
Tailoring the linking patterns of polypyrene cathodes for high-performance aqueous Zn dual-ion batteries†
Chong
Zhang
ab,
Wenyan
Ma
a,
Changzhi
Han
a,
Lian-Wei
Luo
a,
Aigerim
Daniyar
b,
Sihui
Xiang
a,
Xianyong
Wu
b,
Xiulei
Ji
 *b and
Jia-Xing
Jiang
*a
*b and
Jia-Xing
Jiang
*a
aKey Laboratory of Applied Surface and Colloid Chemistry (Shaanxi Normal University), Ministry of Education, Key Laboratory for Macromolecular Science of Shaanxi Province, Shaanxi Key Laboratory for Advanced Energy Devices, School of Materials Science and Engineering, Shaanxi Normal University, Xi’an 710062, P. R. China. E-mail: jiaxing@snnu.edu.cn
bDepartment of Chemistry, Oregon State University, Corvallis, Oregon 97331-4003, USA. E-mail: david.ji@oregonstate.edu
First published on 9th December 2020
Abstract
Although the utilization of mildly acidic electrolyte can enhance the stability and reversibility of Zn anode, it is still challenging to achieve long cycling life for aqueous Zn batteries at low currents, due to the structural instability of cathode materials during the charge/discharge processes. Herein, we report a series of polypyrenes with different linking patterns and electronic structures as Cl−-hosting organic cathodes for aqueous Zn dual-ion batteries (AZDIBs). The comparative study demonstrates that the electronic structures are pivotal to the redox activity of the polypyrenes, which can be tuned by altering the linking patterns on the pyrene unit. Owing to the high surface area, the highly delocalized HOMO distribution, the high HOMO level and the narrow band gap, the polymer CLPy with 1,3,6,8-linking pattern delivers a much higher capacity of 180 mA h g−1 than the two linear counterparts (24 mA h g−1 for LPy-1 and 44 mA h g−1 for LPy-2). Impressively, CLPy exhibits ultra-stable cyclabilities with the capacity retentions of 97.4% after 800 cycles at 50 mA g−1 and 96.4% after 38![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles at 3 A g−1. CLPy also shows a low self-discharge rate with around 90% capacity retention after resting for 28 days. The excellent electrochemical performance demonstrates that CLPy can be a promising cathode material for high-performance AZDIBs.
000 cycles at 3 A g−1. CLPy also shows a low self-discharge rate with around 90% capacity retention after resting for 28 days. The excellent electrochemical performance demonstrates that CLPy can be a promising cathode material for high-performance AZDIBs.
Broader contextThe inherent safety and low cost make aqueous Zn battery (AZB) an ideal candidate for the stationary energy storage. Although the usage of mildly acidic electrolyte can enhance the stability and reversibility of Zn anode, it is still challenging for realizing long life AZBs at low current rates because of the structural instability of the cathodes with repeated Zn2+ or Zn2+/H+ insertion/deinsertion. Dual-ion battery offers a promising opportunity for high performance AZBs, wherein the anion functions as the charge carrier in the cathode. However, few aqueous Zn dual-ion batteries (AZDIBs) can achieve both stable cyclability and high capacity at low current rates. Herein, we developed a series of polypyrene cathodes with different linking patterns for AZDIBs. The results demonstrate the large influence of electronic property and porous structure, which could be finely tuned by altering the linking patterns of the pyrene unit, on the redox activity of polypyrene cathode. The polypyrene cathode with optimized structure shows both high redox activity and stable cycling performance. The revealed profound structure effect offers an in-depth insight into the Cl−-hosting mechanism by conductive polymers and provides an efficient structure design strategy for the development of polymer cathode materials for high-performance AZDIBs. |
Introduction
The global demand for sustainable development promotes the progress of renewable energy technologies such as solar, wind, and tidal energy. What follows are the ever-increasing requirements for developing efficient and reliable grid/mini-grid scale energy storage systems, wherein the safety, cost, and service life are the top concerns.1 Especially, prolonging the service life of energy storage devices can significantly decrease the levelized energy cost.2 Aqueous Zn battery (AZB) represents a unique opportunity for the grid or other stationary storage applications, which relates to the intrinsic safety of aqueous electrolytes and the advantages of Zn-metal anode, including appropriate deposition/stripping potentials, high capacity (820 mA h g−1) and low cost.3,4 Currently, the leading cathodes for AZBs are mainly based on inorganic materials including transition metal oxides or sulfides,3,5 polyanionic frameworks6,7 and Prussian blue analogues.8 However, the issues of the dissolution and structural instability for most cathodes during the repeated Zn2+ intercalation or the H+/Zn2+ co-intercalation commonly leads to a fast capacity fading, resulting in a short cycling life.1 It will be intriguing to avoid the reliance on the Zn-ion intercalation in the cathode, where the cathode can host anions during battery charge, thus constituting an aqueous zinc-metal dual-ion battery (AZDIB).9 A dual-ion battery (DIB) with the electrolyte as the sole source of ionic charge carriers operates in an “accordion” fashion, where anions and cations migrate into/from the cathode and anode in concert, respectively, during charge/discharge processes.The traditional DIBs commonly employ graphite as the anion-hosting cathode material in organic electrolytes or ionic liquids, which could deliver a high specific capacity around 140 mA h g−1 with high operating voltage up to 5 V vs. Li+/Li, endowing the DIBs with high energy densities.10–13 However, there are very few studies reporting the application of graphite cathode in aqueous DIBs,9,14–16 since the conventional aqueous electrolytes with low anodic stability generally cannot afford the high operating voltage for graphite cathode.9,14 Albeit not typically referred to as anion-hosting cathode in DIBs, recent study showed that conductive polymers (CPs) with reversible p-type doping ability could be an alternative of graphite as DIB cathodes with lower operating potentials, which allows organic CPs to operate in aqueous electrolytes without triggering the oxygen evolution reaction.17 In the past years, significant advances have been achieved in the designing and preparation of CP cathodes for AZDIBs.18 For instance, polyaniline,19–23 polyindole,24 and the 9,10-di(1,3-dithiol-2-ylidene)-9,10-dihydroanthracene (exTTF)-based polymer25 showed high redox activities for the storage of anions in AZDIBs. The exTTF-based polymer and poly(1,5-naphthalenediamine) cathodes also exhibited high cycling stabilities with cyclability of 10000 cycles,20,25 demonstrating that CPs would be a promising class of electrode materials for AZDIBs. However, few AZDIBs can achieve both stable cyclability and high capacity at low current rates, possibly due to the large size of anions. Meanwhile, the lack of comprehensive understanding on the structure-performance relationship seriously hinders the development of polymer-based batteries.
As a sub-class of CPs, polypyrenes could be cost-effectively produced on a large scale due to the high yield and low cost of the pyrene monomer.26 The coplanar molecular structure of the pyrene unit and the extended π-conjugated skeleton endow polypyrenes with highly reversible doping/dedoping capability, making polypyrenes as promising electrode materials for rechargeable batteries. Indeed, polypyrenes has been reported as cathodes for Li/Na/Al-metal DIBs.27–31 However, the dense-packed aggregation structure of the linear polypyrene arising from the strong π–π interaction between the polymer chains would hinder the contact between electrolytes and the active sites, and block the diffusion of charge carriers, commonly leading to a low redox activity and sluggish kinetics.28,31 Several strategies have been employed to enhance the electrochemical performance of polypyrene electrodes. For instance, Kovalenko et al. reported that the introduction of nitro group onto a linear polypyrene could improve the storage capability of AlCl4−, since the enlarged space between polymer chains promotes the transport of charge carriers in the polypyrene cathode.28 Pyo et al. achieved enhanced capability for the ClO4− storage in oligopyrene cathode by transforming the crystalline phase into amorphous structure due to the faster kinetics of ClO4− in the amorphous oligopyrene.31 Considering the highly conjugated polymer skeleton and appropriate potential for p-type doping, polypyrene might also be a good candidate cathode material for AZDIBs. In addition, there are multiple active sites on the pyrene monomer, which allows us to control the polymer structures and physical properties to boost the redox activity and electrochemical performance of AZDIBs.
With this in mind, we designed and synthesized a series of polypyrene cathodes with different linking patterns to uncover their structure-performance correlations for AZDIBs. The comparative investigation demonstrated the significant influence of electronic structures, which can be tuned by altering the linking patterns on the pyrene unit, on the redox activity of polypyrene cathode for AZDIBs. The porous polypyrene CLPy with 1,3,6,8-linking pattern delivers a high specific capacity of 180 mA h g−1 with stable cyclabilities of 800 cycles at 50 mA g−1 and 38000 cycles at 3000 mA g−1. The CLPy cathode also exhibits excellent rate performance as well as ultra-low self-discharge rate. This work highlights the key point of structure design of polypyrene on achieving high performance AZDIBs.
Results and discussion
Three polypyrenes were synthesized by the homopolymerization of bromated pyrenes with different substituted positions using nickel-catalyzed Yamamoto coupling reactions (Scheme 1). The linear polypyrenes of LPy-1 and LPy-2 were respectively produced from 2,7-dibromopyrene and 1,6-dibromopyrene, while the cross-linked polypyrene of CLPy as the previous reported YPy32 was generated from 1,3,6,8-tetrabromopyrene. The resulting polymers exhibit different bulk colors ranging from gray (LPy-1) to yellow (LPy-2) and to orange (CLPy) (Scheme 1), although they have the same chemical component of pyrene unit, demonstrating that the linking pattern has a large influence on the physical property and electronic structure for these polypyrenes. Almost no Br element could be detected by the energy dispersive X-ray (EDX) spectrum for all of the polymers (Fig. S1, ESI†), implying all of the functional bromine groups have been consumed by C–C coupling reaction. Fourier transform infrared spectra (FT-IR) showed the peaks at around 704 and 1150 cm−1 from the out-of-plane vibrations of adjacent C–H of the pyrene unit, the signals at around 866 and 1607 cm−1 from the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C vibration of the pyrene ring (Fig. 1a).28,33 LPy-1 and LPy-2 show very different FT-IR spectra as the marked peaks in Fig. 1a due to the difference of linking pattern, while LPy-2 and CLPy with the similar linking positions on pyrene unit show very similar FT-IR spectra. The solid state 13C MAS NMR spectra also confirmed the influence of the linking pattern on the polymer structure, as evidenced by the obvious difference in the chemical shift of the substituted and unsubstituted carbon atoms in the pyrene ring (Fig. 1b). Powder X-ray diffraction patterns demonstrated that LPy-1 has a relatively ordered aggregation structure due to the π–π stacking of the polymer chains (Fig. 1c), while the twisted structure between pyrene units in LPy-2 and the cross-linked polymer structure in CLPy would inhibit the ordered aggregation of the polymer chains, as thus both LPy-2 and CLPy are amorphous in nature. All the polypyrenes show excellent thermal stability with a high onset decomposition temperature of 400 °C under N2 atmosphere (Fig. S2, ESI†). UV/Vis absorption spectra revealed that the 1,6-linked polymer LPy-2 shows an obvious red shift in the absorption spectrum compared to the 2,7-linked polymer LPy-1 (Fig. 1d), implying the extended conjugation degree along the polymer chain in LPy-2 due to the enhanced electronic coupling on the node of the two adjacent pyrene units.34 The cross-linked polymer CLPy with the similar linking pattern as LPy-2 shows a broader light adsorption range, demonstrating that the cross-linked polymer structure could further enhance the conjugation degree, which is in well line with the previous reported results.32,35
C vibration of the pyrene ring (Fig. 1a).28,33 LPy-1 and LPy-2 show very different FT-IR spectra as the marked peaks in Fig. 1a due to the difference of linking pattern, while LPy-2 and CLPy with the similar linking positions on pyrene unit show very similar FT-IR spectra. The solid state 13C MAS NMR spectra also confirmed the influence of the linking pattern on the polymer structure, as evidenced by the obvious difference in the chemical shift of the substituted and unsubstituted carbon atoms in the pyrene ring (Fig. 1b). Powder X-ray diffraction patterns demonstrated that LPy-1 has a relatively ordered aggregation structure due to the π–π stacking of the polymer chains (Fig. 1c), while the twisted structure between pyrene units in LPy-2 and the cross-linked polymer structure in CLPy would inhibit the ordered aggregation of the polymer chains, as thus both LPy-2 and CLPy are amorphous in nature. All the polypyrenes show excellent thermal stability with a high onset decomposition temperature of 400 °C under N2 atmosphere (Fig. S2, ESI†). UV/Vis absorption spectra revealed that the 1,6-linked polymer LPy-2 shows an obvious red shift in the absorption spectrum compared to the 2,7-linked polymer LPy-1 (Fig. 1d), implying the extended conjugation degree along the polymer chain in LPy-2 due to the enhanced electronic coupling on the node of the two adjacent pyrene units.34 The cross-linked polymer CLPy with the similar linking pattern as LPy-2 shows a broader light adsorption range, demonstrating that the cross-linked polymer structure could further enhance the conjugation degree, which is in well line with the previous reported results.32,35
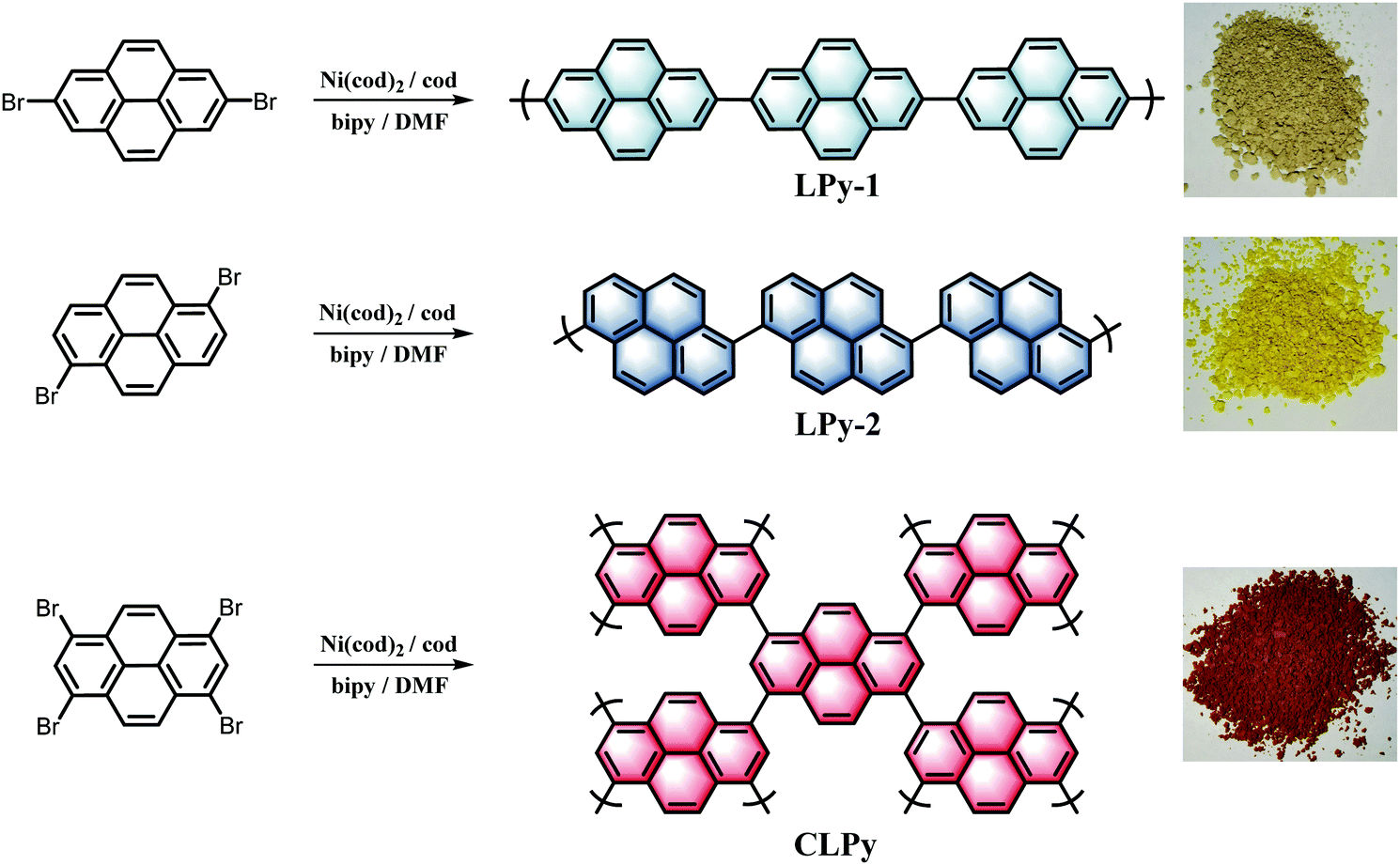 | ||
| Scheme 1 The preparation routes for the polypyrenes and photographs of the as-synthesized polypyrenes. | ||
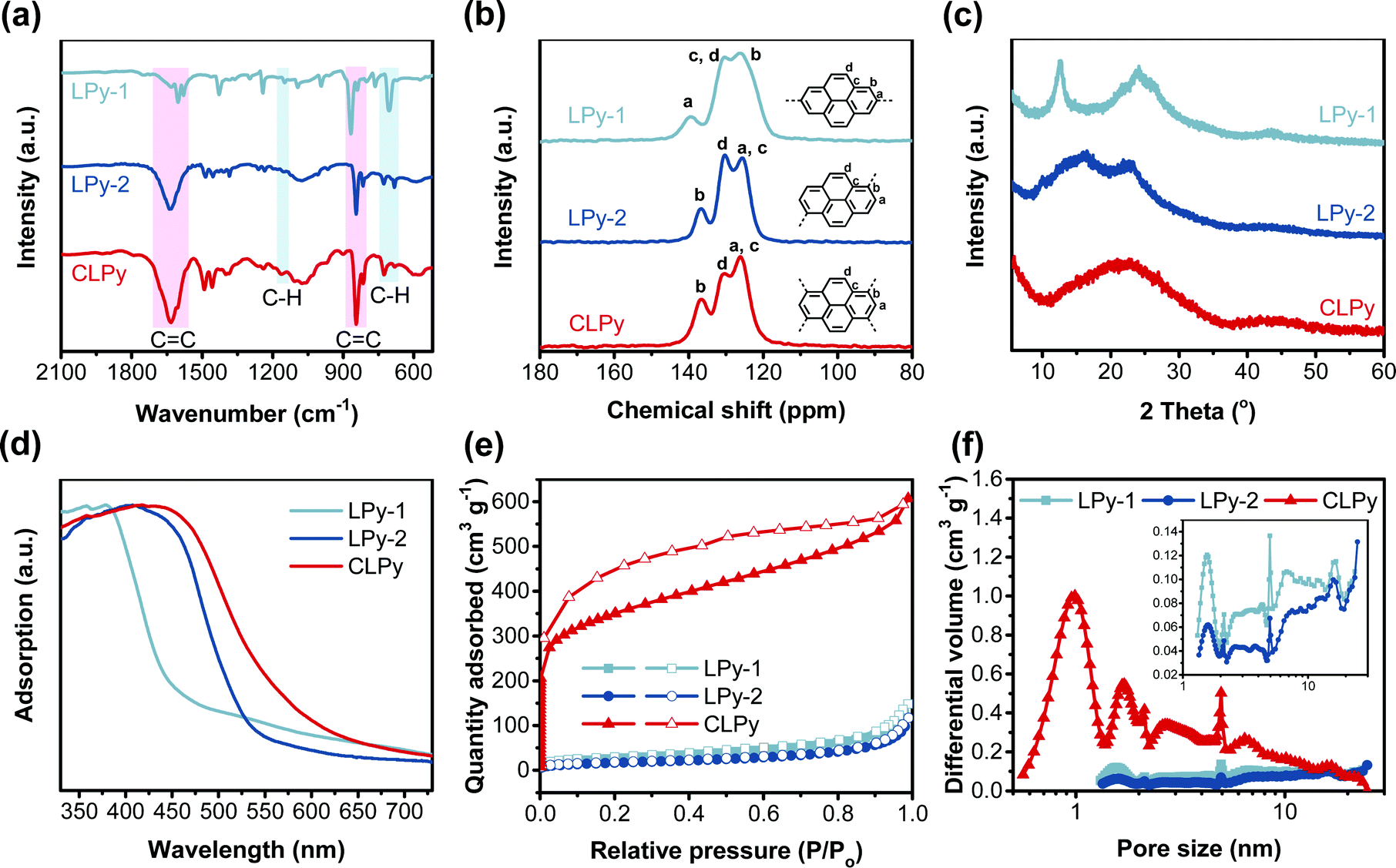 | ||
| Fig. 1 The characterization of polypyrenes. (a) The FT-IR spectra. (b) The solid-state NMR spectra. (c) The XRD patterns. (d) UV/Vis absorption spectra. (e) The N2 adsorption (filled symbols)/desorption (open symbols) isotherms. (f) The PSD curves calculated by NL-DFT (Inset: the PSD curves of LPy-1 and LPy-2). | ||
The three polypyrenes show different N2 adsorption and desorption isotherms (Fig. 1e), indicative of the different porous structures in the samples. The cross-linked polymer CLPy shows a high N2 uptake at the low relative pressures (P/P0 < 0.001), demonstrating the presence of abundant micropores in CLPy. In contrast, both the linear polypyrenes exhibit ultra-low N2 absorption at low relative pressures, indicating the micropore-free structures. The steep rise of N2 adsorption at high relative pressures (P/P0 > 0.9) for the three polypyrenes reveals the existence of meso/macropores from the voids among the nanoparticles. The calculated Brunauer–Emmett–Teller (BET) surface areas are 90, 61 and 1216 m2 g−1 for LPy-1, LPy-2 and CLPy, respectively. The pore size distribution (PSD) calculated by nonlocal density functional theory (NL-DFT) showed that the cross-linked polymer CLPy contains abundant micropores with the pore diameter of around 1.3 and 1.7 nm (Fig. 1f), while the two linear polypyrenes mainly show some mesopores. Scanning electron microscopy revealed that all of the three polypyrenes are obtained as nanospheres, which agglomerate to form larger aggregation nanosphere structures (Fig. S3, ESI†).
The cyclic voltammetry (CV) measurement of the three polypyrenes for AZDIBs was conducted in three-electrode cells using the polymers as cathodes, Zn metal as both the reference and counter electrodes, and ZnCl2 aqueous solution as the electrolyte. The galvanostatic charge/discharge (GCD) performance of the polypyrenes for AZDIBs was investigated in two-electrode cells using the polymers as cathodes, Zn metal as the anode and ZnCl2 aqueous solution as the electrolyte. We previously demonstrated that 30 M ZnCl2 aqueous electrolyte can suppress the formation of Zn dendrite and enhance the reversibility of Zn anode.36 Moreover, the high concentration of electrolyte is crucial to obtain high energy density for DIBs. Therefore, 30 M ZnCl2 “water-in-salt” electrolyte (WiSE) was employed in this work. Fig. 2a and b respectively show the configuration of the polypyrene-based AZDIBs and the diagram of the polypyrene cathode during the charge/discharge processes, the charge storage mechanism of the polypyrene cathode will be discussed in detail below. Fig. 2c shows the CV curves of the three polypyrene cathodes recorded at a scan rate of 0.5 mV s−1. All the polypyrene cathodes show similar redox peaks but different current response in the range of 0.6–1.3 V vs. Zn2+/Zn (∼0.12–0.82 V vs. SHE), corresponding to the typical p-type doping/dedoping reaction of polypyrene electrodes.27,28,30,31 The increased current response demonstrates the enhanced redox activity from LPy-1 to LPy-2 and to CLPy. The reduction potential slightly decreases from 0.88 to 0.82 and then to 0.81 V for LPy-1, LPy-2 and CLPy, respectively, which is in line with the GCD curves showing a slightly decreased average discharge voltage from 1.06, to 1.01 and to 0.94 V for LPy-1, LPy-2 and CLPy, respectively (Fig. 2d). The difference in the average discharge voltage for the three polypyrenes could be attributed to the different capacity contribution from the conductive additive (Ketjen black), which are 62.5%, 34.1% and 8.3% for LPy-1, LPy-2 and CLPy, respectively (Fig. S4, ESI†). The higher the capacity contribution from the Ketjen black in the total capacity of polypyrenes, the higher the average discharge voltage is, since the pure Ketjen black shows a higher average discharge voltage of ∼1.2 V than the polypyrene cathodes (∼0.9 V).
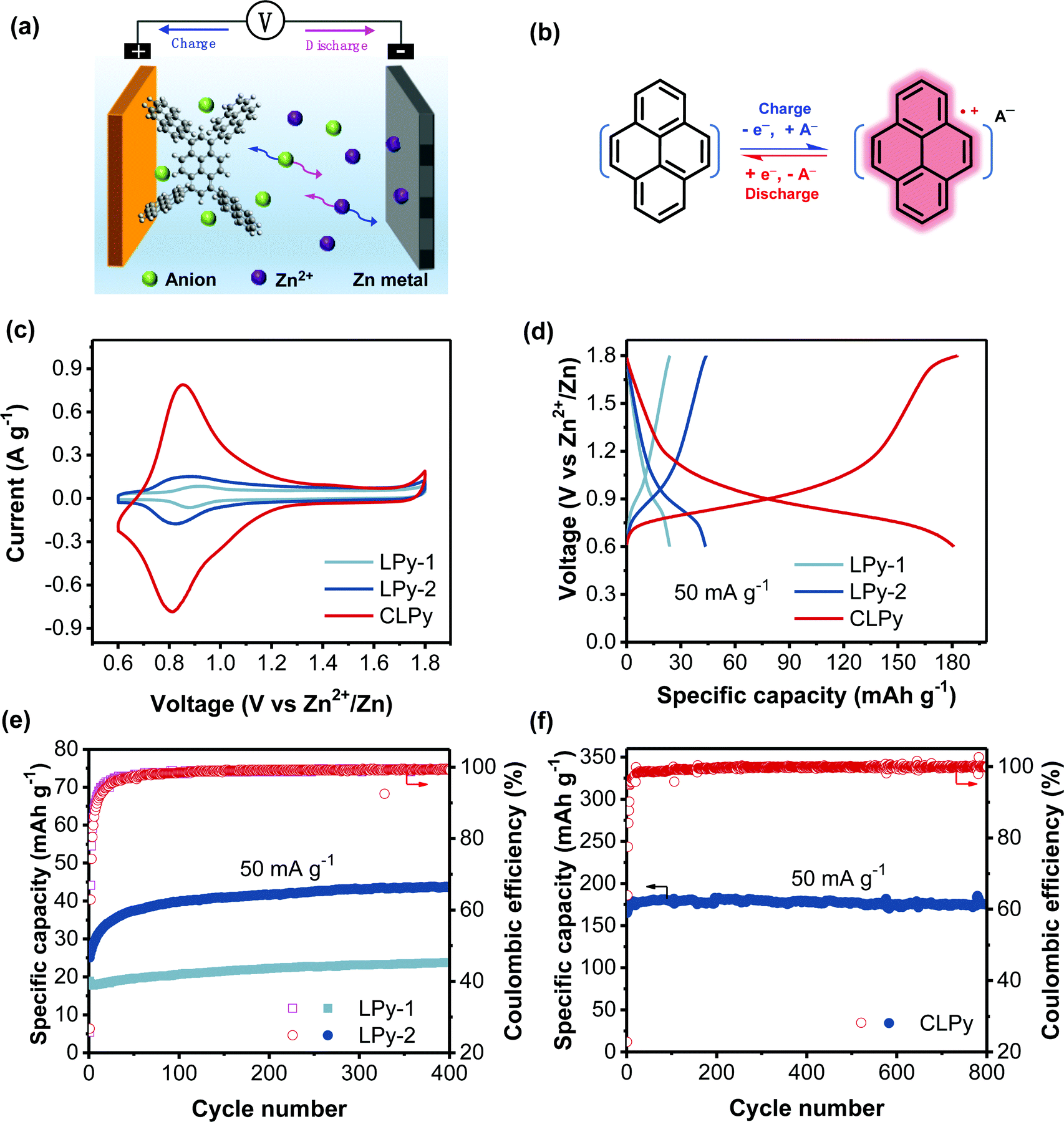 | ||
| Fig. 2 (a) The operation configuration of the polypyrene-based AZDIBs with CLPy as cathode. (b) The charge storage mechanism of polypyrene cathode. (c) The CV curves of polypyrenes at 0.5 mV s−1. (d) GCD potential curves for the three polypyrene cathodes at 50 mA g−1 at the 400th cycle. The cycling performance of (e) LPy-1, LPy-2 and (f) CLPy at 50 mA g−1. | ||
Large difference in the specific capacity was observed in the three polypyrene cathodes, in which CLPy shows a much higher specific capacity of 180 mA h g−1 than LPy-1 (24 mA h g−1) and LPy-2 (44 mA h g−1) at ambient temperature, suggesting the great influence of the linking pattern on the electrochemical performance. After subtracting the capacity contribution from the conductive additive (Fig. S4, ESI†), CLPy can still deliver a high capacity of 165 mA h g−1, corresponding to 1.24 charge-storage per pyrene. This unexpected anion-storage capacity for CLPy exceeds that of all the reported p-type polypyrene cathodes27,28,30,31 and many other anion-hosting organic cathodes (Table S1, ESI†). In previous works, the theoretical capacity of polypyrene cathode was calculated based on the assumption that each pyrene unit can host one charge (anion), delivering a theoretical capacity of 133 mA h g−1.28,31 Recently, Want et al. reported that the pyrene-based conjugated polymer (PPYS) cathode could achieve the storage of 1.25 charge in a pyrene unit.30 In this work, CLPy achieves the storage of 1.24 charge per pyrene unit. Therefore, the charge-storage capability of polypyrene cathode varies with the structure, and thus it is difficult to define an accurate theoretical capacity. This can be attributed to that the electroactivity of conjugated polymers originates from their reversible doping/dedoping reaction. Therefore, the charge-storage capability strongly depends on the doping level of the polymer chains, on which the polymer structure shows a significant influence. One of the advantages of conjugated polymer electrode is to tune the electrochemical performance by tailoring the conjugated polymer structure. In addition, the operation temperature may affect the electroactivity of polypyrene. This was evidenced by the CLPy cathode at higher temperature, which can deliver a higher specific capacity of 204 mA h g−1 at a higher temperature of 50 °C (Fig. S5, ESI†), corresponding to 1.34 charge-storage per pyrene unit.
Fig. 2e shows the cycling performance of the linear polymers at a low current rate of 50 mA g−1. Both LPy-1 and LPy-2 show an obvious increase in the discharge capacity during the first 80 cycles, which could be ascribed to the continuous infiltration of the electrolyte into the polymer cathodes because of the low surface areas of LPy-1 and LPy-2. Benefiting from the high surface area and porous structure, however, the cross-linked polymer CLPy shows a capacity increase only in the first 15 cycles and keeps a stable capacity afterward (Fig. 2f). CLPy can maintain 97.4% capacity retention after 800 cycles at a low current rate of 50 mA g−1. Such excellent cycling performance of CLPy is rarely achieved by other cathodes for AZBs at a low current rate.1,37 We also evaluated the stability of Zn anode in a symmetric cell using 30 m ZnCl2 as the electrolyte and Zn metal as the both electrodes. The Zn electrode delivers a high stability over 300 h without any distinct overpotential fluctuation at a high current density of 2 mA cm−2 with an areal capacity of 4 mA h cm−2 (Fig. S6, ESI†). Therefore, the outstanding cycling stability of the CLPy-based AZDIBs can be ascribed to the high stabilities of both the CLPy cathode and the Zn anode in 30 m ZnCl2 electrolyte.
We need to point out that the three polypyrenes possess the same chemical component of pyrene, but large difference in the redox activity was observed for the polymers, indicating the linking pattern of these polypyrenes has a great influence on the electrochemical performance. It is well-known that the porous structure with high surface area of the cathode can effectively enhance the contact between cathode and electrolyte, and promote the transportation of charge carriers, leading to a high redox activity and excellent rate performance.28,38 Therefore, the cross-linked polymer CLPy with a high surface area of 1216 m2 g−1 shows a high redox activity. However, the linear polymers of LPy-1 and LPy-2 show a reverse trend since LPy-2 with a lower surface area exhibits a higher redox activity than LPy-1 (Fig. 1e and 2c), suggesting that there are some other structure factors affecting the charge-storage capability apart from porosity.
To further insight into the structure-dependence of the polypyrene-based cathodes, we comparatively studied the electronic structures of the polymers, since the anion-storage behavior of CP-based electrodes originates from their reversible p-type doping/dedoping reactions, thereby electronic structure might be a significant factor determining the redox activity.39,40 The combination of CV curve and UV/Vis spectrum were employed to study the electronic structures of the three polypyrenes (Fig. 1d and Fig. S7, ESI†). According to the frontier molecular orbital theory (FMOT), a high the highest occupied molecular orbital (HOMO) level represents a high electron-donating ability with a high p-doping activity, and a narrow band gap indicates a high conductivity.27,40,41 As shown in Fig. 3a, the HOMO level continuously increases from LPy-1 to LPy-2 and to CLPy, indicating the increased electron-donating ability from LPy-1 to CLPy. The decreased band gap from 2.63 eV for LPy-1 to 2.3 eV for LPy-2 and to 2.08 eV for CLPy also suggests the improved electronic conductivity of the polymers, which promotes the electron transport along the conjugated polymer chain during the p-doping reaction. In addition, the HOMO orbital distribution will also affect the p-doping ability of the polypyrenes since the electrons are first extracted from the HOMO orbital (p-doping) of conjugated polymer cathodes during the charge process according to FMOT.
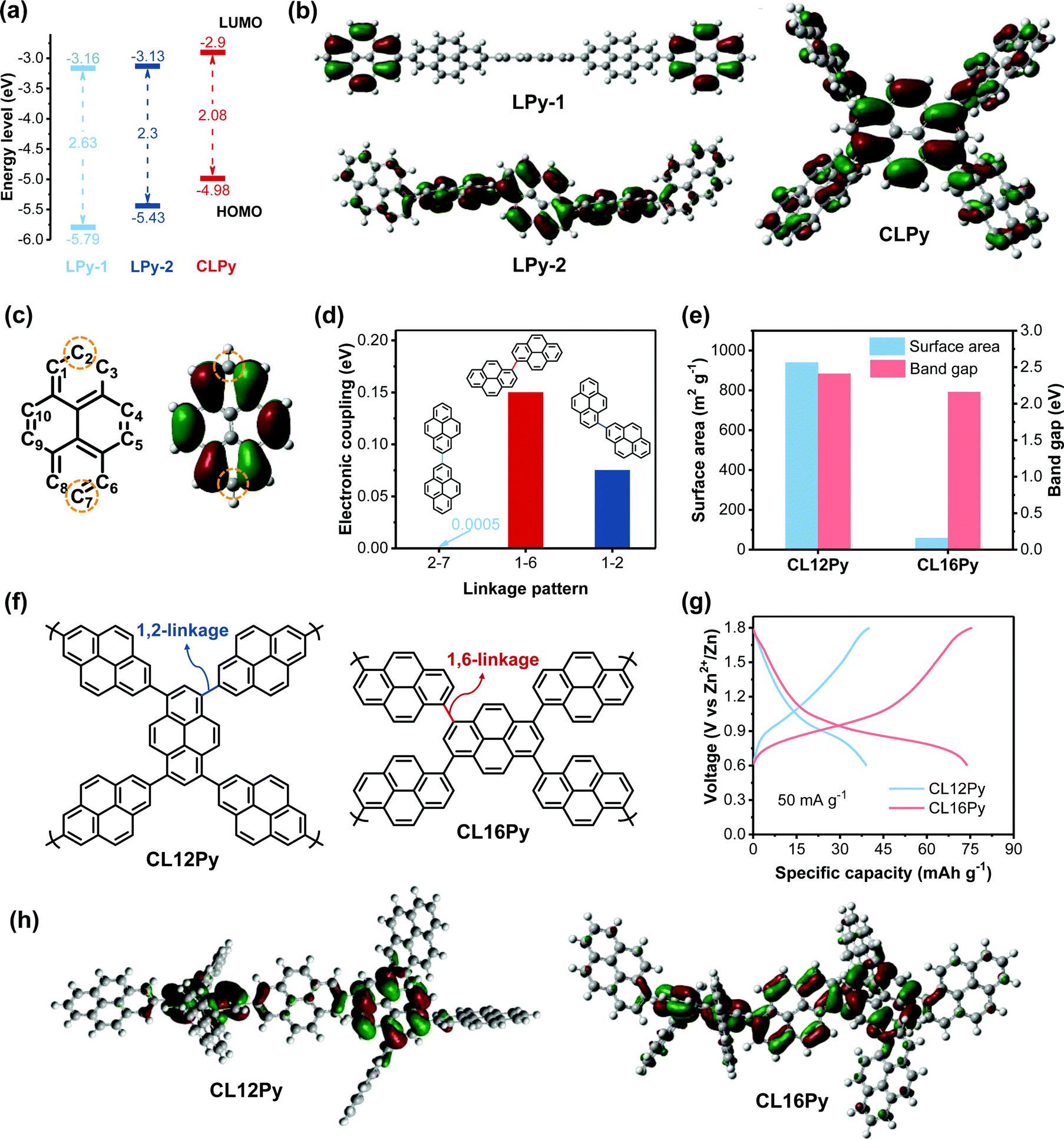 | ||
| Fig. 3 (a) The energy levels and (b) the HOMO diagram of LPy-1, LPy-2 and CLPy. (c) The structure with numbered carbon (left) and the HOMO diagram (right) for the pyrene unit. (d) The electronic coupling for different linking patterns of bipyrene. (e) The surface area and band gaps of CL12Py and CL16Py. (f) The polymer structures of CL12Py and CL16Py. (g) The GCD profiles of CL12Py and CL16Py at 50 mA g−1. (h) The HOMO diagrams of CL12Py and CL16Py. | ||
Density functional theory (DFT) calculation was performed by using Gaussian 09 at the B3LYP/6-311G(d,p) level to simulate the HOMO orbital distribution of the polymers. The simulation results revealed that the HOMO orbital of LPy-1 is mostly distributed on the two separated pyrene units without any overlap, while LPy-2 shows a good overlap of the HOMO orbital spreading on three continuous pyrene units, and the cross-linked CLPy exhibits a further expansion of the HOMO orbital spreading along all the pyrene units (Fig. 3b). The overlap and extension of the HOMO orbital distribution implies the enhanced electron delocalization along the conjugated polymer chain, resulting in the increased HOMO level and decreased band gap from LPy-1 to CLPy.42–45 The different HOMO orbital delocalization can be attributed to the different linkage patterns leading to different orbital overlap degrees. The HOMO diagram of the monomer pyrene showed that the HOMO orbital is distributed on all of the numbered carbon atoms except for C2 and C7 (Fig. 3c). The absence of the HOMO orbital on C2 and C7 leads to the separated HOMO orbital distribution for LPy-1 with 2,7-linkage pattern, as evidenced by the low electronic coupling of 0.0005 eV between C2 and C7 of the two adjacent pyrene units (Fig. 3d), demonstrating that pyrene units are almost electrons isolated in LPy-1.34,43 In contrast, LPy-2 with 1,6-linkage pattern shows a good overlap of the HOMO orbital distribution with enhanced electronic coupling energy of 0.15 eV between C1 and C6 of the two adjacent pyrene units. As for the cross-linked polymer CLPy with 1,3,6,8-linkage pattern, an extended HOMO orbital delocalization was obtained due to the enhanced HOMO orbital overlap from C1, C3, C6 and C8. As a result, CLPy shows a high specific capacity since the high HOMO level, narrow band gaps and the extended HOMO orbital distribution enhance the p-type doping ability of the polymer.
To further verify the above-revealed structure-performance relationships, the other two cross-linked polypyrenes of CL12Py and CL16Py were designed and synthesized from the copolymerization of 1,3,6,8-tetrakis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrene with 2,7-dibromopyrene and 1,6-dibromopyrene, respectively (Fig. 3f). The structure characterizations were shown in Fig. S8 and S9 (ESI†). CL12Py has the 1,2-linkage pattern, while CL16Py possesses 1,6-linkage pattern. As shown in Fig. 3d, the 1,2-linkage pattern shows a higher electronic coupling of 0.075 eV than that of the 2,7-linkage pattern (0.0005 eV) but lower than that of the 1,6-linkage pattern (0.15 eV). Therefore, the electron delocalization can extend over more pyrene repeating units in CL12Py than that in LPy-1 but less than that in CL16Py, which is also supported by the simulated HOMO orbital distribution and the band gap. Compared with CL12Py, CL16Py shows an extended HOMO orbital distribution with a narrower band gap of 2.16 eV (Fig. 3e and h). The band gap of CL16Py is smaller than that of LPy-2 (2.3 eV) but larger than that of CLPy (2.08), indicating that the cross-linking degree also affects the electronic structures besides the linkage pattern. As expected, CL12Py delivers a higher capacity of 38 mA h g−1 than LPy-1 (24 mA h g−1) but much lower than CL16Py (74 mA h g−1), while the capacity of CL16Py lies between LPy-2 (44 mA h g−1) and CLPy (180 mA h g−1) (Fig. 2d, 3g and Fig. S10, ESI†). Although CL16Py shows a much lower surface area of 59 m2 g−1 than CL12Py (940 m2 g−1), it delivers a higher specific capacity of 74 mA h g−1 than CL12Py (38 mA h g−1), which could be attributed to the extended conjugation degree, the expanded HOMO orbital delocalization and the narrower band gap of CL16Py (Fig. 3e–h). These results proved that the electronic structures of polypyrenes could be finely tuned by the linkage pattern and synthetic control, and thus the redox activity of the polymer cathodes for AZDIBs could be efficiently improved by the rational design of the polymer structure. To clearly show the difference in the polymer structures and electrochemical performances of the five developed polypyrenes, we summarized the relevant data in Fig. S11 (ESI†).
The high redox activity of CLPy inspired us to further explore its electrochemical performance. As shown in Fig. 4a and b, CLPy exhibits an excellent rate performance with a slow capacity fading upon the increase in the current rate. Even at a high current rate of 3000 mA g−1, CLPy can still deliver a high reversible capacity of 105 mA h g−1. Remarkably, the CLPy cathode shows an ultra-stable cyclability with a high capacity retention of 96.4% after 38000 charge/discharge cycles at 3000 mA g−1 (Fig. 4c), demonstrating the ultra-stable cyclabilities of the cathode. In fact, such a stable cycling performance represents the state-of-the-art of the cathodes for AZBs. The excellent cycling performance of CLPy should be attributed to its rigid aromatic polymer skeleton and the highly cross-linked porous structure, which render CLPy insoluble nature in electrolyte and provide enough free space and ion diffusion channels for the transport and storage of anions. Additionally, the extended conjugation structure, as discussed above, enhances the delocalization of positive charges (holes) along the 3D polymer chains, which reduces the charge density of active carbon atoms on pyrene unit, leading to high stability for the positively charged polymer chains during the charge–discharge process.46 As a result, highly stable cycling performance was achieved by the CLPy cathode.
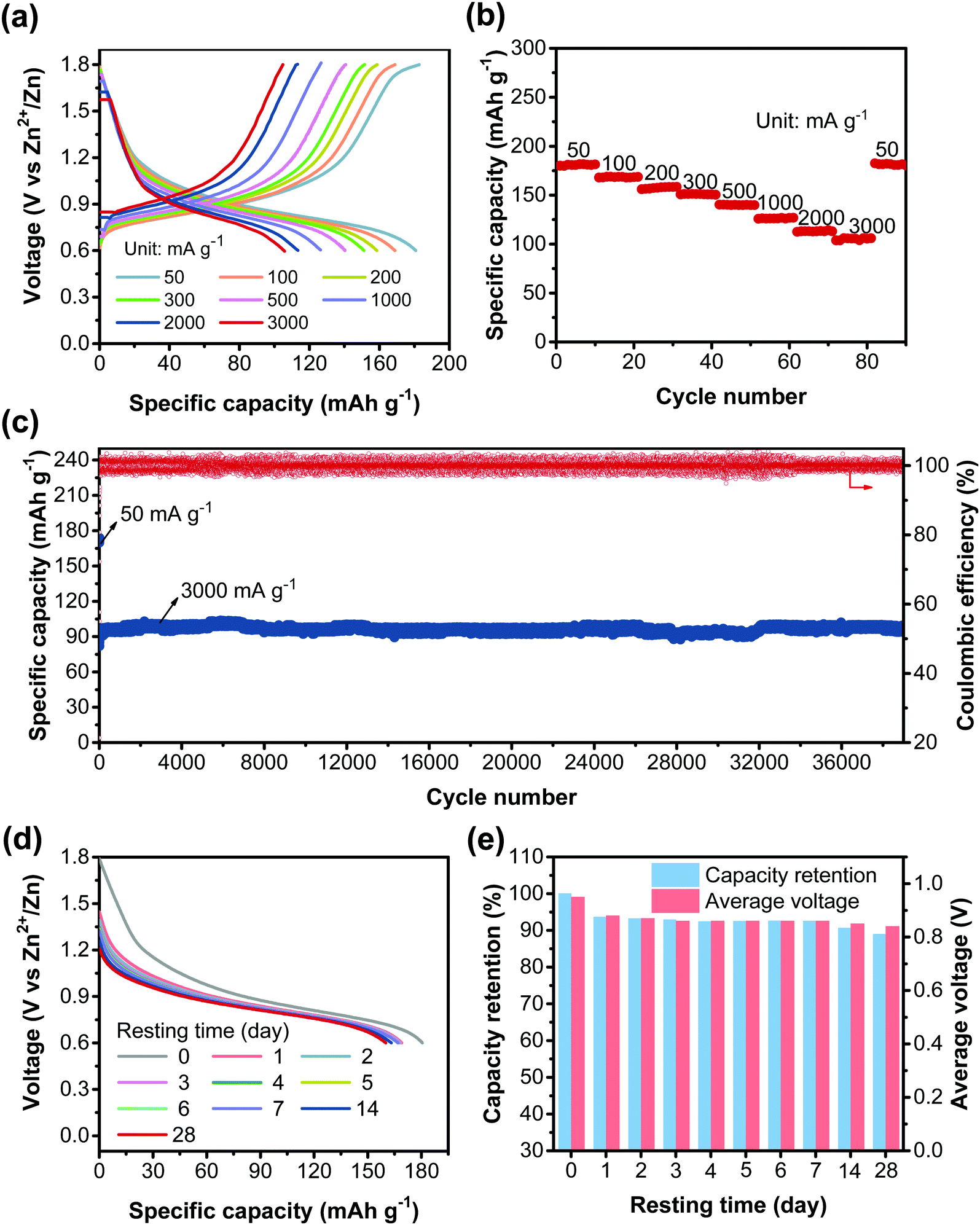 | ||
| Fig. 4 Electrochemical performance of CLPy in AZDIBs. (a) GCD potential curves at different current rates. (b) Rate performance at currents from 50 to 3000 mA g−1. (c) Cycling performance at 3000 mA g−1. (d) Discharge curves after resting different times. (e) The capacity retention and average voltage vs. Zn2+/Zn after resting different times. | ||
As for the real-world applications, the self-discharge performance of the battery is very important, and high self-discharge rate is a showstopper for the application of aqueous batteries.2 Particularly for p-type CP-based cathodes that commonly show poor self-discharge performance with pretty low capacity retention after resting a short time.47,48 Surprisingly, the CLPy cathode exhibits an outstanding self-discharge performance with an ultralow self-discharge rate. It can maintain a high capacity retention of around 90% with an average discharge voltage of 0.84 V after resting the battery for 28 days (Fig. 4d, e and Fig. S12, ESI†). The high capacity retention and energy retention (80%) indicate the ultra-high stability of the CLPy cathode in this AZDIB system.
The charge-storage behavior of the CLPy cathode for AZDIBs was also explored since the cross-linked structure and the high surface area of CLPy might store different anions from the electrolyte. The electrolyte of 30 m ZnCl2 WiSE contains different anion species, in which both Cl− and [ZnCl4]2− have been reported as the charge carriers stored in the cathodes of DIBs.36,49,50 To identify the charge carrier species in the CLPy cathode, EDX measurement was performed. The strong Cl signal was observed, while almost no Zn signal could be detected in the CLPy cathode at the charged state, and the content of Cl decreases at the discharged state (Fig. S13, ESI†). This is also evidenced by the element mapping shown in Fig. 5e and f. The calculated binding energy revealed that Cl− has a higher binding energy with CLPy than [ZnCl4]2− (Fig. 5g and Fig. S14, ESI†), indicating the strong interaction between Cl− and the CLPy cathode. In addition, the [ZnCl4]2− with larger size than Cl− could also inhibit the insertion of [ZnCl4]2− into the pores of the CLPy cathode. Therefore, the charge carrier in the CLPy cathode should be Cl− rather than [ZnCl4]2−. Considering the charge carriers are from the electrolyte in the DIB system, the storage of Cl− enables the battery to generate higher energy density than [ZnCl4]2−. It should be noted that there are very few cathode materials being reported to host Cl− in aqueous batteries (Table S1, ESI†).24,50,51
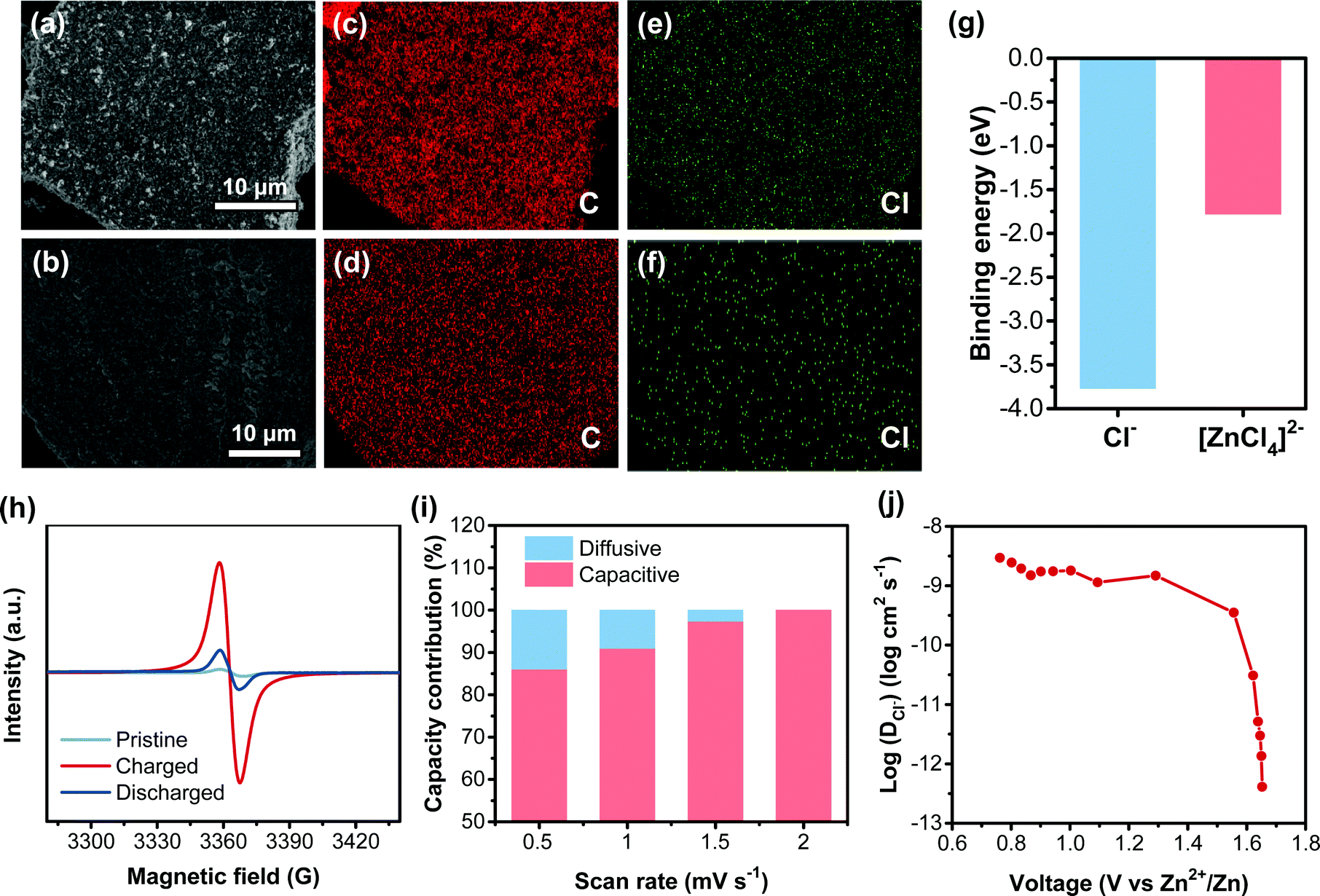 | ||
| Fig. 5 The SEM images of CLPy at cathode (a) charged and (b) discharged states. The elemental mapping of C at (c) charged and (d) discharged states. The elemental mapping of Cl at (e) charged and (f) discharged states. (g) The calculated binding energy of Cl− and [ZnCl4]2− in CLPy. (h) The EPR spectra of CLPy cathode at different states. (i) The capacity composition at different scan rates. (j) The calculated diffusivity of Cl− in CLPy cathode based on GITT. | ||
As for DIBs, it is well known that the electrolyte concentration varies during the charge/discharge processes, because the electrolyte is the only source for the charge carriers of both anions and cations.10 The anions and cations from the electrolyte are stored in the cathode and anode, respectively, during the charge process, which dilutes the electrolyte. The electrolyte concentration will be recovered after the discharge process, since both the stored anions in the cathode and the cations in the anode will be released back to the electrolyte. However, the sharp concentration change may affect the electrochemical performance of the electrode materials for DIBs, especially for the aqueous DIBs.52 Although it is an efficient strategy to stabilize the electrolyte concentration in an appropriate range and thus obtain high battery performance by using excess electrolyte, which commonly leads to a decreased total energy density for a full cell in practical applications.4,53 Accordingly, the high tolerance of the electrode materials on the change of the electrolyte concentration may allow to decrease the usage of electrolyte, thus achieving high energy density for full cells. To evaluate the tolerance of the CLPy cathode on the change of the electrolyte concentration, we directly measured the electrochemical performance of CLPy in ZnCl2 electrolytes with different concentrations. The results showed that the specific capacity of CLPy decreases with diluting the electrolyte concentration from 30 to 15 m (Fig. S15, ESI†), which can be ascribed to the deteriorating contact between the CLPy cathode and the electrolyte (Fig. S16, ESI†). However, it should be noted that CLPy can still deliver a high specific capacity of 140 mA h g−1 with a capacity retention of 92.9% after 500 cycles at 50 mA g−1 in a low ZnCl2 concentration of 15 m, demonstrating a high adaptability of CLPy for high energy density battery systems.
To further study the charge-storage behavior, ex situ electron paramagnetic resonance (EPR) measurement for the CLPy cathode at different states was conducted. The results showed the increased EPR signal for the CLPy cathode at the charged state (1.8 V) (Fig. 5h), which can be attributed to the formation of pyrene radical resulting from the p-type doping reaction of CLPy. Afterwards, the EPR signal becomes very weak at the discharged state (0.6 V), corresponding to the reduction reaction of the positive charged pyrene radical during the discharge process.27,28 Combining the characterization of EDX, EPR and the DFT calculation, the charge-storage process in CLPy is based on the typical reversible p-type doping reaction.54,55 During the charge process, the polypyrene would be oxidized by losing the delocalized π-electron to form positive radical on the conjugated polymer chains. Meanwhile, the anion of Cl− as the charge-compensating ion would be inserted into the CLPy cathode to ensure the electroneutrality of CLPy.54,55 The interaction between the inserted Cl− and the positively charged CLPy might be a metastable chemical bonding.10,56 This is different from electrostatic adsorption in the electrochemical double-layer capacitor, as evidenced by the strong redox peaks in the CV curves (Fig. 2c). During the discharge process, the positive CLPy radical would be reduced to the neutral state by accepting the electron from the Zn anode, and the inserted Cl− would be released back to electrolyte (Fig. 2b).
The Cl− storage kinetic behavior of the CLPy cathode was analyzed by the CV measurement at different scan rates (Fig. S17a, ESI†). According to the equation of i = avb, where i is the current response of CV curves, v represents the scan rate, a and b are parameters.57 The obtained b values for the redox peaks are around 0.9 (Fig. S17b, ESI†), suggesting the storage of Cl− in the CLPy cathode is dominated by a capacitive process since the porous structure and high surface area shorten the diffusion distance of Cl− in the solid-state electrode. The calculated capacity contribution revealed that the capacitive capacity accounts for 86.1% of the total capacity at 0.5 mV s−1, which increased to almost 100% when the scan rate was increased to 2 mV s−1 (Fig. 5i and Fig. S17, ESI†). The galvanostatic intermittent titration technique (GITT) was employed to evaluate the migration rate of Cl− in the CLPy cathode.58,59 The high diffusion coefficient of 9.3 × 10−9–1.3 × 10−12 cm2 s−1 for the storage of Cl− indicates a fast kinetic process (Fig. 5j and Fig. S18, ESI†). Noting that the diffusion coefficient is at the level of 10−9 cm2 s−1 in the voltage range of 0.6–1.6 V, in which CLPy almost achieved 90% charge-storage capacity (160 mA h g−1). This value is comparable with that of Zn2+ in some vanadium oxides, such as Ca0.25V2O5·nH2O (10−8–10−9 cm2 s−1)60 and LiV2O5·nH2O (10−8–10−9 cm2 s−1).61
Conclusions
In summary, we reported a series of polypyrenes as the cathode materials for aqueous Zn dual-ion batteries. The comparative study demonstrated that the electronic structure of the polypyrene cathodes could be finely controlled by the linking pattern. The 1,3,6,8-linking pattern endows the cross-linked polypyrene CLPy with an extended conjugation degree, a highly porous structure, a high HOMO level and the expanded HOMO orbital distribution. As a result of the synergistic effect from these multiple factors, the CLPy cathode for AZDIBs exhibits outstanding electrochemical performances, including high reversible capacity, superior rate capability, stable cyclability, and ultralow self-discharge rate. These results demonstrate that there is a wealth of opportunity to design conjugated porous polymer cathode materials for high-performance AZDIBs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J.-X. Jiang thanks the financial support from National Natural Science Foundation of China (21574077). X. Ji thanks National Science Foundation Award Number: CBET-1551693. C. Zhang is supported by a fellowship from China Scholarship Council (201706870033) and the Fundamental Research Funds for the Central Universities (GK202003045).References
- L. E. Blanc, D. Kundu and L. F. Nazar, Joule, 2020, 4, 771–799 CrossRef CAS.
- X. Ji, Energy Environ. Sci., 2019, 12, 3203–3224 RSC.
- H. Pan, Y. Shao, P. Yan, Y. Cheng, K. S. Han, Z. Nie, C. Wang, J. Yang, X. Li, P. Bhattacharya, K. T. Mueller and J. Liu, Nat. Energy, 2016, 1, 16039 CrossRef CAS.
- F. Wang, O. Borodin, T. Gao, X. Fan, W. Sun, F. Han, A. Faraone, J. A. Dura, K. Xu and C. Wang, Nat. Mater., 2018, 17, 543–549 CrossRef CAS.
- P. He, M. Yan, G. Zhang, R. Sun, L. Chen, Q. An and L. Mai, Adv. Energy Mater., 2017, 7, 1601920 CrossRef.
- F. Wang, E. Hu, W. Sun, T. Gao, X. Ji, X. Fan, F. Han, X.-Q. Yang, K. Xu and C. Wang, Energy Environ. Sci., 2018, 11, 3168–3175 RSC.
- H.-Y. Shi, Y. Song, Z. Qin, C. Li, D. Guo, X.-X. Liu and X. Sun, Angew. Chem., Int. Ed., 2019, 58, 16057–16061 CrossRef CAS.
- L. Zhang, L. Chen, X. Zhou and Z. Liu, Adv. Energy Mater., 2015, 5, 1400930 CrossRef.
- I. A. Rodríguez-Pérez, L. Zhang, J. M. Wrogemann, D. M. Driscoll, M. L. Sushko, K. S. Han, J. L. Fulton, M. H. Engelhard, M. Balasubramanian, V. V. Viswanathan, V. Murugesan, X. Li, D. Reed, V. Sprenkle, M. Winter and T. Placke, Adv. Energy Mater., 2020, 10, 2001256 CrossRef.
- X. Zhou, Q. Liu, C. Jiang, B. Ji, X. Ji, Y. Tang and H.-M. Cheng, Angew. Chem., Int. Ed., 2020, 59, 3802–3832 CrossRef CAS.
- M. Wang and Y. Tang, Adv. Energy Mater., 2018, 8, 1703320 CrossRef.
- S. Rothermel, P. Meister, G. Schmuelling, O. Fromm, H.-W. Meyer, S. Nowak, M. Winter and T. Placke, Energy Environ. Sci., 2014, 7, 3412–3423 RSC.
- K. Beltrop, S. Beuker, A. Heckmann, M. Winter and T. Placke, Energy Environ. Sci., 2017, 10, 2090–2094 RSC.
- J. M. Wrogemann, S. Künne, A. Heckmann, I. A. Rodríguez-Pérez, V. Siozios, B. Yan, J. Li, M. Winter, K. Beltrop and T. Placke, Adv. Energy Mater., 2020, 10, 1902709 CrossRef CAS.
- K. Kim, Q. Guo, L. Tang, L. Zhu, C. Pan, C. Chang, J. Razink, M. M. Lerner, C. Fang and X. Ji, Angew. Chem., Int. Ed., 2020, 59, 19924–19928 CrossRef CAS.
- C. Yang, J. Chen, X. Ji, T. P. Pollard, X. Lü, C. Sun, S. Hou, Q. Liu, C. Liu, T. Qing, Y. Wang, O. Borodin, Y. Ren, K. Xu and C. Wang, Nature, 2019, 569, 245–250 CrossRef CAS.
- S. Muench, A. Wild, C. Friebe, B. Häupler, T. Janoschka and U. S. Schubert, Chem. Rev., 2016, 116, 9438–9484 CrossRef CAS.
- Y. Lu and J. Chen, Nat. Rev. Chem., 2020, 4, 127–142 CrossRef CAS.
- H.-Y. Shi, Y.-J. Ye, K. Liu, Y. Song and X. Sun, Angew. Chem., Int. Ed., 2018, 57, 16359–16363 CrossRef CAS.
- Y. Zhao, Y. Wang, Z. Zhao, J. Zhao, T. Xin, N. Wang and J. Liu, Energy Storage Mater., 2020, 28, 64–72 CrossRef.
- J. Zhang, D. Shan and S. Mu, J. Power Sources, 2006, 161, 685–691 CrossRef CAS.
- C. Chen, Z. Gan, C. Xu, L. Lu, Y. Liu and Y. Gao, Electrochim. Acta, 2017, 252, 226–234 CrossRef CAS.
- P. Li, Z. Fang, Y. Zhang, C. Mo, X. Hu, J. Jian, S. Wang and D. Yu, J. Mater. Chem. A, 2019, 7, 17292–17298 RSC.
- C. Zhijiang and H. Chengwei, J. Power Sources, 2011, 196, 10731–10736 CrossRef.
- B. Häupler, C. Rössel, A. M. Schwenke, J. Winsberg, D. Schmidt, A. Wild and U. S. Schubert, NPG Asia Mater., 2016, 8, e283 CrossRef.
- T. M. Figueira-Duarte and K. Müllen, Chem. Rev., 2011, 111, 7260–7314 CrossRef CAS.
- J. C. Bachman, R. Kavian, D. J. Graham, D. Y. Kim, S. Noda, D. G. Nocera, Y. Shao-Horn and S. W. Lee, Nat. Commun., 2015, 6, 7040 CrossRef.
- M. Walter, K. V. Kravchyk, C. Böfer, R. Widmer and M. V. Kovalenko, Adv. Mater., 2018, 30, 1705644 CrossRef.
- C.-J. Yao, J. Xie, Z. Wu, Z. J. Xu, S. Zhang and Q. Zhang, Chem. – Asian J., 2019, 14, 2210–2214 CrossRef CAS.
- M. Tang, C. Jiang, S. Liu, X. Li, Y. Chen, Y. Wu, J. Ma and C. Wang, Energy Storage Mater., 2020, 27, 35–42 CrossRef.
- S. C. Han, E. G. Bae, H. Lim and M. Pyo, J. Power Sources, 2014, 254, 73–79 CrossRef CAS.
- J.-X. Jiang, A. Trewin, D. J. Adams and A. I. Cooper, Chem. Sci., 2011, 2, 1777–1781 RSC.
- X.-G. Li, Y.-W. Liu, M.-R. Huang, S. Peng, L.-Z. Gong and M. G. Moloney, Chem. – Eur. J., 2010, 16, 4803–4813 CrossRef CAS.
- M. V. Ivanov, K. Thakur, A. Boddeda, D. Wang and R. Rathore, J. Phys. Chem. C, 2017, 121, 9202–9208 CrossRef CAS.
- T. M. Figueira-Duarte, S. C. Simon, M. Wagner, S. I. Druzhinin, K. A. Zachariasse and K. Müllen, Angew. Chem., Int. Ed., 2008, 47, 10175–10178 CrossRef CAS.
- C. Zhang, J. Holoubek, X. Wu, A. Daniyar, L. Zhu, C. Chen, D. P. Leonard, I. A. Rodríguez-Pérez, J.-X. Jiang, C. Fang and X. Ji, Chem. Commun., 2018, 54, 14097–14099 RSC.
- W. Yang, X. Du, J. Zhao, Z. Chen, J. Li, J. Xie, Y. Zhang, Z. Cui, Q. Kong, Z. Zhao, C. Wang, Q. Zhang and G. Cui, Joule, 2020, 4, 1557–1574 CrossRef CAS.
- C. Zhang, Y. He, P. Mu, X. Wang, Q. He, Y. Chen, J. Zeng, F. Wang, Y. Xu and J.-X. Jiang, Adv. Funct. Mater., 2018, 28, 1705432 CrossRef.
- W. Ma, C. Zhang, X. Gao, C. Shu, C. Yan, F. Wang, Y. Chen, J. H. Zeng and J.-X. Jiang, J. Power Sources, 2020, 453, 227868 CrossRef CAS.
- Y. Ding, Y. Li and G. Yu, Chem, 2016, 1, 790–801 CAS.
- Y. Tsuji and R. Hoffmann, Angew. Chem., Int. Ed., 2014, 53, 4093–4097 CrossRef CAS.
- B. G. Kim, X. Ma, C. Chen, Y. Ie, E. W. Coir, H. Hashemi, Y. Aso, P. F. Green, J. Kieffer and J. Kim, Adv. Funct. Mater., 2013, 23, 439–445 CrossRef CAS.
- T. Iwamoto, E. Kayahara, N. Yasuda, T. Suzuki and S. Yamago, Angew. Chem., Int. Ed., 2014, 53, 6430–6434 CrossRef CAS.
- K. Fukui, Science, 1982, 218, 747–754 CrossRef CAS.
- M. V. Ivanov, D. Wang and R. Rathore, J. Am. Chem. Soc., 2018, 140, 4765–4769 CrossRef CAS.
- W. Duan, J. Huang, J. A. Kowalski, I. A. Shkrob, M. Vijayakumar, E. Walter, B. Pan, Z. Yang, J. D. Milshtein, B. Li, C. Liao, Z. Zhang, W. Wang, J. Liu, J. S. Moore, F. R. Brushett, L. Zhang and X. Wei, ACS Energy Lett., 2017, 2, 1156–1161 CrossRef CAS.
- F. Trinidad, J. Electrochem. Soc., 1991, 138, 3186 CrossRef CAS.
- A. Guerfi, J. Trottier, I. Boyano, I. De Meatza, J. A. Blazquez, S. Brewer, K. S. Ryder, A. Vijh and K. Zaghib, J. Power Sources, 2014, 248, 1099–1104 CrossRef CAS.
- Q. Guo, K.-i. Kim, H. Jiang, L. Zhang, C. Zhang, D. Yu, Q. Ni, X. Chang, T. Chen, H. Xia and X. Ji, Adv. Funct. Mater., 2020, 30, 2002825 CrossRef CAS.
- H. Jiang and X. Ji, Carbon Energy, 2020, 2, 437–442 CrossRef CAS.
- K. Koshika, N. Sano, K. Oyaizu and H. Nishide, Macromol. Chem. Phys., 2009, 210, 1989–1995 CrossRef CAS.
- X. Dong, H. Yu, Y. Ma, J. L. Bao, D. G. Truhlar, Y. Wang and Y. Xia, Chem. – Eur. J., 2017, 23, 2560 CrossRef CAS.
- L. Xiang, X. Ou, X. Wang, Z. Zhou, X. Li and Y. Tang, Angew. Chem., Int. Ed., 2020, 59, 17924–17930 CrossRef CAS.
- P. Novak, K. Muller, K. S. V. Santhanam and O. Haas, Chem. Rev., 1997, 97, 207–282 CrossRef CAS.
- P. J. Nigrey, D. M. Jr., D. P. Nairns, A. G. MacDiarmid and A. J. Heeger, J. Electrochem. Soc., 1981, 128, 1651–1654 CrossRef CAS.
- L. Fan, Q. Liu, Z. Xu and B. Lu, ACS Energy Lett., 2017, 2, 1614–1620 CrossRef CAS.
- J. Wang, J. Polleux, J. Lim and B. Dunn, J. Phys. Chem. C, 2007, 111, 14925–14931 CrossRef CAS.
- W. Weppner and R. A. Huggins, J. Solid State Chem., 1977, 22, 297–308 CrossRef CAS.
- P. P. Prosini, M. Lisi, D. Zane and M. Pasquali, Solid State Ionics, 2002, 148, 45–51 CrossRef CAS.
- C. Xia, J. Guo, P. Li, X. Zhang and H. N. Alshareef, Angew. Chem., Int. Ed., 2018, 57, 3943–3948 CrossRef CAS.
- Y. Yang, Y. Tang, G. Fang, L. Shan, J. Guo, W. Zhang, C. Wang, L. Wang, J. Zhou and S. Liang, Energy Environ. Sci., 2018, 11, 3157–3162 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ee03356a |
| This journal is © The Royal Society of Chemistry 2021 |