
The apple dihydrochalcone phloretin suppresses growth and improves chemosensitivity of breast cancer cells via inhibition of cytoprotective autophagy†
Ming
Chen
ab,
Vemana
Gowd
 ab,
Mingfu
Wang
ac,
Feng
Chen
ade and
Ka-Wing
Cheng
*ad
ab,
Mingfu
Wang
ac,
Feng
Chen
ade and
Ka-Wing
Cheng
*ad
aShenzhen Key Laboratory of Marine Microbiome Engineering, Institute for Advanced Study, Shenzhen University, Shenzhen 518060, China. E-mail: kwcheng@szu.edu.cn
bKey Laboratory of Optoelectronic Devices and Systems of Ministry of Education and Guangdong Province, College of Optoelectronic Engineering, Shenzhen University, Shenzhen 518060, China
cSchool of Biological Sciences, The University of Hong Kong, Pokfulam Road, Hong Kong 999077, P.R. China
dInstitute for Innovative Development of Food Industry, Shenzhen University, Shenzhen 518060, China
eInstitute for Food and Bioresource Engineering, College of Engineering, Peking University, Beijing 100871, China
First published on 26th November 2020
Abstract
The inhibitory effect and mechanism of the apple dihydrochalcone, phloretin, on breast cancer cell growth were evaluated in in vitro conditions simulating complete nutrition and glucose-restriction, respectively. In two breast cancer cell lines with different histological backgrounds, phloretin consistently exhibited much stronger activity against cell growth in glucose-limiting than in full media. RNA-seq analysis showed that key autophagy-related genes were downregulated upon phloretin treatment in both estrogen-receptor-positive MCF7 and triple-negative MDA-MB-231 cells. Immunoblotting verified significantly decreased expression of LC3B-II by phloretin in low-glucose and glucose-free media, but not in full medium. Together with the use of two pharmacological autophagy inhibitors, chloroquine and 3-methyladenine, and confocal microscopy of breast cancer cell lines transfected with GFP-LC3B, phloretin demonstrated a strong capability to suppress autophagic flux, which was likely mediated through downregulation of mTOR/ULK1 signaling, whereas the expression of canonical autophagy regulators ATG5 and ATG7 was not significantly affected. Phloretin also reversed tamoxifen- and doxorubicin-induced cytoprotective autophagy in the breast cancer cell lines, and this was manifested in its synergistic growth inhibitory effect with these chemotherapeutic agents. Furthermore, it was able to restore or enhance the chemosensitivity of a tamoxifen-resistant cell line. Taken together, our study has, for the first time, revealed that phloretin could effectively suppress glucose-starvation- and chemotherapeutic-induced cytoprotective autophagy in breast cancer cell lines likely through downregulation of mTOR/ULK1 signaling.
1. Introduction
Breast cancer remains a worldwide public health concern. It is the most prevalent cancer in women and it is only second to lung cancer overall.1 According to the World Health Organization, 521![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 women died in 2012 worldwide due to breast cancer with nearly 1.7 million new cases diagnosed each year.2 Based on histological features, breast cancer can be classified into three subtypes, including human epidermal growth factor receptor-2 (HER2+) overexpressing, hormone (estrogen) receptor positive (ERα+) and triple-negative breast cancer (TNBC).3 Among them, ERα+ patients account for at least 70% of all breast cancer cases. TNBC is the most aggressive form and is often refractory to therapy. Endocrine therapies usually referred to as ERα selective estrogen-receptor modulators, including tamoxifen and fulvestrant, and aromatase inhibitors (AIs) that block the ER signaling are extensively used for ERα+ patients.2 Although most of the patients initially respond well to chemotherapeutic therapies, many of them eventually suffer from relapse and metastasis due to occurrence of drug resistance.4
000 women died in 2012 worldwide due to breast cancer with nearly 1.7 million new cases diagnosed each year.2 Based on histological features, breast cancer can be classified into three subtypes, including human epidermal growth factor receptor-2 (HER2+) overexpressing, hormone (estrogen) receptor positive (ERα+) and triple-negative breast cancer (TNBC).3 Among them, ERα+ patients account for at least 70% of all breast cancer cases. TNBC is the most aggressive form and is often refractory to therapy. Endocrine therapies usually referred to as ERα selective estrogen-receptor modulators, including tamoxifen and fulvestrant, and aromatase inhibitors (AIs) that block the ER signaling are extensively used for ERα+ patients.2 Although most of the patients initially respond well to chemotherapeutic therapies, many of them eventually suffer from relapse and metastasis due to occurrence of drug resistance.4
Macroautophagy, herein referred to as autophagy, is an evolutionarily conserved process which involves progressive sequestration of cytoplasmic materials by autophagosomes that ultimately fuse with lysosomes for cargo recycling in order to maintain cellular homeostasis.5 Autophagy has a complex role in cancer. Initially, it was thought to be tumor-suppressive, as allelic loss of an essential autophagy gene Beclin1 resulted in hepatocellular carcinoma.6 In contrast to its role in suppressing tumor initiation, accumulating evidence implicates autophagy as a tumor promoter in late stages of many cancer types, especially under stress conditions such as nutrient limitation, hypoxia, oxidative stress and chemotherapy.7–9 Meanwhile, autophagy has also been shown to play a role in the development of chemotherapeutic resistance in ERα, HER2+ and TNBC, and targeting autophagy has thus been considered to be a promising approach to restore therapeutic sensitivity.10–12
Epidemiological studies have amply demonstrated that alteration in dietary patterns, obesity, lack of physical activity and hormonal factors are intimately associated with the increased breast cancer morbidity during the past decades.13 Diets rich in fruits and vegetables have been frequently correlated with decreased risk of many chronic diseases including breast cancer.14 Hence, phytochemicals derived from fruits and vegetables have attracted enormous attention from investigators dedicated to the development of efficacious anticancer strategies.15,16 Phloretin (PH), a major bioactive polyphenol in apples, has been widely studied for its beneficial effects such as anti-inflammation, anti-oxidation, and anti-carcinogenesis.17 In ERα+ MCF7 cells, apple extract was found to inhibit the activation of NF-κB, which plays a key role in mediating inflammation and cell proliferation.18 It was also reported to inhibit migration and proliferation of TNBC cells (MDA-MB-231) via inhibition of type 2 glucose transporter, which implies its role in regulating signaling pathways associated with cellular nutrient/energy status.19 However, whether PH plays a role in breast cancer autophagy is yet to be revealed.
In the present study, the effect of PH on the growth of two breast cancer cell lines (MCF7, MDA-MB-231) was evaluated in full versus glucose-limiting media. The underlying mechanism was also examined focusing on its role in the regulation of autophagic flux in these cell lines. Finally, the potential of PH to help overcome resistance to conventional chemotherapeutics (4OH-tamoxifen (4OH-Tam) and doxorubicin (Dox)) through the modulation of autophagy in breast cancer cells was also explored.
2. Materials & methods
2.1. Antibodies and reagents
Chloroquine (CQ), 3-methyladenine (3-MA), crystal violet, N-acetyl-L-cysteine (NAC) and 4OH-Tam were purchased from Sigma-Aldrich (Saint Louis, MO, USA). Dox and PH were from Selleck (Shanghai, China). Antibodies against LC3B, ATG5, ATG7, ULK1, p-MTOR (ser2448), MTOR, p-AKT (Ser473), AKT, p-AMPK (Thr172) and AMPK were from Cell Signaling Technology (Beverly, MA, USA). SQSTM1/p62 antibody was from Abcam (Cambridge, MA, USA). β-Actin was obtained from Sigma-Aldrich (Saint Louis, MO, USA). Secondary anti-rabbit and anti-mouse antibodies were from Santa Cruz Biotechnology (Shanghai, China). DCFH-DA was from Solarbio (Beijing, China). BCA protein assay kit and enhanced chemiluminescence substrate (ECL) were obtained from Pierce (Rockford, IL, USA). Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), antibiotics, PureLink HiPure Plasmid FP (Filter and Precipitator) Maxiprep Kit and Lipofectamine 2000 were from Life Technologies (Shanghai, China). ATG7 siRNA and ULK1 siRNA were obtained from Ribobio. RIPA lysis buffer was from Beyotime (Shanghai, China). CellTiter-Glo Luminescent cell viability assay kit was purchased from Promega Corporation (Madison, WI, USA). Cell Counting Kit (CCK)-8 was purchased from Dojindo (Kumamoto, Japan). PE-Annexin V plus 7-AAD cell apoptosis kit was obtained from BD Pharmingen (San Jose, CA, USA).2.2. Cell culture
Human breast cancer cell lines MCF7, MDA-MB-231 and a normal breast cell line MCF 10A were obtained from the Cell Bank of Type Culture Collection of Chinese Academy of Sciences and were authenticated using short tandem repeat multi-amplification and tested to be mycoplasma negative. Breast cancer cells were maintained in DMEM supplemented with 10% FBS, 100 units per ml penicillin and 100 μg ml−1 streptomycin at 37 °C in a humidified incubator with 5% CO2. MCF 10A cells were maintained in DMEM/F12 supplemented with 10 μg ml−1 insulin, 20 ng ml−1 EGF and 0.5 μg ml−1 hydrocortisone.2.3. siRNA transfection
MCF7 4OH-Tam resistant cells were cultured in 96-well plates and transfected with ATG7 siRNA, ULK1 siRNA (100 nM) or their scrambled control using lipofectamine 2000 for 4–6 h before drug treatment.2.4. Establishment of 4OH-Tam-resistant cells
4OH-Tam-resistant MCF7 cells were established as previously described with slight modifications.2,20 Briefly, MCF7 cells were challenged with escalating doses of 4OH-Tam in phenol red-free DMEM supplemented with 10% charcoal-stripped FBS (Life Technologies) as follows: 1 μM for 2 weeks, 2 μM for 2 weeks and then 4 μM until no appreciable growth inhibition was observed.2.5. RNA processing
Breast cancer cells were treated with PH in 1 mM glucose media for 24 h. A total amount of 1 μg RNA per sample was used as input material for the RNA sample preparation. Sequencing libraries were generated using NEBNext® UltraTM RNA Library Prep Kit for Illumina® (NEB, USA) following manufacturer’ recommendations. Sequencing was performed on an Illumina Novaseq platform and 150 bp paired-end reads were generated. Gene expression indicated by RPKM (Reads Per Kilobases per Million reads) was calculated using feature Counts v1.5.0-p3. Differentially expressed genes (DEG) were tested using the DESeq2 R package (1.16.1).2.6. Intracellular reactive oxygen species (ROS) measurement
For measurement of intracellular ROS in cultured breast cancer cells, cells were detached using 0.25% trypsin with 1 mM EDTA, and stained with DCFH-DA. Data were acquired by flow cytometry (CytoFLEX, Becman Coulter, CA, USA) and analyzed using FlowJo software (Ashland, OR, USA).2.7. Cell viability assay
For cell viability assay, following drug treatment, breast cancer cells were fixed with 4% paraformaldehyde and stained with 0.1% crystal violet for 30 min at room temperature. Excess dye was washed away with water and the remaining dye was extracted with 10% acetic acid. Sample absorbance was measured at 590 nm using a microplate reader (Tecan). Additionally, breast cancer cells were seeded into 96-well plates at a density of 5 × 103 cells per well. Next day, the cells were incubated with PH alone, or in combination with an autophagy inhibitor, 4OH-Tam or Dox in full media or glucose-restricted media for designated time periods. CCK-8 reagent was added according to the manufacturer's instructions, and absorbance was measured using a Tecan microplate reader.2.8. Clonogenic growth assay
Breast cancer cells (1000 per well) were seeded in 12-well plates. After overnight incubation, the cells were cultured in full media, 1 mM glucose media or 0 mM glucose media and treated with different concentrations of PH for 5 days. Then, the media were changed into full media for another 14 days. Colonies were washed with PBS, fixed and stained with a 0.5% crystal violet solution in 20% methanol.2.9. Analysis of apoptotic cell death
Breast cancer cells were seeded in 6-well plates at a density of 5 × 105 cells per well. The cells were incubated overnight and treated with PH for 24 h. Following treatment, the cells were trypsinized and stained with PE-Annexin V and 7-AAD according to the manufacturer's instructions. Cell apoptosis was detected using flow cytometry (Becman Coulter).2.10. Western blot
Breast cancer cells were lysed with RIPA buffer supplemented with protease and phosphatase inhibitors and protein concentrations were determined using a BCA protein assay kit. The samples were resolved by SDS-PAGE and then transferred to PVDF membranes. The blots were blocked, washed and incubated overnight with primary antibodies at 4 °C and then with secondary antibodies for 1 h at room temperature. After incubation with ECL, the blots were visualized in an automated chemiluminescent detection system (Bio-Rad). The intensity of bands was quantified using ImageJ.2.11. Immunofluorescence and confocal imaging
Plasmids GFP-LC3B (Addgene, 11546; deposited by Karla Kirkegaard), mRFP-GFP-LC3B (Addgene, 21074; deposited by Tamotsu Yoshimori) and pBabe-mCherry-GFP-LC3B (Addgene, 22418; deposited by Jayanta Debnath) were amplified in DH5α and extracted using PureLink HiPure Plasmid FP (Filter and Precipitator) Maxiprep Kit according the manufacturer's recommendations. Purified plasmids were dissolved in TE buffer and quantified on a Nanodrop (Thermo Fisher). For transfection experiments, breast cancer cells were plated and transfected with 2 μg plasmids before PH treatment. Cells were then fixed with 4% paraformaldehyde in phosphate-buffered saline and observed by confocal microscopy (Leica SP5). Cells with GFP-LC3B+ puncta (green), mRFP+ (red) or GFP+ mRFP+ (yellow) puncta were analyzed using ImageJ.2.12. Animal study
MDA-MB-231 cells (2 × 106 per mouse) were mixed with matrigel (1:1, v/v) (Corning) and inoculated into the right flank of 6-week-old female Balb/c nude mice. After tumor size reached ∼50 mm3, the mice were divided into four groups (n = 5–7 mice per group): vehicle (0.5% sodium carboxymethyl cellulose (CMC-Na), PH (100 mg kg−1 d−1 in 0.5% CMC-Na, oral gavage), Dox (5 mg kg−1 in 0.9% saline, 2× per wk, intraperitoneal injection), and PH + Dox. Tumor size was measured using a caliper and calculated according to the following formula: tumor volume (mm3) = L × W2/2, where L is the length and W is the width. The treatment period was 21 days and at the end of the experiment, the animals were sacrificed by intraperitoneal injection of chloral hydrate. The protocol of animal care and use was approved by the Animal Care and Use Committee of Shen Zhen University.
2.13. Statistical analysis
The data are presented as mean ± standard error of the means of at least three independent experiments. Statistical analyses were performed using the Student's t-test for comparisons between 2 groups or one-way analysis of variance (ANOVA) for more than 3 groups. The results were considered significantly different at p < 0.05.3. Results
3.1. PH selectively represses breast cancer cell growth in glucose-limiting media
To investigate the effect of PH on breast cancer cell growth, ERα+ MCF7 and TNBC MDA-MB-231 cell lines were cultured in either full (25 mM glucose), 1 mM, or 0 mM glucose media, and treated with increasing concentrations of PH. As shown in Fig. 1A–C, when cultured in full media, PH treatment at all tested concentrations had minimal impact on the growth of both cell lines. However, in 1 mM glucose and glucose-free media, PH effectively suppressed their growth and the effect was dramatically enhanced at high concentrations (≥200 μM). The growth inhibitory effect of PH in glucose-limiting conditions was also evident in the clonogenic assay at both medium and high doses (100 and 200 μM) (Fig. 1D). Additionally, even at high concentrations, PH had minimal impact on the growth of a normal breast cell line (MCF 10A) under full or 1 mM glucose conditions (Fig. 1E and F). These data suggest that PH possesses highly selective growth inhibitory effect towards breast cancer cells under glucose-limiting conditions.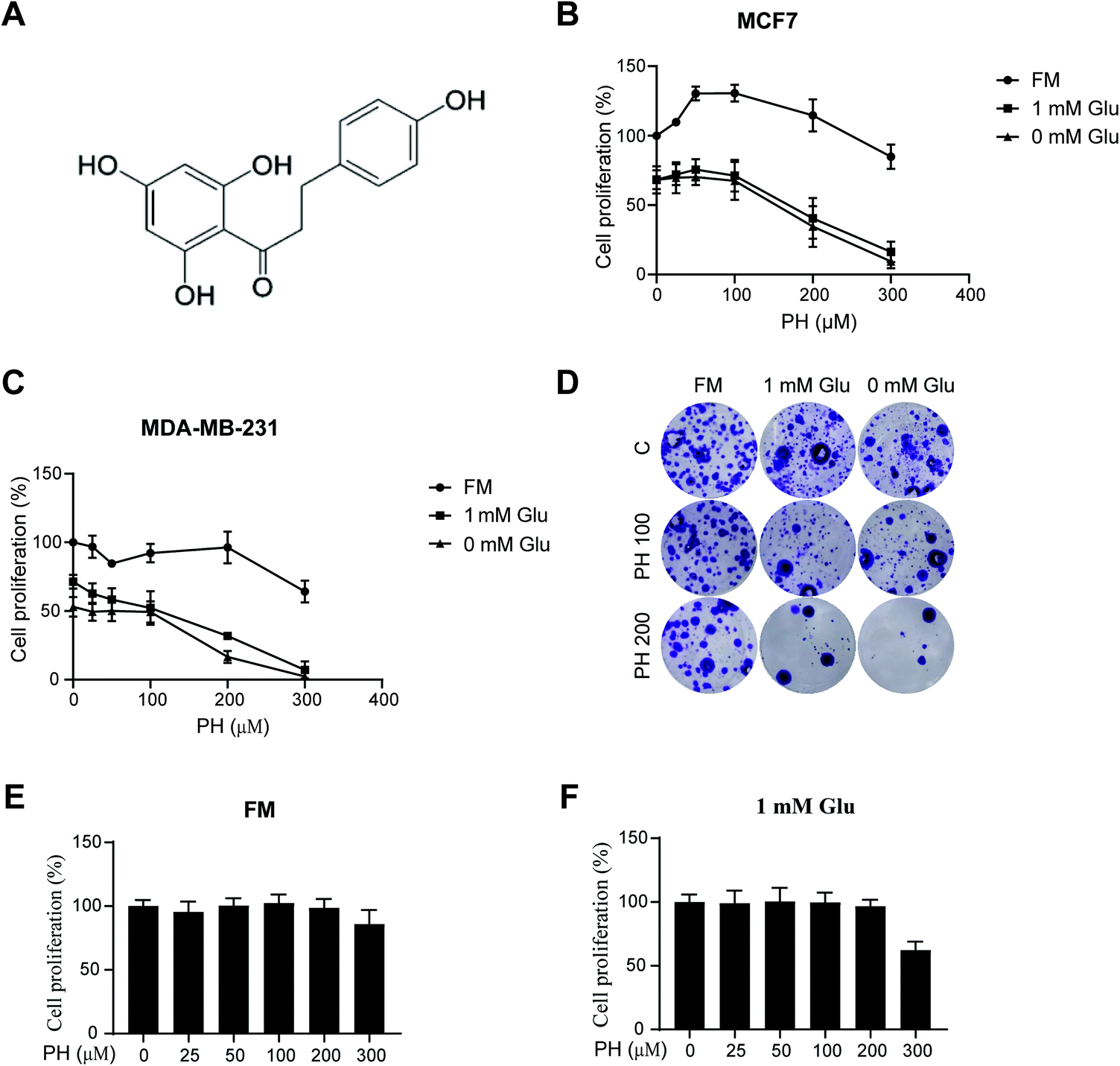 | ||
| Fig. 1 Effect of PH on breast cancer cell growth. (A) Chemical structure of PH. MCF7 (B), MDA-MB-231 cells (C) were treated with increasing concentrations of PH (0, 10, 50, 100, 200, 300 μM) in full, 1 mM glucose and 0 mM glucose media, respectively. Cell viability assay was performed 48 h post treatment. (D) Clonogenic assay of MDA-MB-231 cells treated with different concentrations of PH in full, 1 mM glucose and 0 mM glucose media, respectively. (E and F) MCF 10A cells were treated with increasing concentrations of PH under full (E) or 1 mM glucose media (F). Cell proliferation was measured using the CCK8 assay. FM, full media; Glu, glucose. | ||
3.2. PH inhibits autophagosome formation in breast cancer cells
Autophagy is a critical process that generally promotes cell survival, especially during periods of metabolic stress induced by nutrient deprivation.8 Having observed PH's selective activity against breast cancer cell growth under glucose-limiting conditions, we next examined whether autophagy played a role in this anticancer effect. RNA-Seq revealed that most of the core autophagy-related genes were downregulated in MCF7 and MDA-MB-231 cells treated with PH under 1 mM glucose condition (Fig. 2). western blotting of the relative expression of an autophagosome formation hallmark LC3B-II corroborated the RNA-Seq finding. As shown in Fig. 3A, in full media, PH exhibited minimal effect on the conversion of LC3B-I to LC3B-II, whereas in glucose-limiting media, PH markedly inhibited it. Similar results were observed in MDA-MB-231 cells (Fig. 3B). Further time-course and dose–response analysis also supported the capability of PH to downregulate LC3B-II expression in breast cancer cells under glucose-limiting condition (Fig. 3C and D). To strengthen this observation, MDA-MB-231 cells were transfected with exogenous GFP-LC3B plasmids and treated with PH in glucose-limiting media. A significant reduction in GFP-LC3B-II puncta was observed in PH-treated cells compared to vehicle control (2-fold reduction in 1 mM glucose, 3-fold reduction in 0 mM glucose media) (Fig. 3E). These data collectively suggest that PH suppresses autophagosome formation in breast cancer cells under glucose starvation conditions. | ||
| Fig. 2 mRNA expression profile analysis of autophagy-related genes in MCF7 (A) and MDA-MB-231 (B) breast cancer cells treated with PH vs. vehicle in 1 mM glucose-limiting media. | ||
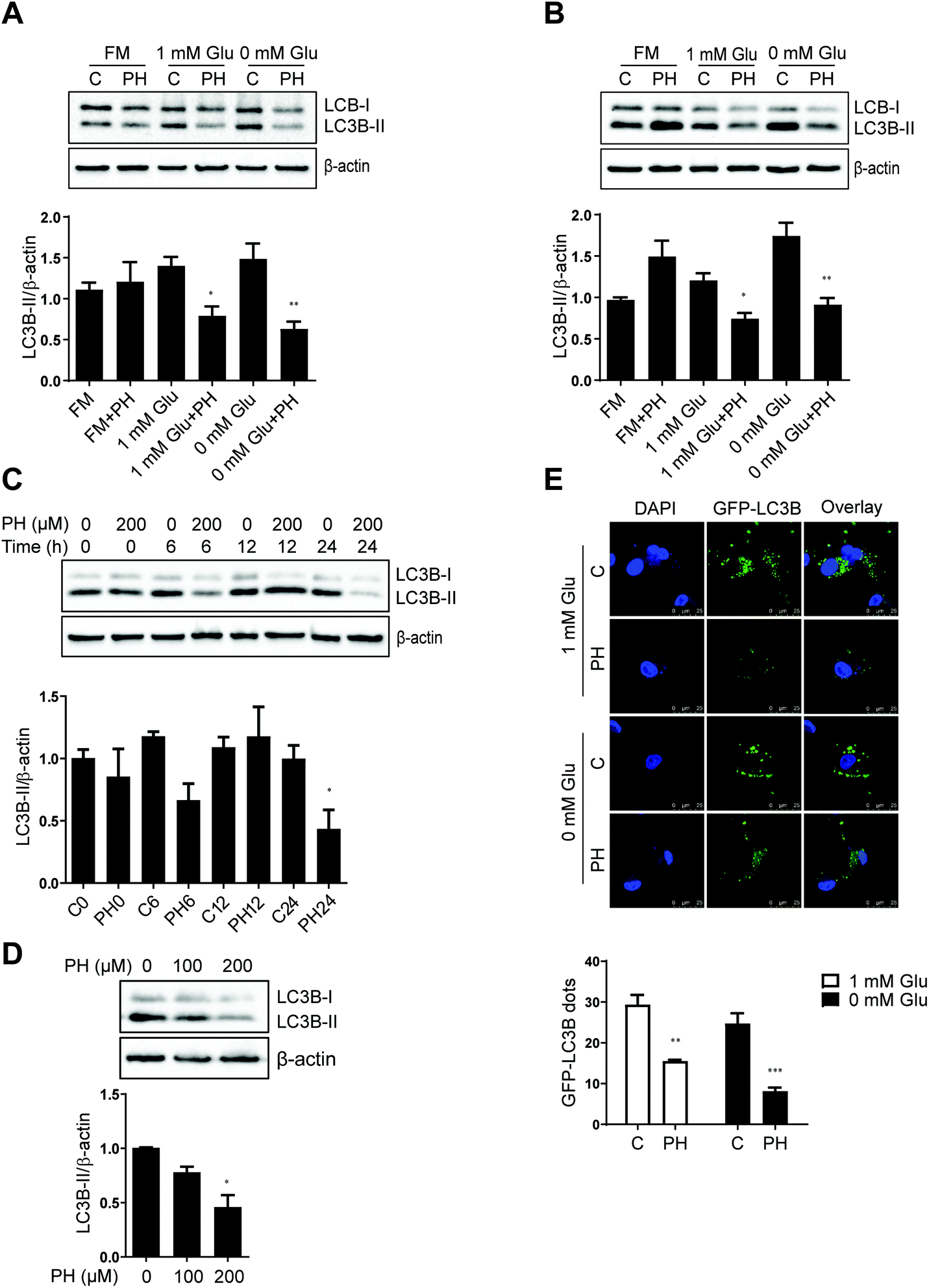 | ||
| Fig. 3 PH inhibits autophagosome accumulation in MCF7 and MDA-MB-231 cells under glucose-limiting conditions. Immunoblots and densitometry data of MCF7 (A) and MDA-MB-231 (B) cells cultured in full, 1 mM Glu and 0 mM Glu media, respectively and treated with PH for 24 h. *p < 0.05 PH vs. control in 1 mM Glu media; **p < 0.01 PH vs. control in 0 mM Glu media. Representative blots and densitometry analysis of LC3B expression in MDA-MB-231 cells challenged with PH for indicated time intervals (C) and doses (D), respectively. *p < 0.05 PH vs. control. (E) Representative images of GFP-LC3B puncta (green) and DAPI (blue) in MDA-MB-231 transfected with the GFP-LC3B plasmid for 24 h and then treated with PH or vehicle in glucose-limiting media for another 24 h. Scale bar: 25 μm. Histogram shows the relative numbers of GFP-LC3B puncta determined using ImageJ software. *p < 0.05, ***p < 0.001 vs. control. | ||
We next evaluated the effect of PH on SQSTM1/p62 expression, an autophagy adaptor that negatively correlates with autophagy activity.21 Surprisingly, a marked decrease in p62 expression was observed in both MCF7 and MDA-MB-231 cells upon PH treatment in glucose-limiting media (Fig. S1A and B†). To resolve this discrepancy, cellular ROS was determined considering the fact that p62 is also known to play a critical role in promoting nuclear translocation and thus activation of the antioxidant defense transcription factor Nrf2.22 In accordance with the downregulation of p62, PH treatment caused a 1.5- to 1.9-fold increase in ROS levels in MCF7 and MDA-MB-231 cells in glucose-limiting media (Fig. S1C and D†). In addition, PH-mediated cell growth inhibition was partially reversed by NAC, a thiol antioxidant and ROS scavenger (Fig. S1E and F†), indicating a role of oxidative stress induction in PH's activity against breast cancer cell growth.
3.3. PH treatment inhibits autophagic flux in breast cancer cells
Having established that PH inhibits autophagosome formation as evidenced by the significantly reduced conversion of LC3B-I to LC3B-II in breast cancer cell lines, we then sought to determine if it represses autophagic flux. To this end, CQ, a lysosomotropic agent that prevents acidification of lysosomes and thus degradation of autophagosome was used. Under 1 mM glucose and glucose-free conditions, CQ pretreatment increased the level of LC3B-II by nearly 1-fold relative to vehicle control in both MCF7 and MDA-MB-231 cells. This accumulation of LC3B-II was significantly attenuated by co-treatment with PH (Fig. 4A–D). These findings were further substantiated by transfection of the two cell lines with an autophagy dual fluorescent reporter mCherry-GFP-LC3B. A robust reduction of mCherry red puncta was observed in both cell lines following treatment with PH in 1 mM glucose media (Fig. 4E). We also transfected both cell lines with another commonly used dual fluorescent reporter plasmid mRFP-GFP-LC3B (Tandem fluorescent-tagged LC3B, tfLC3B) and assessed the number of red versus yellow puncta. Yellow signal was generated by overlaying GFP fluorescence and RFP fluorescence, whereas the “red-only” puncta were generated from the RFP fluorescence due to the quenching of GFP fluorescence. As depicted in Fig. 4F, PH treatment led to a significant decrease (42%–70%) in both yellow (autophagosomes) and red (autolysosomes) puncta in MCF7 cells under 1 mM glucose condition, while under glucose-free condition, it caused a more dramatic decrease (70%) in the number of red puncta. Similar results were also observed for MDA-MB-231 cells (Fig. 4G). Taken together, the above results demonstrate that PH strongly inhibits autophagic flux in breast cancer cells by impairing the formation of autophagosomes and their fusion with lysosomes to form autolysosomes under glucose-limiting conditions.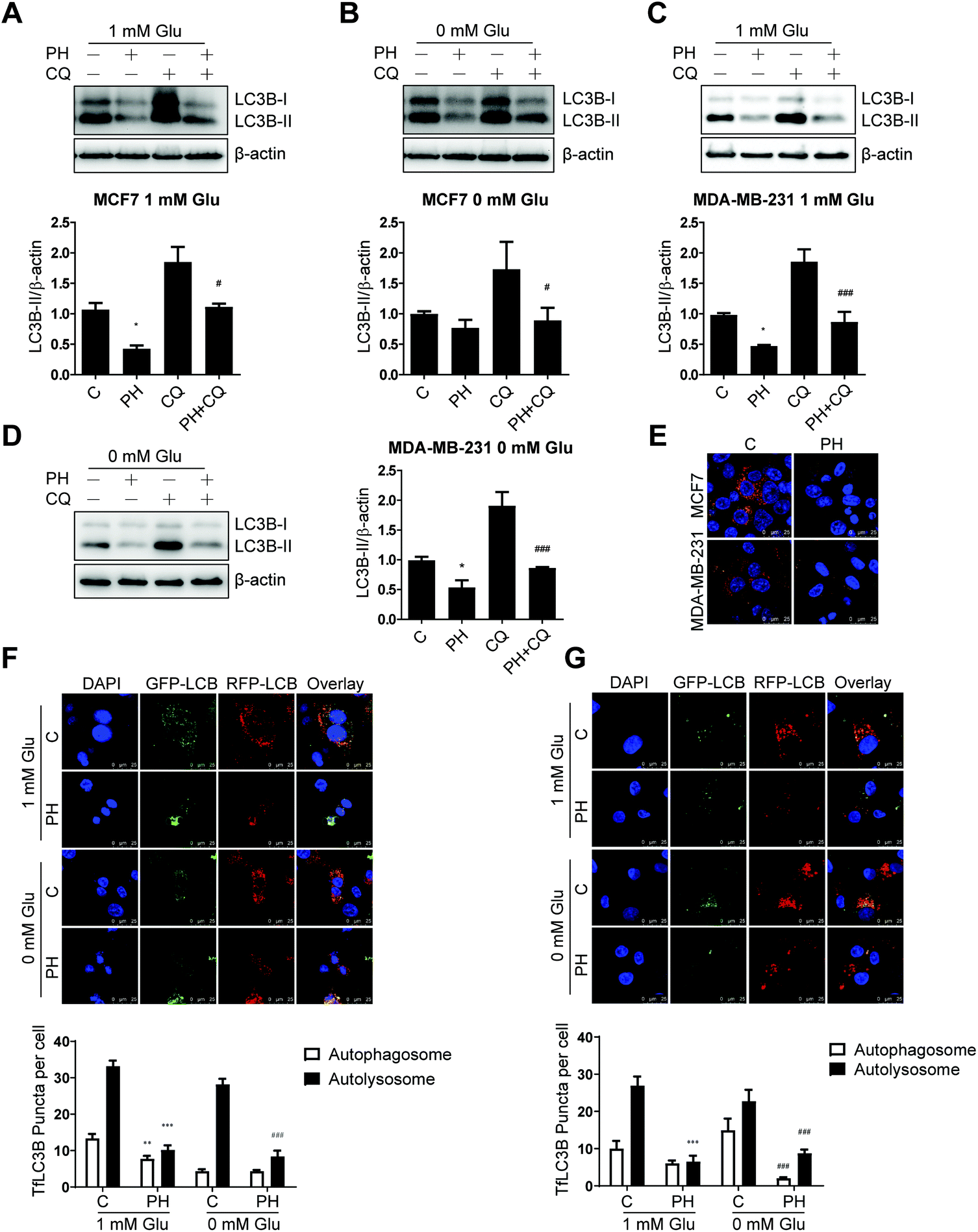 | ||
| Fig. 4 PH suppresses autophagic flux in breast cancer cells cultured in glucose-limiting media. Immunoblots and densitometry data of LC3B expression in MCF7 (A and B) and MDA-MB-231 cells (C and D) pretreated with CQ for 4 h, followed by treatment with PH for 24 h. *p < 0.05, PH vs. vehicle; #p < 0.05, ###p < 0.001, PH vs. CQ. (E) Representative images of breast cancer cells transfected with mCherry-GFP-LC3B vector and then treated with PH in 1 mM Glu media. Scale bar: 25 μm. Representative confocal microscopic images of MCF7 (F) and MDA-MB-231 cells (G) transfected with tfLC3B plasmid and then treated with PH for 24 h. Scale bar: 25 μm. Quantitative analyses of autophagosomes (yellow dots) and autolysosomes (red dots) using ImageJ software are shown in the histograms. **p < 0.01, ***p < 0.001 vs. control in 1 mM Glu media; ###p < 0.001 vs. control in 0 mM Glu media. | ||
3.4. PH inhibits the expression of upstream autophagy regulator ULK1 in breast cancer cells
To understand the underlying molecular mechanism whereby PH restrains autophagy in breast cancer cells, we firstly compared their expression of canonic autophagy regulators ATG5-ATG12 and ATG7 cultured under full, 1 mM glucose and glucose-deprived media. The results showed PH did not cause any significant change in their expression (Fig. 5A and B). Next, we determined the expression of ULK1, an important upstream regulator of autophagy that functions by forming a heterotetrameric complex with ATG13, RB1CC1 and ATG101, and thus plays a critical role in autophagy initiation. A robust reduction in the expression of ULK1 was observed in both MCF7 (3- to 10-fold) and MDA-MB-231 (2- to 7-fold) cells treated with PH in glucose-limiting media (Fig. 5C and D). mTOR has been well established to be a negative regulator of ULK1 through phosphorylation-mediated inactivation, whereas AMPK-mediated phosphorylation promotes its activity.23 We therefore treated MCF7 cells with increasing concentrations of PH in 1 mM glucose media and detected the phosphorylation levels of mTOR and AMPK. PH treatment led to significantly increased phosphorylation of both mTOR (Ser2448) and AMPK (Thr172) while suppressing the expression of LC3B-II (Fig. 6A and B). This pattern was also evident in a time-course (0, 6, 12 and 24 h) analysis (Fig. 6C–F). The induction of AMPK phosphorylation by PH could be attributed to increased ratio of AMP/ATP and ADP/ATP in MCF7 cells.24,25 These findings suggest that mTOR/ULK1 signaling likely mediates PH's anti-autophagic activity in breast cancer cells.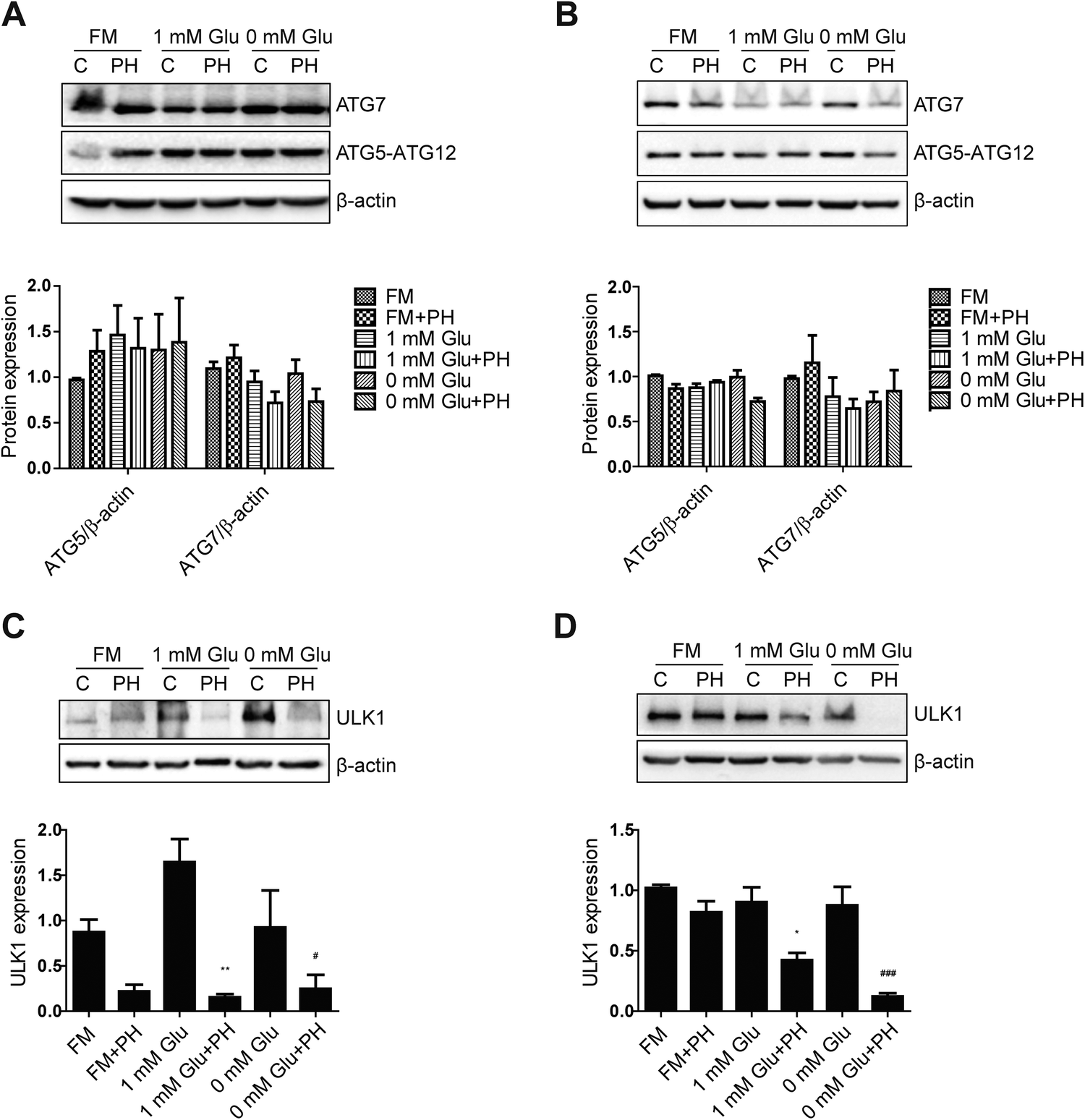 | ||
| Fig. 5 PH inhibits ULK1 expression in breast cancer cells. Immunoblot analysis of MCF7 and MDA-MB-231 cells for ATG5-ATG12 and ATG7 expression (A and B) and ULK1 expression (C and D) treated with PH for 24 h in full, 1 mM Glu and 0 mM Glu media, respectively. Densitometry data are presented in the histograms. *p < 0.05, **p < 0.01 vs. control in 1 mM Glu media; #p < 0.05, ###p < 0.001 vs. control in 0 mM Glu media. | ||
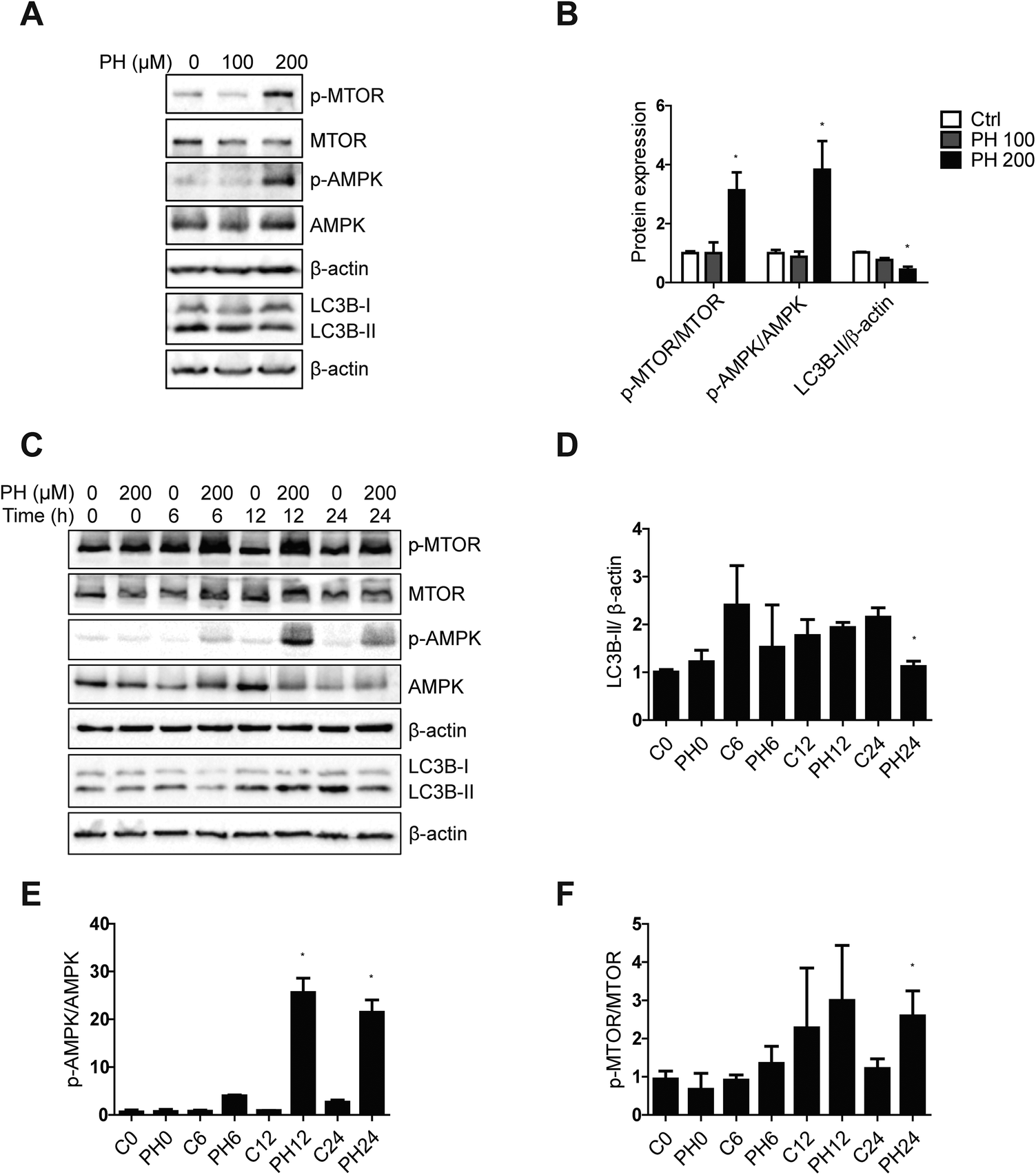 | ||
| Fig. 6 PH downregulates LC3B-II expression through the induction of MTOR phosphorylation. Immunoblot and densitometry analysis of p-MTOR, MTOR, p-AMPK, AMPK and LC3B protein expression in MCF7 cells exposed to different concentrations of PH for 24 h (A and B), and to 200 μM PH for designated time periods (C–F) in 1 mM Glu media. *p < 0.05 vs. control (of the corresponding time point). | ||
3.5. Autophagy inhibition further promotes PH's suppressive effect on breast cancer cell growth
It has been established that autophagy could help sustain metabolic and redox homeostasis to promote cancer cell growth.26 Our data so far strongly indicated an important role of autophagy inhibition in the growth inhibitory effect of PH on breast cancer cells. This also highlighted a close association between autophagy flux and breast cancer cell growth. To further clarify this link, two pharmacological autophagy inhibitors, CQ and 3-MA, were used to examine whether they could further enhance the growth inhibitory effect of PH on breast cancer cells. Consistent with the hypothesis, co-treatment of PH with CQ or 3-MA resulted in a significantly stronger inhibitory effect on cell viability than PH alone under glucose-limiting conditions in MCF7 cells (Fig. 7A and B). Moreover, flow cytometry analysis showed that PH in combination with CQ induced significantly more cell deaths than either agent alone (Fig. 7C). In crystal violet assay, a dose-dependent enhancement in the inhibitory effect of PH plus CQ on cell viability was also observed in MDA-MB-231 cells (Fig. 7D). Finally, using CCK-8 cell proliferation assay, we demonstrated a synergistic growth suppressive effect of PH combined with CQ on MDA-MB-231 cells (Fig. S2†). These data thus reinforce the notion that autophagy is an important survival mechanism in breast cancer cells under glucose-limiting conditions and that inhibition of autophagy plays an important role in the activity of PH against breast cancer cell growth.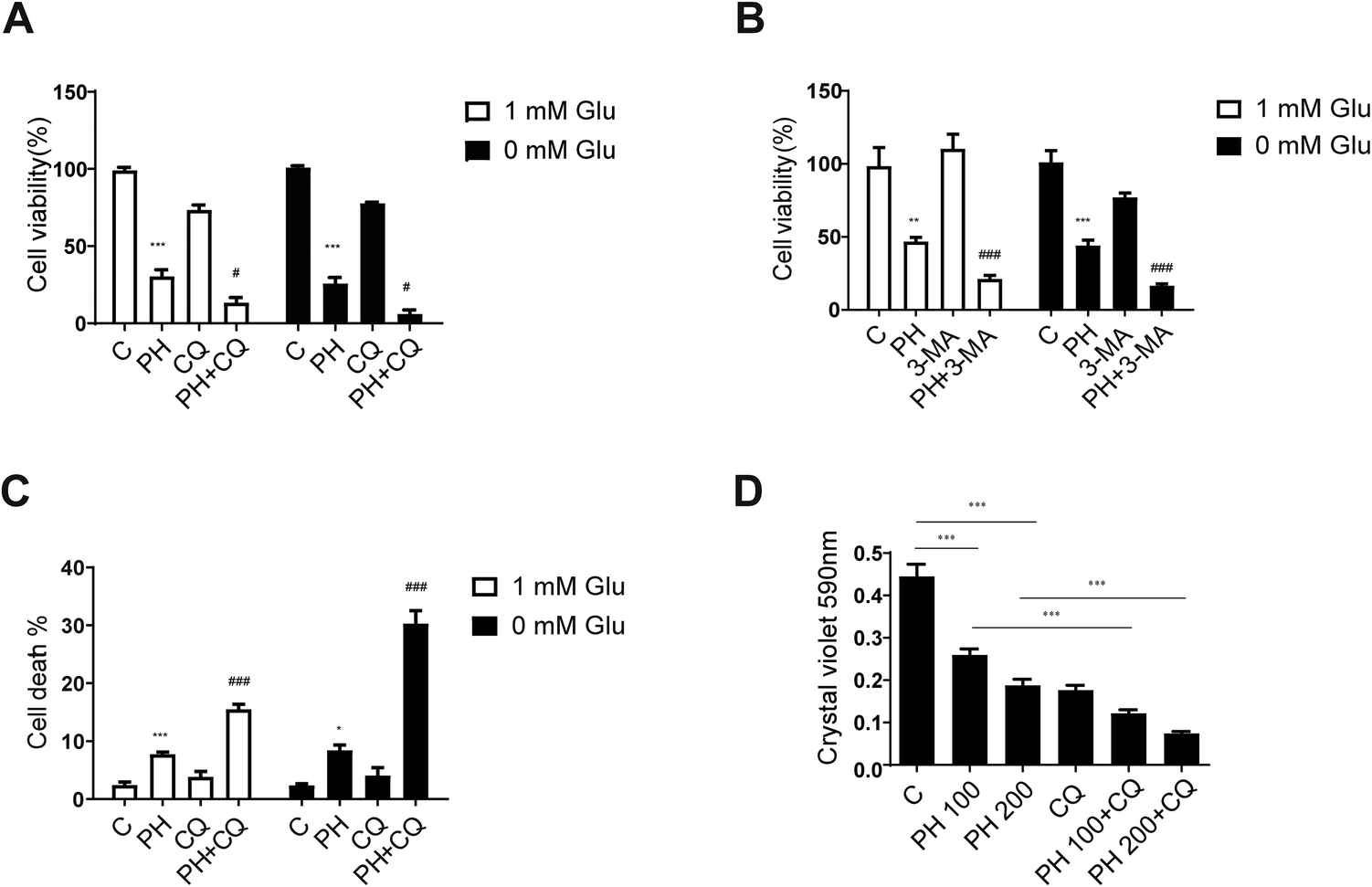 | ||
| Fig. 7 Autophagy inhibitors enhance PH-mediated growth inhibition of breast cancer cells. MCF7 cells were co-treated with PH and an autophagy inhibitor, 20 μM CQ (A) or 3 mM 3-MA (B) in 1 mM and 0 mM Glu media, respectively. Cell viability was analyzed using the CellTiter Glo Luminescent cell viability assay kit. **p < 0.01, ***p < 0.001, PH vs. control; #p < 0.05, PH + CQ vs. PH; ###p < 0.001, PH + 3-MA vs. PH. (C) Flow cytometry analysis of MCF7 cells exposed to PH with or without CQ in 1 mM and 0 mM Glu media, respectively. *p < 0.05, ***p < 0.001, PH vs. control; ###p < 0.001, PH + CQ vs. PH. (D) MDA-MB-231 cells were cultured and treated with PH (100 μM, 200 μM) with or without CQ in 1 mM Glu media. Cell viability was analyzed using the crystal violet assay. ***p < 0.001 vs. indicated group. | ||
3.6. PH improves the sensitivity of breast cancer cells to chemotherapeutic agents
Chemotherapeutic agents such as selective estrogen-receptor modulator 4OH-Tam and Dox have been widely used as a first-line chemotherapy for ERα+ and TNBC, respectively. However, the development of drug resistance during the course of treatment has limited their clinical benefits. In the meantime, emerging evidence reveals that induction of autophagy plays an important role in mediating the development of chemotherapy resistance in several types of cancers including breast cancer 7. For instance, inhibition of autophagy has been shown to restore chemosensitivity of Tam-resistant MCF7 cells.12 As PH was found in our study to significantly reduce autophagic flux in MCF7 cells, we then sought to better understand its potential for aiding in the management of breast cancer by testing its effectiveness against a 4OH-Tam-resistant MCF7 cell line (MCF7 re). MCF7 re was validated by its much higher resistance to 4OH-Tam than its parental counterpart (MCF7 pa) (Fig. 8A). To determine whether PH could attenuate MCF7 re chemoresistance to 4OH-Tam, various combinations of PH and 4OH-Tam were tested versus single agents. It was found that PH alone had similar growth inhibitory effect on MCF7 re compared with MCF7 pa, reducing cell proliferation by 50% at a concentration of 200 μM. As expected, MCF7 re cells were resistant to 4OH-Tam, whereas 200 μM PH was able to restored their sensitivity (82% growth inhibition by their combination relative to 6 μM 4OH-Tam alone) (Fig. 8B). 4OH-Tam boosted the expressions of ULK1 (1.9-fold) and LC3B-II (1.8-fold) under glucose-limiting condition, whereas combined treatment with PH reversed them, suggesting PH was capable of overcoming 4OH-Tam-induced upregulation of cellular protective autophagy in MCF7 re breast cancer cells (Fig. 8C). To investigate the mechanism whereby PH enhanced the chemosensitivity of MCF7 re cells to 4OH-Tam, we treated MCF7 re cells with PH, 4OH-Tam or their combination. The results indicated that PH plus 4OH-Tam treatment significantly increased the ratio of p-mTOR/mTOR and p-AKT/AKT (Fig. S3†), suggesting that the mTOR/AKT pathway, a critical negative regulator of autophagy, was likely involved in PH-mediated suppression of autophagy and enhancement of MCF7 re cells to chemotherapeutic agents. Interestingly, although PH inhibited 4OH-Tam-induced upregulation of LC3B-II, it failed to enhance the sensitivity of MCF7 parental cells to 4OH-Tam treatment (Fig. S4†). We also explored whether PH could sensitize MDA-MB-231 cells to Dox, one of the most commonly used chemotherapeutic agents in TNBC patients. As expected, Dox (1 μM) markedly upregulated ULK1 and LC3B-II expression in MDA-MB-231 cells in glucose-limiting media, while co-treatment with PH attenuated this effect (Fig. 8D). This was also evidenced by a synergistic cell growth inhibitory effect achieved with their combination in vitro (Fig. 8E) and in vivo (Fig. S5†). While PH and Dox alone only modestly (p > 0.05) suppressed tumor growth in Balb/c nude mice implanted subcutaneously with MDA-MB-231 cells compared with control, their combination exhibited a significant growth inhibitory effect. Moreover, knockdown of key autophagy genes ATG7 or ULK1 in MCF re cells further increased their already higher sensitivity to the cytotoxic effect of PH plus 4OH-Tam compared to 4OH-Tam alone (Fig. S6† and Fig. 8F, G), indicating that autophagy was involved in PH-mediated enhancement of sensitivity of MCF7 re cells towards 4OH-Tam. These data demonstrate that PH could sensitize breast cancer cells to conventional chemotherapy through the inhibition of autophagy induced by chemotherapeutic agents.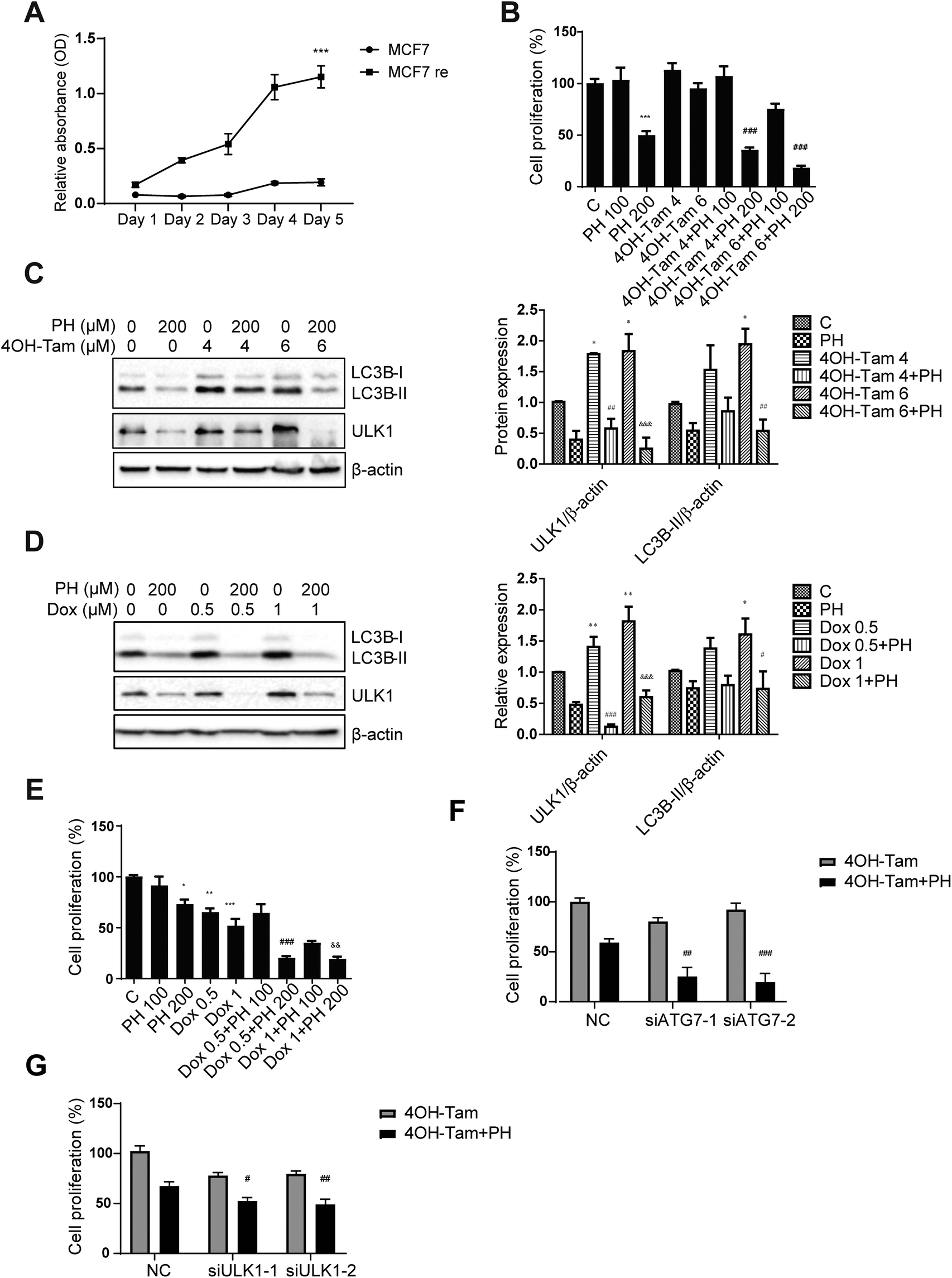 | ||
| Fig. 8 PH enhances sensitivity of breast cancer cells to chemotherapeutic drugs in glucose-limiting media. (A) Growth curve of parental and 4OH-Tam resistant MCF7 cells exposed to 4 μM 4OH-Tam for 5 days. ***p < 0.001. (B) MCF7 re cells were treated with PH (100, 200 μM), 4OH-Tam (4, 6 μM) or their combinations in 1 mM Glu media and cell proliferation was measured by the CCK-8 assay. ***p < 0.001, PH 200 vs. control; ###p < 0.001, 4OH-Tam 4 + PH 200 vs. 4OH-Tam 4 μM, and 4OH-Tam 6 + PH 200 and 4OH-Tam 6 μM. Immunoblot and densitometry analyses of LC3B-II and ULK1 expression in MCF7 re cells treated with 4OH-Tam (C) and MDA-MB-231 cells treated with Dox (D) with or without cotreatment with PH. *p < 0.05, **p < 0.01, 4OH-Tam 4, 4OH-Tam 6, Dox 0.5 or Dox 1 vs. control; ##p < 0.01, 4OH-Tam 4 + PH vs. 4OH-Tam 4; ###p < 0.001, Dox 0.5 + PH vs. Dox 0.5; &p < 0.05, &&p < 0.01, &&&p < 0.001, 4OH-Tam 6 + PH vs. 4OH-Tam 6, Dox 1 + PH vs. Dox 1. (E) CCK-8 assay of MDA-MB-231 cells treated with PH (100, 200 μM), Dox (0.5, 1 μM) or their combinations. *p < 0.05, **p < 0.01, ***p < 0.001, PH 200, Dox 0.5 and Dox 1 vs. control; ###p < 0.001, Dox 0.5 + PH 200 vs. Dox 0.5; &&p < 0.01, Dox 1 + PH 200 vs. Dox 1. Cell proliferation assay of MCF7 re cells transfected with ATG7 siRNA (F), ULK1 siRNA (G) or their scrambled control followed by 4OH-Tam, PH or their combination treatment. #p < 0.05, ##p < 0.01, ###p < 0.001, ATG7 siRNA or ULK1 siRNA vs. NC in PH plus 4OH-Tam treatment. | ||
4. Discussion
The present study has demonstrated that PH, a dihydrochalcone flavonoid found predominantly in apples, possesses promising inhibitory activity against the growth of both ERα+ and TNBC cell lines. This anticancer effect is mediated mainly through the inhibition of autophagy via mTOR/ULK1 signaling pathway. Additionally, PH improves the sensitivity of breast cancer cells to chemotherapeutic drugs 4OH-Tam and Dox.Autophagy is a conserved catabolic process whereby intracellular components are degraded and recycled.27 It is also known for its role in cellular resilience under adverse conditions such as glucose starvation, one of the most common stresses experienced by tumors.7,28,29 The induction of autophagy has thus emerged to be an important mechanism mediating the development of resistance to conventional chemotherapy. Although significant advances have been made in the treatment of breast cancer in recent decades, many patients unavoidably develop resistance to their chemotherapy option(s) in later stages. Bioactive phytochemicals have attracted increasing attention as promising natural agents for the treatment and/or prevention of cancer through targeting various signaling pathways, in particular redox signaling, apoptosis, and autophagy.30,31 PH was reported to inhibit cancer cell growth through multiple mechanisms, including induction of mitochondria-mediated apoptosis, blockade of cyclins and cyclin-dependent kinases, as well as inhibition of type 2 glucose transporter.17,19,32,33 However, it remains unclear whether it could modulate autophagic activity in breast cancer cells or not. In this study, PH preferentially reduced viability of breast cancer cells in glucose-limiting media, suggesting its capability in suppressing glucose starvation-induced cytoprotective autophagy in these cells.
Autophagy has a complex role in breast cancer progression, and it could promote or inhibit breast cancer development depending on factors such as tumor subtype, mutation status, as well as technical aspects, such as the model system used to investigate its function.8 On the one hand, autophagy hyperactivation could induce autophagic cell death, thus inhibiting cancer progression; on the other hand, autophagy inhibition might enhance the effect of chemotherapeutic drugs by re-sensitizing resistant cancer cells.34 Previous studies showed that monoallelic depletion of a core autophagy component Beclin1 led to spontaneous mammary hyperplasia, which remarkably resembled the allelic losses found in human breast cancer.6,35 In TNBC cell models, glycolysis restriction triggered AMPK-mediated upregulation of autophagy, subsequently promoting degradation of colony-stimulating factor and T cell-mediated anti-tumor immunity.36 Adiponectin, a hormone closely associated with nutrient/energy status, induced cytotoxic autophagy in MCF7 and MDA-MB-231 breast cancer cells via the regulation of LKB1-AMPK signaling.25 In the meantime, accumulating evidence also supports a pro-survival role of autophagy in breast cancer. For instance, mouse mammary-specific ablation of Fip200, one of the core components of ULK1 protein complex, led to defective autophagy and suppression of oncogene-driven mammary carcinogenesis and progression.37 Additionally, IFNβ-induced autophagy counteracted IFNβ-mediated apoptosis in MCF7 and MDA-MB-231 cells.38 Our results showed a pro-survival role of autophagy in breast cancer cell lines (MCF7 and MDA-MB-231) under glucose-limiting stress conditions, and are in alignment with the widely accepted notion that autophagy is an important mechanism adopted by tumor cells to cope with stresses in their micro-environments, particularly during advanced stages. This finding was strengthened with the use of an FDA approved autophagy inhibitor CQ, which interrupted autophagic flux (evidenced by the accumulation of LC3B-II) and reduced breast cancer cell viability. Further to this finding, we demonstrated that PH downregulated autophagy under the stress condition, and thus limited the growth potential of breast cancer cells. Meanwhile, co-treatment of PH with CQ resulted in a significantly greater degree of cytotoxicity than either agent alone. Similar synergistic growth inhibitory effect was also observed between PH and another commercially available autophagy inhibitor 3-MA.
Mechanistic evaluation revealed PH suppressed autophagy in breast cancer cell lines through mTOR/ULK1-dependent signaling pathway. The mTOR/AKT pathway played a central role in cell proliferation, survival, and growth and has been frequently reported to be overactivated in breast cancer.39,40 Meanwhile, the mTOR/AKT cascade, a major negative regulator of autophagy, can inhibit autophagy initiation by suppressing ULK1.41 PH induced mTOR/AKT/activation which subsequently led to autophagy inhibition, indicating a complex close relationship between mTOR/AKT signaling, autophagy and breast cancer. The ULK1, a key component of the core autophagic apparatus, forms a protein complex with ATG13, RB1CC1 and ATG101 and plays a crucial role in autophagy initiation.26 Under glucose-limiting conditions, we found that PH had minimal impact on ATG5 and ATG7 expression, but strongly reduced the expression of ULK1, highlighting the importance of ULK1 in the regulation of breast cancer autophagy in this specific setting. The Mtor-AKT cascade is considered as a major negative regulator of autophagy in multiple types of cancer. Our data showed PH induced phosphorylation of mTOR in MCF7 cells, implicating the involvement of mTOR-ULK1 in mediating the inhibitory effect of PH on autophagy under the experimental conditions.
4OH-Tam and Dox are two frequently used first-line chemotherapeutic agents for breast cancer. Unfortunately, resistance to conventional chemotherapeutics remains a major challenge. Tremendous efforts have been made to investigate the mechanism of tamoxifen resistance and several mechanisms have been proposed, including activation of PI3K/AKT-mediated cell growth and proliferation pathway,20 alteration in CDK4/6 cell cycle signaling,42,43 and induction of autophagy.12 Among these, induction of pro-survival autophagy has attracted increasing attention. In fact, clinical trials are currently investigating the possibility of enhancing the efficacy of conventional chemotherapies with various types of autophagy inhibitors.27 Our study has provided multiple lines of evidence supporting the capability of PH to inhibit cytoprotective autophagy in breast cancer cell lines. Furthermore, PH helped overcome resistance of MCF7 and MDA-MB-231 cells to 4OH-Tam and Dox, respectively. Noticeably, 4OH-Tam and Dox both significantly induced the expression of LC3B-II and ULK1, which was completely reversed by co-treatment with PH. These results have, for the first time, revealed that PH could restore sensitivity of breast cancer cells to chemotherapeutic agents through the suppression of cytoprotective autophagy via targeting ULK1.
5. Conclusion
PH has demonstrated great potential as a natural inhibitor of cytoprotective autophagy in breast cancer cells under glucose-limiting conditions. This was supported by the data obtained from an array of assays including RNA-seq, western blotting, immunofluorescence, and cell viability. The anticancer effect and mechanism were further manifested in the ability of PH to restore the sensitivity of MCF7 and MDA-MB-231 cells to conventional chemotherapeutic agents, highlighting its value both as a promising stand-alone pharmaceutical, and as an adjuvant to standard chemotherapy for breast cancer.Abbreviations
| PH | Phloretin |
| LC3/MAP1LC3 | Microtubule-associated protein 1A/1B-light chain 3 |
| tfLC3 | Tandem fluorescent mRFP-GFP-LC3 |
| TNBC | Triple negative breast cancer |
| ERα+ | Estrogen receptor α positive |
| Dox | Doxorubicin |
| 4-OH Tam | 4-Hydroxytamoxifen |
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Special National Key Research and Development Plan of China [2016YFD0400204], the Key-Area Research and Development Program of Guangdong Province [2019B020212001], and the Postdoctoral Science Foundation of China [2019M663081].References
- C. DeSantis, J. Ma, L. Bryan and A. Jemal, Breast cancer statistics, 2013, CA-Cancer J. Clin., 2014, 64, 52–62 CrossRef.
- R. Lu, X. Hu, J. Zhou, J. Sun, A. Z. Zhu, X. Xu, H. Zheng, X. Gao, X. Wang, H. Jin, P. Zhu and L. Guo, COPS5 amplification and overexpression confers tamoxifen-resistance in ERalpha-positive breast cancer by degradation of NCoR, Nat. Commun., 2016, 7, 12044 CrossRef CAS.
- K. Polyak, Breast cancer: origins and evolution, J. Clin. Invest., 2007, 117, 3155–3163 CrossRef CAS.
- E. A. Musgrove and R. L. Sutherland, Biological determinants of endocrine resistance in breast cancer, Nat. Rev. Cancer, 2009, 9, 631–643 CrossRef CAS.
- M. D. Rybstein, J. M. Bravo-San Pedro, G. Kroemer and L. Galluzzi, The autophagic network and cancer, Nat. Cell Biol., 2018, 20, 243–251 CrossRef CAS.
- X. Qu, J. Yu, G. Bhagat, N. Furuya, H. Hibshoosh, A. Troxel, J. Rosen, E. L. Eskelinen, N. Mizushima, Y. Ohsumi, G. Cattoretti and B. Levine, Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene, J. Clin. Invest., 2003, 112, 1809–1820 CrossRef CAS.
- K. L. Cook, A. N. Shajahan and R. Clarke, Autophagy and endocrine resistance in breast cancer, Expert Rev. Anticancer Ther., 2011, 11, 1283–1294 CrossRef CAS.
- A. C. Kimmelman and E. White, Autophagy and Tumor Metabolism, Cell Metab., 2017, 25, 1037–1043 CrossRef CAS.
- E. White, Deconvoluting the context-dependent role for autophagy in cancer, Nat. Rev. Cancer, 2012, 12, 401–410 CrossRef CAS.
- S. Lefort, C. Joffre, Y. Kieffer, A. M. Givel, B. Bourachot, G. Zago, I. Bieche, T. Dubois, D. Meseure, A. Vincent-Salomon, J. Camonis and F. Mechta-Grigoriou, Inhibition of autophagy as a new means of improving chemotherapy efficiency in high-LC3B triple-negative breast cancers, Autophagy, 2014, 10, 2122–2142 CrossRef CAS.
- F. Lozy, X. Cai-McRae, I. Teplova, S. Price, A. Reddy, G. Bhanot, S. Ganesan, A. Vazquez and V. Karantza, ERBB2 overexpression suppresses stress-induced autophagy and renders ERBB2-induced mammary tumorigenesis independent of monoallelic Becn1 loss, Autophagy, 2014, 10, 662–676 CrossRef CAS.
- J. S. Samaddar, V. T. Gaddy, J. Duplantier, S. P. Thandavan, M. Shah, M. J. Smith, D. Browning, J. Rawson, S. B. Smith, J. T. Barrett and P. V. Schoenlein, A role for macroautophagy in protection against 4-hydroxytamoxifen-induced cell death and the development of antiestrogen resistance, Mol. Cancer Ther., 2008, 7, 2977–2987 CrossRef CAS.
- L. Fan, K. Strasser-Weippl, J.-J. Li, J. St Louis, D. M. Finkelstein, K.-D. Yu, W.-Q. Chen, Z.-M. Shao and P. E. Goss, Breast cancer in China, Lancet Oncol., 2014, 15, e279–e289 CrossRef.
- J. Quero, I. Marmol, E. Cerrada and M. J. Rodriguez-Yoldi, Insight into the potential application of polyphenol-rich dietary intervention in degenerative disease management, Food Funct., 2020, 11, 2805–2825 RSC.
- H. S. Aiyer, A. M. Warri, D. R. Woode, L. Hilakivi-Clarke and R. Clarke, Influence of berry polyphenols on receptor signaling and cell-death pathways: implications for breast cancer prevention, J. Agric. Food Chem., 2012, 60, 5693–5708 CrossRef CAS.
- Y. S. Chiou, S. Li, C. T. Ho and M. H. Pan, Prevention of Breast Cancer by Natural Phytochemicals: Focusing on Molecular Targets and Combinational Strategy, Mol. Nutr. Food Res., 2018, 62, e1800392 CrossRef.
- B. Y. Choi, Biochemical Basis of Anti-Cancer-Effects of Phloretin-A Natural Dihydrochalcone, Molecules, 2019, 24(2), 278 CrossRef.
- H. Yoon and R. H. Liu, Effect of selected phytochemicals and apple extracts on NF-kappaB activation in human breast cancer MCF-7 cells, J. Agric. Food Chem., 2007, 55, 3167–3173 CrossRef CAS.
- K. H. Wu, C. T. Ho, Z. F. Chen, L. C. Chen, J. Whang-Peng, T. N. Lin and Y. S. Ho, The apple polyphenol phloretin inhibits breast cancer cell migration and proliferation via inhibition of signals by type 2 glucose transporter, J. Food Drug Anal., 2018, 26, 221–231 CrossRef CAS.
- Y. Zhu, Y. Liu, C. Zhang, J. Chu, Y. Wu, Y. Li, J. Liu, Q. Li, S. Li, Q. Shi, L. Jin, J. Zhao, D. Yin, S. Efroni, F. Su, H. Yao, E. Song and Q. Liu, Tamoxifen-resistant breast cancer cells are resistant to DNA-damaging chemotherapy because of upregulated BARD1 and BRCA1, Nat. Commun., 2018, 9, 1595 CrossRef.
- T. Jiang, B. Harder, M. R. De la Vega, P. K. Wong, E. Chapman and D. D. Zhang, p62 links autophagy and Nrf2 signaling, Free Radicals Biol. Med., 2015, 88, 199–204 CrossRef CAS.
- J. Moscat, M. Karin and M. T. Diaz-Meco, p62 in Cancer: Signaling Adaptor Beyond Autophagy, Cell, 2016, 167, 606–609 CrossRef CAS.
- J. Kim, M. Kundu, B. Viollet and K. L. Guan, AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1, Nat. Cell Biol., 2011, 13, 132–141 CrossRef CAS.
- S. C. Lin and D. G. Hardie, AMPK: Sensing Glucose as well as Cellular Energy Status, Cell Metab., 2018, 27, 299–313 CrossRef CAS.
- S. J. Chung, G. P. Nagaraju, A. Nagalingam, N. Muniraj, P. Kuppusamy, A. Walker, J. Woo, B. Gyorffy, E. Gabrielson, N. K. Saxena and D. Sharma, ADIPOQ/adiponectin induces cytotoxic autophagy in breast cancer cells through STK11/LKB1-mediated activation of the AMPK-ULK1 axis, Autophagy, 2017, 13, 1386–1403 CrossRef CAS.
- V. Lahiri, W. D. Hawkins and D. J. Klionsky, Watch What You (Self-) Eat: Autophagic Mechanisms that Modulate Metabolism, Cell Metab., 2019, 29, 803–826 CrossRef CAS.
- J. M. M. Levy, C. G. Towers and A. Thorburn, Targeting autophagy in cancer, Nat. Rev. Cancer, 2017, 17, 528–542 CrossRef CAS.
- J. Debnath, The multifaceted roles of autophagy in tumors-implications for breast cancer, J. Mammary Gland Biol. Neoplasia, 2011, 16, 173–187 CrossRef.
- A. Walker, A. Singh, E. Tully, J. Woo, A. Le, T. Nguyen, S. Biswal, D. Sharma and E. Gabrielson, Nrf2 signaling and autophagy are complementary in protecting breast cancer cells during glucose deprivation, Free Radicals Biol. Med., 2018, 120, 407–413 CrossRef CAS.
- M. F. Akhtar, A. Saleem, A. Rasul, M. M. Faran Ashraf Baig, M. Bin-Jumah and M. M. Abdel Daim, Anticancer natural medicines: An overview of cell signaling and other targets of anticancer phytochemicals, Eur. J. Pharmacol., 2020, 888, 173488 CrossRef CAS.
- S. Deng, M. K. Shanmugam, A. P. Kumar, C. T. Yap, G. Sethi and A. Bishayee, Targeting autophagy using natural compounds for cancer prevention and therapy, Cancer, 2019, 125, 1228–1246 CrossRef.
- S. T. Lin, S. H. Tu, P. S. Yang, S. P. Hsu, W. H. Lee, C. T. Ho, C. H. Wu, Y. H. Lai, M. Y. Chen and L. C. Chen, Apple Polyphenol Phloretin Inhibits Colorectal Cancer Cell Growth via Inhibition of the Type 2 Glucose Transporter and Activation of p53-Mediated Signaling, J. Agric. Food Chem., 2016, 64, 6826–6837 CrossRef CAS.
- M. Xu, W. Gu, Z. Shen and F. Wang, Anticancer Activity of Phloretin Against Human Gastric Cancer Cell Lines Involves Apoptosis, Cell Cycle Arrest, and Inhibition of Cell Invasion and JNK Signalling Pathway, Med. Sci. Monit., 2018, 24, 6551–6558 CrossRef CAS.
- Y. Jing, W. Liang, J. Liu, L. Zhang, J. Wei, J. Yang, Y. Zhang and Z. Huang, Autophagy-mediating microRNAs in cancer chemoresistance, Cell Biol. Toxicol., 2020, 36, 517–536 CrossRef CAS.
- Z. Yue, S. Jin, C. Yang, A. J. Levine and N. Heintz, Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 15077–15082 CrossRef CAS.
- W. Li, T. Tanikawa, I. Kryczek, H. Xia, G. Li, K. Wu, S. Wei, L. Zhao, L. Vatan, B. Wen, P. Shu, D. Sun, C. Kleer, M. Wicha, M. Sabel, K. Tao, G. Wang and W. Zou, Aerobic Glycolysis Controls Myeloid-Derived Suppressor Cells and Tumor Immunity via a Specific CEBPB Isoform in Triple-Negative Breast Cancer, Cell Metab., 2018, 28, 87–103.e6 CrossRef CAS.
- H. Wei, S. Wei, B. Gan, X. Peng, W. Zou and J. L. Guan, Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis, Genes Dev., 2011, 25, 1510–1527 CrossRef CAS.
- M. Ambjorn, P. Ejlerskov, Y. Liu, M. Lees, M. Jaattela and S. Issazadeh-Navikas, IFNB1/interferon-beta-induced autophagy in MCF-7 breast cancer cells counteracts its proapoptotic function, Autophagy, 2013, 9, 287–302 CrossRef CAS.
- S. E. Nunnery and I. A. Mayer, Targeting the PI3K/AKT/mTOR Pathway in Hormone-Positive Breast Cancer, Drugs, 2020, 80, 1685–1697 CrossRef CAS.
- P. S. Ong, L. Z. Wang, X. Dai, S. H. Tseng, S. J. Loo and G. Sethi, Judicious Toggling of mTOR Activity to Combat Insulin Resistance and Cancer: Current Evidence and Perspectives, Front. Pharmacol., 2016, 7, 395 CAS.
- Q. Xia, M. Xu, P. Zhang, L. Liu, X. Meng and L. Dong, Therapeutic Potential of Autophagy in Glioblastoma Treatment With Phosphoinositide 3-Kinase/Protein Kinase B/Mammalian Target of Rapamycin Signaling Pathway Inhibitors, Front. Oncol., 2020, 10, 572904 CrossRef.
- C. K. Osborne and R. Schiff, Mechanisms of endocrine resistance in breast cancer, Annu. Rev. Med., 2011, 62, 233–247 CrossRef CAS.
- R. Clarke, J. J. Tyson and J. M. Dixon, Endocrine resistance in breast cancer–An overview and update, Mol. Cell. Endocrinol., 2015, 418(Pt 3), 220–234 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Effect of PH on oxidative stress in breast cancer cells under glucose-limiting condition, effect of co-treatment of PH and autophagy inhibitor CQ on MDA-MB-231 cells growth, PH inhibited autophagy by modulating mTOR/AKT signaling in MCF7 re cells, PH failed to enhance sensitivity of MCF7 parental cells to 4OH-Tam in glucose-limiting media, and PH synergized with Dox in suppressing breast cancer xenograft growth in mice. See DOI: 10.1039/d0fo02362k |
| This journal is © The Royal Society of Chemistry 2021 |