
Engineering the interface between LiCoO2 and Li10GeP2S12 solid electrolytes with an ultrathin Li2CoTi3O8 interlayer to boost the performance of all-solid-state batteries†
Chuan-Wei
Wang‡
a,
Fu-Cheng
Ren‡
a,
Yao
Zhou
 a,
Peng-Fei
Yan
b,
Xiao-Dong
Zhou
c,
Shao-Jian
Zhang
a,
Wen
Liu
d,
Wei-Dong
Zhang
e,
Ming-Hua
Zou
e,
Lei-Ying
Zeng
e,
Xia-Yin
Yao
f,
Ling
Huang
g,
Jun-Tao
Li
*a and
Shi-Gang
Sun
g
a,
Peng-Fei
Yan
b,
Xiao-Dong
Zhou
c,
Shao-Jian
Zhang
a,
Wen
Liu
d,
Wei-Dong
Zhang
e,
Ming-Hua
Zou
e,
Lei-Ying
Zeng
e,
Xia-Yin
Yao
f,
Ling
Huang
g,
Jun-Tao
Li
*a and
Shi-Gang
Sun
g
aCollege of Energy, Xiamen University, Xiamen 361005, China. E-mail: jtli@xmu.edu.cn
bBeijing Key Laboratory of Microstructure and Properties of Solids, Institute of Microstructure and Properties of Advanced Materials, Beijing University of Technology, Beijing 100124, China
cDepartment of Chemical Engineering, Institute for Materials Research and Innovation, University of Louisiana at Lafayette, Lafayette, LA 70504, USA
dState Key Laboratory of Chemical Resource Engineering College of Chemistry Beijing University of Chemical Technology, Beijing 100029, China
eXTC New Energy Materials (Xiamen) Co., Ltd, Xiamen 361026, China
fNingbo Institute of Materials Technology and Engineering, Chinese Academy of Sciences, Ningbo 315201, China
gState Key Lab of Physical Chemistry of Solid Surface, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China
First published on 21st November 2020
Abstract
Sulfide-based all-solid-state lithium-ion batteries (ASSLIBs) are promising candidates in the next generation of energy storage technology; the voltage mismatch and the resulting side reactions at the interface between the cathode and the solid electrolyte, however, dramatically deteriorate their cycling performance. Herein, for the first time, we report that the chemical interaction between LiCoO2 (LCO) and TiO2 can be regulated by two additives, carbon and Li2CO3, which in situ form a continuous ultrathin pure-phase Li2CoTi3O8 (LCTO) layer with a stable 3D network of spinel structures, relatively low electronic conductivity (2.5 × 10−8 S cm−1) and high lithium diffusion coefficient (DLi+ = 8.22 × 10−7 cm2 s−1) on the surface of LCO. When assembled in ASSLIBs, such an LCTO layer functions as an interlayer between the LCO and the Li10GeP2S12 solid electrolyte (LGPS). As a consequence, the original interface LCO/LGPS is substituted by two new interfaces LCO/LCTO and LCTO/LGPS. DFT calculations indicate that, compared with the LCO/LGPS, the new interfaces are not only thermodynamically and electrochemically more compatible, but also have higher interfacial affinity. Therefore, the relevant ASSLIB exhibits evidently reduced interfacial impedance, and it also displays a high initial capacity of 140 mA h g−1 and a reversible discharge specific capacity of 116 mA h g−1 after 200 cycles at room temperature (0.1C). In comparison, the ASSLIB assembled without the LCTO interlayer delivers an initial capacity of 98 mA h g−1 and only retains 22.4% capacity after 100 cycles (0.1C). Even at a high cutoff voltage (4.5 V vs. Li/Li+), the ASSLIB with the LCTO interlayer could also exhibit a high initial capacity of 180 mA h g−1 and a remarkable retention of 132 mA h g−1 after 100 cycles.
Broader contextA sulfide-based solid supporting electrolyte (e.g., LGPS) exhibits high Li+ conductivity at room temperature; however, when used in the all-solid-state lithium-ion batteries (ASSLIBs), severe interfacial deterioration would occur between the cathode and solid electrolyte, especially for a LiCoO2-based cathode (LCO). Herein, a novel material, Li2CoTi3O8 (LCTO), with an ultrathin layer around 1.5 nm in thickness was introduced on the surface of LCO through the redox reaction between LCO microparticles and TiO2 nanoparticles; such LCTO is characterized with a stable 3D network of spinel structures, relatively low electronic conductivity and high lithium diffusion coefficients. With the introduction of such an interlayer, the original fragile interface of LCO–LGPS is replaced by two new interfaces (i.e., the LCO–LCTO and the LGPS–LCTO), both of which demonstrate much improved interfacial thermodynamic and electrochemical stability as well as enhanced chemical affinity; in particular, the LCTO interlayer could create a reasonable potential drop from the LCO side to the LGPS side, which thus provides a solution to the voltage mismatch issue harassing the LCO–LGPS interface; the resulting ASSLIBs exhibit significantly improved cycling stability. The formation of such an LCTO interlayer is easily scalable, which is expected to contribute to the development of high-performance all-solid-state lithium-ion batteries. |
Introduction
Rechargeable lithium-ion batteries (LIBs) have experienced rapid growth in the last few decades, widely applied to various markets, such as traditional consumer electronics, electric vehicles and energy storage power stations.1 Conventional lithium ion batteries, however, do face overarching challenges such as flammability, limited electrochemical stability, and an unstable solid–electrolyte interphase (SEI) layer against Li metal.2 To circumvent these issues, all-solid-state lithium-ion batteries (ASSLIBs) have gained increasing attention for their inherent safety and potentially high energy density, providing a promising option to replace conventional LIBs that have flammable organic electrolytes.3Various all-solid-state energy storage systems have been developed during the past few years, which can be summarized into three categories: oxide-based electrolyte systems, polymer electrolyte systems and sulfide-based electrolyte systems.3 The sulfide-based solid electrolytes with a Li+ conductivity comparable to organic liquid electrolytes at room temperature are especially attractive for the next generation of energy storage technology.4 However, sulfide-based ASSLIBs often suffer from interface mismatch between the cathode and solid electrolyte. As a typical example, undesirable and low-conductivity decomposition products (Li3PS4, S, GeS2, etc.) would inevitably be generated at the interface between LiCoO2 (LCO) and Li10GeP2S12 (LGPS), due to the mismatch between the LCO work potential (3.9 V vs. Li/Li+) and the narrow electrochemical window range of sulfide-based electrolytes (1.7–2.1 V vs. Li/Li+).5 Moreover, a space-charge layer (SCL) can be formed at the interface between LCO and LGPS, due to the mismatch of their electrochemical potential. Such an interfacial instability often leads to a large interfacial resistance, causing severe deterioration of cycling performance.
It is hence essential to achieve a stable interface between the sulfide-based solid state electrolyte and the oxide cathode for ASSLIBs. One strategy is to encapsulate the oxide cathode in an ideal coating material that exhibits high electrochemical stability, structural stability, chemical stability (limited reactivity with solid state electrolytes and oxide cathode) and high lithium ion conductivity.6 In this regard, LiNbO3 is the most commonly used and effective coating material.7 Other coating materials, including Li4Ti5O12,8 Li2CO3,9 LiAlO2,10 Li2SiO3,11 Al2O3,10 Li3PO4,12etc., also show the potential to suppress an increase in the interfacial resistance.
When assembled in ASSLIBs, the coating layer actually serves as an interlayer between the active cathode material and the solid electrolyte; the original cathode–electrolyte interface would be replaced by two new ones; in this situation, the chemical and electrochemical properties of such an interlayer are extremely important, as it provides a chance to engineer the fragile interface between the oxide cathode and the sulfide-based solid electrolyte. Specifically, such an interlayer should have high ion conductivity and be thermodynamically stable with both the solid electrolyte and the cathode. Moreover, to reduce the solid–solid interface impedance in ASSLIBs, an ideal interlayer should also have high chemical affinity with both the cathode and the solid electrolyte. Meanwhile, regarding the electronic conductivity, there exists a dilemma. On the one hand, for efficient transfer of electrons within the cathode, such interlayer (which is also a coating layer of the oxide cathode) with relatively high electronic conductivity is intuitively preferred; on the other hand, however, to address the aforementioned voltage mismatch issue, the interlayer should be able to create a suitable potential drop from the oxide cathode down to the sulfide-based electrolyte; that is, the interlayer shall have relatively low electronic conductivity.5,13
In this work, as shown in Scheme 1, for the first time the chemical reaction between LCO and TiO2 with carbon and Li2CO3 as critical additives under high temperature has been explored to form a highly pure layer of Li2CoTi3O8 (LCTO) on the surface of LCO via an easily scalable solid-phase method. The Li2CoTi3O8 layer is ultrathin (∼1.5 nm) and is characterized with a stable 3D network of spinel structures,14 relatively low electronic conductivity (2.5 × 10−8 S cm−1), and high lithium diffusion coefficients (DLi+ = 8.22 × 10−7 cm2 s−1). When such an LCTO@LCO cathode is assembled in an ASSLIB with LGPS as the solid electrolyte, the LCTO layer serves as a stable interlayer which buffers the unfavorable voltage difference between the cathode LCO with high oxidative potential and the LGPS with low oxidation limit; with the presence of such an LCTO interlayer, both the interfacial impedance and stability between the cathode and the solid electrolyte were found evidently improved. Our LCTO@LCO exhibits a high initial capacity of 140 mA h g−1 and delivers a reversible discharge specific capacity of 116 mA h g−1 after 200 cycles at room temperature (0.1C, 1C = 150 mA h g−1); in comparison, the bare LCO delivers an initial capacity of 98 mA h g−1 and only retains 22.4% capacity after 100 cycles (0.1C). With the increase of cutoff voltage (From 3.7 V to 3.9 V vs. Li–In anode), the LCTO@LCO could also exhibit a high initial capacity of 180 mA h g−1 and a considerable retention of 73.3% after 100 cycles. Moreover, the LCTO@LCO also demonstrates enhanced rate capability and reduced polarization during ASSLIB cycling. The new interfaces arising from the introduction of the LCTO interlayer between the cathode and solid electrolyte (LCO/LGPS), namely the LCO/LCTO interface and the LCTO/LGPS interface, were evaluated via DFT calculations and material project analysis to understand the interfacial structure as well as the interfacial chemical affinity and chemical stability.
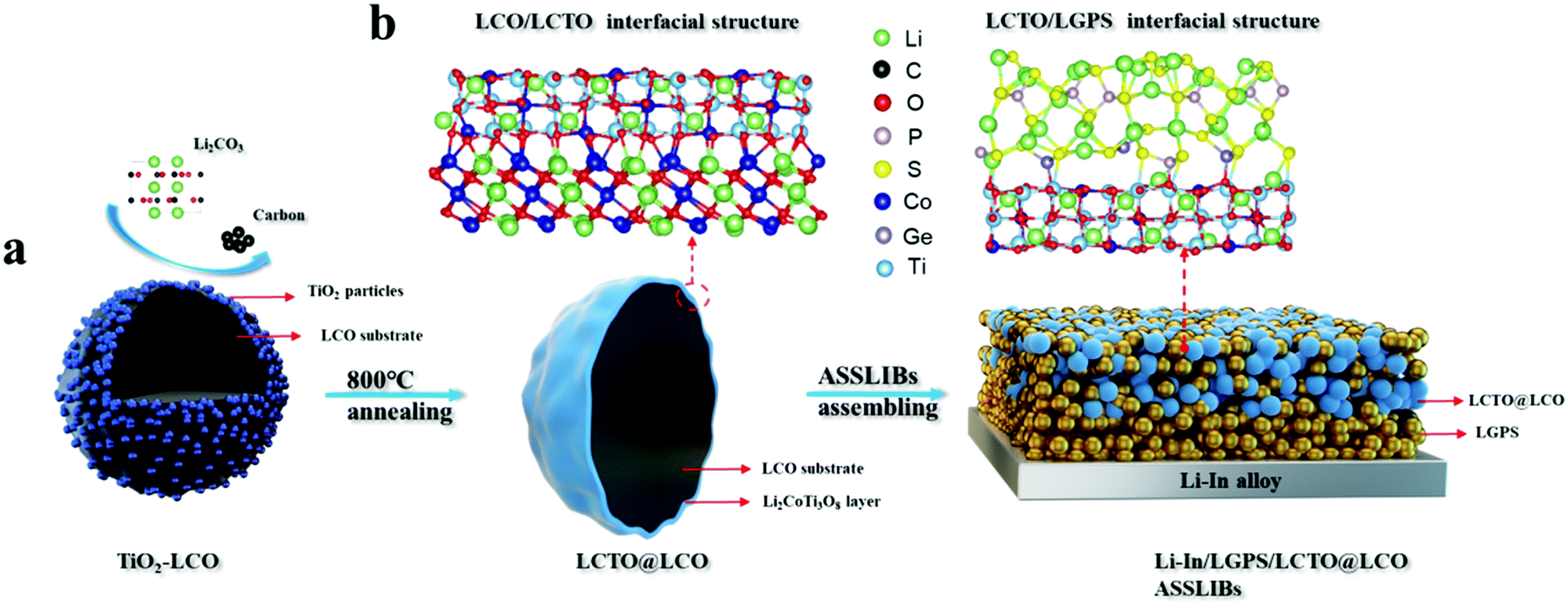 | ||
| Scheme 1 A schematic flowchart to show the in situ formation of the LCTO coating layer on the LCO core. (a) The loading and the annealing of TiO2 on the surface of LiCoO2 to form the LCTO@LCO core–shell structure at high temperatures. (b) The interfacial structure of LCO/LCTO and LCTO/LGPS at an atomic level obtained from DFT calculations. | ||
Results and discussion
Modulation of the chemical reaction between LCO and TiO2
Ti-containing oxides have been widely used and proved to be effective in the surface modification of LCO in both liquid and solid state batteries;15,16 in those coating processes, thermal post-treatments are often required to enhance the solid–solid interfacial interaction between the coating layer and the LCO substrate. A series of chemical reactions, however, have been often overlooked, which occur on the interface because of the inter-diffusion of the elements between the Ti-containing oxides and the LCO substrate during the high temperature treatment (e.g., 800 °C, a temperature adopted in this work).Initially, we noticed that there exists a strong chemical interaction between the TiO2 solids and the LCO spheres at high temperatures; and more importantly, such chemical interaction could be modulated by the presence of carbon and Li2CO3. A control experiment was then carried out. We found that when the mixture of TiO2 and LCO with a molar ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (note that such a ratio is much larger than the real ratio employed for the synthesis of LCTO@LCO cathode materials discussed in the following section) was subjected to a thermal treatment (without the presence of carbon or Li2CO3), the XRD pattern of the end-product (Fig. 1a) shows the formation of Li2CoTi3O8 and Co3O4 (2θ = 31.2, 36.8, 59.3) as well as the presence of some residual TiO2 (2θ = 27.4, 36.1, 54.3) (Fig. 1b), indicating the partial reaction between LCO and TiO2 (eqn (1)). On the other hand, when TiO2 and LCO were treated under the same conditions with the presence of carbon and Li2CO3, Li2CoTi3O8 with a P4332 space group was obtained as the only solid product, as confirmed by XRD analysis (JCPDS card No. 89-1309) (Fig. 1a and eqn (2)). The optical images of the products reveal additional differences (inset of Fig. 1a). The color of the product from the control experiment (i.e., without carbon and Li2CO3) is dark green, but the powder obtained with the presence of carbon and Li2CO3 is bright blue.17 Considering the reducing nature of carbon, it is speculated that the presence of carbon could accelerate the reduction of cobalt cations in the LCO at the early annealing process, and the presence of an appropriate amount of Li2CO3 serves as an additional source of Li+, which can help maintain a stoichiometric ratio of Li2CoTi3O8, as shown in eqn (2).
:1 (note that such a ratio is much larger than the real ratio employed for the synthesis of LCTO@LCO cathode materials discussed in the following section) was subjected to a thermal treatment (without the presence of carbon or Li2CO3), the XRD pattern of the end-product (Fig. 1a) shows the formation of Li2CoTi3O8 and Co3O4 (2θ = 31.2, 36.8, 59.3) as well as the presence of some residual TiO2 (2θ = 27.4, 36.1, 54.3) (Fig. 1b), indicating the partial reaction between LCO and TiO2 (eqn (1)). On the other hand, when TiO2 and LCO were treated under the same conditions with the presence of carbon and Li2CO3, Li2CoTi3O8 with a P4332 space group was obtained as the only solid product, as confirmed by XRD analysis (JCPDS card No. 89-1309) (Fig. 1a and eqn (2)). The optical images of the products reveal additional differences (inset of Fig. 1a). The color of the product from the control experiment (i.e., without carbon and Li2CO3) is dark green, but the powder obtained with the presence of carbon and Li2CO3 is bright blue.17 Considering the reducing nature of carbon, it is speculated that the presence of carbon could accelerate the reduction of cobalt cations in the LCO at the early annealing process, and the presence of an appropriate amount of Li2CO3 serves as an additional source of Li+, which can help maintain a stoichiometric ratio of Li2CoTi3O8, as shown in eqn (2).
| LiCo(III)O2(s) + 3TiO2(s) → 1/2Li2Co(II)Ti3O8(s) + 1/6Co3O4(s) + 3/2TiO2(s) + 1/6O2(g) | (1) |
| LiCo(III)O2(s) + 3TiO2(s) + 1/2Li2CO3(s) + 1/4C(s) → Li2Co(II)Ti3O8(s) + 3/4CO2(g) | (2) |
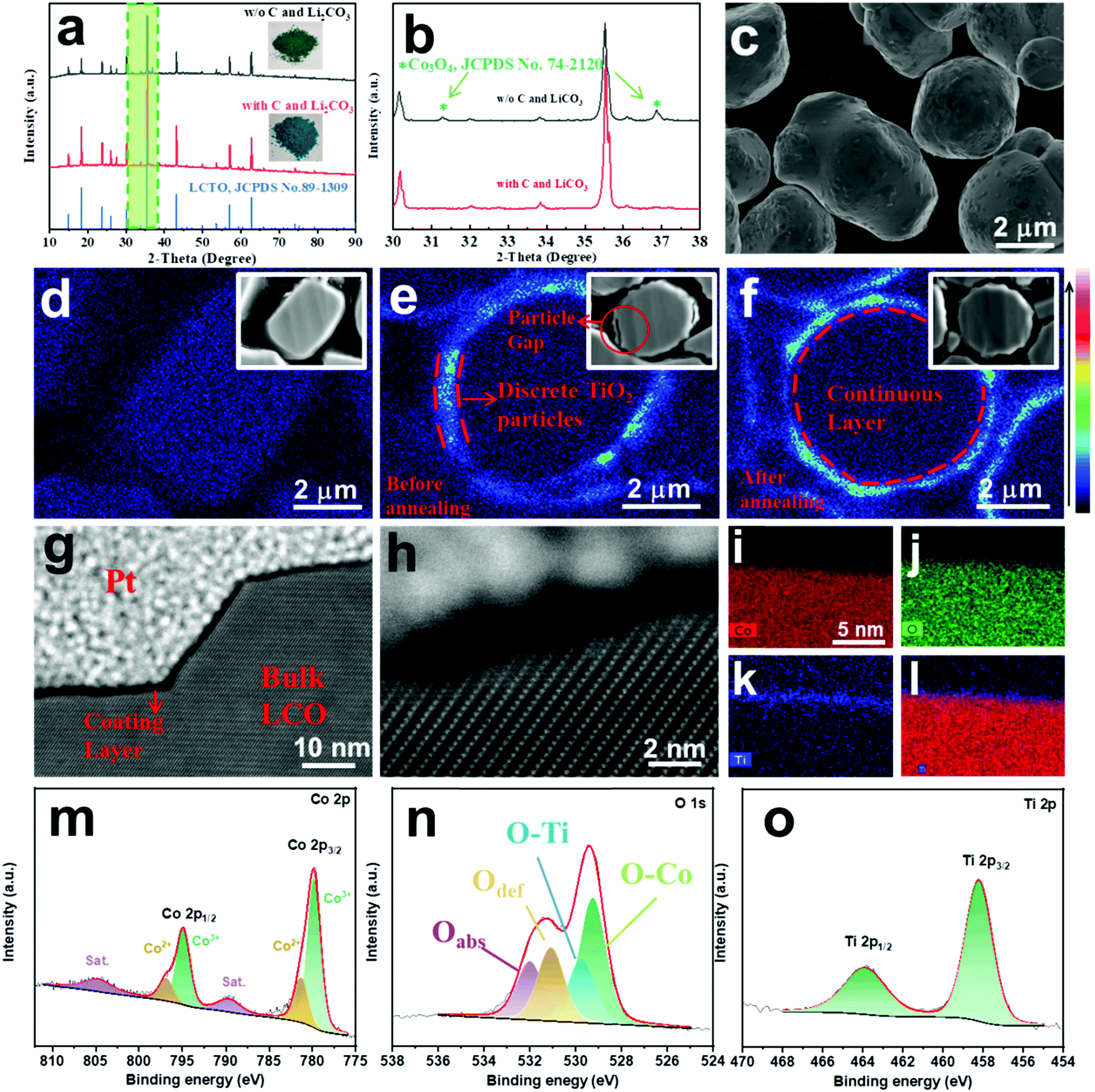 | ||
| Fig. 1 (a) XRD patterns and optical images of the products from thermal treatment of LCO and TiO2 mixture with and without the presence of carbon and Li2CO3 and (b) the enlarged XRD patterns framed in (a); (c) FESEM image of LCTO@LCO; (d–f) EPMA mapping images of the cross section of: (d) the bare LCO, (e) the intermediate sample TiO2–LCO (LCO loaded with discrete TiO2 particles before annealing), (f) LCTO@LCO; (g and h) STEM-HAADF images, (i–l) EDS elemental maps of LCTO@LCO prepared by FIB and (l) overall element distribution of Co, O and Ti; (m–o) XPS spectra of the pristine LCTO@LCO: (m) Co 2p, (n) O 1s and (o) Ti 2p. | ||
In situ formation of a pure-phase LCTO ultrathin layer on the LCO
Based on the above reaction between the TiO2 and LCO, an ultrathin layer of pure-phase LCTO was formed on site on the commercial bare LCO microspheres, resulting in the LCTO@LCO electrode material. As depicted in Scheme 1, the individual TiO2 nanoparticles were firstly deposited in a discrete manner on the LCO microsphere via a high speed mixing process; the resulting 0.5% wt TiO22013LCO composite was then annealed with the addition of carbon and Li2CO3 at 800 °C for 3 hours, leading to the LCTO@LCO composite. For comparison, one more control sample (TiO2@LCO) was obtained by annealing the intermediate sample (TiO2–LCO) under the same conditions but without the addition of carbon and Li2CO3; note that, in this situation (namely with no carbon and Li2CO3), the TiO2 nanoparticles would partially react with the surficial LCO species, according to eqn (1).As shown in Fig. S1 (ESI†) and Fig. 1c, the surface of pristine LCO particles is generally smooth with an average diameter of 3–4 μm (Fig. S1a, ESI†). The intermediate sample TiO2–LCO (Fig. S1c, ESI†) exhibits a slightly rough surface, on which numerous TiO2 nanoparticles are found (Fig. S1b, ESI†); more importantly, the surface morphology integrity of the LCO particles was well maintained after such a high speed mixing process, as supported by the relevant SEM images. The LCTO@LCO, which was obtained from high-temperature annealing of TiO2–LCO, displays a smooth surface (Fig. 1c), suggesting the reaction between TiO2 nanoparticles and LCO, as shown in eqn (2).
To further verify the occurrence of the LCTO coating layer on the LCO particles, the samples were polished by ion beam and were observed by Electron Probe Micro-Analysis (EPMA) at the cross sections. As shown in Fig. 1d, the pristine LCO shows no signals of Ti at the edge of the cross sections. For the intermediate sample TiO2–LCO, the cross sectional EPMA (Fig. 1e) and SEM image (Fig. S1c, ESI†) reveal discrete green signals of Ti on the boundary of the LCO sphere. After a thermal treatment at 800 °C, the distribution of Ti occurs in a continuous manner on the boundary of the LCO particles, supporting the formation of a Ti-containing continuous coating layer on the LCO surface in the LCTO@LCO composite (Fig. 1f). In addition, the XRD patterns of such TiO2–LCO and LCTO@LCO composites reveal little signals of TiO2 and LCTO crystals, due to the low content of the coating material (Fig. S2, ESI†). Since observing the LCO cross-section through EPMA will not only obtain the same horizontal signal, it is hard to infer specifically the thickness of the LCTO layer.
To investigate the exact thickness of LCTO@LCO, a thin slice of LCTO@LCO sample was prepared by Focused Ion Beam (FIB) and observed by High-Resolution Transmission Electron Microscope (HRTEM). As shown in Fig. 1g–l, a continuous and compact layer with a thickness of ∼1.5 nm can be observed on the LCTO@LCO surface, even on the uneven staircase-like location (Fig. 1g), clearly proving the occurrence of the LCTO layer on the LCO. The relevant elemental mapping reaffirms the presence of the element Ti on the surface of the particle (Fig. 1i–l); the overlapping of the distribution of Co, Ti and O also suggests the formation of LCTO.
The samples were then analyzed by X-ray Photoelectron Spectroscopy (XPS) (Fig. 1 and Fig. S3, ESI†). The Ti 2p spectrum of our LCTO@LCO exhibits strong peaks at around 458.5 eV and 464.2 eV, corresponding to the Ti 2p3/2 and Ti 2p1/2 of LCTO species (Fig. 1o),18 supporting the successful formation of the LCTO coating layer in this composite (Fig. S3i, ESI†). Meanwhile, the O 1s spectra vary evidently among different samples. In general, as shown in Fig. 1n and Fig. S3 (ESI†), the peak at 532.0 eV (the one shaded in purple) is associated with chemisorbed or dissociated oxygen species and/or hydroxide (Oabs); the peak (the one shaded in yellow) at 531.0 eV is assigned to O2− in the surface oxygen deficient regions (Odef).19,20 We found that, when the bare LCO is calcined alone at 800 °C for 3 hours in air (namely the sample LCO-800) (Fig. S3e, ESI†), the total fraction of Oabs and Odef would decrease significantly from 59% to 49.8% (Table S1, ESI†); similarly, our LCTO@LCO (Fig. 1n) exhibited significantly decreased Oabs (20.4%) and Odef (26.0%), which is even lower than those in the bare LCO-800 or the control TiO2@LCO (Oabs ∼24.3%, Odef ∼33.1%, Fig. S3k, ESI†), as shown in Table S1 (ESI†). As a reference, it shall be mentioned that the pure LCTO alone (which was prepared by calcination of TiO2 and LCO with a molar ratio of 3:1 with the presence of carbon and Li2CO3 according to eqn (2) has a small portion of Oabs (7.8%) and Odef (21.8%) (Table S1 and Fig. S3h, ESI†). Considering the fact that XPS analysis mainly provides insight into a surface down to a depth of a few nanometers, the relatively low portion of Oabs and Odef in our LCTO@LCO suggests that the LCO core in our LCTO@LCO was better wrapped up by the LCTO coating layer, which has higher crystallinity and structural integrity than the control sample TiO2@LCO.
In addition to the Oabs and Odef species, the peak at 529.2 eV (the one shaded in green) and the peak at 529.8 eV (the one shaded in blue) correspond to Co–O bonds and Ti–O bonds in the lattice,19 respectively (Fig. 1o). As shown in Table S1 (ESI†), the fraction of Ti–O species in the LCTO@LCO is 16.5%, evidently higher than that of the TiO2@LCO, which is 10.2%; such a comparative result further suggests higher structural integrity of the LCTO coating layer in our LCTO@LCO sample.
Furthermore, it has been reported that the Co 2p spectrum which displays intense satellites and large 2p1/2–2p3/2 energy separation suggests an effect of exchange splitting of the 2p levels by the unpaired valence electrons.21 Accordingly, the XPS Co 2p spectrum of the aforementioned pure LCTO (Fig. S3g, ESI†) shows two strong main peaks at 796.5 eV and 780.7 eV, corresponding to the Co(II) 2p3/2, and Co(II) 2p1/2 spin–orbit peaks; such wide 2p1/2–2p3/2 peak separation (15.72 eV) indicates typical high spin cobalt(II) species. Similar to the pure LCTO crystals, the Co 2p spectrum of LCTO@LCO displays larger satellite areas and wider 2p1/2–2p3/2 peak separation than the bare LCO, further confirming the generation of an LCTO coating layer (Table S2, ESI†). Meanwhile, in addition to the Co2+ species, the Co 2p spectra of both TiO2@LCO and LCTO@LCO exhibit a peak at 794.9 eV for Co 2p3/2 and 779.8 eV for Co 2p1/2, which could be assigned to Co3+ species (Fig. 1m). As compared in Table S2 (ESI†), the relative surface atomic ratio of Co2+/Co3+ for TiO2@LCO is calculated as 0.57 (using the peak area of Co 2p1/2) or 0.46 (using the peak area of 2p3/2), while for LCTO@LCO it is 0.62 (2p1/2) or 0.54 (2p3/2). Note that the cobalt species in LCTO have bivalent valence charge while those in LCO are trivalent. Therefore, such a result further supports the formation of more LCTO species in the LCTO@LCO, which was prepared with the presence of carbon, and Li2CO3.
The above comparison between the LCTO@LCO and TiO2@LCO revealed the important role of carbon and Li2CO3 in regulating the reaction between the LCO and TiO2, which determines the eventual purity and the structural integrity of the as-formed LCTO coating layer. It shall be mentioned that, in addition to the driving force from the chemical redox reaction between the LCO and the TiO2, the successful conversion of the discrete TiO2 with a diameter of 10 nm into an ultrathin LCTO coating layer through the above solid–solid interfacial reaction depends also on the size and the homogeneous distribution of TiO2 nanoparticles. In our work, TiO2 particles with a small size (10 nm on average) are homogeneously deposited on the surface of the LCO micro particles, which therefore could provide extensive solid–solid interface and enable the occurrence of the redox reaction. Meanwhile, the high annealing temperature is also necessary; in this situation, the chemical interaction as well as the diffusion behavior across the solid–solid interface would be largely enhanced between the LCO and TiO2. It shall also be emphasized that, as depicted in Scheme 1a, the continuous LCTO coating layer with a thickness of ∼1.5 nm connects intimately with the LCO substrate; as it is formed through the in situ chemical interaction between TiO2 nanoparticles and LCO at high temperatures, it is expected that extensive interfacial oxygen bridge bonding, Ti–O–Co, occurs between the as-formed LCTO coating layer and the LCO core (Scheme 1b). The formation of such an external LCTO coating layer can be explained by the relatively low total energy of Ti which tends to stay on the external surface rather than diffusing inward into the deep bulk interior.22
Evaluation of LCTO as an interlayer between LCO and LGPS
To understand the role of the LCTO coating layer introduced between the cathode material LCO and the solid electrolyte LGPS, the above-mentioned pure LCTO powder with a size of ∼1 μm for the primary particles (Fig. S1e, ESI†) was further investigated. Fig. 2a shows cyclic voltammetry (CV) curves of the LCTO bulk particles with different scan rates (0.1–0.7 mV s−1) in the voltage range of 0.05–3.0 V. The 1st cycle at 0.1 mV s−1 (Fig. S5b, ESI†) varies widely from the subsequent cycles (Fig. 2a), which can be attributed to initial structural changes and activation of electrodes. As shown in the CV curves after the initial activation, an intense anodic peak at 2.09 V and a cathodic peak below 0.5 V were observed, which were attributed to the lithium ion insertion and extraction from different sites of the spinel structure.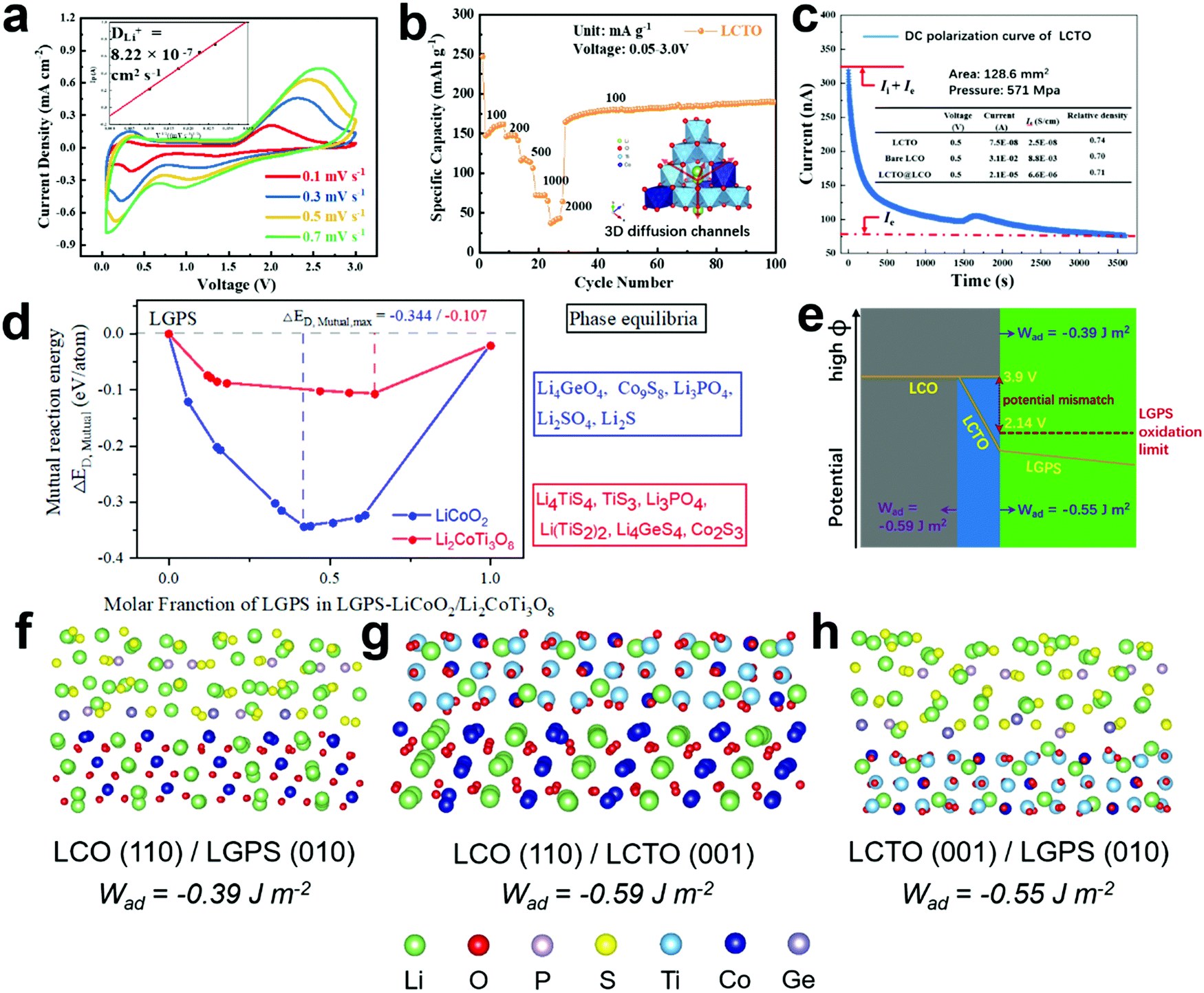 | ||
| Fig. 2 (a) CV profiles of the LCTO electrode with varying sweeping rates and Ip–v1/2 plots; (b) rate performance of LCTO; (c) direct-current (DC) polarization curve of LCTO and the calculated electronic conductivity of LCTO, bare LCO and LCTO@LCO; (d) calculated mutual reaction energy of LCO/LGPS and LTCO/LGPS as a function of the mixing ratio of LGPS and phase equilibria (in box) with the largest magnitude of decomposition enthalpy; (e) schematic illustrations of the potential profile near the solid electrolyte/LCO interface with and without the LCTO interlayer; (f–h) Simulation of the interfacial structure and the work of adhesion (Wad) of LCO/LGPS, LCO/LCTO and LCTO/LGPS. | ||
The CV profiles were also used to calculate the lithium diffusion coefficients (DLi+), based on the Randles−Sevcik equation,23 which was obtained as 8.22 × 10−7 cm2 s−1 for LCTO, around 104 times larger than pure LCO and commercial titanium based electrode materials (LiCoO2, DLi+ = 10−11–10−13 cm2 s−1;14 Li4Ti5O12, DLi+ = 3.2 × 10−11 cm2 s−1).24 Such a great DLi+ in LCTO can provide a fast pathway for Li ions in the LCTO@LCO electrodes. As shown in Table S7 (ESI†), the crystal structure information and the lithium diffusion coefficients of various coating materials under different temperatures are compared. Compared with those typical coating materials such as LiNbO3, our LCTO indeed was observed with superior ionic conductivity.
Accordingly, the pure LCTO displays remarkable cycling performance and rate capability (Fig. 2b and Fig. S5a, ESI†). When tested with the current density of 100, 200, 500, 1000 and 2000 mA g−1, a reversible discharge specific capacity of 162, 148, 119, 72 and 43 mA h g−1 could be obtained, respectively; when the current density returned to 100 mA g−1, an excellent cycling reversibility was observed, with a specific capacity of 191 mA h g−1 after 100 cycles.
The excellent cycling stability of LCTO at both 1000 mA g−1 (Fig. S5a, ESI†) and 100 mA g−1 (Fig. 2b) indicates its inherently stable structural characteristics. This stable 3D network of spinel structures provides a fast and stable pathway for the migration of lithium ions (as shown in the inset of Fig. 2b). The electrochemical behaviors of the pure LCTO were further evaluated under relatively high voltage up to the charging cut-off voltage of LCO (i.e., 4.5 V vs. Li/Li+). As shown in Fig. S5d–e (ESI†), there is no obvious redox peak in the CV curves within the working voltage of LCO, and the charge–discharge curves during long-term cycling are also highly reversible. Such results suggest that the spinel LCTO crystal stays stable under even relatively high voltage and provided a fast diffusion channel for Li ions.
As mentioned previously, the chemical stability of the interface between the cathode and the solid electrolyte is crucial for the cycling performance of the relevant all solid state batteries (ASSLIBs). Particularly for sulfide-based solid electrolytes, taking the LGPS for example, with an oxidation limit as low as 2.1 V vs. Li/Li+, the voltage mismatch issue between the LCO cathode material (with working potential at 3.9 V vs. Li/Li+) and the LGPS often leads to continuous interface side reactions that facilitate the degradation of cycling performance of ASSLIBs.25–27 When assembled in an ASSLIB, with the introduction of the LCTO interlayer, the original interface LCO/LGPS is substituted by two new ones, namely the LCO/LCTO and the LCTO/LGPS. Herein, the mutual reaction energy, ΔED,mutual, at different interfaces is calculated and employed as an effective criterion to evaluate the strength of interfacial chemical reactions, as shown in the Fig. 2d. The details about phase equilibria and decomposition energies of the LGPS/LCO and LGPS/LCTO interfaces are provided in Table S3 (ESI†). It is clear that the decomposition enthalpy of LGPS/LCTO is much lower than that of LGPS/LCO, indicating that the interface between LGPS and LCTO is more thermodynamically stable. Thus, theoretically, the introduction of the ultrathin LCTO interlayer could block the undesirable but thermodynamically favorable chemical reaction between the LGPS and LCO.28–30
Moreover, the relatively insulating nature of LCTO provides a solution to the inherent voltage mismatch issue between the LCO and the LGPS, thus ensuring the interfacial electrochemical stability between the LCTO and LGPS during the charging and discharging process. Pure LCTO is characterized with a large band gap (2.315 eV, from Material Project Data basis, id: mp-1177906), and the electronic conductivity of LCTO, bare LCO and LCTO@LCO were also measured through the method of direct-current (DC) polarization.31 DC polarization was performed on the dense pellets of LCO and LCTO, which were applied at a constant voltage of 0.5 V at 30 °C. As shown in Fig. 2c, pure LCTO exhibits low electronic conductivity of 2.5 × 10−8 S cm−1, which corresponds to its large band gap; the bare LCO shows electronic conductivity of 8.8 × 10−3 S cm−1, which is much higher than LCTO; the electronic conductivity of LCTO@LCO composites decreases by an order of magnitude (6.6 × 10−6 S cm−1) compared with that of the bare LCO. As depicted in Fig. 2e, the profile of electronic conductivity strongly supports that the LCTO layer can make sufficient potential drop and thus solve the potential mismatch issue long harassing the interface of LCO/LGPS; such a hypothesis could also be supported by relevant DFT calculations and experimental observation reported previously.5,13
In addition to the reasonable Li-ion mobility, chemical stability and low electronic conductivity, the LCTO layer could also enhance interface contact between the cathode and the solid electrolyte. As depicted in Fig. 2f–h, the interfacial compatibility of LCO in contact with LGPS, before and after coating by LCTO, that is, the three interfaces LCO/LGPS, LCO/LCTO and LCTO/LGPS, were further studied by modeling the interfacial structures at atomic levels. The calculated values of the work of adhesion (Wad) for interfaces, which is defined as the reversible work of creating the relevant free surfaces from a connected interface, are presented in Fig. 2f–h under each interfacial structure; a larger Wad is believed to imply higher chemical affinity between the two surfaces of concern. The Wad of the relevant interface LCO/LGPS is calculated to be −0.39 J m−2. After the introduction of the LCTO interlayer, two interfaces, i.e., LCO/LCTO and LCTO/LGPS, display Wad values as −0.59 J m−2 and −0.55 J m−2, respectively, both of which are evidently larger than that of the LCO/LGPS interface. The larger Wad of the interfaces between LCO/LCTO or LCTO/LGPS proves the affinity between the LGPS with both the cathode materials and the solid electrolyte; in this scenario, our ultrathin LCTO interlayer provides a favorable bridge that connects tightly with LCO and LGPS during cycling, preventing generation of gaps to increase impedance. With enhanced solid–solid interfacial contact between the cathode and the solid electrolyte, the interfacial impedance of ASSLIBs thus would be effectively reduced.
Electrochemical performance of ASSLIBs assembled with the LCTO interlayer
The performance of the ASSLIBs with the presence of the LCTO interlayer (i.e., our LCTO@LCO cathode) between the LCO and LGPS was evaluated at 25 °C and was compared with that of the control samples including the bare LCO, TiO2–LCO, and TiO2@LCO cathodes, as shown in Fig. 3a. At 0.1C (1C = 150 mA h g−1), the bare LCO delivers an initial capacity of 98 mA h g−1 and only retains 22.4% of the initial capacity after 100 cycles. The TiO2–LCO cathode exhibits a slightly enhanced initial capacity of 105 mA h g−1 and 31.9% retention after 100 cycles; this improvement can be attributed to the presence of TiO2 nanoparticles, which prevents direct contact between LCO and LGPS, thus partly reducing the side reactions. The TiO2@LCO delivers an initial capacity of 132 mA h g−1, but the capacity still fades rapidly, with a retention of only 61.4% after 100 cycles. Compared with the above three control samples, the cycling stability of our LCTO@LCO is notably improved, which yields a higher initial capacity of 140 mA h g−1 with a retention of 82.9% after 200 cycles.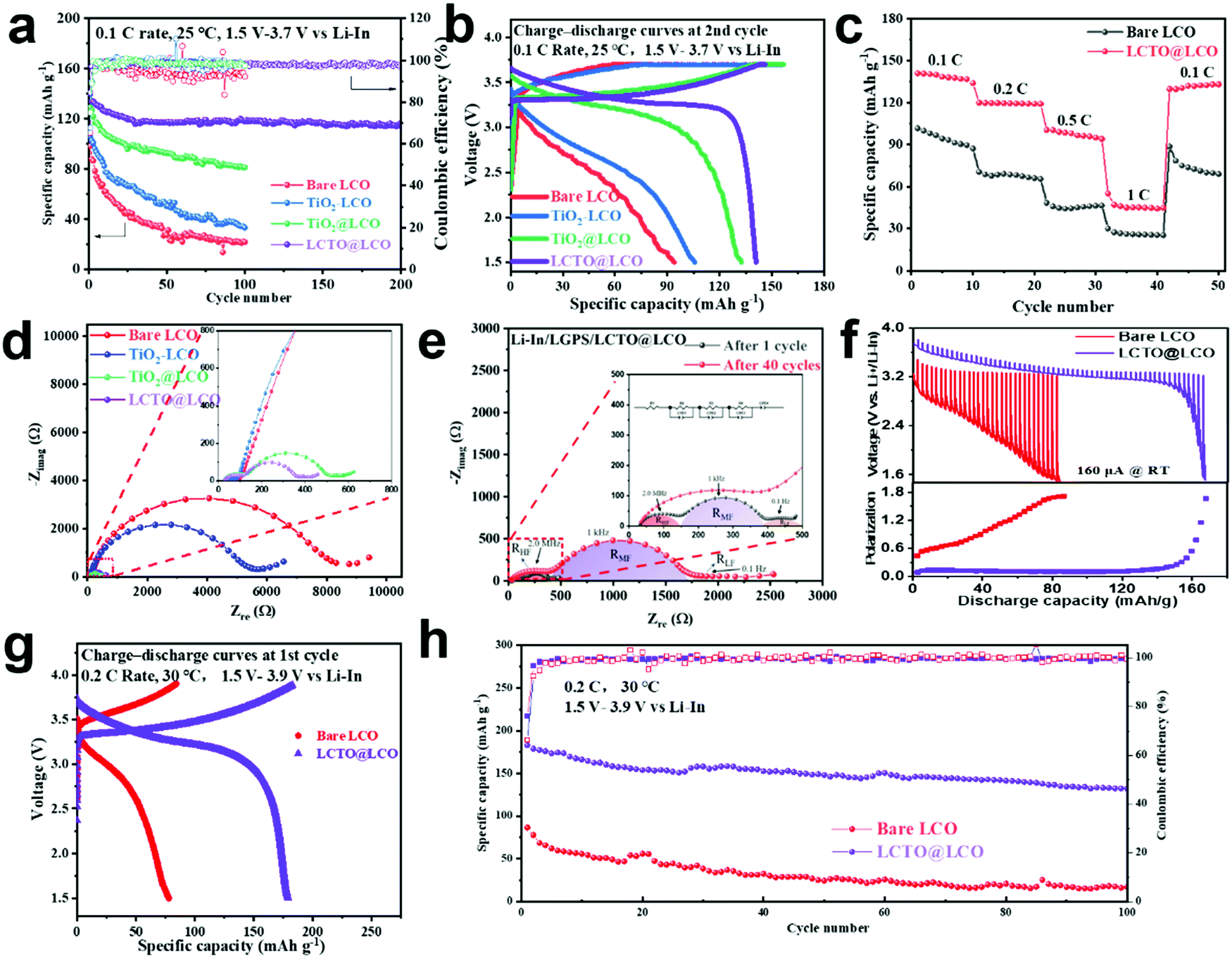 | ||
| Fig. 3 Electrochemical performance of ASSLIBs with the bare LCO, TiO2–LCO, TiO2@LCO, and LCTO@LCO: (a) cycling stability and coulombic efficiency, (b) charge–discharge curves at the 2nd cycle, and (c) rate performance; (d) Nyquist plots of the four ASSLIBs after the first charging; (e) Nyquist plots of the LCTO@LCO after the 1st and 40th cycles; (f) Galvanostatic Intermittent Titration Technique (GITT) curves and battery polarization of the bare LCO and LCTO@LCO. High-voltage electrochemical performance of ASSLIBs with the bare LCO and LCTO@LCO: (g) charge–discharge curves at the 2nd cycle; (h) cycling stability and Coulombic efficiency. | ||
The rate capability of the cathodes was then evaluated (Fig. 3c). The LCTO@LCO indicates a higher capacity and smaller polarization than the bare LCO cathodes at different current densities (Fig. S6, ESI†). When the current density increased from 0.1C to 0.2C, the capacity of the bare LCO was 70 mA h g−1, transforming into a sharp decrease of 30%, while the LCTO@LCO cathode retained a capacity of 120 mA h g−1, which corresponds to a slight decrease of only 10%. The LCTO@LCO also delivers higher capacity than the bare LCO at relatively high current density. High-rate cycling tests were also performed at 1C (150 mA g−1). As shown in Fig. S6 (ESI†), when the current density increased to 1C, there is an obvious increase in polarization of ASSLIBs. However, our LCTO@LCO still exhibits good cycling stability with a highly reversible capacity of 71.4 mA h g−1 after 50 cycles (95.3% retention), indicating that the LCTO interlayer could efficiently suppress the side reaction between LCO and LGPS, even at a high current density. In comparison, the bare LCO electrode shows larger polarization and faster capacity fading than that in LCTO@LCO.
The difference in the interfacial compatibility is reflected in the charge–discharge curves and Electrochemical Impedance Spectroscopy (EIS) tests. The voltage profiles clearly suggest a remarkable decrease in the reaction polarization for LCTO@LCO in the charge–discharge curves, compared with the bare LCO electrode. The LCTO@LCO cathode has a stable plateau, higher discharge voltage and lower overpotential (Fig. 3b). EIS tests can explain these results from the aspect of interfacial kinetics. The relevant impedance spectra are shown in Fig. 3d. Additionally, the EIS spectra were fitted with the equivalent circuit (the inset of Fig. 3e). The semicircular intercept at high-frequency (∼MHz) represents the interfacial impedance (RHF) of the cathode material and the solid electrolyte.13 After the charging process in the first cycle, RHF in the LCTO@LCO cathode drops to 110 Ω cm−2, while RHF in the bare LCO is 8363 Ω cm−2 and RHF in TiO2–LCO is 5414 Ω cm−2. Moreover, as compared in Fig. 3e and Fig. S6c, the RHF of LCTO@LCO increases slightly to 454 Ω cm−2 after the 40th cycle, while the RHF of the bare LCO increases to over 40000 Ω cm−2. A large increase in RHF can be explained through the decomposition of solid electrolytes into substances with a low ionic conductivity. Such comparative EIS tests indicate severe interfacial side reactions occurring upon the solid–solid interface for the battery constructed with the bare LCO cathode. The middle frequency region, which corresponds to the cathode/solid electrolyte interface, also increased from 231 Ω cm2 to 1320 Ω cm2 after 40 cycles, indicating slight degradation of the interface stability with the ASSLIBs cycling.
The electrochemical performances of the relevant ASSLIBs at high cutoff voltage (3.9 V vs. Li–In, i.e., 4.5 V vs. Li/Li+) are also tested at 30 °C with a rate of 0.2C. Increasing the charging cutoff voltage of LCO means that the potential mismatch between the LCO and the LGPS electrolyte will be further aggravated, which thus sets even higher requirement on the stability of the cathode–electrolyte interface. Encouragingly, the ASSLIB with the LCTO interlayer exhibits a higher initial capacity of 180 mA h g−1 with a retention of 73.3% after 100 cycles; whereas the bare LCO can only deliver an initial capacity of 86 mA h g−1 with a retention of 19.9% after 100 cycles (Fig. 3h). There are also obvious differences between these two samples in the early charge–discharge curves (Fig. 3g). Due the aggravating potential mismatch at such high cutoff voltage, the battery with the bare LCO shows dramatical polarization and the plateau almost disappeared in the charge–discharge, whereas the ASSLIB with the LCTO@LCO cathode has a stable plateau, relatively high discharge voltage and low overpotential. The Galvanostatic Intermittent Titration Technique (GITT) was carried out to evaluate the battery polarization of the bare LCO and LCTO@LCO (Fig. 3f). Obviously, the bare LCO battery shows much larger polarization than that of LCTO@LCO, indicating the better kinetic properties of the LCTO@LCO battery, which is also consistent with previous EIS results. Such comparative results prove that the presence of the LCTO interlayer can significantly enhance the interfacial compatibility between the LCO and the LGPS. Moreover, to balance the electronic conductivity and interfacial stability at high cutoff voltage, the loading and the thickness of the LCTO coating layer have been optimized. As shown in Fig. S7 (ESI†), under high loading of LCTO, e.g., 2% wt, the ASSLIB would experience severe polarization due to poor electronic conductivity; nevertheless, when the coating layer is too thin, e.g., with a mass fraction of 0.25% wt for LCTO, the cycling stability would be impaired due to the occurrence of interfacial side reactions.
Compositional and structural analysis of the cathode–electrolyte interface after electrochemical cycling
To evaluate the interfacial stability, the surface of the bare LCO cathode and the LCTO@LCO after 40 cycles were investigated by XPS analysis; the elements S, P and Ge that originate from the decomposition of the solid electrolyte LGPS were analyzed (Fig. 4). For comparison, the XPS spectra of S 2p, P 2p and Ge 3d of the pristine LGPS are shown in Fig. 4a–c. In the S 2p spectra of the LCTO@LCO cathode, similar to the LGPS, it exhibits two major peaks at 161.2 eV and 162.3 eV, which are ascribed to the S 2p3/2 and S 2p1/2 of the (P/Ge) –S–Li species in LGPS.32 Compared with those of the pristine LGPS, the S 2p spectrum of the bare LCO and LCTO@LCO cathode shows two additional strong peaks at 163.0 eV and 164.1 eV, resulting from –S–S– or CoSx species generated from the decomposition of the solid electrolyte,33 while the peak density associated with the decomposition product is much less in LCTO@LCO than that in the bare LCO; there are also more SOx species detected at a high binding energy (∼169.5 eV) on the surface of the cycled bare LCO cathode.13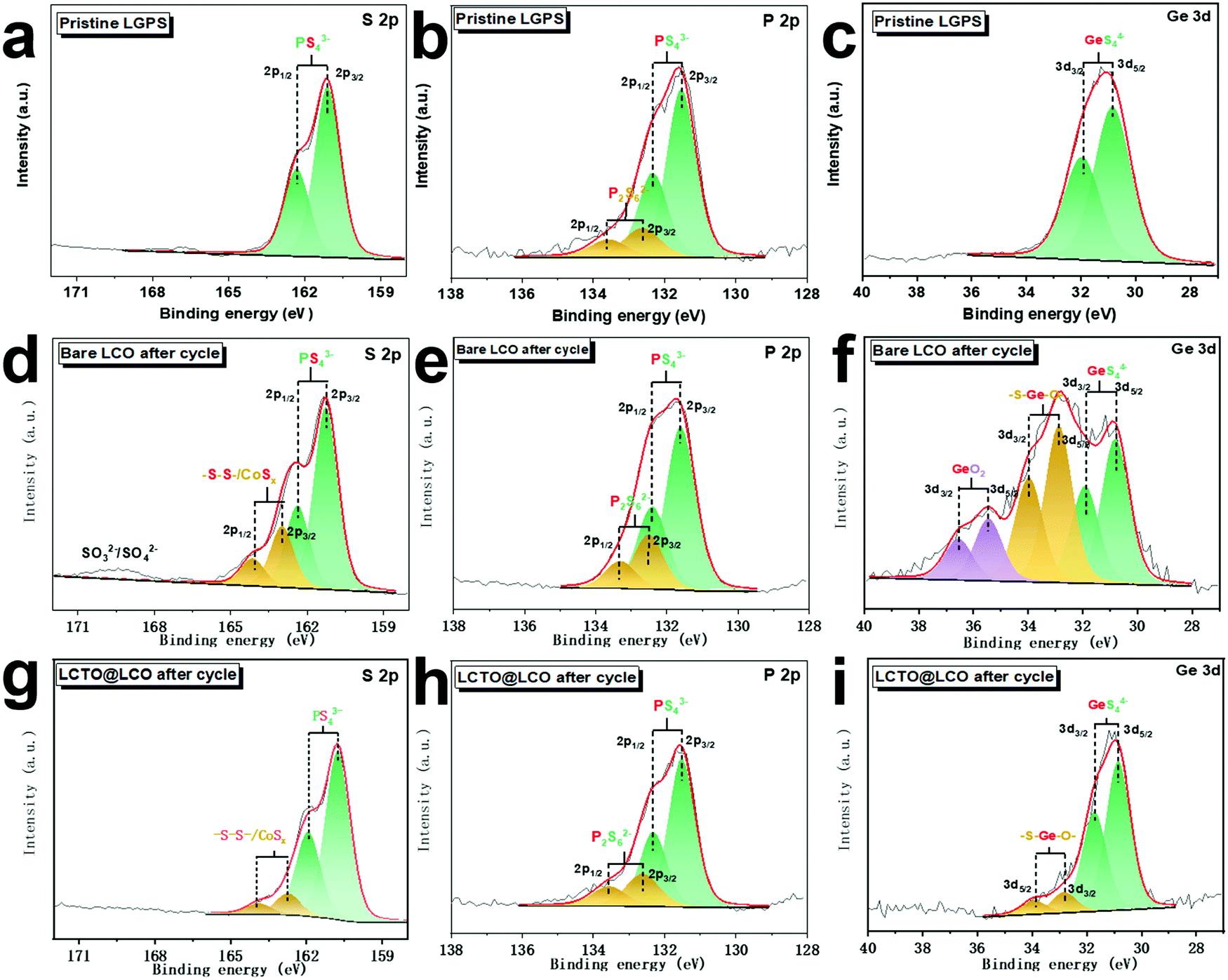 | ||
| Fig. 4 XPS spectra of S 2p, P 2p and Ge 3d for the (a–c) pristine LGPS, (d–f) the bare LCO after 40 cycles and (g–i) LCTO@LCO after 40 cycles. | ||
The differentiated oxidation degree of the LGPS solid electrolyte between the bare LCO and the LCTO@LCO were also reaffirmed by comparing their P 2p and Ge 3d spectra. As shown in Fig. 4b, e and h, the two major peaks from the deconvolution of the P 2p signals at 132.3 eV and 131.5 eV could be assigned to PS43− species; in addition to these two peaks, two more bands centered at 132.6 eV ∼133.6 eV were found, which could be assigned to P2S62− species where P is highly oxidized during the cycling process. Among the three samples, the bare LCO after 40 cycles exhibited the highest fraction of P2S62− species.34 Such a comparative result reconfirmed that the decomposition reaction of the LGPS is effectively suppressed by the LCTO coating layer during the charging and discharging process in the LCTO@LCO cathode. Similarly, in the Ge 3d spectra, the pristine LGPS shows the main chemical state of GeS44− at 30.2 eV, while the LCTO@LCO and the bare LCO samples display additional peaks at higher binding energy due to the oxidation of the solid electrolyte. Specifically, in the bare LCO sample, partially oxidized GeS44− with binding energy at 31.5 eV35 and GeO2 at 32.4 eV36 were found. In comparison, in the LCTO@LCO sample, only oxidized GeS44−(–S–Ge–O–) is found and the relevant peak has weaker intensity than that of the LCO.
The X-ray absorption fine structure (XAFS) technique was employed to reveal the finer local structure.37,38 Co K-edge XAFS spectra of bare LCO, LCTO@LCO and LCTO@LCO pellets after 100 cycles were investigated to extract quantitative structural parameters. Fig. 5a shows the extended X-ray absorption fine structure (EXAFS) spectrum and Fig. 5b shows their fitting Fourier transforms of Co K-edge EXAFS. EXAFS fitting parameters at the Co K-edge of various samples are collected in Table S4 (ESI†).
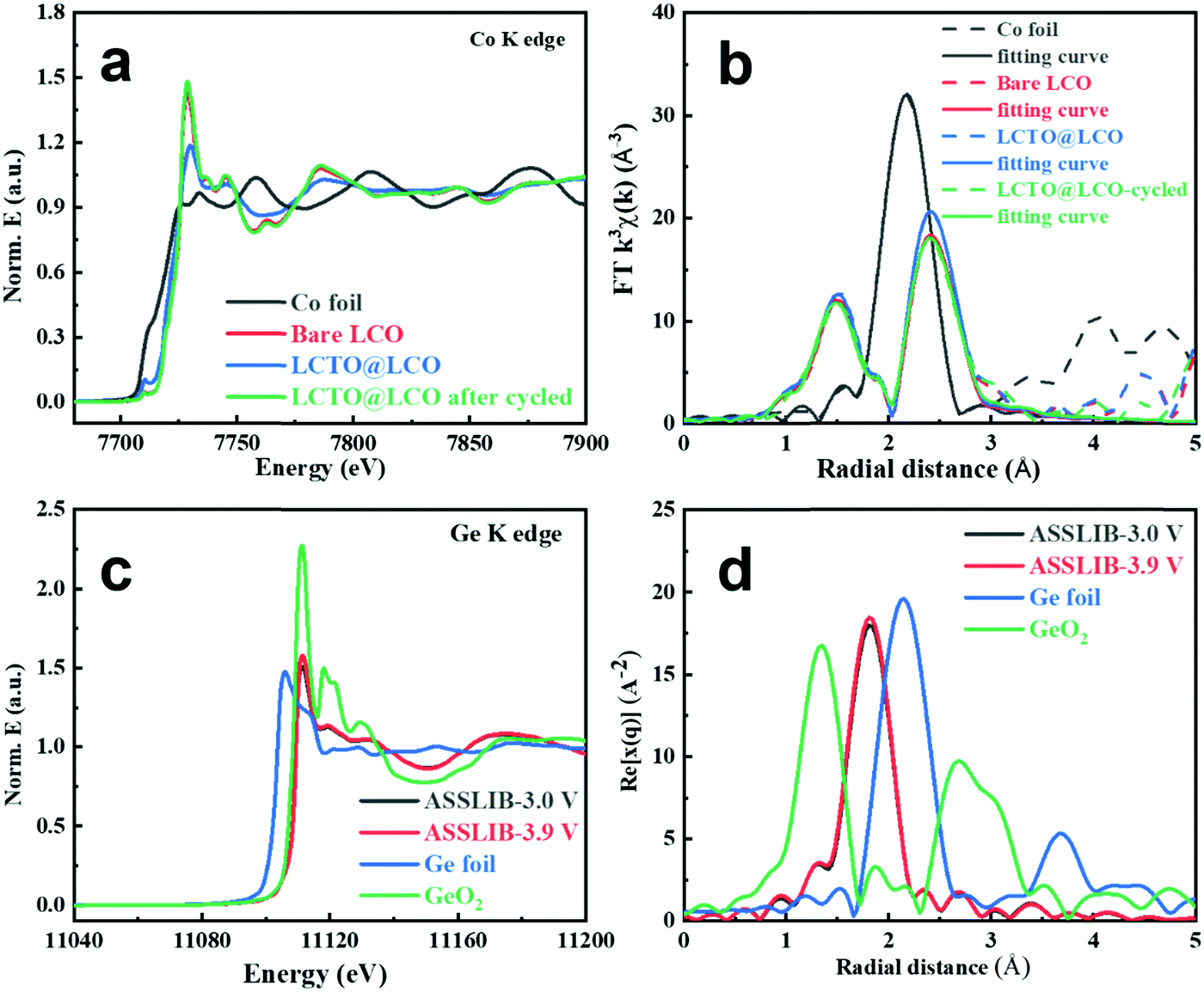 | ||
| Fig. 5 XAFS analysis of Co foil, the bare LCO, the pristine LCTO@LCO and LCTO@LCO after 40 cycles: (a) normalized Co K-edge XANES spectra, (b) Fourier transforms of the Co K-edge and fitting results; (c) normalized Ge K-edge XANES spectra of Ge foil, GeO2, and LCTO@LCO cathode pellets of all-solid-state batteries (ASSLIBs) at different charging voltages, and (d) Fourier transforms of Ge foil, GeO2, and ASSLIBs with the LCTO@LCO cathode at different charging voltages. | ||
From the fitting results of the Co K-edge, the bond distances of Co–O and Co–Co are similar among the three samples including the bare LCO, LCTO@LCO and LCTO@LCO-cycled (Co–O = 1.91 Å and Co–Co = 2.81 Å, respectively), indicating that the bond distance of the cathode materials does not change significantly before and after coating and cycling. More importantly, the fitting results of the bond distance illustrates that there is no significant CoSx (i.e., Co–S bonds) generation after LCTO@LCO being cycled.
In sulfide-based ASSLIBs, due to the instability of the solid–solid electrolyte interface, it is easy to generate CoSx and GeOx at the SE–cathode interface (as investigated in XPS analyses), which greatly hinders the lithium-ion diffusion at the cathode–electrolyte interface, and further deteriorates the performance of ASSLIBs. The signal of Ge in the solid electrolyte for the battery constructed with LCTO@LCO after being charged at different potentials (namely ASSLIB-3.0 V and ASSLIB-3.9 V) were obtained and compared with those of Ge and GeO2 as the reference (Fig. 5c). The valence of Ge in the solid electrolyte is 4+, and its oxidative state exhibits no change at different potentials, proving the excellent interface stability owing to the introduction of the LCTO coating layer. Accordingly, as shown in Fig. 5d, the LCTO@LCO cathode pellets charging to 3.0 V and 3.9 V both only show the occurrence of the Ge–S coordination; no presence of Ge–O and Ge–Ge coordination could be found, which indicates the absence of GeOx in the battery being charged under different potentials.
Thus, the absence of CoSx as well as the GeOx in the LCTO@LCO cathode indicates that the LCTO layer can effectively suppress the side reactions between LCO and LGPS both in the repetitive cycles and at different charging potentials. The XAFS investigation supports our findings from the perspective of local electronic environment, which is consistent with the XPS analysis and supports the enhanced electrochemical performance (including cycling stability, the charge–discharge curves and the impedance spectra before and after cycling) of the LCTO@LCO cathode.
The structural stability of the cathode active materials is also of great concern. A comparison among the mechanical properties of LCO, LCTO and other well-known coating materials is listed in Table S8 (ESI†), which shows that our LCTO possesses strong mechanical strength; thus, it could tolerate the volume variation and prevent the occurrence of structural fractures for the LCO core during the cycling process. The bare LCO and LCTO@LCO after cycling were also subjected to cross-sectional SEM observation. As shown in Fig. 6, the cycled bare LCO electrode exhibited cracks and fractures, whereas most LCO particles protected by the LCTO interlayer remained intact. The analysis of the elemental distributions at the cross section of ASSLIBs further revealed that the distribution of Ti was still uniform on the encapsulated LCO particles, separating the LCO cathode from LGPS, while the existence of massive S was found along the cracks deep into the bare LCO. Such comparative results proved the important role of LCTO as a coating layer in stabilizing the LCO active materials.
 | ||
| Fig. 6 Cross-sectional SEM images of (a and b) the bare LCO and (d and e) LCTO@LCO; the relevant EPMA mapping images of various elements of (c) the bare LCO and (f) LCTO@LCO. | ||
Conclusions
A spinel Li2CoTi3O8 layer, with excellent structural stability, high lithium diffusion coefficient (DLi+ = 8.22 × 10−7 cm2 s−1) and relatively low electronic conductivity (2.5 × 10−8 S cm−1), is for the first time formed in situ at the LCO surface via the chemical reaction between LCO and TiO2 with carbon and Li2CO3 as critical additives. With the introduction of such an ultrathin LCTO interlayer (∼1.5 nm) into the interface of LCO cathode materials and LGPS solid electrolyte, the performance of the sulfide-based all-solid-state lithium-ion batteries has significantly improved. A high initial capacity of 140 mA h g−1 and a reversible discharge specific capacity of 116 mA h g−1 after 200 cycles at room temperature (0.1C) were delivered. At a high cutoff voltage (e.g., 4.5 V vs. Li/Li+), the battery could also exhibit a high initial capacity of 180 mA h g−1 and a considerable retention of 73.3% after 100 cycles. DFT calculations revealed that the new interface of LCTO/LGPS arising from the introduction of the LCTO interlayer is endowed with inherently high chemical affinity in comparison with the pristine LCO/LGPS interface, which significantly reduces the interface impedance. The LCTO/LGPS interface also has higher interfacial compatibility which offers an effective solution to solve the inherent thermodynamic and electrochemical mismatch issue between the LCO and the LGPS. Our finding puts an emphasis on the controlled synthesis and the favorable interfacial engineering effect of LCTO as an effective interlayer between the cathode and sulfide-based solid electrolyte for high-performance sulfide-based ASSLIBs.Methods
Reaction between LiCoO2 and TiO2
The LCTO, namely the stoichiometric Li2CoTi3O8 powder, was synthesized by a solid-state method. TiO2 (20 g, 99.8%, 5-10nm, anatase, Aladdin), LiCoO2 (8.16 g, 99.5%, Xiamen Tungsten Co., Ltd), Li2CO3 (3.08 g) and acetylene black (8 g) were mixed, followed by a heat treatment at 800 °C in a muffle furnace for 3 h. A control experiment without the presence of carbon and Li2CO3 was also performed. TiO2 (20 g) and LiCoO2 (8.16 g) were mixed, followed by heat treatment at 800 °C in a muffle furnace for 3 h.Synthesis of LiCoO2 materials
The bare commercial LiCoO2 powder (bare LCO) employed in this work was provided by Xiamen Tungsten Co., Ltd (XTC). The precursor TiO2–LiCoO2 (TiO2–LCO) was prepared by mixing TiO2 (1.5 g) with bare LCO powders (300 g) in a high-speed solid-phase mixing machine (NOBILTA-130, Hosokawa Micron, Japan) at 5000 rpm min−1 for 20 minutes. The LCTO@LCO, namely, was obtained by mixing as-prepared TiO2-LCO (50 g) mixture with Li2CO3 (0.24 g) and acetylene black (0.1 g), followed by annealing at 800 °C in a muffle furnace for 3 h. The TiO2@LCO, was obtained by annealing the as-prepared TiO2–LCO at 800 °C in a muffle furnace for 3 h (without the presence of carbon and Li2CO3).Electrochemical measurements
Li-In alloy/LGPS/LCO ASSLIBs were assembled to examine the electrochemical performance of the LCO materials. The cathode mixtures consisted of LCO, LGPS and acetylene black in a weight ratio of 60:38:2. The cathode mixture was prepared by hand grinding for 10 min. Then 10 mg cathode mixtures and 80 mg LGPS were pressed into the pellet under 360 MPa (φ = 10 mm, with 10 mg cm−2 active material) for 10 min. 10 mg Li–In alloy (Li:In = 1:6) was placed on the cathode and solid electrolyte pellets followed by 20 MPa cold pressing to form ASSLIBs. All the preparation processes of the ASSLIBs mentioned above were carried out in an Ar-filled glovebox.
2025-Type-coin-cells were assembled in an Ar-filled glove-box with lithium metal as the anode and 1 M LiPF6 in ethylene carbonate (EC)/diethyl carbonate (DEC) (1:1, v/v) as an electrolyte. The prepared LCTO powder was mixed with acetylene black and polyvinylidene fluoride binder with a weight ratio of 8:1:1 in N-methyl pyrrolidone solvent and then cast on a clean Al foil, which was dried under vacuum at 100 °C overnight.
The prepared ASSLIBs were assembled in a test mold with good tightness and the cycling performance was evaluated at the voltage range of 1.5–3.7 V (vs. Li–In), which corresponds to 4.3 V vs. Li/Li+, with a current density of 0.1C rate (i.e. 15 mA g−1, 1C = 150 mA h g−1) at 25 °C. High voltage performance of ASSLIBs was evaluate at the voltage range of 1.5–3.9 V (vs. Li–In), with a current density of 0.2C rate (i.e. 30 mA g−1,1C = 150 mA h g−1) at 30 °C. The coin cells of LCTO were tested on a LAND-2001A battery tester (Land, Wuhan, China) with a cut-off voltage of 0.05–3.0 V (vs. Li/Li+) at 25 °C. Cyclic voltammetry (CV) tests were carried out on a CHI 660D electrochemical workstation (Shanghai, China) using a coin cell with a different scan rate of 0.1 mV s−1, 0.3 mV s−1, 0.5 mV s−1, and 0.7 mV s−1. The electrochemical impedance spectroscopies (EIS) of solid and liquid batteries were all tested on a CHI 660D electrochemical workstation (Shanghai, China) at an AC perturbation signal of 50 mV and frequencies from 200 MHz to 0.01 Hz. The GITT was performed with a 5 min discharge at 160 μA followed by 2 hours of relaxation.
Material characterization
XRD, SEM and XPS characterization: X-ray diffraction (XRD, Rigaku D/Max-IV) was used to determine the phase structure of the samples. The morphology of the materials was investigated via field emission scanning electron microscopy (FESEM, S4800, Hitachi). X-ray photoelectron spectroscopy (XPS, PHI 500 VB III) was used to analyze the surface of the electrodes with Al Kα (14866.6 eV) radiation as the primary excitation source operated at 150 W.EPMA and TEM characterization: To characterize the interface of the entire LCO particles and the elemental distributions of the cross sections, electron probe microanalysis (EPMA, JXA8530F, JEOL Co.) was used on the polished LCO cross sections. Cross section samples for EPMA were prepared by ion beam polished with Cross Section Polisher (IB-09010CP, JEOL Co.,). A high-resolution transmission electron microscope (HRTEM) test was used to investigate the exact thickness and surficial structure of the samples. TEM samples was prepared by focused ion beam (FIB) conducted on an FEI Helios Dual Beam FIB.
X-ray absorption fine structure (XAFS) characterization: the X-ray absorption spectra were used to analyze the local electronic structure of LCTO@LCO before and after electrochemical cycling at Shanghai Synchrotron Radiation Facility (SSRF). The obtained XAFS data were processed in Athena (version 0.9.25) for background, pre-edge line and post-edge line calibrations. Then Fourier transformed fitting was carried out in Artemis (version 0.9.25). The k3 weighting, k-range of 2–12 Å−1 and R range of 1–3 Å were used for the fitting. The models of bulk Co and CoO2 were used to calculate the simulated scattering paths. The four parameters, coordination number, bond length, Debye–Waller factor and E0 shift (CN, R, σ2, ΔE0) were fitted without any one being fixed, constrained, or correlated.
First-principle calculations
The density functional theory (DFT) calculations were carried out by using the Vienna ab initio Simulation Package.39 The projector-augmented wave (PAW) approach was used to take the electron–ion interaction into account,40 and the electron exchange–correlation interactions were described using the generalized gradient approximation (GGA) and parameterized by Perdew–Burke–Ernzerhof (PBE) formula.41 The kinetic cut-off energy of the plane-wave basis was set to 500 eV. Brillouin-zone integrations were sampled using special k-point sampling of the Monkhorst–Pack scheme42 with a k-point mesh of 1 × 4 × 1. The self-consistency convergency criterion for the energy was set to be 10−4 eV and the geometric relaxation convergency criterion was set to be less than 0.05 eV Å−1 for all forces.The interfacial structures of LCO/LGPS, LCO/LCTO and LCTO/LGPS were built by using the fast-ion-transport surface of LCO (110), LCTO (001) and LGPS (010),43–45 respectively. 3 × 1 × 1, 2 × 1 × 1 and 5 × 3 × 1 super cells of LCTO (001), LGPS (010) and LCO (110) were built to construct the interface layers. Only the one interface is contained in each supercell, and a vacuum region of 20 Å was added to avoid the interaction between the adjacent interface layers. The optimized structure parameters are listed in Tables S5 and S6 (ESI†).
The work of adhesion for the interface (Wad) was calculated by the following equation:46
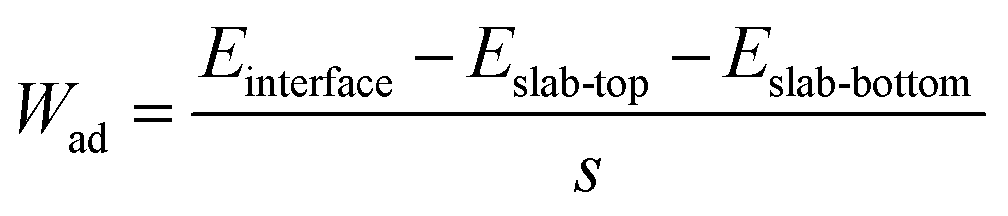
Material project analysis
Using the scheme proposed by Mo et al.,47 the solid electrolyte (SE)-cathode can be considered as a pseudo-binary composition| Cinterface(CSE,Celectrode,x) = x·CSE + (1 − x)·Celectrode |
| Einterface(SE,electrode,x) = x·E(SE) + (1 − x)·E(electrode) |
| ΔED(phase) = Eeq(C) − E(phase) |
| ED(SE, electrode, x) = Eeq(Cinterface(CSE, Celectrode, x)) − Einterface(SE, electrode, x) |
| ΔED, mutual(SE, electrode, x) = ΔED(SE, electrode, x) − x·ΔED(SE) − (1 − x)ΔED(electrode) |
Author contributions
Chuan-Wei Wang conceived and designed the experiments. Chuan-Wei Wang synthesized the materials, did the material characterization and performed the electrochemical measurements. Fu-Cheng Ren carried out DFT calculations and material project analysis. Chuan-Wei Wang, Yao Zhou and Xiao-Dong Zhou wrote the paper. Peng-Fei Yan prepared the FIB sample and did STEM. Xia-Yin Yao performed the electrochemical measurements of ASSLIBs. All authors analyzed the data, discussed the results and commented on the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was financially supported by the Xiamen Science and Technology Project (3502Z20201012) and the National Key Research and Development of China (2016YFB0100202).References
- J. B. Goodenough and Y. Kim, Chem. Mater., 2009, 22, 587–603 CrossRef.
- E. Quartarone and P. Mustarelli, Chem. Soc. Rev., 2011, 40, 2525–2540 RSC.
- L. Fan, S. Wei, S. Li, Q. Li and Y. Lu, Adv. Energy Mater., 2018, 8, 1702657 CrossRef.
- N. Kamaya, K. Homma, Y. Yamakawa, M. Hirayama, R. Kanno, M. Yonemura, T. Kamiyama, Y. Kato, S. Hama and K. Kawamoto, Nat. Mater., 2011, 10, 682 CrossRef CAS.
- F. Han, Y. Zhu, X. He, Y. Mo and C. Wang, Adv. Energy Mater., 2016, 6, 1501590 CrossRef.
- Y. Xiao, L. J. Miara, Y. Wang and G. Ceder, Joule, 2019, 3, 1252–1275 CrossRef CAS.
- N. Ohta, K. Takada, I. Sakaguchi, L. Zhang, R. Ma, K. Fukuda, M. Osada and T. Sasaki, Electrochem. Commun., 2007, 9, 1486–1490 CrossRef CAS.
- N. Ohta, K. Takada, L. Zhang, R. Ma, M. Osada and T. Sasaki, Adv. Mater., 2006, 18, 2226–2229 CrossRef CAS.
- J. Kim, M. Kim, S. Noh, G. Lee and D. Shin, Ceram. Int., 2016, 42, 2140–2146 CrossRef CAS.
- K. Okada, N. Machida, M. Naito, T. Shigematsu, S. Ito, S. Fujiki, M. Nakano and Y. Aihara, Solid State Ionics, 2014, 255, 120–127 CrossRef CAS.
- K. Takada, Acta Mater., 2013, 61, 759–770 CrossRef CAS.
- K. Takahashi, K. Hattori, T. Yamazaki, K. Takada, M. Matsuo, S. Orimo, H. Maekawa and H. Takamura, J. Power Sources, 2013, 226, 61–64 CrossRef CAS.
- W. Zhang, F. H. Richter, S. P. Culver, T. Leichtweiss, J. G. Lozano, C. Dietrich, P. G. Bruce, W. G. Zeier and J. R. Janek, ACS Appl. Mater. Interfaces, 2018, 10, 22226–22236 CrossRef CAS.
- L. Wang, Q. Xiao, Z. Li, G. Lei, L. Wu, P. Zhang and J. Mao, Electrochim. Acta, 2012, 77, 77–82 CrossRef CAS.
- A. Sakuda, A. Hayashi and M. Tatsumisago, J. Power Sources, 2010, 195, 599–603 CrossRef CAS.
- A. Zhou, X. Dai, Y. Lu, Q. Wang, M. Fu and J. Li, ACS Appl. Mater. Interfaces, 2016, 8, 34123–34131 CrossRef CAS.
- S. Kimura and Y. Suzuki, Ceram. Int., 2019, 45, 12602–12607 CrossRef CAS.
- R. Laskowski and P. Blaha, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 205104 CrossRef.
- J. Zhang, P. Tang, T. Liu, Y. Feng, C. Blackman and D. Li, J. Mater. Chem. A, 2017, 5, 10387–10397 RSC.
- J. Qian, L. Liu, J. Yang, S. Li, X. Wang, H. L. Zhuang and Y. Lu, Nat. Commun., 2018, 9, 4918 CrossRef.
- D. C. Frost, C. A. Mcdowell and I. S. Woolsey, Mol. Phys., 1974, 27, 1473–1489 CrossRef CAS.
- J.-N. Zhang, Q. Li, C. Ouyang, X. Yu, M. Ge, X. Huang, E. Hu, C. Ma, S. Li and R. Xiao, Nat. Energy, 2019, 1 Search PubMed.
- J. Xie, T. Tanaka, N. Imanishi, T. Matsumura, A. Hirano, Y. Takeda and O. Yamamoto, J. Power Sources, 2008, 180, 576–581 CrossRef CAS.
- J. Sugiyama, I. Umegaki, T. Uyama, R. M. McFadden, S. Shiraki, T. Hitosugi, Z. Salman, H. Saadaoui, G. D. Morris and W. A. MacFarlane, Phys. Rev. B, 2017, 96, 094402 CrossRef.
- Q. Zhao, S. Stalin, C.-Z. Zhao and L. A. Archer, Nat. Rev. Mater., 2020, 5, 229–252 CrossRef CAS.
- T. Krauskopf, F. H. Richter, W. G. Zeier and J. Janek, Chem. Rev., 2020, 120, 7745–7794 CrossRef CAS.
- Y. Gao, A. M. Nolan, P. Du, Y. Wu, C. Yang, Q. Chen, Y. Mo and S. H. Bo, Chem. Rev., 2020, 120, 5954–6008 CrossRef CAS.
- J. Haruyama, K. Sodeyama, L. Han, K. Takada and Y. Tateyama, Chem. Mater., 2014, 26, 4248–4255 CrossRef CAS.
- J. Haruyama, K. Sodeyama and Y. Tateyama, ACS Appl. Mater. Interfaces, 2017, 9, 286–292 CrossRef CAS.
- Y. Tateyama, B. Gao, R. Jalem and J. Haruyama, Curr. Opin. Electrochem., 2019, 17, 149–157 CrossRef CAS.
- S. Wang, M. Yan, Y. Li, C. Vinado and J. Yang, J. Power Sources, 2018, 393, 75–82 CrossRef CAS.
- S. Wenzel, S. Randau, T. Leichtweiß, D. A. Weber, J. Sann, W. G. Zeier and J. R. Janek, Chem. Mater., 2016, 28, 2400–2407 CrossRef CAS.
- W. Zhang, T. Leichtweiß, S. P. Culver, R. Koerver, D. Das, D. A. Weber and W. G. Zeier, and J. r. Janek, ACS Appl. Mater. Interfaces, 2017, 9, 35888–35896 CrossRef CAS.
- C. Wang, X. Li, Y. Zhao, M. N. Banis, J. Liang, X. Li, Y. Sun, K. R. Adair, Q. Sun and Y. Liu, Small Methods, 2019, 1900261 CrossRef CAS.
- P. R. Abel, K. C. Klavetter, K. Jarvis, A. Heller and C. B. Mullins, J. Mater. Chem. A, 2014, 2, 19011–19018 RSC.
- V. Mitsa, R. Holomb, O. Kondrat, N. Popovych, N. Tsud, V. Matolín, K. C. Prince, G. Lovas, S. Petretskiy and S. Tóth, J. Non-Cryst. Solids, 2014, 401, 258–262 CrossRef CAS.
- M. Balasubramanian, X. Sun, X. Yang and J. McBreen, J. Power Sources, 2001, 92, 1–8 CrossRef CAS.
- A. Deb, U. Bergmann, S. Cramer and E. J. Cairns, Electrochim. Acta, 2005, 50, 5200–5207 CrossRef CAS.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and D. P. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- M. Stamatakis, J. Phys.: Condens. Matter, 2015, 27, 013001 CrossRef.
- M. Murayama, R. Kanno, Y. Kawamoto and T. Kamiyama, Solid State Ionics, 2002, 154, 789–794 CrossRef.
- Y. Fang, X. Kong, Y. Yu, X. Chen, T. Gao, C. Xiao and T. Lu, J. Phys. Chem. C, 2019, 123, 6477–6486 CrossRef CAS.
- Y. Liu and X. Ning, Comput. Mater. Sci., 2014, 85, 193–199 CrossRef CAS.
- Y. Zhu, X. He and Y. Mo, J. Mater. Chem. A, 2016, 4, 3253–3266 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ee03212c |
| ‡ Chuan-Wei Wang and Fu-Cheng Ren contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |