the observed inequalities in terms of catalytic activity.
It is well known that the presence of micropores in heterogeneous catalysts can limit the diffusion of reagents to the active sites and/or the release of the products from the active sites. The presence of micropores in the walls of Ga-P-0 and Ga-F-0 was here evaluated by the t-plot method. In the case of Pluronic templated solids, the formation of micropores occurs by aggregation of the hydrophilic blocks of two micelles within the pre-formed silica walls. This phenomenon leads to microporosity in the final material once the templated agents are eliminated by calcination.41 The analysis of the t-plot allows accessing the microporous area and micropore volume of our solids (Fig. S14†). The microporous surface area of Ga-F-0 and Ga-P-0 was 250 m2 g−1 and 170 m2 g−1, respectively. Ga-F-0 and Ga-P-0 show micropore volumes of 0.08 cm3 g−1 and 0.04 cm3 g−1, respectively. The analysis via the mathematical modeling using density functional theory (DFT) allows evaluating the diameter of the micropores that is located in the 1 to 2 nm range. Hence, the decreased activity of the Ga-F-0 could be at least partially ascribed to a limited accessibility of the active sites located in the micropores.
However, to shed more light on the different catalytic behavior, FT-IR measurements of ammonia adsorption on Ga-P-0 and Ga-F-0 solids were conducted as well. This analysis allows investigating the acidic properties and provides support for an interpretation of the observed catalytic performances. For this study, ammonia (a basic molecular probe), which can interact with both Brønsted (surface hydroxyls) and Lewis (unsaturated Ga(III)) sites, was selected.
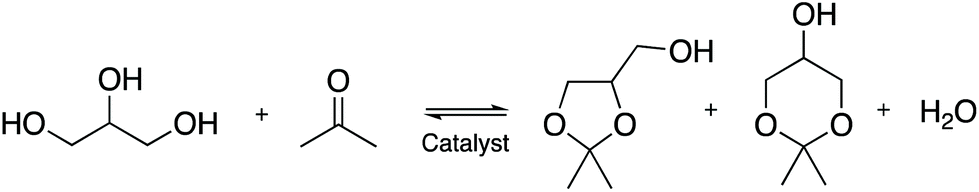
FT-IR difference spectra of ammonia adsorption on both Ga-P-0 and Ga-F-0 are shown in Fig. 5(a and b), where the negative bands indicate species that disappear upon ammonia dosage. In the high frequency region (3800–2000 cm−1), the adsorption of ammonia generates the formation of several negative bands due to the various hydroxyl groups interacting with NH3. In particular, both samples show a negative band at about 3745 cm−1 due to free silanols and two negative bands at about 3720 cm−1 together with a broad shoulder at 3550 cm−1 related to terminal and H-bonded silanols in hydroxyls chains, respectively.42 The positive signals at around 3400 and 3316 cm−1 are ascribed to the N–H stretching modes and the broad envelope in the high frequency range is due to the hydroxyl species engaged in H-bonding with ammonia molecules.
 | ||
| Fig. 5 Difference FT-IR spectra of NH3 dosages on (a) Ga-P-0, (b) Ga-F-0 and (c) outgassed at room temperature. | ||
In the low frequency region (1800–1300 cm−1), for both samples the absorption band at 1635 cm−1 corresponds to the bending mode of NH3 molecules H-bonded to surface OH groups, whereas the broad signal at around 1460 cm−1 is due to the bending mode of NH4+ ions formed by a proton-transfer reaction from Brønsted sites. The formation of ammonium ions on both solids indicates that the incorporation of Ga within the silica framework provides Brønsted sites that are acidic enough to protonate NH3, at variance with the surface hydroxyls of unsubstituted mesoporous silica which only establish hydrogen bonding. The blue shift of the ammonium band observed with increasing NH3 equilibrium pressure is generally ascribed to the multiple lateral interactions of NH4+ ions with surface oxygen atoms and/or OH groups. As expected from 71Ga NMR both Ga-F-0 and Ga-P-0 display similar acid properties. However, a direct comparison between the difference spectra of Ga-P-0 and Ga-F-0 outgassed at room temperature after ammonia dosage (Fig. 5c) demonstrates some differences. It is important to highlight that the observed residual signals correspond to species formed on the strongest acidic sites and thus irreversible upon ammonia removal. In the high frequency region, the fully outgassed samples show some dissimilarities, in particular for Ga-P-0 the residual broad signal at around 2800 cm−1 is more intense than that found on Ga-F-0, suggesting a larger population of residual H-bonded silanols in interaction with ammonia. Furthermore, a close inspection of the residual negative hydroxyl bands of the two samples evidences more intense residual free silanol bands (3744 cm−1) for Ga-F-0 compared to Ga-P-0, which at variance shows higher negative components at 3720 cm−1 and at 3500 cm−1. Moreover, a negative shoulder at about 3692 cm−1 (labelled with an asterisk in the inset) can be discerned on Ga-P-0, which based on the literature can be ascribed to hydroxyl linked to gallium sites, in analogy to that observed on hydroxylated gallium oxide.43
For both fully outgassed samples the signal of NH4+ ions became less intense upon outgassing (reversible proton transfer) and for the two samples a very similar residual intensity can be claimed. The absorption band attributed to NH3 physisorbed on silanols (1635 cm−1) decreased in intensity and was red-shifted up to 1621 cm−1 for the fully outgassed samples (residual pressure <1.10−3) ascribed to ammonia molecules adsorbed on Ga3+ acting as Lewis sites, in analogy to amorphous aluminosilicates.42 Since this signal appears slightly more intense for Ga-P-0 than for Ga-F-0, it can be considered as evidence of a little higher concentration of exposed surface Lewis sites.
Taken together, these spectroscopic data reveal very similar surface features in terms of acidic sites for the two samples. However, some differences regarding the hydroxyl populations and Lewis site concentration have been evidenced, suggesting an overall higher acidity of the Ga-P-0 surface, most likely due to a Ga-enriched surface compared to Ga-F-0. This slightly higher acidity of the Ga-P-0 solid together with the increased accessibility of the active sites (lower contribution of micropores) may constitute a synergistic combination of features contributing to the enhanced catalytic performances.
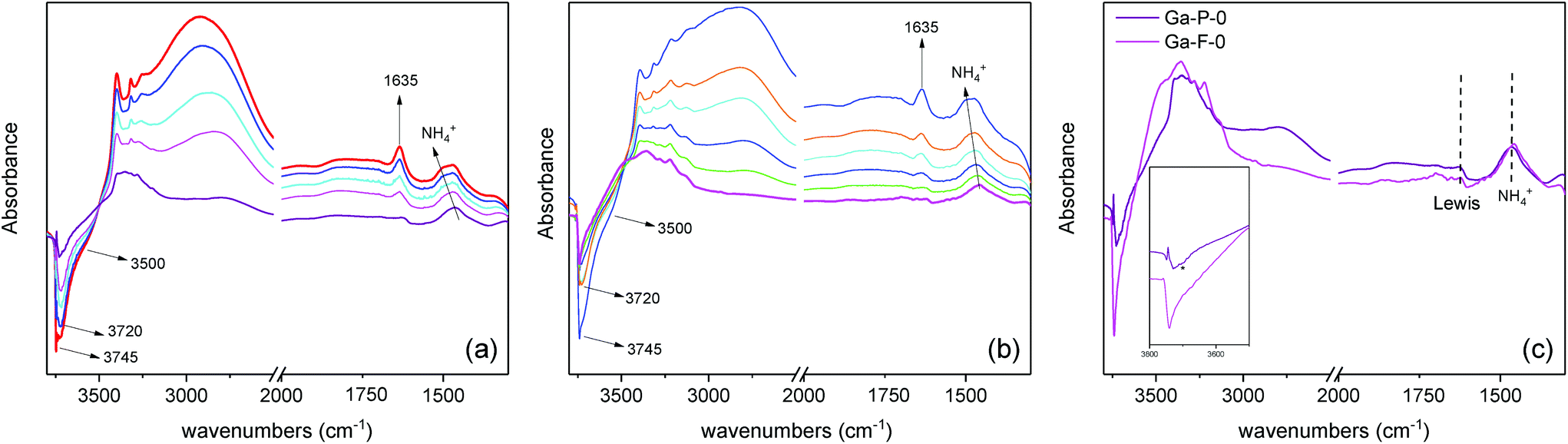
The catalytic activity of the best materials in each series was also evaluated over time (Fig. 6). Regardless of the catalyst considered, the TON increased rapidly during the first 8 h of reaction followed by a plateau (the conversion values at 8 h are reported in Table S4†). The functionalized materials display a higher TON compared to non-functionalized materials. Moreover, the difference observed between non-functionalized and functionalized materials remains constant over time.
 | ||
| Fig. 6 Evaluation of the catalytic activity over time. Comparison between Ga-P-5-Me and Ga-P-0 (left), and Ga-F-5-Me and Ga-F-0 (right). Conditions of the catalytic tests: 10 mg of catalyst, 50 °C, glycerol:acetone = 1 (0.01 mol):4. | ||
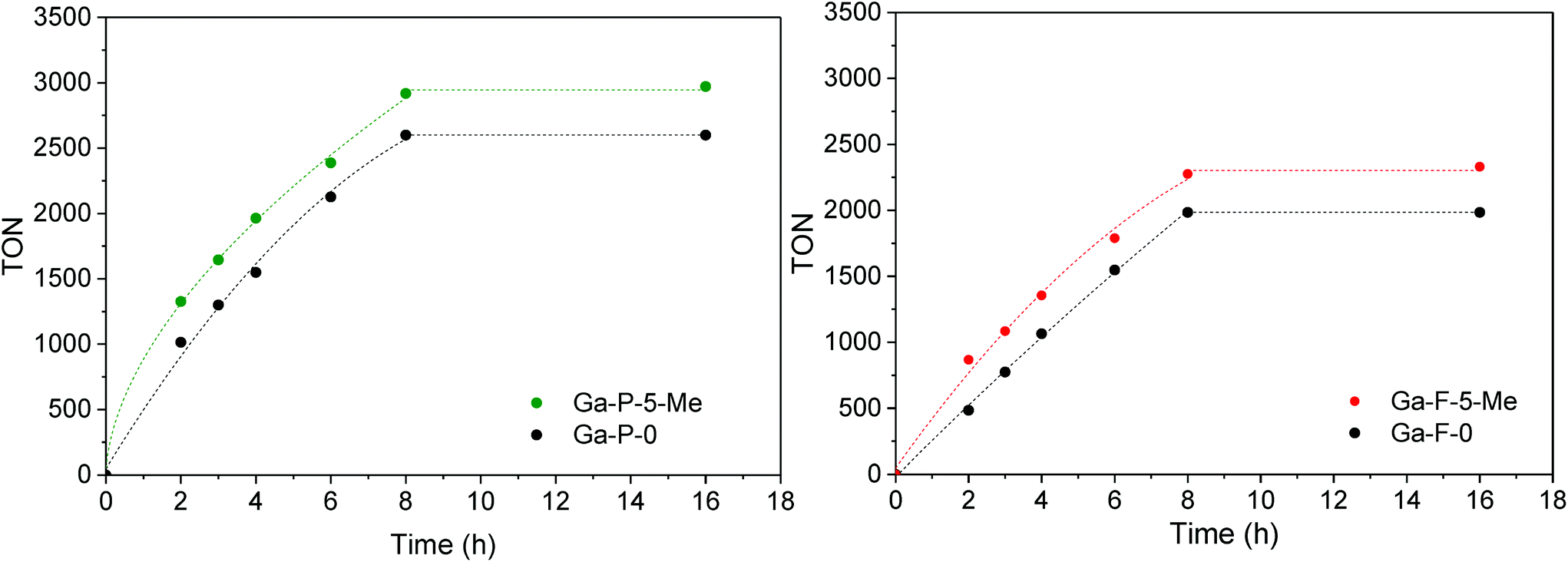
In order to have a meaningful notion of the performances of our best catalyst (Ga-P-5-Me), an additional catalytic test was performed in the presence of t-butanol, a solvent commonly used in the literature to convert glycerol into solketal. The comparison with reference materials given in Fig. 7 evidences the outstanding catalytic performance of Ga-P-5-Me. The high activity of our solid can be ascribed to a combination of aspects such as the incorporation of gallium mainly as a single site, its open mesostructure or even the functionalization of its surface with methyl groups.
 | ||
| Fig. 7 Comparison of the catalytic activity of Ga-P-5-Me with metal-containing silicates reported in the literature for the conversion of glycerol to solketal: (a),19 (b).21 Conditions of the catalytic test: 25 mg of catalyst, 1.48 g of tert-butanol, 80 °C, 2 hours, glycerol:acetone = 1 (0.01 mol):4. | ||
From the perspective of sustainable processes promoted by heterogeneous catalysts, a pivotal role is played by the robustness of the catalyst under the selected reaction conditions, which determines the possibility to reuse it in multiple catalytic runs. The reusability of our most promising catalyst – Ga-P-5-Me – was evaluated in consecutive cycles (Fig. 8, left). At the end of each cycle, the catalyst was recovered by centrifugation, washed with ethanol and thermally regenerated at 300 °C for two hours. It is important to highlight that the selected thermal treatment allows the preservation of the initial number of methyl groups in our catalyst as confirmed by quantitative 29Si MAS measurement (Fig. S14†). The calcination treatment is of fundamental importance since it was already reported that the activity gradually decreases upon reuse if the catalyst is only washed with the solvent. From this study it emerged that the good activity and selectivity of the catalyst were preserved at least for four catalytic cycles.
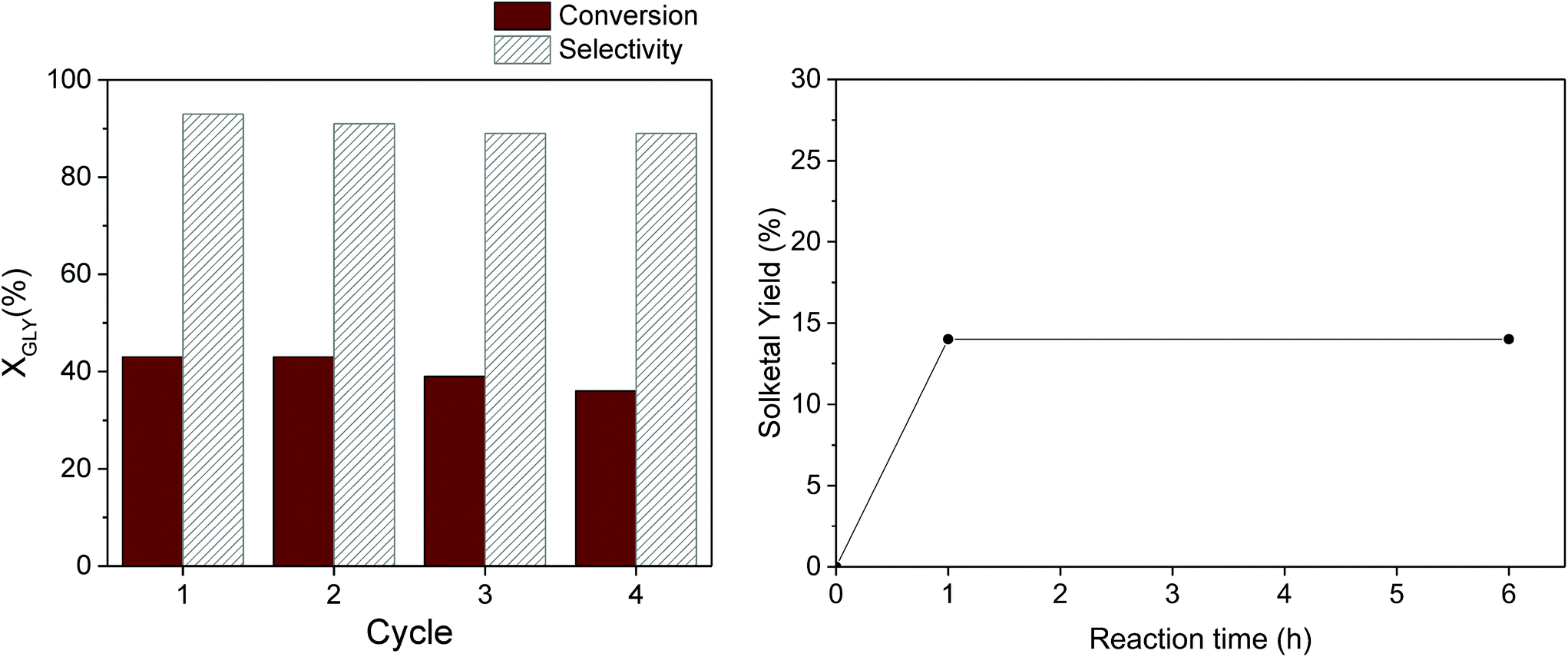 | ||
| Fig. 8 Recycling test with Ga-P-5-Me (left). Conditions: 50 mg of catalyst, 50 °C, 1 hour, glycerol:acetone = 1 (0.017 mol):4. Hot filtration experiment with Ga-P-5-Me (right). Conditions: 25 mg of catalyst, 0.70 mL of ethanol, 50 °C, 1 hour, glycerol:acetone = 1 (0.01 mol):4. | ||
In order to investigate the stability of Ga-P-5-Me, a hot filtration experiment was performed as well (Fig. 8, right). The solketal yield was measured after one hour of reaction after which the catalyst was removed by hot filtration (50 °C). The filtrate was then reacted for 5 additional hours. The hot filtration experiment shows that the solketal yield remains constant indicating the absence of active species in the filtrate.
To further evaluate the sustainability of our process, the associated E-Factor was calculated, selecting a reaction time of 8 hours (to reach the maximum conversion). After the reaction, the catalyst was removed by centrifugation and acetone was recovered by distillation. The E-Factor was calculated according to the following formula: E-Factor = [(4.60 g glycerol + 11.6 g acetone − 2.41 g acetone recovered by distillation − 3.50 g solketal)/3.50 g solketal] = 2.9. Such a low E-factor value is in the same range as typical values reported for the industrial production of bulk chemicals,44 and supports the idea that this catalytic process is promising. Moreover, the optimization of the distillation setup used to recover acetone could certainly allow further improvement of the E-Factor of the process.
As mentioned in the beginning of this work, the importance of preserving the organic functionalities on the surface of the solids while removing efficiently the structuring agent determined the adoption of a mild thermal treatment at 250 °C. However, in the literature, analogous non-functionalized metal-silicates are usually subjected directly to a thermal treatment ranging from 500 °C to 600 °C to effectively remove the templating agent.21,45,46 It is important to highlight that the calcination temperature may affect not only the physicochemical properties of the materials (e.g. surface area), but also the surface hydroxyl content, responsible for the hydrophilic character of the surface.
For this reason, we investigated the catalytic behavior of Ga-P-0 at different calcination temperatures. As reported in Fig. 9, the increase of the calcination temperature from 250 °C to 550 °C had a beneficial impact on the catalytic performance of the non-functionalized solid. If we compare the physico-chemical properties of the material calcined at 250 °C and 550 °C, a decrease of the BET surface area (from 440 m2 g−1 to 350 m2 g−1) is observed and could be ascribed to the increasing condensation of neighboring silanols (vide infra). The loss of BET surface area with the increase of the calcination temperature (until 550 °C) appears incoherent with the improvement of the catalytic activity. In fact, if the TON is normalized by the surface area (Fig. S15†), this improvement is even more evident. No other major differences were observed in terms of pore size distribution or gallium insertion (Fig. S16†). Therefore, these observations could be ascribed to the beneficial role played by the calcination temperature in the modification of the surface hydrophobicity.
 | ||
| Fig. 9 Study of the influence of the calcination temperature on the catalytic activity of Ga-P-0. Conditions of the catalytic tests: 10 mg of catalyst, 50 °C, 2 hours, glycerol:acetone = 1 (0.01 mol):4. | ||
As the temperature of the thermal treatment increases, the silica surface is dehydrated causing the removal of the adsorbed water and the thermal dehydration reaction, where two adjacent silanols condense into a siloxane bridge. Experimental data reported in the literature demonstrated that by increasing the temperature of the thermal treatment the silica surface becomes more hydrophobic due to the condensation of surface hydroxyls and the formation of siloxane bridges.47–49 The reduction of the hydroxyl group population was here proved by the higher degree of condensation. The latter was estimated by deconvolution analysis using Gaussian functions of the nQ contributions on quantitative direct excitation 29Si MAS NMR spectra (Fig. 10). This last procedure could represent an interesting alternative to modify the hydrophilic/hydrophobic balance of the surface proving again its beneficial role in particular in the selected reaction.
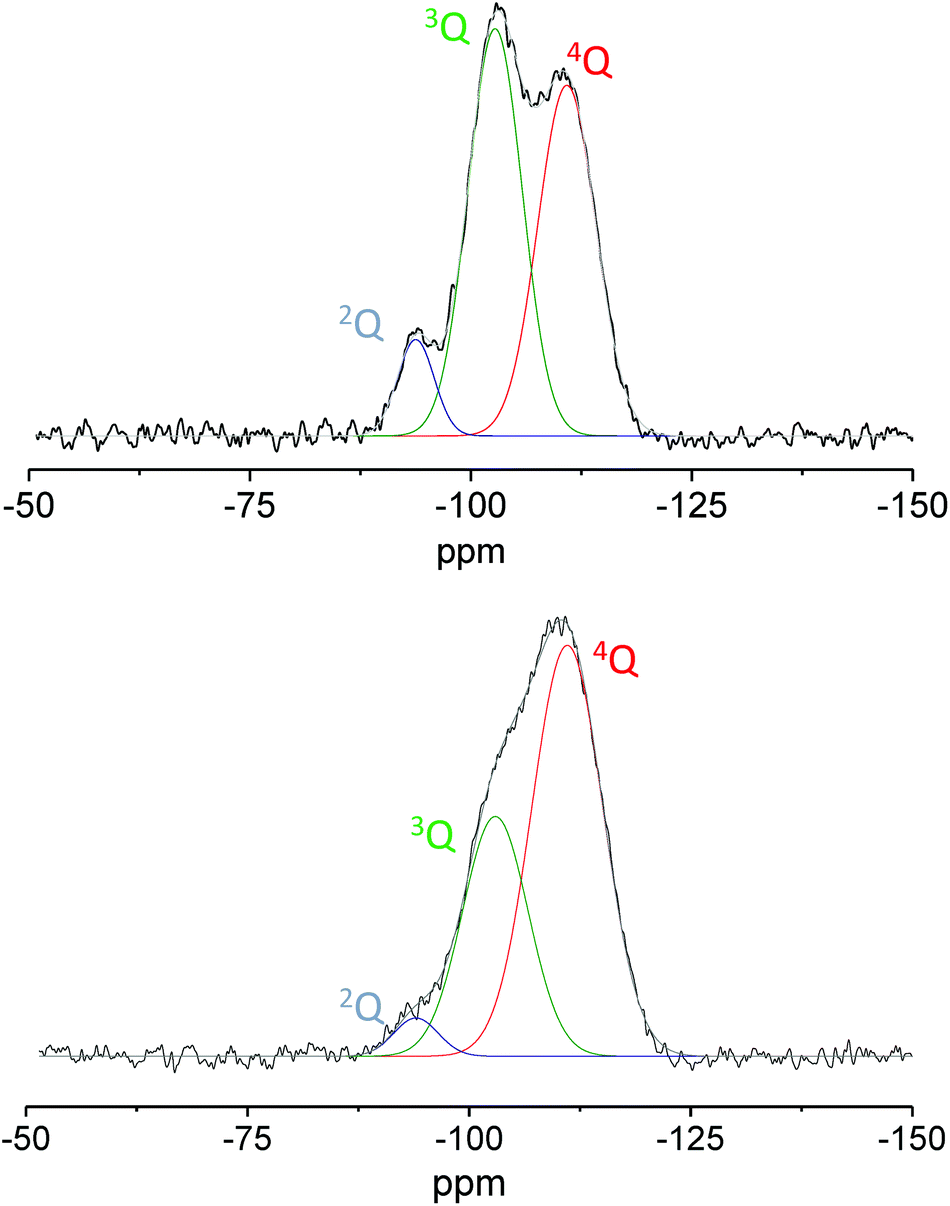 | ||
| Fig. 10 29Si direct excitation MAS NMR spectra of Ga-P-0 calcined at 250 °C (top) and 550 °C (bottom). | ||
Finally, raising the calcination temperature to 650 °C resulted in a rather remarkable loss of catalytic activity that in this case could be ascribed to a major loss of surface area (around 200 m2 g−1), probably caused by the collapse of the catalyst structure.
Conclusions
The aerosol-assisted sol–gel process was used to synthesize, in a straightforward one-pot procedure and with low environmental impact, a series of functionalized Ga-silicates. The efficient incorporation of isolated gallium as a single site within the silica framework was confirmed via71Ga solid state NMR and the stability of the organic moieties was assessed using 29Si solid-state MAS NMR. The materials exhibited record activity in the acetalization of glycerol to yield solketal at low temperature and under solventless conditions. In particular, the materials synthesized using P123 as the templating agent demonstrated the best catalytic activity. A positive influence of the functionalization on the catalytic performances of the materials emerged in the presence of methyl moieties. In addition, it was observed that only a low degree of methylation was effective to enhance the catalytic performance of the materials, shifting locally the reaction to the production of solketal. The best catalyst was successfully used in multiple catalytic runs, thus proving its robustness under the selected reaction conditions. To further evaluate the sustainability of our process, the associated E-factor was calculated and can be compared with the E-factor reported for the industrial production of bulk chemicals. Moreover, a systematic study evidenced the importance of a careful tuning of surface polarity, achievable not only with surface functionalization, but also with different thermal treatments applied to non-functionalized solids. The control of the hydroxyl group population on the surface, possible with a control on the calcination temperature, also allows enhancement of the catalytic activity, thus further supporting the pivotal role of the surface polarity. Nevertheless, considering the balance between sustainability and performances as a key aspect, functionalized solids constitute the best option as they present the advantage of a less energy-consuming thermal treatment at mild temperature.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge the “Communauté française de Belgique” for the financial support – including the PhD fellowship of A. Vivian – through the ARC programme (15/20-069). L. Soumoy thanks the FNRS for the financial support in the context of her FRIA PhD grant. Damien Debecker thanks the Francqui Foundation for the “Francqui Research Professor” chair. F. Devred is thanked for the technical and logistical support. This research used the resources of the PC2 (Plateforme Technologique Physico-Chemical Characterization) and the MORPH-IM (Morphology & Imaging) platforms located at the University of Namur.References
- Transforming our world: the 2030 Agenda for Sustainable Development, https://sustainabledevelopment.un.org/post2015/transformingourworld#, (accessed 06/05/2020).
- A European Green Deal, https://ec.europa.eu/info/strategy/priorities-2019-2024/european-green-deal_en, (accessed 06/05/2020).
- Renewable Energy – Recast to 2030 (RED II), https://ec.europa.eu/jrc/en/jec/renewable-energy-recast-2030-red-ii, (accessed 06/05/2020).
- I. M. Atadashi, M. K. Aroua and A. A. Aziz, Renewable Sustainable Energy Rev., 2010, 14, 1999–2008 CrossRef CAS.
- T. Issariyakul and A. K. Dalai, Renewable Sustainable Energy Rev., 2014, 31, 446–471 CrossRef CAS.
- M. Pagliaro, R. Ciriminna, H. Kimura, M. Rossi and C. Della Pina, Angew. Chem., Int. Ed., 2007, 46, 4434–4440 CrossRef CAS.
- P. T. Anastas and J. C. Warner, 12 Principles of Green Chemistry. Green Chemistry: Theory and Practice, Oxford Univ. Press, New York, 1998 Search PubMed.
- A. Behr, J. Eilting, K. Irawadi, J. Leschinski and F. Lindner, Green Chem., 2008, 10, 13–30 RSC.
- J. I. García, H. García-Marín and E. Pires, Green Chem., 2014, 16, 1007–1033 RSC.
- M. R. Nanda, Y. Zhang, Z. Yuan, W. Qin, H. S. Ghaziaskar and C. Xu, Renewable Sustainable Energy Rev., 2016, 56, 1022–1031 CrossRef CAS.
- A. Cornejo, I. Barrio, M. Campoy, J. Lázaro and B. Navarrete, Renewable Sustainable Energy Rev., 2017, 79, 1400–1413 CrossRef.
- R. A. Sheldon, ACS Sustainable Chem. Eng., 2018, 6, 32–48 CrossRef CAS.
- T. N. Pham, T. Sooknoi, S. P. Crossley and D. E. Resasco, ACS Catal., 2013, 3, 2456–2473 CrossRef CAS.
- F. D. L. Menezes, M. D. O. Guimaraes and M. J. da Silva, Ind. Eng. Chem. Res., 2013, 52, 16709–16713 CrossRef CAS.
- R. Gérardy, D. P. Debecker, J. Estager, P. Luis and J.-C. M. Monbaliu, Chem. Rev., 2020, 120(15), 7219–7347 CrossRef.
- L. Roldán, R. Mallada, J. M. Fraile, J. A. Mayoral and M. Menéndez, Asia–Pac. J. Chem. Eng., 2009, 4, 279–284 CrossRef.
- P. Ferreira, I. M. Fonseca, A. M. Ramos, J. Vital and J. E. Castanheiro, Appl. Catal., B, 2010, 98, 94–99 CrossRef CAS.
- J. Kowalska-Kus, A. Held, M. Frankowski and K. Nowinska, J. Mol. Catal. A: Chem., 2017, 426, 205–212 CrossRef CAS.
- L. Li, T. I. Korányi, B. F. Sels and P. P. Pescarmona, Green Chem., 2012, 14, 1611–1619 RSC.
- G. Vicente, J. A. Melero, G. Morales, M. Paniagua and E. Martín, Green Chem., 2010, 12, 899–907 RSC.
- X. Collard, L. Li, W. Lueangchaichaweng, A. Bertrand, C. Aprile and P. P. Pescarmona, Catal. Today, 2014, 235, 184–192 CrossRef CAS.
- A. Vivian, L. Soumoy, L. Fusaro, P. Louette, A. Felten, S. Fiorilli, D. P. Debecker and C. Aprile, Synthesis and In-Depth Characterization of Ga-Based Structured Catalysts: Enhancing Glycerol Conversion, 2020, ChemRxiv, DOI: DOI:10.26434/chemrxiv.12612926.v1.
- D. P. Debecker, S. Le Bras, C. Boissière, A. Chaumonnot and C. Sanchez, Chem. Soc. Rev., 2018, 47, 4112–4155 RSC.
- C. Wang and H. Bai, Ind. Eng. Chem. Res., 2011, 50, 3842–3848 CrossRef CAS.
- L. Kuai, J. Geng, C. Chen, E. Kan, Y. Liu, Q. Wang and B. Geng, Angew. Chem., Int. Ed., 2014, 53, 7547–7551 CrossRef CAS.
- V. Smeets, C. Boissière, C. Sanchez, E. M. Gaigneaux, E. Peeters, B. F. Sels, M. Dusselier and D. P. Debecker, Chem. Mater., 2019, 31, 1610–1619 CrossRef CAS.
- E. Kan, L. Kuai, W. Wang and B. Geng, Chem. – Eur. J., 2015, 21, 13291–13296 CrossRef CAS.
- A. Vivian, L. Fusaro, D. P. Debecker and C. Aprile, ACS Sustainable Chem. Eng., 2018, 6, 14095–14103 CrossRef CAS.
- F. Colbeau-Justin, C. Boissière, A. Chaumonnot, A. Bonduelle and C. Sanchez, Adv. Funct. Mater., 2014, 24, 233–239 CrossRef CAS.
- D. P. Debecker, M. Stoyanova, F. Colbeau-Justin, U. Rodemerck, C. Boissière, E. M. Gaigneaux and C. Sanchez, Angew. Chem., Int. Ed., 2012, 51, 2129–2131 CrossRef CAS.
- A. Kim, C. Sanchez, B. Haye, C. Boissière, C. Sassoye and D. P. Debecker, ACS Appl. Nano Mater., 2019, 2, 3220–3230 CrossRef CAS.
- V. Smeets, L. Ben Mustapha, J. Schnee, E. M. Gaigneaux and D. P. Debecker, Mol. Catal., 2018, 452, 123–128 CrossRef CAS.
- X. Ji, Q. Hu, J. E. Hampsey, X. Qiu, L. Gao, J. He and Y. Lu, Chem. Mater., 2006, 18, 2265–2274 CrossRef CAS.
- M. Kruk, M. Jaroniec, C. H. Ko and R. Ryoo, Chem. Mater., 2000, 12, 1961–1968 CrossRef CAS.
- R. M. Grudzien and M. Jaroniec, Chem. Commun., 2005, 1076–1078 RSC.
- C. R. Bayense, A. P. Kentgens, J. W. De Haan, L. J. Van de Ven and J. H. Van Hooff, J. Phys. Chem., 1992, 96, 775–782 CrossRef CAS.
- M. Chatterjee, T. Iwasaki, Y. Onodera, T. Nagase, H. Hayashi and T. Ebina, Chem. Mater., 2000, 12, 1654–1659 CrossRef CAS.
- N. Godard, A. Vivian, L. Fusaro, L. Cannavicci, C. Aprile and D. P. Debecker, ChemCatChem, 2017, 9, 2211–2218 CrossRef CAS.
- M. R. Nanda, Z. Yuan, W. Qin, H. S. Ghaziaskar, M.-A. Poirier and C. C. Xu, Fuel, 2014, 117, 470–477 CrossRef CAS.
- D. H. Olson, W. O. Haag and W. S. Borghard, Microporous Mesoporous Mater., 2000, 35–36, 435–446 CAS.
- W. J. J. Stevens, K. Lebeau, M. Mertens, G. Van Tendeloo, P. Cool and E. F. Vansant, J. Phys. Chem. B, 2006, 110, 9183–9187 CrossRef CAS.
- B. Bonelli, B. Onida, J. D. Chen, A. Galarneau, F. Di Renzo, F. Fajula and E. Garrone, Microporous Mesoporous Mater., 2004, 67, 95–106 CrossRef CAS.
- C. O. Areán, A. López Bellan, M. P. Mentruit, M. R. Delgado and G. T. Palomino, Microporous Mesoporous Mater., 2000, 40, 35–42 CrossRef.
- R. A. Sheldon, Green Chem., 2017, 19, 18–43 RSC.
- R. Luque, J. M. Campelo, T. D. Conesa, D. Luna, J. M. Marinas and A. A. Romero, Microporous Mesoporous Mater., 2007, 103, 333–340 CrossRef CAS.
- K. Okumura, K. Nishigaki and M. Niwa, Microporous Mesoporous Mater., 2001, 44–45, 509–516 CrossRef CAS.
- V. Bolis, B. Fubini, L. Marchese, G. Martra and D. Costa, J. Chem. Soc., Faraday Trans., 1991, 87, 497–505 RSC.
- E. F. Vansant, P. Van Der Voort and K. C. Vrancken, Characterization and chemical modification of the silica surface, Elsevier, 1995 Search PubMed.
- A. Matsumoto, T. Sasaki, N. Nishimiya and K. Tsutsumi, Colloids Surf., A, 2002, 203, 185–193 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc02562c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |