
Urolithin A protects dopaminergic neurons in experimental models of Parkinson's disease by promoting mitochondrial biogenesis through the SIRT1/PGC-1α signaling pathway
Jia
Liu†
a,
Jingjing
Jiang†
a,
Jingru
Qiu
a,
Liyan
Wang
b,
Jing
Zhuo
c,
Baozhu
Wang
a,
Deqing
Sun
d,
Shuyan
Yu
 e and
Haiyan
Lou
*a
e and
Haiyan
Lou
*a
aDepartment of Pharmacology, School of Basic Medical Sciences, Cheeloo College of Medicine, Shandong University, Jinan, Shandong 250012, China. E-mail: louhaiyan@sdu.edu.cn; Tel: +86 531 88380515
bCenter for Experimental Nuclear Medicine and Electron Microscopy, School of Basic Medical Sciences, Cheeloo College of Medicine, Shandong University, Jinan, Shandong 250012, China
cDepartment of Ultrasound, Qilu Hospital of Shandong University, Jinan, Shandong 250012, China
dDepartment of Pharmacy, The Second Hospital, Cheeloo College of Medicine, Shandong University, Jinan, Shandong 250012, China
eDepartment of Physiology, School of Basic Medical Sciences, Cheeloo College of Medicine, Shandong University, Jinan, Shandong 250012, China
First published on 23rd November 2021
Abstract
Mitochondrial dysfunction contributes to the pathogenesis of neurodegenerative diseases such as Parkinson's disease (PD). Therapeutic strategies targeting mitochondrial dysfunction hold considerable promise for the treatment of PD. Recent reports have highlighted the protective role of urolithin A (UA), a gut metabolite produced from ellagic acid-containing foods such as pomegranates, berries and walnuts, in several neurological disorders including Alzheimer's disease and ischemic stroke. However, the potential role of UA in PD has not been characterized. In this study, we investigated the underlying mechanisms for role of UA in 6-OHDA-induced neurotoxicity in cell cultures and mice model of PD. Our results revealed that UA protected against 6-OHDA cytotoxicity and apoptosis in PC12 cells. Meanwhile, administration of UA to 6-OHDA lesioned mice ameliorated both motor deficits and nigral-striatal dopaminergic neurotoxicity. More important, UA treatment significantly attenuated 6-OHDA-induced mitochondrial dysfunction in PC12 cells accompanied by enhanced mitochondrial biogenesis. Mechanistically, we demonstrated that UA exerts neuroprotective effects by promoting mitochondrial biogenesis via SIRT1-PGC-1α signaling pathway. Taken together, these data provide new insights into the novel role of UA in regulating mitochondrial dysfunction and suggest that UA may have potential therapeutic applications for PD.
1. Introduction
Parkinson's disease (PD) is the second most common neurodegenerative disease characterized by the loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc). While the pathophysiology of PD is complex, mitochondrial dysfunction is a prominent pathological hallmark of both sporadic and familial PD.1 Mitochondria are the primary source of energy in the brain, and their impairment has been implicated in the aging process and neurodegenerative diseases. Thus, targeting mitochondrial dysfunction is a promising approach for the treatment of PD.2 Over the past decade, the identification of natural products that target mitochondrial function in the central nervous system has become an emerging field in drug discovery.3Mitochondrial quality control is regulated by the process of mitochondrial biogenesis and mitophagy. Mitochondrial biogenesis is a complex process that results in the generation of new mitochondria, the maintenance of which is critical for the normal function of neurons. Several reports indicate that peroxisome proliferator activated receptor gamma coactivator 1-alpha (PGC-1α) and the NAD+-dependent deacetylase SIRT1 are the two major cooperating players in mitochondrial biogenesis.4 Because mitochondrial biogenesis is impaired with aging and in neurodegenerative diseases,5,6 identifying therapeutic compounds that induce mitochondrial biogenesis through the modulation of the SIRT1-PGC-1α pathway may be considered as a novel therapeutic approach for PD.
Urolithin A (UA) is the major metabolite of ellagic acid (EA) and ellagitannins (ETs), which are abundant in pomegranates, berries, and nuts.7,8 Several studies have demonstrated that UA possesses antioxidant and anti-inflammatory properties.9 Recent studies have also revealed that UA exhibits neuroprotective effects in an APP/PS1 mice model of AD and in an experimental model of ischemic stroke.10–13 Moreover, UA was shown to be bioavailable in the plasma of elderly individuals and resulted in the modulation of mitochondrial gene expression in skeletal muscles.14,15 However, it is unknown whether UA might attenuate dopaminergic neurotoxicity by modulating mitochondrial function in PD. Therefore, in the present study, we investigated whether UA was effective in experimental models of PD and the possible mechanisms involved. Our findings revealed that UA attenuated 6-OHDA-induced neurotoxicity in PC12 cells as well as in 6-OHDA lesioned C57BL/6 mice. Moreover, the beneficial effect of UA appears to be associated with SIRT1-PGC-1α signaling-mediated mitochondrial biogenesis.
2. Materials and methods
2.1 Cell viability assessment and apoptosis analysis
The PC-12 cell line was cultured at 37 °C in DMEM supplemented with 10% fetal bovine serum in a humidified atmosphere of 5% CO2. The cells were differentiated by treating with nerve growth factor (NGF) (50 ng ml−1) every other day for six days. Cells were pretreated with UA (S5312, Selleck) for 2 h and were then subjected to 6-OHDA (162957, Sigma-Aldrich) stimulation for different hours. Cell viability was measured using the CCK-8 assay according to the manufacturer's instruction (CK-04, DOJINDO). Briefly, cells were incubated with 10 μl of CCK-8 solution at 37 °C for 30 min, then the absorbance was measured at 450 and 600 nm on a plate reader.6-OHDA-induced cell apoptosis was detected with an annexin V-FITC/propidium iodide (PI) double staining kit (FXP018-100, 4A Biotech) with flow cytometry according to the manufacturer's instruction. Briefly, cells were treated with UA for 2 h before being exposed to 6-OHDA for an additional 24 h. The cells were harvested, washed with ice-cold PBS, and then evaluated for apoptosis by double staining with PI and annexin V-FITC in binding buffer using a Cytoflex flow cytometer. TUNEL staining was also performed to detect apoptotic cells using an in situ cell death detection kit (12156792910, Roche Diagnostic). Briefly, the slides were fixed with 4% paraformaldehyde at 4 °C for 1 h and incubated in 3% H2O2 for 10 min. After incubation in 0.1% Triton X-100 for 15 min, the slides were incubated in a TUNEL reaction mixture at 37 °C for 1 h. Finally, the slides were counterstained with DAPI and observed with a fluorescence microscope.
2.2 6-OHDA-induced mice model of PD and behavioral tests
All animal experiments were performed according to the National Institutes of Health Guide for the Care and Use of Laboratory animals (NIH publications no. 8023, revised 1978) and approved by the Animal Ethics Committee of Shandong University. Male C57BL/6J mice (8–10 weeks old) were anesthetized with 30 mg kg−1 sodium pentobarbital and placed in a stereotaxic device, and 9 μg of 6-OHDA was then injected into two different sites of the right striatum of the brain using a Hamilton syringe. The stereotaxic coordinates of the two different sites of the right STR, measured in millimeters, were as follows: bregma +1.0, lateral 2.1, and ventral −2.9 and bregma −0.3, lateral 2.3, and ventral −2.9. Mice were decapitated for biochemical or histological assessments at different time points (1, 7, 14, and 21 days) following 6-OHDA injection. For UA treatment, mice were intraperitoneally administered 10 mg kg−1 UA for 7 days before 6-OHDA injection. Rotarod test, pole test, suspension test and apomorphine-induced rotation tests were performed as previously described.16,172.3 Mitochondrial function
Mitochondrial membrane potential (MMP) was detected by the mitochondrial membrane potential assay kit with a fluorescent probe JC-1 following the manufacturer's protocol. ATP content was measured with a commercial kit (S0026, Beyotime, China) according to the manufacturer's instructions. Reactive oxygen species (ROS) levels were assessed using the dichloro-dihydro-fluorescein diacetate (DCFH-DA) fluorescence assay as previously described.172.4 Quantification of mitochondrial DNA content
The relative copy number of mitochondrial DNA was detected by real-time quantitative PCR assay, which was performed on a Bio-Rad iCycler system (Bio-Rad, Hercules, CA) using SYBR Green methods. Total cellular DNA was extracted using a genomic DNA preparation kit (D0061, Beyotime, China) following the manufacturer's protocol. DNA primers were designed to detect Cyt b and cytochrome c oxidase subunit II (COII) as markers for mitochondrial DNA (mtDNA), and β-actin as a marker for nuclear DNA (nDNA). The specific primer sequences were as follows: Cytb: forward (5′-ACAACGCAGCTTAACATTCCG-3′) and reverse (5′-TTCTACTGGTTGGCCTCCGA-3′); COII: forward (5′-TACAAGACGCCA CATCACCT) and reverse (5′-TTCTTGGGCGTCTATTGTGCT-3′); β-actin: forward (5′-CTCTGTGTGGATTGGTGGCT-3′) and reverse (5′-CGCAGCTCAGTAACAGTCCG-3′).2.5 Transmission electron microscopy (TEM)
Electron microscopic sample handling and detection were performed with the electron microscopic core lab of Shandong University as described. 106 PC-12 cells and 1 mm3 brain tissue of mouse SN were prepared for TEM.2.6 Western blot
Protein extracts were prepared from PC-12 cells or brain tissues by homogenization in RIPA buffer with protease inhibitors and then were centrifugated to eliminate particulates at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 10 min at 4 °C. Protein concentration was measured by the BCA method. Samples were separated on SDS-PAGE and transferred to nitrocellulose membranes. Immunoblotting was carried out with the primary antibodies specific for TOM20 (11802-1-AP, Proteintech), Tim23 (11123-1-AP, Proteintech), TFAM (22586-1-AP, Proteintech), PGC-1α (66369-1-lg, Proteintech), SIRT1 (66369-l-lg, Abcam), and tyrosine hydroxylase (TH, MAB318, Millipore). The primary antibody against β-actin (66009-1-lg, Proteintech) was used to correct the sample loading. Densitometry analysis was performed using the AlphaEaseFC software.
000g for 10 min at 4 °C. Protein concentration was measured by the BCA method. Samples were separated on SDS-PAGE and transferred to nitrocellulose membranes. Immunoblotting was carried out with the primary antibodies specific for TOM20 (11802-1-AP, Proteintech), Tim23 (11123-1-AP, Proteintech), TFAM (22586-1-AP, Proteintech), PGC-1α (66369-1-lg, Proteintech), SIRT1 (66369-l-lg, Abcam), and tyrosine hydroxylase (TH, MAB318, Millipore). The primary antibody against β-actin (66009-1-lg, Proteintech) was used to correct the sample loading. Densitometry analysis was performed using the AlphaEaseFC software.
2.7 Immunohistochemical staining
Animals were anesthetized with sodium pentobarbital, transcardially perfused with 0.9% normal saline, followed by 4% paraformaldehyde in 0.1 M PBS (pH 7.4). Brains were dissected out, postfixed in 4% paraformaldehyde for 24 h, and cryopreserved in 30% sucrose for 48 h. Frozen brains were then coronally sectioned at 20 μm thickness on a cryomicrotome and sections were mounted on slides. For immunohistochemistry, samples were incubated with an antibody against TH, followed by horseradish peroxidase (HRP) conjugated secondary antibody. The sections were then visualized using diaminobenzidine (DAB). Digital images were collected in a bright field microscope. To verify the specificity of the TH antibody, control sections were processed with the same protocol, except that the primary antibody was omitted.For TH+ neuron quantification, we used unbiased stereology. The total number of TH+ cells in the SN was counted using the optical fractionator probe (MicroBrightfield) by investigators blinded to the experimental design. Samples from every fifth section were included in the procedure, covering the entire study regions. The data were expressed as a percentage of the corresponding area from the control group. The density of TH+ fibers in the striatum was measured by densitometry using the ImageJ software, and the data are expressed as a percentage of the corresponding area from the control group.
2.8 Bioinformatic analysis
Genes associated with PD (PD-related genes) were retrieved from the Kyoto Encyclopedia of Genes and Genomes (KEGG), Online Mendelian Inheritance in Man (OMIM) and Phenotype-Genotype Integrator (PheGenI) database. Genes from the three lists were then mapped onto the official gene symbols of humans reported in HUGO Gene Nomenclature Committee (HGNC) to obtain the PD-related genes. To elucidate the potential molecular functions (MF), biological process (BP) and cellular components (CC) associated with the PD-related genes, gene ontology (GO) enrichment analysis was performed utilizing the Database for Annotation, Visualization and Integrated Discovery (DAVID, version 6.8). Likewise, the Swiss Target Prediction database was used to screen the pharmacological targets of UA. All primary targets of UA and PD were evaluated using Venn diagrams to identify the potential targets for UA against PD.2.9 Statistic analysis
Statistical significance between multiple groups was examined by one-way ANOVA followed by Tukey's post hoc testing. All data are expressed as mean ± SEM with p < 0.05 considered statistically significant.3. Results
3.1 UA attenuates 6-OHDA-induced neurotoxicity in PC12 cells
The present study was aimed to determine the potential protective effects of UA in the context of 6-OHDA-mediated neurotoxicity. Therefore we chose to use differentiated PC12 cells as an in vitro model and challenged the cells with 6-OHDA. We first assessed the effect of UA itself on cell viability. As shown in Fig. 1A, there were no adverse effects on the growth of PC12 cells up to 10 μM of UA. To test the ability of UA to prevent 6-OHDA-induced cell death, we performed CCK-8 assay after challenging cells with 150 μM of 6-OHDA. Compared to vehicle-treated control cells, pretreatment of cells with UA significantly reduced 6-OHDA-associated cell death as measured by CCK-8 (Fig. 1B). To further determine the neuroprotective effect of UA against 6-OHDA neurotoxicity, we utilized flow cytometric analysis to measure the relative numbers of Annexin V and PI stained cells. As shown in Fig. 1C, 6-OHDA significantly increased cell apoptotic cell death. However, pretreatment with UA decreased the apoptotic rate. In addition, the neuroprotective effects of UA was confirmed by TUNEL staining (Fig. 1D).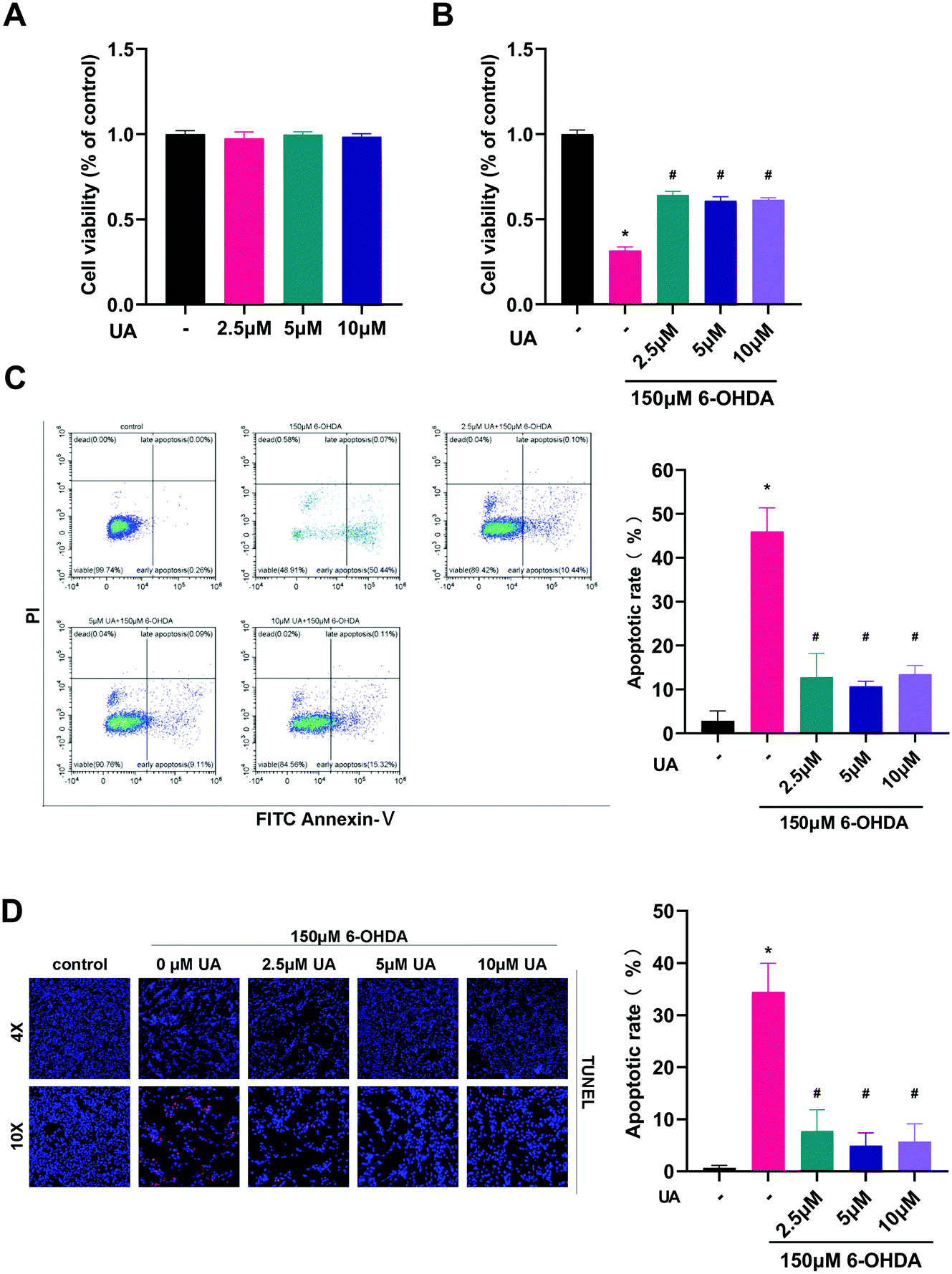 | ||
| Fig. 1 UA attenuates 6-OHDA-induced PC12 cell death. (A) PC-12 cells were incubated with UA at indicated concentrations for 24 h, and cell viability was determined by a CCK-8 assay. (B) Cells were pretreated with UA for 2 h before exposure to 150 μM 6-OHDA for an additional 24 h. The effects of UA on cell viability were measured by a CCK-8 assay. (C) Cells were pretreated with UA for 2 h before exposure to 150 μM 6-OHDA for an additional 24 h. The effects of UA on apoptotic cells were determined by annexin-V/PI staining. (D) The effects of UA on 6-OHDA-induced cell apoptosis were further measured by the TUNEL assay. Data are presented as mean ± SEM. One-way analysis of variance. *p < 0.05 vs. control group; #p < 0.05 vs. 6-OHDA group. | ||
3.2 UA prevents dopaminergic neurodegeneration in 6-OHDA-induced PD mice model
Following our in vitro observations described above, we next undertook studies to investigate whether UA protect mice from 6-OHDA-induced neurotoxicity in vivo. The administration of 6-OHDA caused a significant impairment of motor activity as determined by the rotarod test, pole test, suspension test and apomorphine-induced rotation test. The 6-OHDA-induced motor dysfunction was significantly improved by UA treatment (Fig. 2A–D). We then observed dopaminergic neuron loss in the nigrostriatal system following 6-OHDA administration by Nissl staining and immunohistochemistry (IHC) with an anti-TH antibody. As shown in Fig. 2E, stereological counts of SN total neurons defined by Nissl staining showed that there were reduced Nissl-positive neurons in 6-OHDA mice, 6-OHDA injection also caused a significant reduction in the TH-positive dopaminergic neurons in the SN region and a similar degree of dopaminergic terminal loss in the striatum. More importantly, UA treatment attenuated 6-OHDA-induced loss of TH-positive dopaminergic neurons in SN and dopaminergic fiber density fibers in the striatum (Fig. 2F and H). These results were further supported by immunoblots of nigral and striatal extracts evaluated using an anti-TH antibody, which showed higher TH protein levels in UA-treated mice (Fig. 2G and I) compared with 6-OHDA mice. Collectively, these findings indicate that UA has protective effects in the nigrostriatal system.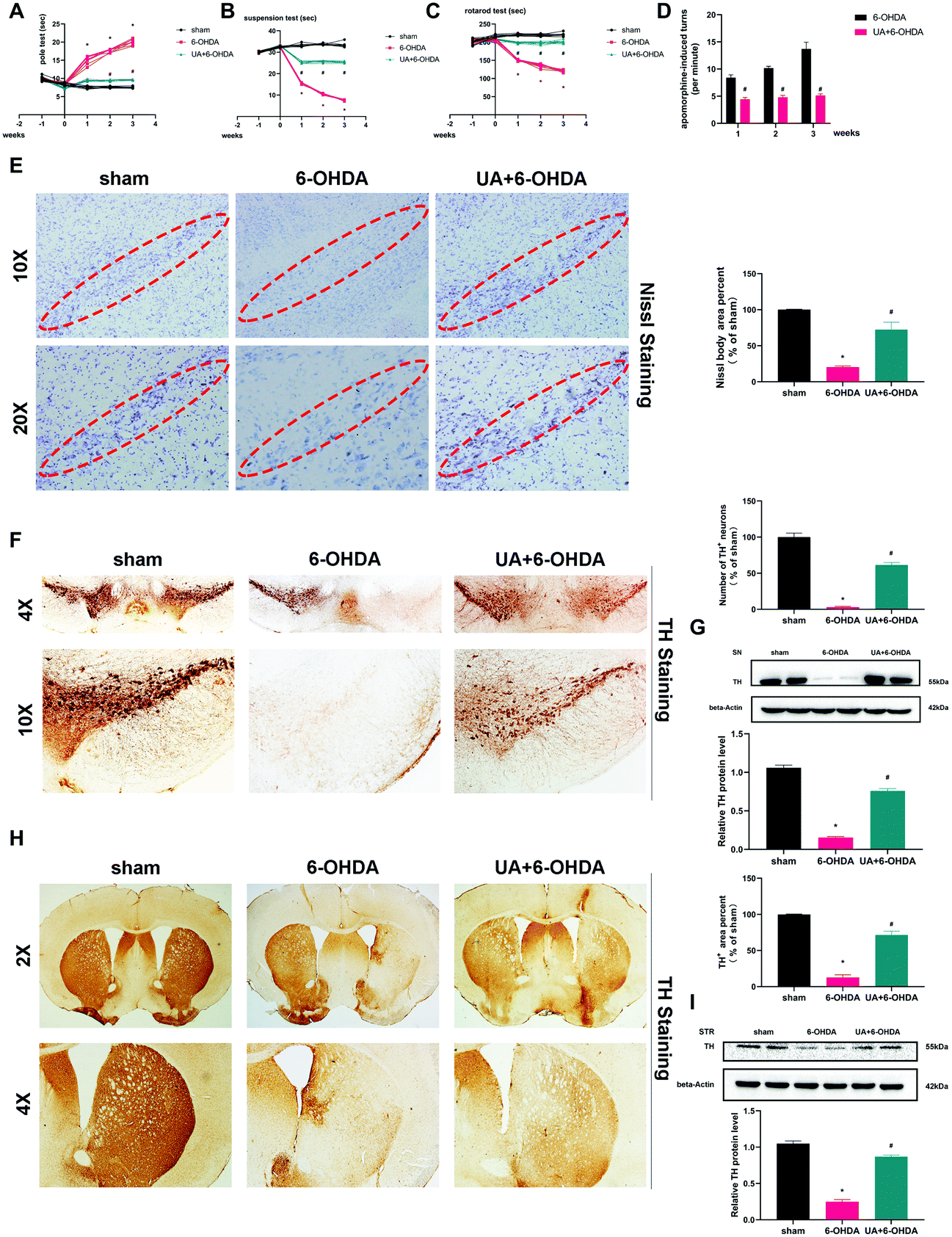 | ||
| Fig. 2 UA improves 6-OHDA-induced motor symptoms and alleviates 6-OHDA-induced dopaminergic neurotoxicity in experimental models of PD. (A)–(D) Pole tests, suspension tests, rotarod tests and apomorphine-induced turns were assessed at 7 days before 6-OHDA injection and 0, 7, 14 and 21 days after 6-OHDA injection. n = 10. Data are presented as mean ± SEM. Repeated measures analysis of variance. *p < 0.05 vs. sham group; #p < 0.05 vs. 6-OHDA group. (E) Representative images and quantitative analysis of Nissl-stained neurons in the SN of mice at 21 days after 6-OHDA injection. n = 3. (F) and (H) TH immunoreactivity in the SN and striatum at 21 days after 6-OHDA injection. n = 6. (G) and (I) Representative western blot showing that UA attenuates 6-OHDA-induced loss of TH in mice SN and striatum. n = 6. Data are presented as mean ± SEM. One-way analysis of variance. *p < 0.05 vs. sham group; #p < 0.05 vs. 6-OHDA group. | ||
3.3 UA rescues 6-OHDA-induced mitochondria damage and dysfunction both in vitro and in vivo
In order to address whether the beneficial effects of UA are related to its ability to improve mitochondrial function, we first assessed mitochondrial function in PC12 cells by measuring MMP using the fluorescence probe JC-1. As measured by immunofluorescence microscopy and flow cytometric analysis, the ratio of JC-1 red to JC-1 green, which reports levels of polarized functional mitochondria, was significantly lower in 6-OHDA-treated cells than in controls, supporting impaired mitochondrial function in 6-OHDA treated cells. However, UA effectively attenuated 6-OHDA-induced decline of MMP (Fig. 3A and B). Mitochondrial dysfunction may lead to oxidative stress and generate ROS. We therefore measured ROS level by flow cytometry using DCFH-DA. As shown in Fig. 3C, compared to controls, 6-OHDA treatment caused a robust increase in ROS production and UA treatment reduced 6-OHDA-indcued increase in ROS levels. Dysfunctional mitochondria are deficient at energy production, resulting in ATP depletion. As expected, cellular ATP levels were also significantly reduced in the 6-OHDA group and UA could restore ATP levels, indicating that UA promotes mitochondrial activity (Fig. 3D). UA treatment also preserved 6-OHDA-induced mitochondrial abnormalities by maintaining the mitochondrial morphology of PC12 cells and dopaminergic neurons in the SN region of PD mice (Fig. 3E and F). These findings show that UA could attenuate 6-OHDA-induced mitochondrial dysfunction and damage both in vitro and in vivo.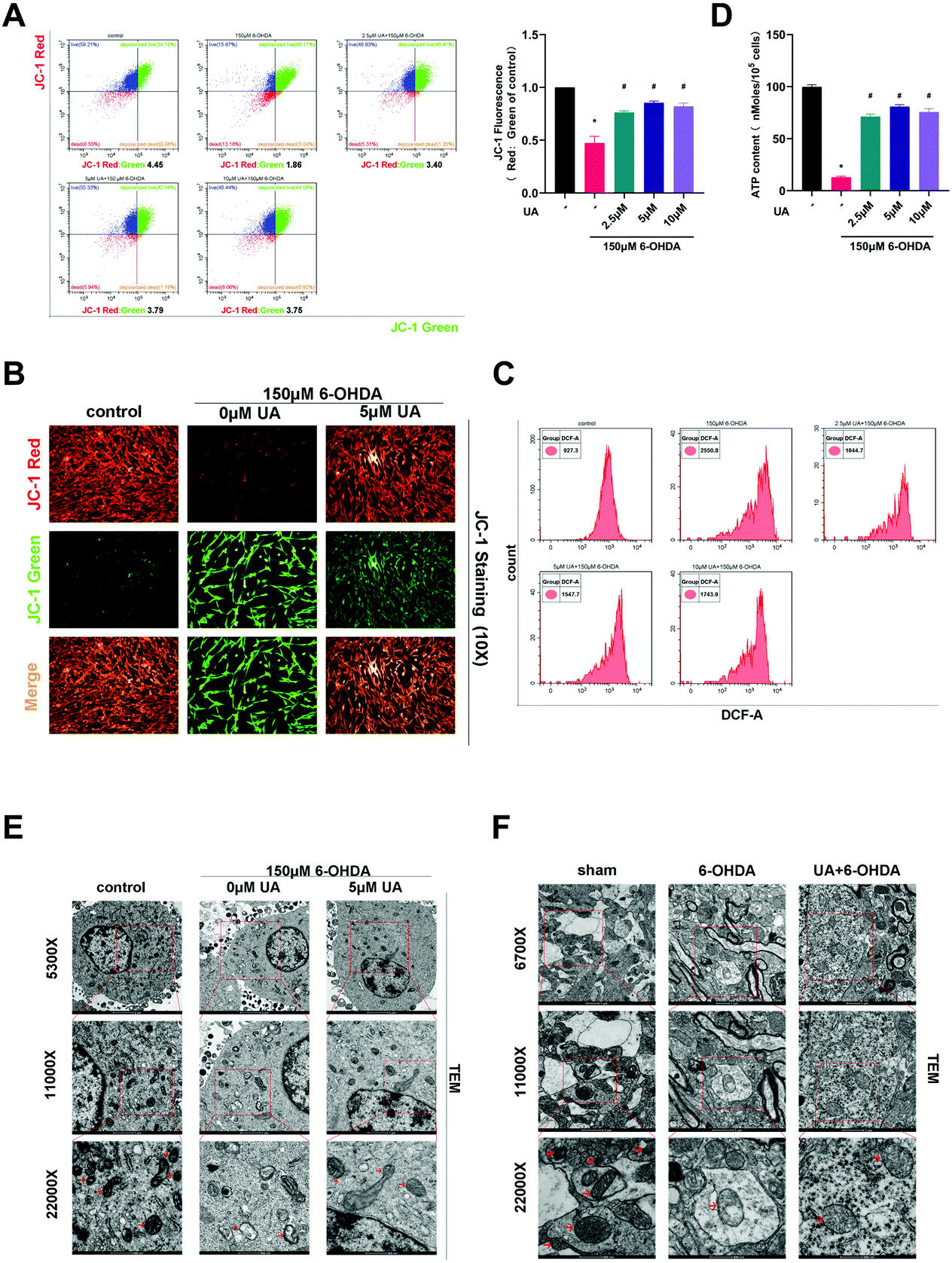 | ||
| Fig. 3 UA rescues 6-OHDA-induced mitochondria damage and dysfunction both in vitro and in vivo. (A and B) PC-12 cells were pretreated with UA for 2 h before exposure to 150 μM 6-OHDA for an additional 18 h. The effects of UA on mitochondrial membrane potential (MMP) was measured by JC-1 probe using flow cytometry analysis (A) and immunofluorescence microscopy (B). (C) Cells were pretreated with UA for 2 h before exposure to 150 μM 6-OHDA for an additional 18 h. Intracellular levels of ROS were measured with DCFH-DA staining and flow cytometry. (D) The effects of UA on OXPHOS was measured by the ATP assay. (E) Representative transmission electron microscopy (TEM) images showing mitochondrial morphology of PC-12 cells with different treatments. (F) Representative TEM images demonstrating mitochondrial morphology of dopaminergic neurons in mice SN with different treatment. Data are presented as mean ± SEM. One-way analysis of variance. *p < 0.05 vs. control group; #p < 0.05 vs. 6-OHDA group. | ||
3.4 UA promotes neuronal mitochondrial biogenesis
To determine if the increase in ATP production observed above were related to mitochondrial content changes, we measured mitochondrial DNA content (mtDNA) by real-time PCR. The results revealed that UA treatment attenuated 6-OHDA-induced decline of the ratio of mitochondrial to nuclear DNA (mtDNA:nDNA) in PC12 cells (Fig. 4A). In addition, treatment of PC12 cells with UA increased the protein expression of outer mitochondrial protein TOM20 and Tim23, two mitochondrial-specific proteins, indicating increased mitochondrial biogenesis (Fig. 4B). To further evaluate the effect of UA on mitochondrial biogenesis, we measured PGC-1α as well as its downstream target TFAM (transcription factor A, mitochondrial), all of which regulate mitochondrial biogenesis. As shown in Fig. 4C–F, both the expression of PGC-1α and TFAM were enhanced by UA treatment in vitro and in vivo. Collectively, these results strongly suggest that UA has a mitochondria protective role by maintaining mitochondrial biogenesis in 6-OHDA-injured PC12 cells and PD mice.
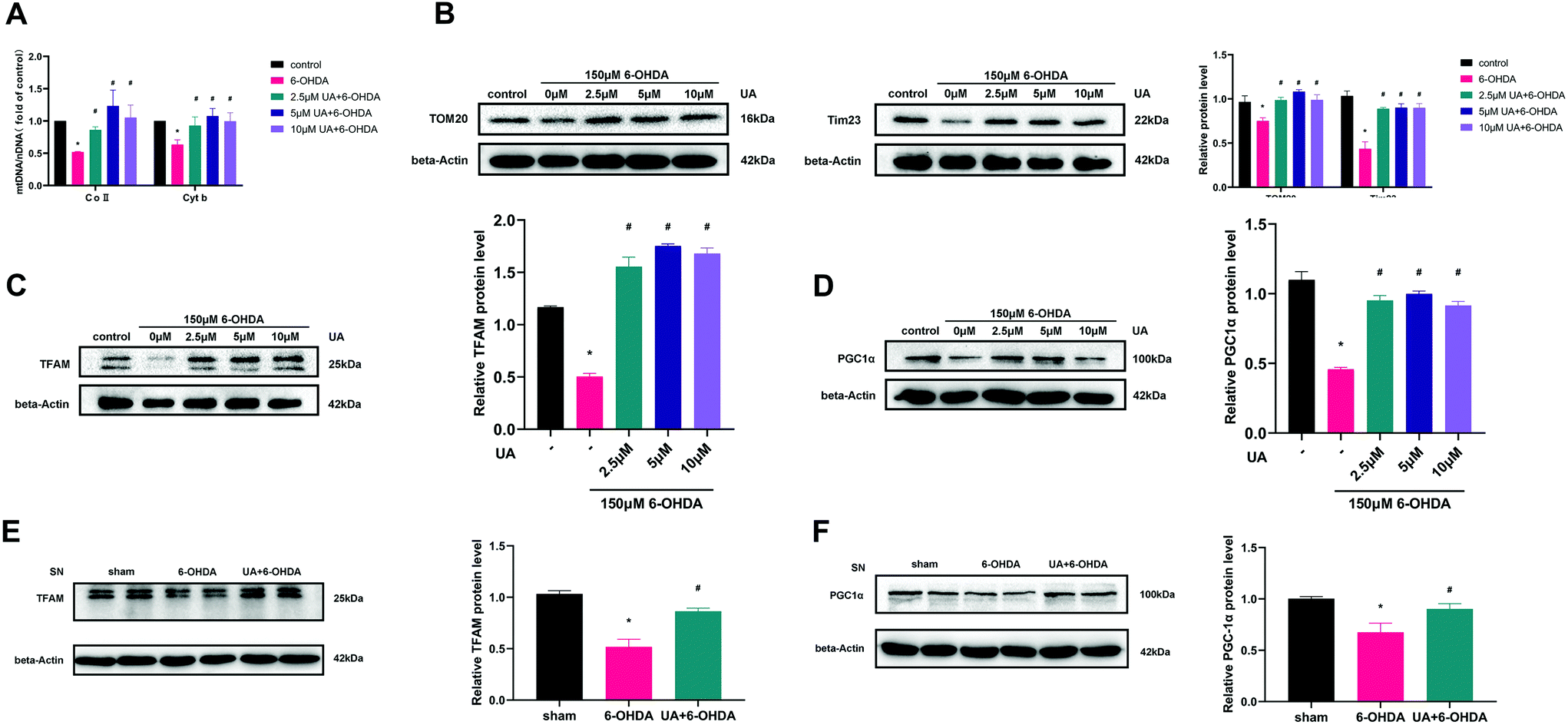 | ||
| Fig. 4 UA promotes mitochondrial biogenesis in PC-12 cells and PD mice. (A) Relative mitochondrial DNA content (mtDNA:nDNA) in PC-12 cells with different treatments. The results were normalized to the ratio of mtDNA:nDNA of control. (B) Representative western blot and summarized data showing the relative TOM20 and Tim23 expression in cells with different treatments. (C and D) Representative western blot and summarized data showing the relative TFAM and PGC1α expression in PC12 cells. Cells were pretreated with UA for 2 h before exposure to 150 μM 6-OHDA for an additional 18 h. (E and F) Representative western blot and summarized data showing the relative TFAM and PGC1α expression at 21 days after 6-OHDA injection in the mice SN. Data are presented as mean ± SEM. One-way analysis of variance. *p < 0.05 vs. sham group; #p < 0.05 vs. 6-OHDA group. | ||
3.5 UA induces the expression of SIRT1 both in vitro and in vivo
SIRT1 is a major regulator of mitochondrial biogenesis, we wondered if SIRT1 was involved in the protective mechanisms of UA on mitochondrial biogenesis. We first performed bioinformatic analysis to predict the potential drug target of UA on PD. As expected, the gene ontology (GO) functional classification of 429 PD-related genes indicated that the pathology of PD was closely associated with mitochondrial dysfunction (Fig. 5A). We then got 96 potential targets of UA using the Swiss Target Prediction database. A Venn diagram of the two targets sets was then used to obtain 8 intersection targets including SIRT1 (Fig. 5B). Together, these bioinformatic results indicate that SIRT1 may be involved in the pathogenesis of PD and the protective effect of UA.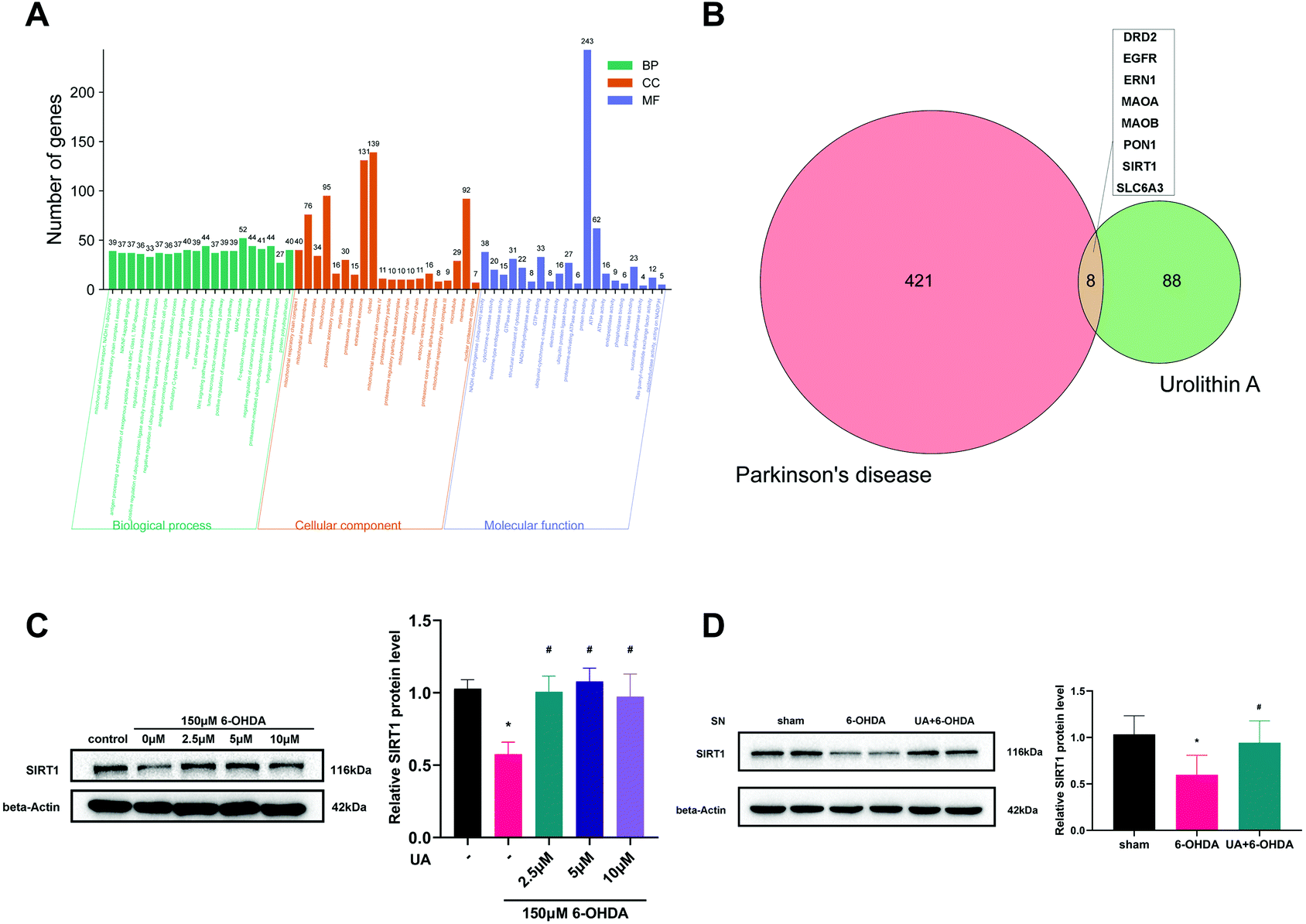 | ||
| Fig. 5 UA induces the expression of SIRT1 both in vitro and in vivo. (A) Gene ontology (GO) functional classification of PD-related genes. The histograms show the top 20 biological process (BP), cellular component (CC) and molecular function (MF) enrichment results. The y-axis represents the number of enriched genes and different colors represent different GO terms. (B) The total targets of Parkinson's disease (PD), UA and 8 intersection targets of UA action against PD were identified via a Venn diagram. (C) Representative western blot and summarized data showing the relative SIRT1 expression in PC12 cells. Cells were pretreated with UA for 2 h before exposure to 150 μM 6-OHDA for an additional 18 h. (D) Representative western blot and summarized data showing the relative SIRT1 expression at 21 days after 6-OHDA injection in the mice SN. Data are presented as mean ± SEM. One-way analysis of variance. *p < 0.05 vs. sham group; #p < 0.05 vs. 6-OHDA group. | ||
Next, we measured the protein levels of SIRT1 in response to UA both in vitro and in vivo. As shown in Fig. 5C, 6-OHDA treatment reduced the levels of SIRT1 in PC12 cells, the expression of SIRT1 was markedly restored by UA treatment. In vivo, a significant decrease of SIRT1 in SN from 6-OHDA PD mice was also observed as compared with sham group, while UA treatment rescued this decline (Fig. 5D).
3.6 Inhibition of SIRT1 counteracts the protective effects of UA in PC12 cells treated with 6-OHDA
SIRT1 is well established to induce mitochondrial biogenesis and function by its ability to regulate transcription involving PGC-1α. To address if UA increases mitochondrial biogenesis in a SIRT1-dependent manner, we treated PC12 cells with UA in the presence of EX-527, a selective chemical inhibitor of SIRT1 activity. As shown in Fig. 6A, EX-527 treatment abrogated the UA-induced increase in PGC-1α and TFAM expression. Functionally, the inhibition of SIRT1 by EX-527 also abrogated the UA-induced increase in mtDNA content in PC12 cells (Fig. 6B). Finally, we examined the contribution of SIRT1 to the neuroprotective action of UA and found that UA-mediated protective effects on mitochondrial function and cell fates of PC12 cells treated with 6-OHDA was abrogated in the presence of EX-527 (Fig. 6C–F). These findings indicate that SIRT1 plays an important role in contributing to the neuroprotective effects of UA.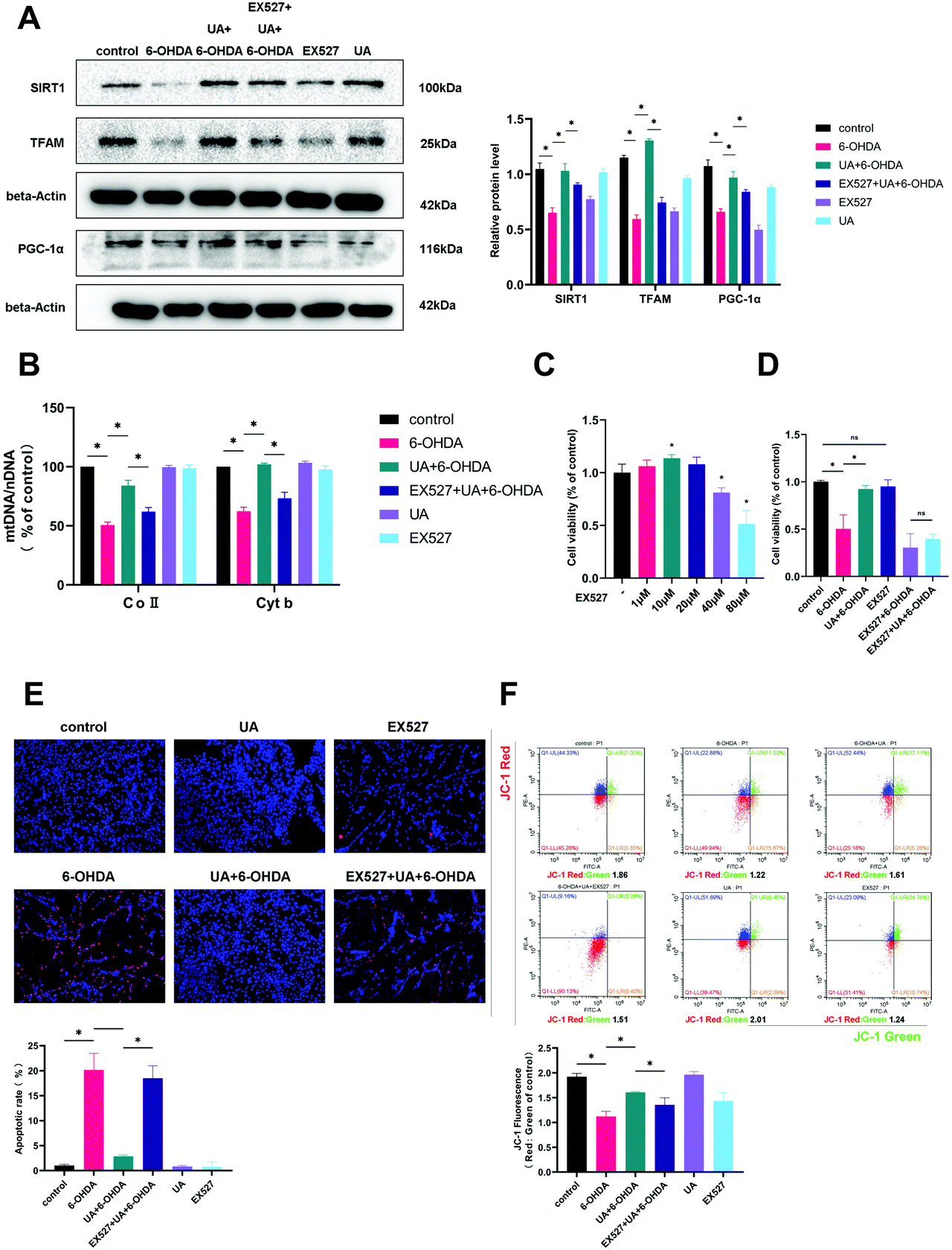 | ||
| Fig. 6 Inhibition of SIRT1 counteracted the protective effects of UA in PC12 cells treated with 6-OHDA. (A) Representative western blot and summarized data showing the relative TFAM, PGC1α and SIRT1 expression in cells with different treatment. (B) Relative mitochondrial DNA content (mtDNA:nDNA) in PC-12 cells with different treatments. The results were normalized to the ratio of mtDNA:nDNA of control. (C and D) The effects of UA on cell viability was measured by CCK-8 assay. (E) The effects of UA on apoptotic cells were determined by the TUNEL assay. (F) The effects of UA on mitochondrial membrane potential (MMP) was measured by JC-1. Data are presented as mean ± SEM. One-way analysis of variance. *p < 0.05 vs. control group; #p < 0.05 vs. 6-OHDA group. | ||
4. Discussion
The major finding of this study is the observation that UA significantly ameliorated 6-OHDA-induced dopaminergic neurodegeneration and mitochondrial dysfunction. Moreover, our results revealed that UA promotes mitochondrial biogenesis in PC12 cells. Further study demonstrated that UA promotes mitochondrial biogenesis and function in a SIRT1-dependent manner. Taken together, our study highlights the pivotal role of UA in protecting against 6-OHDA neurotoxicity by stimulating mitochondrial biogenesis (Fig. 7).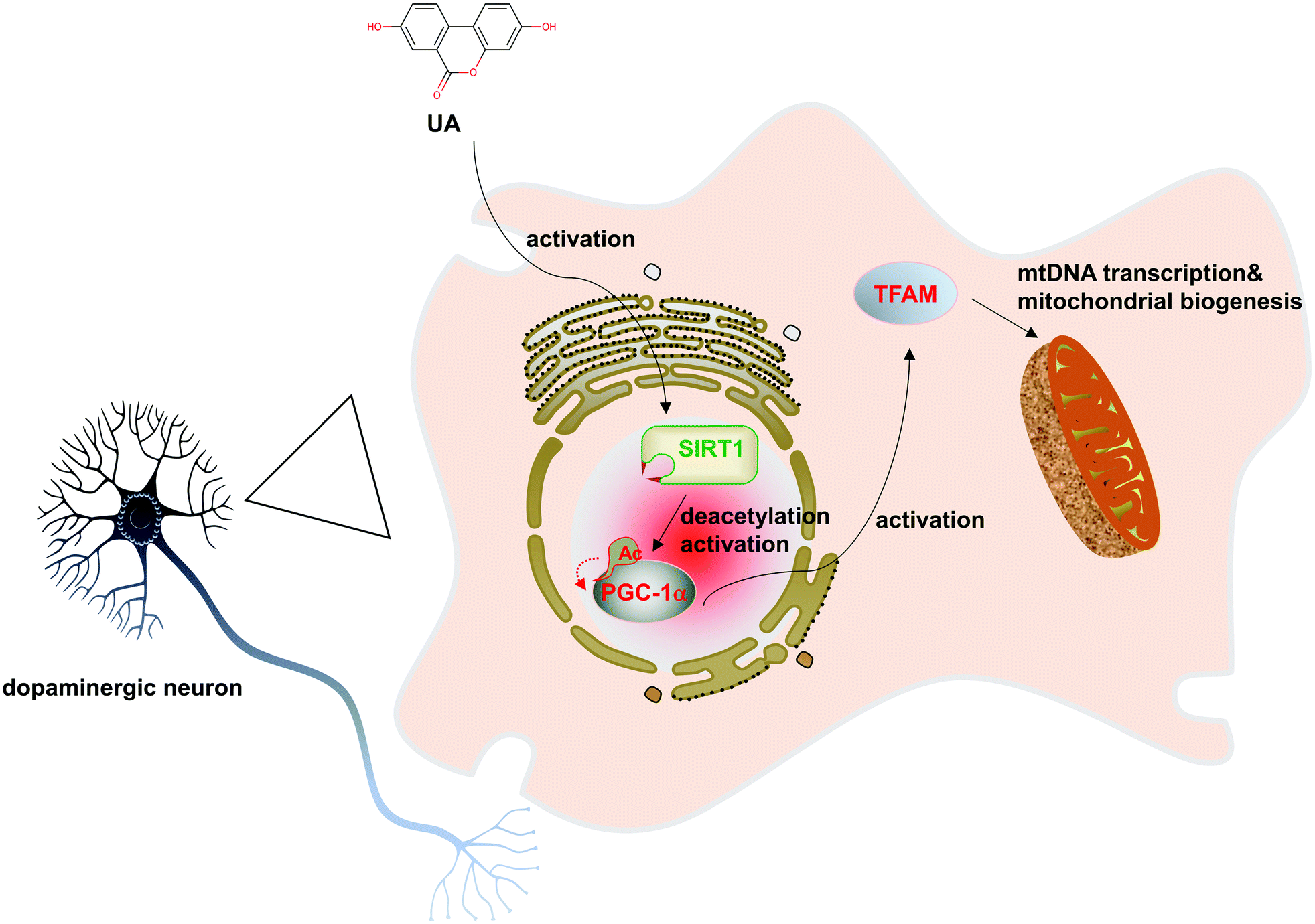 | ||
| Fig. 7 Schematic illustration of the putative mechanism for UA effects on mitochondrial biogenesis. UA increases SIRT1 expression, which is a key regulator of mitochondrial biogenesis. SIRT1 may then modulate PGC-1α expression, and in turn, enhance mitochondrial transcription factor TFAM expression, thus mediating effects of UA on mitochondrial biogenesis. This enhancement in mitochondrial biogenesis may contribute to the neuroprotective effects of UA from 6-OHDA toxicity. | ||
Neuroprotective effects of UA have been demonstrated in several neurological disorders including ischemic stroke, MS and Alzheimer's disease.10–13,18 Thus far, few studies have examined the effects of UA on dopamine neurons. Here, we show for the first time that UA regulates mitochondrial biogenesis in nigral dopaminergic neurons and exerts neuroprotective effects in experimental models of PD. The results of our study suggested that UA may represent a possible pharmacological intervention for treating patients in the early stages of PD. In support of our findings, works by Kujawska et al. showed that pomegranate juice could protect against PD in a rat model of parkinsonism induced by rotenone.19 They speculated that the neuroprotective effects of pomegranate juice were attributed to the pomegranate ellagitannin-derived metabolite UA. Our findings extended those of previous studies by directly measuring the protective role of UA in a 6-OHDA-induced mice model of PD. Furthermore, the direct intake of UA instead of ellagitannins may be a more effective way for the body to absorb and utilize UA.20
Our results also revealed a previously unidentified role of UA in promoting mitochondrial biogenesis. A recent study revealed that in human DA neurons lacking PARKIN, the mitochondrial deficits are primarily due to defects in mitochondrial biogenesis that are driven by the upregulation of PARIS (PARKIN interacting substrate) and the subsequent downregulation of PGC-1α, highlighting the importance of mitochondrial biogenesis in the pathogenesis of PD.21 Promotion of mitochondrial biogenesis could restore mitochondrial function and protect against mitochondrial insults in PD.22 Mitochondrial biogenesis is a complicated process that requires coordinated regulation of multiple proteins. As an important transcriptional regulator of mitochondrial function, PGC-1α promotes mitochondrial biogenesis by upregulating the expression levels of TFAM. Previous studies have found that knocking down the expression of PGC-1α would result in an obvious PD phenotypic feature in mice. Downregulated PGC-1α and other mitochondrial markers are also observed in clinical patients with PD.23 In the present study, the improvement of mitochondrial biogenesis by UA was verified by elevated mtDNA/nDNA and upregulated mitochondrial specific proteins. UA treatment also restored 6-OHDA-induced decrease of PGC-1α and TFAM levels both in vivo and in vitro. Furthermore, our results revealed that the beneficial effects of UA on mitochondrial biogenesis and function were dependent on the presence of SIRT1, pointing to an essential role for SIRT1 in the molecular pathway mediating the effects of UA.
While our current research focused on the role of UA on mitochondrial biogenesis, the beneficial effects of UA on PD may involve mechanisms that are yet to be elucidated. Further studies using multiple experimental models of PD are warranted to gain a better understanding of the mechanisms underlying the potential protective effects of UA in the setting of PD pathology. Another limitation of our study is that we used PC12 cells in our in vitro experiment, and additional studies using primary dopaminergic neurons are required to assess the effects of UA on neurodegeneration and mitochondrial function.
In summary, we provided new evidence that the food-derived compound UA greatly ameliorates the motor dysfunction and neuropathology in experimental models of PD, as well as improves mitochondrial dysfunction in a cell-based model of PD. We report for the first time that these beneficial effects may involve the promotion of mitochondrial biogenesis via a SIRT1-PGC-1α-mediated mechanism. The neuroprotective property of this compound found in the present study, together with the favorable safety profile and the favorable pharmacokinetic profile observed in clinical trials,24 indicates that UA may serve as a valuable and alternative therapeutic approach that can be transferred into clinical practice in PD in the near future.
Author contributions
Haiyan Lou planned experiments, interpreted data, and approved the manuscript for publication. Haiyan Lou wrote the article. Jia Liu and Jingjing Jiang performed most of the experiments and analyzed data. Jingru Qiu and Jing Zhuo participated in the animal experiment. Shuyan Yu and Deqing Sun participated in the experiment design and data interpretation. Baozhu Wang participated in the cell experiment. Liyan Wang performed the TEM experiment. All authors have read and approved the final article.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
This work was supported by grants from the Shandong Natural Science Foundation (no. ZR2021MH400 and ZR2020MH413).Notes and references
- A. B. Malpartida, M. Williamson, D. P. Narendra, R. Wade-Martins and B. J. Ryan, Mitochondrial Dysfunction and Mitophagy in Parkinson's Disease: From Mechanism to Therapy, Trends Biochem. Sci., 2021, 46, 329–343 CrossRef CAS PubMed.
- J. Prasuhn, R. L. Davis and K. R. Kumar, Targeting Mitochondrial Impairment in Parkinson's Disease: Challenges and Opportunities, Front. Cell Dev. Biol., 2021, 8, 615461 CrossRef PubMed.
- Z. Liang, A. Currais, D. Soriano-Castell, D. Schubert and P. Maher, Natural products targeting mitochondria: emerging therapeutics for age-associated neurological disorders, Pharmacol. Ther., 2021, 221, 107749 CrossRef CAS PubMed.
- L. D. Popov, Mitochondrial biogenesis: An update, J. Cell. Mol. Med., 2020, 24, 4892–4899 CrossRef CAS PubMed.
- M. Uittenbogaard and A. Chiaramello, Mitochondrial Biogenesis: A Therapeutic Target for Neurodevelopmental Disorders and Neurodegenerative Diseases, Curr. Pharm. Des., 2014, 20, 5574–5593 CrossRef CAS PubMed.
- M. Golpich, E. Amini, Z. Mohamed, R. Azman Ali, N. Mohamed Ibrahim and A. Ahmadiani, Mitochondrial Dysfunction and Biogenesis in Neurodegenerative diseases: Pathogenesis and Treatment, CNS Neurosci. Ther., 2017, 23, 5–22 CrossRef PubMed.
- J. C. Espín, M. Larrosa, M. T. García-Conesa and F. Tomás-Barberán, Biological Significance of Urolithins, the Gut Microbial Ellagic Acid-Derived Metabolites: The Evidence So Far, J. Evidence-Based Complementary Altern. Med., 2013, 2013, 1–15 CrossRef PubMed.
- F. A. Tomás-Barberán, A. González-Sarrías, R. García-Villalba, M. A. Núñez-Sánchez, M. V. Selma, M. T. García-Conesa and J. C. Espín, Urolithins, the rescue of “old” metabolites to understand a “new” concept: Metabotypes as a nexus among phenolic metabolism, microbiota dysbiosis, and host health status, Mol. Nutr. Food Res., 2017, 61, 1500901 CrossRef PubMed.
- H. Ishimoto, M. Shibata, Y. Myojin, H. Ito, Y. Sugimoto, A. Tai and T. Hatano, In vivo anti-inflammatory and antioxidant properties of ellagitannin metabolite urolithin A, Bioorg. Med. Chem. Lett., 2011, 21, 5901–5904 CrossRef CAS PubMed.
- X. Lin, X. Ye, Q. Li, Z. Gong, X. Cao, J. H. Li, S. T. Zhao, X. D. Sun, X. S. He and A. G. Xuan, Urolithin A Prevents Focal Cerebral Ischemic Injury via Attenuating Apoptosis and Neuroinflammation in Mice, Neuroscience, 2020, 448, 94–106 CrossRef CAS PubMed.
- E. F. Fang, Y. Hou, K. Palikaras, B. A. Adriaanse, J. S. Kerr, B. Yang, S. Lautrup, M. M. Hasan-Olive, D. Caponio, X. Dan, P. Rocktäschel, D. L. Croteau, M. Akbari, N. H. Greig, T. Fladby, H. Nilsen, M. Z. Cader, M. P. Mattson, N. Tavernarakis and V. A. Bohr, Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer's disease, Nat. Neurosci., 2019, 22, 401–412 CrossRef CAS PubMed.
- Z. Gong, J. Huang, B. Xu, Z. Ou, L. Zhang, X. Lin, X. Ye, X. Kong, D. Long, X. Sun, X. He, L. Xu, Q. Li and A. Xuan, Urolithin A attenuates memory impairment and neuroinflammation in APP/PS1 mice, J. Neuroinflammation, 2019, 16, 62 CrossRef PubMed.
- A. Ahsan, Y. R. Zheng, X. L. Wu, X. L. Wu, W. D. Tang, M. R. Liu, S. J. Ma, L. Jiang, W. W. Hu, X. N. Zhang and Z. Chen, Urolithin A–activated autophagy but not mitophagy protects against ischemic neuronal injury by inhibiting ER stress in vitro and in vivo, CNS Neurosci. Ther., 2019, 25, 976–986 CrossRef CAS PubMed.
- P. Luan, D. D'Amico, P. A. Andreux, P. P. Laurila, M. Wohlwend, H. Li, T. Imamura de Lima, N. Place, C. Rinsch, N. Zanou and J. Auwerx, Urolithin A improves muscle function by inducing mitophagy in muscular dystrophy, Sci. Transl. Med., 2021, 13, eabb0319 CrossRef CAS PubMed.
- D. Ryu, L. Mouchiroud, P. A. Andreux, E. Katsyuba, N. Moullan, A. A. Nicolet-Dit-Félix, E. G. Williams, P. Jha, G. Lo Sasso, D. Huzard, P. Aebischer, C. Sandi, C. Rinsch and J. Auwerx, Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents, Nat. Med., 2016, 22, 879–888 CrossRef CAS PubMed.
- Y. Qin, J. Qiu, P. Wang, J. Liu, Y. Zhao, F. Jiang and H. Lou, Impaired autophagy in microglia aggravates dopaminergic neurodegeneration by regulating NLRP3 inflammasome activation in experimental models of Parkinson's disease, Brain, Behav., Immun., 2021, 91, 324–338 CrossRef CAS PubMed.
- S. Yan, X. Wei, W. Jian, Y. Qin, J. Liu, S. Zhu, F. Jiang, H. Lou and B. Zhang, Pharmacological Inhibition of HDAC6 Attenuates NLRP3 Inflammatory Response and Protects Dopaminergic Neurons in Experimental Models of Parkinson's Disease, Front. Aging Neurosci., 2020, 12, 78 CrossRef CAS PubMed.
- P. Shen, X. Li, S. Deng, L. Zhao, Y. Y. Zhang, X. Deng, B. Han, J. Yu, Y. Li, Z. Z. Wang and Y. Zhang, Urolithin A ameliorates experimental autoimmune encephalomyelitis by targeting aryl hydrocarbon receptor, EBioMedicine, 2021, 64, 103227 CrossRef CAS PubMed.
- M. Kujawska, M. Jourdes, M. Kurpik, M. Szulc, H. Szaefer, P. Chmielarz, G. Kreiner, V. Krajka-Kuźniak, P.Ł. Mikołajczak, P. L. Teissedre and J. Jodynis-Liebert, Neuroprotective Effects of Pomegranate Juice against Parkinson's Disease and Presence of Ellagitannins-Derived Metabolite-Urolithin A-In the Brain, Int. J. Mol. Sci., 2020, 21, 202 CrossRef CAS PubMed.
- A. Singh, D. D. Amico, P. A. Andreux, G. Dunngalvin, T. Kern, W. Blanco-Bose, J. Auwerx, P. Aebischer and C. Rinsch, Direct supplementation with Urolithin A overcomes limitations of dietary exposure and gut microbiome variability in healthy adults to achieve consistent levels across the population, Eur. J. Clin. Nutr., 2021 DOI:10.1038/s41430-021-00950-1.
- M. Kumar, J. Acevedo-Cintron, A. Jhaldiyal, H. Wang, S. A. Andrabi, S. Eacker, S. S. Karuppagounder, S. Brahmachari, R. Chen, H. Kim, H. S. Ko, V. L. Dawson and T. M. Dawson, Defects in Mitochondrial Biogenesis Drive Mitochondrial Alterations in PARKIN-Deficient Human Dopamine Neurons, Stem Cell Rep., 2020, 15, 629–645 CrossRef CAS PubMed.
- K. Hasegawa, T. Yasuda, C. Shiraishi, K. Fujiwara, S. Przedborski, H. Mochizuki and K. Yoshikawa, Promotion of mitochondrial biogenesis by necdin protects neurons against mitochondrial insults, Nat. Commun., 2016, 7, 10943 CrossRef CAS PubMed.
- H. Jiang, S. Kang, S. Zhang, S. Karuppagounder, J. Xu, Y. K. Lee, B. G. Kang, Y. Lee, J. Zhang, O. Pletnikova, J. C. Troncoso, S. Pirooznia, S. A. Andrabi, V. L. Dawson and T. M. Dawson, Adult Conditional Knockout of PGC-1α Leads to Loss of Dopamine Neurons, eNeuro, 2016, 3, 116–183 CrossRef PubMed.
- P. A. Andreux, W. Blanco-Bose, D. Ryu, F. Burdet, M. Ibberson, P. Aebischer, J. Auwerx, A. Singh and C. Rinsch, The mitophagy activator urolithin A is safe and induces a molecular signature of improved mitochondrial and cellular health in humans, Nat. Metab., 2019, 1, 595–603 CrossRef CAS PubMed.
Footnote |
| † These two authors contribute equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |