
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltrahigh solar-driven atmospheric water production enabled by scalable rapid-cycling water harvester with vertically aligned nanocomposite sorbent†
Jiaxing
Xu‡
 ,
Tingxian
Li‡
*,
Taisen
Yan‡
,
Si
Wu
,
Minqiang
Wu
,
Jingwei
Chao
,
Xiangyan
Huo
,
Pengfei
Wang
and
Ruzhu
Wang
*
,
Tingxian
Li‡
*,
Taisen
Yan‡
,
Si
Wu
,
Minqiang
Wu
,
Jingwei
Chao
,
Xiangyan
Huo
,
Pengfei
Wang
and
Ruzhu
Wang
*
Research Center of Solar Power & Refrigeration, Institute of Refrigeration and Cryogenics, Shanghai Jiao Tong University, Shanghai 200240, China. E-mail: litx@sjtu.edu.cn; rzwang@sjtu.edu.cn
First published on 28th September 2021
Abstract
Freshwater scarcity is a globally significant challenge threatening the development of human society. Sorption-based atmospheric water harvesting offers an appealing way to solve this challenge by extracting clean water from the air. However, the weak ability of sorbents to capture water from dry air and the low water productivity of devices are two long-standing bottlenecks for realizing efficient atmospheric water harvesting. Here, we report a vertically aligned nanocomposite sorbent, LiCl@rGO–SA, by confining lithium chloride (LiCl) in a reduced graphene oxide (rGO) and sodium alginate (SA) matrix. The sorbent shows high water uptake, as high as thrice its weight, by integrating the chemisorption of LiCl, deliquescence of monohydrate LiCl·H2O, and absorption of LiCl aqueous solution. Moreover, LiCl@rGO–SA exhibits fast sorption–desorption kinetics enabled by the vertically aligned and hierarchical pores of the rGO–SA matrix as water vapor transfer channels. We further engineered a scalable solar-driven rapid-cycling continuous atmospheric water harvester with synergetic heat and mass transfer enhancement. The water harvester using LiCl@rGO–SA realized eight continuous water capture-collection cycles per day and ultrahigh water productivity up to 2120 mLwater kgsorbent−1 day−1 from dry air without any other energy consumption. Our demonstration of the high-performance nanocomposite sorbent and scalable atmospheric water harvester offers a low-cost and promising strategy for efficiently extracting water from the air.
Broader contextMany regions suffering from water shortage are generally far away from liquid water sources, and thus traditional water harvesting technologies are inaccessible. The possible route to solving the water shortage is to capture water from dry air by atmospheric water harvesting using sorbents. However, the sorption-based water harvesting technologies show low water production ability in this stage. Here, we comprehensively make substantial improvements on sorbents, devices, and operation strategies to realize a significant lift in the performance of water production. The synthesized vertically aligned nanocomposite sorbent, LiCl@rGO–SA, shows fast kinetics and record water harvesting capacity from dry air, and the rationally designed continuous semi-active water harvesting device exhibits the distinct capability of rapid cycling of water capture and water release. The impressive high-productivity of the water generator makes the atmospheric water harvesting technology one step closer to practical personal/household utilization. |
Introduction
Clean water scarcity is an urgent global challenge threatening the survival and development of human beings, especially in arid regions where people are undergoing water shortages.1 It is estimated that more than half of the worldwide population will face clean water scarcity by 2050s with the growth of the population and the worsening of pollution.2 Fortunately, atmospheric water is regarded as a potential water source due to its huge amount and accessibility anytime and anywhere through extracting clean water from the air.3 The major atmospheric water harvesting (AWH) technologies are foggy fix enabled by a hydrophilic interface,4 dew water collection by cooling technologies, and water capture-release by sorbents,5 wherein the fog collection technology highly depends on the high relative humidity (RH) of air and the dew water collection technology is energy-intensive when it operates at low humidity or even loses its ability when the dew point is too low, triggering freeze.6 Benefiting from the high affinity of sorbents with water vapor, sorption-based AWH (SAWH) is one of the most promising technologies for realizing water extraction from dry air at low RH.7 More importantly, the operation of SAWH can be directly driven by low-grade thermal sources such as solar energy, making it more attractive for low-cost operation and thus economy-friendly for wide application in poverty-stricken regions.8The working performance of the SAWH system is strongly dependent on the water sorption capacity of sorbents; however, traditional porous sorbents (e.g., silica gels and zeolites) provide low water productivity because of their poor water sorption performance at low RH.9 Therefore, many novel sorbents such as metal–organic frameworks (MOFs),10–14 hydrogels,15–17 liquid solutions,18,19 and composite sorbents20,21 with extraordinary performance were designed and exploited by efforts from materials scientists in past years. Lately, these novel sorbents with high water sorption performance and special thermal/electrical effects also inspired energy-related applications such as energy storage,22,23 thermal management,24–27 electricity/fuel generations,28,29 or co-applications.30–32 The MOF-801-based solar-driven SAWH prototype was first reported in 2017 with an all-in-one design by integrating light absorber, sorbent, and condenser in one chamber;33 afterwards, the SAWH prototype was improved by introducing radiative cooling to promoting the water sorption at night.34 The Yaghi research team from UC Berkeley further showed the feasibility of solar-driven SAWH in a practical desert,35 then developed a solar PV-driven SAWH system using the cheaper MOF-303 but achieved better working performance.36 Apart from MOFs, hydrogels have also attracted much attention as promising sorbents for SAWH, wherein ZnO hydrogel37 and PPy-Cl-PNIPAM hydrogel38 were first reported for achieving super high SAWH capacity exceeding 4 g g−1 at high RH. The recently reported hydrogels not only present high-water capture capacity but also have many other desirable superiorities of thermo-response,15,38–40 fast sorption–desorption kinetics,41 and low-cost, indicating promising application in agricultural irrigation.21,42
In comparison with the above state-of-the-art sorbents, the hygroscopic sorbents of lithium chloride (LiCl), calcium chloride (CaCl2), and ion solutions have strong competitiveness for SAWH in arid regions due to their higher water uptake at low RH. In our previous work, we elaborated on the multi-step water sorption mechanism of hygroscopic salts that integrate chemisorption, deliquescence, and solution absorption processes, obtaining a high-water uptake at low RH.43 To address the challenges of slow kinetics and weak stability of such hygroscopic materials, many kinds of porous materials such as hydrogels,31,44–46 MOFs,43 porous carbons,47 and agricultural wastes48 were employed as the matrix to confine salts, named composite sorbents, which have the advantages of high-water uptake at low RH, satisfactory stability, and easy to synthesis. The composite sorbents are also named “composite salt in porous matrix” (CSPM), first proposed by Yuri Aristov et al., and are applied in sorption-based refrigeration, heat pumps, and thermal energy storage devices.49 However, the composite sorbents generally have the risk of solution leakage if they are exposed to a moist environment for a long time where the captured water is too much to be stored inside the pores. It is hard to conclude from cycling tests using weighting methods for milligram-scale samples because the leaked solution may adhere to the external surface of samples or remain in the sample crucible. It is worth noting that although composite sorbents with solution leakage may show very high water uptake at high RH, they are not the true composite sorbent as a result of irreversible salt losses and agglomeration of salts once they are applied in large-scale SAWH systems.50 Therefore, it is highly valuable to develop a porous matrix with ultrahigh pore volume to load a high content of salts and store a large amount of captured water to prevent the risk of liquid leakage.
Although various novel sorbents have been designed for SAWH in the past three years, there is still a large gap between the materials and the practical applications of SAWH devices due to the insufficient design and optimization of devices. Generally, the reported conventional SAWH devices only realize one water capture-collection cycle per day, and the prototypes are incompact and heavy when scaled up to meet personal water demand (including food sources) of 3700 and 2700 mL day−1 for men and women, respectively, aged 19–30 years.51 To realize a practical portable AWH device, it is vital to design the SAWH device from the materials level to the system level, including sorbent design, thermal design, and water vapor transfer optimization.3,52 For example, researchers from Stanford University demonstrated water sorption experiments using the fluidization of MOF-801, which showed at least one order of magnitude higher water sorption rates than the previous packed MOF-801.53 Also, researchers from MIT designed a special dual-stage SAWH device using a traditional aluminophosphate sorbent of AQSOA-Z01, successfully achieving higher water productivity of 0.77 L m−2 day−1.54 Besides, it was reported that the active SAWH system can be more compact and efficient,36,55,56 where forced convection air is introduced and the sorbent can be packed layer by layer, while the sorbent in the passive SAWH system is employed as a single-layer for solar collection, resulting in a relatively low water productivity. Therefore, to realize a highly efficient SAWH system, the sorbent materials, heat and mass transfer, components of the device, and the operating strategies of the system must be rationally designed together.
Herein, we report a strategy of synthesizing a vertically aligned nanocomposite sorbent (LiCl@rGO–SA) by confining lithium chloride (LiCl) in a reduced graphene oxide (rGO) and sodium alginate (SA) matrix, achieving record water harvesting capacities of 1.01 g g−1 and 1.52 g g−1 at low RH of 15% and 30%, respectively. Induced by ice crystal growth, the formed GO–SA matrix has an ultrahigh pore volume of 20.8 cm3 g−1 and desirable vertically aligned and hierarchical pores, working as water vapor transfer channels, allowing a high salt loading of 78 wt% and fast water sorption–desorption kinetics. To maximize the SAWH potential of this vertically aligned nanocomposite sorbent, we have demonstrated a semi-active solar-driven SAWH prototype with synergetic heat and mass transfer enhancement. Four layers of sorbent sheets were integrated to realize rapid-cycling continuous atmospheric water harvesting, wherein one releases water and the other three capture water from the air. Forced convection air generated by a solar PV-powered fan is introduced to simultaneously accelerate the water sorption of sorbents and the water condensation for fast water collection. Through alternately switching the positions of four sorbent sheets, the solar-driven SAWH device can realize eight continuous sorption–desorption cycles for water harvesting per day, yielding ultrahigh water productivity up to 2120 mLwater kgsorbent−1 under low humidity conditions without any other energy input. More importantly, the LiCl@rGO–SA-based rapid-cycling continuous SAWH prototype exhibits much higher water harvesting capacity than the reported devices at the system level, showing the global daily SAWH capacity up to 33 mL per kilogram of device (mLwater kgdevice−1), 18 mL per liter of device (mLwater Ldevice−1), and 1407 mL per square solar collection area (mLwater msolar−2), which makes the sorption-based AWH technology one step closer to practical personal/household utilization.
Results and discussion
Synthesis and characterization of vertically aligned nanocomposite sorbents
Hydrogels have attracted much attention for SAWH with extraordinary sorption capacity;57,58 however, most of them suffer from low sorption kinetics and thus require a long time to reach sorption equilibrium, ascribed to their high internal water diffusion resistance caused by the tortuous pore structures. Therefore, to realize fast and highly efficient SAWH, the structures of hydrogels should be well designed and constructed. Inspired by the aligned porous structures of ceramics and freeze cast materials,59 we developed a strategy of synthesizing vertically aligned nanocomposite sorbents by confining LiCl in a reduced graphene oxide (rGO) and sodium alginate (SA) matrix using a directional freezing casting method (Fig. 1A). The vertically aligned graphene oxide (GO)–sodium alginate (SA) matrix was first prepared by directional ice crystal growth and vacuum drying. By using different sizes of directional freezing molds (Fig. S1, ESI†), the shape and size of the GO–SA matrix can be easily tuned according to different demands (Fig. 1B). We then evaluated the impact of the concentrations of GO and SA suspensions on the morphology of the porous matrix and optimized the concentrations of SA and GO suspensions as 2.0 wt% and 0.25 wt% with both excellent pore structure and structure stability (Fig. S2, ESI†). The concentration of GO governs the pore sizes and the self-assembled GO nanosheets play the role of skeletons in this GO–SA matrix (Fig. S2b, ESI†). Moreover, we found that the samples with higher SA content showed thicker pore walls, and SA was coated on the GO nanosheets (Fig. S2a, ESI†), indicating that SA contributes to creating a hydrophilic surface and strengthening the structure of the pores. For a direct comparison, we prepared a pure SA matrix and a pure GO matrix using the same methods, but we did not observe the vertically aligned pores in the pure SA matrix, caused by flexible organic chains (Fig. S3a, ESI†), which also has the risk of drying to form a xerogel after the thermal drying process at high temperature. For the pure GO matrix without the assistance of SA, although vertically aligned pores were observed, the walls of the pores seemed discontinuous and fragile (Fig. S3b, ESI†). Particularly, the pure GO matrix will destroy its pore structure by self-disassembly if touched with liquid (Fig. S3c, ESI†). Consequently, the combination of GO and SA integrates their respective advantages to form a desirable robust porous matrix.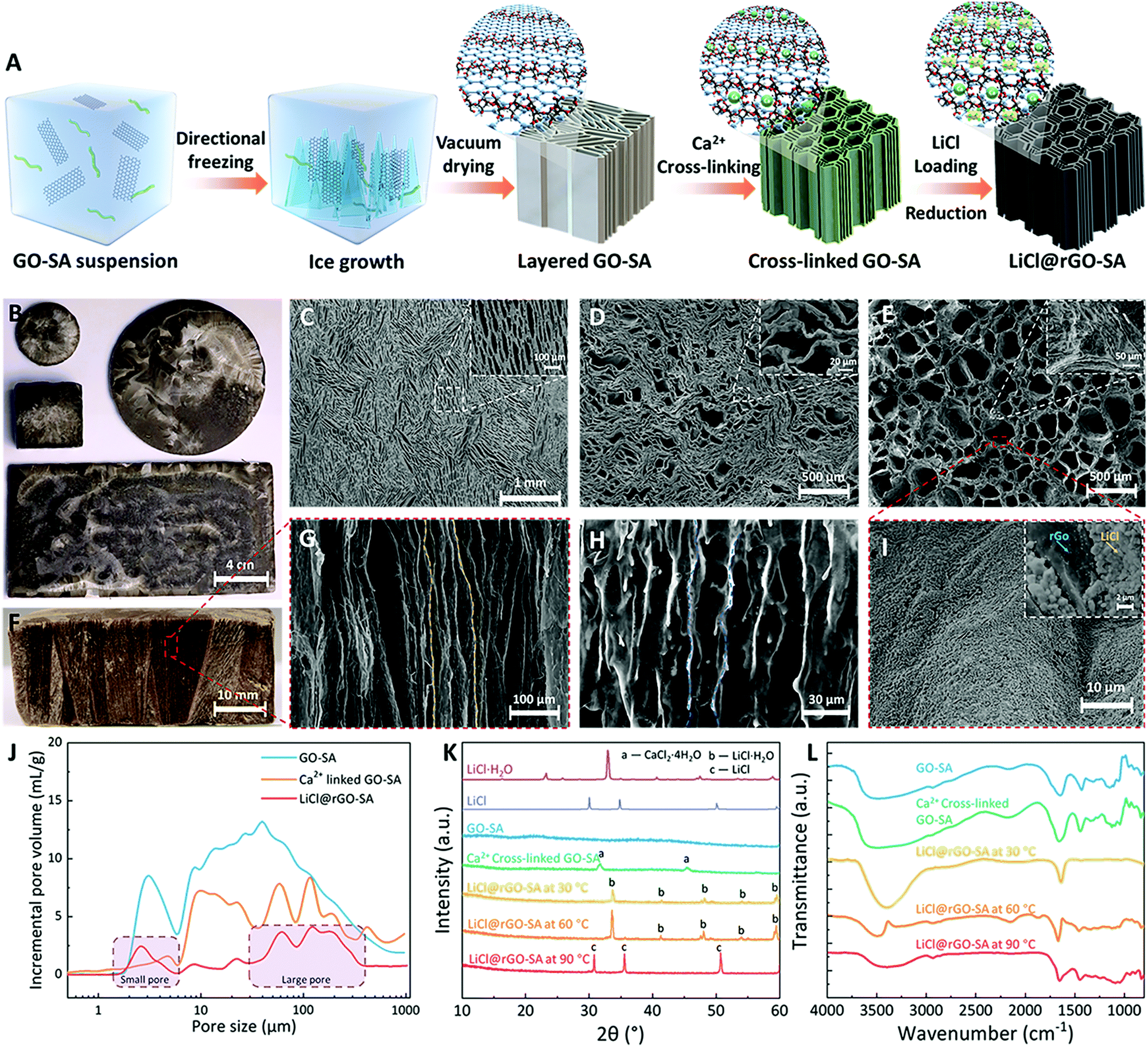 | ||
| Fig. 1 The design, synthesis, and characterization of vertically aligned nanocomposite sorbents (LiCl@rGO–SA). (A) A schematic of the synthesis route. The GO (black sheets) and SA (green chains) suspensions were first mixed together in an optimal ratio inside a designed mold, and then the directional self-assembly of the GO sheets occurred by ice-growth-induced directional freezing. The layered GO–SA matrix was obtained after vacuum drying. Afterwards, the SA chains were cross-linked by Ca2+ and LiCl crystals were loaded on the surface of the GO–SA matrix by salt solution soaking and drying. (B) Optical images of the GO–SA matrix with different sizes and shapes. (C–E) The SEM images of layered GO–SA, cross-linked GO–SA, and LiCl@rGO–SA from the top view, respectively. (F and G) The optical image and SEM images of the GO–SA matrix in the vertical sectional view showing its well-shaped vertically aligned pore structures. (H) SEM image of LiCl@rGO–SA in the vertical sectional view indicating that the vertically aligned structure is well maintained after salt loading. (I) SEM image of LiCl@rGO–SA showing the LiCl crystals uniformly distributed on the surface of the matrix; more interestingly, the LiCl crystals did not agglomerate but appeared as many individual 200–900 nm-scale cubic particles. (J) The pore size distributions of the GO–SA matrix, Ca2+-linked GO–SA, and LiCl@rGO–SA, obtained by the mercury intrusion method, showing that the matrix and sorbent have large pore volumes. LiCl@rGO–SA shows two different pores, 100 μm-scale large pores and 2 μm-scale small pores. (K) Variable temperature XRD patterns of the porous matrix and LiCl@rGO–SA showing the desorption of LiCl·H2O to LiCl during the heating process. (L) Variable temperature FTIR patterns of LiCl@rGO–SA showing the decrease in hydrogen bonds during the dehydration process, indicating the water release from the sample during the heating process. | ||
Fig. 1F and G show that the prepared GO–SA porous matrix has the expected vertically aligned and hierarchical pores, where the pores are continuous throughout the sample from the top surface to the bottom surface. The structural stability of GO–SA was further enhanced by the Ca2+ cross-linking of SA. Fig. S4 (ESI†) shows the stress–strain curve of the matrixes and composite sorbent under a compression process, and the results confirmed that the cross-linked SA makes a strong contribution to improving the mechanical strength of the porous matrix. The top view SEM images show that the GO–SA matrix has long and narrow pore structures (Fig. 1C), and the walls of the pores partly bend and deform after cross-linking (Fig. 1D), making some pores become large and others become smaller. Finally, the vertically aligned nanocomposite sorbent (LiCl@rGO–SA) was obtained after loading lithium chloride by salt solution soaking and drying, wherein the GO was partly reduced to form rGO at the desorption temperature (see detailed synthesis information in the Experimental methods section). By this one-dimensional (1D) directional ice growth, the pores of the GO–SA matrix show ordered alignments in the vertical direction while pores in the horizontal direction are random (Fig. 1C). Since the diffusion of water vapor in sorbent mostly moves along the vertical direction, this 1D pore structure design of the porous matrix is enough for accelerating the sorption kinetics for fast SAWH.
The leakage risk of salt solution in ambient moisture is one of the most serious challenges of composite sorbents. To avoid this risk, we have proposed a synthesis guideline for the preparation of composite sorbents by choosing suitable concentrations of salt solution during the salt impregnation process. The salt solution with low concentration can ensure the stability of composite sorbents under high RH, but it is realized at the expense of low salt content, corresponding to low water sorption capacity. However, benefiting from the ultra-large pore volume and high porosity of the GO–SA matrix, we successfully prepared the vertically aligned nanocomposite sorbents with high salt content using LiCl solution at a low concentration of only 10 wt% (Fig. S5, ESI†). The nanocomposite sorbents can avoid the risk of solution leakage theoretically, even at high RH, as high as 90%, covering most of the climatic conditions of arid regions.60 After salt loading, some large pores were generated by the bending and deformation of SA (Fig. 1E). Although a slight shrinking of GO–SA matrix was observed during this preparation process (Fig. S6, ESI†), the vertically aligned pores maintained continuous channels from the top surface to the bottom surface of the rGO–SA matrix (Fig. 1H).
The packed agglomeration of salts in the conventional composite sorbent is the key issue that limits water vapor transport during sorption–desorption processes, and this causes the difficulty of realizing several water capture-release cycles per day for SAWH.7 The slow water sorption–desorption cycle of sorbents further results in the low water productivity of SAWH devices. Therefore, avoiding the packed agglomeration of salts inside the porous matrix is the common objective for all composite sorbents using hygroscopic salts.20,22,23,31,43,45,55,61 The elemental mapping with EDX of the LiCl@rGO–SA sorbent showed LiCl crystals uniformly distributed on the surface of pores inside the rGO–SA matrix (Fig. 1I and Fig. S7, ESI†), remarkably appearing as many individual 200–900 nm cubic salt particles, which is in favor of faster water capture due to large sorption interface areas and less reaction resistance (see SEM image of cubic salt particles in Fig. S8, ESI†). This special phenomenon attributes to the low concentration of the salt solution (10%), weak nucleation resistance of crystallization on the moderately hydrophilic rGO–SA surface, and slow crystallization process during the soaking-drying process. We observed that the cubic salt particles may partly agglomerate at the violent crystallization stage during a practical SAWH process (Fig. S9, ESI†), but the coating thickness of LiCl can be always kept at a low value, and thus can ensure a fast water sorption rate.
We further quantitatively evaluated the pore properties of the GO–SA matrix and the vertically aligned nanocomposite sorbent by the mercury intrusion method (Fig. 1J). The GO–SA matrix showed hierarchical pores with a wide size distribution ranging from 2 μm to 100 μm, featuring an ultra-large pore volume of 20.78 cm3 g−1, an order of magnitude higher than other reported porous matrixes.22,43,61 This extraordinary large pore volume is ascribed to the light weight and thin GO walls of the high-porosity 3D architectures, formed during the freeze-casting process, where GO sheets are rejected from the forming ice and entrapped between neighboring ice crystals to form a continuous network.62 Consequently, the synthesized nanocomposite sorbents show a record salt content of 78 wt%, even using a low concentration (10 wt%) of LiCl solution in the soaking process. Remarkably, the high salt-content nanocomposite sorbent also showed a high pore volume of 4.55 cm3 g−1 after loading salts, which provided enough space for storing water during the water capture process. The pore size distribution of LiCl@rGO–SA (Fig. 1J) also confirmed that the vertically aligned nanocomposite sorbent has a hierarchical pore distribution at sizes of ∼2 μm and ∼100 μm, coinciding with the SEM images (Fig. 1E). In comparison with the GO–SA matrix, there was an apparent decrease of the pores at the diameters of 5–30 μm for the nanocomposite sorbent, indicating that most of the salts were confined inside these pores. We further confirmed the Ca2+ cross-linking and LiCl loading by X-ray diffraction (XRD) (Fig. 1K), and the desorption of LiCl·H2O to LiCl was observed when the sample was heated to 90 °C. The disappearance of diffraction peaks of CaCl2 hydrates in the nanocomposite sorbent indicated that the remaining Ca2+ was exchanged out by Li+ during the subsequent salt loading process. The Fourier transform infrared spectroscopy (FTIR) patterns also illustrated the water release during the heating process, corresponding to the weak peak of the hydrogen bonding at high temperatures (Fig. 1L).
Water sorption–desorption performance evaluation
LiCl@rGO–SA showed excellent water vapor sorption performance for water uptake capacity, kinetics, and stability. As shown in Fig. 2A, a three-step water uptake mechanism was observed in water sorption isotherms, where the chemisorption of LiCl, the deliquescence of LiCl·H2O, and the absorption of LiCl solution were integrated to realize an ultrahigh water uptake capacity. Since the porous matrix of rGO–SA makes a negligible contribution to the water sorption, the salt content of LiCl@rGO–SA was estimated to be as high as 78 wt% according to the water sorption capacity by the following equation: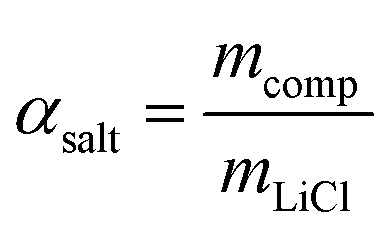 | (1) |
 | (2) |
 | ||
| Fig. 2 Water sorption–desorption performance of LiCl@rGO–SA. (A) Water sorption isotherms at 30 °C, 50 °C, and 70 °C respectively. (B) Water desorption isobars at water vapor pressures of 1.27 kPa and 4.25 kPa. (C) The pressure–temperature phase diagram showing the multi-step desorption transition mechanism and water sorption–desorption quasi-equilibrium characteristics of LiCl@rGO–SA. (D) TGA curves showing the dynamic water sorption process of LiCl@rGO–SA at different working conditions of 15% RH, 30% RH, and 60% RH at 30 °C and the dynamic water desorption process under 6.0% RH at 90 °C. (E) TGA curves showing the dynamic water sorption process of pure LiCl under the same testing conditions. (F) TGA curves showing the dynamic water desorption process of LiCl@rGO–SA at the same water vapor pressure of 4.25 kPa but different temperatures of 60 °C, 70 °C, 80 °C, and 90 °C. (G) 10 water sorption–desorption cycling tests of the nanocomposite sorbent at 30 °C, 1.2 kPa (30% RH) for sorption and 90 °C, 4.2 kPa for desorption, showing satisfactory water sorption–desorption cycling stability. (H) The water sorption capacity comparison of recently reported state-of-the-art sorbents,10,21,33,36,38,41,43,47,63 showing the record water sorption capacity of LiCl@rGO–SA under typical working conditions of sorption-based atmospheric water harvesting. (I) The water sorption kinetics comparison of recently reported state-of-the-art composite sorbents,20,43–45,47,48,61,64 showing the faster water sorption rates of LiCl@rGO–SA and confirming the great contribution of vertically aligned pore structures to accelerating the water sorption kinetics. For a fair comparison, we compared the LiCl@rGO–SA with other typical SAWH composite sorbents under similar testing conditions of arid climates under 20–40% RH (see details in Table S1, ESI†). | ||
Apart from the water sorption, desorption characteristics also determine the final water harvesting potentials, thus we evaluated the water desorption isobars of the LiCl@rGO–SA nanocomposite sorbent (Fig. 2B). Considering some arid regions distributed at high latitudes (e.g., Tibet in China) where the local temperature is generally low, we evaluated the water desorption isobars at two typical water vapor pressures of 1.2 kPa and 4.2 kPa, corresponding to condensation temperatures of 10 °C and 30 °C. The water desorption isobars also showed the three-step desorption mechanism: the water evaporation from LiCl solution, the crystallization of LiCl·H2O, and the chemical desorption of LiCl·H2O, successively. Fig. 2C shows the pressure–temperature quasi-equilibrium phase diagram according to the experimental data, further elaborating the multistep water desorption process of LiCl@rGO–SA. The difference between measured quasi-equilibrium lines at different water contents and theoretical equilibrium lines of three-step desorption (Fig. S10, ESI†) is ascribed to the incomplete water desorption at the experimental quasi-equilibrium point and needs a necessary driving force (temperature/pressure difference between testing conditions and theoretical equilibrium conditions) for the total release of water. The total regeneration (desorption) temperatures of the LiCl@rGO–SA nanocomposite sorbent are 85 °C and 69 °C at these two typical water vapor pressures of 4.2 kPa and 1.2 kPa. Such low regeneration temperatures of the LiCl@rGO–SA nanocomposite sorbent indicate the accessibility of employing solar energy or low-grade waste heat to drive the water sorption–desorption cycle.
The water sorption–desorption kinetics of the nanocomposite sorbent was evaluated by thermogravimetric analysis (TGA) (see the schematics of the measuring device in Fig. S11, ESI†). To simulate the water capture-release in typical dry regions all over the world (see the climates of typical arid regions in Fig. S12, ESI†), we carried out the experiments under three typical air humidity conditions: extremely arid climate (15% RH), semi-arid climate (30% RH), and moderate climate (60% RH) (Fig. 2D). The nanocomposite sorbent showed fast water sorption–desorption kinetics and can finish water capture within three hours (under 30% RH), as well as water desorption within two hours (see pictures of the dynamic water capture process in Fig. S13, ESI†). To directly show the superiority of the nanocomposite sorbent, we also measured the dynamic sorption–desorption process of pure LiCl under the same air conditions. The pure salt showed much slower sorption–desorption kinetics and it could not reach sorption equilibrium within eight hours. Besides, the pure salt cannot release the coordinated water of LiCl·H2O at 90 °C due to the serious agglomeration effects (Fig. 2E). Consequently, the vertically aligned nanocomposite sorbent not only shows a faster water harvesting rate but also shows a higher water capture-release capacity when compared to the pure salts. We measured the water sorption–desorption performance of pure porous matrixes before and after cross-linking with Ca2+. The results showed that the porous matrix and residual CaCl2 make little contribution to the water sorption capacity of the composite sorbent (Fig. S14, ESI†), while major water sorption ability comes from the hydration of LiCl.
To show the SAWH potential of LiCl@rGO–SA under different solar thermal collection conditions, we evaluated the water release ability of this sorbent at different desorption temperatures (60 °C, 70 °C, 80 °C, and 90 °C) under the same water vapor pressure of 4.2 kPa (Fig. 2F). The results showed that a desorption temperature higher than 80 °C is necessary to exploit the potential of this composite sorbent on SAWH. Importantly, the LiCl@rGO–SA still has a moderate SAWH capacity of ∼0.5 g/g, even at a low desorption temperature of 60 °C, indicating its strong climate adaptability and working ability at different regions with relatively low sunlight intensity. We also measured the heat flow during the water sorption–desorption process, which showed that the nanocomposite sorbent has extraordinary exothermic/endothermic effects of 3260–3487 kJ kg−1 of sorbent due to its large amount of water uptake/release (Fig. S15, ESI†). This large amount of heat release/storage indicates that the nanocomposite sorbent is also one of the potential candidates for sorption-based heat transformation.
Since the cycling stability of composite sorbents is one of the most important characteristics and needs to be well measured, we evaluated the water sorption–desorption stabilities of the nanocomposite sorbent by repeating sorption–desorption cycling tests at 30% RH for water sorption using a commercial thermogravimetric analyzer (STA 449, Netzsch). The results showed that the nanocomposite sorbent has satisfactory working stability for repeated sorption–desorption (Fig. 2G). Less than a 5% performance decrease was found during the first several sorption–desorption cycles due to the loss of very little salts on the surface of the matrix, while major salts can remain inside the pores of the matrix and thus maintain a stable sorption capacity. We further evaluated the water sorption–desorption stability of this nanocomposite sorbent at a high RH of 70%, showing that the sample can maintain excellent cycling ability under conditions of moderate humidity (Fig. S16, ESI†). Moreover, the water solution leakage phenomenon was not observed even at RH as high as 90% (Fig. S17, ESI†). Considering the possibility of the salt solution leakage along the vertical pore walls of the composite sorbent under vibration conditions, we tested the water storage ability of the composite sorbent by strongly shaking to determine whether the water would drop out, or not, before the sorbent captured the water under 60% RH overnight. Video S1 (ESI†) shows that the water was not shaken out from LiCl@rGO–SA, indicating the potential for desirable stability. This desirable stability is majorly ascribed to the strong capillary force of water inside the porous matrix with relatively small pores with a diameter of 2–200 μm (Fig. 1J) and strong hydrophilic characteristics of the GO–SA surface (see contact angles in Fig. S18, ESI†). The capillary force is strong enough to overcome the gravity force (Fig. S19, ESI†). According to the static force balance, we provided design targets of pore size and hydrophilic characteristics for preventing the solution from dropping out at different sorbent thicknesses (Fig. S20, ESI†).
Fig. 2H shows the comparison of the water sorption capacity of the LiCl@rGO–SA and other reported state-of-the-art sorbents for SAWH, revealing that the LiCl@rGO–SA has performance superiority for SAWH under a wide RH range of arid climates. Apart from the record water sorption amount, the nanocomposite sorbent also showed a sharper stepwise transition at the RH near 11% as compared to other sorbents (e.g., MOF-801 and MOF-303), which is highly desirable for realizing high water harvesting within a small temperature/pressure swing. Apart from the superiority of the water sorption capacity, the LiCl@rGO–SA showed faster kinetics than other composite sorbents (Fig. 2I). As discussed above, the fast sorption kinetics is contributed by the vertically aligned pore structure of the rGO–SA matrix. To verify this mechanism, we prepared a similar rGO–SA matrix but using a undirectional freezing method, and encapsulated LiCl inside this matrix, named LiCl@rGO–SA_unordered (see pictures of this sample in Fig. S21, ESI†). In comparison with LiCl@rGO–SA_unordered, the faster water sorption kinetics of LiCl@rGO–SA confirmed the effect of the vertically aligned pore structure on accelerating water sorption rates. More detailed tests, analysis, and simulation on water sorption kinetics will be discussed in the following.
To realize the high water productivity of SAWH, more researchers have started to focus on multiple water capture-release cycles during the daytime,36,41,47 thus the sorbents with fast sorption–desorption kinetics become more important for accelerating the SAWH cycle and improving water productivity. However, the lack of a rational and standard evaluation method for water sorption kinetics is a long-time bottleneck for the SAWH materials. Generally, researchers carry out dynamic water sorption tests using a very small number of sorbents using a commercial thermogravimetric analyzer (TGA), vapor sorption instrument or using constant temperature and humidity equipment. Although these methods are valuable for micro-/milligram-scale loose sorbent particles, they are not accurate enough to evaluate the sorption performance of packed massive sorbents for SAWH, ascribed to the uncontrollable heat and mass transfer conditions and deviation of sample states (e.g., mass, thickness, and shape) from the actual state in SAWH devices. Here, we provide a simple, low-cost, and accurate measuring strategy to evaluate the water sorption kinetics of gram-scale sorbents in view of SAWH (Fig. S22, ESI†). The SAWH device is located in a chamber with constant humidity and temperature, or connected to a humidity generator, and can measure the sorption kinetics of sorbents under a specific climatic condition and a controllable air flow rate.
The water sorption kinetics of packed sorbents (e.g., MOF, zeolites) generally depends on the inter-crystalline vapor diffusion and intra-crystalline vapor diffusion,52 as illustrated in Fig. 3A. Since the packed sorbent particles are generally unordered, they usually show low sorption rates due to the high mass transfer resistance for water vapor transport, especially for massive sorbents with high packing densities and high packing thicknesses. For the water sorption process of the LiCl@rGO–SA nanocomposite sorbent, however, the water molecules are first transferred from ambient air (water concentration of ambient air, Camb) to the external surface of the sorbent (Csurf), then diffuse into the vertically aligned pores of the rGO–SA matrix to the internal pore surface of the salts (Csorb), and are finally captured by salts until sorption equilibrium (Csorb,eq) is reached (Fig. 3B). Therefore, its sorption kinetics are mainly dominated by three parts: the forced/natural convection transfer resistance on the external surface (Rsurf), the diffusion transfer resistance inside the pores (Rdiff), and the apparent reaction resistance for the salt-gas sorption interface (Rreact) (Fig. 3C). By using the sorption kinetics testing device, we first measured the dynamic water sorption process of the nanocomposite sorbent under different air flow conditions (Fig. 3D). For a concise expression, the obtained data for water uptake were normalized to the dimensionless fractional transient water uptakes, mt/m∞, where mt represents the water uptake at a certain time and m∞ represents the equilibrium water uptake. The forced air convection can efficiently improve the water sorption rate as compared to natural air flow, where the sorption duration can be shorted from above nine hours to five hours for this gram-scale sorbent. We would like to emphasize here that our packed sorbents in this kinetics test have scaled-up sizes in comparison to the sorbent powders/particles in TGA tests, thus the mass and heat transfer are closer to those in practical SAWH systems.
 | ||
| Fig. 3 Water sorption–desorption kinetics of the LiCl@rGO–SA sorbent. (A) Schematic of water vapor transport from ambient air to traditional packed sorbents with unordered arrangement. (B) Schematic of water vapor transport pathways from ambient air to the vertically aligned nanocomposite sorbent. The enlarged view shows the water sorption interface of salts and water vapor, where several layers of salts coat the surface of the rGO–SA matrix. (C) The mass transfer resistance analysis for water vapor during the water sorption process. The total transfer resistance consists of three parts: the forced/natural convection transfer resistance on the external surface (Rsurf), the diffusion transfer resistance inside the pores (Rdiff), and the apparent reaction resistance for the salt-gas sorption interface (Rreact). (D) The dynamic water sorption process of the nanocomposite sorbent with a thickness of 3.1 mm under different external air flow conditions: natural air convection, forced air convection at flow rate of 2 m s−1, and forced air convection at a flow rate of 3 m s−1. (E) The calculated real-time vapor transfer resistance during the sorption tests under different external air flow conditions. (F) The dynamic water sorption process of nanocomposite sorbents with different thicknesses of 1.7 mm, 3.1 mm, and 5.0 mm, under the same testing condition of 30% RH at 30 °C, and the same forced air flow at a rate of 2 m s−1. (G) The calculated real-time vapor transfer resistance of the samples with different thicknesses during the sorption tests. (H) The dynamic water desorption process of nanocomposite sorbents with different thicknesses of 1.7 mm, 3.1 mm, and 5.0 mm, without forced air convection. (I) The calculated real-time vapor transfer resistance during the desorption tests. | ||
To reveal the inherent mechanism of kinetics enhancement, we further theoretically analyzed the mass and heat transfer process of the vertically aligned nanocomposite sorbents by developing mathematical models using the commercial software of COMSOL Multiphysics (see detailed simulation calculations in Supplementary Note 1, ESI†). The simulated results are in good agreement with the experimental results, confirming that the mathematical model is credible for predicting the performance of the nanocomposite sorbents. According to the simulated evolutions of water sorption rates (Fig. S23, ESI†) and water vapor concentrations (Fig. S24, ESI†), the real-time Rsurf, Rdiff, and Rreact were calculated and compared, as shown in Fig. 3E. It was observed that Rsurf always dominates the whole sorption process for the water sorption under natural air convection. Rsurf decreased when forced air convection was introduced, even if the air flow rate was as low as 2 m s−1. This is because the introduction of forced air convection not only accelerates the diffusion of water molecules, but also efficiently takes away the sorption heat released by LiCl@rGO–SA (Fig. S25, ESI†). The low sorption temperature improves the sorption capacity of LiCl@rGO–SA according to sorption equilibrium characteristics, appearing as the further enhanced sorption kinetics. These findings suggest that introducing forced air convection is highly necessary to enhance the sorption kinetics, even providing a low air flow rate.
We investigated the influence of the packed thickness of the sample on the sorption kinetics of LiCl@rGO–SA (Fig. 3F). The tests were carried out by using samples with different packed thicknesses under the same air flow conditions of 2 m s−1. We found the sorption kinetics increased with decreasing the packed thickness of the sample. The comparison of mass transfer resistances at different thicknesses of samples showed that Rdiff was four times higher than Rsurf for the sample with a thickness of 5.0 mm (Fig. 3G), indicating that the diffusion transfer resistance will dominate the whole water sorption process. Moreover, the diffusion transfer resistance Rdiff can be reduced by adjusting the packed thickness of the sample. For example, the Rdiff decreased from 190 m s−1 to 68 m s−1 when the packed thickness ranged from 5.0 mm to 1.7 mm. Similarly, we also carried out the water desorption evaluation of the nanocomposite sorbent with different packed thicknesses to simulate the water desorption process in passive SAWH devices under natural air convection (Fig. 3H). Without the forced air convection, the water desorption kinetics was dominated by Rsurf for all samples with different thicknesses at the initial desorption stage (Fig. 3I). However, we found that the water reaction resistance was higher than the other resistances during the end phase of water desorption, indicating that the water reaction resistance of chemical desorption (releasing coordinated water) dominated the water desorption rate during the water desorption due to the strong bonding energy of the chemical decomposition reaction. The above results and analyses further revealed that the release of coordinated water by chemical desorption of the composite sorbent not only needs a higher desorption temperature but also continues for a longer time than the other water evaporation and crystallization steps because of the slow desorption kinetics. The failure to release coordinated water in the limited desorption time will result in a lower SAWH capacity than the predicted values based on water isotherms of composite sorbents.
According to the above results, we optimized the packed thickness of the sample and air flow rates for achieving fast water capture. For nearly all of the measuring conditions for water sorption and desorption, the results showed that the apparent reaction resistance Rreact always had the lowest value among these resistances (Fig. 3E, G and I), indicating that the reaction between LiCl and water is fast enough due to the low loading thickness of the salt on the surface of the pores inside matrix and the good distribution of the salt in the form of individual 200–900 nm-scale cubic particles as shown in the SEM image (Fig. S8, ESI†). Consequently, the LiCl@rGO–SA nanocomposite sorbent exhibited fast water sorption–desorption kinetics as compared to the reported packed composite sorbents20,44–47 due to the following innovations: (i) the low convection transfer resistance on the external surface (Rsurf) by introducing forced air convection; (ii) the low diffusion resistance (Rdiff) inside the LiCl@rGO–SA nanocomposite sorbent, enabled by the vertically aligned and hierarchical pores as water transport channels; (iii) the low reaction resistance (Rreact) between LiCl and water vapor on the interface of the sorption reaction, enabled by the low loading thickness and good distribution of nano-sized salt particles in the matrix.
One-sun, indoor SAWH demonstration
To promote the development of portable solar-driven SAWH devices, many active SAWH devices/systems have been proposed in recent years to realize high water productivity.36,55 However, these SAWH devices generally have discontinuous operations using one sorption bed by performing sorption and desorption modes alternately. However, the water production ability can be further exploited if the water collection works continuously during the daytime by employing well-designed SAWH devices.Inspired by continuous sorption-based refrigeration systems,65 where two sorption beds are employed for realizing continuous cooling by switching the operation modes of two beds alternately, we designed a solar-powered rapid-cycling continuous SAWH device, as shown in Fig. 4A. According to the analysis of the sorption–desorption kinetics of the nanocomposite sorbent (Fig. 3), the sorption duration is approximately three times longer than that of desorption for LiCl@rGO–SA. This indicates that the traditional experiences of the continuous sorption-based refrigerator with two beds cannot be directly used for the continuous SAWH device where the sorption time of one bed is equal to the desorption time of the other bed. Here, we propose a synergetic enhancement strategy for engineering a rapid-cycling continuous SAWH device with optimized heat and mass transfer conditions (Fig. 4B). First, four sorption beds fabricated by LiCl@rGO–SA sorbent sheets are employed in this device to achieve continuous SAWH, wherein one releases water and the other three capture water, simultaneously (Fig. 4C). For every sorption bed, it has triple the duration for water capture as compared to its water release duration, corresponding to the kinetics characteristics of the faster desorption rate than the sorption rate. To verify the feasibility of rapid cycling SAWH in this demonstration, we chose a simple switching method by manually adjusting the locations of samples. To get a compact SAWH device with as small space as possible, these four sorbent beds were combined to form a rectangular array (see photos in Fig. S26, ESI†). Secondly, the thermal design of the sorption beds with synergetic heat and mass transfer enhancement was proposed to realize rapid-cycling continuous SAWH by introducing forced air convection. A 6 W solar PV-driven fan was employed to create forced air convection flow (for more information see Fig. S27, ESI†), which provides a relatively strong air flow of 1.7 m s−1 to 3.7 m s−1 under different solar irradiation intensities (Fig. S28, ESI†). Considering that the external forced air convection may bring potential contaminants of dirt and bugs on the sorbent, the installation of air filter meshes on the inlet and outlet of the air duct is necessary for practical long-term utilization.
 | ||
| Fig. 4 The design, optimization, and performance of the rapid-cycling continuous water harvester under standard one sun irradiation. (A) Structure schematics of the water harvester. Four sorbent units are assembled together where one of them faces the sun for solar collection to perform water release while the other three face the ground and are exposed to air for capturing water at ambient temperature. A solar PV-powered fan is placed on the top of the device to create a forced air convection for accelerating both the water condensation and water capture by removing heat in a timely manner (both condensation heat and water sorption heat) and bringing water vapor from ambient air to the sorbents. (B) The energy balance of the SAWH device during the water release-condensation process. The input heat source only comes from solar irradiation; a part is transferred for driving the water release of sorbent and other parts are lost to ambient air through the top acrylic cover or bottom condenser. To reduce the heat loss through the bottom condenser, an insulation panel was installed between the sorbent and condenser. (C) Operation principle of this rapid-cycling continuous water harvester. (D) The temperature evolutions of sorbent, condenser, and ambient air after installing an insulated panel between the sorbent and condenser in the SAWH under a stable solar irradiation of 950 W m−2. (E) The infrared temperature mapping showing the temperature evolutions of sorbents during the cooling-sorption process with/without forced air convection. (F) The temperature evolutions of the condenser (inside chamber) and ambient air with closing/opening the air fan. (G) The temperature evolutions of different parts of the SAWH device during a typical water collection process under standard one sun irradiation (950–1050 W m−2). (H) The temperature evolutions of different parts of SAWH devices during a continuous 8 time water collection process under standard one sun irradiation. (I) The masses of collected water, desorbed water, and the accumulated collected water over the 8 time water collection process within 10 hours. | ||
The LiCl@rGO–SA nanocomposite sorbent showed a high solar light absorbance of 96.1%, but it also showed a high emittance during the range from the near- to middle-infrared wavebands (Fig. S29, ESI†). To prevent large heat loss by infrared radiation during the water collection process, we selected a commercial solar selective absorber integrated with the LiCl@rGO–SA sheet to collect the heat from sunlight, which has satisfactory solar absorbance at solar spectrum wavebands and a low emittance at near- to middle-infrared wavebands (Fig. S30, ESI†). To achieve fast heat transfer during the SAWH process, we directly stuck the nanocomposite sorbent sheet on the back side of the selective absorber by using a thermal interface material (TIM) of high thermal conductive tape (see photos in Fig. S31, ESI†). In addition, to realize efficient water collection and lower the condensation temperature of water vapor, two strip aluminum fin radiators were assembled together to enhance the heat exchange between ambient air and condenser for the timely removal of condensation heat (Fig. S32, ESI†).
For a SAWH device, the high heating temperature of the sorbent and low condensation temperature of water vapor are desirable for realizing water collection. However, we found that the convective heat exchange between the nanocomposite sorbent and the aluminum condenser brings a large amount of heat loss (see detailed analysis in Supplementary Note 2, Fig. S33 and Table S2, ESI†). This large heat loss is caused by the compact design of our SAWH device, wherein the distance between the sorbent and the condenser is less than 5 cm. To avoid the heat loss from the sorbent to the condenser, an insulated panel (impermeable extruded polystyrene foam, thermal conductivity of 0.028 W m−1 k−1) was inserted between them. After installing the insulated panel, the sorbent temperature during the water desorption process increased significantly. In particular, the temperature difference between the sorbent and the condenser increased from 18.5 °C (without the insulated panel, Fig. S34, ESI†) to 27.2 °C (with the insulated panel, Fig. 4D). To evaluate the effects of the insulation panel on the water desorption, we numerically simulated the temperature, velocity, and water concentration distributions in the device during the desorption-condensation process via COMSOL (Fig. S35, ESI†). The results showed that the addition of the insulation panel reduced the air convection velocity near the sorbent, and thus exhibited a higher heating temperature. As a result, the estimated diffusion flux with the insulating panel was about 24% higher than the diffusion flux without the insulating panel (Fig. S36, ESI†). Furthermore, we predicted that the permeable insulating panel can reduce the vapor pressure difference of the air near the condenser and sorbent, bringing lower desorption temperature requirements. The above results confirmed that our strategy is effective for reducing the heat loss between the high-temperature sorbent and the low-temperature condenser, especially for a compact all-in-one SAWH device where the light absorber, sorbent, and condenser are integrated into one chamber.
Before the water harvesting tests, we evaluated the contribution of forced air convection on the water sorption of the LiCl@rGO–SA sheet. A similar phenomenon to the simulated results above was found, where the sorption heat can be quickly taken away with the assistance of forced air flow, while the contrasting experiment without forced air flow showed a much longer time for cooling down (Fig. 4E) (see detailed temperature evolutions of center points at the sorbent in Fig. S37, ESI†). Different from the previously reported water harvester with a large water condensation surface, we designed a much smaller condensation surface (6 cm×20 cm) to fabricate the all-in-one SAWH device as compact as possible. The water vapor desorbed from the LiCl@rGO–SA sheet condensed in this area and created real flowing liquid water for freshwater collection (see the flow of water droplets in Video S2, ESI†). Benefiting from the enhanced heat exchange of the condenser by forced air convection, the cooling power can well cover the condensation heat released by the water vapor, appearing as an apparent temperature decrease in the condenser from Tamb + 16.5 °C under natural air flow to Tamb + 5.1 °C under forced air flow (Fig. 4F).
We carried out the indoor SAWH tests by using a xenon lamp as the solar simulator (Fig. S38, ESI†) for creating a stable irradiation intensity of 950–1050 W m−2. The spectra of the sunlight and xenon lamp, as well as the transmissivity of the acrylic cover for SAWH were compared (Fig. S39, ESI†). During the water collection process, we found that a part of the condensed water droplets hardly flowed down due to the poor hydrophobicity of the aluminum surface, thus, a commercial waterproof membrane (PET) with superhydrophilic character was coated on the surface of the aluminum to accelerate the flow of the water droplets (Fig. S40, S41 and Video S3, ESI†). Recently, a hydrophobic CuO nanostructure was reported in an interfacial solar vapor generation device for accelerating condensation, which can be introduced into SAWH devices in the future.66Fig. 4G shows the temperature changes of different components of the SAWH device under typical indoor conditions, wherein a maximum heat collection temperature of 90 °C was achieved due to the good insulation of the device. Interestingly, we found that the temperature difference between the selective light absorber and nanocomposite sorbent increased as water desorption occurred, especially in the desorption phase during 20 to 50 minutes, indicating that the fast water desorption brought a strong endothermic effect, where a large amount of water vapor was desorbed from the nanocomposite sorbent. Afterwards, the sorbent temperature gradually increased until water release was complete, together with a gradually decreased condensation temperature.
For most reported SAWH devices, only one water capture-collection cycle can be realized during the daytime due to the discontinuous operation and long-time water capture/release caused by slow sorption/desorption kinetics. By optimizing the structural design of the vertically aligned nanocomposite sorbent and synergetic heat and mass transfer enhancement of the SAWH device, we successfully carried out rapid-cycling continuous SAWH eight times during the daytime (Fig. 4H) by alternately switching the working modes of the four sorbent units (see detailed information of these four sorbent units in Table S3, ESI†). The sunshine duration per day is about 10 hours for many arid regions; thus, we let each sorbent sheet take 70–80 minutes for releasing water to the condenser and 210–240 minutes for capturing water from the air. Remarkably, the total amount of 46.3 g water was collected after these eight continuous water release processes, corresponding to an ultrahigh SAWH capacity of up to 2120 mLwater kgsorbent−1 (Fig. 4I). The total desorbed water reached as high as 65.6 g, showing a water collection coefficient of 70.6%. According to the amount of water desorbed, we calculated the thermal efficiency of this SAWH device as 22.8%, higher than the major reported SAWH devices. In addition, the thermal efficiency can be further improved by reducing the sensible heat consumption of the SAWH device during the water sorption–desorption processes (see theoretical analysis in Supplementary Note 2, ESI†).
Practical outdoor AWH demonstration
To confirm the feasibility of the rapid-cycling continuous SAWH device, we conducted the outdoor tests under the irradiation of natural sunlight on the roof of our lab, on the campus of Shanghai Jiao Tong University, on August 17, 2020. The SAWH device was put on an inclined plane with a tunable angle set at 30° by considering the latitude of Shanghai (31° N) (Fig. 5A). Two shading panels were used to prevent the sunlight directly shining on the other sorbents from undergoing the water capture process, and a pyranometer was used to record the solar irradiation intensity. We would like to emphasize here that this cuboid-shaped SAWH unit is modular and scalable, and can be easily scaled up by assembling many such units in series and parallel connections, and these two shading panels can be removed if SAWH units were installed close together and in parallel, where the sunlight cannot shine on the sorbents that are undergoing water capture. | ||
| Fig. 5 Outdoor tests of the rapid-cycling continuous SAWH device under practical solar irradiation at Shanghai, China. (A) A photo of the SAWH device on the rooftop of our lab. (B) The temperature evolutions of different positions of the SAWH device during the first water collection cycle in the morning on August 17, 2020. (C) The temperature evolutions of sorbent, condenser, and ambient during eight times water collection on August 17, 2020. (D) The masses of collected water, desorbed water, and the accumulated collected water over eight times outdoor water collection. | ||
At 7:00 in the morning, we put these four sorbent units into the SAWH device, where the adsorbed one was put into the sealed water collection chamber for water release and the others were located at the other three sides to realize further water capture from the air. Once the selective solar absorber is exposed to the sunlight, it only takes 15 minutes to increase its temperature from ambient temperature (∼30 °C) to a high temperature. Such a fast heating rate indicates a high thermal collection efficiency and relatively low heat capacity of this compact all-in-one SAWH device (Fig. 5B). To show the dynamic water collection process, we recorded the volume changes in the collected water during a typical outdoor water collection cycle (Fig. S42, ESI†). After the first cycle of water collection, the water-released sorbent unit was taken out from the water collection chamber and exchanged with another one. By cooling through forced air flow, the temperature of the sorbent unit quickly dropped to the water sorption temperature within several minutes, starting to capture water from air for the next water release (Fig. S43, ESI†). The solar irradiation intensity gradually increased from 500 W m−2 to the peak value of 980 W m−2 at midday, resulting in the increase in the sorbent temperature from 55 °C to 74 °C (Fig. 5C). Moreover, benefiting from the fast heat dissipation by forced air convection, the temperature of the condenser was always maintained below 44 °C. This condensation temperature is higher than the other reported testing conditions in literatures,33,34,37,38 due to the relatively high ambient temperature of up to 38 °C in the summer, but more closely to the practical climatic conditions in hot arid regions (see RH changes during the whole day in Fig. S44, ESI†). This relatively high condensation temperature indicates that the water harvesting rate is controlled by the condensation rate rather than the water desorption rate or internal water vapor transport rate (see more details in Fig. S45, ESI†). Hence, seeking a more efficient condensation strategy is still one of the most important research targets in the future. Although the condensation temperature was as high as 44 °C, we realized an impressive water collection amount of 22.8 g using only 21.8 g nanocomposite sorbent (Fig. 5D), corresponding to the specific water harvesting capacity of 1050 mLwater kgsorbent−1. The high AWH performance confirmed the flexible climatic adaptation of our vertically aligned nanocomposite sorbent and compact all-in-one SAWH device, even under extreme climatic conditions. After repeating the water sorption–desorption operation about 10 times for each sorbent unit in several days, we checked the shape of sorbents and found that they showed satisfactory stability with a constant length of ∼18 cm and thickness of ∼3 mm; meanwhile, salt was not observed agglomerating on the top surface (Fig. S46, ESI†).
We comprehensively evaluated the SAWH performance in view of the weight, space, and solar collection surface of the compact all-in-one device, as listed in Table S4 (ESI†). Due to the daily water collection eight times under one solar collection surface, the specific SAWH capacity per surface realized high values of 1407 mLwater m−2 day−1 and 692 mLwater m−2 day−1 under indoor and outdoor testing conditions. Considering the potential of individual portable SAWH, we also showed the SAWH capacity in the view of the weight and space of the whole device, as high as 16–33 mL per kilogram of device (mLwater kgdevice−1 day−1) and 9–18 mL per liter of device (mLwater Ldevice−1 day−1) under outdoor and indoor conditions (see detailed information about surface area, weight, and space of SAWH device in Table S4, ESI†). These figures are much higher than the previously reported state-of-the-art sunlight-driven portable SAWH devices.35,36 Encouragingly, more attention will be paid to designing more compact and lighter SAWH devices to bridge the gap between the materials and portable applications in our future work. Considering that the facile operation of SAWH is desirable, there is the prospect of the automatic switching of water sorption and water desorption by adding mechanical rotation in this continuous SAWH device (Fig. S47, ESI†). The rotation components preliminarily illustrated that a PV-powered DC motor was employed to drive the rotation of gears, and the gears drive the rotation of spokes and shells of the SAWH device (Fig. S48, ESI†). We believe a similar automation design can be easily realized and be quickly optimized in the commercial iteration process. To accelerate the development of portable SAWH technologies, we appeal to researchers to consider evaluating the SAWH performance from the perspectives of devices in future studies. Finally, we detected the concentrations of the possible metal and ions in the collected water, and the results showed that the quality of the water extracted from the air meets the requirements for drinking-water quality by the WHO (Tables S5 and S6, ESI†).
Conclusions
In this study, we have made a comprehensive reformation of solar-driven atmospheric water harvesting from the design and synthesis of materials to the construction and optimization of devices. First, through an object-oriented approach, we developed a high-performance atmospheric water harvesting sorbent with record water harvesting capacity under arid climates, together with fast water sorption–desorption kinetics, and desirable cycling stability. We showed that the graphene hydrogel-based nanocomposite sorbent of LiCl@rGO–SA can effectively capture water from the air even under extremely low RH conditions, and achieve ultrahigh water sorption capacities up to 1.01 g g−1, 1.52 g g−1, and 2.76 g g−1 under RH of 15%, 30%, and 60%. The specially designed vertically aligned pore structures of the nanocomposite sorbent offer the significant advantages of lower water transfer resistance, enabling faster water sorption–desorption kinetics. To make full use of the superiority of fast sorption–desorption kinetics, we demonstrated a rapid-cycling continuous atmospheric water harvester with eight times water capture-release ability per day. After the synergetic optimization of heat and mass transfer of the device, the rapid-cycling continuous water harvester showed an extraordinary working performance of 1050–2120 mLwater kgsorbent−1 day−1, 692–1407 mLwater m−2 day−1 in indoor and outdoor experiments under low humidity conditions. We anticipate that this study will pave the way towards a compact, low-cost, scalable, and high-productive water harvesting strategy, accelerating the development and practical applications of portable SAWH devices.Experimental methods
Synthesis of LiCl@rGO–SA
Graphene oxide (GO) was first prepared by a modified Hummers’ method.67 The GO aqueous suspension (10 mg mL−1) was then prepared by distributing GO into DI water under ultrasonication (100 W for 30 min). The sodium alginate (SA) solution (4 wt%) was prepared by adding SA to DI water and magnetically stirred at 80 °C for several hours until completely dissolved. Afterwards, GO aqueous suspension, deionized water, and SA solution were mixed together in a weight ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2 and stirred for three hours. The GO–SA mixture was poured into the specially designed molds (see Fig. S1, ESI†), which were used to realize one-dimensional directional cooling from the bottom to the top. After directional ice crystal growth driven by liquid nitrogen, the ice was removed by vacuum drying in a vacuum freeze-dryer (<1 Pa pressure) for 2 days to get the vertically aligned GO–SA matrix. Then, the porous matrix was immersed in CaCl2 solution (2 wt%) for 24 hours to make the SA form cross-linked networks. Subsequently, the cross-linked GO–SA porous matrix was treated at 120 °C and thermally reduced to rGO–SA. Finally, the LiCl@rGO–SA was obtained by soaking the rGO–SA matrix in LiCl solution (10 wt%) overnight and dried completely. By using different sizes of molds in the directional freeze-drying step, we synthesized several small-sized blocks (3 cm × 3 cm × different thicknesses) for characterization, several middle-sized circular tablets (diameter, 9 cm × different thicknesses) for the sorption–desorption kinetics tests, and several large-sized rectangular sheets (18 cm × 9 cm × ∼3 mm) for the demonstration of atmospheric water harvesting.
:1:2 and stirred for three hours. The GO–SA mixture was poured into the specially designed molds (see Fig. S1, ESI†), which were used to realize one-dimensional directional cooling from the bottom to the top. After directional ice crystal growth driven by liquid nitrogen, the ice was removed by vacuum drying in a vacuum freeze-dryer (<1 Pa pressure) for 2 days to get the vertically aligned GO–SA matrix. Then, the porous matrix was immersed in CaCl2 solution (2 wt%) for 24 hours to make the SA form cross-linked networks. Subsequently, the cross-linked GO–SA porous matrix was treated at 120 °C and thermally reduced to rGO–SA. Finally, the LiCl@rGO–SA was obtained by soaking the rGO–SA matrix in LiCl solution (10 wt%) overnight and dried completely. By using different sizes of molds in the directional freeze-drying step, we synthesized several small-sized blocks (3 cm × 3 cm × different thicknesses) for characterization, several middle-sized circular tablets (diameter, 9 cm × different thicknesses) for the sorption–desorption kinetics tests, and several large-sized rectangular sheets (18 cm × 9 cm × ∼3 mm) for the demonstration of atmospheric water harvesting.
Characterization of LiCl@rGO–SA
The morphologies and structures of GO–SA, rGO–SA and LiCl@rGO–SA were characterized by a field emission scanning electron microscope (FESEM, Sirion 200 instrument, FEI) equipped with an energy dispersive X-ray spectrometer (EDS, INCA X-Act attachment, Oxford). The X-ray diffraction (XRD) patterns were measured by a poly-functional X-ray diffractometer (D8 ADVANCE Da Vinci, Bruker) at different temperatures (30 °C, 60 °C, and 90 °C). Fourier-transform infrared (FT-IR) spectra of sorbents were measured using an FT-IR spectrometer (Nicolet 6700, ThermoFisher Scientific) at different temperatures (30 °C, 60 °C, and 90 °C). The pore properties of the porous matrix and sorbents were measured by an automatic mercury porosimeter (Micromeritics AutoPore Iv 9510). The contact angles between samples and water droplets were measured by using a contact angle instrument (DSA100, Kruss). The compression strengths of the samples were evaluated by a dynamic mechanical analyzer (DMA Q800, TA Instrument).Water sorption tests of LiCl@rGO–SA
Water sorption isotherms of the graphene hydrogel-based nanocomposite sorbent were measured by a commercial gas sorption apparatus (ASAP 2020 plus, Micromeritics) under different temperatures. The water sorption isobaric curves were measured by using a self-constructed testing system,43,68 testing at 1.2 kPa and 4.2 kPa. The dynamic water sorption–desorption tests were performed on a thermogravimetric analyzer (STA 449C, Netzsch), equipped with a moisture humidity generator (MHG 32, ProUmid). The LiCl@rGO–SA samples were completely dried at 90 °C, then the samples were placed in the thermogravimetric analyzer and kept at 30 °C under different humidities (30%, 60%, and 90% RH) for 12 hours for water sorption. After sorption, the samples were heated from 30 °C to 90 °C for desorption under a vapor pressure of 4200 Pa. The sorption–desorption cycling stability tests were carried out by switching the temperature and vapor pressure between 30 °C, 1.2 kPa and 90 °C, 4.2 kPa for 10 cycles. The water sorption/desorption kinetics tests were conducted by a self-constructed device (see Fig. S22, ESI†), with a controlled temperature and humidity environment using a constant temperature and humidity chamber.Fabrication and testing of the rapid-cycling continuous atmospheric water harvester
Four solar collection-sorbent units were first fabricated by coating the rectangular sorbent (18 cm × 9 cm × ∼3 mm) on the back side of a solar selective absorber sheet (20 cm × 10 cm × 0.3 mm) using a thermally conductive adhesive tape (see Fig. S31 and Table S3, ESI†). Then, we designed and constructed a square tubular acrylic skeleton of the SAWH device (10 cm × 10 cm × 20 cm, thickness of 3 mm), where one surface (solar collection surface) was covered by an acrylic cover and the other three were empty (only prisms), and slots were grooved on every prism for the assembly of four sorbent units (see Fig. S26, ESI†). The square tubular device was divided into two regions after installing a condenser (see Fig. S26 and S32, ESI†); one is the closed chamber for the water collection and the other is the open channel for the water capture from the air. To collect the liquid water, a silicone tube was connected to the water collection chamber at the bottom of the device. A solar PV-driven air fan was put in the top of the device to create a forced air flow (see Fig. S27, ESI†), accelerating both the water condensation and water sorption process through the enhanced heat and mass transfer. At last, the device was sealed with rubber rings, flanges, and seal gums. To measure the temperatures of different components, we placed several temperature sensors (PT-100 platinum resistance temperature sensors) into the device to monitor the temperature changes during the water collection process. We also constructed an aluminum alloy supporter with an adjustable incline angle to allow sunlight to shine perpendicularly at the SAWH device. The water harvesting experiments were first carried out under laboratory conditions under artificial lighting generated by a solar simulator with an irradiation intensity of 0.95–1.05 kW m−2 (TRM-PD, Jinzhou Sunshine Technology, spot diameter of 0.2 m), with uniformity of 10%, temporal instability of 10%, and spectral match classification of Class B. The outdoor water harvesting experiments were implemented on the roof of our laboratory on August 17, 2020. The solar flux was measured by a pyranometer (TBQ-DL, Jinzhou Sunshine Technology). The real-time signals from these sensors were collected using a data collector (34970A, Agilent) and recorded on a computer. The mass of the collected water was obtained by directly weighing the harvested liquid water using an electronic balance; the mass of desorbed water was obtained by measuring the mass change of sorbent before and after the water release for each cycle. The concentrations of possible metals and ions present were measured by Inductively Coupled Plasma-Optical Emission Spectroscopy (ICP-OES, iCAP7600, Thermo Fisher Scientific) and Ion Chromatography (ICS-5000, Thermo Dionex).Author contributions
J. X., T. Y., and T. L. conceived and designed the experiment. J. X. and T. Y. designed, synthesized, and characterized the nanocomposite sorbent. T. Y. carried out the water sorption–desorption kinetics simulation. J. X. designed and engineered the atmospheric water harvester. J. X. and T. Y. tested the working performance of the water harvester. J. X. and T. L. wrote the manuscript. All the authors discussed the results and commented on the manuscript. T. L. and R. W. supervised the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China under the contract No. 51876117 and the National Key R&D Program of China under the contract No. 2018YFE0100300. Part of this work was funded by the Innovative Research Group Project of National Natural Science Foundation of China under the contract No. 51521004.References
- M. M. Mekonnen and A. Y. Hoekstra, Sci. Adv., 2016, 2, e1500323 CrossRef PubMed.
- A. Boretti and L. Rosa, npj Clean Water, 2019, 2, 15 CrossRef.
- W. Xu and O. M. Yaghi, ACS Cent. Sci., 2020, 6, 1348–1354 CrossRef CAS PubMed.
- Y. Zheng, H. Bai, Z. Huang, X. Tian, F. Q. Nie, Y. Zhao, J. Zhai and L. Jiang, Nature, 2010, 463, 640–643 CrossRef CAS PubMed.
- Y. Tu, R. Wang, Y. Zhang and J. Wang, Joule, 2018, 2, 1452–1475 CrossRef CAS.
- R. Peeters, H. Vanderschaeghe, J. Rongé and J. A. Martens, Environ. Sci.: Water Res. Technol., 2020, 6, 2016–2034 RSC.
- X. Zhou, H. Lu, F. Zhao and G. Yu, ACS Mater. Lett., 2020, 2, 671–684 CrossRef CAS.
- J. H. Humphrey, J. Brown, O. Cumming, B. Evans, G. Howard, R. N. Kulabako, J. Lamontagne, A. J. Pickering and E. N. Wang, Lancet Planetary Health, 2020, 4, e91–e92 CrossRef PubMed.
- T. Pan, K. Yang and Y. Han, Chem. Res. Chin. Univ., 2020, 36, 33–40 CrossRef CAS.
- A. J. Rieth, S. Yang, E. N. Wang and M. Dincă, ACS Cent. Sci., 2017, 3, 668–672 CrossRef CAS PubMed.
- M. J. Kalmutzki, C. S. Diercks and O. M. Yaghi, Adv. Mater., 2018, 30, 1704304 CrossRef PubMed.
- N. Hanikel, M. S. Prévot and O. M. Yaghi, Nat. Nanotechnol., 2020, 15, 348–355 CrossRef CAS PubMed.
- M. Nemiwal and D. Kumar, Inorg. Chem. Commun., 2020, 122, 108279 CrossRef CAS.
- G. Mouchaham, F. S. Cui, F. Nouar, V. Pimenta, J. S. Chang and C. Serre, Trends Chem., 2020, 2, 990–1003 CrossRef CAS.
- K. Matsumoto, N. Sakikawa and T. Miyata, Nat. Commun., 2018, 9, 2315 CrossRef PubMed.
- D. K. Nandakumar, S. K. Ravi, Y. Zhang, N. Guo, C. Zhang and S. C. Tan, Energy Environ. Sci., 2018, 11, 2179–2187 RSC.
- L. Yang, D. K. Nandakumar, L. Miao, L. Suresh, D. Zhang, T. Xiong, J. V. Vaghasiya, K. C. Kwon and S. C. Tan, Joule, 2020, 4, 176–188 CrossRef CAS.
- X. Wang, X. Li, G. Liu, J. Li, X. Hu, N. Xu, W. Zhao, B. Zhu and J. Zhu, Angew. Chem., Int. Ed., 2019, 58, 12054–12058 CrossRef CAS PubMed.
- H. Qi, T. Wei, W. Zhao, B. Zhu, G. Liu, P. Wang, Z. Lin, X. Wang, X. Li, X. Zhang and J. Zhu, Adv. Mater., 2019, 31, 1903378 CrossRef CAS PubMed.
- P. A. Kallenberger and M. Fröba, Commun. Chem., 2018, 1, 28 CrossRef.
- J. Yang, X. Zhang, H. Qu, Z. G. Yu, Y. Zhang, T. J. Eey, Y. W. Zhang and S. C. Tan, Adv. Mater., 2020, 32, 2002936 CrossRef CAS PubMed.
- J. X. Xu, T. X. Li, J. W. Chao, T. S. Yan and R. Z. Wang, Energy, 2019, 185, 1131–1142 CrossRef CAS.
- K. Yang, Y. Shi, M. Wu, W. Wang, Y. Jin, R. Li, M. W. Shahzad, K. C. Ng and P. Wang, J. Mater. Chem. A, 2020, 8, 1887–1895 RSC.
- S. Pu, J. Fu, Y. Liao, L. Ge, Y. Zhou, S. Zhang, S. Zhao, X. Liu, X. Hu, K. Liu and J. Chen, Adv. Mater., 2020, 32, 1907307 CrossRef CAS PubMed.
- R. Li, Y. Shi, M. Wu, S. Hong and P. Wang, Nat. Sustainability, 2020, 3, 636–643 CrossRef.
- C. Wang, L. Hua, H. Yan, B. Li, Y. Tu and R. Wang, Joule, 2020, 4, 435–447 CrossRef CAS.
- J. Xu, J. Chao, T. Li, T. Yan, S. Wu, M. Wu, B. Zhao and R. Wang, ACS Cent. Sci., 2020, 6, 1542–1554 CrossRef CAS PubMed.
- T. Xu, X. Ding, Y. Huang, C. Shao, L. Song, X. Gao, Z. Zhang and L. Qu, Energy Environ. Sci., 2019, 12, 972–978 RSC.
- X. Zhang, J. Yan, R. Borayek, H. Qu, D. K. Nandakumar, Q. Zhang, J. Ding and S. C. Tan, Nano Energy, 2020, 75, 104873 CrossRef CAS.
- X. Zhang, J. Yang, H. Qu, Z. G. Yu, D. K. Nandakumar, Y. Zhang and S. C. Tan, Adv. Sci., 2021, 8, 2003939 CrossRef CAS PubMed.
- K. Yang, T. Pan, I. Pinnau, Z. Shi and Y. Han, Nano Energy, 2020, 78, 105326 CrossRef CAS.
- Y. Zhang, D. K. Nandakumar and S. C. Tan, Joule, 2020, 4, 2532–2536 CrossRef.
- H. Kim, S. Yang, S. R. Rao, S. Narayanan, E. A. Kapustin, H. Furukawa, A. S. Umans, O. M. Yaghi and E. N. Wang, Science, 2017, 356, 430–434 CrossRef CAS PubMed.
- H. Kim, S. R. Rao, E. A. Kapustin, L. Zhao, S. Yang, O. M. Yaghi and E. N. Wang, Nat. Commun., 2018, 9, 1191 CrossRef PubMed.
- F. Fathieh, M. J. Kalmutzki, E. A. Kapustin, P. J. Waller, J. Yang and O. M. Yaghi, Sci. Adv., 2018, 4, eaat3198 CrossRef PubMed.
- N. Hanikel, M. S. Prévot, F. Fathieh, E. A. Kapustin, H. Lyu, H. Wang, N. J. Diercks, T. G. Glover and O. M. Yaghi, ACS Cent. Sci., 2019, 5, 1699–1706 CrossRef CAS PubMed.
- D. K. Nandakumar, Y. Zhang, S. K. Ravi, N. Guo, C. Zhang and S. C. Tan, Adv. Mater., 2019, 31, 1806730 CrossRef PubMed.
- F. Zhao, X. Zhou, Y. Liu, Y. Shi, Y. Dai and G. Yu, Adv. Mater., 2019, 31, 1806446 CrossRef PubMed.
- A. Karmakar, P. G. Mileo, I. Bok, S. B. Peh, J. Zhang, H. Yuan, G. Maurin and D. Zhao, Angew. Chem., Int. Ed., 2020, 59, 11003–11009 CrossRef CAS PubMed.
- G. Yilmaz, F. L. Meng, W. Lu, J. Abed, C. K. N. Peh, M. Gao, E. H. Sargent and G. W. Ho, Sci. Adv., 2020, 6, eabc8605 CrossRef CAS PubMed.
- H. Yao, P. Zhang, Y. Huang, H. Cheng, C. Li and L. Qu, Adv. Mater., 2020, 32, 1905875 CrossRef CAS PubMed.
- X. Zhou, P. Zhang, F. Zhao and G. Yu, ACS Mater. Lett., 2020, 2, 1419–1422 CrossRef CAS.
- J. X. Xu, T. X. Li, J. W. Chao, S. Wu, T. S. Yan, W. C. Li, B. Y. Cao and R. Z. Wang, Angew. Chem., Int. Ed., 2020, 59, 5202–5210 CrossRef CAS PubMed.
- R. Li, Y. Shi, M. Alsaedi, M. Wu, L. Shi and P. Wang, Environ. Sci. Technol., 2018, 52, 11367–11377 CrossRef CAS PubMed.
- A. Entezari, M. Ejeian and R. Wang, ACS Mater. Lett., 2020, 2, 471–477 CrossRef CAS.
- F. Ni, N. Qiu, P. Xiao, C. Zhang, Y. Jian, Y. Liang, W. Xie, L. Yan and T. Chen, Angew. Chem., Int. Ed., 2020, 59, 19237–19246 CrossRef CAS PubMed.
- R. Li, Y. Shi, M. Wu, S. Hong and P. Wang, Nano Energy, 2020, 67, 104255 CrossRef CAS.
- F. Gong, H. Li, Q. Zhou, M. Wang, W. Wang, Y. Lv, R. Xiao and D. V. Papavassiliou, Nano Energy, 2020, 74, 104922 CrossRef CAS.
- Y. I. Aristov, G. Restuccia, G. Cacciola and V. N. Parmon, Appl. Therm. Eng., 2002, 22, 191–204 CrossRef CAS.
- Y. Sun, A. Spieß, C. Jansen, A. Nuhnen, S. Gökpinar, R. Wiedey, S. J. Ernst and C. Janiak, J. Mater. Chem. A, 2020, 8, 13364–13375 RSC.
- J. J. Otten, J. P. Hellwig and L. D. Meyers, Dietary reference intakes: the essential guide to nutrient requirements, National Academies Press, 2006 Search PubMed.
- A. LaPotin, H. Kim, S. R. Rao and E. N. Wang, Acc. Chem. Res., 2019, 52, 1588–1597 CrossRef CAS PubMed.
- A. Terzis, A. Ramachandran, K. Wang, M. Asheghi, K. E. Goodson and J. G. Santiago, Cell Rep. Phys. Sci., 2020, 1, 100057 CrossRef.
- A. LaPotin, Y. Zhong, L. Zhang, L. Zhao, A. Leroy, H. Kim, S. R. Rao and E. N. Wang, Joule, 2021, 5, 166–182 CrossRef CAS.
- J. Y. Wang, R. Z. Wang, Y. D. Tu and L. W. Wang, Energy, 2018, 165, 387–395 CrossRef.
- W. Wang, S. Xie, Q. W. Pan, Y. J. Dai, R. Z. Wang and T. S. Ge, Renewable Sustainable Energy Rev., 2021, 141, 110802 CrossRef CAS.
- Y. Guo, J. Bae, Z. Fang, P. Li, F. Zhao and G. Yu, Chem. Rev., 2020, 120, 7642–7707 CrossRef CAS PubMed.
- F. Zhao, Y. Guo, X. Zhou, W. Shi and G. Yu, Nat. Rev. Mater., 2020, 5, 388–401 CrossRef.
- G. Shao, D. A. Hanaor, X. Shen and A. Gurlo, Adv. Mater., 2020, 32, 1907176 CrossRef CAS PubMed.
- K. M. Willett, P. D. Jones, N. P. Gillett and P. W. Thorne, J. Clim., 2008, 21, 5364–5383 CrossRef.
- A. Entezari, M. Ejeian and R. Z. Wang, Mater. Today Energy, 2019, 13, 362–373 CrossRef.
- L. Qiu, J. Z. Liu, S. L. Chang, Y. Wu and D. Li, Nat. Commun., 2012, 3, 1241 CrossRef PubMed.
- L. Garzón-Tovar, J. Pérez-Carvajal, I. Imaz and D. Maspoch, Adv. Funct. Mater., 2017, 27, 1606424 CrossRef.
- M. Wang, T. Sun, D. Wan, M. Dai, S. Ling, J. Wang, Y. Liu, Y. Fang, S. Xu, J. Yeo and H. Yu, Nano Energy, 2021, 80, 105569 CrossRef CAS.
- T. X. Li, R. Z. Wang and H. Li, Prog. Energy Combust. Sci., 2014, 40, 1–58 CrossRef CAS.
- F. Wang, N. Xu, W. Zhao, L. Zhou, P. Zhu, X. Wang, B. Zhu and J. Zhu, Joule, 2021, 5, 1602–1612 CrossRef CAS.
- Y. Xu, K. Sheng, C. Li and G. Shi, ACS Nano, 2010, 4, 4324–4330 CrossRef CAS PubMed.
- T. S. Yan, T. X. Li, J. X. Xu and R. Z. Wang, Int. J. Heat Mass Transfer, 2019, 139, 990–999 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ee01723c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |