
The role of atomic carbon in directing electrochemical CO(2) reduction to multicarbon products†
Hongjie
Peng‡
 ab,
Michael T.
Tang‡
bc,
Xinyan
Liu
ab,
Philomena
Schlexer Lamoureux
ab,
Michal
Bajdich
a and
Frank
Abild-Pedersen
*a
ab,
Michael T.
Tang‡
bc,
Xinyan
Liu
ab,
Philomena
Schlexer Lamoureux
ab,
Michal
Bajdich
a and
Frank
Abild-Pedersen
*a
aSUNCAT Center for Interface Science and Catalysis, SLAC National Accelerator Laboratory, California 94025, USA. E-mail: abild@slac.stanford.edu
bSUNCAT Center for Interface Science and Catalysis, Department of Chemical Engineering, Stanford University, California 94305, USA
cDepartment of Material Science and Engineering, Stanford University, California 94305, USA
First published on 23rd November 2020
Abstract
Electrochemical reduction of carbon-dioxide/carbon-monoxide (CO(2)R) to fuels and chemicals presents an attractive approach for sustainable chemical synthesis, but it also poses a serious challenge in catalysis. Understanding the key aspects that guide CO(2)R towards value-added multicarbon (C2+) products is imperative in designing an efficient catalyst. Herein, we identify the critical steps toward C2 products on copper through a combination of energetics from density functional theory and micro-kinetic modeling. We elucidate the importance of atomic carbon in directing C2+ selectivity and how it introduces surface structural sensitivity on copper catalysts. This insight enables us to propose two simple thermodynamic descriptors that effectively identify C2+ selectivity on metal catalysts beyond copper and hence it defines an intelligible protocol to screen for materials that selectively catalyze CO(2) to C2+ products.
Broader contextCarbon dioxide (CO2) is a major by-product of many important reactions that make modern life possible. Still, it also poses as a risk to anthropogenic climate change. Thus, the resolution of how to store or reuse CO2 becomes relevant with ever-increasing energy demand. In lieu of simply sequestering CO2 underground, an alternative strategy is to recycle CO2 back into higher-valued fuels. Some advantages to such an approach is the creation of a neutral carbon cycle, as well as the capability of using carbon-based fuels in areas where batteries are a less practical solution. As the levelized cost of electricity of renewable energy decreases, it raises the question whether we can leverage electrochemistry to convert CO2 into fuels, especially those value-added multicarbon products. As such, fundamental questions regarding the catalytic mechanisms involved in electrochemical CO2 reduction have to be addressed. |
Introduction
The electrochemical reduction of CO2 and CO paves a promising pathway towards sustainable chemicals and fuels.1,2 The catalytic features that drives the generation of high-value multicarbon (C2+) products is of particular interest but despite many years of research it still remains elusive.3 So far elemental copper (Cu) and Cu-based compounds are the only materials that can produce C2(+) hydrocarbons and oxygenates of any significance albeit at high overpotential and with poor selectivity.4 To optimize Cu-based catalysts or find alternative materials for selective C2(+) production from CO(2), in-depth mechanistic insight is needed in order to unravel the complexities of CO(2)R.5Recent experimental efforts have focused on improving the selectivity towards C2(+) products on Cu by tailoring catalyst composition,6–10 the surface morphology,11–14 the reaction conditions at the catalyst/electrode interface,15–18 and by engineering the electrochemical reactors.19–22 To identify key intermediates and tie that into theoretical efforts, in situ or operando characterization tools have been employed,23–25 but the precise mechanism of the first C–C bond formation remains inconclusive. Inspired by an experimentally observed larger shift in onset potential with pH for C2 than single-carbon (C1) products,26–28 theoretical work has concentrated on coupling steps early in the reduction pathway, in particular CO dimerization.29–34 The CO dimerization step is strongly affected by solvation and the electric field present at the electrochemical interface,30,35,36 which introduces extra complexity that hinders descriptor-based materials discovery beyond Cu-based catalysts.10
In this work, we investigate the critical steps of CO reduction (COR) toward C2 products with density functional theory (DFT) based reaction and activation energies and introduce an electrochemical microkinetic model that appropriately describes the experimental trends in activity and selectivity. Our model (exemplified on Cu(100)) identifies a potential, U0, at which the reduction of CO to atomic carbon (C*) via the COH* intermediate exhibits higher rate than both the hydrogen evolution reaction (HER) and the reduction of CO to CHO*. Subsequently, the surface C* enables thermodynamically favorable coupling with CO at the interface. In comparison with other pathways including CO dimerization, this process is found to be the dominant C2 pathway at more reducing potentials, i.e., U < −0.5 V vs. the reversible hydrogen electrode (RHE) at pH = 7. This enables a characterization of the C2 selectivity relative to the C1 selectivity through the energetic difference between barriers for CCO* and CH* formation, which further rationalizes the facet dependency of C2 selectivity on Cu. Finally, this insight allows us to identify two simple descriptors that traces the C2 selectivity on different metal surfaces at varying potentials: the adsorption free energies of CO* and C* (GCO* and GC*).
Results and discussion
Reaction pathways for COR
In this paper, we evaluate the CO2R reaction to C1 and C2(+) products involving more than two proton-electron transfer steps with water as the proton donor. It has been verified that CO* is the key intermediate in COR and CO2R leading to further reduced products,4,26 we therefore focus on CO as the initial reactant. Since Cu is the only catalyst with significant C2(+) production from CO(2) and its (100) surface has been identified as the major exposed facet under reaction conditions,12,37 we select Cu(100) as the model surface. All the computational details and the simplifications of the electrochemical models are shown in Supplementary Note with corresponding data and justification present in Fig. S1–S6 and Tables S1–S9 (ESI†).Fig. 1 shows the most relevant reactions considered in this work. Clearly, CO* is initially reduced to either CHO* or COH*. In a recent study,38 we showed that the formation of CHO* is a chemical step preceded by surface hydrogenation whereas COH* is formed through an electrochemical reduction step. CH* leading to CH4 forms as an intermediate in both pathways, either via COH reduction to C* + H2O or through CHO reduction to CHOH*. A number of intermediates present in the CH4 pathway are considered as seeds for C2(+) production: CO* dimerization to OCCO* and subsequent reduction to OCCOH* (OCCOH pathway), CO* coupling with C* (OC–C pathway), CO* coupling with CH* (OC–CH pathway), and CH2* dimerization to ethylene (C2H4) (CH2–CH2 pathway). In accordance with previous studies, we only consider OCCOH* as the reduced product of OCCO*.29,32,33,35 Potential coupling reactions of CO with either CHO* or COH* are compared with the OCCOH pathway in the next section. The above considered C2 pathways except for the CH2 dimerization lead to CHCO* and based on previous thermodynamic analyses,29 all subsequent reaction steps are assumed to be downhill in energy.
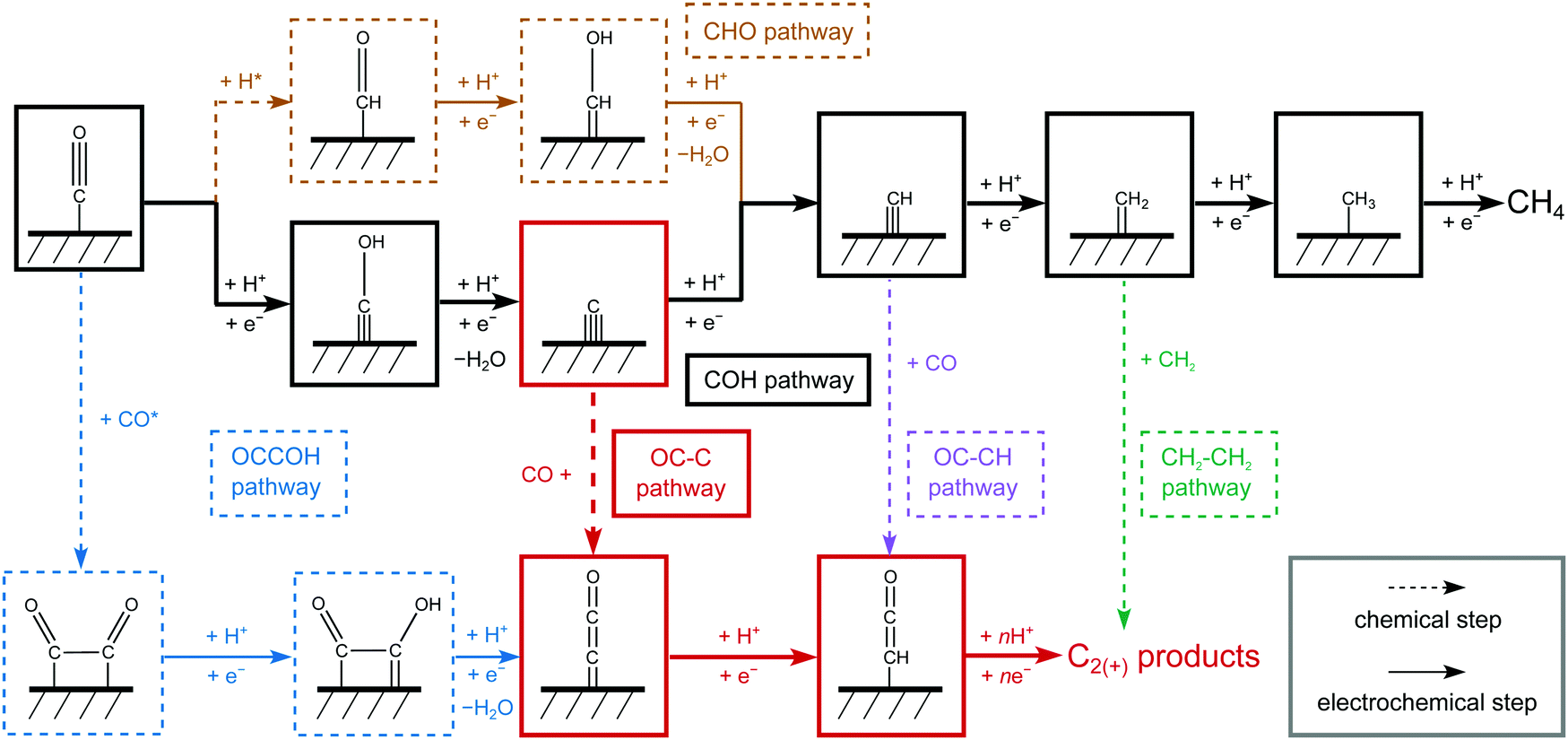 | ||
| Fig. 1 Schematic diagram of reaction steps beyond CO. Pathways toward C1 (CH4 as the main product) and C2 products beyond CO are shown as different colored branches: yellow (CHO pathway), black (COH pathway), blue (OCCOH pathway), red (OC–C pathway), violet (OC–CH pathway), and green (CH2–CH2 pathway). The number of involved reduction steps are increasing from left to right. | ||
Fig. 2a–e depicts the Gibbs free energetics of competing pathways, as proposed in Fig. 1, at two applied potentials vs. RHE on Cu(100). URHE = −0.13 V was chosen as it corresponds to the zero-charge potential of Cu(100) at pH = 7, showing the energetics un-affected by fields; while URHE = −0.73 V was chosen as it is the value at which substantial C2(+) products begin forming on Cu in neutral-pH CO(2)R.26,27,39 Cation-induced interfacial fields have been suggested to significantly stabilize C2 species like OCCO*.13,30,36 Given these previous insights, we have built a simple model (see details in Supplementary Note 4, ESI†) to correlate the field-affected energetics to the potentials vs. the standard hydrogen electrode (SHE), which we denote as USHE. For all chemical steps the adsorption energies at URHE = 0 V and barriers were calculated in vacuum and all electrochemical barriers were obtained using an explicit solvent model combined with the charge-extrapolation method.40 Free-energy and solvation corrections were applied to adsorption energies for all important intermediate species (Tables S3 and S4, ESI†). The relevant transition state (TS) structures are shown in Fig. 2f.
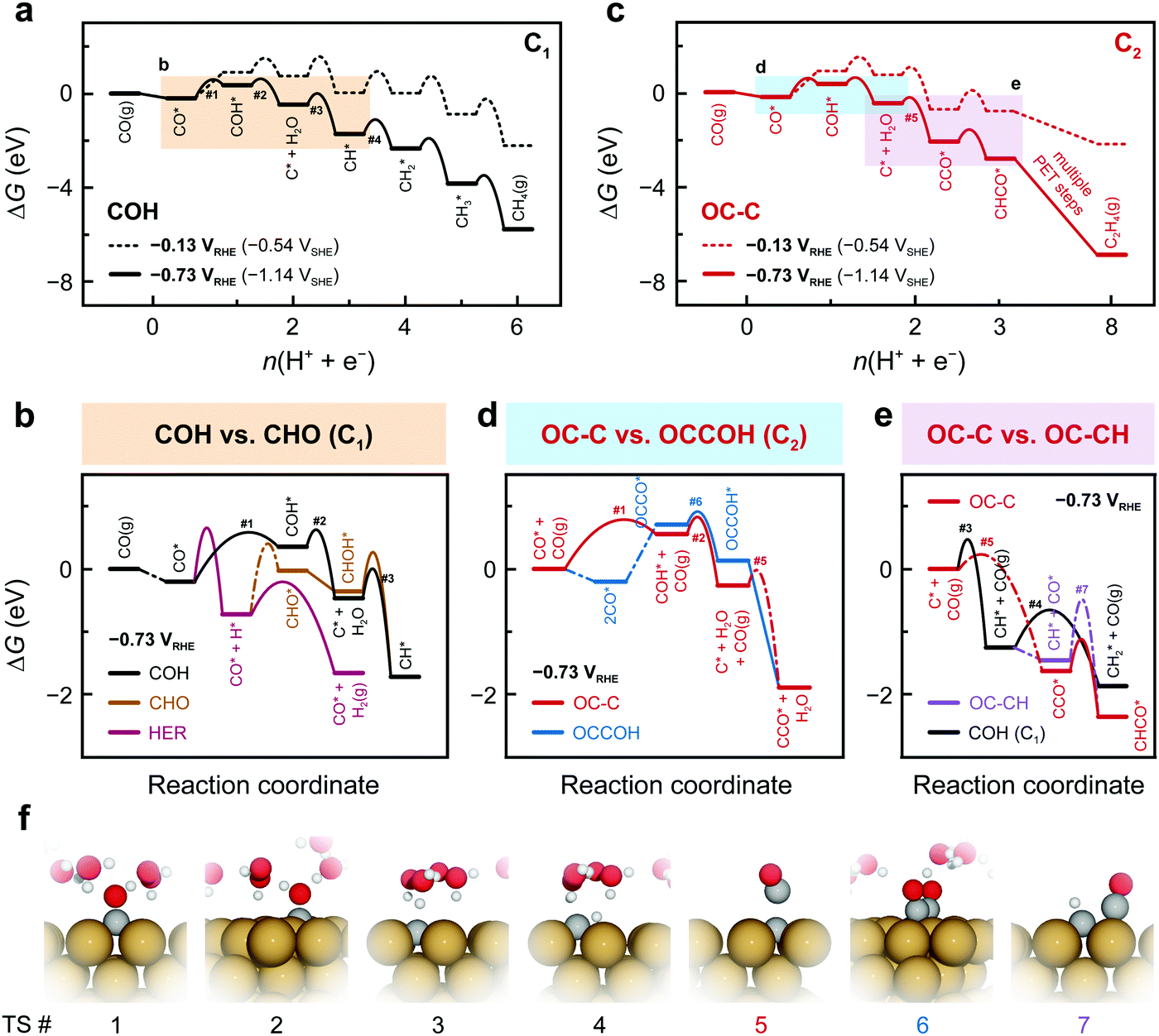 | ||
| Fig. 2 Free energy diagrams (FEDs) of COR on Cu(100) at pH 7. FEDs at potentials URHE = −0.13 V and URHE = −0.73 V, showing (a) the COH pathway toward CH4 and (c) the OC–C pathway toward C2 pathway where C2H4 is used as the representative product. The colored squares highlights the major steps competing with other pathways at potential URHE = −0.73 V: (b) COH (black), CHO (brown), and HER (wine); (d) OC–C (red) and OCCOH (blue); (e) OC–CH (violet) and COH toward C1 (black). Note that the scale of the x-axes in (a) and (c) are non-uniform due to the presence of chemical steps. These chemical steps are shown as the dash-dotted lines in (b, d, and e). (f) TS structures of key elementary steps with the indexes indicated in (a)–(e): 1. CO–H to COH*, (2) COH–H to C* + H2O, (3) C–H to CH*, (4) CH–H to CH2*, (5) C–CO coupling, (6) OCCO–H to OCCOH*, and (7) OC–CH coupling. | ||
At low overpotentials, the formation of C* + H2O through sequential CO–H and COH–H protonation is identified to control the overall rate of the COH pathway (Fig. 2a). With increasing overpotential, the CO–H protonation is shown as the rate-determining step (RDS) with a lower barrier than both the HER and the CHO pathway (Fig. 2b) and thus the COH pathway is more favorable within a wide potential window (Fig. S7, ESI†). Therefore, C* becomes available on the surface under these conditions, opening up pathways leading to C1 products through further protonation (Fig. 2a) or C2 products via coupling with CO (Fig. 2c). On Cu(100) CO binds at least −1.25 eV stronger to C* than the surface throughout the common potential range (Fig. S8, ESI†). In addition, C* coupling to gas-phase CO possesses a barrier of 0.31 eV, which is lower than the 0.73 eV for the surface mediated coupling (Fig. S9, ESI†). The energetics agrees well with observed facile low-temperature CO dissociation on Cu induced by C–CO coupling.41 With an additional stabilization of C–CO TS by the interfacial field, the CCO formation from C* is more favorable at low overpotentials than the CH formation, thus resolving the earlier onset potentials for C2 than for C1 in CO(2)R.26,27,39
As mentioned in Fig. 1, several possible competing carbon-coupling pathways are considered. The rate of the OCCOH pathway is identified to be largely controlled by CO dimerization and subsequent protonation (Fig. S10, ESI†). Despite the significant field stabilization of OCCO* and OCCOH*, this conventional pathway is found to possess higher activation energies than the COH/OC–C pathway, thereby being less dominant at sufficiently negative potentials (Fig. 2d). In addition, the OCCOH pathway is found to be more predominant than other coupling reactions early in the reduction pathway, such as OC–COH and OC–CHO coupling (Fig. S11, ESI†). Owing to the higher coupling barriers than the protonation barriers at negative potentials, the OC–CH and CH2 dimerization pathways cannot compete with their protonation counterparts of CH–H and CH2–H, respectively (Fig. 2e and Fig. S12, ESI†).
Microkinetic model of COR
To further illustrate the role of C* as a potential bifurcating intermediate for C1/C2+ products, we have developed a mean-field microkinetic model that consider adsorbate–adsorbate interactions.35 Given the intrinsic DFT errors (±0.15 eV) and the uncertainties brought by the parameterization and the variations in the solvent structure, the microkinetic model only serves as a tool for qualitative comparison with experimental trends in activity and selectivity.Fig. 3a shows the product distribution from the microkinetic model at bulk pH = 7 across a wide range of potentials vs. RHE. The distribution aligns well with the trends seen in experiments.27 The partial contributions from each pathway as shown in Fig. S13 (ESI†) is a reflection of the free energetics shown in the last section, identifying the COH pathway and the OC–C pathway as the two dominant pathways that lead to C1 and C2 products, respectively.
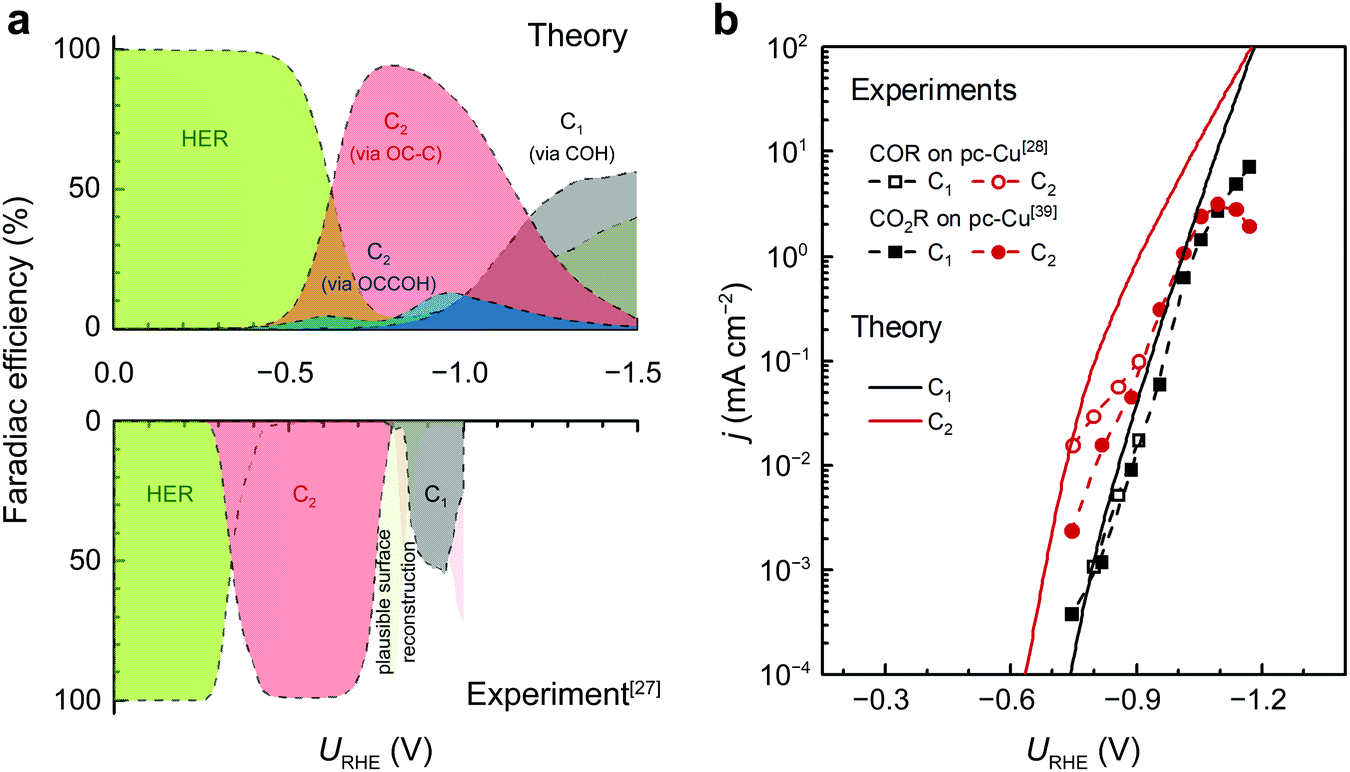 | ||
| Fig. 3 Product distributions and polarization curves of CO(2)R on Cu(100) (bulk pH7). (a) Theoretical (top) and experimental (bottom) distributions of products. The experimental product distribution for COR on a Cu(100) single-crystal electrode was from Koper et al.27 The HER and C2 activity at URHE < −0.7 V is hypothesized to result from potential-driven surface reconstruction.27 (b) The comparison between theoretical COR and experimental CO(2)R polarization curves. The experimental curves for COR and CO2R are obtained on pc-Cu from ref. 28 and 39, respectively. | ||
Further comparison with the experimental polarization curves of COR and CO2R on polycrystalline Cu (pc-Cu) electrodes underscores the ability of our proposed model to accurately predict the potential-dependent variations in activity and selectivity (Fig. 3b). The observed experimental downward trend of C2 formation seen in Fig. 3b can be attributed to the CO depletion induced by mass transport limitation.35
We note that under steady-state conditions, CO* is the major surface species (Fig. S14, ESI†). Therefore, stronger CO* adsorption on step sites combined with a low step formation energy on Cu could drive possible surface reconstruction of Cu(100).42 Nevertheless, a comparison of our microkinetic model with previously established ones on stepped surfaces35,43 show similar accuracy in describing the CO(2)R trends on pc-Cu. While the C* intermediate has been proposed previously,26,31,44 none of the previous models have revealed the particular OC–C coupling mechanism leading to C2+.
pH effects
To understand the observed correlations between C2+ selectivity and bulk pH, we also consider COR under experimental alkaline conditions (0.1 M KOH, pHbulk = 13;18,27,28 and 5 M KOH, pHbulk = 14.722). Our model shows that the dominant region for the OCCOH mechanism expands on the USHE scale with increasing alkalinity (Fig. 4a). Previous understanding of the alkaline protonation process revealed that electrochemical barriers are dependent on the USHE while reaction energies are URHE-dependent.35 Therefore, the potential required to drive C* formation shifts more negative with increasing pHbulk due to the additional reduction step needed than for OCCOH* formation. A detailed degree-of-rate-control analysis45 also reveals such a transition in the dominant reaction pathways (Fig. S15, ESI†). In short, from a neutral pHbulk to an alkaline pHbulk of around 13, we suggest that the experimentally observed large shifts in pH of C2+ activity/selectivity in Fig. 4b and c can be rationalized by the OCCOH pathway being dominant at low overpotentials, whereas the COH/OC–C pathway only become relevant at high overpotential.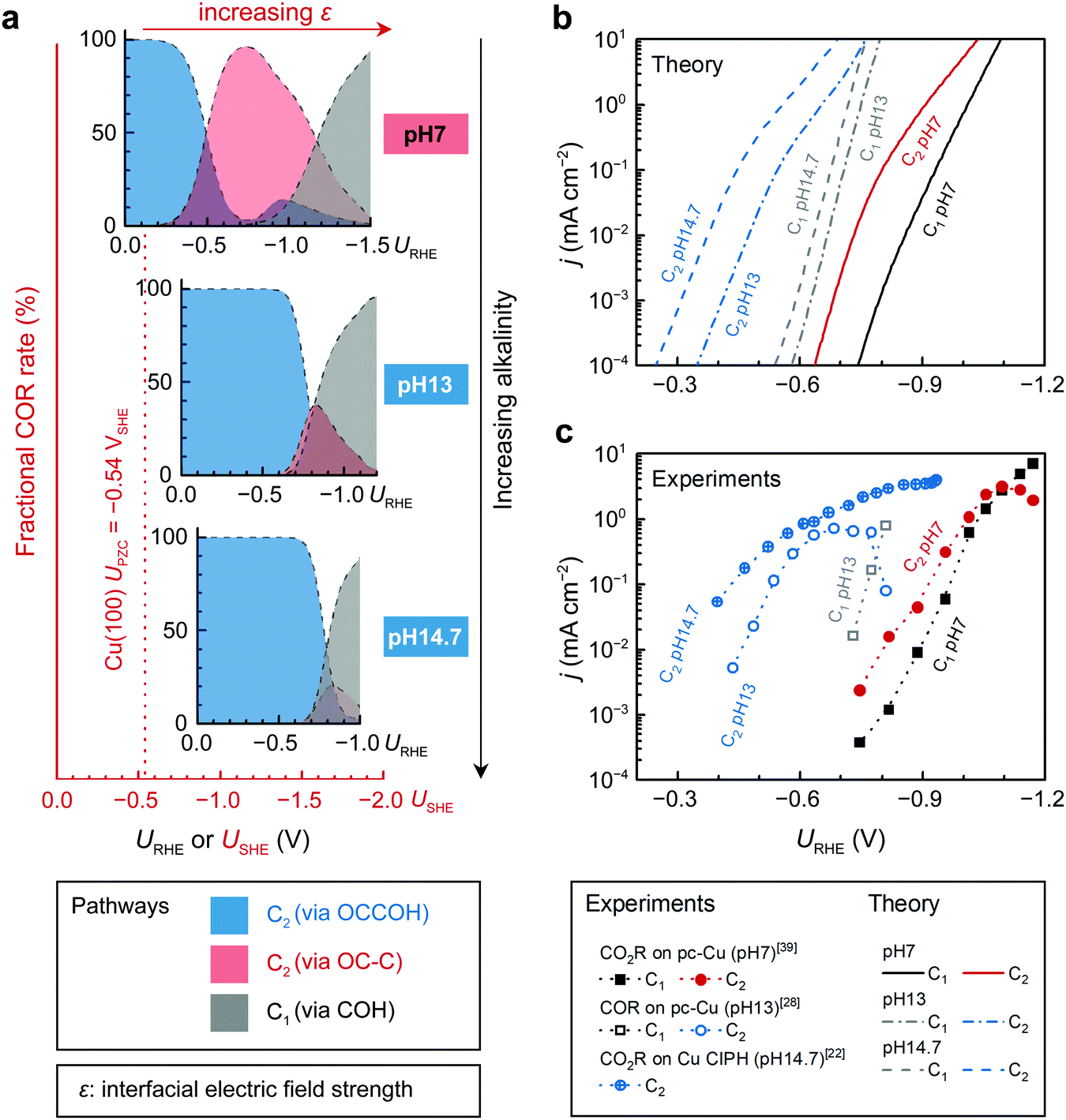 | ||
| Fig. 4 Product distributions and polarization curves of CO(2)R on Cu(100) under different bulk pH (pHbulk) conditions. (a) Fractional COR rates (by normalizing to the total COR rate) from three pathways: OCCOH to C2 (blue), OC–C to C2 (red), and COH to C1 (gray). The pHbulk increases from top to the bottom. (b) Theoretical polarization curves. (c) Experimental polarization curves of COR and CO2R are obtained on pc-Cu from ref. 39 (0.1 M KOH), ref. 28 (0.1 M KHCO3), and on Cu catalyst-ionomer planar heterojunction (CIPH) from ref. 22 (5 M KOH), respectively. Note that the current densities obtained from ref. 22 were normalized to electrochemical specific area. | ||
Recent developments in CO(2)R reactors has promoted CO(2)R current densities to 102–103 mA cm−2, which generate a high local pH of around 10–11.1,14,19–22 To show the reliability of our model for such systems, we also simulated COR at a pHbulk of 7 and local pH (pHinterface) of 9 and 11 according to recent efforts in modeling14,46 and characterization.25 Due to the independency of absolute potential at the working electrode on local pH, the high local pH only destabilizes the final state of each reduction step by 0.059ΔpH at 298 K (Fig. S16a, ESI†). As long as the RDS is the first protonation step (CO–H and OCCO–H protonation identified in Fig. S15, ESI†), the reaction rate remains the same. Therefore, similar to the change in pHbulk, the increased local alkalinity only results in more negative potentials required to switch the dominant C2 reaction pathways from the OCCOH to the OC–C (Fig. S16b and c, ESI†); whereas the overall trends of COR within the potential window of interest (usually URHE < −0.8 V to obtain a sufficiently high pHinterface of > 9 in neutral electrolytes) still reveals a dominance of the OC–C pathway for C2 production and hence consistent with the neutral-pHbulk model (Fig. S17, ESI†). The observed dependency of C2+ selectivity on the pHinterface could also be attributed to different CO2 concentrations.15,25 Regardless of local pH effects, C* becomes the key intermediate that directs C1 and C2 selectivity on Cu below a certain potential.
Through the above analysis, we clarify that the proposed OC–C pathway is not in conflict with the CO dimerization pathway at low overpotentials. In fact, both the OC–C and CO dimerization steps are important for the mechanistic understanding and the development of practical solutions. The local-field-dependency on C2+ selectivity, rationalized by the electric field effect on the CO dimerization barrier, still holds in the OC–C pathway due to the field stabilization to C–COTS.15,16 Engineering parameters in our field model such as the zero-charge potential of the metal electrode and the thickness of electrochemcial double layer could be feasible strategies to increase the C2 selectivity.36 In brief, by combining competing reaction pathways and specific pH/interface conditions our model provides reasonable agreement with experimental observation and it shows the importance of including the OC–C pathway in the overall CO(2)R mechanism.
Facet dependent selectivity of C2 on Cu
Our approach and microkinetic modeling enables us to identify four key reaction steps that determines the CO(2)R activity and selectivity:(i) CO* (or CO(g) + *) + H+ + e− → COH* (CO–H protonation)
(ii) CO* (or CO(g) + *) + 2(H+ + e−) → C* +H2O (C formation)
(iii) CO(g) (or CO*) + C* → CCO* (C–CO coupling)
(iv) C* + H+ + e− → CH* (C–H protonation)
Here reaction (i) and (ii) determine the overall rate which explicitly accounts for the shift in RDS with applied potential. The competition between reaction (iii) and (iv) determine the selectivity.
Based on our microkinetic model and a simple quasi-equilibrium assumption, surface C* will become accessible at a certain potential, U0, defined by the condition; ΔG(ii)rxn = 0. According to the computational hydrogen electrode model,48U0 is defined as (GC* − GCO* (or GCO(g)))/(−2e). When U < U0, the forward rates of reaction (iii) and reaction (iv) are given by:
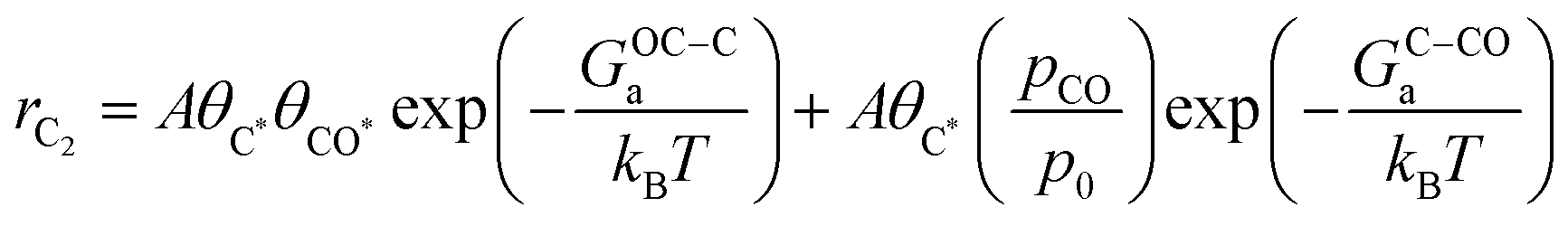
where A is a pre-exponential factor, kB Boltzmann's constant, T the absolute temperature, θC* and θCO* are surface coverages of C* and CO*, respectively, (pCO/p0) is the partial pressure of gas-phase CO, and GOC–Ca, GC–COa, and GC–Ha are forward activation energies of OC–C surface coupling, C–CO gas-phase coupling, and C–H protonation at URHE = 0 V, respectively. The charge transfer coefficients for the C–H protonation step, β, vary from 0.45 to 0.60 depending on the surface orientation (see Table 1). Since both ΔG(iii)rxn and ΔG(iv)rxn are considerably downhill in energy when U < U0, only the forward rates are taken into account. According to a previous analysis,49 we only regard molecular water as the proton donor within the relevant pH range (pH > 4), at which [H2O] = 1 is a reasonable assumption. In this study, all facets Cu(100), Cu(111), Cu(511), Cu(310), except for Cu(211) exhibit much larger GOC–Ca than GC–COa (see Table 1), hence the expression for the C2 rate is simplified when omitting the first term such that:
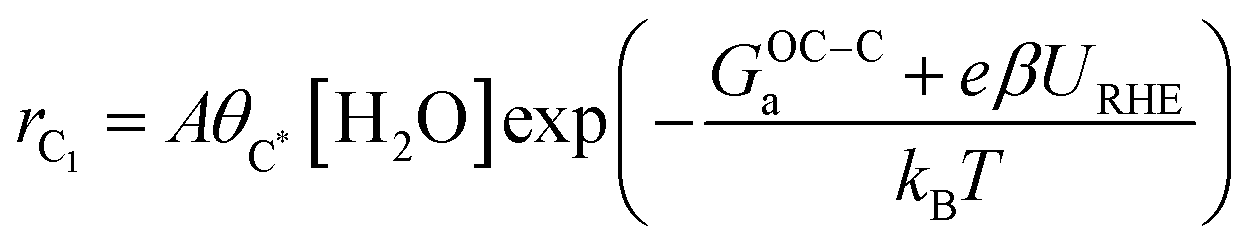
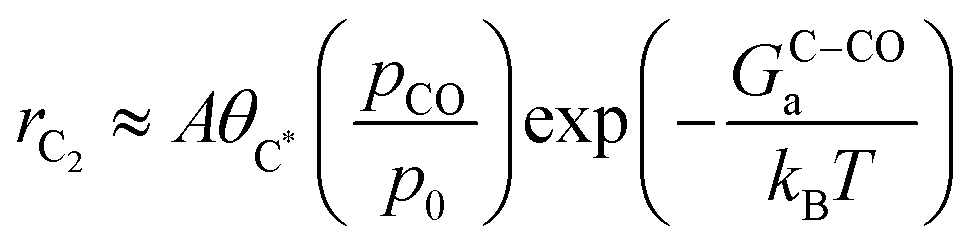
| Cu facets | Site ensembles | G OC–Ca (eV) | G C–COa (eV) | G C–Ha (eV) | β C–H |
|---|---|---|---|---|---|
| Note: the OC–C barrier calculation on Cu(111) and C–CO barrier calculation on Cu(211) automatically relax to the C–CO and OC–C mechanism, respectively. For simplicity, the energetics considered for facet dependency are without field correction. | |||||
| (100) | (8-8-8-8) | 0.73 | 0.31 | 0.91 | 0.60 |
| (111) | (9-9-9) | — | 0.15 | 0.63 | 0.45 |
| (211) | (7-7-10-10-9′) | 0.46 | — | 0.80 | 0.50 |
| (511) | (7-7-8-8) | 0.49 | 0.27 | 0.84 | 0.50 |
| (310) | (6-8-8-9) | 0.69 | 0.30 | 0.85 | 0.50 |
If we assume similar pre-exponential factors for the two reaction steps, the selectivity of C2 over C1 can be expressed as:
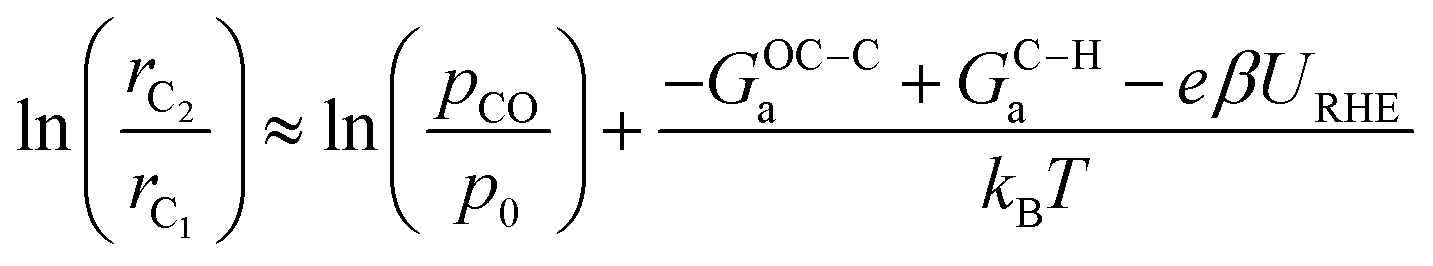
This result enables a quantitative assessment of the slope and intercept of ln(rC2/rC1) on a certain type of active site as well as the mapping of selectivity as a function of potential (Fig. 5a). Note that CO partial pressure is assumed to be 0.05 bar for all Cu facets, the same as what is applied during the microkinetic modeling for consistency. The accurate determination of the CO partial pressure at the reaction interface requires additional multiscale simulations explicitly considering mass transport. The typical site ensembles of different Cu facets are shown in Fig. 5b based on their local coordination number (CN).
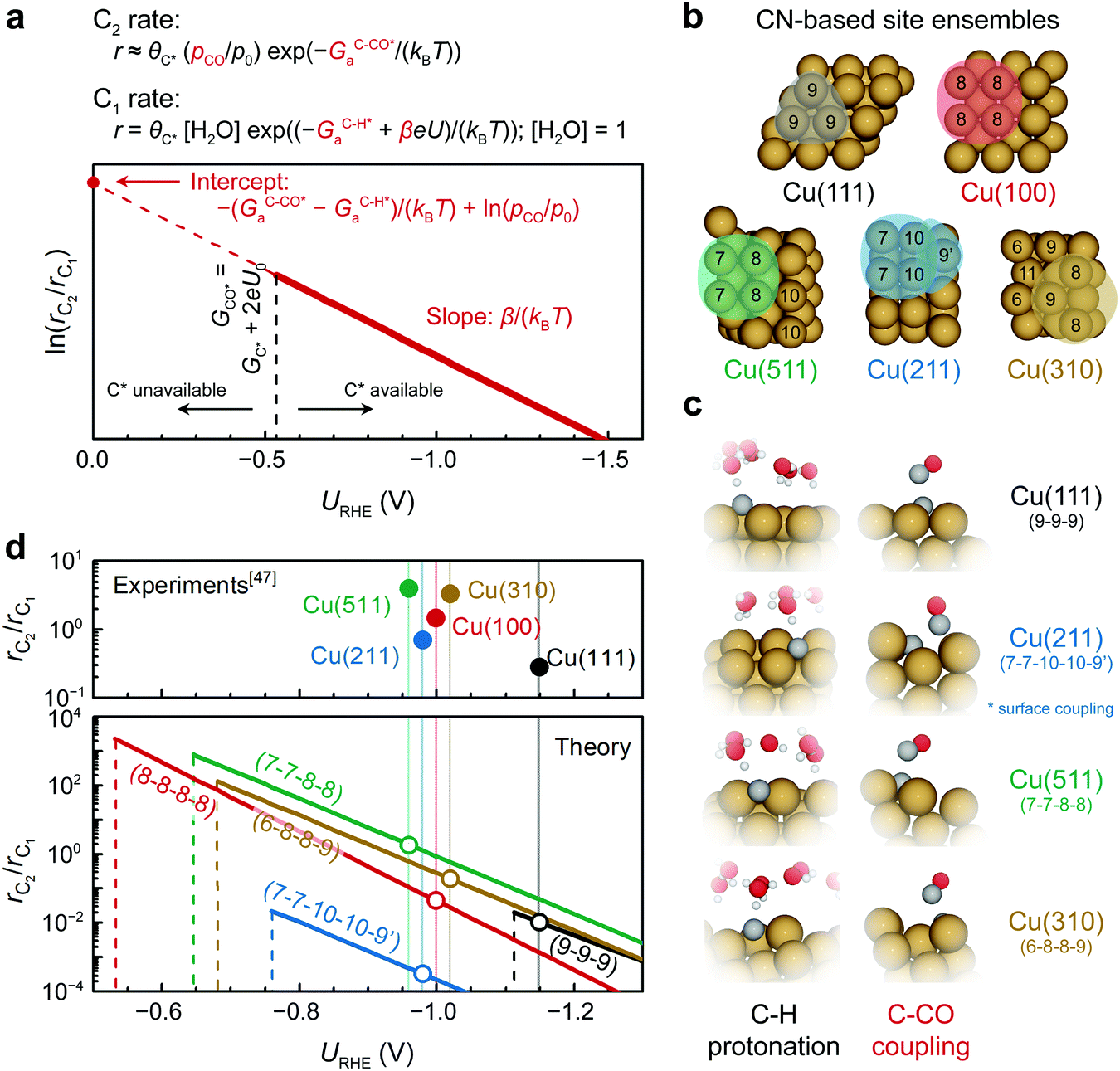 | ||
Fig. 5 Facet dependent selectivity of C2 on Cu (pH7). (a) Schematic illustration of the potential-dependent C2 selectivity over a certain facet, showing the expression for onset potential U0, slope, and intercept. (b) Site ensembles defined by their local CNs on (111), (211), (511), (100), and (310) facets of Cu. (c) TS structures of C–H protonation and C–CO coupling on the site ensembles shown in b. (d) Potential-dependent C2 selectivity on the representative site ensemble of (111), (211), (511), (100), and (310) facets of Cu. Vertical dash lines indicate the onset potential U0 for CO protonation and hence the formation of C* on the surface. The experimental numbers for rC1/rC2 are obtained from ref. 47 on various single-crystal Cu electrodes. The light vertical lines indicate the experimental applied potential for long-term electrolysis experiments. (* On Cu(211), CO* couples with C* via a surface mediated mechanism due to a geometric constraint; thus, the intercept in (a) is expressed as 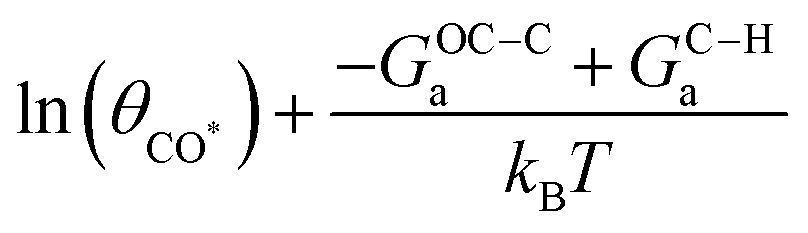 for Cu(211). 9′ in the site ensemble of 7-7-10-10-9′ represents the binding site of CO*.). for Cu(211). 9′ in the site ensemble of 7-7-10-10-9′ represents the binding site of CO*.). | ||
Since the TS structures shown in Fig. 5c are very similar on different site ensembles, the above expression possesses certain generality. Fig. 5d depicts the theoretical trends in C2 selectivity on different Cu site ensembles. Typical four-fold hollow sites such as (8-8-8-8), (7-7-8-8), and (6-8-8-9) are very selective towards C2 at low overpotentials (−0.7 to −0.9 V). The (7-7-10-10-9′) site is seen to have a higher barrier, GOC–Ca, compared to the other facets and therefore it is less selective, whereas the typical three-fold hollow site of (9-9-9) possesses a low GC–COa which automatically result in a favored C2 selectivity. However, the instability of C* limits the (9-9-9) site, which results in a much lower U0 compared to the other site ensembles and thus an inferior C2-selectivity. Due to the weak binding strength of C* on Cu(111) an additional C1 pathway becomes possible in which the protonation of COH* to CHOH* is favored over C* formation thus providing an increased C1 activity on the Cu(111) surface and an overall lower experimental rC2/rC1.32 In general, the observed facet dependency is rationalized by the greater stabilization of C* on Cu (100)-like sites. This characteristic four-fold geometry combined with the unique electronic structures of Cu is what causes the favored selectivity of C2 over C1 products. These trends are in good agreement with experimentally observed facet dependencies by Hori et al.,47 demonstrating the ability of our model to describe structure sensitivity for CO(2)R catalysts.
The facet dependency insight achieved through the simple expression of ln(rC2/rC1) above, also offers several strategies to enhance the C2+ selectivity by tuning the effective CO pressure/concentration, including system pressurization,28 CO/CO2 co-feeding,50 and doing the electrocatalysis in tandem.9,50
Selectivity maps with GCO* and GC* as descriptors
Selectivity trends can be qualitatively revealed through energetic analysis of reactions (i)–(iv). Assuming that the reaction energies are adequately described through scaling by GCO* and GC* (Fig. S18, ESI†), we can form a selectivity map across various metals. We also highlight the importance of four-fold sites in stabilizing C* by including the (100), (111), (211) facets on close-packed metals. Similar maps using binding strengths of CO and OH as descriptors for selectivity towards C1 products have been introduced recently.38As shown in Fig. 6, the selectivity map uses simple thermodynamic conditions based on the reaction energies (ΔGrxn) for reactions (i)–(iv):
| CO reduction to COH* at an acceptable rate (TOF 1 s−1 site−1 at 300 K): ΔG(i)rxn < 0.75 eV | (1) |
| CO reduction to C* is kinetically accessible: ΔG(ii)rxn = 0 | (2) |
| C–CO coupling should be more favorable than CO adsorption: ΔG(iii)rxn < GCO* | (3) |
| C–CO coupling should be more favorable than C–H protonation: ΔG(iv)rxn < ΔG(iii)rxn | (4) |
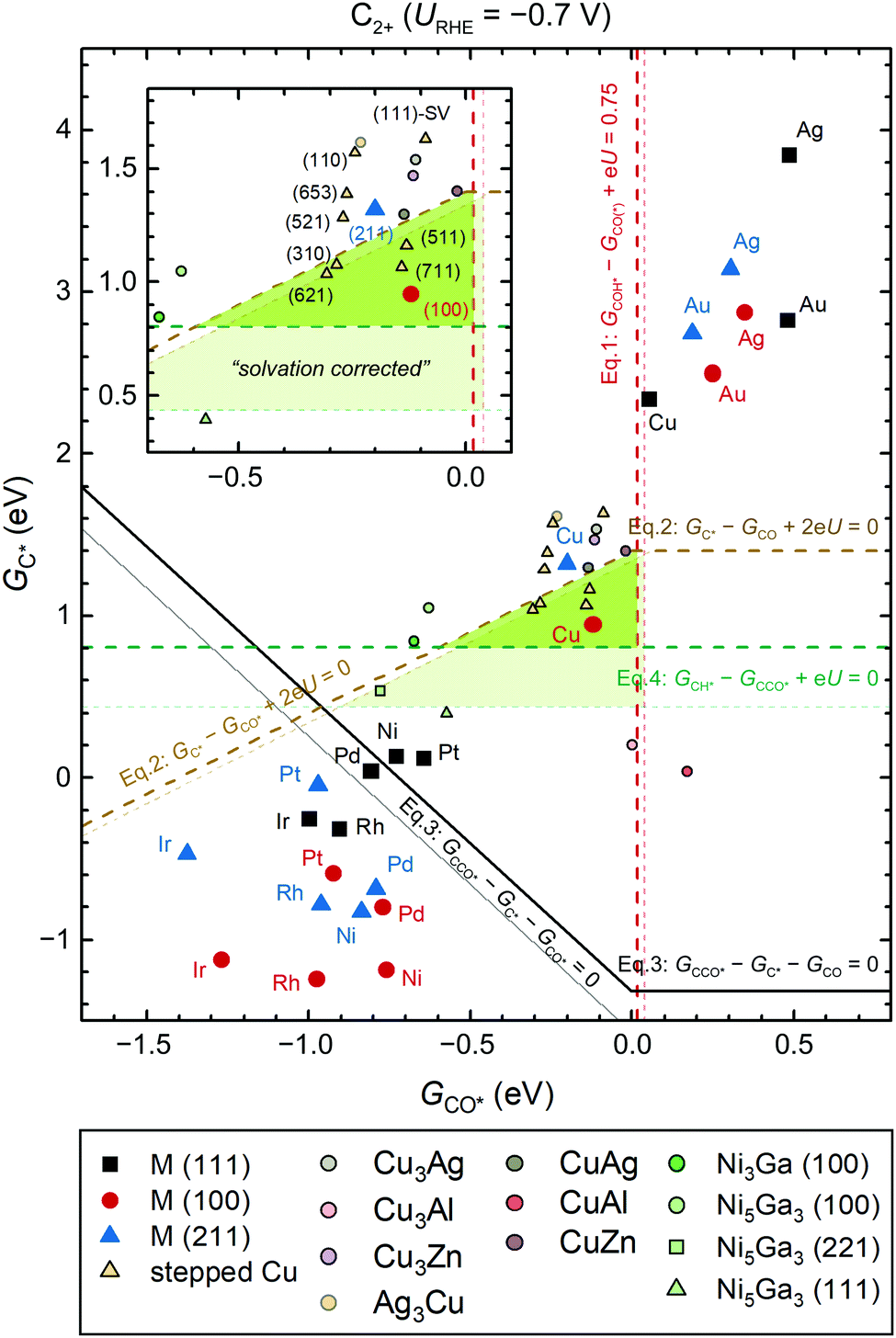 | ||
| Fig. 6 (GCO*, GC*) selectivity map at URHE = −0.7 V (pH7). The GCO* and GC* used herein are obtained through vacuum-level calculations for easy implementation. The C2-selective region is highlighted in green. Various metallic and intermetallic systems are included with symbols as indicated in the legend. The detailed surface orientations and computational details can be found in the ESI.† Square, circular, and triangular symbols correspond to surfaces with three-fold hollow sites, four-fold hollow sites, and under-coordinated step sites, respectively. Electrochemically driven processes are shown as dashed lines and the potential-independent C–CO coupling process is indicated with solid lines. The thin lines in light colors, as well as corresponding highlighted region in light green are shown to refer to solvation effects. Note that the U in the equations is referenced to RHE. | ||
Since CO–H protonation eventually becomes the RDS of CO(2)R (Fig. S15, ESI†), we therefore use ΔG(i)rxn as an estimate of the barrier for the RDS and assign a turnover frequency of ∼1 s−1 per site at 300 K as the lowest acceptable overall CO(2)R rate. Such a rate corresponds to the condition defined by eqn (1). To accurately describe the effect of CO adsorption, we replace GCO* in eqn (1)–(3) with GCO = 0 when the adsorption of CO on the catalytic surface is unfavorable (GCO* > 0).
Clearly, the C2 selectivity changes with applied potential and at −0.7 V vs. RHE, the above thermodynamic conditions form a triangular region (marked in green in Fig. 6) where a decent overall CO(2)R rate to C2 products can be obtained and where CCO* is thermodynamically favored over CH* formation. This map presents a powerful tool, as it is capable of qualitatively discerning C1 and C2 product selectivity across all metals. Remarkably, all known C2 selective Cu facets fall near the center of the region at this potential. It is noteworthy that Cu(211) sits on the edge of the region, whereas Cu(111) is unable to catalyze CO(2)R to C2 or C1 at such low overpotential. Despite the simple approach based only on thermodynamic arguments, the depicted trend is in agreement with the sophisticated kinetics analysis used in the previous sections. In short, the thermodynamic selective map is sufficient to qualitatively describe the CO(2)R selectivity across a large span of materials space.
Furthermore, the maps at various potentials also shows the narrow potential window of opportunity to form C2 products (Fig. S19 and S20, ESI†). (100)-like facets are found to be essential for improving C2 selectivity since the above narrow window does not include materials with dominant (111)-like facets (Fig. S21 and S22, ESI†). These effects accentuate the challenge in identifying C2-selective catalysts beyond Cu, Cu-based alloys, and intermetallics. Ag and Au do not form C2 because of their poor C* binding energies. Strong-binding metals can easily reduce CO to C* but on these surfaces the C* is not as reactive, thus favoring CO adsorption over the C–CO coupling step. Very few candidates relevant from experimental results fall in the C2-selective region, including Cu alloys with Zn,7 Ag,8 and Al,10 Ni-Ga intermetallics,51 as well as defective Cu52 (modeled as Cu(111) with a single vacancy, denoted as (111)-SV).
While the CO dimerization pathway has been successful in describing the pH independent behavior of C2 rates on Cu surfaces, we note in the following several observations that cannot easily be understood from a pathway involving CO dimerization, which on the other hand can be explained by the COH and OC–C pathway:
• Surface-carbon-induced deactivation of Cu catalysts during CO(2)R4,53 can only be attained through coupling of atomic carbon and not through decomposition of CHx species. This is because of the inability of Cu to break the C–H bond at room temperature,54 hence, atomic carbon must be present as an intermediate during CO(2)R, thus supporting the COH pathway.
• CO dimerization on Au exhibits a ΔGrxn of only 0.50 eV (∼0.50 eV lower than Cu) considering the same solvation and electric field conditions as on Cu. Hence, Au should in principle be a particularly selective catalyst toward C2 products through the CO dimerization. This, however, has never been experimentally validated.
• Ni-Rich Ni3Ga(100) and (111) surfaces were found unable to stabilize OCCO*. Hence, we do not expect CO dimerization to account for the observed C2 production on Ni–Ga intermetallics.51 The observed earlier onset potential for C2 products on Ni-Ga intermetallics than found on Cu, can however be well understood based on the OC–C mechanism as shown in Fig. S19 (ESI†).
Thus, the OC–C mechanism offers the ability to rationalize the above experimental observations with regards to material screening under neutral pH conditions, whereas the CO dimerization mechanism is more relevant for alkaline conditions. This suggests that understanding the role of atomic carbon in the CO(2)R provides a necessary insight into the reaction mechanism and paves the way for discovery of new materials. The insights developed with the OC–C mechanism enables us to propose several pathways for enhancing the C2(+) selectivity from CO(2)R: (1) precise control of micro-/nanostructures of Cu catalysts to increase the number of sites resembling the local environment on highly selective single-crystal Cu electrodes such as the Cu(511) and Cu(310) surfaces; (2) engineering of reaction microenvironments that increases the local availability of CO/CO2, regulate the charge and electric field distribution, and modulate the water activity at the interface; (3) design of alloy/intermetallic catalysts with the desirable CO and C binding strengths on geometrically well-defined surface structures.
Conclusion
We have identified the relevant reaction pathways for CO(2)R towards further reduced C1 (methane) and C2+ based on first principles reaction energetics and micro-kinetic modeling. We elucidated the role of atomic carbon as the key surface intermediate that directs the C1/C2+ selectivity through two distinct competing reaction pathways. Our model enables quantification of experimentally observed activity/selectivity trends for CO(2)R on Cu at varying potentials and changes in surface orientation. We also demonstrated that with the two simple thermodynamic descriptors, CO and C binding strengths, a number of experimental observations can be rationalized across a range of metal and metal alloy catalysts. In particular, four-fold hollow sites on Cu-like materials were identified as strongly C2+-selective, however only within a narrow potential window. These insights enable us to identify the immense challenges associated with the search for new materials that are similar or even surpass Cu in terms of activity and selectivity.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The material for all the energetic computation and the microkinetic models is based upon work performed by the Joint Center for Artificial Photosynthesis, a DOE Energy Innovation Hub, supported through the Office of Science of the U.S. Department of Energy under Award Number DE-SC0004993. The material for reaction kinetics with the analysis on proton sources is based on work performed by the Liquid Sunlight Alliance, which is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Fuels from Sunlight Hub under Award Number DE-SC0021266. We acknowledge the use of the computer time allocation for the Material Simulations in Joint Center for Artificial Photosynthesis (JCAP) at the National Energy Research Scientific Computing Center, a DOE Office of Science User Facility supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. P. S. L. gratefully acknowledges the Alexander von Humboldt Foundation (AvH) for financial support. The authors thank Dr Tao Wang and Dr Alan C. Luntz for insightful discussions and helpful suggestions.References
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef CAS.
- M. B. Ross, P. De Luna, Y. F. Li, C. T. Dinh, D. Kim, P. Yang and E. H. Sargent, Nat. Catal., 2019, 2, 648–658 CrossRef CAS.
- D. F. Gao, R. M. Aran-Ais, H. S. Jeon and B. R. Cuenya, Nat. Catal., 2019, 2, 198–210 CrossRef CAS.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Y. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS.
- Y. Y. Birdja, E. Perez-Gallent, M. C. Figueiredo, A. J. Gottle, F. Calle-Vallejo and M. T. M. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- C. W. Li, J. Ciston and M. W. Kanan, Nature, 2014, 508, 504–507 CrossRef CAS.
- D. Ren, B. S. H. Ang and B. S. Yeo, ACS Catal., 2016, 6, 8239–8247 CrossRef CAS.
- E. L. Clark, C. Hahn, T. F. Jaramillo and A. T. Bell, J. Am. Chem. Soc., 2017, 139, 15848–15857 CrossRef CAS.
- C. G. Morales-Guio, E. R. Cave, S. A. Nitopi, J. T. Feaster, L. Wang, K. P. Kuhl, A. Jackson, N. C. Johnson, D. N. Abram, T. Hatsukade, C. Hahn and T. F. Jaramillo, Nat. Catal., 2018, 1, 764–771 CrossRef CAS.
- M. Zhong, K. Tran, Y. M. Min, C. H. Wang, Z. Y. Wang, C. T. Dinh, P. De Luna, Z. Q. Yu, A. S. Rasouli, P. Brodersen, S. Sun, O. Voznyy, C. S. Tan, M. Askerka, F. L. Che, M. Liu, A. Seifitokaldani, Y. J. Pang, S. C. Lo, A. Ip, Z. Ulissi and E. H. Sargent, Nature, 2020, 581, 178–183 CrossRef CAS.
- F. S. Roberts, K. P. Kuhl and A. Nilsson, Angew. Chem., Int. Ed., 2015, 54, 5179–5182 CrossRef CAS.
- D. Kim, C. S. Kley, Y. F. Li and P. D. Yang, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 10560–10565 CrossRef CAS.
- K. Jiang, R. B. Sandberg, A. J. Akey, X. Y. Liu, D. C. Bell, J. K. Nørskov, K. R. Chan and H. T. Wang, Nat. Catal., 2018, 1, 111–119 CrossRef CAS.
- J. J. Lv, M. Jouny, W. Luc, W. L. Zhu, J. J. Zhu and F. Jiao, Adv. Mater., 2018, 30, 1803111 CrossRef.
- M. R. Singh, Y. Kwon, Y. Lum, J. W. Ager and A. T. Bell, J. Am. Chem. Soc., 2016, 138, 13006–13012 CrossRef CAS.
- J. Resasco, L. D. Chen, E. Clark, C. Tsai, C. Hahn, T. F. Jaramillo, K. Chan and A. T. Bell, J. Am. Chem. Soc., 2017, 139, 11277–11287 CrossRef CAS.
- D. F. Gao, I. T. McCrum, S. Deo, Y. W. Choi, F. Scholten, W. M. Wan, J. G. G. Chen, M. J. Janik and B. R. Cuenya, ACS Catal., 2018, 8, 10012–10020 CrossRef CAS.
- J. Li, D. H. Wu, A. S. Malkani, X. X. Chang, M. J. Cheng, B. J. Xu and Q. Lu, Angew. Chem., Int. Ed., 2020, 59, 4464–4469 CrossRef CAS.
- S. C. Ma, M. Sadakiyo, R. Luo, M. Heima, M. Yamauchi and P. J. A. Kenis, J. Power Sources, 2016, 301, 219–228 CrossRef CAS.
- C. T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. G. de Arquer, A. Kiani, J. P. Edwards, P. De Luna, O. S. Bushuyev, C. Q. Zou, R. Quintero-Bermudez, Y. J. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783–787 CrossRef CAS.
- M. Jouny, W. Luc and F. Jiao, Nat. Catal., 2018, 1, 748–755 CrossRef CAS.
- F. Pelayo García de Arquer, C. T. Dinh, A. Ozden, J. Wicks, C. McCallum, A. R. Kirmani, D. H. Nam, C. Gabardo, A. Seifitokaldani, X. Wang, Y. C. Li, F. W. Li, J. Edwards, L. J. Richter, S. J. Thorpe, D. Sinton and E. H. Sargent, Science, 2020, 367, 661–666 CrossRef.
- A. D. Handoko, F. X. Wei, Jenndy, B. S. Yeo and Z. W. Seh, Nat. Catal., 2018, 1, 922–934 CrossRef CAS.
- E. L. Clark and A. T. Bell, J. Am. Chem. Soc., 2018, 140, 7012–7020 CrossRef CAS.
- K. L. Yang, R. Kas and W. A. Smith, J. Am. Chem. Soc., 2019, 141, 15891–15900 CrossRef CAS.
- Y. Hori, R. Takahashi, Y. Yoshinami and A. Murata, J. Phys. Chem. B, 1997, 101, 7075–7081 CrossRef CAS.
- K. J. P. Schouten, Z. S. Qin, E. P. Gallent and M. T. M. Koper, J. Am. Chem. Soc., 2012, 134, 9864–9867 CrossRef CAS.
- L. Wang, S. A. Nitopi, E. Bertheussen, M. Orazov, C. G. Morales-Guio, X. Y. Liu, D. C. Higgins, K. R. Chan, J. K. Nørskov, C. Hahn and T. F. Jaramillo, ACS Catal., 2018, 8, 7445–7454 CrossRef CAS.
- F. Calle-Vallejo and M. T. M. Koper, Angew. Chem., Int. Ed., 2013, 52, 7282–7285 CrossRef CAS.
- J. H. Montoya, C. Shi, K. Chan and J. K. Nørskov, J. Phys. Chem. Lett., 2015, 6, 2032–2037 CrossRef CAS.
- W. J. Luo, X. W. Nie, M. J. Janik and A. Asthagiri, ACS Catal., 2016, 6, 219–229 CrossRef CAS.
- H. Xiao, T. Cheng and W. A. Goddard, J. Am. Chem. Soc., 2017, 139, 130–136 CrossRef CAS.
- T. Cheng, H. Xiao and W. A. Goddard, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1795–1800 CrossRef CAS.
- A. J. Garza, A. T. Bell and M. Head-Gordon, ACS Catal., 2018, 8, 1490–1499 CrossRef CAS.
- X. Y. Liu, P. Schlexer, J. P. Xiao, Y. F. Ji, L. Wang, R. B. Sandberg, M. Tang, K. S. Brown, H. J. Peng, S. Ringe, C. Hahn, T. F. Jaramillo, J. K. Nørskov and K. R. Chan, Nat. Commun., 2019, 10, 32 CrossRef CAS.
- S. Ringe, E. L. Clark, J. Resasco, A. Walton, B. Seger, A. T. Bell and K. Chan, Energy Environ. Sci., 2019, 12, 3001–3014 RSC.
- Y. G. Kim, A. Javier, J. H. Baricuatro, D. Torelli, K. D. Cummins, C. F. Tsang, J. C. Hemminger and M. P. Soriaga, J. Electroanal. Chem., 2016, 780, 290–295 CrossRef CAS.
- M. Tang, H. Peng, P. S. Lamoureux, M. Bajdich and F. Abild-Pedersen, Appl. Catal., B, 2019, 279, 119384 CrossRef.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC.
- K. Chan and J. K. Nørskov, J. Phys. Chem. Lett., 2015, 6, 2663–2668 CrossRef CAS.
- M. L. Ng, F. Abild-Pedersen, S. Kaya, F. Mbuga, H. Ogasawara and A. Nilsson, Phys. Rev. Lett., 2015, 114, 246101 CrossRef CAS.
- M. T. Tang, Z. W. Ulissi and K. R. Chan, J. Phys. Chem. C, 2018, 122, 14481–14487 CrossRef CAS.
- X. Y. Liu, J. P. Xiao, H. J. Peng, X. Hong, K. Chan and J. K. Nørskov, Nat. Commun., 2017, 8, 15438 CrossRef CAS.
- J. J. Kim, D. P. Summers and K. W. Frese, J. Electroanal. Chem., 1988, 245, 223–244 CrossRef CAS.
- C. T. Campbell, ACS Catal., 2017, 7, 2770–2779 CrossRef CAS.
- B. Zijlstra, X. Zhang, J. X. Liu, I. A. W. Filot, Z. Y. Zhou, S. G. Sun and E. J. M. Hensen, Electrochim. Acta, 2020, 335, 135665 CrossRef CAS.
- Y. Hori, I. Takahashi, O. Koga and N. Hoshi, J. Mol. Catal. A: Chem., 2003, 199, 39–47 CrossRef CAS.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jonsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- P. S. Lamoureux, A. R. Singh and K. R. Chan, ACS Catal., 2019, 9, 6194–6201 CrossRef CAS.
- X. Wang, J. Ferreira de Araújo, W. Ju, A. Bagger, H. Schmies, S. Kühl, J. Rossmeisl and P. Strasser, Nat. Nanotechnol., 2019, 14, 1063–1070 CrossRef CAS.
- D. A. Torelli, S. A. Francis, J. C. Crompton, A. Javier, J. R. Thompson, B. S. Brunschwig, M. P. Soriaga and N. S. Lewis, ACS Catal., 2016, 6, 2100–2104 CrossRef CAS.
- M. Liu, M. X. Liu, X. M. Wang, S. M. Kozlov, Z. Cao, P. De Luna, H. M. Li, X. Q. Qiu, K. Liu, J. H. Hu, C. K. Jia, P. Wang, H. M. Zhou, J. He, M. Zhong, X. Z. Lan, Y. S. Zhou, Z. Q. Wang, J. Li, A. Seifitokaldani, C. T. Dinh, H. Y. Liang, C. Q. Zou, D. L. Zhang, Y. Yang, T. S. Chan, Y. Han, L. Cavallo, T. K. Sham, B. J. Hwang and E. H. Sargent, Joule, 2019, 3, 1703–1718 CrossRef CAS.
- Z. Weng, X. Zhang, Y. S. Wu, S. J. Huo, J. B. Jiang, W. Liu, G. J. He, Y. Y. Liang and H. L. Wang, Angew. Chem., Int. Ed., 2017, 56, 13135–13139 CrossRef CAS.
- S. Wang, V. Petzold, V. Tripkovic, J. Kleis, J. G. Howalt, E. Skulason, E. M. Fernandez, B. Hvolbaek, G. Jones, A. Toftelund, H. Falsig, M. Bjorketun, F. Studt, F. Abild-Pedersen, J. Rossmeisl, J. K. Nørskov and T. Bligaard, Phys. Chem. Chem. Phys., 2011, 13, 20760–20765 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ee02826f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |