
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The product selectivity zones in gas diffusion electrodes during the electrocatalytic reduction of CO2†
Tim
Möller
 ,
Trung
Ngo Thanh
,
Xingli
Wang
,
Wen
Ju
,
Zarko
Jovanov
and
Peter
Strasser
*
,
Trung
Ngo Thanh
,
Xingli
Wang
,
Wen
Ju
,
Zarko
Jovanov
and
Peter
Strasser
*
The Electrochemical Energy, Catalysis, and Materials Science Laboratory, Department of Chemistry, Chemical Engineering Division, Technical University Berlin, Berlin, Germany. E-mail: pstrasser@tu-berlin.de
First published on 13th October 2021
Abstract
Here we report on the most prominent factors influencing the performance of a Cu-based CO2 electrolyzer operating at high currents. Using a flow-electrolyzer design where CO2 gas feed passes directly through the electrode interacting with the Cu catalyst layer, we observed that the selectivity of the electrochemical CO2 reduction in (bulk) pH neutral media can greatly be influenced by adjusting the structure of the electrode. In this, the variations in catalyst loading and ionomer content can profoundly affect the selectivity of CO2RR. We explore the hypothesis that this originates from the overall mass transport variations within the porous catalytic layer of the gas diffusion electrode. As further evidence for this, apart from the CO2 electrolysis results, we propose a special method to benchmark the reactant mass transport in flow-cells using oxygen reduction reaction (ORR) limiting current measurements. Our analysis suggests that a restriction of mass transport is highly desirable due to its connection to a local alkalization and corresponding suppression of pH-dependent reaction products, given the absence of local CO2 concentration limitations. We further show how the electrode structure can be used to push the observed catalytic CO2 reduction selectivity either towards C1 or C2+ products, dependent on the ionomer content and catalyst loading in a cathodic current range of 50 to 700 mA cm−2. Measurements at various KHCO3 electrolyte concentrations agree with the notion of the local pH dictating the overall selectivity and point towards the presence of pronounced concentration gradients within the system. Overall, our work suggests that the differences in electrocatalytic CO2 reduction selectivity at high currents (in a range of pH neutral buffering electrolytes) largely originate from the local concentration gradients defined by the initial catalyst ink formulation and architecture of the catalytic layer, both of which represent a powerful tool for optimization in the production of selected value-added products.
Broader contextThe electrocatalytic CO2 reduction reaction (CO2RR) on Cu-based catalysts has the potential to enable the sustainable production of commodity chemicals and fuels by upcycling waste-CO2 into value-added compounds. Over the recent years, much progress has been made by fabrication of catalysts into so-called gas diffusion electrodes (GDEs) to tackle the issue of low CO2 solubility in aqueous electrolytes and to advance towards commercially viable current densities of >300 mA cm−2. Though the selectivity towards CO2RR could be largely improved over the competing HER using GDEs, the underlying factors for an efficient production of the individual CO2RR products remain elusive. In the present study, we deploy a flow-electrolyzer system to investigate structure–selectivity-interrelations of a Cu-based GDE in a buffering electrolyte. By systematic investigation of three parameters: (i) particle catalyst loading, (ii) ionomer to catalyst ratio and (iii) buffer capacity, we set out to understand the structural properties that dictate the spatial variation of the selectivity across the catalyst layer at high reaction rates. Thereby, we describe a sequence of zones of distinct selectivity within the catalyst layer (“selectivity zones”), which arise from the inhomogeneous distribution of the reactants (CO2 and pH) and control the overall selectivity of the system. |
Introduction
The direct electrochemical CO2 reduction reaction (CO2RR) has been emerging as a potential technology for storage of renewable energy and sustainable production of carbon-based chemicals.1 In this area, the catalyst research has mainly been focused on Cu due to its capability to reduce CO2 towards value-added, post-CO products, such as hydrocarbons and alcohols.2 The control over the electrocatalytic selectivity has been the focal point of Cu-based catalyst research, due to parallel reaction pathways and the resulting mixture of products.3 Various modifications of Cu have been investigated in order to steer the selectivity towards distinct products, as for instance exposure of specific facets through nanostructuring and altered catalyst composition through oxidative treatments or addition of second metals.4–7 Apart from the catalyst itself, the reaction conditions have been repeatedly reported to influence the selectivity of the CO2RR. For instance, local pH value gradients establishing at the electrode electrolyte interface, also described as alkalization, were shown to have a decisive impact on reaction selectivity by suppression of mechanistic pathways dependent on the proton transfer as a rate-limiting step.8–10 Studies performed in very commonly used KHCO3 electrolytes with neutral bulk pH, show that the local pH gradients near electrode surface essentially arise from a deficient mass transport unable to buffer the cathodic production of OH−.11–13 For H-type setups, effects of variations in the structure of the catalytic layer have been repeatedly reported to influence the mass transport, critically altering the reaction environment and therefore determining the observed selectivity of the system during electrochemical CO2RR. Here, the mass transport can be divided into a transport of reactants directed towards the surface and a transport of products leaving the surface. On the reactant side, in the aqueous electrolyte dissolved, CO2 is transported towards the catalytic sites, where abundantly available water molecules supply the protons to form reduced carbon compounds under production of OH−. The generated OH− can readily react with the commonly used HCO3− electrolyte acting as a buffer. However, HCO3− transport from the bulk towards the surface is not sufficiently fast to keep up with the OH− production rate, which locally shifts the pH and bicarbonate-equilibrium near the reaction sites. This effect of local alkalization during CO2RR was observed experimentally at already moderate potentials by surface enhanced IR and Raman spectroscopy, simulated based on bicarbonate equilibria and often suggested to be the origin of a high selectivity for multi-carbon products during electrochemical CO2RR.12,14–18 On the products side, carbon compounds need to be transported away from the surface to allow for further reactants to fill in. However, the sustained presence of reactive intermediates such as CO have also been reported to show a beneficial effect on production of ethylene and oxygenates, and was also suggested to show a suppression for the competing HER from water reduction due to the occupation of the reactive sites.19–23 Generally, an increasing thickness of the catalytic layer, together with the associated electrochemically active surface area (ECSA), increases the mean path of transportation for the reactants and products. This has been proposed to impede a homogeneous through-plane mass transport within the catalytic layer, which results in more pronounced concentration gradients throughout the system.12,15,24 Accordingly, it has been suggested for mesoporous Ag electrodes of various thicknesses that the CO2RR selectivity is sensitive to the concentration gradients and shows the strongest suppression of the competing HER for the thickest electrode at the lowest part of the catalytic layer, where the effect of alkalization was most pronounced.25,26Recently, the field of CO2RR has progressively moved towards studies of the high current regime in pH neutral and alkaline, gas-feed flow-electrolyzers to approach technological rates.27–29 As the cathodic production of OH− is a function of the current, flow-electrolyzers in buffered electrolytes have been suggested to be particularly affected by local alkalization due to insufficient HCO3− transport and the observed high selectivity for C2+ products has been, at least in part, associated to that effect.30–32 Additionally, the depletion of CO2 near the catalyst surface has also been reported to limit the accessible CO2RR currents and greatly influence the observed catalytic selectivity.33–35 Despite those indications for the crucial role of the electrode structure for high-current CO2RR, correlations similar to the impact of mass transport in low-current H-cells remain underexplored for gas diffusion electrodes in flow-electrolyzers. It is plausible that high rate CO2RR on GDEs in flow-cell electrolyzers shows the same fundamental correlations of mass transport and catalytic selectivity as those reported for H-cell setups. We propose that the catalytic selectivity of Cu during high rate CO2RR in pH buffering KHCO3 electrolyte is largely controlled by the mass transport, which can be rationalized by a discussion of CO2 transport and transport of pH buffering HCO3− anions.
Flow-cell electrolyzer setup
We employed a flow-cell electrolyzer with three distinct compartments to investigate CO2RR electrolysis at high currents and catalytic rates (Fig. 1a). Two different solutions of 1 M KHCO3 were used separately as anolyte and catholyte and continuously looped through the two liquid compartments divided by an anion conducting membrane. Anodic and cathodic reaction products were transported out of the respective compartments with the flow of the electrolytes. A convective stream of CO2 was fed from the third compartment through the backside of the porous cathode towards the catalytic layer, which allowed a fast supply of reactant to the cathodic reaction sites.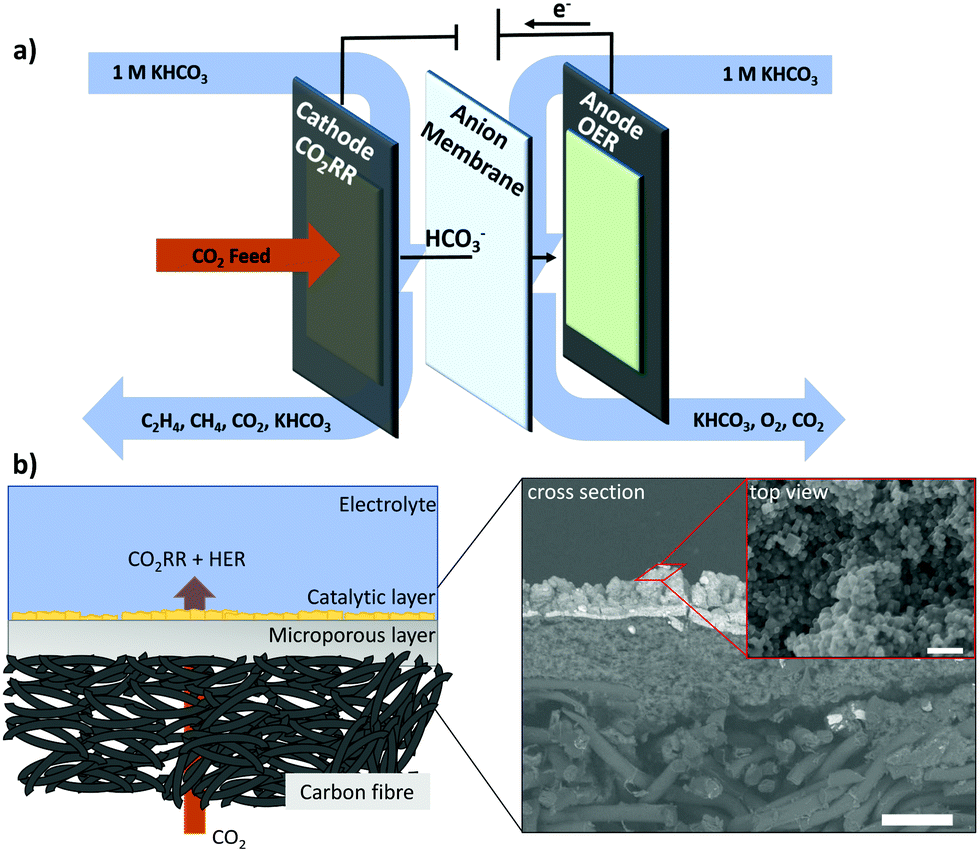 | ||
| Fig. 1 Schematic representation of a 3-compartment flow-cell electrolyzer with indications of the transport directions for reactants, products and electrolyte (a). Schematic representation of carbon-based gas diffusion electrode with cross-section (50 μm scalebar) and top view SEM images (400 nm scalebar) of a GDE prepared by spray-coating a dispersion of a cubic Cu2O catalyst with Nafion binder onto a carbon gas diffusion layer (b). | ||
Cathode electrode design and architecture
The cathode catalyst layer was deposited by airbrushing an ink containing a mixture of Nafion and cubic Cu2O nanoparticles on a carbon-based Freudenberg H23C2 gas diffusion layer serving as substrate. As anode, a commercially available dimensionally stable electrode composed of a Ti sheet covered by an Ir-MMO was routinely used. Throughout this study, we will refer to the uncoated Freudenberg H23C2 as the gas diffusion layer, “GDL”, and to the particle-coated Freudenberg H23C2 as gas diffusion electrode, “GDE”. Fig. 1b shows the schematic structure of a GDE prepared by spraycoating. In this system, the different elements of the GDE are spatially separated and can be assigned to different layers. The lowest layer consists of carbon fibers and acts as mechanical and conductive backbone of the entire structure. Next, the microporous layer (MPL) functions as conductive and hydrophobic substrate, which allows the electronic and CO2 transport towards the adjoining catalytic layer. The hydrophobicity, gas permeability and electrical conductivity of the GDL are important parameters that influence the CO2RR performance and stability of the GDE. Especially, the hydrophobicity has proven decisive to maintain a sufficient gas (CO2) transport towards the catalytically active sites by preventing a “flooding” that is an extensive wetting of the porous GDE structure. For carbon-based GDLs an instability of the wetting characteristics has been observed under application of cathodic potentials and correlated to the catalytic activity towards HER of the carbon structure.36 To circumvent this effect, PTFE membranes have been investigated as GDL materials, but can suffer from issues of electrical conductivity and mechanical instability.29 In this study, we use a “flow-through” approach, depicted in Fig. 1(b), to minimize flooding issues connected to the use of a carbon-based substrate and focus exclusively on properties of the catalytic layer. The catalytic layer represents the uppermost element of the GDE and is the interface between gaseous transports of CO2 and liquid transport of electrolyte. Within the catalytic layer, the electronic, ionic and reactant transport intersect and define the reaction environment of the electrocatalytic CO2RR and the reactivity of the system. The morphology of the catalytic layer obtained by spraycoating is not trivial and showed a porous, 3-dimensional appearance with a rough surface, visible in cross-section and top-view SEM images of Fig. 1b.In this paper, we use a previously reported nanocubic Cu2O catalyst (see Fig. S1, ESI†) with an edge length of 35 nm to systematically alter the structure of the catalytic layer of a gas diffusion electrode (GDE) for tests in a flow-electrolyzer.37 By changing macroscopic parameters, as the particle catalyst loading and the ionomer to particle catalyst ratio (Nafion content), we influence the accessibility of catalytically active sites and the through-plane mass transport in the porous structure of the catalytic layer. In this, we correlate the electrode structure with restriction in mass transport, manifesting in local concentration gradients and alkalization, which in turn influences the selectivity of CO2RR during a high current density operation in a bulk neutral bicarbonate electrolyte. We chose our previously reported and well-characterized cubic Cu2O catalyst as a model catalyst system to investigate the influence of concentration gradients within a flow-cell electrolyzer by variation of three macroscopic parameters:
• Particle catalyst loading: first, we varied the amount of deposited catalyst particles to obtain GDEs with various catalyst loadings that show differences in layer thickness, roughness and ECSA. In doing so, we affect the mean path of transportation from the bulk sources of CO2 and KHCO3 towards the catalytic sites.
• Ionomer to particle catalyst ratio: to introduce an impediment for the mass transport, we varied the Nafion content of the ink formulation to obtain GDEs that show different ionomer to catalyst ratios for a constant absolute catalyst loading. Nafion acts as a strong adhesive to introduce a mechanical stability to the catalytic layer, however, the ionomer distribution is a known issue in fuel cell research due to its influence on local reactant transportation and associated mass (oxygen) transport resistances.38,39 Likewise, the transport of reactants in CO2RR towards the active sites should also be sensitive to the local distribution of the ionomer, acting as a barrier for the CO2 and HCO3− transport.
• Buffer capacity: to induce various buffer capacities, we varied the concentration of the KHCO3 electrolyte to alter the buffer capacity within our system. A higher concentration of HCO3− offers more buffer capacity, in turn resulting in decreased pH gradients at the interface and counteracting the effect of alkalization.
The present work is not about setting new number records in product selectivity, but to pinpoint controlling factors for the product selectivity in a technological GDE under more realistic reaction conditions. To characterize the mass transport properties of CO2RR GDEs, we employ oxygen reduction limiting current measurements typically used in the hydrogen fuel cell community as diagnostic tool in the field of CO2 reduction, the results of which reveal how closely GDE structure and the resulting catalytic selectivity are connected. The conclusions of this work are consistent with previous reports on the importance of near electrode surface pH and CO2 concentration, but expand our understanding of the relationships between catalyst layer structure of a GDE and reaction selectivity during CO2RR.12,29,31,33,40,41 The systematic investigation of the effects within our work prompt us to propose the notion of layered “selectivity zones”, which has not been, to the best of our knowledge, previously described for a Cu-based GDE in a flow-electrolyzer. We believe that our selectivity zone model will aid the future design of efficient Cu-based GDEs for CO2 electrolyzers in buffered electrolytes of near-neutral pH.
Results
Effect of particle catalyst loading on CO2 electrolysis
In Fig. 2, we present the distributions of four important products observed during CO2RR on Cu: C2H4, CH4, H2 and HCOO−, obtained at fixed applied current densities on electrodes containing varying catalyst loadings. The influence of catalyst loading on the product distribution has been tested at fixed concentration of 1.0 M KHCO3 and at fixed binder content (10 wt% Nafion). Here, the FE of C2H4, CH4, H2 and HCOO− is displayed as a function of the applied current density for catalyst mass loadings ranging from 0.3 mg cm−2 to 2.0 mg cm−2. We specifically focus on this set of products, as they have distinctly different rate-limiting reaction steps, which show either a strong pH-dependence (CH4, HCOO− and H2) or primarily independence (C2H4) and can therefore provide insight in the local reaction environment.42,43 A first key observation is the clear suppression of all pH-dependent products (CH4, HCOO− and H2) at high mass loadings of the catalyst, which was invariably linked to a favored production of ethylene. Note how the FE of CH4, which is a preferred product during CO2RR at high proton concentration, plummeted to below 1%, as soon as catalyst loading was increased to 1.3 mg cm−2 and higher. In contrast, in experiments using very low catalysts loadings of below 0.4 mg cm−2 the FE for CH4 raised to above 20%, see Fig. 2b. While this trend was qualitatively in line with the production of H2 and HCOO− (Fig. 2c and d) the change in FE for C2H4 showed the opposite behavior, see Fig. 2a. Here, the decrease of catalyst loading below 0.4 mg cm−2 resulted in a FE for C2H4 of below 20%, whereas higher loadings allowed for a more selective C2H4 production and increased FE to around 30%. Due to the well-known sensitivity of CO2RR selectivity to the electrode potential, we plotted the product FE against IR-free RHE potentials in Fig. S2 (ESI†). Note that as the shift in selectivity is also visible on an IR-free potential scale, we thereby succeed in excluding variations in electrode potential as the sole origin of the observed effect. Additional information of FE for the remaining CO2RR products (EtOH, PrOH and CO) are given in Fig. S3 (ESI†) and agree with the observed favorable production C2+ compounds at high catalyst loadings. We also plotted the FE of gas products as a function of time, see Fig. S4 (ESI†), which confirmed the applied current density as cause for the observed change in selectivity over the investigated testing time of 18 hours. Only for the low loading samples of 0.3 mg cm−2 and 0.4 mg cm−2 a decline in CO2RR selectivity could be observed at constant current, which suggests a decreasing stability with decreasing particle catalyst loading. Morphological investigation of the GDEs prepared by deposition of various catalyst loadings showed a clear dependence on the absolute catalyst loading. Here, top-view SEM images after CO2 electrolysis are given in Fig. S5a and b (ESI†). We observed a progressive roughening and introduction of pores to the surface upon increase of particle loading. Such structural changes suggest an increase in electrochemically accessible surface area (ECSA), which we have investigated by quantification of the electrochemical charging of the double layer (see Fig. S6, ESI†). As a measure of the ECSA, we calculated the double-layer capacitance, shown in Fig. S5c (ESI†), which showed an increase with particle loading and agrees with our observation of a progressive surface roughening in SEM images. In accordance with an increased ECSA, we noticed a higher catalytic activity during CO2RR for the high particle loadings visible in the polarization curves of Fig. S5d (ESI†). Our observations suggest that the catalytic selectivity during high-rate CO2RR is sensitive to the structure of the catalytic layer, which can be affected by variation of the catalyst loading. We suspect that the correlation between catalytic selectivity and catalyst loading originates in changes of the mass transport (e.g. transport of CO2 and HCO3−), which in turn affect the local pH and reactant concentration near the electrode.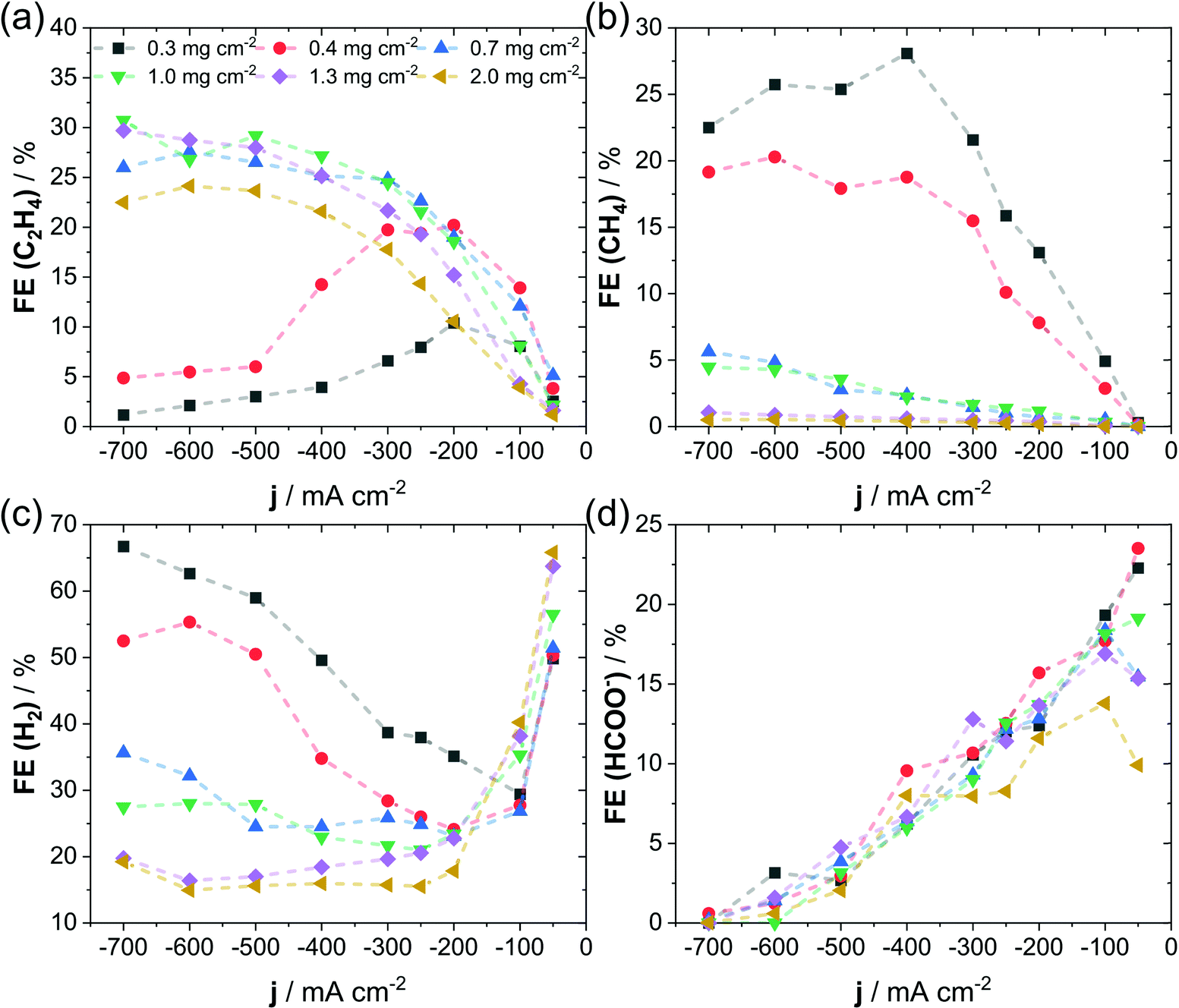 | ||
| Fig. 2 Faradaic efficiency as a function of applied current density at varying catalyst mass loadings for C2H4 (a), CH4 (b), H2 (c) and HCOO− (d). Reactions were conducted under following conditions: 3 cm2 of geometric surface area of cathode, 1 M KHCO3 and 10 wt% of Nafion used as binder in catalyst ink. Additional products (CO, EtOH, and PrOH) are given in Fig. S3 of the ESI.† Dashed lines are shown to guide the eye. | ||
Effect of particle catalyst to ionomer ratio (Nafion content) on CO2 electrolysis
In order to further investigate the mass transport as potential origin of shifts in catalytic selectivity, we varied the binder content within the catalytic layer. Inspired by fuel cell research, we suspect that increasing amounts of ionomer within the catalytic layer represents a barrier for the mass transport of reactive species as dissolved CO2 and HCO3− towards reaction sites. Fig. 3 shows the effect of an increase in Nafion content within the catalytic layer of a GDE on the selectivity during CO2RR. A suppression of CH4 and HCOO− is evident in samples with high Nafion content, see Fig. 3b and d, similar to what we have observed at high catalyst mass loadings. Interestingly, samples with high Nafion content (30 and 50 wt%) show a suppression of HER and increased C2H4 FE only at a relatively low current density of smaller than 300 mA cm−2 (Fig. 3a and c). A further increase of current density, results in a strong rise of FE for HER at the cost of total CO2RR FE. We suspect that this change in selectivity at high currents is caused by an excessive content of Nafion, which reduces the mass transport to a point, where the reactant (CO2) transport becomes limiting for CO2RR and causes the HER to dominate in the process. While such a correlation between the Nafion content and the limited reactant transport is restricting for CO2RR selectivity for a current density larger than 300 mA cm−2, it shows an increase at lower rates. Excessive amounts of ionomer influence potentially the electrical accessibility of the catalyst particles, which is why we investigated the change in electrochemical double layer capacitance and catalytic activity. Here, we only observed minor changes in double layer capacitance with varied Nafion loading, but a lower catalytic activity of the 50 wt% Nafion sample was apparent (see Fig. S7, ESI†). Again, to exclude differences in electrode overpotential as cause of the observed trend, we plotted IR-free RHE electrode potentials against the product FE in Fig. S8 (ESI†), which still clearly shows the ionomer induced shift in selectivity. In agreement with our previous observation, the FE for gas products as a function of time showed only small changes at constant current for the electrodes prepared with different Nafion loadings, see Fig. S9 (ESI†), suggesting a stability of the system over the investigated testing time. Furthermore, we have also investigated the change in surface structure at different Nafion contents by SEM, shown in Fig. S10 (ESI†). Here, an intermediate content of Nafion (10 and 30 wt%) resulted in a rough surface of the catalytic layer, whereas the extreme cases of 0 wt% and 50 wt% showed a relatively smooth surface. Noteworthy is the pronounced coverage of the Cu2O cubes supposedly by the ionomer in the 50 wt% Nafion sample, visible in images of higher magnification, consistent with the idea of representing a distinct barrier for the mass transport. Additional CO2RR products (CO, EtOH and PrOH) are given in Fig. S11 (ESI†), which showed a higher selectivity for EtOH with increased Nafion content at moderate currents. Our observations on the effects of increased Nafion content are qualitatively in line with the effect of particle catalyst loading. In both cases, we generally observed a higher selectivity for C2+ products when either binder content or particle loading was increased. It seems feasible that a similar origin causes the observed selectivity shifts, given their high qualitative agreement. | ||
| Fig. 3 Faradaic efficiency as a function of applied current density with varying Nafion contents for C2H4 (a), CH4 (b), H2 (c) and HCOO− (d). Conditions were as follows: 3 cm2 of geometric surface area, 0.7 mg cm−2 catalyst mass loading and 1 M KHCO3. Additional products (CO, EtOH, and PrOH) are given in Fig. S11 of the ESI.† Dashed lines are shown to guide the eye. | ||
Effect of buffer capacity (KHCO3 concentration) on CO2 electrolysis
Fig. 4a–f shows the influence of the bulk KHCO3 concentration on the C2+ (C2H4, EtOH and PrOH) and C1 (CH4, HCOO−) selectivity during CO2 electrolysis in the flow-cell. Here, we used different Nafion contents of 10 wt% to 50 wt% and a constant catalyst loading of about 0.7 mg cm−2. For the 50 wt% Nafion sample we observed no significant change in catalytic selectivity, when increasing electrolyte concentration from 0.1 M to 1.0 M KHCO3, see Fig. 4a and d. Both concentrations showed the highest C2+ selectivity of roughly 40% FE at 200 mA cm−2, whereas a larger current density resulted in the increase of the competing HER (Fig. S12a, ESI†). Further increase of KHCO3 electrolyte to 3 M lowered the C2+ FE and favored HER over the whole investigated current range. In contrast, the FE for C1 products seemed largely unaffected by the change in KHCO3 concentration. Next, the 30 wt% Nafion sample showed a clear dependence of C2+ and C1 FE on the KHCO3 concentration during CO2RR, see Fig. 4b and e. While we were able to achieve a combined C2+ FE of almost 70% at 600 mA cm−2, the use of higher KHCO3 concentrations of 1 and 3 M led to a decreased C2+ FE to around 50% and 30%, respectively. For the FE of C1 products we observed an inverse behavior, here 3 M KHCO3 showed the highest combined C1 FE of roughly 20%, while the use of 1 M and 0.1 M KHCO3 led to a subsequent decrease. Our observations during CO2RR, using a 10 wt% Nafion sample were quite similar to the case of a 30 wt% Nafion sample, see Fig. 4c and f. Again, we achieved the highest C2+ selectivity at the lowest KHCO3 concentration of 0.1 M, which enabled a combined FE of almost 60% over a broad range of cathodic currents. The use of an increased KHCO3 concentration of 1 and 3 M resulted in a dramatically decreased C2+ FE of 40% and 5%, respectively. At the same time, the highest C1 FE observed on the 10 wt% Nafion sample increased from a maximum of 13%, towards 17% and roughly 23% for respective KHCO3 concentrations of 0.1 M, 1.0 M and 3.0 M. In parallel to the CO2RR, the HER also proved sensitive to KHCO3 concentration and showed an overall increase with KHCO3 concentration, whereas the 50 wt% sample seemed to be generally less sensitive to the buffer concentration (Fig. S12a–c, ESI†). At all Nafion contents we observed a higher selectivity for C2+ products and generally lower HER and C1 FE, when we employed KHCO3 electrolytes of a lower concentration during electrolysis, however, this effect seemed to be dependent on Nafion content and was most pronounced for the lowest Nafion content of 10 wt%. | ||
| Fig. 4 Effect of variations in KHCO3 concentration on the CO2RR selectivity towards C2+ products using 50 wt% (a), 30 wt% (b), and 10 wt% (c) of Nafion. Effect of variations in KHCO3 concentration on the CO2RR selectivity towards C1 products using 50 wt% (d), 30 wt% (e), and 10 wt% (f) of Nafion. In all cases Cu2O loading was const. at 0.7 mg cm−2. Additional information on FE of H2 and CO are given in Fig. S12 (ESI†). | ||
Implementation of the ORR as a diagnostic tool for benchmarking the mass transport limitations within a complex porous electrode system
As the effects of the local pH, the mass transport and the catalyst kinetics are superimposed and, therefore, often difficult to be unambiguously deconvoluted, especially due to parallel reaction pathways of CO2RR on Cu and the competing HER, we resorted to the oxygen reduction reaction (ORR) for further discussion of the correlation between binder content and mass transport. In doing so, we were able to exploit the more positive standard reduction potential of the ORR, with respect to the CO2RR, to fully avoid the HER. Hence, we obtained data that we can interpret with overall less complexity, schematically depicted in Fig. 5a. We take the view that such an approach allows for a more direct investigation of the mass transport limitations within our system. To probe the mass transport towards the catalytically active centers of the Cu2O particles, we varied the partial pressure of O2 in an N2 feed at a constant electrode potential of −0.45 VRHE. We chose this potential to achieve the highest possible rate of ORR, while avoiding a region of considerable HER activity, indicated in Fig. 5b. By tracing the change in ORR current as a function of O2 partial pressure at a fixed electrode potential, we can directly access changes in mass transport caused by variations in binder content. In Fig. 5c, we can see a rise in ORR current with increase in partial pressure of O2 until around 0.3 bar, where the ORR current of the 10 wt% Nafion sample approaches a plateau. This transition supposedly originates in a change from a primarily mass transport dominated regime, towards one, were activation resistances are denying higher reduction rates. The regimes of mass transport and catalytic kinetics are indicated by different levels of shading in Fig. 5c, but should be rather understood as a visual orientation and not as a strict border between the two. A comparison of three samples with Nafion loadings ranging from 10 to 50 wt% shows an increased ORR current in the mass transport domain of below 0.3 bar of O2 partial pressure for lower loadings of the ionomer, however, this difference seemed to decrease progressively with a higher partial O2 pressure (Fig. S13, ESI†). The observed behavior suggests that the mass transport remains limiting even at higher O2 partial pressure for samples of high Nafion content, whereas samples of reduced binder content showed primarily kinetic limitations at a similar partial pressure of O2 and constant electrode potential. Our results suggest that the binder content of the catalytic layer of a GDE interferes with the reactant transport. Here, a high binder content induces larger resistances for the reactant transport and can lead to significant mass transport limitations.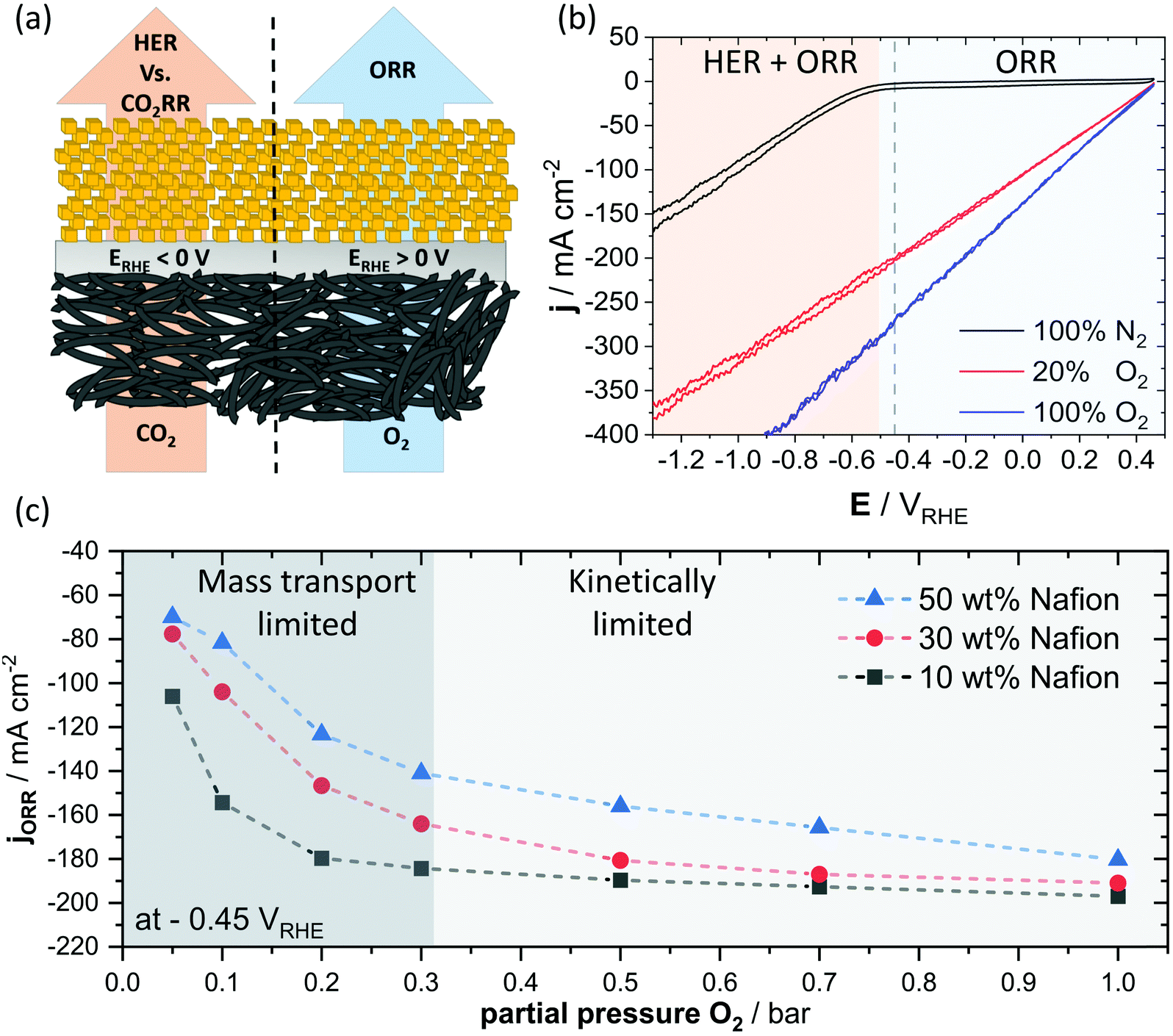 | ||
| Fig. 5 Schematic representation of CO2RR and ORR on the Cu surface of a GDE (a). Cyclic voltammetry under different ratios of O2/N2 saturated gas atmosphere in 2 M KHCO3 for a 10 wt% of Nafion GDE. Different colors are indicating a potential regime of pure ORR or mixed regime of HER and ORR (b). ORR current as a function of partial O2 pressure from O2/N2 mixtures in 2 M KHCO3 for different Nafion contents in the catalytic layer at a constant electrode potential of −0.45 VRHE with dashed lines to guide the eye. Different levels of shading indicate the primary limitation, either mass transport or reaction kinetics (c). | ||
Discussion
Electrode structure determines concentration gradients during CO2 electrolysis
Combining our observations, we can schematically depict the proposed influence of the catalytic layer structure on the selectivity during high-rate CO2RR electrolysis in a pH-neutral buffering media. Fig. 6a, shows a schematic GDE and illustrates the relevant reactions, as well as the through-plane transport of reactants and products in the catalytic layer during CO2RR. While CO2 is fed in a gaseous state to the GDE, we take the view that CO2 is highly likely to physically dissolve in the electrolyte prior to its reduction on the catalyst surface due to the presumable presence of a native solvent layer caused by the hydrophilicity of charged electrodes. Accordingly, the transport of reactive species (CO2 and HCO3−) and products has to occur through a diffusion layer of finite thickness to reach or leave the electrode surface. On the catalyst, OH− is being produced during the reductive reaction of CO2 and H2O. The OH− can readily react with the present buffering HCO3− anions that are diffusively transported from the bulk electrolyte, which reduces the local pH increase near the surface and creates CO32− in the process. The change of the local pH is a function of the current density that is a measure of the OH− production rate and the molar flux of HCO3− anions transported towards the electrode surface in a diffusive manner. The generated CO32− is being transported towards the bulk of the electrolyte, where it can equilibrate with excess CO2 to regenerate the buffer by formation of HCO3−, therefore, preventing larger changes in bulk pH. It is important to note that in GDEs the source of CO2 and buffering HCO3− anions are on opposing sides as indicated by opposed directions of arrows in Fig. 6a. This phase asymmetric feed implies a largely decoupled transport of dissolved CO2 and HCO3− towards the electrode surface of GDEs in flow-electrolyzers, which is in contrast to conventional electrodes that are fully submerged in liquid electrolytes during CO2RR in H-cell setups.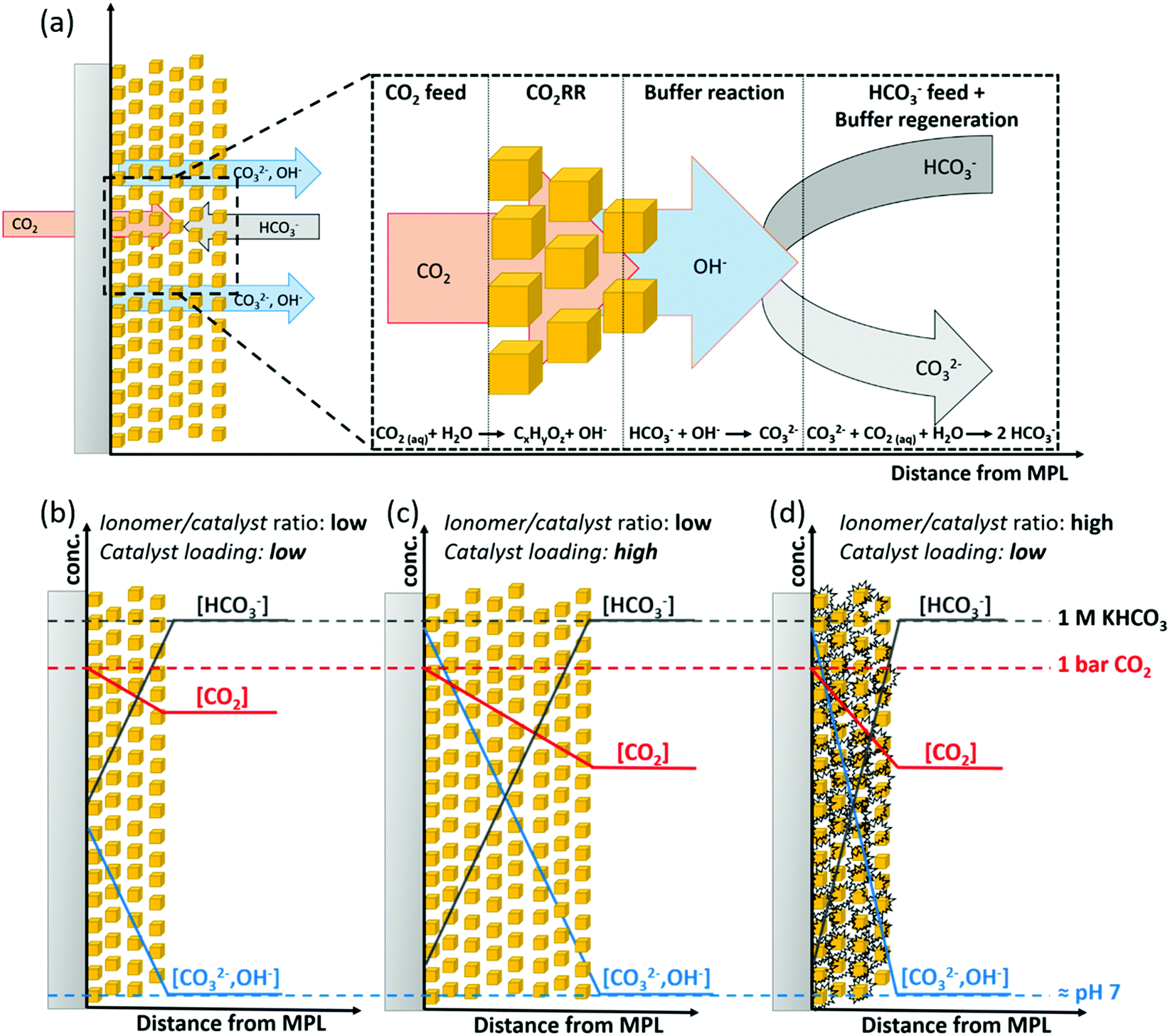 | ||
| Fig. 6 Scheme qualitatively visualizing the flux of reactive species and products with indications of their respective transport directions throughout the structure of a Cu-GDE during CO2RR in KHCO3 (a). Schematic representation of the proposed influence of variations in the particle catalyst loading (b and c) and the ionomer to catalyst ratio (b and d) on the concentration gradients of HCO3− (grey), CO2 (red) and OH− (blue) throughout the catalytic layer. | ||
The proposed effect for variation of ionomer to catalyst ratio (Nafion content) and particle catalyst loading is depicted in Fig. 6b–d and is largely associated to the through-plane concentration gradients of CO2 and HCO3− within the catalytic layer. Here, the effect of increasing the particle catalyst loading can be rationalized by a larger mean path of transportation for CO2 and HCO3− through the thicker catalytic layer. This longer distance of transportation results in a further decrease along the concentration gradients for both species, see Fig. 6b and c. This would in turn cause regions of high alkalinity and depletion of CO2, respectively. We suspect that the increase in Nafion content causes a quite similar drop in reactant concentration throughout the layer. Here, as we have shown from our ORR measurements, an extensive content of Nafion in the catalytic layer presents a barrier for the mass transport of reactants, e.g. O2 or CO2, towards the Cu sites. Such impediment of the mass transport can be rationalized by a slower through-plane transport of reactants (CO2 and HCO3−) and steeper associated concentration gradients perpendicular to the catalytic layer, see Fig. 6b and d. Essentially, the outcome is similar to the increased particle loading and results in regions of high alkalinity and depletion of CO2 at high ionomer content. Additionally, both, the Nafion content and particle catalyst loading influenced the apparent morphology of the catalyst layer (see Fig. S5 and S10, ESI†), which has often been reported in literature to change the catalytic selectivity. However, given the comparatively small change in double layer capacitance (see Fig. S6 and S7, ESI†), we take view that such morphological changes did not critically influence the overall performance. A more detailed discussion on the change in morphology can be found in the ESI† of this work.
Finally, the effect of an increased KHCO3 bulk concentration can also be rationalized by a discussion of the HCO3− concentration gradient. Here, the generally higher HCO3− concentration offers a higher buffer capacity and can in turn reduce the pH gradient throughout the catalytic layer. An additional effect could be caused by the sensitivity of the physical solubility of CO2 for the ionic strength, which could lead to a depletion of CO2 reactant at high KHCO3 concentrations.
In line with the proposed emergence of concentration gradients, as suggested within the present work and visualised in Fig. 6, previous studies reported on considerable local deviations from bulk reactant concentrations during CO2RR based on calculations from transport models. Here, most studies focused on the electrode to catholyte interface and described a depletion in CO2 concentration and an increase in OH− and CO32− concentration in this near-electrode surface region. The magnitude of the emerging concentration gradients was suggested to be heavily dependent on the diffusion layer thickness, which is highly sensitive to convective interferences through effects such as buoyancy.12,14,31,44 Studies that additionally included the thickness of an extended catalyst layer in their transport models reported on an inhomogeneous reactant concentration throughout the catalyst layer during high-rate electrolysis, which is qualitatively in agreement with our proposition depicted in Fig. 6.26,32,45
Concentration gradients establish “selectivity zones”
As a consequence of the through-plane concentration gradients for CO2 and pH within the catalytic layer, we propose zones of distinctly different CO2RR selectivity as a function of distance from the MPL substrate in direction of the catholyte, shown in Fig. 7. Here, a schematic cross section of a GDE with indications for the suggested change in local reaction environment and catalytic selectivity dependent on the spatial location within the catalytic layer is depicted. The zone closest to the MPL shows the highest proximity to the gaseous CO2 feed and furthest distance from the bulk KHCO3 electrolyte, which results in a region of high pH and CO2 concentration. With increasing distance from the MPL, those conditions reverse and can result in a CO2-deficient zone with lower pH value. Our observed experimental catalytic CO2 selectivities directly correlate and support the zone model. Here, the region of high pH and CO2 concentration directly adjacent to the MPL gives optimal conditions for selective production of pH independent CO2RR (C2+) products, as ethylene. With further distance from the MPL, the pH value decreases and the catalytic selectivity can shift towards CO2RR products, which are preferred at higher proton concentration (C1), e.g. CH4. Finally, in the outermost region of the catalytic layer CO2 concentration might become deficient, which shifts the catalytic selectivity towards an increased competition by HER. The proposed “selectivity zone” model is qualitatively in line with recent computational studies that report on pronounced differences in spatial production rates for individual CO2RR products throughout the extended structure of the catalyst layer. Based on micro kinetic calculations, the studies revealed pronounced differences in local reaction rates that arise from a non-uniform reactant distribution. The reactant concentration gradients have been described to establish in-plane as well as through-plane of the catalyst layer, causing a complex spatial distribution in CO2RR and HER activity.26,32,45,46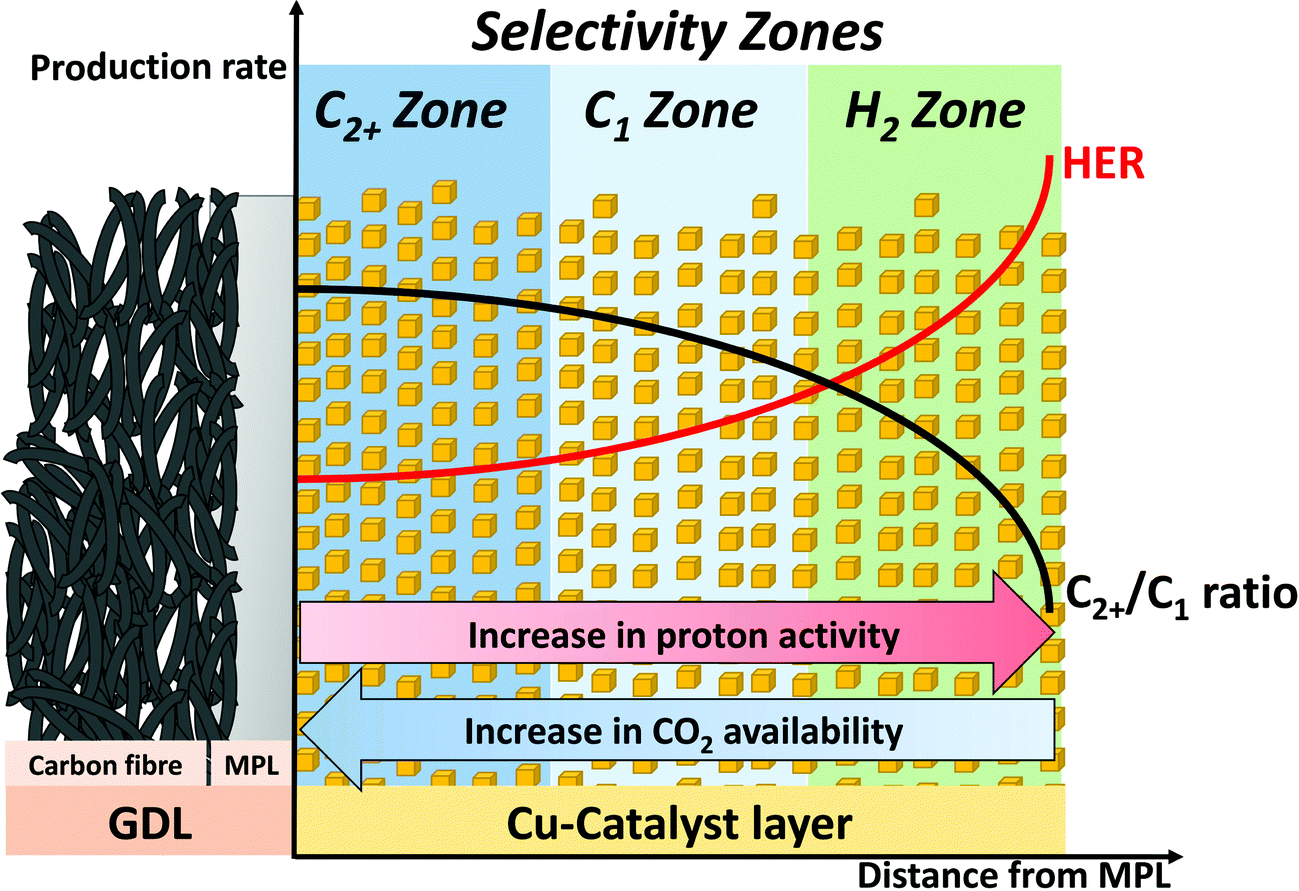 | ||
