
Arsenite oxyanions affect CeO2 nanoparticle dissolution and colloidal stability†
Chelsea W.
Neil‡
 ,
Xuanhao
Wu§
,
Doyoon
Kim¶
,
Haesung
Jung||
,
Yanzhe
Zhu**
,
Jessica R.
Ray††
and
Young-Shin
Jun
*
,
Xuanhao
Wu§
,
Doyoon
Kim¶
,
Haesung
Jung||
,
Yanzhe
Zhu**
,
Jessica R.
Ray††
and
Young-Shin
Jun
*
Department of Energy, Environmental & Chemical Engineering, Washington University in St. Louis, One Brookings Drive, Campus Box 1180, St. Louis, MO 63130, USA. E-mail: ysjun@wustl.edu
First published on 25th November 2020
Abstract
While highly reactive cerium oxide nanoparticles (CeO2 NPs) are widely used in industry, their transport in aquatic systems is not well understood. To fill this knowledge gap, the interactions of CeO2 NPs with arsenite (As3+), a toxic metalloid and potential co-present contaminant, were investigated with respect to CeO2 NP colloidal stability, dissolution, and surface redox reactions. Arsenite showed distinctive effects at different concentrations, with a high As3+ concentration (10−4 M) inducing 90% of CeO2 NPs to settle from solution after 8 hours, while lower As3+ concentrations (10−5 or 10−6 M) led to only 20% of CeO2 NPs settling. The dissolution of NPs was most significant in the 10−5 M As3+ system owing to a lesser extent of aggregation, exposing more CeO2 surface for dissolution. In the three As3+ concentration systems, >97% of aqueous arsenic remained as As3+ over 6 hours. On the NP surface, adsorbed AsIII was oxidized to AsV, resulting in 58–70% of the adsorbed arsenic remaining as AsIII. Simultaneously CeIV was reduced to CeIII, increasing CeIII on the CeO2 NP surface from 17% (without arsenite) to 21–25% (with arsenite). Further mechanistic analyses revealed that the adsorption of arsenite was the main contributor to neutralizing the CeO2 NP surface potential, enhancing particle sedimentation. These findings suggest that the fate and transport of CeO2 NPs in our experimental systems are strongly affected by arsenite concentration and its adsorption on NPs. The results also highlight the importance of the interplay between NP aggregation, oxidation, and dissolution in predicting the behaviors of CeO2 NPs and associated toxic elements in aquatic systems.
Environmental significanceDue to growing industrial applications, cerium oxide nanoparticles (CeO2 NPs) are an emerging environmental contaminant of increasing concern. In this study, we elucidated the effects of aqueous arsenite, a potential co-present contaminant, on CeO2 NP aggregation, settling, and redox reactivity. We found that at lower arsenite concentrations, NPs remained suspended in solution while adsorbing high percentages of arsenite, allowing NPs to transport arsenite over long distances. At high arsenite concentrations, CeO2 NPs aggregated and settled from solution. Furthermore, the effect of arsenite concentration on NP dissolution encompasses interplay between redox interactions and NP aggregation, complicating risk assessment. These findings have important implications for predicting the behavior of engineered nanomaterials in water and wastewater treatment plants and in industrial waste streams. |
Introduction
Cerium oxide nanoparticles (CeO2 NPs) are widely used as fuel additives and catalysts, as well as in pharmaceutical and cosmetic applications and in semiconductor production.1,2 The annual production of CeO2 NPs in the U.S. is estimated to be 35–700 tons per year,3 and global CeO2 NP production is predicted to increase from approximately 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons per year in 2014 to 58000 tons per year by 2020.4 Increasingly widespread applications of engineered CeO2 NPs will result in their increasing presence in natural and engineered aquatic systems, posing a challenge to water and wastewater treatment as well as to risk management.5 Thus, a better understanding of CeO2 NP behavior is needed, particularly in aqueous systems where interactions with other compounds can significantly alter their fate and transport.
000 tons per year in 2014 to 58000 tons per year by 2020.4 Increasingly widespread applications of engineered CeO2 NPs will result in their increasing presence in natural and engineered aquatic systems, posing a challenge to water and wastewater treatment as well as to risk management.5 Thus, a better understanding of CeO2 NP behavior is needed, particularly in aqueous systems where interactions with other compounds can significantly alter their fate and transport.
The highly catalytic activity of CeO2 NPs is attributed to oxygen defects in their structure, which allow for oxygen storage and reversible transformation between CeIV and CeIII.6,7 Because there can be multiple oxidation states of cerium in our experimental system—both in solution and in the solid phase—the oxidation states will be denoted by Roman numerals for solid phases (e.g., CeIV and CeIII) and Arabic numerals for aqueous phases (e.g., Ce4+ and Ce3+) throughout the manuscript. Regarding these different oxidation state moieties, CeIV is less soluble than CeIII (Ksp = 5.0 × 10−60 for CeIVO2,8 and Ksp = 1.6 × 10−20 for CeIII(OH)39–11). The redox reversibility evidenced by the multiple oxidation states co-present in CeO2 NPs has been linked to cytotoxicity in organisms.1 For example, the oxidative stress caused by the reduction of CeIV to CeIII and the dissolution of Ce3+ can induce chronic toxicity to E. coli and adverse cell responses in human lung epithelial cells (BEAS-2B).12,13 In addition, CeO2-containing suspensions used for chemical mechanical planarization have been shown to inhibit the proliferation and viability of human cells.2 To predict environmental risks to ecosystems and human health, it is important to improve our understanding of CeO2 NPs' behavior and their transformation in aquatic systems.
Because CeO2 NPs are an emerging environmental contaminant, there is little data regarding CeO2 in the environment or in water treatment facilities. One field-based measurement found that sewage sludge ash in Japan has a mean Ce element concentration of 35.4 ppm.14 It was also estimated in 2010 that 0.4–7% of the annual global production of 260000–309000 metric tons of engineered nanomaterials was released directly into water bodies.15 As the use of these NPs in industrial applications increases, it becomes more likely that NPs such as CeO2 will coexist with other aqueous constituents. Among such constituents of aquatic systems, arsenic is of particular interest due to its own inherent toxicity, as well as its active redox reactivity. Arsenic enters aqueous environments through both natural geochemical processes, such as the dissolution of arsenic-bearing minerals,16–18 and anthropogenic activities, such as leaching from municipal solid waste.19 Toxic and carcinogenic, arsenic can cause acute and chronic adverse health effects such as tumors through various pathways.20 Aqueous arsenic usually exists in two forms: arsenite (AsO33−, pKa = 9.23, 12.13, and 13.4, abbreviated to As3+ in this manuscript), which is more toxic and mobile, or arsenate (AsO43−, pKa = 2.22, 6.98, and 11.53, abbreviated to As5+), which is less toxic and adsorbs more easily on common mineral surfaces in the environment.21 Furthermore, As3+ can exist in naturally reducing environments such as anoxic lake sediments,22 and can persist during water treatment processes.23
Physicochemical interactions between CeIVO2(s) and arsenic are also of particular interest in the field of chemical mechanical planarization.24 Along with silica (SiO2) and alumina (Al2O3) NPs, CeO2 NPs are frequently used in aqueous slurries to polish wafers during semiconductor manufacturing.25,26 Gallium arsenide (GaAs), for example, is a III–V group semiconductor which is important in the manufacturing of high-efficiency solar cells.27,28 Waste from polishing GaAs wafers can contain high concentrations of dissolved arsenic (1800–2400 mg L−1), along with CeO2 NPs.29 Chemical reactions between arsenic and CeO2 NPs in this waste stream can impact the efficacy of wastewater treatment processes to remove these contaminants and may also affect the polishing process by changing NP aggregation.24,30 The co-existence of high levels of arsenic and CeO2 NPs in this waste stream has been the impetus for recent studies on interactions between CeO2 NPs and arsenic.24,30,31
In addition to these natural and industrial situations where arsenic and CeO2 NPs can coexist, there is increasing interest in applying CeO2 NPs as novel sorbents for the removal of arsenic species.32–36 However, while these studies have proven the sorption capacity of CeO2 NPs, little effort has been spent on understanding how sorption may affect the surface chemistry of NPs after water treatment.37 This is of particular importance because separation of these NPs from solution after treatment requires a thorough knowledge of the nature of NPs. For example, the identity and aggregate size of NPs will determine the selectivity of cross-flow membrane filtration, a commonly used method for nanoparticle separation from an aqueous solution.38 As CeO2 NPs find increasing application as novel sorbents, it is even more vital to characterize how interactions with target adsorbates, such as arsenic, will affect the surface chemistry of NPs.
Adsorption isotherms of aqueous As5+ and As3+ onto CeO2 NPs have been investigated previously, with a particular emphasis on arsenic remediation.39 However, this study did not consider the possibility of redox reactions between arsenic species and CeO2 NPs and its consequential effect on the stability and surface chemistry of CeO2 NPs. Another recent study also reported the adsorption isotherms of As5+ and As3+ on CeO2 NPs at pH 3.6, and found that the adsorption of As5+ and As3+ onto CeO2 NPs inhibited the NP surface reactivity.24 While this study provided useful information on surface reactivity, a better understanding of the surface redox interactions between arsenite and CeO2 NPs and their impacts on aggregation and dissolution of CeO2 NPs will help to predict how these interactions affect the fate and transport of CeO2 NPs and their associated hazards.
The fate, transport, and associated risk posed by CeO2 NPs in aquatic systems are largely determined by their dissolution and colloidal stability. As mentioned previously, the mobilization of Ce from solid CeO2 NPs to the aqueous phase generally results from the reduction of CeIV to CeIII.40 In addition, Ce3+ has been shown to have a higher toxicity than Ce4+.41 For risk reduction, it is vital to understand the reductive dissolution of CeO2 NPs. To predict the environmental risk of CeO2 NPs, their colloidal stability must also be assessed because it is an indicator of the potential quantities that can be transported downstream. A previous study on NP transport in a model wastewater treatment plant found that up to 6% of the original quantity of CeO2 NPs were present in the secondary effluent streams.42 This large amount can be attributed to the high colloidal stability of these NPs in aqueous systems, owing to their surface coating with surfactants during NP preparation,42 as well as to surface charge alteration by the adsorption of ions (e.g., Fe2+),10,43 natural organic matter44 and proteins45,46 present in wastewater, which increase the electrostatic repulsive forces between NPs.
Moreover, the surface chemistry of colloidal NPs can also be altered by redox reactions, which can form additional solid coatings. With regard to the potential redox reactions between CeO2 NPs and As3+, the half reactions and overall reaction are listed below:47,48
| AsO33− + H2O → AsO43− + 2H+ + 2e−, E01/2 = −0.56 V | (1) |
| CeO2(s) + e− + 4H+ → Ce3+ + 2H2O, E01/2 = 1.66 V | (2) |
| 2CeO2(s) + AsO33− + 6H+ → 2Ce3+ + AsO43− + 3H2O E0cell = 1.10 V | (3) |
The purpose of this study is, therefore, to investigate the effects of As3+ on the colloidal stability, fate, and transport of CeO2 NPs in a model aqueous system. First, the effects of As3+ on the colloidal stability of CeO2 NPs were examined for three different As3+concentrations, then the dissolution of CeO2 NPs was compared for the same systems. Trends in dissolution and colloidal stability were next systematically investigated to delineate the mechanisms governing each system. This paper, for the first time, reports how As3+ adsorption and redox surface reactions on CeIVO2 NPs impact CeO2 NP colloidal stability, dissolution, and the corresponding implications for the fate of CeO2 NPs in aqueous environments. Hence, this study provides important information that can support more accurate risk assessment of CeO2 NPs in natural and engineered aquatic systems.
Experimental section
Materials
In this study, commercial CeO2 NPs (<25 nm, Sigma-Aldrich, MO) were used to simulate industrially manufactured NPs. X-ray photoelectron spectroscopy (XPS) analysis of the starting CeO2 NP material showed that the powder contained 15.5% CeIII (Fig. 1A), which is consistent with the CeIII starting content measured by a previous study using CeO2 NPs.49 Reagents used included sodium nitrate (NaNO3, ACS grade, J.T. Baker, PA), sodium arsenite (NaAsO2, ≥90%, Sigma Aldrich, MO), and 67–70% nitric acid (HNO3, BDH, PA). To preclude the effect of oxygen on redox reactions, all preparations and reaction procedures were performed in an anaerobic chamber (Coy vinyl type-B, MI). All water used to generate the reaction solutions for anaerobic experiments was degassed de-ionized (DI) water. The resistivity of the DI water was equal to or higher than 18.2 MΩ cm. DI water was deoxygenated by boiling it in an electric kettle and cooling it overnight to room temperature in the anaerobic chamber.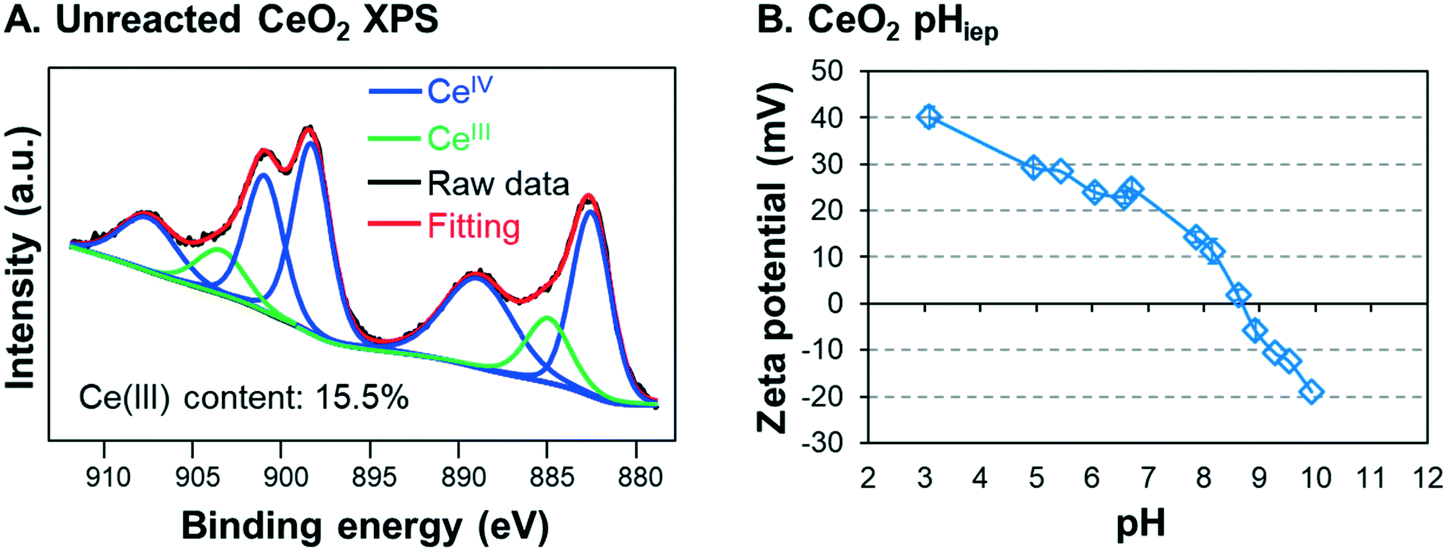 | ||
| Fig. 1 (A) XPS spectra for unreacted CeO2 nanoparticles (NPs) and (B) measurement of the isoelectric point pH for ceria nanoparticles in 10 mM sodium nitrate. | ||
Because dissolved oxygen is present in real aquatic systems, we also tested the settling trends of CeO2 NPs under aerobic conditions for comparison (Fig. S1–S3, ESI†). For those experiments, DI water equilibrated with atmospheric O2 was used, and the experiments were performed under atmospheric O2 conditions. While these experiments helped to determine whether atmospheric oxygen affects interactions between CeO2 NPs and As3+ oxyanions, they also complicated our exploration of the mechanistic interactions between CeO2 NPs and As3+ oxyanions. To clearly elucidate the surface redox interactions between As3+ and CeO2 NPs, we thus limited further investigations to anaerobic systems only.
Sedimentation experiments
For anaerobic experiments, stock solutions, including CeO2 NP dispersions, As3+, and NaNO3 solutions (for ionic strength adjustment), were prepared in the anaerobic chamber. First, a 50 mg L−1 CeO2 NP dispersion was created by adding commercial CeO2 NPs to deoxygenated DI water. The suspension was sonicated for 60 minutes to break up aggregates. A 0.005 M sodium arsenite (As3+) stock solution and a 0.5 M NaNO3 stock solution were also prepared in the anaerobic chamber. The 50 mg L−1 CeO2 dispersion was separated into 50 mL test tubes. Aliquots of As3+ and NaNO3 stock solutions were added to the dispersions to create four systems: a control system, which contained only 10 mM NaNO3, and three different arsenite systems, which contained 10 mM NaNO3 with 10−6, 10−5, or 10−4 M As3+. Arsenic concentrations as high as 10000 ppb (1.4 × 10−4 M) have been observed in environmental systems.50–52 By adding dilute HNO3, the As systems and control system were adjusted to pH 5, a value which is relevant to acidic aquatic systems, such as acid mine drainage sites or sites impacted by acid rain. In addition, this pH is observed in water treatment systems during coagulation with iron (pH 4.5–5.5) or aluminum (pH 5–6).53 At this system pH, As3+ will exist primarily as H3AsO3, while As5+, if it were to form, would exist primarily as H2AsO4−.
Next, the solutions were allowed to settle in the anaerobic chamber. Starting immediately after pH adjustment (i.e., 0 h), approximately 1 mL samples were taken at 2 hour intervals for the first 8 hours of reaction, and after 24 h. To avoid oxygen exposure, these samples were placed in 3 mL polyethylene cuvettes and capped before being removed from the chamber. Then, the absorbance was immediately measured at a wavelength of 305 nm using a UV-visible spectrometer (UV-vis, Varian Cary 50 Bio, CA). The 305 nm wavelength was chosen because the absorbance of CeO2 NPs is at its peak there, while that of As is negligible.40 The linear relationship between CeO2 concentration and the 305 nm wavelength absorption was confirmed by creating a calibration curve (Fig. S4 in the ESI†). Samples were taken from the same vertical depth (approximately 1–2 cm below the surface) in the test tube to ensure that settling was monitored accurately. The design of these experiments was based on our previously reported colloidal stability studies,10,43,44 and all reaction systems were performed in triplicate. After 2 hours, the particle size and surface charge were measured for the particle dispersion using a Zetasizer (Malvern ZEN3600, U.K.). Because all systems came from the same 50 mg L−1 CeO2 NP dispersion stock, the initial particle size and zeta potential of CeO2 NPs were assumed to be the same for all systems. To determine the isoelectric point (pHiep) of unreacted CeO2 NPs, the solution was separated into four test tubes, the pH of each was adjusted to values between 3 and 10, and the zeta potential of each pH system was measured. The pHiep was calculated by interpolation between the measured zeta potentials over the pH range.
Dissolution experiments
The stock solutions for CeO2 NP dissolution experiments were prepared following the same procedure as for the sedimentation experiments. However, rather than allowing the CeO2 NPs to settle, 5 mL aliquots of each system were divided into test tubes. The test tubes were placed in a tube rotator (VWR 10136-084, PA) and rotated at 18 rpm for 24 hours, during which time samples were taken at 0, 2, 4, 6, 8, and 24 hours. This mixing allowed for uniform reaction between the CeO2 NPs and solution. To remove CeO2 NPs, samples were centrifuged at 40000 rpm for 30 minutes using an ultracentrifuge (Thermo Scientific 46900WX80, NC). Based on a study by Tsao et al.,54 this ultracentrifugation speed and time are sufficient to settle 25 nm NPs from solution, and we expect most NPs in our systems exist as >200 nm aggregates, as measured after reaction. This method has also been widely applied to separate CeO2 NPs from solution in previous studies.55–57 The supernatant was then filtered through a 0.22 μm polypropylene syringe filter and acidified to 1% v/v HNO3. If this ultracentrifuge/filtration method did not sufficiently remove CeO2 NPs from solution, we would expect increased Ce concentrations at 0 h, where the aggregate size is expected to be smallest. Instead, we found that the early time points had lower concentrations. In particular, for the control system at 0 h, where there were no reactants to accelerate dissolution, there was negligible Ce in solution (Fig. 4A). This conclusively shows that all NPs which were in the solid state were separated from solution using our ultracentrifuge/filtration method.
Concentrations of aqueous Ce and As in the supernatant were measured by inductively coupled plasma mass spectroscopy (ICP-MS, Agilent 7500 series, CA). As the solubility of CeIII is 3.2 × 1039 times higher than that of CeIV, we assumed all soluble Ce ions were Ce3+. It has also been shown in recent studies that dissolution of CeO2 leads to surface depletion of CeIII.58–60 All samples for settling and dissolution experiments were collected in triplicate. Reported error bars give the standard deviation between triplicate samples.
To quantify As speciation (As3+ or As5+), additional samples containing arsenic were measured for As speciation after 6 hours and 24 hours. For this test, samples were centrifuged and filtered as described above. Next, their pH was adjusted to 3.5, and 10 mL of sample was passed through an ion-exchange column packed with resin (Dowex 1 × 8 in chloride form, Sigma Aldrich, MO), which allowed only As3+ to pass.61 The first 5 mL were discarded and the next 5 mL were collected and measured using ICP-MS. These samples gave the amount of As3+ in solution, while the samples which were not passed through the column gave the total As.
Characterizations of solid phases and surface complexation
Solid phase characterization was carried out using several complementary techniques. First, transmission electron microscopy (TEM, JEOL 2100F, MA) was used to image the morphologies and aggregated patterns of CeO2 NPs. Electron diffraction patterns for selected areas were obtained to examine secondary precipitation in our reaction systems. As described above, four systems were created for settling experiments. After 2 hours of settling, approximately 50 μL of solution from each reaction system was placed on a 300 mesh Cu formvar–carbon grid, and the four grids were dried in a desiccator in the anaerobic chamber. After drying, the grids were placed in a storage box and taken out of the anaerobic chamber for analysis.To determine the oxidation states of cerium and arsenic, XPS (PHI 5000 VersaProbe II, Ulvac-PHI with monochromatic Al Kα radiation (1486.6 eV)) was used. High resolution scans were taken at 0.1 eV steps and a pass energy of 23.5 eV. For XPS sample preparation, four 1 L batches of samples were created for the same reaction conditions described above and reacted for 24 hours. The solutions were then ultra-centrifuged in small batches for 30 minutes at 40000 rpm. After removal of the supernatant, the solids in the test tubes were collected and dried in a desiccator inside the anaerobic chamber. Ce 3d, As 3d, and O 1s spectra were analyzed and fitted using MultiPak software (Physical Electronics) with the Gaussian–Lorentzian fitting function, using the C 1s (284.8 eV) spectrum as the energy reference. In fitting of spectra of different samples, the binding energies were fixed with 0.1 eV variation. For example, 44.25 eV to 44.34 eV were considered as 44.3 eV. The full width at half maximum (FWHM) of the peaks were fixed with no variation. The peak heights and areas were variables to be fitted. The area percentage of an oxidation state was used to represent its amount percentage among different oxidation states. Note that during the fitting, small changes of peak binding energies (<0.1 eV) would lead to a percentage error of ±2%. The reference binding energy peaks for Ce 3d were 884.3 and 902.6 eV for CeIII, and 907.0, 900.6, 898.1, 888.7, 882.1 eV for CeIV.44,62,63 The reference binding energy peaks for As 3d were 44.3 eV for AsIII and 45.3 eV for AsV.64 The reference binding energy peaks for O 1s were 529.3 eV for lattice oxygen in CeO2, 530.9 eV for As–O bond, 531.4 eV for H–O bond, and 533.4 eV for residual adsorbed H2O.44
To investigate how surface reactions might influence CeO2 NP stability in the presence of arsenite, Fourier transform infrared spectroscopy (FTIR, Thermo Nicolet Nexus 470, NC) examined arsenite surface complexation with CeO2 NPs. For these experiments, large batches were prepared identically to those for XPS experiments. Once samples were dried in the anaerobic chamber, they were mixed with KBr at a 10:1 ratio. Samples were measured immediately at a resolution of 0.1, and 1000 scans were taken.
Results and discussion
Fastest settling in the 10−4 M As3+ system
To identify surface chemistry changes of CeO2 NP in the presence of arsenite, we first measured the aggregation and settling rates of CeO2 NP under different aqueous conditions. Fig. 2A shows the sedimentation trends for the As3+-containing systems and the control system as a function of time. Over the first eight hours of reaction, settling trends were similar in the 10−5 M As3+, 10−6 M As3+, and control systems. On the other hand, for the 10−4 M As3+ system, settling occurred very quickly. By eight hours, less than 10% of the CeO2 NPs remained in solution for the 10−4 M As system, while around 80% remained in solution for the other two As3+-containing systems.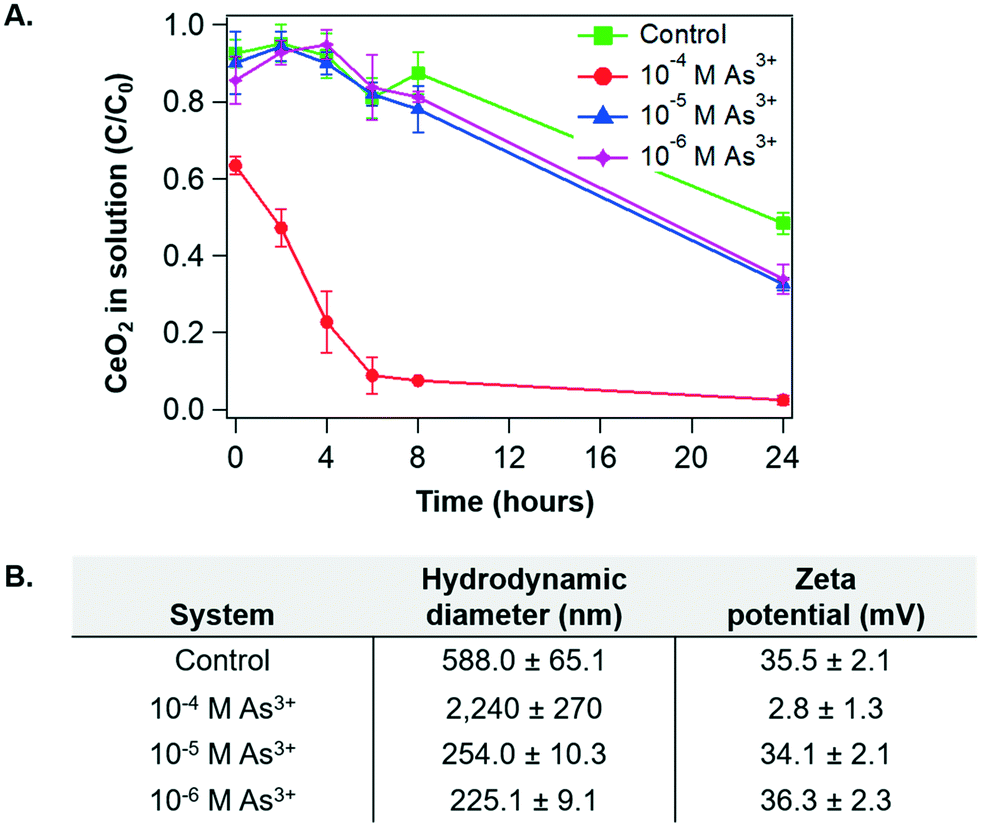 | ||
| Fig. 2 (A) Sedimentation of CeO2 NPs in 10 mM NaCl at pH 5 for control, 10−4 M, 10−5 M, and 10−6 M As3+ systems as a function of time. Absorbances of CeO2 NPs were measured at wavelength of 305 nm, where the highest absorbance by CeO2 NP was obtained.40 (B) Hydrodynamic diameter and zeta potential measurements for CeO2 NP colloids in the control, 10−4 M, 10−5 M, and 10−6 M As3+ systems, measured after 2 hours. Error bars are calculated from triplicate measurements. | ||
To better understand these trends, the particle sizes and zeta potentials of CeO2 NPs were measured for the four systems after 2 hours of reaction, at which point the settling differences had become defined. Aggregate sizes and zeta potentials for the four systems are shown in Fig. 2B. Note that the pHiep of unreacted CeO2 NPs is 8.7 with 10 mM NaNO3 (Fig. 1B). For the 10−5 M As3+, 10−6 M As3+, and control systems, the zeta potentials were highly positive, leading to strong electrostatic repulsive forces which prevented extensive aggregation. Therefore, smaller aggregate sizes and higher colloidal stability in solutions were observed. Moreover, because the zeta potentials were similar for these systems, we speculate that the smaller size in the 10−5 M and 10−6 M As3+ systems than that in the control resulted from changes in the surface hydrophilicity by arsenite adsorption. In other words, the CeO2 NP surface is intrinsically hydrophobic40 due to the unique electronic structure of cerium. Thus, adsorption of arsenite that is easily solvated can make the NP surface less hydrophobic, decreasing the tendency of these NPs to aggregate without significantly altering the zeta potential. For the 10−4 M As3+ system, on the other hand, the zeta potential decreased to close to zero, suggesting that electrostatic repulsive forces between NPs became significantly weaker. As a result, CeO2 NPs in this system aggregated quickly, resulting in a large hydrodynamic diameter and fast settling.
The differences in NP morphology between NPs in the 10−4 M As3+ system and the other systems were examined using TEM. As shown in Fig. 3, CeO2 NPs in the 10−4 M As3+ system were more heavily aggregated than those in other systems, which was consistent with particle size and settling trends. It is noteworthy that the drying process on the TEM grid can cause aggregation, but the extent of the effect of drying on aggregation should be similar across all reaction systems. On the other hand, there was no distinct difference in the morphology of individual particles in the four systems, and we found no evidence of secondary mineral phase formation from the high-resolution images and lattice fringe analyses of CeO2 NPs (Fig. S5 and S6 in the ESI†).
 | ||
| Fig. 3 Representative TEM images of dried CeO2 NP aggregates taken after 2 hours of reaction for the (A) control, (B) 10−6 M As3+, (C) 10−5 M As3+, and (D) 10−4 M As3+ systems. | ||
Fastest dissolution in the 10−5 M As3+ system
To obtain additional insight into the settling and size trends of CeO2 NPs, the dissolution of CeO2 NPs was measured over a 24-hour reaction period. As shown in Fig. 4A, the greatest dissolution of CeO2 NPs occurred in the 10−5 M As3+ system, followed by the 10−4 M As3+ system. Very little dissolution of CeO2 NPs occurred in the 10−6 M As3+ system or in the control system. The total aqueous arsenic concentrations in these systems were also measured, and the results can be found in Fig. S2A in the ESI.† Such dissolution trends are interesting because the greatest dissolution of CeO2 NPs occurred in the median As3+ concentration of 10−5 M, rather than in the 10−4 M As3+ system, which would be expected if As3+ enhanced dissolution through redox interactions. This dissolution trend instead appears to be more related to aggregation. In particular, the 10−4 M As3+ system had a larger degree of aggregation than the 10−5 M As3+ system, which had an aggregate size similar to the 10−6 M As3+ system. As these systems contained the same initial quantities of CeO2 NPs, we expect that the larger aggregates in the 10−4 M As3+ system will result in less exposed surface area for dissolution than the 10−5 M As3+ system, and thus less Ce dissolution can occur. A discussion of aggregation effects on the nanoparticle surface area can be found in section S4 in the ESI.†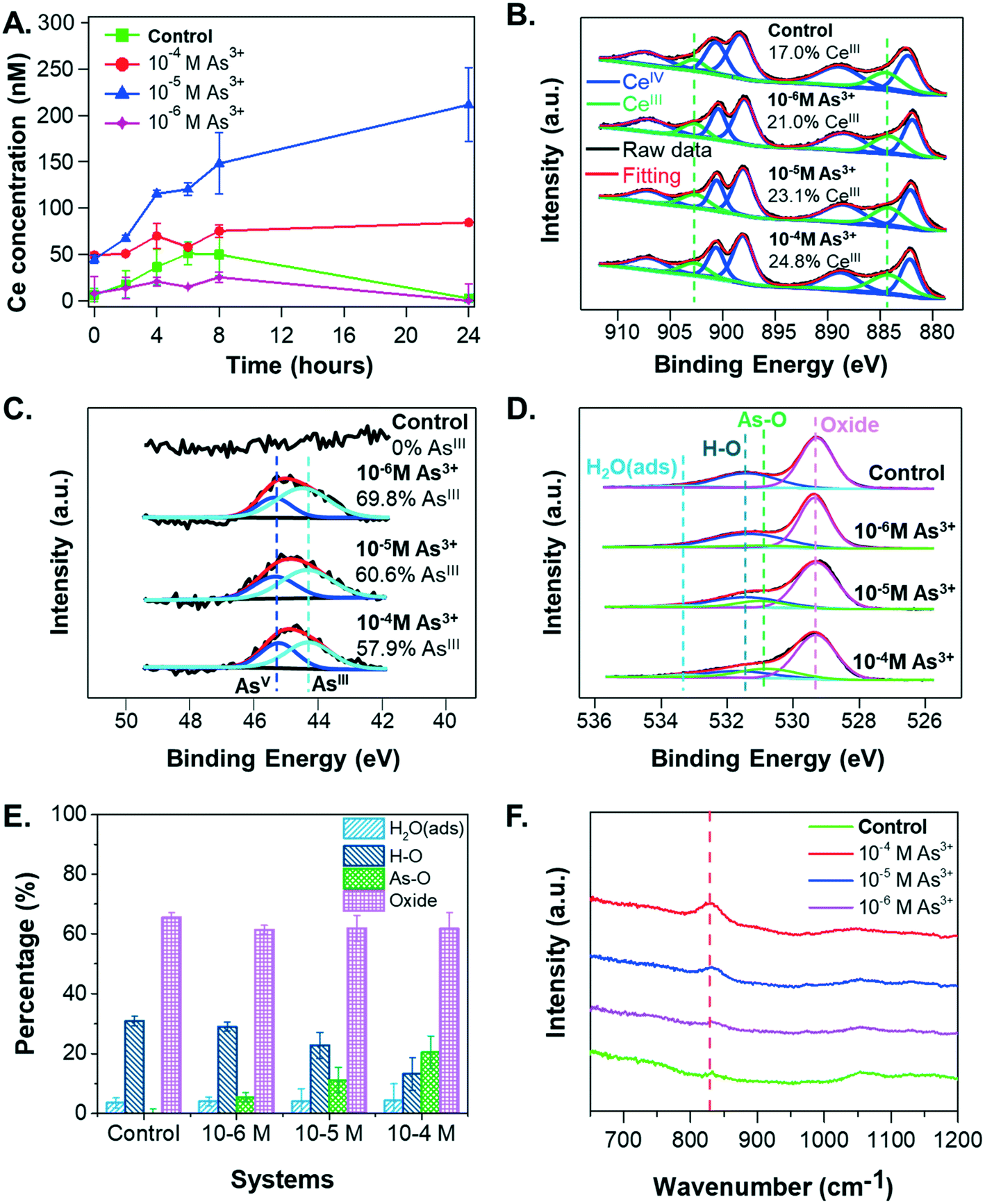 | ||
| Fig. 4 (A) Dissolved cerium concentrations for the control, 10−4 M, 10−5 M, and 10−6 M As3+ systems over the 24 hours reaction period measured using ICP-MS. Error bars are calculated from triplicate measurements. XPS spectra of NPs' surfaces for (B) cerium (Ce 3d), (C) adsorbed arsenic (As 3d), and (D) oxygen (O 1s) in the control, 10−4 M, 10−5 M, and 10−6 M As3+ systems after 24 hour reaction. (E) Area percentages (%) of each oxygen bond based on the XPS analyses of four systems. (F) FTIR spectra for control CeO2 NP samples and CeO2 NP samples reacted with different concentrations of arsenite. | ||
Although the dissolution trend of CeO2 NPs is interesting, it does not explain why aggregation occurred heavily with 10−4 M As3+, leading to fast settling in this system. Considering that dissolution of CeO2 NPs can be indicative of redox interactions, due to the low solubility of CeIV compared to CeIII, we hypothesized that the oxidation state of Ce and/or adsorbed As on the CeO2 NP surface could change the zeta potential and subsequent aggregation of CeO2 NPs.
Redox interactions of CeIV and As3+
To test this hypothesis, we considered the oxidation states of Ce and As in the solid phase and As speciation in the aqueous phase. In the presence of CeO2 NPs, the percentages of aqueous As3+ were 99.7%, 97.7%, and 99.5% for the 10−6 M As3+, 10−5 M As3+, and 10−4 M As3+ systems after 6 hours, respectively, confirming that the reaction condition was anaerobic during experiments and that no significant arsenite oxidation occurred in the aqueous phase within the experimental period. Considering that the starting arsenite salt itself had an assay value of ≥90% per the manufacturer, the slight differences among three systems could not be attributed conclusively to the extents of redox interactions between CeO2 NPs and As3+.Because the speciation of redox-active species can differ depending on the phase (i.e., in solution or on solid surfaces), we also used XPS to monitor the speciation of Ce (Fig. 4B) and As (Fig. 4C) on the CeO2 NP surfaces. Ce 3d spectra showed increases in CeIII content with increasing As3+ concentrations, with the CeIII percentage increasing from 17.0% in the control sample to 21.0%, 23.1%, and 24.8% in the 10−6 M, 10−5 M, and 10−4 M As3+ systems, respectively (Fig. 4B). The As 3d spectra also showed increasing extents of redox reactions with increasing As3+ concentrations. In particular, a clear difference can be observed between the 10−6 M As3+ system and 10−4 M As3+ system (Fig. 4C). Surface arsenic in the 10−6 M As3+ system was 69.8% AsIII, indicating that 30.2% of arsenite adsorbed on CeO2 NP surfaces had been oxidized to AsV. For the 10−4 M As3+ systems, the percentages of AsIII oxidized to AsV were 42.1%, 11.9% higher than that in the 10−6 M As3+ system, which was significant, considering the fitting error (±2%). Therefore, these results indicate that (1) the co-occurrence of CeO2 NPs and As3+ will trigger redox interactions between CeIV and AsIII, forming CeIII and AsV, as predicted by thermodynamic calculations (eqn (3)); and (2) increasing aqueous arsenite concentrations will trigger a higher extent of arsenite oxidation on NP surfaces. The AsV percentages on CeO2 NP surfaces were also significantly higher than the AsV percentages in solutions, indicating that the oxidation of arsenite happened predominantly on the CeO2 NP surfaces.
Interestingly, the increased surface CeIII percentages do not account for the aqueous cerium concentration trends in the different arsenite concentration systems. The CeIII percentages on the NP surfaces increased with higher aqueous arsenite concentrations, whereas the dissolved cerium concentration (Ce3+) was the highest in the 10−5 M As3+ system. Although redox interactions are expected to increase Ce solubility, the higher extent of aggregation of CeO2 NPs in the 10−4 M As3+ system appears to have prevented more dissolution of the formed CeIII from the CeO2 surface, contributing to the higher surface CeIII percentages measured with XPS. In terms of the effects of redox interactions on the NP surface charge, redox interactions alone cannot explain the significantly lower zeta potential in the 10−4 M system. For instance, while the observed redox interaction extents from XPS were similar between the 10−5 M and 10−4 M As3+ systems (Fig. 4B and C), the zeta potentials for these two systems varied greatly. We therefore further hypothesized that aqueous As3+ adsorption contributes to the observed change in NP surface charge. We proceeded to test this hypothesis using both XPS results and the literature, as described in the following section.
A proposed mechanism for CeO2–As3+ interactions
To test whether As3+ adsorption onto CeO2 NPs could be the underlying mechanism for the observed settling trends, we examined the XPS O1s spectra of the samples. These spectra provide additional information about the extent of arsenic adsorption onto CeO2 NP surfaces (Fig. 4D and E). The peak at 531.0 eV was attributed to the As–O bond, observed on the CeO2 NP surface in the presence of arsenite.65 With increasing arsenic concentrations, the relative intensity of this peak increases, indicating a larger arsenic sorption extent. For example, when As3+ concentration increased from 10−6 M to 10−4 M, the area percentage of the As–O bonds increased from 5.5% to 20.5% (Fig. 4E), while the area percentage of the H–O bonds decreased from 29.0% to 13.3%. This observation suggests that the adsorption of arsenite onto CeO2 NPs might replace the original hydroxyl groups on the surface. Previous studies have also reported ligand exchange of the hydroxyl group of metal oxides with arsenate or arsenite during arsenic adsorption.66,67 Furthermore, FTIR results (Fig. 4F) show a peak at ∼830 cm−1 for CeO2 NP samples from arsenite-containing systems, which was attributed to the stretching of As–O bonds in arsenite.68 The intensity of this peak increased with increasing arsenite concentrations, indicating a higher extent of arsenite adsorption on CeO2 NP surfaces. Because the peak position did not change, arsenite in these systems are thought to be sorbed in the same fashion, and the differences in CeO2 NP sedimentation could result from the quantity sorbed rather than changes in the sorbing mechanism.Jain and Ali21 have shown that As3+ adsorption on Fe-containing minerals can decrease their zeta potentials owing to surface complexation between As3+ and mineral surfaces, which replaces surface hydroxyl groups, thus decreasing the surface charge. We propose that a similar mechanism is responsible for the lower colloidal stability of CeO2 NPs in arsenic systems. Prior to arsenite adsorption, a high degree of surface protonation leads to a high positive zeta potential value for CeO2 NP surfaces at pH 5, as the pHiep of CeO2 NPs is 8.7 in 10 mM sodium nitrate (Fig. 1B). The CeO2 surface was also reported to be positively charged due to protonated surface hydroxyl groups, –OH2+, at pH lower than pHiep.44,69 After As3+ adsorbs onto CeO2 NPs in the primary form of H3AsO3 at pH 5, it decreases the surface charge of NPs by replacing –OH2+ groups from the surface with arsenite during surface complexation—which is confirmed by the XPS O 1s results (Fig. 4E). With more arsenite molecules adsorbed onto the CeO2 NP surfaces, there is a net loss of protons,21 further decreasing the positive surface charge and lowering the zeta potential, despite the fact that arsenite is uncharged at pH 5. The surface charge alteration of CeO2 NPs owing to the release of surface protons has also been reported for the interaction of CeO2 NPs with natural organic matter.44 Moreover, the adsorbed arsenate anions (H2AsO4− at pH 5) could contribute partially to the more neutralized surface charge at higher As3+ concentrations. However, considering there was no difference in the size or zeta potential for the 10−6 M and 10−5 M As3+ systems, despite the 10−5 M As3+ system having both more sorption and significantly more oxidation to As(V), the adsorption of As5+ may not be a main contributor to CeO2 NPs' colloidal stability.
To further confirm our hypothesis, we estimated the quantities of adsorbed arsenic molecules on the CeO2 NP surface. Using the arsenic concentrations in solution measured by ICP-MS (Fig. S2A, ESI†)—which accounts for the net aqueous arsenic concentration after adsorption—we calculated the number of arsenic molecules adsorbed on the NP surface by subtracting these values from the total arsenic concentrations added. For the 10−6 M, 10−5 M, and 10−4 M As3+ systems, the adsorbed arsenic amounts were 1.1 × 1019, 6.8 × 1019, and 6.7 × 1020 As molecules per g CeO2, respectively. These values correspond with an As loading of 1.3 to 83.4 mg g−1 for 0.075 to 7.5 mg L−1 As3+, which aligns well with reported values for As3+ sorption by CeO2 NPs.33 With higher aqueous arsenite concentrations, the adsorbed arsenic molecules on CeO2 NPs increased, further neutralizing the surface charge and decreasing the electrostatic repulsive forces between NPs. Therefore, we conclude that arsenite adsorption is the dominant mechanism responsible for the fastest aggregation and settling in the 10−4 M As3+ system.
Conclusions and environmental implications
This study reports important physicochemical interactions between As3+ and CeO2 NPs, which are summarized and presented in Fig. 5. First, in the 10−4 M As3+ system, the fastest aggregation and settling occurred due to neutralization of the surface potential of NP surfaces by arsenite adsorption. In higher As3+ concentration systems (10−5 M and 10−4 M As3+), we observed oxidation of adsorbed AsIII to AsV, triggering the reduction of CeVI to CeIII and the dissolution of Ce3+ from the NP surface. However, dissolution was more prominent in the 10−5 M As3+ system than in the 10−4 M As3+ system, because less CeO2 NP aggregation led to a higher exposed surface area.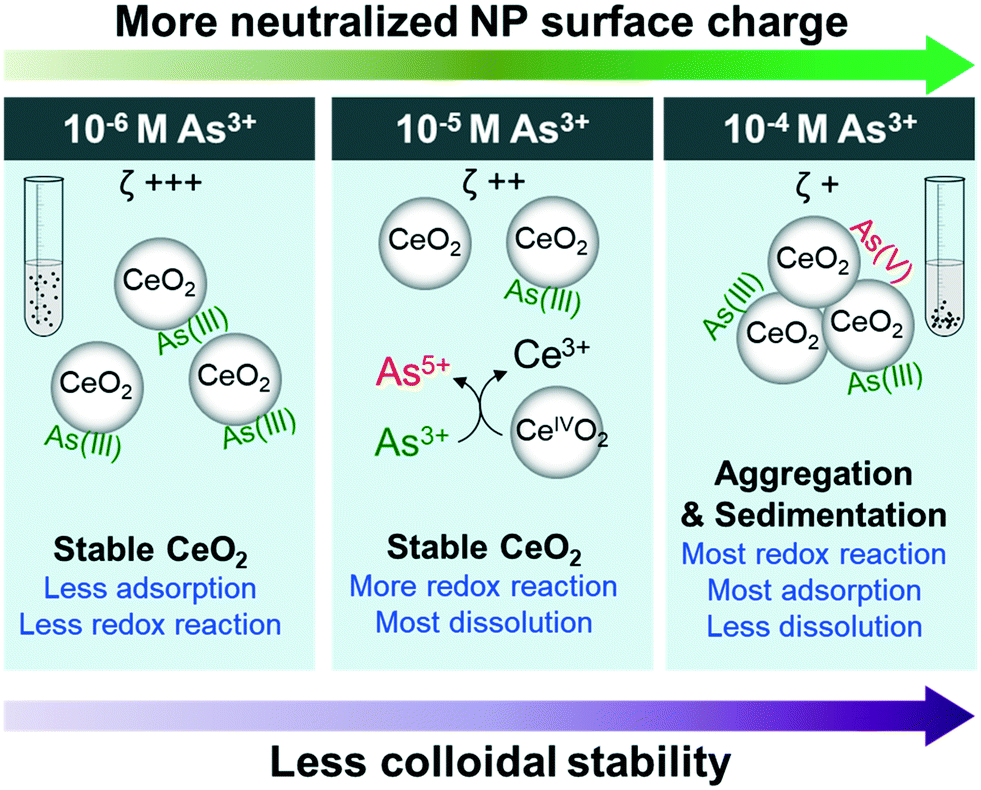 | ||
| Fig. 5 Proposed mechanisms of As3+ interactions with CeO2 NPs. Increasing As3+ concentrations lead to a more neutralized surface, and thus more aggregation and settling. As a result, the most dissolution occurs in the median concentration (10−5 M As3+), where increased redox reactions coincide with maintained colloidal stability, leading to increases in reactive surface area for dissolution relative to the 10−4 M As3+ system. | ||
This study deepens our current understanding of the hazards posed by widely applied CeO2 NPs. As demonstrated, in the presence of 10−4 M As3+, CeO2 NPs aggregated and settled more quickly due to As3+ adsorption. Thus, less settlement time is required to remove CeO2 NPs by sedimentation under this condition. The altered surface charge can also affect the bioaccumulation of CeO2 NPs. A previous study found that positively charged CeO2 NPs, which were observed in our control, 10−6 M, and 10−5 M As3+ systems, were significantly more toxic to Caenorhabditis elegans than neutrally charged CeO2 NPs, which were observed only in our 10−4 M As3+ system.70 Lastly, increased aggregation of CeO2 NPs led to less Ce3+ mobilization in the 10−4 M As3+ system than in the 10−5 M As3+ system, which directly affects the risk posed by CeO2 NPs because Ce3+ is toxic to organisms. However, it is also important to consider how other environmentally abundant water constituents, such as natural organic matter, sulfate, phosphate, and nitrate, will influence interactions between CeO2 NPs and arsenic species. To gain a more detailed molecular scale understanding of such dynamic systems, more studies on complexation (e.g., inner or outer-sphere complexation), and surface electron transfer between CeIV and AsIII to cause the redox reactions can be good future research directions.
The high colloidal stability of CeO2 NPs, which was observed in the control system and systems with lower initial As3+ concentrations, indicated that NPs can stay suspended for longer times, thus either more settlement time should be allowed before discharge in water treatment plants, or additional treatment may be required to remove these NPs. More importantly, this study shows that CeO2 NPs also have a high adsorption capacity for arsenic, with 90–95% of As3+ adsorbed on the particle surface in the 10−6 M As3+ system. Therefore, in systems with low As3+ concentrations, As3+ coexisting with CeO2 NPs may pose additional challenges, as it tends to stay on CeO2 NP surfaces and travel for long distances, requiring more comprehensive risk assessment and waste management. These new insights into CeO2 NPs transport and reactivity, along with their increasing industrial use, provide an impetus for future study of additional factors impacting these emerging NPs, such as the effects of pH, ionic strength, and the presence of additional redox-reactive compounds.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Science Foundation's Environmental Chemical Science Program (CHE-1214090). J. R. R. was supported by an Environmental Protection Agency STAR Fellowship, and C. W. N. was supported by a Mr. and Mrs. Spencer T. Olin Fellowship. We wish to thank our Environmental NanoChemistry Group members for their valuable discussions. The authors acknowledge Professor James C. Ballard for reviewing the manuscript, and Washington University's Institute of Materials Science & Engineering (IMSE) for the use of XPS and TEM.Los Alamos National Laboratory, an affirmative action/equal opportunity employer, is operated by Triad National Security, LLC, for the National Nuclear Security Administration of U.S. Department of Energy (contract no. 89233218CNA000001). By approving this article, the publisher recognizes that the U.S. Government retains nonexclusive, royalty-free license to publish or reproduce the published form of this contribution, or to allow others to do so, for U.S. Government purposes. Los Alamos National Laboratory requests that the publisher identify this article as work performed under the auspices of the U.S. Department of Energy. Los Alamos National Laboratory strongly supports academic freedom and a researcher's right to publish; as an institution, however, the Laboratory does not endorse the viewpoint of a publication or guarantee its technical correctness.
References
- H. Zhang, X. He, Z. Zhang, P. Zhang, Y. Li, Y. Ma, Y. Kuang, Y. Zhao and Z. Chai, Nano-CeO2 Exhibits Adverse Effects at Environmental Relevant Concentrations, Environ. Sci. Technol., 2011, 45(8), 3725–3730 CrossRef CAS PubMed.
- D. Speed, P. Westerhoff, R. Sierra-Alvarez, R. Draper, P. Pantano, S. Aravamudhan, K. L. Chen, K. Hristovski, P. Herckes, X. Bi, Y. Yang, C. Zeng, L. Otero-Gonzalez, C. Mikoryak, B. A. Wilson, K. Kosaraju, M. Tarannum, S. Crawford, P. Yi, X. Liu, S. V. Babu, M. Moinpour, J. Ranville, M. Montano, C. Corredor, J. Posner and F. Shadman, Physical, chemical, and in vitro toxicological characterization of nanoparticles in chemical mechanical planarization suspensions used in the semiconductor industry: towards environmental health and safety assessments, Environ. Sci.: Nano, 2015, 2(3), 227–244 RSC.
- F. Piccinno, F. Gottschalk, S. Seeger and B. Nowack, Industrial production quantities and uses of ten engineered nanomaterials in Europe and the world, J. Nanopart. Res., 2012, 14(9), 1–11 CrossRef.
- Z. M. Milani, F. Charbgoo and M. J. C. I. Darroudi, Impact of physicochemical properties of cerium oxide nanoparticles on their toxicity effects, Ceram. Int., 2017, 43(17), 14572–14581 CrossRef.
- F. Gomez-Rivera, J. A. Field, D. Brown and R. Sierra-Alvarez, Fate of cerium dioxide (CeO2) nanoparticles in municipal wastewater during activated sludge treatment, Bioresour. Technol., 2012, 108, 300–304 CrossRef CAS PubMed.
- I. Celardo, M. De Nicola, C. Mandoli, J. Z. Pedersen, E. Traversa and L. Ghibelli, Ce3+ ions determine redox-dependent anti-apoptotic effect of cerium oxide nanoparticles, ACS Nano, 2011, 5(6), 4537–4549 CrossRef CAS PubMed.
- T. Pirmohamed, J. M. Dowding, S. Singh, B. Wasserman, E. Heckert, A. S. Karakoti, J. E. King, S. Seal and W. T. Self, Nanoceria exhibit redox state-dependent catalase mimetic activity, Chem. Commun., 2010, 46(16), 2736–2738 RSC.
- T. V. Plakhova, A. Y. Romanchuk, S. N. Yakunin, T. Dumas, S. Demir, S. Wang, S. G. Minasian, D. K. Shuh, T. Tyliszczak, A. A. Shiryaev, A. V. Egorov, V. K. Ivanov and S. N. Kalmykov, Solubility of Nanocrystalline Cerium Dioxide: Experimental Data and Thermodynamic Modeling, J. Phys. Chem. C, 2016, 120(39), 22615–22626 CrossRef CAS.
