 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Spiers Memorial Lecture
Carbon nanostructures by macromolecular design – from branched polyphenylenes to nanographenes and graphene nanoribbons
Zijie
Qiu
 a,
Akimitsu
Narita
a and
Klaus
Müllen
*ab
a,
Akimitsu
Narita
a and
Klaus
Müllen
*ab
aMax Planck Institute for Polymer Research, Ackermannweg 10, Mainz, Germany. E-mail: muellen@mpip-mainz.mpg.de
bDepartment of Chemistry, University of Cologne, Greinstr. 4-6, Cologne, Germany
First published on 5th March 2020
Abstract
Nanographenes (NGs) and graphene nanoribbons (GNRs) are unique connectors between the domains of 1D-conjugated polymers and 2D-graphenes. They can be synthesized with high precision by oxidative flattening processes from dendritic or branched 3D-polyphenylene precursors. Their size, shape and edge type enable not only accurate control of classical (opto)electronic properties, but also access to unprecedented high-spin structures and exotic quantum states. NGs and GNRs serve as active components of devices such as field-effect transistors and as ideal objects for nanoscience. This field of research includes their synthesis after the deposition of suitable monomers on surfaces. An additional advantage of this novel concept is in situ monitoring of the reactions by scanning tunnelling microscopy and electronic characterization of the products by scanning tunnelling spectroscopy.
1. Introduction
The elucidation of the benzene structure and the discussion of the aromaticity concept are classic topics in organic chemistry.1–3 Benzene is a fascinating species, not only as a model for spectroscopic and theoretical studies, but also as a starting compound for chemical functionalization and the subsequent synthesis of natural products and as a versatile building block for conjugated π-systems. One of the oldest challenges of organic chemistry, the synthesis of colourants, would not have been possible without benzene chemistry. These areas might appear to be distinct at first glance, but a very good case for their association can be made by relating two major breakthroughs in materials science, conjugated “conducting” polymers as synthetic metals4–6 and graphenes as new carbon allotropes,7,8 to structural and functional concepts derived from the study of benzene compounds. Going from benzene to biphenyl, terphenyl and higher oligophenyls builds up a “box” of delocalized, polarizable electrons, and this property enables the chain molecules to interact with light and serve as chromophores or to undergo electron transfer and serve as “electrophores”. One can even argue that key processes in modern energy technology, such as the transformation of light energy into electrical energy in solar cells9–12 or the storage of charge in batteries,13–15 would have remained impossible without the concepts developed during the study of benzene and conjugated π-systems.While oligophenyls are chain structures, the benzene π-system can be extended in another way, by fusing the hexagons to naphthalene, anthracene and higher acenes, or by creating other topologies such as those in phenanthrene, triphenylene and higher polycyclic aromatic hydrocarbons (PAHs). The description of the resulting rod- and disc-type π-systems based on molecular orbital theory and spectroscopy has been instrumental for modern organic chemistry. Notably, however, a comprehensive discussion of such conjugated systems cannot be restricted to molecular features but must also consider supramolecular chemistry.2,16,17 By attaching different aliphatic chains at the peripheries of a PAH, various packing modes and phases can be realized in the solid state, depending on the arrangement of chains and discs, which determines the kinetics of intermolecular processes such as exciton or electron transfer. Equally important are interfacial structures, such as the edge-on or face-on arrangement of conjugated molecules on surfaces.
Two characteristics are crucial for the molecular and supramolecular description of π-systems and provide powerful design concepts: size and dimensionality. Building homologous series by the stepwise addition of another repeating unit is one of the key principles when trying to “synthesize” desired properties.2,18,19 Additionally, plotting a measurable quantity, such as the wavelength of an absorption band or the redox potential, against the number n of repeat units and extrapolating to infinite n offers deep insight into the nature of the corresponding polymers.20 Rigid-rod-type poly-para-phenylene (PPP) with an “infinite” chain can be regarded as a prototype of a conjugated polymer (CP). A similar argument holds for 2D-polymer graphene, which is thus the infinite form of a PAH.
The organic chemistry of small benzene-based unsaturated molecules and the materials science of materials such as PPP and graphene have largely advanced as separate disciplines, often kept further apart by different nomenclatures. Two main criteria distinguish an organic material from an organic compound. For a material, (i) one examines a complex function as part of a complex system, e.g. a device, which cannot be achieved by just recording a spectrum, and (ii) one must create a defined macroscopic state of matter such as a fibre or a thin film. One readily concludes that organic chemistry would have much synthetic expertise to contribute if it were not restricted to focusing on dilute solutions.
Chemistry has become more interdisciplinary over recent years, and while classical topics such as synthetic method development will always be important, the challenges defined by materials science have opened new horizons. A similar stimulus came from another emerging discipline – nanoscience. Two achievements have helped to bring about nanoscience, namely the ability in physics to “see” small single molecules or particles, and the ability in chemistry to fabricate molecules or particles extending over different length scales. The example of graphene is instructive. Graphene can be fabricated by exfoliating a single layer from bulk graphite.21,22 The critical features of such a “top-down” protocol are the size of the resulting sheet and the avoidance of multilayers. In an alternative approach, described as “bottom-up”, one could consider synthesizing increasingly larger PAHs, and ultralarge PAHs have been coined nanographenes (NGs).23–25
Compared with the hot field of graphene, the study of CPs has already seen several ups and downs. Following research on the science and technology of synthetic metals, based on the occurrence of electrical conductivity due to partial electron transfer and lost momentum, the light emission from CPs in electroluminescent devices attracted attention for use in displays and lighting systems,26–28 followed by the application of CPs in solar cells as light absorbers and electron donors.9–12 One should be careful not to prematurely describe a research field as mature. CPs offer nearly unlimited opportunities for structural variations and thus synthesis-driven property control. However, problems of a fundamental and technological nature remain. One drawback relates to the stability of devices, which may depend not only on the prevailing structure but also on the occurrence of defects and impurities. Another drawback relates to the special situation in a device such as a field-effect transistor (FET). The charge-carrier transport, an important performance criterion, is dependent on the supramolecular order between the molecules and their arrangement with respect to the substrate. The resulting morphologies can thus be influenced by the molecular structures and by the applied processing techniques, and the fabrication of thin-film devices by unavoidable trial and error may lead to undesirable packing modes.29,30 This uncertainty has strengthened the desire for electronic materials with either high intrinsic charge-carrier mobility or robust, controllable packing motifs. Such materials can potentially be realized with graphene and NG molecules. Graphene was shown in the pioneering work of Geim and Novoselov to possess an ultrahigh charge-carrier mobility, which qualifies it for use as an active material in FETs.31–33 However, the problem is that graphene also features a vanishing electronic bandgap, which would never allow an off state of the current flowing in an FET. A switch that is never off is not practical, so the bandgap must be opened. As suggested by theory, geometric confinement of the graphene layer, such as that in a graphene nanoribbon (GNR), is the method of choice to open the bandgap.34,35 A GNR is a quasi-1D π-system, as opposed to 2D-graphene, and thus bears a close resemblance to the classical 1D-CPs described above (Scheme 1). Double-stranded CPs (ribbon polymers) have already played an important role in polymer science and can pave the way to GNRs.36–39 From a synthetic point of view, the transition from CPs to GNRs is far from trivial, and the underlying protocols are one of the key considerations of this text.
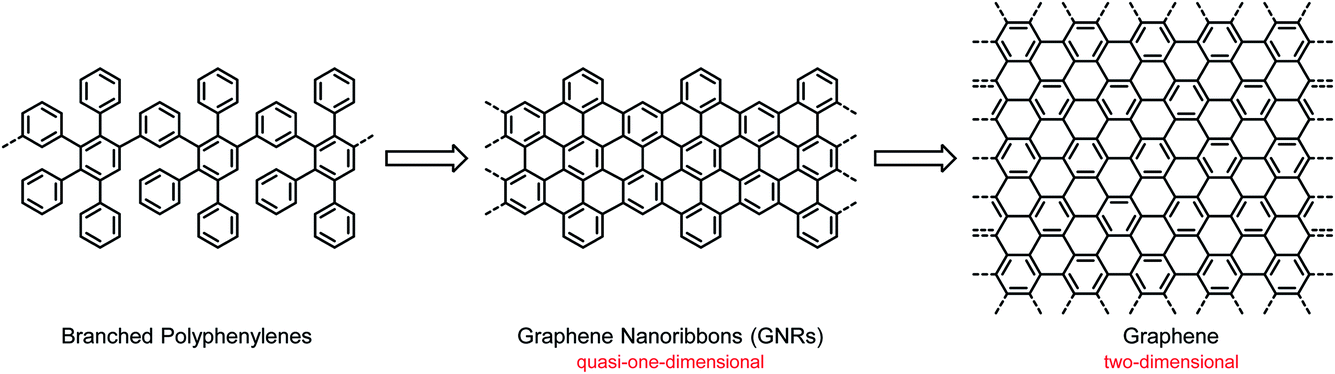 | ||
| Scheme 1 From branched polyphenylenes to GNRs and infinite graphene sheets. | ||
The reader might thus consider GNRs as bridges connecting the graphene and CP fields, and this view appears to have some merits. Nevertheless, the present text is not restricted to simply making CPs slightly better. Instead of only looking at classical cases of charge-carrier transport or light emission, the focus will be on where GNRs and their NG model systems open new avenues for fundamental research and technology. We anticipate that the bandgaps of GNRs can be tailored by the width of the polymer – and by the nature of the periphery.24,40–42 A zigzag periphery is particularly relevant since it holds promise for creating edge-localized electronic states. These, in turn, are the origin of exotic quantum states giving rise to high-spin systems and topological insulators.43,44
Stimulated by pioneering theoretical work, the importance of GNRs has been recognized by physicists and materials scientists. However, the methods of GNR fabrication, such as graphene lithography or slicing open carbon nanotubes, suffer from a lack of control over the edge structures,45–50 which is necessary to truly enter the unknown territory just mentioned. Herein, we therefore dwell on the power of precision polymer synthesis, which is initially a challenge for solution chemistry. An additional, closely connected concept is GNR synthesis after the immobilization of monomeric building blocks on metal surfaces. In both cases, the expertise in synthesizing unique polymers and uncovering their unconventional electronic properties must be closely connected. Understandably, this text is a tribute to both the synthetic chemists from our group and the physicists from our partner groups.
2. From PAHs to NGs
2.1 Solution synthesis
A pioneer of PAH chemistry is Erich Clar, who has contributed not only to the synthesis of PAHs but also to their electronic and spectroscopic characterization.51 After a long time during which PAHs were mainly considered from a toxicological perspective, their chemistry has experienced a remarkable renaissance since the mid-1990s. This has been further stimulated by the rise of graphene and the fact that both the chemical and physical properties of PAHs are of strong significance for the graphene field. In this spirit, we described the emergence of “PAHs in the Graphene Era” in a recent review that the reader may want to consult.52 Herein, ultralarge PAHs, which we name NGs, serve as model cases for the synthesis and structural elucidation of GNRs.Extending the size of PAHs to the nanoscale becomes possible by “dimerization” via the fusion of biaryl precursors under oxidative conditions in the Scholl reaction. A known case is the transition from 1,1′-binaphthyl to perylene, while the fusion of perylene to quaterrylene can be regarded as an intermolecular version. More recently, we used bichrysenyl 1 and tetrachrysenyl 3 as starting compounds to achieve fusion to realize short segments of the cove-edge GNRs 2 and 4, respectively (Scheme 2A).53,54 A more elegant concept that allows a much steeper increase in size comprises the synthesis of a polyphenylene precursor whose topology allows full flattening of the benzene rings into the PAH plane by cyclodehydrogenation, namely the Scholl reaction. The synthesis of hexa-peri-hexabenzocoronene (HBC) 5 from hexaphenylbenzene is the most prominent example. HBC, with a large hexagonal aromatic structure, can be well described as a superbenzene. Interestingly, supernaphthalene and other higher homologues of HBC with varying symmetries, including C72 6, C96 7, and C138 8, have become accessible starting from appropriately designed polyphenylene precursors (Scheme 2B).55,56 Astonishingly, polyphenylene 9 can be transformed into C222-hexagon 10 (Scheme 2C).57 The crystal structure of the soluble precursor 9 nicely exhibits the presence of twisted, tightly packed and interlocked benzene rings. Molecule 9 is just a dendrimer made only from benzene rings with intra- and intermolecular voids ready for the uptake of guest molecules. In a separate strand of our research, increasingly larger polyphenylene dendrimers with molecular weights up to 1.9 MDa could be synthesized by a divergent route and characterized as monodisperse, structurally perfect 3D polyphenylenes. What makes these dendrimers different from all other dendrimers is that they are semi-rigid, since their arms do not undergo back-bending. Therefore, the incorporation of functional groups such as chromophores at the core, in the scaffold or on the rim of the dendrimers leads to perfect nanosite definition and provides, for example, unique light harvesting systems with controlled transfer of the excitation energy. Another issue is important in the present context: multiply branched or dendritic polyphenylenes with suitable structures are carbon reservoirs for the formation of graphenic molecules by oxidative 3D- to 2D-transformations.
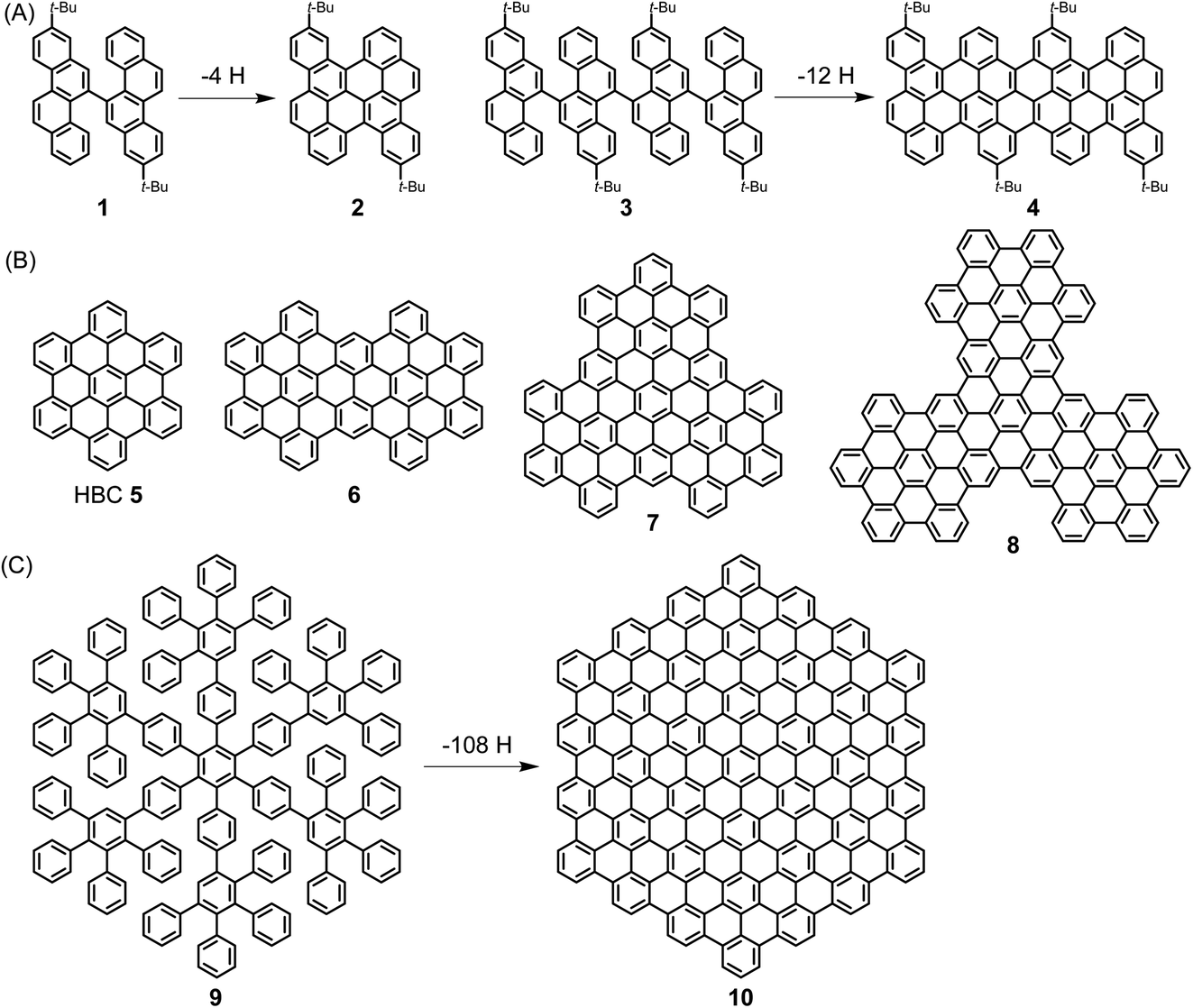 | ||
| Scheme 2 (A) Syntheses of PAHs 2 and 4 by cyclodehydrogenation. (B) Structures of HBC homologues 5–8 with varying symmetries. (C) Synthesis of C222-hexagon 10 by cyclodehydrogenation. | ||
The synthesis and structural characterization of PAHs and their larger NG congeners is severely hampered by their limited solubility in organic solvents, which arises from the pronounced tendency of the disc-type molecules to aggregate. A surprisingly simple way of solubilizing rigid discs is exhaustive edge chlorination, by treating suspensions of the hydrocarbons with iodine chloride (Fig. 1A).58 The sterically encumbered chloro substituents cause strong ring puckering; thus, sufficient solubility for crystallization is induced (Fig. 1B and C). Structural analysis gives information of crucial importance to describe the bonding situation in NGs, such as bond length alternation and packing in the solid. Attaching long alkyl chains and thus affording a higher solvation entropy is another way of dealing with the solubility issue. This structural variation, except when using very bulky substituents, does not avoid aggregate formation in solution and even promotes the formation of discotic mesophases with a columnar liquid-crystalline order in the melt.59–61 Notably, this phase formation under stacking of the discs does not necessarily result from attractive π–π interactions, but rather from nanophase separation between the “hard” aromatic cores and the “soft” aliphatic shells. For a long time, hexa-alkoxy triphenylenes have been regarded as the main model systems for discotic mesophase formation.62–64 The HBC congeners have much more stable phases, which allow systematic studies of charge-carrier transport along the columnar superstructures. Furthermore, mesophases can persist at temperatures at which thermal carbon–carbon cleavage within the alkyl chains and subsequent crosslinking occur. This is reminiscent of carbomesophase formation during the graphitization of pitch. Similar “connections” are possible, such as in the fabrication of carbon nanofibres by pyrolysis of either polyacrylonitrile or pitch.25,65–69 Again, complex crosslinking reactions occur in a preorganized phase, but the detailed chemical processes are often described in rather speculative terms. Model reactions under controlled conditions are urgently needed. A helpful case is the use of poly(methyl vinyl ketone) as the starting compound because acene formation can proceed by an aldol condensation rather than thermolytically.70–72 Revitalized PAH chemistry can branch out into many other areas since PAHs are much-needed model compounds, for example, in astrophysics and the analysis of asphaltenes.73,74
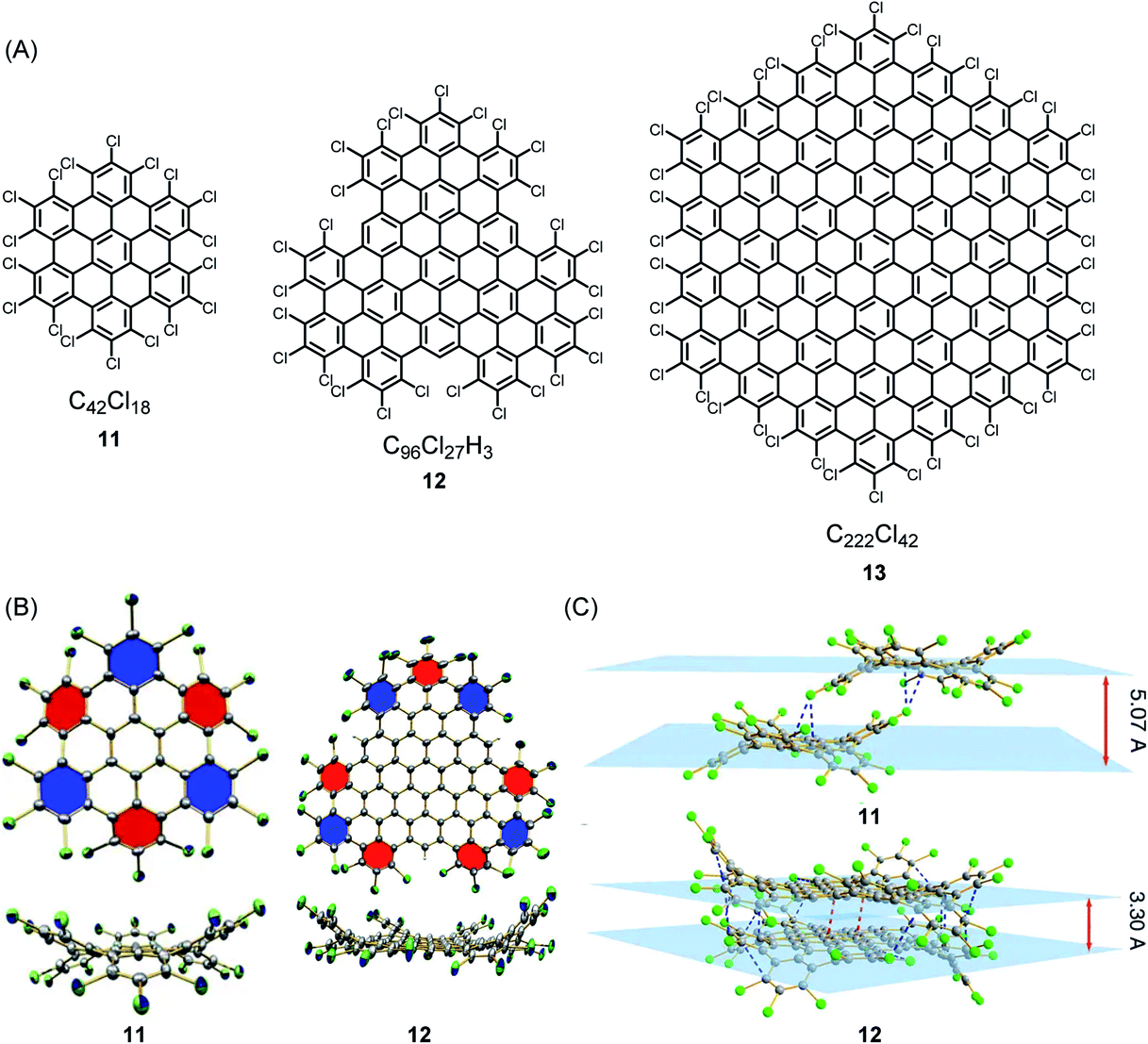 | ||
| Fig. 1 (A) Examples of chlorinated NGs 11–13. (B) Crystal structures and (C) crystal packing of chlorinated NGs 11 and 12.58 Copyright 2013. Adapted with permission from Macmillan Publishers Ltd: Nature Communications. | ||
Importantly, PAHs play a key role as part of the emerging nanoscience field. PAHs could be deposited into ordered monolayers on conducting surfaces such as metal or highly oriented pyrolytic graphite (HOPG), and the resulting patterns could be visualized by scanning tunnelling microscopy (STM), often with atomic resolution. Ground-breaking work in this direction was performed by Roman Fasel and his group. While, for example, HBC and π-extended HBC with two additional rings, or K-regions, could be sublimed under ultrahigh vacuum (UHV) conditions, higher NGs escaped similar treatment due to the higher sublimation point and thermal decomposition.75 Additionally, having soluble, alkyl-substituted NGs available allowed their deposition from solution. The pioneering experiments of Jürgen Rabe and Paolo Samorí demonstrated the formation of an ordered monolayer by physisorption of alkyl-substituted HBC 14, visualizing single molecules with the STM tip immersed in the supernatant solution on the substrate (Fig. 2A and B).76,77 While the crystallization of large NGs is hardly possible and their NMR spectra are severely broadened due to the strong aggregation tendency, STM provided an additional possibility for their characterization, visibly revealing their structures. One can thus conclude that the synthesis of large NGs and their visualization by STM were closely connected. Even more important from an electronic point of view was a subsequent scanning tunnelling spectroscopy (STS) experiment that allowed one to record separate current–potential curves for the aromatic and aliphatic domains of individual molecules, thus disclosing their electronic differences. Later, even more sophisticated electronic functions of a single molecule could be resolved by the Rabe group for dyad molecule 15 (Fig. 2C).78,79 After physisorption, HBC cores can be identified at the corners as bright circular features with central spots corresponding to high tunnelling currents, together with four anthraquinone (AQ) moieties in between (Fig. 2D). The AQ moiety was subjected to donor–acceptor formation with an electron-rich dimethoxyanthracene (DMA) added to the solution (Fig. 2E and F). The dipole arising within the donor–acceptor complex acted as a kind of gate to modulate the current through the HBC. This design could therefore be regarded as a single-molecule FET with source and drain electrodes formed by the STM tip and the conductive substrate.
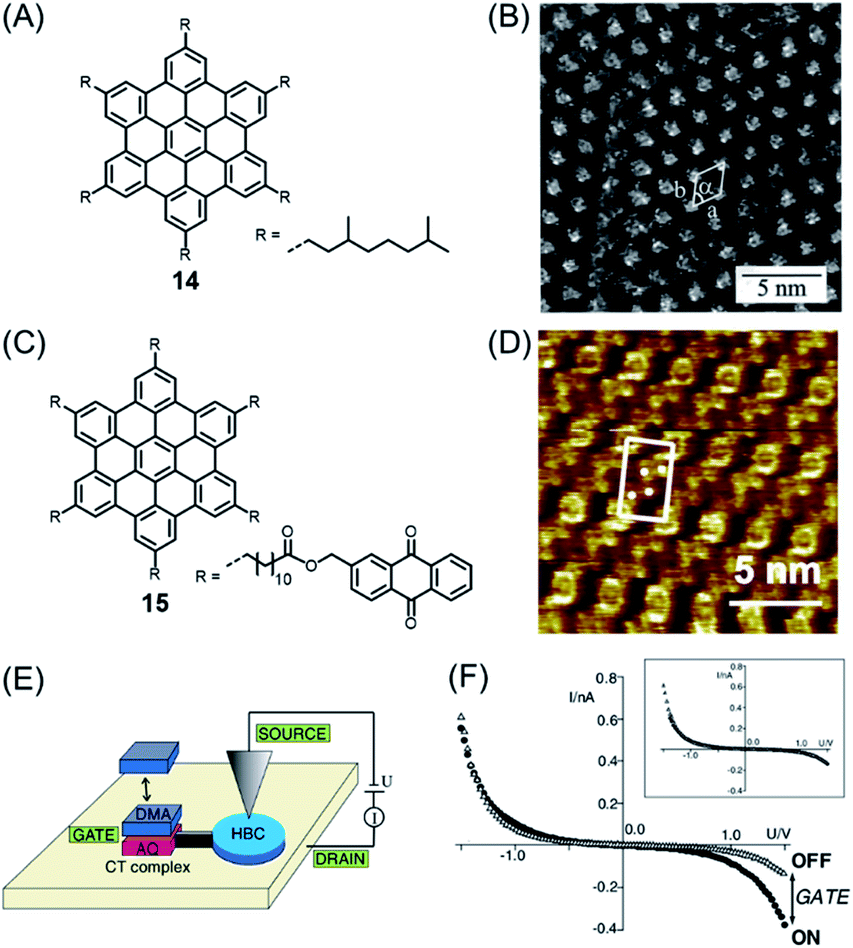 | ||
| Fig. 2 (A) Chemical structure of studied HBC derivative 14. (B) STM current image of 14 at the 1,2,4-trichlorobenzene–HOPG interface. HOPG = highly oriented pyrolytic graphite.77 Copyright 2001. Adapted with permission from the American Chemical Society. (C) Chemical structure of HBC decorated with six AQs 15. (D) STM current images of highly ordered monolayers of 15 on highly oriented pyrolytic graphite. Unit cells and AQs are indicated. (E) Schematic representation of a prototypical single-molecule chemical FET. (F) I–V through HBC cores in domains of 15 and DMA where charge-transfer (CT) complexes are adsorbed (solid circles) or not adsorbed (open triangles). Inset: Shifted and normalized data.79 Copyright 2004. Adapted with permission from the American Physical Society. | ||
Since its beginning, the application of the Scholl reaction for HBC synthesis has offered many surprises.80 The stepwise planarization could be stopped by isolating the dibenzo[fg,ij]phenanthro[9,10,1,2,3-pqrst]pentaphene with a single twisted phenyl substituent.81 Additionally, planarization of the hexaphenylbenzene precursors worked even in the case of pronounced steric hindrance and thus nonplanarity of the new π-system. Thus, dodecamethyl hexaphenylbenzene 16 gave HBC 17, while octadecamethoxy derivative 18 yielded 19, which, due to its doubly concave structure, could bind C60 fullerene, yielding the crystalline columnar structure displayed in Fig. 3.82,83
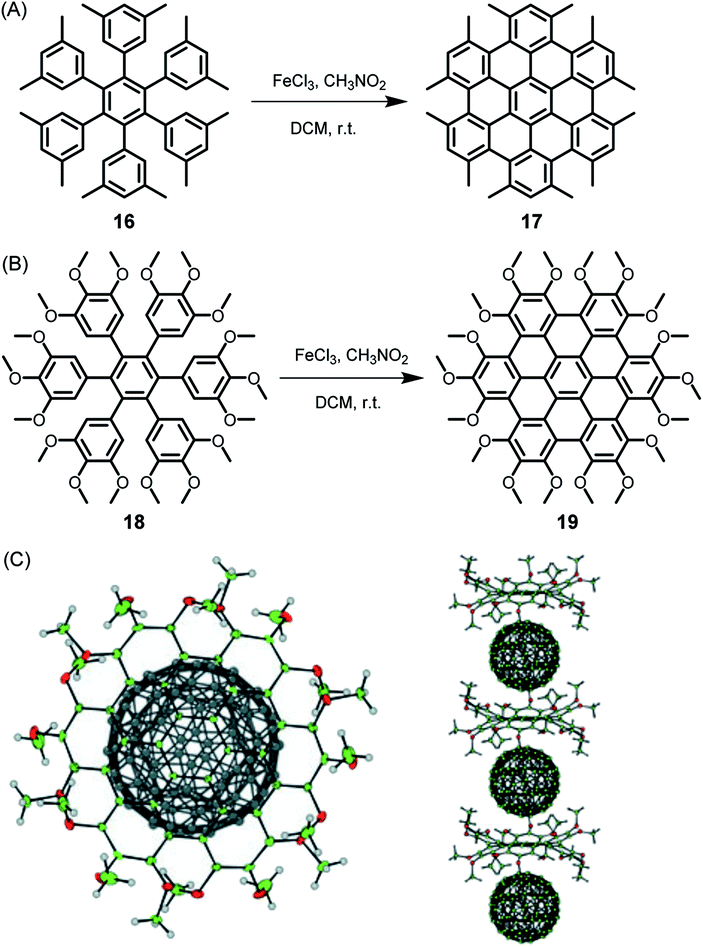 | ||
| Fig. 3 Synthetic route to (A) dodecamethyl HBC 17 and (B) octadecamethoxy HBC 19; DCM = dichloromethane. (C) Single-crystal structure of the complex formed between fullerene and 19: top view and side view of the columnar packing arrangement.82 Copyright 2005. Adapted with permission from WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
The electronic structure of HBC can be described as consisting of only “Clar sextets” and thus as belonging to a family of fully benzenoid PAHs with large highest occupied molecular orbital (HOMO)–lowest unoccupied molecular orbital (LUMO) gaps and pronounced chemical stability.84–87 This situation changes when additional rings are formed at the periphery. To understand the rich opportunities of this extension, introducing the different PAH peripheries of graphenic sheets is important (see Scheme 3). While hexa-peri-hexabenzocoronene (yellow) features armchair edges, its analogue hexa-cata-hexabenzocoronene (violet) possesses coves at the edges. The “upper” part of perylene (sky blue) has a zigzag edge, whereas its bay area represents an armchair. From a chemical point of view, it is relevant that the bay region can act as a diene for Diels–Alder cycloaddition with reactive dienophiles to furnish a new six-membered ring, affording ovalene 21 from bisanthene 20, as displayed in Scheme 4A.88 This scheme also presents an example of PAH modification using the ring closure of an ethynyl unit attached to a bay area (Scheme 4B).89 Depending on the topology of the twisted oligophenylenes and oligoarylenes, six-membered ring formation has also been accomplished by Friedel–Crafts reactions, as exemplified by the synthesis of 25 (Scheme 4C).90
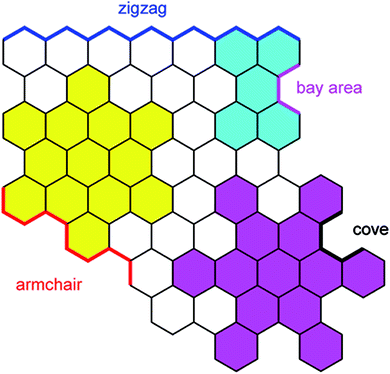 | ||
| Scheme 3 Schematic representation of typical edge structures of PAHs. | ||
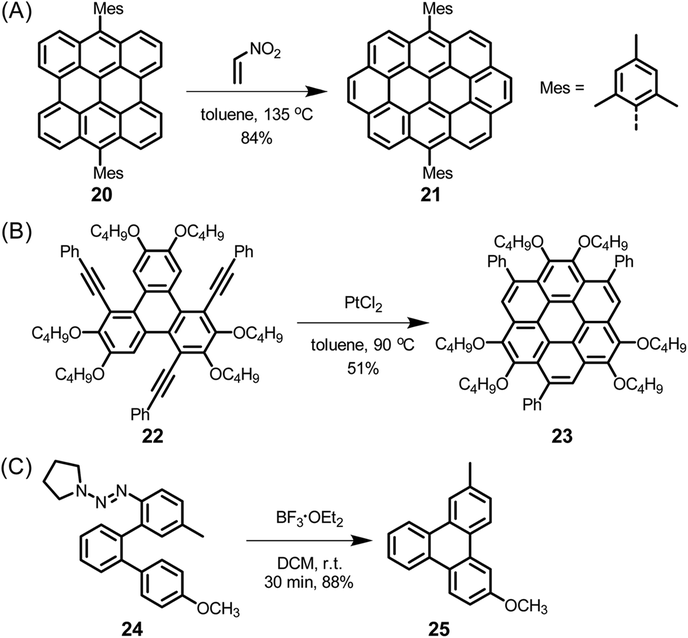 | ||
| Scheme 4 (A) Diels–Alder reaction between bisanthene 20 and nitroethylene. (B) Intramolecular cyclization to realize functionalized coronene 23. (C) Friedel–Crafts reaction to construct triphenylene derivative 25. | ||
In the meantime, HBC and its oligomers have found widespread use as building blocks to synthesize various NGs, and this manifold of hydrocarbons includes nonplanar and chiral structures.91–94 However, a full account of this rich chemistry is beyond the scope of this text.
The stepwise extension of HBCs with extra rings, or K-regions, has been accomplished within the series of 26–29, where the successive lowering of the HOMO–LUMO gap was reflected by the bathochromic shift of the electron absorption bands (Fig. 4A).95–97 A recent case is 29, where HBC was extended by adding four K-regions, which formed extended zigzag edges.98 The HOMO–LUMO gap of 29 was drastically reduced to 2.5 eV in comparison to the value of 3.6 eV for pristine HBC based on density functional theory (DFT) calculations. Zethrenes 30/31, developed by Jishan Wu,99–102 and higher anthenes 32/33 (n = 1 or 2), pioneered by Takashi Kubo,103–105 are representative NGs with zigzag edges, which demonstrated smaller HOMO–LUMO gaps and open-shell biradical character (Fig. 4B).
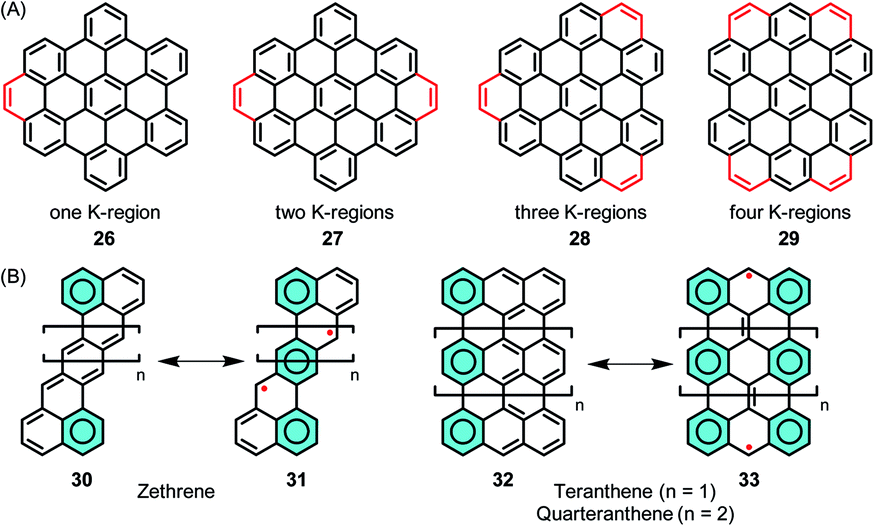 | ||
| Fig. 4 (A) Stepwise extension of HBCs with extra K-regions. (B) Structures of zethrenes and higher anthenes, and their resonance structures. | ||
While such open-shell NGs are typically unstable, dibenzo[hi,st]ovalene (DBOV) 35, which also has zigzag edges, displays closed-shell properties with very high stability (Fig. 5A).106–108 DBOV was revealed to have sharp absorption and emission peaks with a small Stokes shift of 12 nm (Fig. 5B). DBOV exhibited strong red emission with a high photoluminescence quantum yield of ∼80%, which could later be improved up to 97% by substituting the bay regions with aryl groups.109 Although aggregation-caused fluorescence quenching, a typical problem for large aromatic compounds, was observed, solid-state emission and stimulated emission were achieved by using a polymer matrix. Moreover, ultrafast transient absorption measurements by the group of Francesco Scotognella elucidated stimulated emission from DBOV at 695 nm, indicating the potential of DBOV as an optical gain material. Amplified spontaneous emission could be further demonstrated, rendering such NGs highly promising not only for organic light emitting diodes but also organic laser applications. Wu, Casado, Díaz-García and their co-workers later demonstrated the fabrication of lasers using NGs, providing a proof of concept.110 Moreover, DBOV and other NGs, including HBC, were revealed by the group of Mischa Bonn to show fluorescence blinking on timescales useful for modern super-resolution microscopy, including single-molecule localization microscopy and minimal emission flux microscopy.111 Remarkably, DBOV continued to blink in various environments, including air, and maintained blinking behaviour after 125 days of storage, in stark contrast to standard organic dyes such as Alexa 647. The latter requires a special buffer and stops blinking after a few hours. NGs thus also have high potential for imaging applications with super-resolution microscopy, both in bio-related studies and materials science. Additionally, we have also regarded DBOV as a new building block for constructing novel and even larger NGs. Thus, π-extension at the two bay regions of DBOV by annulation of diarylacetylene led to circumpyrene 37 with multiple zigzag edges (Fig. 5C and D), which represents an exciting NG structure studied theoretically but never previously achieved.112
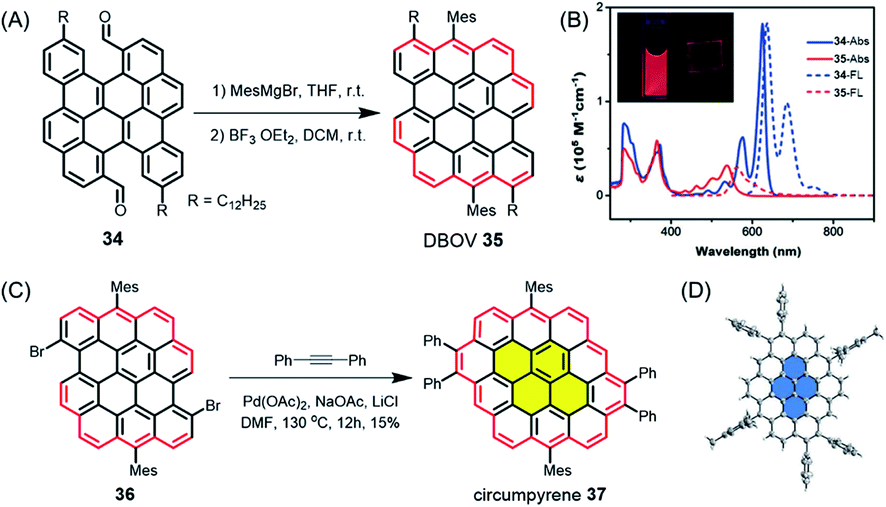 | ||
| Fig. 5 (A) Synthetic route to DBOV 35. (B) UV/Vis absorption and fluorescence spectra of precursor 34 and DBOV 35. Inset: Images of DBOV 35 in solution and 1 wt% polystyrene composite film under UV irradiation.107 Copyright 2017. Adapted with permission from WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. (C) Synthesis of circumpyrene 37via transition metal-catalysed benzannulation; DMF = dimethylformamide. (D) Single-crystal structure of circumpyrene 37.112 Copyright 2019. Adapted with permission from the American Chemical Society. | ||
2.2 On-surface synthesis
As early as 1999, the group of Christof Wöll deposited a hexaphenylbenzene derivative with alkoxy groups on copper surfaces and heated the resulting layers to temperatures above 710 K.113 The oxygen atoms were thought to serve as anchors to prevent the organic compound from desorption under UHV and upon heating. X-ray photoelectron spectroscopy (XPS) and near-edge X-ray absorption fine structure analysis of the product proved the successful planarization of the HBC species. The formation of PAHs by thermally induced dehydrogenation of oligophenylene precursors on a metal substrate, the on-surface variant of the above-described Scholl reaction in solution, has achieved great importance, leading to a variety of NGs and GNRs with fascinating properties in the last decade. While the resolution of STM images was limited in early studies, Roman Fasel demonstrated monitoring of the ring-closing processes of cyclophane 38 to triangular PAH 39 by high-resolution STM, providing unambiguous proof of the structure with atomic resolution (Fig. 6A and B).114 The detailed mechanism of the reaction is still unknown, although its core process can be formally described as an electrocyclic ring closure of an o-terphenyl to a triphenylene moiety accompanied by the loss of hydrogen, which is also reminiscent of the stilbene-(dihydro)phenanthrene transformation.115,116 Photochemical cyclization of 40 to coronene 41 in solution is formerly the same as the conversion of 38 to 39 on surface.117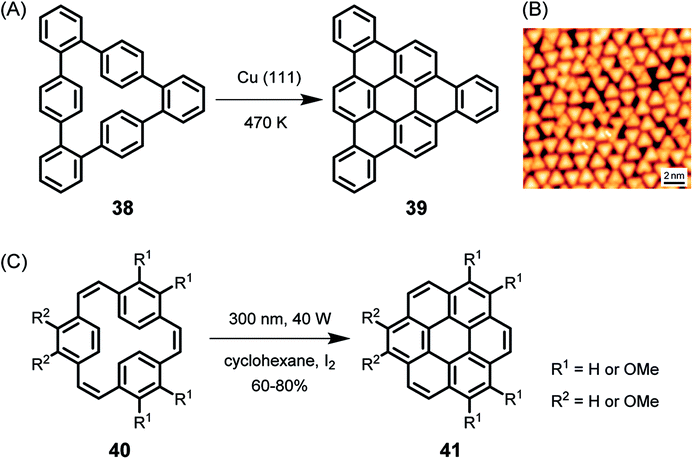 | ||
| Fig. 6 (A) Cyclodehydrogenation of 38 to tribenzo[a,g,m]coronene 39. (B) STM image of 39 on Cu(111); Vs = −0.6 V, I = 50 pA, 5 K.114 Copyright 2010. Adapted with permission from Macmillan Publishers Ltd: Nature Chemistry. (C) Synthesis of coronene compounds via photochemical cyclization of 40. | ||
These PAH syntheses are intramolecular processes. Later, we will consider diradicals as reactive intermediates for polymerization on surfaces, leading to GNRs (see Section 3.2). A kind of preparatory case is the reaction of cation 42 (Scheme 5A).118 In solution, if subjected to deprotonation upon treatment with amine, this cation gives rise to polycyclic azomethine ylide 43. This species can be described as either a 1,3-dipole or a diradical and undergoes a typical 3+2-cycloaddition with olefins as dipolarophiles, providing a protocol to construct nitrogen-containing PAHs (Scheme 5B).119 Furthermore, Ito, Nozaki and their co-workers demonstrated the annulation of 43 with fullerene and even corannulene,120,121 highlighting the unique reactivity of 43. This is a recent example of the famous Huisgen chemistry. Moreover, as demonstrated by Carlos-Andres Palma, 43 can also undergo dimerization and oxidation in solution to realize pyrazine-incorporated hexabenzoperylene (HBP) 47 (Scheme 5C), or dimerization on a metal surface followed by cyclodehydrogenation to yield 48 (Scheme 5D and E).122 Notably, both 47 and 48 are antiaromatic diaza-hexacycles.
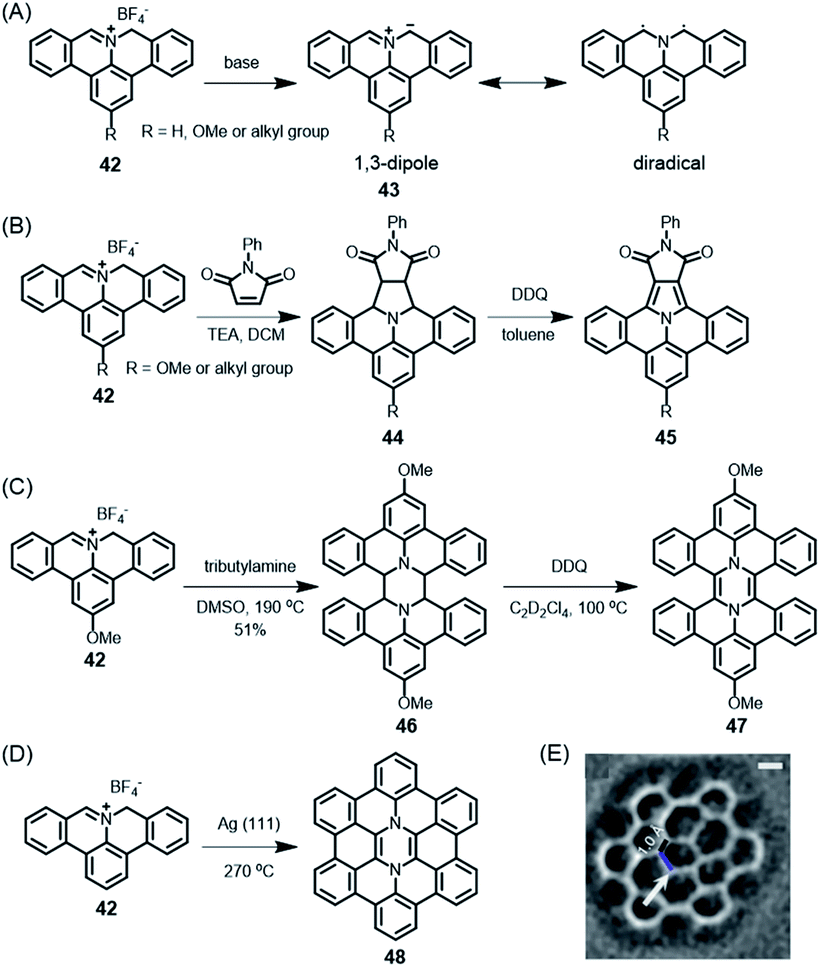 | ||
Scheme 5 (A) Generation of polycyclic azomethine ylide 43 and its resonance structures. (B) N-Phenylmaleimide as an example of a dipolarophile for the 1,3-dipolar cycloaddition reaction with 42; DDQ = 2,3-dichloro-5,6-dicyano-1,4-benzoquinone. (C) Synthesis of diaza-HBP 47 in solution; TEA = triethylamine, DMSO = dimethyl sulfoxide. (D) Synthesis of internally N-doped diaza-HBC 48 on a Ag(111) surface. (E) Laplace-filtered frequency-modulated atomic force microscopy (AFM) data revealing details of the molecular structure; apparent C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C distance of 1.0 Å (black), and N–C distance of 1.8 Å (blue).122 Copyright 2017. Adapted with permission from Macmillan Publishers Ltd: Nature Communications. C distance of 1.0 Å (black), and N–C distance of 1.8 Å (blue).122 Copyright 2017. Adapted with permission from Macmillan Publishers Ltd: Nature Communications. | ||
One might conclude that thermal on-surface reactions basically replicate the solution reactions, but this is not true. Oxidative cyclodehydrogenation in solution is a complex process that depends on the conformation of the polyphenylene precursor and the prevailing spin and charge density distributions within the intermediate (radical) cations. Thus, tetranaphthyl-p-terphenyl 49 affords double [7]helicenes with racemic (50) and meso (51) forms, which can be separated by crystallization utilizing their distinct solubilities (Fig. 7A).123 The enantiomeric forms of chiral isomer 50 can be separated by chiral high-performance liquid chromatography and do not reveal any racemization at room temperature due to the very high isomerization barrier, calculated to be 46 kcal mol−1 by DFT. Assuming a radical cation species as an intermediate for ring closure, the spin density at the α-position of its naphthyl group is much higher than that at the β-position, so carbon–carbon bond formation occurs for the former. In contrast, as proven by Lifeng Chi, an on-surface reaction of the same precursor 49, when thermally induced after adsorption on a metal surface, yields the “fully collapsed” benzo-fused perihexacene 52 (Fig. 7B).124 The interaction between the π-system and the metal appears to favour planarization.
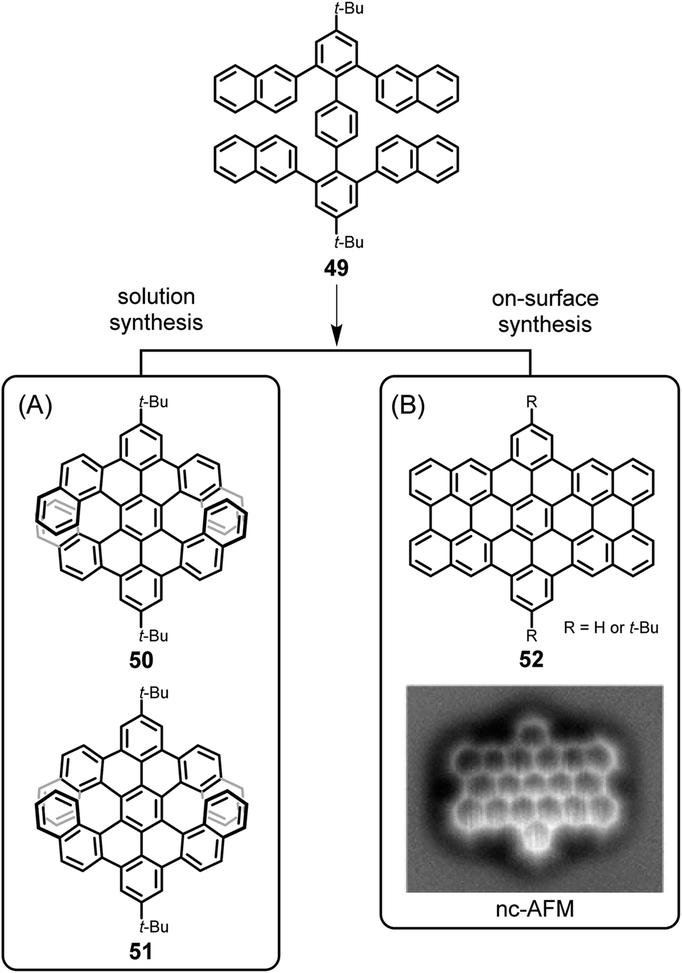 | ||
| Fig. 7 Different cyclodehydrogenation pathways for solution and on-surface syntheses: (A) double [7]helicenes 50 and 51, and (B) benzo-fused periacene 52. Inset in (B): constant-height noncontact AFM (nc-AFM) image of 52 on Cu(110).124 Copyright 2019. Adapted with permission from the American Chemical Society. | ||
One can thus change the course of a reaction in solution by immobilizing a precursor on a surface; more importantly, one can obtain access to molecular structures that cannot be made or are too unstable in solution, such as extended acenes,125,126 peripentacene,127,128 and triangulenes.129–131 NGs thus appear to be ideal model cases for the new chemistry of GNRs. This holds for the (i) synthesis, (ii) spectroscopic and microscopic methods of structure proof, and (iii) understanding of their electronic structures.
3. From NGs to GNRs
3.1 Solution synthesis
The synthesis of a CP, which has been amply described in previous dedicated reviews,4,10,132–134 requires a suitably functionalized building block from which two new bonds, usually carbon–carbon bonds, are formed for incorporation into a chain. Often, transition metal catalysis is involved, such as in the polymerization of acetylenes to realize polyacetylenes or the polycondensation of functionalized arenes to obtain polyarylenes. In these cases, the connection of the repeating units is accomplished via single bonds. Cases can also be found, for example, in the synthesis of poly(phenylene vinylene)s or their nitrogen-incorporating analogues, where the coupling process directly furnishes double bonds.135,136 The synthesis of GNRs is more complex, both conceptually and experimentally, since the growth of the polymer requires the formation of multiple bonds in sequential processes. Referring again to NG synthesis as a test case, one would have to first synthesize polyarylenes (e.g., with naphthalene or anthracene units) or polyphenylenes with additional oligophenylene side arms as precursor polymers. These would then be subjected to a planarization step, possibly mild graphitization, via additional carbon–carbon bond formation by either dehydrogenation or the loss of hydrogen halides or halogens. The prime target is then the synthesis of precursors, such as branched polymers like 55 and 58. Two critical issues thus arise, namely the synthesis of the precursor polymers, for example, 55 and 58 by Suzuki and Yamamoto-type coupling, respectively, and the subsequent dehydrogenation (Scheme 6A and B).137–139 Regarding the first step, the molecular weights are known to be limited by side reactions, especially the loss of functional groups. The possibility of synthesizing multiply branched polyphenylene 61 by repetitive Diels–Alder cycloadditions of AB-type building block 60 has therefore been a major breakthrough (Scheme 6C).140 This approach has two main advantages: the formation of each new benzene ring is “driven” by the extrusion of carbon monoxide, and no side reaction occurs as in the above protocols. Remarkably, polymer 61 could be obtained as a soluble material that, according to analysis by size-exclusion chromatography and laser light scattering, had an average molecular weight of ∼600 kDa. Size, as shown for NGs, is of course a critical issue, not only for electronic reasons but also for when one wants to bridge the channel of an FET with GNRs as semiconductors. Notably, the concept of repetitive Diels–Alder cycloadditions has also been extended to the synthesis of polyphenylene dendrimers.141–144 The diene and dienophile functions have been combined with the AB2 moiety, which serves as a branching unit. A second critical issue in GNR synthesis is cyclodehydrogenation by the Scholl reaction. The fact that giant NGs can be made from polyphenylene precursors by the loss of more than a hundred hydrogen atoms through the formation of multiple new carbon–carbon bonds is amazing. In the case of GNRs, this fusion has even been extended to polymers with ultrahigh molecular weights. | ||
| Scheme 6 (A) Suzuki- and (B) Yamamoto-type coupling and (C and D) Diels–Alder cycloadditions for realizing GNRs in solution. | ||
Does this synthetic protocol leading to GNRs meet the strict requirements of CP synthesis in terms of structural perfection, processability and scalability? The question regarding failure of the flattening process cannot be set aside. First, electron absorption spectroscopy results reflect the formation of extended π-systems without being able to rigorously exclude residual 3D units. Raman and IR spectroscopy serve as additional tools to detect possible defects resulting from incomplete dehydrogenation. STM visualization, after the deposition of polymers from solution, provides additional microscopic evidence – however, without being able to exclude the possibility that defective molecules have gone undetected. An advantage of precursor protocols for polymer synthesis is often that the precursors are more soluble and can be processed from solution, followed by transformation into the target polymer.145 In the case of GNR 62, surprisingly, solution liquid-phase processing is still possible. As described for NGs in Section 2.1, this is due to the attached alkyl chains. In both cases, however, one expects pronounced aggregation. The synthesis of GNRs can be modified for the incorporation of functional substituents to build in chromophores or stable free radicals, which will be useful later.146–150 Additionally, GNRs can be subjected to further transformation by employing chlorination58 and potentially hydrogenation,151 as already established for NGs. Regarding scalability, the above-described synthesis has been performed on a gram scale and can certainly be developed for even larger quantities. Furthermore, an advantage of this repetitive cycloaddition is that the AB-type moieties can be modified to make the ribbons increasingly wider – without ever giving up the strict requirement of structural perfection. One approach uses the somewhat larger tetraaryl cyclopentadienone 63, according to Scheme 6D,152 and another employs Diels–Alder additions not only for “longitudinal” polymer growth but also for “lateral” broadening of the ribbons.153 The key trick in the latter method is to avoid “spatial overlap” of benzene rings, since they must all fit into one plane upon cyclodehydrogenation.
Structural perfection and processability are mandatory requirements for studies on nanoscience and device fabrication. Solution deposition of GNR 62 allowed the fabrication of FETs.154–156 Therefore, the active components were present as a network of ribbons whose contact resistance was the limiting factor for the charge-carrier mobility. The fabrication of single-GNR FETs will be described below. A preliminary experiment aimed at the electronic characterization of single GNRs was performed that consisted of depositing 62 onto a channel formed from metal electrodes and recording a current–potential curve (Fig. 8A and B).157
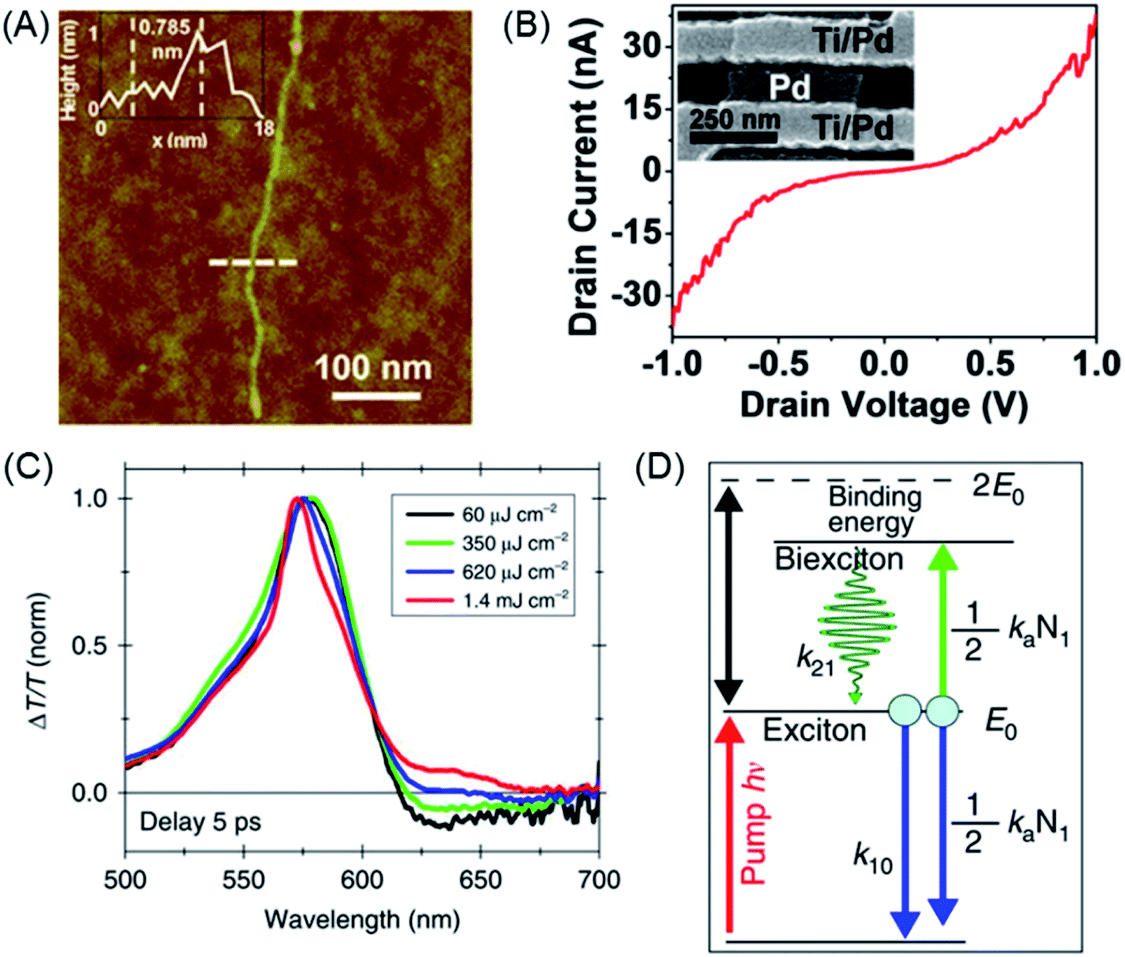 | ||
| Fig. 8 (A) AFM tapping-mode height image of GNR 62 with length >500 nm. The inset shows a height profile of GNR 62 (at the dashed line), revealing a height of 0.785 nm. (B) I–Vd characteristics of an individual GNR device after metal angle deposition to obtain a channel length of ∼20 nm. The inset shows a scanning electron microscope image of a 20 nm gap between Ti/Pd and angle-deposited Pd.157 Copyright 2014. Adapted with permission from the American Chemical Society. (C) Normalized ΔT/T spectra of GNR 62 for different excitation fluences at a fixed pump–probe delay of 5 ps. (D) Sketch of the energetic levels and the kinetic model.160 Copyright 2016. Adapted with permission from Macmillan Publishers Ltd: Nature Communications. | ||
GNRs provide new prospects in many other areas of functional studies.158,159 The exciton dynamics of GNR 62 were investigated by ultrafast transient absorption spectroscopy (Fig. 8C).160 Stimulated emission could be detected, which could be assigned to biexciton formation, which possessed a surprisingly high binding energy (Fig. 8D). We will further exemplify the power of solution synthesis of GNRs when we discuss even more complex chemical modifications in Section 4, but introducing the complementary role of on-surface techniques as a new route to NG chemistry is helpful at this point.
3.2 On-surface synthesis
On-surface reactions of reactive monomers and polymerizations of their reactive intermediates were studied by Bent in 1993,161 followed by many ingenious attempts at obtaining, for example, polyfluorenes after thermally induced carbon–halogen cleavage of fluorene species and subsequent polymerization.162 The reaction could be monitored by STM. We could utilize the concept proposed by Roman Fasel and his group for the on-surface synthesis of polyanthrylene 66 at 200 °C, followed by thermal dehydrogenation to realize the target GNR 68 at 400 °C (Fig. 9A).163 Visualization of the new GNR and the perfection of its edges by STM is impressive (Fig. 9D). This ribbon, along with its width, is denoted 7-AGNR, where “A” indicates the armchair periphery and 7 is the number of carbons counted across the ribbon. As we have described in detail elsewhere, the width of the ribbon allows synthetic control of the finite bandgap in an atomically precise way.24,52 To make GNRs wider, one can just use a different monomer design. As can be seen from Fig. 9B and E, dibromo-o-terphenyl 69 serves as a precursor to fabricate a 9-AGNR.164 The synthesis of this monomer might appear straightforward at first glance. However, its purification by repeated crystallization is crucial, since even traces of the monobromo compound would be preferentially sublimed and act as endcappers, thus limiting the achievable molecular weight. The narrower counterpart, 5-AGNR 74, predicted to have a particularly low bandgap and thus be favourable for achieving high charge-carrier mobility, can be obtained from functionalized naphthalene or perylene precursors. Thus, as has been shown by the group of Lifeng Chi, the commercially available tetrabromonaphthalene 72 gives rise to 5-AGNR 74 without requiring dehydrogenation (Fig. 9C and F).165 The same product can be obtained from, for example, a dibromoperylene, following thermal dehydrogenation of the twisted precursor polymer.166 Access to 5-AGNRs becomes possible by on-surface and solution polymer chemistry.167 The tetrabromoperylene is obtained by Hunsdiecker decarboxylation of the perylene tetracaboxiimide.168 This opens an interesting pathway from colourant to graphene chemistry. We also developed the perylene-based chromophore into a homologous series of dyes with absorption bands far into the near-infrared (NIR) range.169–171 These structures comprise 5-AGNRs.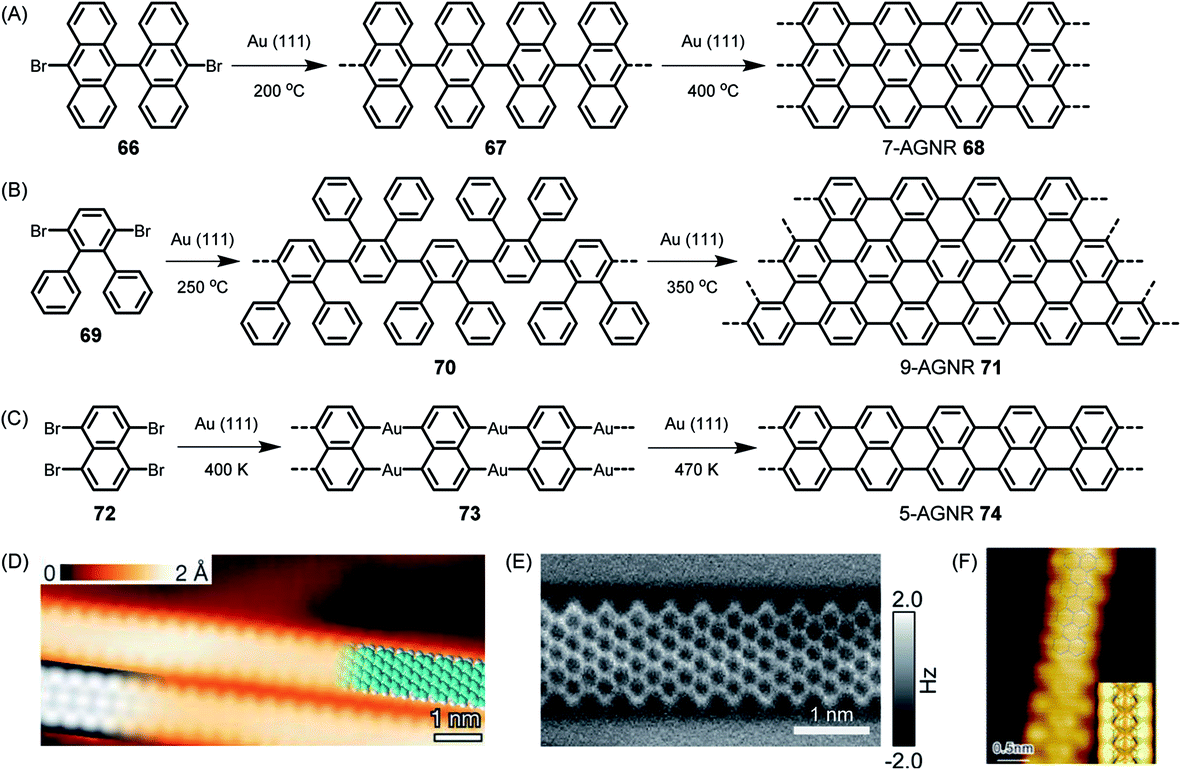 | ||
| Fig. 9 Reaction schemes for (A) 7-AGNR 68, (B) 9-AGNR 71, and (C) 5-AGNR 74. (D) High-resolution STM image with a partly overlaid molecular model (blue) of 68. At the bottom left is a DFT-based STM simulation of 68, shown as a greyscale image.163 Copyright 2010. Adapted with permission from Macmillan Publishers Ltd: Nature. (E) High-resolution nc-AFM frequency-shift image of 71.164 Copyright 2017. Adapted with permission from the American Chemical Society. (F) High-resolution STM image of 5-AGNR 74. The chemical structure of GNR 74 is overlaid on the image for better illustration. Inset: DFT simulated STM image of GNR 74 on Au(111) surface.165 Copyright 2015. Adapted with permission from the American Chemical Society. | ||
Other sources of diradical formation can be utilized in addition to the most commonly employed carbon–halogen cleavages, such as the Bergman cyclization of diethinyl arene species.172–174 In an on-surface process, this ring closure is followed by polymerization. Remarkably, to be practically useful in solution, this ring closure is performed in the presence of a cyclohexadiene as a hydrogen source to avoid coupling between radicals. Currently, the ever-growing number of studies on on-surface syntheses are tackling an increasing number of chemical issues, such as phthalocyanine formation from phthalodinitrile, head-to-tail polymerization starting from nonsymmetric monomers, or copolymerization of different monomers, with many more to come.172,175–178
The above cases, while extremely useful, rely on carbon–carbon bond formation via aryl–aryl coupling. Under the same conditions, if the precursor polymer contains a methyl group in the spatial neighbourhood of another aryl unit, then a kind of CH-bond activation occurs, leading to bond closure between the aryl and methyl groups. This affords five-membered rings to yield fluorene-type structures, such as 77 in Fig. 10 (ref. 179 and 180), or new six-membered rings, as in the case of 80 in Fig. 11.181 This case deserves special attention because it gives rise to a GNR with zigzag edges. Polymer 80 is unique since, unlike other GNRs, its synthesis has no equivalent in solution synthesis. More importantly, DFT calculations predict extremely low bandgaps and the presence of edge-localized electronic states. This was proven by Roman Fasel et al. via STS. For this purpose, the polymer had to be lifted from the metal surface by shifting it with the tip onto a sodium chloride monolayer, thus weakening the electronic interaction between the metal and adsorbate. This interaction is one of the problematic aspects of the metal surface, although it is highly advantageous for the synthesis (see below).
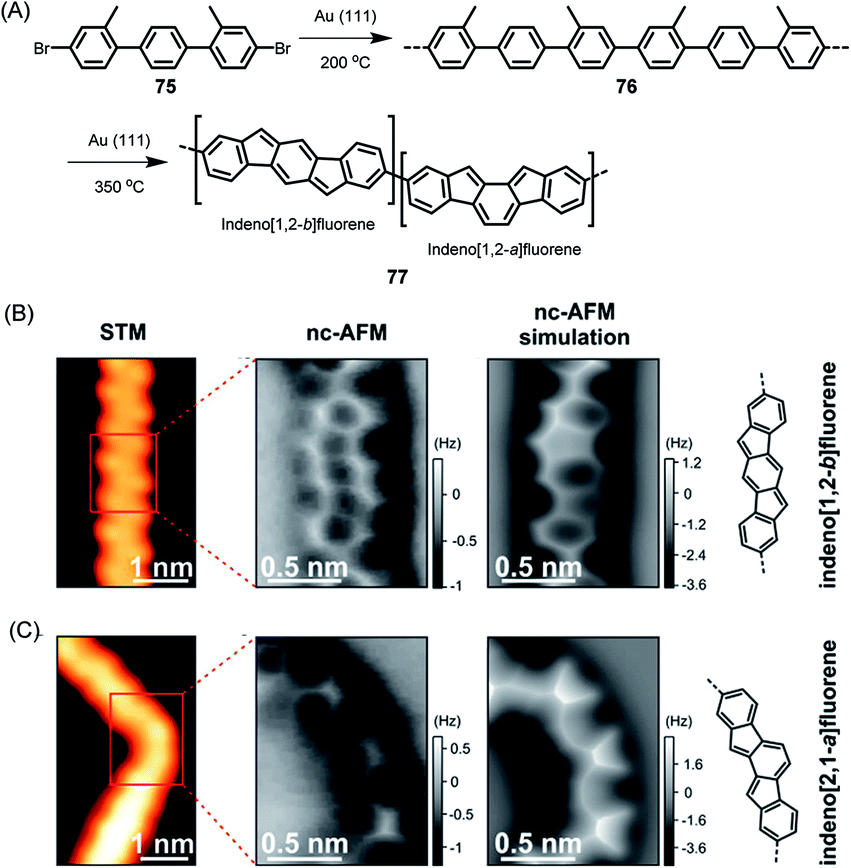 | ||
| Fig. 10 (A) On-surface synthesis of indenofluorene-based polymer 77. (B and C) STM images, constant-height frequency-shift nc-AFM images, simulated nc-AFM images and chemical structures of the straight and bent segments of 77.180 Copyright 2017. Adapted with permission from the American Chemical Society. | ||
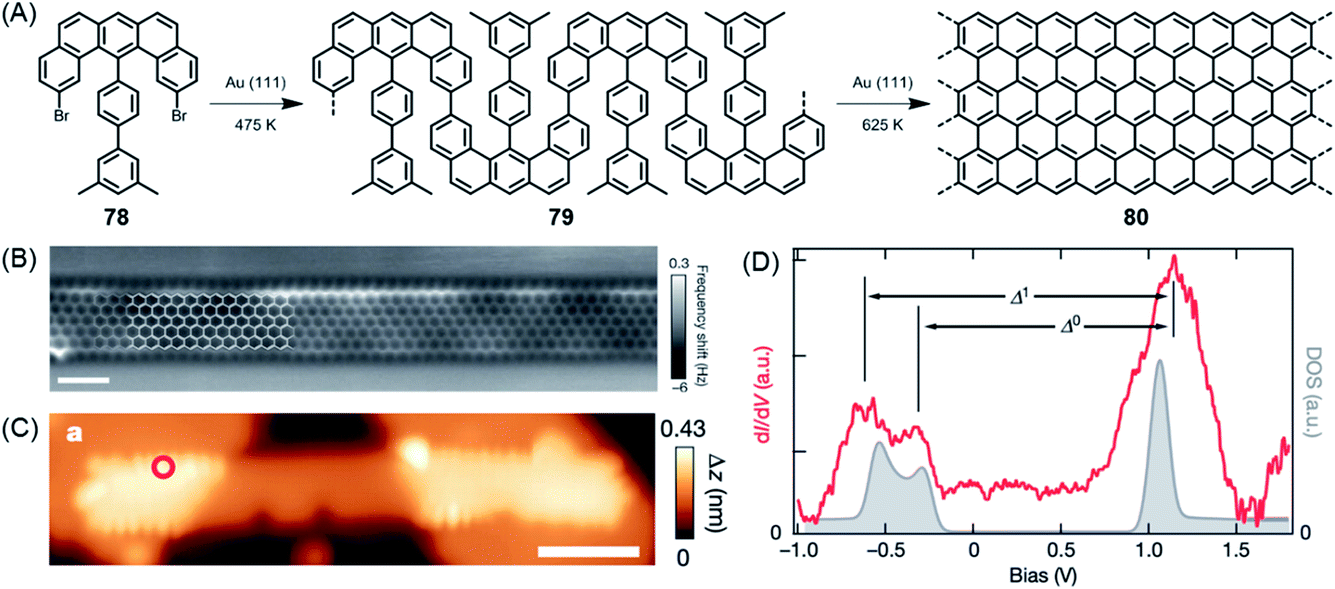 | ||
| Fig. 11 (A) Surface-assisted synthesis of zigzag-edged GNR 80. (B) Constant-height nc-AFM image of GNR 80 on Au(111). (C) STM topography image of GNR 80 bridging two NaCl monolayer islands. (D) Differential conductance (dI/dV) spectrum (red) taken at the zigzag edge marked by the red circle in (C), and the quasiparticle density of states (DOS; grey).181 Copyright 2016. Adapted with permission from Macmillan Publishers Ltd: Nature. | ||
While the on-surface synthesis of NGs and GNRs appears to be a fascinating branch of current nanoscience, its scope and limitations deserve further discussion. First, scanning probe methods, preferably STM and nc-AFM due to the accessible atomic resolution, allow one to precisely analyse the resulting structures and follow the mechanistic details of product formation. Under the prevailing UHV conditions, (di)radical intermediates can be made to couple without side reactions involving solvent molecules, which could quench the radical in solution chemistry. Second, apart from the beautiful visualization of the target polymers as part of the proof of the structure, these methods allow detailed electronic characterizations (see below). Third, NGs and GNRs can be synthesized that are not accessible by solution chemistry or that escape further characterization due to the limited solubility and/or stability. Fourth, feeding different monomers into the UHV chamber provides access to more complex GNR structures, such as by the site-selective incorporation of heteroatoms and the formation of molecularly defined p–n-junctions.182 However, characteristic conceptual and experimental drawbacks exist. First, the reactions require the assistance of a metal to stabilize either the radical intermediates or the target structures, as well as to catalyse the cyclodehydrogenation to form C–C bonds. The π-systems thus obtained may well owe their stability to the electronic interactions with the metal. This can obscure the intrinsic properties of the adsorbate, such as the bandgaps. Second, the subsequent utilization of thus-formed GNRs in FET devices requires transfer from the metal to insulating substrates. This step is certainly an additional complication, but suitable lift-off techniques are available.183–186 Even if the visualization becomes more demanding, it might be desirable to perform similar GNR syntheses on insulating surfaces with the use of metal atoms. Third, deposition of the precursor molecules by sublimation in UHV must be possible, which, as already mentioned, imposes restrictions on the molecular size. Alternative deposition techniques are currently being studied, such as electrospray ionization deposition.156 Fourth, upscaling is certainly an issue in terms of the small amounts of material obtained. One might respond, however, that making a structurally precise monolayer with designed electronic characteristics or with a precise pore size is an extreme form of atom efficiency. Working FET devices can be fabricated by transferring a monolayer film of GNRs synthesized on a surface. Furthermore, chemical vapour deposition (CVD) allows not only graphene fabrication from carbon precursors such as methane,187–189 but also GNR synthesis using the same dihalogenated precursors as in the on-surface synthesis described above, even under atmospheric pressure (Fig. 12A).190,191 The structure of CVD-grown GNR has been characterized by Raman spectroscopy and XPS, the results of which are both in excellent agreement with those obtained by the UHV method (Fig. 12B and C).
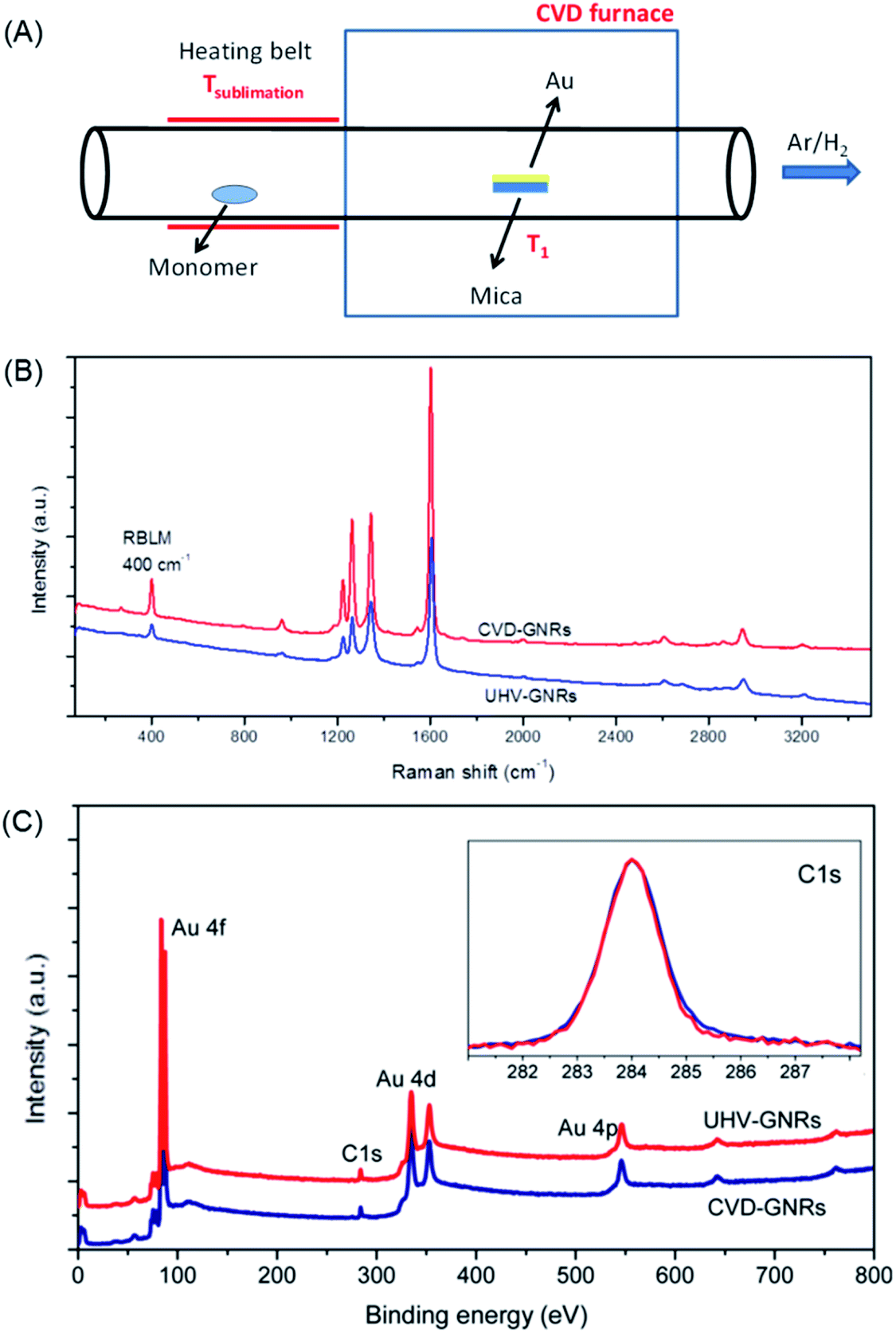 | ||
| Fig. 12 (A) Schematic illustration of GNR synthesis by CVD. (B) Raman and (C) XPS spectra of the UHV-grown and CVD-grown GNRs for comparison. In the inset of (C), the C 1s core-level spectra are presented.190 Copyright 2016. Adapted with permission from the American Chemical Society. | ||
As is the case for all new chemical procedures, many challenges remain for mechanistic studies and structural modifications. Regarding the former aspect, the coupling of dihaloarene molecules on metal surfaces has been described as an Ullmann-type reaction, which is known in organic chemistry as the biaryl formation of haloarenes upon treatment with copper powder.192,193 This reaction implies electron transfer, such that the halogen is removed as a halide anion, whereas in the above case, carbon–halogen cleavage occurs homolytically. Bromine or iodine atoms can be detected by STM but may readily evaporate upon heating. This swift removal does not prevent halogen from reacting with growing chain ends and acting as an endcapper. Further chain growth may also be inhibited by the premature removal of hydrogen from the branched or dendritic precursors forming C–C bonds.194 Reactions under UHV conditions will thus not mean that no side reaction occurs. Nevertheless, the use of diiodinated monomers was found to reproducibly provide longer polymers compared with their dibrominated counterparts, which could be explained by the lower temperature of the homolytic cleavage of the carbon–halogen bonds, suppressing the undesired release of hydrogen.195 Another essential feature is connected to the concept of “immobilization”. One may expect some stabilization of diradicals by interactions with the metal or by the intermediate formation of covalently bound carbon–metal species. Dimerization or polymerization, however, can only occur if the intermediate radical species can still diffuse on the surface. Here, a brief look at Carothers’ equation is instructive; it describes the molecular weight achievable by step-growth polycondensation as a function of the degree of conversion.196 High molecular weights can only be obtained at high degrees of conversion, that is, when higher oligomers begin to condense. In on-surface synthesis, however, larger oligomers may no longer be able to migrate, so growth is expected to mainly arise from the capture of fresh monomers.
The main motivation for GNR synthesis was to obtain FETs with pronounced on–off ratios, while maintaining the high charge-carrier mobility of graphene. The groups of Jeff Bokor and Roman Fasel could lift-off 9-AGNR 71 from the metal after on-surface synthesis, achieve deposition on a substrate with a hafnium oxide layer as the gate dielectric, and fabricate single-ribbon FETs with a remarkable on–off ratio of 105 and a high on-current of >1 μA at a drain voltage of −1 V (Fig. 13).197 However, the challenge of contact resistance with the metal electrodes hindered demonstration of the expected high mobilities of the GNRs. Close collaboration between chemists, physicists and electrical engineers is essential for further improvement of FET devices using synthesized GNRs. This might be achieved through further optimization of (1) GNR synthesis, in terms of a lower bandgap and a larger length, (2) the GNR transfer process, and (3) device fabrication, including the choice of electrode and gate dielectric materials.
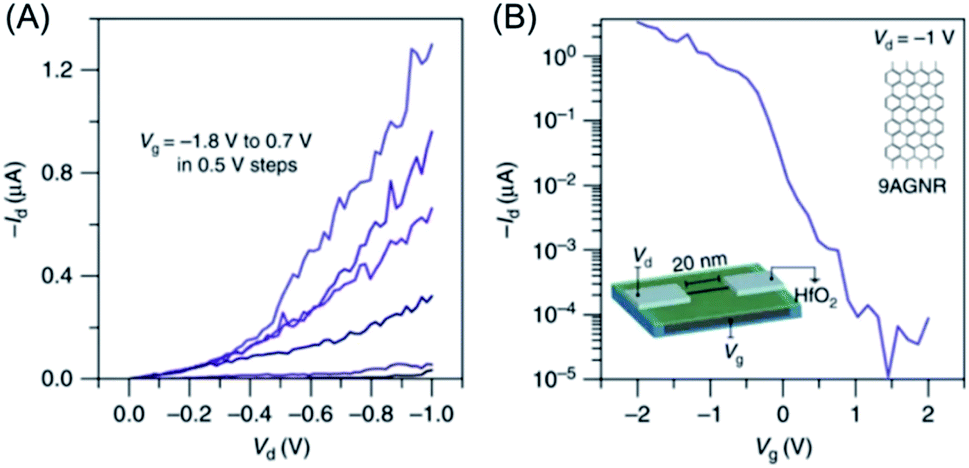 | ||
| Fig. 13 (A) Id–Vd characteristics of the 9-AGNR FET. (B) Id–Vg of the device showing high Ion > 1 μA for a 0.95 nm wide 9-AGNR and a high Ion/Ioff of ∼105.197 Copyright 2017. Adapted with permission from Macmillan Publishers Ltd: Nature Communications. | ||
4. Synthesis and opportunities in physics
Does a chemist target small or polymeric molecules only because the underlying structure could be imagined, and is a successful synthesis sufficient justification for this research? GNRs, as a new generation of CPs, are certainly a challenge for precision polymer synthesis, but they have been fabricated for their extremely promising electronic, optical and even magnetic functions. This is why the above text, while focusing on synthetic concepts, also includes brief excursions into physics that are hopefully not too superficial. In this section, we focus on future opportunities in materials science, even if a very large gap remains between the initial synthesis and robust technologies. We combine organic and macromolecular, as well as solution and on-surface chemistry.The role of surface physics in achieving the final step towards realizing zigzag GNR (ZGNR) 80 is not obscured in noting the basic chemical issues. The nature of the periphery can be controlled by the design of the monomeric building blocks, but the monomer must first be synthesised. Without going into detail, the 12-step synthesis of the monomer 78 needed for the achievement of the ZGNR 80 can be said to be far from trivial PAH chemistry. A closely connected field of research in which all the above synthetic aspects must be combined is the formation of high-spin structures. The creation, manipulation and transport of spins define special challenges in graphenic structures due to their importance for future spintronics and quantum technologies.198–201 Defects in semiconductor materials can give rise to high-spin structures, but the challenge is to form these defects in a controllable fashion.202–204 This so-called defect engineering, a well-established protocol for inorganic materials such as steel and ceramics, has also recently been applied to organic materials.205 How can one achieve deviations from the regular “honeycomb” lattice of graphene in a rational way? Extending PAHs by the formation of additional rings, as described above, can produce not only six-membered, but also five- and seven-membered rings. A remarkable case is compound 85 (Fig. 14A), which has been made available by Xinliang Feng and Junzhi Liu.206 Five-membered rings are first formed after 1,2-phenyl migration upon oxidative treatment of 81; then, seven-membered rings result from Friedel–Crafts ring closures. Dehydrogenation of 84 towards the realization of fully unsaturated NG 85 incorporating two azulene units is accompanied by a drastic bathochromic shift of the absorption band of 435 nm. The reason for this shift is the occurrence of biradical structure 86, which can be corroborated by electron paramagnetic resonance (EPR) spectroscopy and superconducting quantum interference device (SQUID) studies (Fig. 14B and C). A related case is that of diindeno-fused bichrysenyl 87, which gives rise to diradical 88 (Fig. 14D and E).207 Pulsed EPR spectroscopy of 88, measured by the group of Lapo Bogani, provides the spin lattice and coherence times as characteristic features of the high-spin structure.208 The significance of high coherence times will be reconsidered for spin-bearing GNRs below.
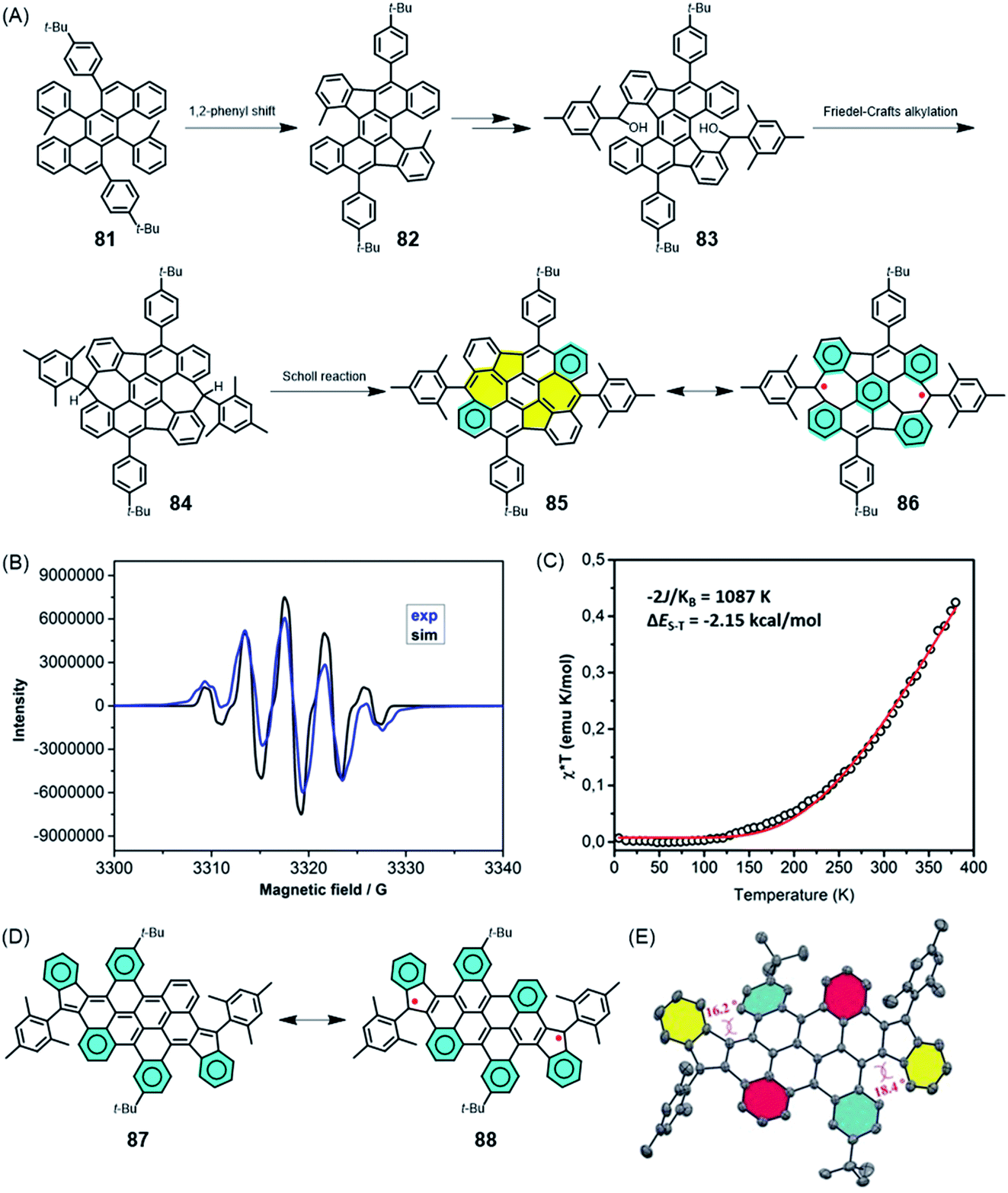 | ||
| Fig. 14 (A) Synthetic route for 85 and its resonance structure 86. (B) EPR spectrum of 85 in toluene solution at 240 K (blue line), and the corresponding simulated spectrum (black line). (C) Experimental χ*T–T curve of a powder sample of 85 measured via SQUID (black circles). The red curve is a fit to the experimental data using the Bleaney–Bowers equation with ge = 2.206 Copyright 2019. Adapted with permission from the American Chemical Society. (D) Resonance structures and (E) crystal structure of 87.207 Copyright 2017. Adapted with permission from Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
An example of a diradical structure that results from topological frustration rather than from five- or seven-membered defects is the case of 90, the so-called Clar goblet with a bowtie shape, which was introduced by Roman Fasel and Xinliang Feng (Fig. 15A).209 It can be fabricated by on-surface thermal treatment of 89 and was studied by STM and spin excitation spectroscopy (Fig. 15B–E). The essential outcome is a robust antiferromagnetic order with an exchange coupling strength of 23 meV, which is much higher than the Landauer limit of minimum dissipation at room temperature.210
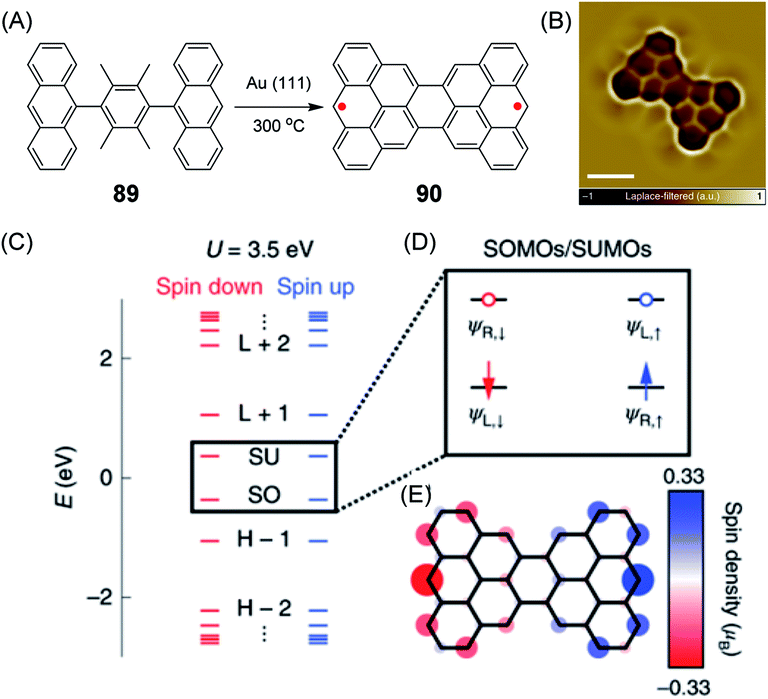 | ||
| Fig. 15 (A) Synthetic route for 90 on a Au(111) surface. (B) Laplace-filtered ultrahigh-resolution STM image of 90; scale bar: 0.5 nm. (C) Energy spectrum of 90; the labels H−2, H−1, SO, SU, L+1 and L+2 denote the HOMO−2, HOMO−1, SOMOs, SUMOs, LUMO+1 and LUMO+2 of 90, respectively. (D) Schematic representation of frontier states; ΨL,↑/↓ and ΨR,↑/↓ indicate spin-polarized wave functions localized on the left and right sides of 90, respectively. Blue/red arrows denote spin up/spin down electrons, respectively. Blue/red open circles denote spin up/spin down unoccupied levels, respectively. (E) Mean-field Hubbard spin density distribution of 90. Blue/red isosurfaces denote spin up/spin down densities, respectively.209 Copyright 2020. Adapted with permission from Macmillan Publishers Ltd: Nature Nanotechnology. | ||
When discussing edge-localized and spin-bearing structures, ZGNR 80 appears to be an ideal model (Fig. 11A).181 The trouble is its high reactivity. When exposed to an oxygen-containing atmosphere, it readily undergoes reactions, presumably under the formation of carbonyl functions, among others, and thus escapes detailed studies. A surprisingly simple way of producing spin density on a GNR is to use the bromo-functionalized GNR 92, prepared in solution as mentioned above, and subject it to palladium-catalysed coupling reactions with the gold complex of the nitronyl nitroxide radical (NIT), affording the connection of stable free radicals at the GNR edges of 94 (Fig. 16A).146 The attached peripheral radicals inject part of their spin density into the π-system of the GNR, which remains chemically stable and becomes susceptible to electron spin resonance (ESR) experiments (Fig. 16B). According to Lapo Bogani, NIT-GNR 94 demonstrates an extremely high spin coherence time of 0.5 μs at room temperature. While this is a fundamental study, a stable phase relation is mandatory before qubits can enter superposition within quantum computing.
 | ||
| Fig. 16 (A) Synthetic route for NIT-polyphenylene 93 and NIT-GNR 94. (B) Multifrequency ESR spectra for NIT-polyphenylene 93 (green) and NIT-GNR 94 (red), along with simulations (black), plotted against the magnetic field from the edge-state resonance.146 Copyright 2018. Adapted with permission from Macmillan Publishers Ltd: Nature. | ||
Equally important for the future of computing is access to topological insulators that have been obtained in 2D- and 3D-inorganic structures. During syntheses of the zigzag-extended 7-AGNRs 95 and 96, utilizing the cyclization involving methyl groups in the synthesis of ZGNR 80 described above, Oliver Gröning and Roman Fasel theoretically revealed that such zigzag edge extension of AGNRs can create topological insulators in quasi-1D-GNRs (Fig. 17A).43 The concept is based on the electronic characterization of a cis-polyacetylene by the Su–Schrieffer–Heeger theory, which uses two coupling parameters to describe the interactions across the double and single bonds.211 This concept is extended to zigzag-extended AGNRs such as 95 and 96 by depicting the intramolecular electronic coupling between the electronic states at the zigzag edges on opposite peripheries. In this theoretical model, GNR 95 was predicted to be a trivial insulator with a reduced bandgap compared to that of the pristine 7-AGNR. STS studies revealed a significantly lower bandgap of 0.65 ± 0.1 eV for GNR 95, 1.75 eV smaller than the 2.4 eV bandgap of the 7-AGNR. Moreover, GNR 96 was characterized as a topological insulator with unique topological end states. Demonstration of this topological end state in GNR 96 was hindered by its hybridization with the end state typical for 7-AGNRs terminated with zigzag edges.212 Nevertheless, the fabrication of heterojunctions of GNR 96 and pristine 7-AGNR allowed unambiguous experimental elucidation of the topological end states at the junctions, which fully agreed with the theoretical prediction (Fig. 17C and D).
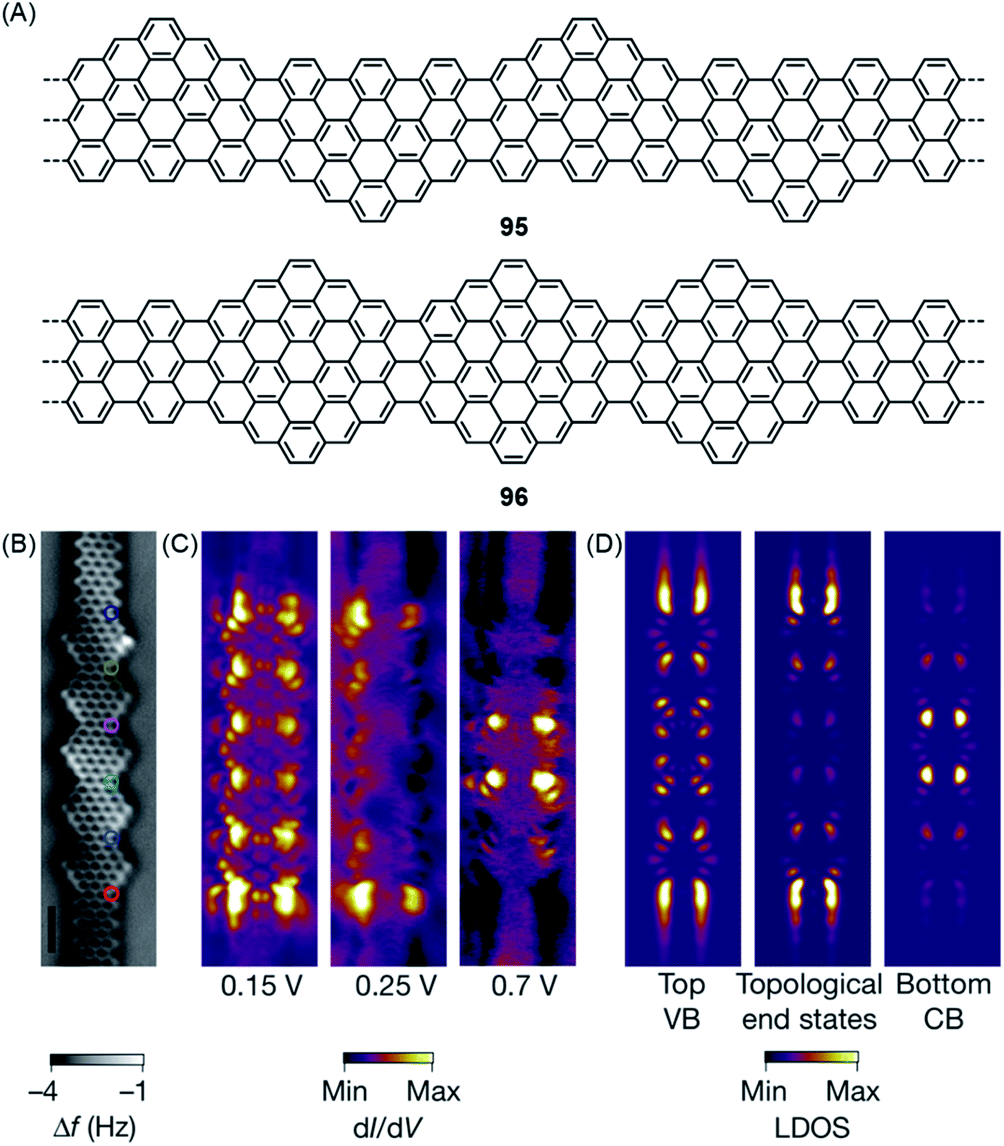 | ||
| Fig. 17 (A) Chemical structures of GNRs 95 and 96 with topological phases. (B) Constant-height nc-AFM image of GNR 96 on Au(111). (C) Experimental and (D) simulated dI/dV maps of GNR 96 on Au(111).43 Copyright 2018. Adapted with permission from Macmillan Publishers Ltd: Nature. | ||
5. Conclusion
NGs continue to present great challenges for synthesis, theory and spectroscopy and will serve as increasingly relevant active components of (opto)electronic devices. Promising examples include applications in photodetection, sensing and organic lasing. Moreover, special importance might also be acquired by chiral NGs in view of the chiral-induced spin-selectivity effect.213–215 This promises control over long-range electron transfer processes with many applications, for example in biorecognition and spintronics. Obtaining enantiomerically pure NGs not only by chromatographic separation but also by enantioselective synthesis would be helpful. Here, synthetic organic chemistry will lead the way.Additionally, NGs have proven to be ideal test cases for GNRs – in terms of synthesis and physical characterization. Above all, GNRs will define further exciting targets for chemistry, including reactions at their surfaces. Edge chlorination and hydrogenation have already been mentioned in the case of NGs, but fluorination or the attachment of auxochromic groups is equally challenging. In another direction, we have recently synthesized the 2:1-conjugated molecule 98 consisting of one porphyrin and two NG subunits, which demonstrated electronic absorption up to ∼1500 nm (Fig. 18).216 Establishing a zigzag edge at the periphery of NG precursor 97 for oxidative fusion with the porphyrin edge was mandatory. From a technical viewpoint, rigorous optimization of the cyclodehydrogenation process was necessary to avoid unwanted chlorination. This is a critical issue in many other cyclodehydrogenation processes and requires careful adjustment of the oxidants and their stoichiometry. Much can be gained, however, since the strong bathochromic shift seen for 98 with absorption far into the NIR domain suggests that despite the synthetic demand, researchers should aim for the related polymer. Special opportunities could arise from the incorporation of metals with a high-spin configuration or binding of axial ligands. Many other hybrid structures await realization. Site-selective heteroatom incorporation, which has already been mentioned, would be among the subjects for further study.151,217 Replacing the carbon centres of a graphenic lattice by boron or nitrogen centres bears a fascinating similarity to the inclusion of main group III or V elements, respectively, in a silicon semiconductor. Unlike CVD techniques (see above), the bottom-up synthesis from monomers immobilized on surfaces holds promise for site-selective doping.
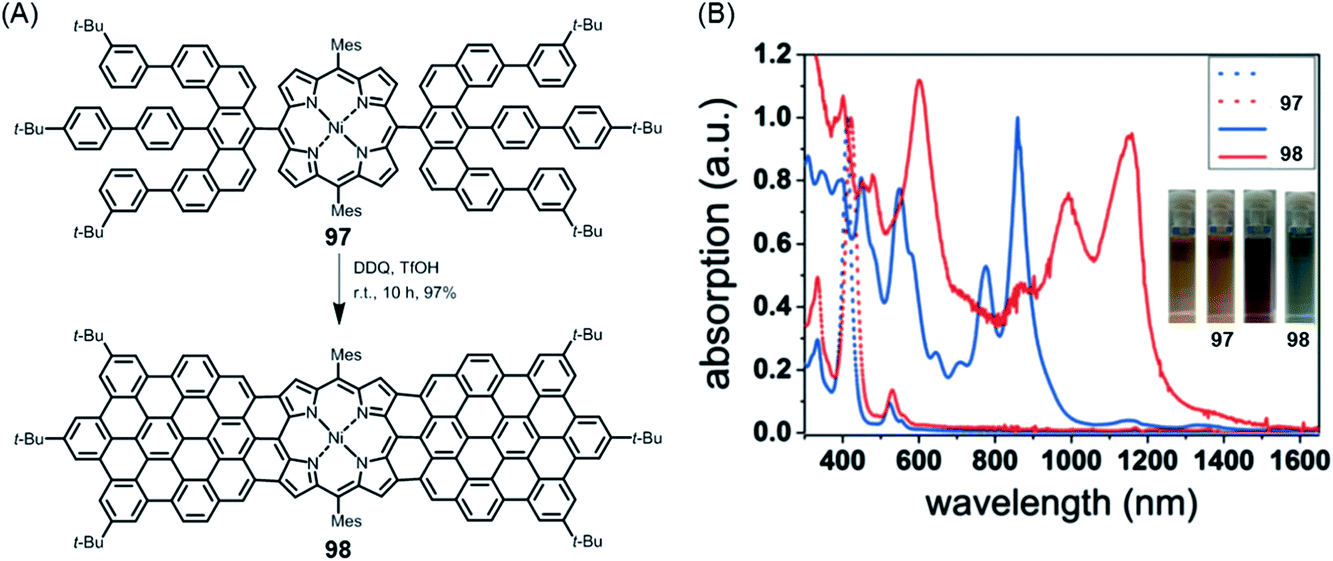 | ||
| Fig. 18 (A) Synthesis of triply fused porphyrin–NG conjugate 98; TfOH = trifluoromethanesulfonic acid. (B) UV-vis-NIR absorption spectra of 97 and 98. The inset photographs show the tetrahydrofuran solutions of 97 and 98.216 Copyright 2018. Adapted with permission from Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
In general, GNRs occupy a core position at the crossroads of materials science since they connect the domains of graphene, CPs and NGs. GNRs will play an important role as semiconductors in ensembles and in the quantum-transport regime for single-molecule devices. Additionally, they open avenues towards “synthesizing” exotic quantum states and thus will become instrumental for future computing technologies. The early history of CP research is rich in polymers with poorly defined structures and undisclosed structural defects, and it has taken the joint effort of the dedicated community to advance precision synthesis together with reliable structure–property relationships. The widely studied graphene oxide (GO) has, in a sense, played a similar role: it comprises a dispersible graphenic structure, can be processed, and offers many opportunities for chemical functionalization by covalent or noncovalent binding of partner molecules. This has stimulated many materials-related studies using commercial samples whose identity and reproducible properties often remain unclear. GO has also served as a precursor for the fabrication of graphene by chemical or thermal reduction, but the quality has been low. GNRs can be obtained by “top-down” or the much more demanding “bottom-up” protocols, and the above text has emphasized the search for unconventional electronic structures and unprecedented functions. What will be the future role of GNRs in the broad context of graphene? While graphene and related 2D materials are nearly omnipresent in the literature, GNRs, by comparison, have attracted much less attention. This is partly due to their limited availability since there is still a high chemical barrier. It falls to synthetic chemists not only to devise new complex structures but also to remove this blockade.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors extend cordial thanks to all their collaborators as well as current and previous co-workers in their group who contributed to some of the results described in this article. We appreciate the financial support from the Max Planck Society, EU Projects GENIUS (ITN-264694), UPGRADE and MoQuaS (FP7 FET-ICT-2013-10 610449), Graphene Flagship (No. CNECT-ICT-604391), ERC-Adv.-Grant 267160 (NANO-GRAPH), the Office of Naval Research BRC Program (molecular synthesis and characterization), DFG Priority Program SPP 1459 and the Alexander von Humboldt Foundation. Open Access funding provided by the Max Planck Society.References
- K. Hafner, Angew. Chem., Int. Ed., 1979, 18, 641–651 CrossRef.
- M. D. Watson, A. Fechtenkötter and K. Müllen, Chem. Rev., 2001, 101, 1267–1300 CrossRef CAS.
- N. Martin and L. T. Scott, Chem. Soc. Rev., 2015, 44, 6397–6400 RSC.
- Z. Qiu, B. A. G. Hammer and K. Müllen, Prog. Polym. Sci., 2020, 100, 101179 CrossRef CAS.
- H. G. Kiess, Conjugated conducting polymers, Springer-Verlag, Berlin, 1992, vol. 102 Search PubMed.
- Y. Olivier, D. Niedzialek, V. Lemaur, W. Pisula, K. Müllen, U. Koldemir, J. R. Reynolds, R. Lazzaroni, J. Cornil and D. Beljonne, Adv. Mater., 2014, 26, 2119–2136 CrossRef CAS.
- M. I. Katsnelson, Mater. Today, 2007, 10, 20–27 CrossRef CAS.
- M. J. Allen, V. C. Tung and R. B. Kaner, Chem. Rev., 2010, 110, 132–145 CrossRef CAS.
- A. Facchetti, Chem. Mater., 2011, 23, 733–758 CrossRef CAS.
- Y.-J. Cheng, S.-H. Yang and C.-S. Hsu, Chem. Rev., 2009, 109, 5868–5923 CrossRef CAS.
- J. L. Segura, N. Martin and D. M. Guldi, Chem. Soc. Rev., 2005, 34, 31–47 RSC.
- S. Günes, H. Neugebauer and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324–1338 CrossRef.
- S. Muench, A. Wild, C. Friebe, B. Haupler, T. Janoschka and U. S. Schubert, Chem. Rev., 2016, 116, 9438–9484 CrossRef CAS.
- J. F. Mike and J. L. Lutkenhaus, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 468–480 CrossRef CAS.
- Y. Liang, Z. Tao and J. Chen, Adv. Energy Mater., 2012, 2, 742–769 CrossRef CAS.
- W. Pisula, Ž. Tomović, C. Simpson, M. Kastler, T. Pakula and K. Müllen, Chem. Mater., 2005, 17, 4296–4303 CrossRef CAS.
- M. Müller, C. Kübel and K. Müllen, Chem.–Eur. J., 1998, 4, 2099–2109 CrossRef.
- K. Müllen and G. Wegner, Electronic materials: the oligomer approach, John Wiley & Sons, 2008 Search PubMed.
- K. Müllen, Pure Appl. Chem., 1993, 65, 89–96 Search PubMed.
- V. Gebhardt, A. Bacher, M. Thelakkat, U. Stalmach, H. Meier, H.-W. Schmidt and D. Haarer, Adv. Mater., 1999, 11, 119–123 CrossRef CAS.
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Sun, S. De, I. T. McGovern, B. Holland, M. Byrne, Y. K. Gun’Ko, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563–568 CrossRef CAS.
- K. S. Novoselov, D. Jiang, F. Schedin, T. J. Booth, V. V. Khotkevich, S. V. Morozov and A. K. Geim, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10451–10453 CrossRef CAS.
- X.-Y. Wang, A. Narita and K. Müllen, Nat. Rev. Chem., 2018, 2, 0100 CrossRef.
- A. Narita, X. Y. Wang, X. Feng and K. Müllen, Chem. Soc. Rev., 2015, 44, 6616–6643 RSC.
- L. Zhi and K. Müllen, J. Mater. Chem., 2008, 18, 1472–1484 RSC.
- J. H. Burroughes, D. D. C. Bradley, A. R. Brown, R. N. Marks, K. Mackay, R. H. Friend, P. L. Burns and A. B. Holmes, Nature, 1990, 347, 539–541 CrossRef CAS.
- A. C. Grimsdale, K. L. Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897–1091 CrossRef CAS.
- J. Bharathan and Y. Yang, Appl. Phys. Lett., 1998, 72, 2660–2662 CrossRef CAS.
- B. J. Schwartz, Annu. Rev. Phys. Chem., 2003, 54, 141–172 CrossRef CAS.
- C.-L. Liu, C.-H. Lin, C.-C. Kuo, S.-T. Lin and W.-C. Chen, Prog. Polym. Sci., 2011, 36, 603–637 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, M. I. Katsnelson, I. V. Grigorieva, S. V. Dubonos and A. A. Firsov, Nature, 2005, 438, 197–200 CrossRef CAS.
- F. Schwierz, Nat. Nanotechnol., 2010, 5, 487–496 CrossRef CAS.
- A. Narita, Z. Chen, Q. Chen and K. Müllen, Chem. Sci., 2019, 10, 964–975 RSC.
- B. Obradovic, R. Kotlyar, F. Heinz, P. Matagne, T. Rakshit, M. D. Giles, M. A. Stettler and D. E. Nikonov, Appl. Phys. Lett., 2006, 88, 142102 CrossRef.
- U. Scherf and K. Müllen, Makromol. Chem., Rapid Commun., 1991, 12, 489–497 CrossRef CAS.
- J. Lee, B. B. Rajeeva, T. Yuan, Z. H. Guo, Y. H. Lin, M. Al-Hashimi, Y. Zheng and L. Fang, Chem. Sci., 2016, 7, 881–889 RSC.
- U. Scherf, J. Mater. Chem., 1999, 9, 1853–1864 RSC.
- K. Chmil and U. Scherf, Makromol. Chem., Rapid Commun., 1993, 14, 217–222 CrossRef CAS.
- Y. W. Son, M. L. Cohen and S. G. Louie, Phys. Rev. Lett., 2006, 97, 216803 CrossRef.
- L. Yang, C. H. Park, Y. W. Son, M. L. Cohen and S. G. Louie, Phys. Rev. Lett., 2007, 99, 186801 CrossRef.
- Y.-C. Chen, D. G. De Oteyza, Z. Pedramrazi, C. Chen, F. R. Fischer and M. F. Crommie, ACS Nano, 2013, 7, 6123–6128 CrossRef CAS.
- O. Gröning, S. Wang, X. Yao, C. A. Pignedoli, G. B. Barin, C. Daniels, A. Cupo, V. Meunier, X. Feng, A. Narita, K. Müllen, P. Ruffieux and R. Fasel, Nature, 2018, 560, 209–214 CrossRef.
- D. J. Rizzo, G. Veber, T. Cao, C. Bronner, T. Chen, F. Zhao, H. Rodriguez, S. G. Louie, M. F. Crommie and F. R. Fischer, Nature, 2018, 560, 204–208 CrossRef CAS.
- X. Li, X. Wang, L. Zhang, S. Lee and H. Dai, Science, 2008, 319, 1229–1232 CrossRef CAS.
- L. Jiao, X. Wang, G. Diankov, H. Wang and H. Dai, Nat. Nanotechnol., 2010, 5, 321–325 CrossRef CAS.
- D. V. Kosynkin, A. L. Higginbotham, A. Sinitskii, J. R. Lomeda, A. Dimiev, B. K. Price and J. M. Tour, Nature, 2009, 458, 872–876 CrossRef CAS.
- L. Jiao, L. Zhang, X. Wang, G. Diankov and H. Dai, Nature, 2009, 458, 877–880 CrossRef CAS.
- X. Wang, Y. Ouyang, X. Li, H. Wang, J. Guo and H. Dai, Phys. Rev. Lett., 2008, 100, 206803 CrossRef.
- L. Tapaszto, G. Dobrik, P. Lambin and L. P. Biro, Nat. Nanotechnol., 2008, 3, 397–401 CrossRef CAS.
- E. Clar and R. Schoental, Polycyclic Hydrocarbons, Springer, Berlin, Heidelberg, 1964, vol. 2 Search PubMed.
- X.-Y. Wang, X. Yao and K. Müllen, Sci. China: Chem., 2019, 62, 1099–1144 CrossRef CAS.
- J. Liu, B. W. Li, Y. Z. Tan, A. Giannakopoulos, C. Sanchez-Sanchez, D. Beljonne, P. Ruffieux, R. Fasel, X. Feng and K. Müllen, J. Am. Chem. Soc., 2015, 137, 6097–6103 CrossRef CAS.
- T. A. Chen and R. S. Liu, Chem.–Eur. J., 2011, 17, 8023–8027 CrossRef CAS.
- R. Rieger and K. Müllen, J. Phys. Org. Chem., 2010, 23, 315–325 CAS.
- V. S. Iyer, M. Wehmeier, J. D. Brand, M. A. Keegstra and K. Müllen, Angew. Chem., Int. Ed., 1997, 36, 1604–1607 CrossRef CAS.
- C. D. Simpson, J. D. Brand, A. J. Berresheim, L. Przybilla, H. J. Räder and K. Müllen, Chem.–Eur. J., 2002, 8, 1424–1429 CrossRef CAS.
- Y. Z. Tan, B. Yang, K. Parvez, A. Narita, S. Osella, D. Beljonne, X. Feng and K. Müllen, Nat. Commun., 2013, 4, 2646 CrossRef.
- A. Fechtenkötter, N. Tchebotareva, M. Watson and K. Müllen, Tetrahedron, 2001, 57, 3769–3783 CrossRef.
- A. M. van de Craats, J. M. Warman, A. Fechtenkötter, J. D. Brand, M. A. Harbison and K. Müllen, Adv. Mater., 1999, 11, 1469–1472 CrossRef CAS.
- J. Wu, W. Pisula and K. Müllen, Chem. Rev., 2007, 107, 718–747 CrossRef CAS.
- S. Kumar, Liq. Cryst., 2004, 31, 1037–1059 CrossRef CAS.
- S. Chandrasekhar, Liq. Cryst., 1993, 14, 3–14 CrossRef CAS.
- M. Cotrait, P. Marsau, M. Pesquer and V. Volpilhac, J. Phys., 1982, 43, 355–359 CrossRef CAS.
- T. N. Hoheisel, S. Schrettl, R. Szilluweit and H. Frauenrath, Angew. Chem., Int. Ed., 2010, 49, 6496–6515 CrossRef CAS.
- L. Zhi, J. Wu, J. Li, U. Kolb and K. Müllen, Angew. Chem., Int. Ed., 2005, 44, 2120–2123 CrossRef CAS.
- B. Y. Yoo, C. K. Huang, J. R. Lim, J. Herman, M. A. Ryan, J. P. Fleurial and N. V. Myung, Electrochim. Acta, 2005, 50, 4371–4377 CrossRef CAS.
- L. Gherghel, C. Kubel, G. Lieser, H.-J. Rader and K. Müllen, J. Am. Chem. Soc., 2002, 124, 13130–13138 CrossRef CAS.
- W. Pisula, M. Kastler, D. Wasserfallen, R. J. Davies, M.-C. Garcia-Gutierrez and K. Müllen, J. Am. Chem. Soc., 2006, 128, 14424–14425 CrossRef CAS.
- N. Grassie and J. N. Hay, Makromol. Chem., 1963, 64, 82–94 CrossRef CAS.
- U. Kador and P. Mehnert, Makromol. Chem., 1971, 144, 37–43 CrossRef CAS.
- J. W. Krumpfer, E. Giebel, E. Frank, A. Müller, L.-M. Ackermann, C. N. Tironi, G. Mourgas, J. Unold, M. Klapper, M. R. Buchmeiser and K. Müllen, Chem. Mater., 2017, 29, 780–788 CrossRef CAS.
- X.-Y. Wang, A. Narita and K. Müllen, AstroPAH, 2018, issue 47, pp. 4–16 Search PubMed.
- K. Hayakawa, Polycyclic Aromatic Hydrocarbons, 2018, pp. 213–223 Search PubMed.
- P. Ruffieux, O. Gröning, R. Fasel, M. Kastler, D. Wasserfallen, K. Müllen and P. Gröning, J. Phys. Chem. B, 2006, 110, 11253–11258 CrossRef CAS.
- P. Samorí and J. P. Rabe, J. Phys.: Condens. Matter, 2002, 14, 9955–9973 CrossRef.
- P. Samorí, A. Fechtenkötter, F. Jäckel, T. Böhme, K. Müllen and J. P. Rabe, J. Am. Chem. Soc., 2001, 123, 11462–11467 CrossRef.
- K. Müllen and J. P. Rabe, Acc. Chem. Res., 2008, 41, 511–520 CrossRef.
- F. Jäckel, M. D. Watson, K. Müllen and J. P. Rabe, Phys. Rev. Lett., 2004, 92, 188303 CrossRef.
- X. Dou, X. Yang, G. J. Bodwell, M. Wagner, V. Enkelmann and K. Müllen, Org. Lett., 2007, 9, 2485–2488 CrossRef CAS.
- C. Kübel, K. Eckhardt, V. Enkelmann, G. Wegner and K. Müllen, J. Mater. Chem., 2000, 10, 879–886 RSC.
- Z. Wang, F. Dotz, V. Enkelmann and K. Müllen, Angew. Chem., Int. Ed., 2005, 44, 1247–1250 CrossRef CAS.
- M. Treier, P. Ruffieux, R. Schillinger, T. Greber, K. Müllen and R. Fasel, Surf. Sci., 2008, 602, L84–L88 CrossRef CAS.
- A. D. Zdetsis, J. Phys. Chem. C, 2018, 122, 17526–17536 CrossRef CAS.
- M. Solà, Front. Chem., 2013, 1, 22 Search PubMed.
- N. Ohkami, A. Motoyama, T. Yamaguchi, H. Hosoya and I. Gutman, Tetrahedron, 1981, 37, 1113–1122 CrossRef CAS.
- I. Gutman, Ž. Tomović, K. Müllen and J. P. Rabe, Chem. Phys. Lett., 2004, 397, 412–416 CrossRef CAS.
- E. H. Fort and L. T. Scott, Angew. Chem., Int. Ed., 2010, 49, 6626–6628 CrossRef CAS.
- D. Wu, H. Zhang, J. Liang, H. Ge, C. Chi, J. Wu, S. H. Liu and J. Yin, J. Org. Chem., 2012, 77, 11319–11324 CrossRef CAS.
- J. Zhou, W. Yang, B. Wang and H. Ren, Angew. Chem., Int. Ed., 2012, 51, 12293–12297 CrossRef CAS.
- M. A. Majewski and M. Stepien, Angew. Chem., Int. Ed., 2019, 58, 86–116 CrossRef CAS.
- M. Gingras, Chem. Soc. Rev., 2013, 42, 968–1006 RSC.
- C. Li, Y. Yang and Q. Miao, Chem.–Asian J., 2018, 13, 884–894 CrossRef CAS.
- S. H. Pun and Q. Miao, Acc. Chem. Res., 2018, 51, 1630–1642 CrossRef CAS.
- M. Kastler, J. Schmidt, W. Pisula, D. Sebastiani and K. Müllen, J. Am. Chem. Soc., 2006, 128, 9526–9534 CrossRef CAS.
- Z. Wang, Z. Tomovic, M. Kastler, R. Pretsch, F. Negri, V. Enkelmann and K. Müllen, J. Am. Chem. Soc., 2004, 126, 7794–7795 CrossRef CAS.
- X. Feng, J. Wu, M. Ai, W. Pisula, L. Zhi, J. P. Rabe and K. Müllen, Angew. Chem., Int. Ed., 2007, 46, 3033–3036 CrossRef CAS.
- T. Dumslaff, B. Yang, A. Maghsoumi, G. Velpula, K. S. Mali, C. Castiglioni, S. De Feyter, M. Tommasini, A. Narita, X. Feng and K. Müllen, J. Am. Chem. Soc., 2016, 138, 4726–4729 CrossRef CAS.
- Z. Sun, S. Lee, K. H. Park, X. Zhu, W. Zhang, B. Zheng, P. Hu, Z. Zeng, S. Das, Y. Li, C. Chi, R. W. Li, K. W. Huang, J. Ding, D. Kim and J. Wu, J. Am. Chem. Soc., 2013, 135, 18229–18236 CrossRef CAS.
- Y. Li, W. K. Heng, B. S. Lee, N. Aratani, J. L. Zafra, N. Bao, R. Lee, Y. M. Sung, Z. Sun, K. W. Huang, R. D. Webster, J. T. Lopez Navarrete, D. Kim, A. Osuka, J. Casado, J. Ding and J. Wu, J. Am. Chem. Soc., 2012, 134, 14913–14922 CrossRef CAS.
- Z. Sun, K. W. Huang and J. Wu, J. Am. Chem. Soc., 2011, 133, 11896–11899 CrossRef CAS.
- Z. Sun, Z. Zeng and J. Wu, Acc. Chem. Res., 2014, 47, 2582–2591 CrossRef CAS.
- A. Konishi, Y. Hirao, K. Matsumoto, H. Kurata, R. Kishi, Y. Shigeta, M. Nakano, K. Tokunaga, K. Kamada and T. Kubo, J. Am. Chem. Soc., 2013, 135, 1430–1437 CrossRef CAS.
- A. Konishi, Y. Hirao, M. Nakano, A. Shimizu, E. Botek, B. Champagne, D. Shiomi, K. Sato, T. Takui, K. Matsumoto, H. Kurata and T. Kubo, J. Am. Chem. Soc., 2010, 132, 11021–11023 CrossRef CAS.
- T. Kubo, Chem. Lett., 2015, 44, 111–122 CrossRef.
- Q. Chen, S. Thoms, S. Stottinger, D. Schollmeyer, K. Müllen, A. Narita and T. Basche, J. Am. Chem. Soc., 2019, 141, 16439–16449 CrossRef CAS.
- G. M. Paterno, Q. Chen, X. Y. Wang, J. Liu, S. G. Motti, A. Petrozza, X. Feng, G. Lanzani, K. Müllen, A. Narita and F. Scotognella, Angew. Chem., Int. Ed., 2017, 56, 6753–6757 CrossRef CAS.
- G. M. Paternò, L. Nicoli, Q. Chen, K. Müllen, A. Narita, G. Lanzani and F. Scotognella, J. Phys. Chem. C, 2018, 122, 25007–25013 CrossRef.
- Q. Chen, D. Wang, M. Baumgarten, D. Schollmeyer, K. Müllen and A. Narita, Chem.–Asian J., 2019, 14, 1703–1707 CrossRef CAS.
- V. Bonal, R. Muñoz-Mármol, F. G. Gámez, M. Morales-Vidal, J. M. Villalvilla, P. G. Boj, J. A. Quintana, Y. Gu, J. Wu, J. Casado and M. A. Díaz-García, Nat. Commun., 2019, 10, 3327 CrossRef.
- X. Liu, S.-Y. Chen, Q. Chen, X. Yao, M. Gelléri, S. Ritz, S. Kumar, C. Cremer, K. Landfester, K. Müllen, S. H. Parekh, A. Narita and M. Bonn, Angew. Chem., Int. Ed., 2020, 59, 496–502 CrossRef CAS.
- Q. Chen, D. Schollmeyer, K. Müllen and A. Narita, J. Am. Chem. Soc., 2019, 141, 19994–19999 CrossRef CAS.
- K. Weiss, G. Beernink, F. Dötz, A. Birkner, K. Müllen and C. H. Wöll, Angew. Chem., Int. Ed., 1999, 38, 3748–3752 CrossRef CAS.
- M. Treier, C. A. Pignedoli, T. Laino, R. Rieger, K. Müllen, D. Passerone and R. Fasel, Nat. Chem., 2011, 3, 61–67 CrossRef CAS.
- F. B. Mallory and C. W. Mallory, Org. React., 2004, 30, 1–456 Search PubMed.
- F. B. Mallory, C. S. Wood and J. T. Gordon, J. Am. Chem. Soc., 1964, 86, 3094–3102 CrossRef CAS.
- R. Rieger, M. Kastler, V. Enkelmann and K. Müllen, Chem.–Eur. J., 2008, 14, 6322–6325 CrossRef CAS.
- R. Berger, A. Giannakopoulos, P. Ravat, M. Wagner, D. Beljonne, X. Feng and K. Müllen, Angew. Chem., Int. Ed., 2014, 53, 10520–10524 CrossRef CAS.
- R. Berger, M. Wagner, X. Feng and K. Müllen, Chem. Sci., 2015, 6, 436–441 RSC.
- S. Ito, Y. Tokimaru and K. Nozaki, Chem. Commun., 2015, 51, 221–224 RSC.
- Y. Tokimaru, S. Ito and K. Nozaki, Angew. Chem., Int. Ed., 2017, 56, 15560–15564 CrossRef CAS.
- X.-Y. Wang, M. Richter, Y. He, J. Björk, A. Riss, R. Rajesh, M. Garnica, F. Hennersdorf, J. J. Weigand, A. Narita, R. Berger, X. Feng, W. Auwärter, J. V. Barth, C.-A. Palma and K. Müllen, Nat. Commun., 2017, 8, 1948 CrossRef.
- Y. Hu, X.-Y. Wang, P.-X. Peng, X.-C. Wang, X.-Y. Cao, X. Feng, K. Müllen and A. Narita, Angew. Chem., Int. Ed., 2017, 56, 3374–3378 CrossRef CAS.
- Q. Zhong, Y. Hu, K. Niu, H. Zhang, B. Yang, D. Ebeling, J. Tschakert, T. Cheng, A. Schirmeisen, A. Narita, K. Müllen and L. Chi, J. Am. Chem. Soc., 2019, 141, 7399–7406 CrossRef CAS.
- J. Kruger, F. Garcia, F. Eisenhut, D. Skidin, J. M. Alonso, E. Guitian, D. Perez, G. Cuniberti, F. Moresco and D. Pena, Angew. Chem., Int. Ed., 2017, 56, 11945–11948 CrossRef.
- R. Zuzak, R. Dorel, M. Krawiec, B. Such, M. Kolmer, M. Szymonski, A. M. Echavarren and S. Godlewski, ACS Nano, 2017, 11, 9321–9329 CrossRef CAS.
- X. Y. Wang, T. Dienel, M. Di Giovannantonio, G. B. Barin, N. Kharche, O. Deniz, J. I. Urgel, R. Widmer, S. Stolz, L. H. De Lima, M. Muntwiler, M. Tommasini, V. Meunier, P. Ruffieux, X. Feng, R. Fasel, K. Müllen and A. Narita, J. Am. Chem. Soc., 2017, 139, 4671–4674 CrossRef CAS.
- C. Rogers, C. Chen, Z. Pedramrazi, A. A. Omrani, H. Z. Tsai, H. S. Jung, S. Lin, M. F. Crommie and F. R. Fischer, Angew. Chem., Int. Ed., 2015, 54, 15143–15146 CrossRef CAS.
- N. Pavlicek, A. Mistry, Z. Majzik, N. Moll, G. Meyer, D. J. Fox and L. Gross, Nat. Nanotechnol., 2017, 12, 308–311 CrossRef CAS.
- S. Mishra, D. Beyer, K. Eimre, J. Liu, R. Berger, O. Gröning, C. A. Pignedoli, K. Müllen, R. Fasel, X. Feng and P. Ruffieux, J. Am. Chem. Soc., 2019, 141, 10621–10625 CrossRef CAS.
- J. Su, M. Telychko, P. Hu, G. Macam, P. Mutombo, H. Zhang, Y. Bao, F. Cheng, Z.-Q. Huang, Z. Qiu, S. J. R. Tan, H. Lin, P. Jelínek, F.-C. Chuang, J. Wu and J. Lu, Sci. Adv., 2019, 5, eaav7717 CrossRef CAS.
- Y. Chujo, Conjugated polymer synthesis, Wiley-VCH, Weinheim, 2010 Search PubMed.
- K. Müllen, J. R. Reynolds and T. Masuda, Conjugated Polymers: A Practical Guide to Synthesis, Royal Society of Chemistry, 2013 Search PubMed.
- Y. Liu, J. W. Y. Lam and B. Z. Tang, Natl. Sci. Rev., 2015, 2, 493–509 CrossRef CAS.
- S. Pfeiffer and H.-H. Hörhold, Macromol. Chem. Phys., 1999, 200, 1870–1878 CrossRef CAS.
- M. Li, S. Berritt, C. Wang, X. Yang, Y. Liu, S. C. Sha, B. Wang, R. Wang, X. Gao, Z. Li, X. Fan, Y. Tao and P. J. Walsh, Nat. Commun., 2018, 9, 1754 CrossRef.
- L. Dossel, L. Gherghel, X. Feng and K. Müllen, Angew. Chem., Int. Ed., 2011, 50, 2540–2543 CrossRef.
- M. G. Schwab, A. Narita, Y. Hernandez, T. Balandina, K. S. Mali, S. De Feyter, X. Feng and K. Müllen, J. Am. Chem. Soc., 2012, 134, 18169–18172 CrossRef CAS.
- X. Yang, X. Dou, A. Rouhanipour, L. Zhi, H.-J. Rader and K. Müllen, J. Am. Chem. Soc., 2008, 130, 4216–4217 CrossRef CAS.
- A. Narita, X. Feng, Y. Hernandez, S. A. Jensen, M. Bonn, H. Yang, I. A. Verzhbitskiy, C. Casiraghi, M. R. Hansen, A. H. Koch, G. Fytas, O. Ivasenko, B. Li, K. S. Mali, T. Balandina, S. Mahesh, S. De Feyter and K. Müllen, Nat. Chem., 2014, 6, 126–132 CrossRef CAS.
- I. C.-Y. Hou, Y. Hu, A. Narita and K. Müllen, Polym. J., 2018, 50, 3–20 CrossRef CAS.
- Z. B. Shifrina, M. S. Averina and A. L. Rusanov, Macromolecules, 2000, 33, 3525–3529 CrossRef CAS.
- B. A. Hammer and K. Müllen, Chem. Rev., 2016, 116, 2103–2140 CrossRef CAS.
- B. A. Hammer, R. Moritz, R. Stangenberg, M. Baumgarten and K. Müllen, Chem. Soc. Rev., 2015, 44, 4072–4090 RSC.
- A. Abdulkarim, F. Hinkel, D. Jansch, J. Freudenberg, F. E. Golling and K. Müllen, J. Am. Chem. Soc., 2016, 138, 16208–16211 CrossRef CAS.
- M. Slota, A. Keerthi, W. K. Myers, E. Tretyakov, M. Baumgarten, A. Ardavan, H. Sadeghi, C. J. Lambert, A. Narita, K. Müllen and L. Bogani, Nature, 2018, 557, 691–695 CrossRef CAS.
- Y. Hu, P. Xie, M. De Corato, A. Ruini, S. Zhao, F. Meggendorfer, L. A. Straaso, L. Rondin, P. Simon, J. Li, J. J. Finley, M. R. Hansen, J. S. Lauret, E. Molinari, X. Feng, J. V. Barth, C. A. Palma, D. Prezzi, K. Müllen and A. Narita, J. Am. Chem. Soc., 2018, 140, 7803–7809 CrossRef CAS.
- A. Keerthi, B. Radha, D. Rizzo, H. Lu, V. Diez Cabanes, I. C. Hou, D. Beljonne, J. Cornil, C. Casiraghi, M. Baumgarten, K. Müllen and A. Narita, J. Am. Chem. Soc., 2017, 139, 16454–16457 CrossRef CAS.
- Y. Huang, Y. Mai, U. Beser, J. Teyssandier, G. Velpula, H. van Gorp, L. A. Straaso, M. R. Hansen, D. Rizzo, C. Casiraghi, R. Yang, G. Zhang, D. Wu, F. Zhang, D. Yan, S. De Feyter, K. Müllen and X. Feng, J. Am. Chem. Soc., 2016, 138, 10136–10139 CrossRef CAS.
- D. Joshi, M. Hauser, G. Veber, A. Berl, K. Xu and F. R. Fischer, J. Am. Chem. Soc., 2018, 140, 9574–9580 CrossRef CAS.
- X. Yao, X. Y. Wang, C. Simpson, G. M. Paterno, M. Guizzardi, M. Wagner, G. Cerullo, F. Scotognella, M. D. Watson, A. Narita and K. Müllen, J. Am. Chem. Soc., 2019, 141, 4230–4234 CrossRef CAS.
- A. Narita, I. A. Verzhbitskiy, W. Frederickx, K. S. Mali, S. A. Jensen, M. R. Hansen, M. Bonn, S. De Feyter, C. Casiraghi, X. Feng and K. Müllen, ACS Nano, 2014, 8, 11622–11630 CrossRef CAS.
- I. C. Y. Hou, A. Narita and K. Müllen, Macromol. Chem. Phys., 2020, 221, 1900374 CrossRef CAS.
- U. Zschieschang, H. Klauk, I. B. Müeller, A. J. Strudwick, T. Hintermann, M. G. Schwab, A. Narita, X. Feng, K. Müllen and R. T. Weitz, Adv. Electron. Mater., 2015, 1, 1400010 CrossRef.
- R. Konnerth, C. Cervetti, A. Narita, X. Feng, K. Müllen, A. Hoyer, M. Burghard, K. Kern, M. Dressel and L. Bogani, Nanoscale, 2015, 7, 12807–12811 RSC.
- P. Fantuzzi, L. Martini, A. Candini, V. Corradini, U. del Pennino, Y. Hu, X. Feng, K. Müllen, A. Narita and M. Affronte, Carbon, 2016, 104, 112–118 CrossRef CAS.
- A. N. Abbas, G. Liu, A. Narita, M. Orosco, X. Feng, K. Müllen and C. Zhou, J. Am. Chem. Soc., 2014, 136, 7555–7558 CrossRef CAS.
- S. Zhao, L. Rondin, G. Delport, C. Voisin, U. Beser, Y. Hu, X. Feng, K. Müllen, A. Narita, S. Campidelli and J. S. Lauret, Carbon, 2017, 119, 235–240 CrossRef CAS.
- I. A. Verzhbitskiy, M. D. Corato, A. Ruini, E. Molinari, A. Narita, Y. Hu, M. G. Schwab, M. Bruna, D. Yoon, S. Milana, X. Feng, K. Müllen, A. C. Ferrari, C. Casiraghi and D. Prezzi, Nano Lett., 2016, 16, 3442–3447 CrossRef CAS.
- G. Soavi, S. Dal Conte, C. Manzoni, D. Viola, A. Narita, Y. Hu, X. Feng, U. Hohenester, E. Molinari, D. Prezzi, K. Müllen and G. Cerullo, Nat. Commun., 2016, 7, 11010 CrossRef CAS.
- M. Xi and B. E. Bent, J. Am. Chem. Soc., 1993, 115, 7426–7433 CrossRef CAS.
- L. Lafferentz, F. Ample, H. Yu, S. Hecht, C. Joachim and L. Grill, Science, 2009, 323, 1193–1197 CrossRef CAS.
- J. Cai, P. Ruffieux, R. Jaafar, M. Bieri, T. Braun, S. Blankenburg, M. Muoth, A. P. Seitsonen, M. Saleh, X. Feng, K. Müllen and R. Fasel, Nature, 2010, 466, 470–473 CrossRef CAS.
- L. Talirz, H. Söde, T. Dumslaff, S. Wang, J. R. Sanchez-Valencia, J. Liu, P. Shinde, C. A. Pignedoli, L. Liang, V. Meunier, N. C. Plumb, M. Shi, X. Feng, A. Narita, K. Müllen, R. Fasel and P. Ruffieux, ACS Nano, 2017, 11, 1380–1388 CrossRef CAS.
- H. Zhang, H. Lin, K. Sun, L. Chen, Y. Zagranyarski, N. Aghdassi, S. Duhm, Q. Li, D. Zhong, Y. Li, K. Müllen, H. Fuchs and L. Chi, J. Am. Chem. Soc., 2015, 137, 4022–4025 CrossRef CAS.
- A. Kimouche, M. M. Ervasti, R. Drost, S. Halonen, A. Harju, P. M. Joensuu, J. Sainio and P. Liljeroth, Nat. Commun., 2015, 6, 10177 CrossRef CAS.
- W. Yang, A. Lucotti, M. Tommasini and W. A. Chalifoux, J. Am. Chem. Soc., 2016, 138, 9137–9144 CrossRef CAS.
- Y. Zagranyarski, L. Chen, D. Jansch, T. Gessner, C. Li and K. Müllen, Org. Lett., 2014, 16, 2814–2817 CrossRef CAS.
- T. Weil, T. Vosch, J. Hofkens, K. Peneva and K. Müllen, Angew. Chem., Int. Ed., 2010, 49, 9068–9093 CrossRef CAS.
- H. Quante, Y. Geerts and K. Müllen, Chem. Mater., 1997, 9, 495–500 CrossRef CAS.
- S. Kaloyanova, Y. Zagranyarski, S. Ritz, M. Hanulova, K. Koynov, A. Vonderheit, K. Müllen and K. Peneva, J. Am. Chem. Soc., 2016, 138, 2881–2884 CrossRef CAS.
- R. Lindner and A. Kuhnle, ChemPhysChem, 2015, 16, 1582–1592 CrossRef CAS.
- Q. Sun, C. Zhang, Z. Li, H. Kong, Q. Tan, A. Hu and W. Xu, J. Am. Chem. Soc., 2013, 135, 8448–8451 CrossRef CAS.
- B. Schuler, S. Fatayer, F. Mohn, N. Moll, N. Pavlicek, G. Meyer, D. Pena and L. Gross, Nat. Chem., 2016, 8, 220–224 CrossRef CAS.
- A. Gourdon, On-Surface Synthesis, Springer International Publishing, Cham, 2016 Search PubMed.
- M. Piantek, D. Serrate, M. Moro-Lagares, P. Algarabel, J. I. Pascual and M. R. Ibarra, J. Phys. Chem. C, 2014, 118, 17895–17899 CrossRef CAS.
- Q. Shen, H.-Y. Gao and H. Fuchs, Nano Today, 2017, 13, 77–96 CrossRef CAS.
- Q. Sun, R. Zhang, J. Qiu, R. Liu and W. Xu, Adv. Mater., 2018, 30, 1705630 CrossRef.
- M. Di Giovannantonio, K. Eimre, A. V. Yakutovich, Q. Chen, S. Mishra, J. I. Urgel, C. A. Pignedoli, P. Ruffieux, K. Müllen, A. Narita and R. Fasel, J. Am. Chem. Soc., 2019, 141, 12346–12354 CrossRef CAS.
- M. Di Giovannantonio, J. I. Urgel, U. Beser, A. V. Yakutovich, J. Wilhelm, C. A. Pignedoli, P. Ruffieux, A. Narita, K. Müllen and R. Fasel, J. Am. Chem. Soc., 2018, 140, 3532–3536 CrossRef CAS.
- P. Ruffieux, S. Wang, B. Yang, C. Sánchez-Sánchez, J. Liu, T. Dienel, L. Talirz, P. Shinde, C. A. Pignedoli, D. Passerone, T. Dumslaff, X. Feng, K. Müllen and R. Fasel, Nature, 2016, 531, 489–492 CrossRef CAS.
- J. Cai, C. A. Pignedoli, L. Talirz, P. Ruffieux, H. Söde, L. Liang, V. Meunier, R. Berger, R. Li, X. Feng, K. Müllen and R. Fasel, Nat. Nanotechnol., 2014, 9, 896–900 CrossRef CAS.
- P. Nemes-Incze, L. Tapasztó, G. Z. Magda, Z. Osváth, G. Dobrik, X. Jin, C. Hwang and L. P. Biró, Appl. Surf. Sci., 2014, 291, 48–52 CrossRef CAS.
- L. Ci, L. Song, D. Jariwala, A. L. Elías, W. Gao, M. Terrones and P. M. Ajayan, Adv. Mater., 2009, 21, 4487–4491 CrossRef CAS.
- J. Kang, D. Shin, S. Bae and B. H. Hong, Nanoscale, 2012, 4, 5527–5537 RSC.
- B. Dlubak, P. R. Kidambi, R. S. Weatherup, S. Hofmann and J. Robertson, Appl. Phys. Lett., 2012, 100, 173113 CrossRef.
- A. Reina, X. Jia, J. Ho, D. Nezich, H. Son, V. Bulovic, M. S. Dresselhaus and J. Kong, Nano Lett., 2009, 9, 30–35 CrossRef CAS.
- X. Li, W. Cai, J. An, S. Kim, J. Nah, D. Yang, R. Piner, A. Velamakanni, I. Jung, E. Tutuc, S. K. Banerjee, L. Colombo and R. S. Ruoff, Science, 2009, 324, 1312–1314 CrossRef CAS.
- K. S. Kim, Y. Zhao, H. Jang, S. Y. Lee, J. M. Kim, K. S. Kim, J. H. Ahn, P. Kim, J. Y. Choi and B. H. Hong, Nature, 2009, 457, 706–710 CrossRef CAS.
- Z. Chen, W. Zhang, C. A. Palma, A. Lodi Rizzini, B. Liu, A. Abbas, N. Richter, L. Martini, X. Y. Wang, N. Cavani, H. Lu, N. Mishra, C. Coletti, R. Berger, F. Klappenberger, M. Klaui, A. Candini, M. Affronte, C. Zhou, V. De Renzi, U. Del Pennino, J. V. Barth, H. J. Rader, A. Narita, X. Feng and K. Müllen, J. Am. Chem. Soc., 2016, 138, 15488–15496 CrossRef CAS.
- Z. Chen, R. Berger, K. Müllen and A. Narita, Chem. Lett., 2017, 46, 1476–1478 CrossRef CAS.
- F. Monnier and M. Taillefer, Angew. Chem., Int. Ed., 2008, 47, 3096–3099 CrossRef CAS.
- C. Sambiagio, S. P. Marsden, A. J. Blacker and P. C. McGowan, Chem. Soc. Rev., 2014, 43, 3525–3550 RSC.
- L. Talirz, H. Söde, J. Cai, P. Ruffieux, S. Blankenburg, R. Jafaar, R. Berger, X. Feng, K. Müllen, D. Passerone, R. Fasel and C. A. Pignedoli, J. Am. Chem. Soc., 2013, 135, 2060–2063 CrossRef CAS.
- M. Di Giovannantonio, O. Deniz, J. I. Urgel, R. Widmer, T. Dienel, S. Stolz, C. Sánchez-Sánchez, M. Muntwiler, T. Dumslaff, R. Berger, A. Narita, X. Feng, K. Müllen, P. Ruffieux and R. Fasel, ACS Nano, 2018, 12, 74–81 CrossRef CAS.
- H. Kricheldorf, Polycondensation: History and New Results, 2014, pp. 35–50 Search PubMed.
- J. P. Llinas, A. Fairbrother, G. Borin Barin, W. Shi, K. Lee, S. Wu, B. Yong Choi, R. Braganza, J. Lear, N. Kau, W. Choi, C. Chen, Z. Pedramrazi, T. Dumslaff, A. Narita, X. Feng, K. Müllen, F. Fischer, A. Zettl, P. Ruffieux, E. Yablonovitch, M. Crommie, R. Fasel and J. Bokor, Nat. Commun., 2017, 8, 633 CrossRef.
- B. Trauzettel, D. V. Bulaev, D. Loss and G. Burkard, Nat. Phys., 2007, 3, 192–196 Search PubMed.
- Y. W. Son, M. L. Cohen and S. G. Louie, Nature, 2006, 444, 347–349 CrossRef CAS.
- O. V. Yazyev and M. I. Katsnelson, Phys. Rev. Lett., 2008, 100, 047209 CrossRef.
- P. Recher and B. Trauzettel, Nanotechnology, 2010, 21, 302001 CrossRef.
- L. Hao, H.-Y. Lu and C. S. Ting, Phys. Rev. Mater., 2019, 3, 024003 CrossRef CAS.
- A. R. Rocha, T. B. Martins, A. Fazzio and A. J. da Silva, Nanotechnology, 2010, 21, 345202 CrossRef CAS.
- S. A. Wolf, D. D. Awschalom, R. A. Buhrman, J. M. Daughton, S. von Molnár, M. L. Roukes, A. Y. Chtchelkanova and D. M. Treger, Science, 2001, 294, 1488–1495 CrossRef CAS.
- K. Müllen, Nat. Rev. Mater., 2016, 1, 15013 CrossRef.
- J. Liu, S. Mishra, C. A. Pignedoli, D. Passerone, J. I. Urgel, A. Fabrizio, T. G. Lohr, J. Ma, H. Komber, M. Baumgarten, C. Corminboeuf, R. Berger, P. Ruffieux, K. Müllen, R. Fasel and X. Feng, J. Am. Chem. Soc., 2019, 141, 12011–12020 CrossRef CAS.
- J. Ma, J. Liu, M. Baumgarten, Y. Fu, Y. Z. Tan, K. S. Schellhammer, F. Ortmann, G. Cuniberti, H. Komber, R. Berger, K. Müllen and X. Feng, Angew. Chem., Int. Ed., 2017, 56, 3280–3284 CrossRef CAS.
- F. Lombardi, A. Lodi, J. Ma, J. Liu, M. Slota, A. Narita, W. K. Myers, K. Müllen, X. Feng and L. Bogani, Science, 2019, 366, 1107–1110 CrossRef CAS.
- S. Mishra, D. Beyer, K. Eimre, S. Kezilebieke, R. Berger, O. Gröning, C. A. Pignedoli, K. Müllen, P. Liljeroth, P. Ruffieux, X. Feng and R. Fasel, Nat. Nanotechnol., 2020, 15, 22–28 CrossRef CAS.
- R. Landauer, IBM J. Res. Dev., 1961, 5, 183–191 Search PubMed.
- W. P. Su, J. R. Schrieffer and A. J. Heeger, Phys. Rev. Lett., 1979, 42, 1698–1701 CrossRef CAS.
- S. Wang, L. Talirz, C. A. Pignedoli, X. Feng, K. Müllen, R. Fasel and P. Ruffieux, Nat. Commun., 2016, 7, 11507 CrossRef CAS.
- R. Naaman and D. H. Waldeck, J. Phys. Chem. Lett., 2012, 3, 2178–2187 CrossRef CAS.
- E. Medina, L. A. González-Arraga, D. Finkelstein-Shapiro, B. Berche and V. Mujica, J. Chem. Phys., 2015, 142, 194308 CrossRef.
- R. Naaman and D. H. Waldeck, Annu. Rev. Phys. Chem., 2015, 66, 263–281 CrossRef CAS.
- Q. Chen, L. Brambilla, L. Daukiya, K. S. Mali, S. De Feyter, M. Tommasini, K. Müllen and A. Narita, Angew. Chem., Int. Ed., 2018, 57, 11233–11237 CrossRef CAS.
- E. Bresó-Femenia, B. Chaudret and S. Castillón, Catal. Sci. Technol., 2015, 5, 2741–2751 RSC.
| This journal is © The Royal Society of Chemistry 2021 |