| Fig. 7 Schematic cross section of a Gas Diffusion Electrode (GDE) of a CO2 electrolyzer comprising the Gas Diffusion Layer, GDL (including its Microporous Layer, MPL) and Cu catalyst layer (not drawn to scale). Focus is on the cross section of the enlarged Cu catalyst layer illustrating the concept of spatial “selectivity zones”. Zones are denoted by their major reduction products (“C2+” blue, “C1” aqua, and “H2” green). Origin of the zones are the spatial through-plane variations in local pH (proton activity, red arrow) and local CO2 concentration (CO2 availability, blue arrow). The red and black spatial distribution curves illustrate schematically the production rate of hydrogen and the production rate ratio of C2+/C1. | ||
Based on the proposed selectivity zones, a selective CO2RR electrolyzer for production of C2+ compounds in buffering electrolytes would require the deliberate introduction of an impediment for the transport of buffering anions. This could be realized by controlling the structure of the GDE through an increased thickness and a higher density of the catalytic layer, if the transport of CO2 remains sufficient. Next to the discussed parameters within this work, further structural control of the catalytic layer could be achieved by variations in the deployed catalyst particle size or introduction of conductive supporting materials of a defined porosity. Additionally, operation conditions could achieve a similar effect and contribute for further increase in C2+ selectivity. Here, lowering electrolyte flow-rates and avoiding bubble formation at the electrode to electrolyte interface could reduce the interfacial convection and in turn lead to creation of an increased hydrodynamic boundary layer thickness, further limiting the transport of buffering anions. We hypothesize that the notion of spatial “selectivity zones” (selectivity zone model) has a broader validity; as a result of this, the structure–selectivity-relationships described here should be also applicable to other (non-Cu) CO2RR systems or even to other reaction processes in GDEs. Similar interrelations of catalyst layer thickness and CO2RR selectivity in buffered electrolytes have been reported for CO evolving systems of Ag and Au catalysts, as well as for systems deploying Sn particles for selective formation of HCOO−.25,47–51 At its core, the selectivity zones model describes a dependency of the HER and CO2RR reaction rates on the spatial reactant concentration due to concentration overpotentials and accordingly should apply to various systems in the field of electrocatalytic CO2RR.
Conclusion
In this study, we have investigated relations between electrode structure, more specifically, catalyst loading and Nafion content, and the resulting product selectivity of Cu-based CO2RR electrolysis at high currents in a bulk pH-neutral flow-electrolyzer. We found that with increasing catalyst loading, that is catalyst layer thickness, or Nafion content the production of pH-sensitive products (e.g. H2, CH4 and HCOO−) could be suppressed and C2+ species were produced more selectively. To explain this, we showed that the Nafion content influences the mass transport using an ORR limiting current analysis at varying oxygen partial pressure as a diagnostic tool. We concluded that such changes in mass transport define and control the local reaction environment in form of pH and CO2 concentration, and hence can be used to deliberately tune the reaction selectivity. Here, concentration gradients in through-plane direction of the porous catalytic layer are more pronounced and shift the observed catalytic selectivity during CO2RR electrolysis. Varying the KHCO3 electrolyte concentration showed that the selectivity of the system is highly sensitive to the concentration of the buffering media and agreed with our proposal that the local pH variations are crucial in determining the CO2RR selectivity. Our study demonstrates how the structure of the catalytic layer is a key parameter to influence local mass transport and provides an effective way to tune the selectivity during pH neutral (bulk) CO2RR electrolysis at high currents.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work received funding by the German Federal Ministry of Education and Research (Bundesministerium für Bildung und Forschung, BMBF) under grant #033RC004E – (“eEthylene”). The research leading to these results has received funding from the European Union's Horizon 2020 research and innovation program under grant agreement no. 851441, SELECTCO2. The research leading to these results has received funding from the European Union's Horizon 2020 research and innovation program under grant agreement no. 101006701, EcoFuel.References
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef CAS PubMed.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC.
- A. Loiudice, P. Lobaccaro, E. A. Kamali, T. Thao, B. H. Huang, J. W. Ager and R. Buonsanti, Angew. Chem., Int. Ed., 2016, 55, 5789–5792 CrossRef CAS PubMed.
- D. Gao, I. Zegkinoglou, N. J. Divins, F. Scholten, I. Sinev, P. Grosse and B. Roldan Cuenya, ACS Nano, 2017, 11, 4825–4831 CrossRef CAS PubMed.
- C. Kim, T. Möller, J. Schmidt, A. Thomas and P. Strasser, ACS Catal., 2018, 9, 1482–1488 CrossRef.
- R. M. Arán-Ais, F. Scholten, S. Kunze, R. Rizo and B. Roldan Cuenya, Nat. Energy, 2020, 5, 317–325 CrossRef.
- H. Xiao, T. Cheng, W. A. Goddard and R. Sundararaman, J. Am. Chem. Soc., 2016, 138, 483–486 CrossRef CAS PubMed.
- X. Liu, P. Schlexer, J. Xiao, Y. Ji, L. Wang, R. B. Sandberg, M. Tang, K. S. Brown, H. Peng, S. Ringe, C. Hahn, T. F. Jaramillo, J. K. Nørskov and K. Chan, Nat. Commun., 2019, 10, 32 CrossRef CAS PubMed.
- K. J. P. Schouten, E. Pérez Gallent and M. T. M. Koper, J. Electroanal. Chem., 2014, 716, 53–57 CrossRef CAS.
- A. S. Varela, M. Kroschel, T. Reier and P. Strasser, Catal. Today, 2016, 260, 8–13 CrossRef CAS.
- K. Yang, R. Kas and W. A. Smith, J. Am. Chem. Soc., 2019, 141, 15891–15900 CrossRef CAS PubMed.
- H. Hashiba, L.-C. Weng, Y. Chen, H. K. Sato, S. Yotsuhashi, C. Xiang and A. Z. Weber, J. Phys. Chem. C, 2018, 122, 3719–3726 CrossRef CAS.
- N. Gupta, M. Gattrell and B. MacDougall, J. Appl. Electrochem., 2006, 36, 161–172 CrossRef CAS.
- K. Klingan, T. Kottakkat, Z. P. Jovanov, S. Jiang, C. Pasquini, F. Scholten, P. Kubella, A. Bergmann, B. Roldan Cuenya, C. Roth and H. Dau, ChemSusChem, 2018, 11, 3449–3459 CrossRef CAS PubMed.
- R. Kas, R. Kortlever, A. Milbrat, M. T. Koper, G. Mul and J. Baltrusaitis, Phys. Chem. Chem. Phys., 2014, 16, 12194–12201 RSC.
- R. Kas, R. Kortlever, H. Yılmaz, M. T. M. Koper and G. Mul, ChemElectroChem, 2015, 2, 354–358 CrossRef CAS.
- D. Ren, Y. Deng, A. D. Handoko, C. S. Chen, S. Malkhandi and B. S. Yeo, ACS Catal., 2015, 5, 2814–2821 CrossRef CAS.
- J. Gao, H. Zhang, X. Guo, J. Luo, S. M. Zakeeruddin, D. Ren and M. Gratzel, J. Am. Chem. Soc., 2019, 141, 18704–18714 CrossRef CAS PubMed.
- Y. Lum and J. W. Ager, Energy Environ. Sci., 2018, 11, 2935–2944 RSC.
- X. Wang, J. F. de Araujo, W. Ju, A. Bagger, H. Schmies, S. Kuhl, J. Rossmeisl and P. Strasser, Nat. Nanotechnol., 2019, 14, 1063–1070 CrossRef CAS PubMed.
- C. G. Morales-Guio, E. R. Cave, S. A. Nitopi, J. T. Feaster, L. Wang, K. P. Kuhl, A. Jackson, N. C. Johnson, D. N. Abram, T. Hatsukade, C. Hahn and T. F. Jaramillo, Nat. Catal., 2018, 1, 764–771 CrossRef CAS.
- H. Ooka, M. C. Figueiredo and M. T. M. Koper, Langmuir, 2017, 37, 9307–9313 CrossRef PubMed.
- R. David, M. Mark, P. J. Ha and W. Chao, J. Electrochem. Soc., 2018, 165, F799–F804 CrossRef.
- Y. Yoon, A. S. Hall and Y. Surendranath, Angew. Chem., Int. Ed., 2016, 55, 15282–15286 CrossRef CAS PubMed.
- S. Suter and S. Haussener, Energy Environ. Sci., 2019, 12, 1668–1678 RSC.
- M. Ma, E. L. Clark, K. T. Therkildsen, S. Dalsgaard, I. Chorkendorff and B. Seger, Energy Environ. Sci., 2020, 13, 977–985 RSC.
- G. L. De Gregorio, T. Burdyny, A. Loiudice, P. Iyengar, W. A. Smith and R. Buonsanti, ACS Catal., 2020, 10, 4854–4862 CrossRef CAS PubMed.
- C.-T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. García de Arquer, A. Kiani, J. P. Edwards, P. De Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783 CrossRef CAS PubMed.
- R. Kas, K. Yang, D. Bohra, R. Kortlever, T. Burdyny and W. A. Smith, Chem. Sci., 2020, 11, 1738–1749 RSC.
- T. Burdyny and W. A. Smith, Energy Environ. Sci., 2019, 12, 1442–1453 RSC.
- L. C. Weng, A. T. Bell and A. Z. Weber, Phys. Chem. Chem. Phys., 2018, 20, 16973–16984 RSC.
- Y. C. Tan, K. B. Lee, H. Song and J. Oh, Joule, 2020, 4, 1104–1120 CrossRef CAS.
- M. Duarte, B. De Mot, J. Hereijgers and T. Breugelmans, ChemElectroChem, 2019, 6, 5596–5602 CrossRef CAS.
- F. P. García de Arquer, C.-T. Dinh, A. Ozden, J. Wicks, C. McCallum, A. R. Kirmani, D.-H. Nam, C. Gabardo, A. Seifitokaldani, X. Wang, Y. C. Li, F. Li, J. Edwards, L. J. Richter, S. J. Thorpe, D. Sinton and E. H. Sargent, Science, 2020, 367, 661–666 CrossRef PubMed.
- K. Yang, R. Kas, W. A. Smith and T. Burdyny, ACS Energy Lett., 2021, 6, 33–40 CrossRef CAS.
- T. Möller, F. Scholten, T. N. Thanh, I. Sinev, J. Timoshenko, X. Wang, Z. Jovanov, M. Gliech, B. Roldan Cuenya, A. S. Varela and P. Strasser, Angew. Chem., Int. Ed., 2020, 59, 17974–17983 CrossRef PubMed.
- S. Ott, A. Orfanidi, H. Schmies, B. Anke, H. N. Nong, J. Hübner, U. Gernert, M. Gliech, M. Lerch and P. Strasser, Nat. Mater., 2020, 19, 77–85 CrossRef CAS PubMed.
- R. Alink, R. Singh, P. Schneider, K. Christmann, J. Schall, R. Keding and N. Zamel, Molecules, 2020, 25, 1523 CrossRef CAS PubMed.
- X. Wang, A. Xu, F. Li, S. F. Hung, D. H. Nam, C. M. Gabardo, Z. Wang, Y. Xu, A. Ozden, A. S. Rasouli, A. H. Ip, D. Sinton and E. H. Sargent, J. Am. Chem. Soc., 2020, 142, 3525–3531 CrossRef CAS PubMed.
- A. Sedighian Rasouli, X. Wang, J. Wicks, G. Lee, T. Peng, F. Li, C. McCallum, C.-T. Dinh, A. H. Ip, D. Sinton and E. H. Sargent, ACS Sustainable Chem. Eng., 2020, 8, 14668–14673 CrossRef CAS.
- A. Bagger, W. Ju, A. S. Varela, P. Strasser and J. Rossmeisl, ACS Catal., 2019, 9, 7894–7899 CrossRef CAS.
- T. Cheng, H. Xiao and W. A. Goddard, J. Am. Chem. Soc., 2016, 138, 13802–13805 CrossRef CAS PubMed.
- T. Burdyny, P. J. Graham, Y. Pang, C.-T. Dinh, M. Liu, E. H. Sargent and D. Sinton, ACS Sustainable Chem. Eng., 2017, 5, 4031–4040 CrossRef CAS.
- L.-C. Weng, A. T. Bell and A. Z. Weber, Energy Environ. Sci., 2020, 13, 3592–3606 RSC.
- R. Kas, A. G. Star, K. Yang, T. Van Cleve, K. C. Neyerlin and W. A. Smith, ACS Sustainable Chem. Eng., 2021, 9, 1286–1296 CrossRef CAS.
- A. Wuttig, M. Yaguchi, K. Motobayashi, M. Osawa and Y. Surendranath, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E4585–E4593 CrossRef CAS PubMed.
- A. S. Hall, Y. Yoon, A. Wuttig and Y. Surendranath, J. Am. Chem. Soc., 2015, 137, 14834–14837 CrossRef CAS PubMed.
- Q. Wang, H. Dong and H. Yu, RSC Adv., 2014, 4, 59970–59976 RSC.
- J. Wu, P. P. Sharma, B. H. Harris and X.-D. Zhou, J. Power Sources, 2014, 258, 189–194 CrossRef CAS.
- D. Kopljar, A. Inan, P. Vindayer, N. Wagner and E. Klemm, J. Appl. Electrochem., 2014, 44, 1107–1116 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ee01696b |
| This journal is © The Royal Society of Chemistry 2021 |