- Z. Jianjun, L. Bingwei, Y. Chenxuan and R. Xiangdong, Application of Ce(IV) in industrial wastewater treatment, J. Rare Earths, 2010, 28, 37–39 Search PubMed.
- X. Liu, J. R. Ray, C. W. Neil, Q. Li and Y.-S. Jun, Enhanced colloidal stability of CeO2 nanoparticles by ferrous ions: adsorption, redox reaction, and surface precipitation, Environ. Sci. Technol., 2015, 49(9), 5476–5483 CrossRef CAS PubMed.
- D. Speed, Environmental aspects of planarization processes, in Advances in Chemical Mechanical Planarization (CMP), Elsevier, 2016, pp. 229–269 Search PubMed.
- A. Thill, O. L. Zeyons, O. Spalla, F. Chauvat, J. M. Rose, M. L. Auffan and A. M. Flank, Cytotoxicity of CeO2 nanoparticles for Escherichia coli. Physico-chemical insight of the cytotoxicity mechanism, Environ. Sci. Technol., 2006, 40(19), 6151–6156 CrossRef CAS PubMed.
- E.-J. Park, J. Choi, Y.-K. Park and K. Park, Oxidative stress induced by cerium oxide nanoparticles in cultured BEAS-2B cells, Toxicology, 2008, 245(1), 90–100 CrossRef CAS PubMed.
- Integrated laboratory Systems (ILS), Chemical Information Profile for Ceric Oxide [CAS No. 1306-38-3], Prepared for the National Toxicology Program, National Institute of Environmental Health Sciences, February, 2006. Available from: http://ntp.niehs.nih.gov/ntp/htdocs/chem_background/exsumpdf/ceric_oxide2_508.pdf.
- A. A. Keller, S. McFerran, A. Lazareva and S. Suh, Global life cycle releases of engineered nanomaterials, J. Nanopart. Res., 2013, 15(6), 1–17 CrossRef.
- C. W. Neil, Y. J. Yang and Y.-S. Jun, Arsenic mobilization and attenuation by mineral-water interactions: implications for managed aquifer recharge, J. Environ. Monit., 2012, 14(7), 1772–1788 RSC.
- X. Wu, B. Bowers, D. Kim, B. Lee and Y.-S. Jun, Dissolved Organic Matter Affects Arsenic Mobility and Iron (III)(hydr) oxide Formation: Implications for Managed Aquifer Recharge, Environ. Sci. Technol., 2019, 53(24), 14357–14367 CrossRef CAS PubMed.
- X. Wu, S. Burnell, C. W. Neil, D. Kim, L. Zhang, H. Jung and Y.-S. Jun, Effects of Phosphate, Silicate, and Bicarbonate on Arsenopyrite Dissolution and Secondary Mineral Precipitation, ACS Earth Space Chem., 2020, 4(4), 515–525 CrossRef CAS.
- A. Ghosh, M. Mukiibi and W. Ela, TCLP underestimates leaching of arsenic from solid residuals under landfill conditions, Environ. Sci. Technol., 2004, 38(17), 4677–4682 CrossRef CAS PubMed.
- H. Shi, X. Shi and K. J. Liu, Oxidative mechanism of arsenic toxicity and carcinogenesis, Mol. Cell. Biochem., 2004, 255(1–2), 67–78 CrossRef CAS PubMed.
- C. Jain and I. Ali, Arsenic: occurrence, toxicity and speciation techniques, Water Res., 2000, 34(17), 4304–4312 CrossRef CAS.
- S. E. Hoeft, F. O. Lucas, J. T. Hollibaugh and R. S. Oremland, Characterization of microbial arsenate reduction in the anoxic bottom waters of Mono Lake, California, Geomicrobiol. J., 2002, 19(1), 23–40 CrossRef CAS.
- W. Driehaus, R. Seith and M. Jekel, Oxidation of arsenate(III) with manganese oxides in water treatment, Water Res., 1995, 29(1), 297–305 CrossRef CAS.
- X. Bi, C. Zeng and P. Westerhoff, Adsorption of arsenic ions transforms surface reactivity of engineered cerium oxide nanoparticles, Environ. Sci. Technol., 2020, 54(15), 9437–9444 CrossRef CAS PubMed.
- D. Speed, P. Westerhoff, R. Sierra-Alvarez, R. Draper, P. Pantano, S. Aravamudhan, K. L. Chen, K. Hristovski, P. Herckes and X. Bi, Physical, chemical, and in vitro toxicological characterization of nanoparticles in chemical mechanical planarization suspensions used in the semiconductor industry: towards environmental health and safety assessments, Environ. Sci.: Nano, 2015, 2(3), 227–244 RSC.
- P. B. Zantye, A. Kumar and A. Sikder, Chemical mechanical planarization for microelectronics applications, Mater. Sci. Eng., R, 2004, 45(3), 89–220 CrossRef.
- K. W. Torrance, H. E. Keenan, A. S. Hursthouse and D. Stirling, Measurement of arsenic and gallium content of gallium arsenide semiconductor waste streams by ICP-MS, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2010, 45(4), 471–475 CrossRef CAS PubMed.
- D. Guimard, R. Morihara, D. Bordel, K. Tanabe, Y. Wakayama, M. Nishioka and Y. Arakawa, Fabrication of InAs/GaAs quantum dot solar cells with enhanced photocurrent and without degradation of open circuit voltage, Appl. Phys. Lett., 2010, 96(20), 3507 CrossRef.
- K. Torrance and H. E. Keenan, In Characterization of arsenic-rich waste slurries generated during gallium arsenide wafer lapping and polishing, 2009 International Conference on Compound Semiconductor MANufacturing TECHnology, 2009 Search PubMed.
- X. Bi and P. Westerhoff, Adsorption of iii/v ions (In(iii), Ga(iii) and As(v)) onto SiO2, CeO2 and Al2O3 nanoparticles used in the semiconductor industry, Environ. Sci.: Nano, 2016, 3(5), 1014–1026 RSC.
- C. Zeng, C. Nguyen, S. Boitano, J. A. Field, F. Shadman and R. J. E. R. Sierra-Alvarez, Cerium dioxide (CeO2) nanoparticles decrease arsenite (As (III)) cytotoxicity to 16HBE14o-human bronchial epithelial cells, Environ. Res., 2018, 164, 452–458 CrossRef CAS PubMed.
- S. Bhattacharya, I. Saha, A. Mukhopadhyay, D. Chattopadhyay and U. Chand, Role of nanotechnology in water treatment and purification: potential applications and implications, Int. J. Chem. Technol., 2013, 3(3), 59–64 Search PubMed.
- R. Li, Q. Li, S. Gao and J. K. Shang, Exceptional arsenic adsorption performance of hydrous cerium oxide nanoparticles: Part A. Adsorption capacity and mechanism, Chem. Eng. J., 2012, 185, 127–135 CrossRef.
- P. K. Mishra, A. Saxena, A. S. Rawat, P. K. Dixit, R. Kumar and P. K. Rai, Surfactant-free one-pot synthesis of low-density cerium oxide nanoparticles for adsorptive removal of arsenic species, Environ. Prog. Sustainable Energy, 2018, 37(1), 221–231 CrossRef CAS.
- L. Yu, Y. Ma, C. N. Ong, J. Xie and Y. Liu, Rapid adsorption removal of arsenate by hydrous cerium oxide–graphene composite, RSC Adv., 2015, 5(80), 64983–64990 RSC.
- Y. Yu, C. Zhang, L. Yang and J. P. Chen, Cerium oxide modified activated carbon as an efficient and effective adsorbent for rapid uptake of arsenate and arsenite: Material development and study of performance and mechanisms, Chem. Eng. J., 2017, 315, 630–638 CrossRef CAS.
- D. Lin, X. Tian, F. Wu and B. Xing, Fate and transport of engineered nanomaterials in the environment, J. Environ. Qual., 2010, 39(6), 1896–1908 CrossRef PubMed.
- I. Ali, New generation adsorbents for water treatment, Chem. Rev., 2012, 112(10), 5073–5091 CrossRef CAS PubMed.
- Q. Feng, Z. Zhang, Y. Ma, X. He, Y. Zhao and Z. Chai, Adsorption and desorption characteristics of arsenic onto ceria nanoparticles, Nanoscale Res. Lett., 2012, 7(1), 1–8 CrossRef PubMed.
- X. Liu, J. R. Ray, C. W. Neil, Q. Li and Y.-S. Jun, Enhanced Colloidal Stability of CeO2 Nanoparticles by Ferrous Ions: Adsorption, Redox Reaction, and Surface Precipitation, Environ. Sci. Technol., 2015, 49(9), 5476–5483 CrossRef CAS PubMed.
- M. L. Auffan, J. R. M. Rose, M. R. Wiesner and J.-Y. Bottero, Chemical stability of metallic nanoparticles: a parameter controlling their potential cellular toxicity in vitro, Environ. Pollut., 2009, 157(4), 1127–1133 CrossRef CAS PubMed.
- L. K. Limbach, R. Bereiter, E. Muller, R. Krebs, R. Galli and W. J. Stark, Removal of Oxide Nanoparticles in a Model Wastewater Treatment Plant: Influence of Agglomeration and Surfactants on Clearing Efficiency, Environ. Sci. Technol., 2008, 42(15), 5828–5833 CrossRef CAS.
- J. R. Ray, X. Wu, C. W. Neil, H. Jung, Z. Li and Y.-S. Jun, Redox chemistry of CeO2 nanoparticles in aquatic systems containing Cr(vi)(aq) and Fe2+ ions, Environ. Sci.: Nano, 2019, 6(7), 2269–2280 RSC.
- X. Wu, C. W. Neil, D. Kim, H. Jung and Y.-S. Jun, Co-effects of UV/H2O2 and natural organic matter on the surface chemistry of cerium oxide nanoparticles, Environ. Sci.: Nano, 2018, 5(10), 2382–2393 RSC.
- L. E. Barton, M. Auffan, M. Bertrand, M. Barakat, C. Santaella, A. Masion, D. Borschneck, L. Olivi, N. Roche and M. R. Wiesner, Transformation of pristine and citrate-functionalized CeO2 nanoparticles in a laboratory-scale activated sludge reactor, Environ. Sci. Technol., 2014, 48(13), 7289–7296 CrossRef CAS.
- L. E. Barton, M. Auffan, L. Olivi, J.-Y. Bottero and M. R. Wiesner, Heteroaggregation, transformation and fate of CeO2 nanoparticles in wastewater treatment, Environ. Pollut., 2015, 203, 122–129 CrossRef CAS PubMed.
- V. Dutré, C. Vandecasteele and S. Opdenakker, Oxidation of arsenic bearing fly ash as pretreatment before solidification, J. Hazard. Mater., 1999, 68(3), 205–215 CrossRef.
- P. Yu, S. A. Hayes, T. J. O'Keefe, M. J. O'Keefe and J. O. Stoffer, The phase stability of cerium species in aqueous systems: II. The systems. Equilibrium considerations and pourbaix diagram calculations, J. Electrochem. Soc., 2005, 153(1), C74 CrossRef.
- K. M. Dunnick, R. Pillai, K. L. Pisane, A. B. Stefaniak, E. M. Sabolsky and S. S. Leonard, The Effect of Cerium Oxide Nanoparticle Valence State on Reactive Oxygen Species and Toxicity, Biol. Trace Elem. Res., 2015, 166(1), 96–107 CrossRef CAS PubMed.
- D. B. Hawkins, Environmental path of arsenic in groundwater: Completion report, Institute of Water Resources, University of Alaska, 1976 Search PubMed.
- B. Hildum, Arsenic speciation and groundwater chemistry at a landfill site: a case study of SHepley's Hill Landfill, Boston College, 2013 Search PubMed.
- D. I. Nornan, Arsenic geochemistry and remediation using natural materials, in Water—Pollution, World Scientific, 2001, pp. 68–87 Search PubMed.
- E. L. Sharp, S. A. Parsons and B. Jefferson, Seasonal variations in natural organic matter and its impact on coagulation in water treatment, Sci. Total Environ., 2006, 363(1–3), 183–194 CrossRef CAS PubMed.
- T. M. Tsao, Y. M. Chen and M. K. Wang, Origin, separation and identification of environmental nanoparticles: a review, J. Environ. Monit., 2011, 13(5), 1156–1163 RSC.
- M. Baalousha, Y. Ju-Nam, P. A. Cole, B. Gaiser, T. F. Fernandes, J. A. Hriljac, M. A. Jepson, V. Stone, C. R. Tyler and J. R. Lead, Characterization of cerium oxide nanoparticles—part 1: size measurements, Environ. Toxicol. Chem., 2012, 31(5), 983–993 CrossRef CAS.
- M. Baalousha, P. Le Coustumer, I. Jones and J. Lead, Characterisation of structural and surface speciation of representative commercially available cerium oxide nanoparticles, Environ. Chem., 2010, 7(4), 377–385 CrossRef CAS.
- N. J. Rogers, N. M. Franklin, S. C. Apte, G. E. Batley, B. M. Angel, J. R. Lead and M. Baalousha, Physico-chemical behaviour and algal toxicity of nanoparticulate CeO2 in freshwater, Environ. Chem., 2010, 7(1), 50–60 CrossRef CAS.
- C. L. Corkhill, D. J. Bailey, F. Y. Tocino, M. C. Stennett, J. A. Miller, J. L. Provis, K. P. Travis and N. C. Hyatt, Role of microstructure and surface defects on the dissolution kinetics of CeO2, a UO2 fuel analogue, ACS Appl. Mater. Interfaces, 2016, 8(16), 10562–10571 CrossRef CAS PubMed.
- R. Mehmood, S. S. Mofarah, W.-F. Chen, P. Koshy and C. C. Sorrell, Surface, Subsurface, and Bulk Oxygen Vacancies Quantified by Decoupling and Deconvolution of the Defect Structure of Redox-Active Nanoceria, Inorg. Chem., 2019, 58(9), 6016–6027 CrossRef CAS PubMed.
- J. Yu, Z. Wang, J. Wang, W. Zhong, M. Ju, R. Cai, C. Qiu, X. Long and S. Yang, The Role of Ceria in a Hybrid Catalyst toward Alkaline Water Oxidation, ChemSusChem, 2020, 13(19), 5273–5279 CrossRef CAS PubMed.
- J. A. Wilkie and J. G. Hering, Rapid Oxidation of Geothermal Arsenic(III) in Streamwaters of the Eastern Sierra Nevada, Environ. Sci. Technol., 1998, 32(5), 657–662 CrossRef CAS.
- P. W. Park and J. S. Ledford, Effect of crystallinity on the photoreduction of cerium oxide: A study of CeO2 and Ce/Al2O3 catalysts, Langmuir, 1996, 12(7), 1794–1799 CrossRef CAS.
- K.-D. Schierbaum, Ordered ultra-thin cerium oxide overlayers on Pt (111) single crystal surfaces studied by LEED and XPS, Surf. Sci., 1998, 399(1), 29–38 CrossRef CAS.
- M. Soma, A. Tanaka, H. Seyama and K. Satake, Characterization of arsenic in lake sediments by X-ray photoelectron spectroscopy, Geochim. Cosmochim. Acta, 1994, 58(12), 2743–2745 CrossRef CAS.
- N. Tian, X. Tian, X. Liu, Z. Zhou, C. Yang, L. Ma, C. Tian, Y. Li and Y. Wang, Facile synthesis of hierarchical dendrite-like structure iron layered double hydroxide nanohybrids for effective arsenic removal, Chem. Commun., 2016, 52(80), 11955–11958 RSC.
- S. Zhang, H. Niu, Y. Cai, X. Zhao and Y. Shi, Arsenite and arsenate adsorption on coprecipitated bimetal oxide magnetic nanomaterials: MnFe2O4 and CoFe2O4, Chem. Eng. J., 2010, 158(3), 599–607 CrossRef CAS.
- Y. Zhang, M. Yang, X.-M. Dou, H. He and D.-S. Wang, Arsenate adsorption on an Fe−Ce bimetal oxide adsorbent: role of surface properties, Environ. Sci. Technol., 2005, 39(18), 7246–7253 CrossRef CAS PubMed.
- N. Rivera-Reyna, L. Hinojosa-Reyes, J. L. Guzman-Mar, Y. Cai, K. O'Shea and A. Hernandez-Ramirez, Photocatalytical removal of inorganic and organic arsenic species from aqueous solution using zinc oxide semiconductor, Photochem. Photobiol. Sci., 2013, 12(4), 653–659 CrossRef CAS PubMed.
- H. Boehm, Acidic and basic properties of hydroxylated metal oxide surfaces, Discuss. Faraday Soc., 1971, 52, 264–275 RSC.
- B. Collin, E. Oostveen, O. V. Tsyusko and J. M. Unrine, Influence of Natural Organic Matter and Surface Charge on the Toxicity and Bioaccumulation of Functionalized Ceria Nanoparticles in Caenorhabditis elegans, Environ. Sci. Technol., 2014, 48(2), 1280–1289 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: ESI includes 8 pages and 6 figures, which describe additional studies on the impact of dissolved oxygen on the experimental system, calibration of UV-vis sedimentation measurements, TEM investigation of secondary mineral formation, and aggregation effects on NP surface area. See DOI: 10.1039/d0en00970a |
| ‡ Current address: Earth and Environmental Sciences Division, Los Alamos National Laboratory, Los Alamos, NM 87545, USA. |
| § Current address: Department of Chemical and Environmental Engineering, Yale University, New Haven, Connecticut, 06510, USA. |
| ¶ Current address: Department of Civil and Environmental Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. |
| || Current address: School of Civil, Environmental and Chemical Engineering, Changwon National University, Changwon-si, Gyeongsangnam-do, 51140, Republic of Korea. |
| ** Current address: Environmental Science & Engineering, California Institute of Technology, Pasadena, CA 91125. |
| †† Current address: Department of Civil & Environmental Engineering, University of Washington, Seattle, WA 98195. |
| This journal is © The Royal Society of Chemistry 2021 |