
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSize-resolved characterization of organic aerosol in the North China Plain: new insights from high resolution spectral analysis†
Weiqi
Xu
a,
Chun
Chen
ab,
Yanmei
Qiu
ab,
Conghui
Xie‡
ab,
Yunle
Chen
c,
Nan
Ma
d,
Wanyun
Xu
e,
Pingqing
Fu
bf,
Zifa
Wang
ab,
Xiaole
Pan
a,
Jiang
Zhu
ab,
Nga Lee
Ng
cgh and
Yele
Sun
 *abi
*abi
aState Key Laboratory of Atmospheric Boundary Layer Physics and Atmospheric Chemistry, Institute of Atmospheric Physics, Chinese Academy of Sciences, Beijing 100029, China. E-mail: sunyele@mail.iap.ac.cn
bCollege of Earth and Planetary Sciences, University of Chinese Academy of Sciences, Beijing 100049, China
cSchool of Earth and Atmospheric Sciences, Georgia Institute of Technology, Atlanta, Georgia 30332, USA
dInstitute for Environmental and Climate Research, Jinan University, Guangzhou 511443, China
eState Key Laboratory of Severe Weather, Key Laboratory for Atmospheric Chemistry, Institute of Atmospheric Composition, Chinese Academy of Meteorological Sciences, Beijing, 100081, China
fInstitute of Surface-Earth System Science, Tianjin University, Tianjin 300072, China
gSchool of Chemical and Biomolecular Engineering, Georgia Institute of Technology, Atlanta, GA 30332, USA
hSchool of Civil and Environmental Engineering, Georgia Institute of Technology, Atlanta, GA 30332, USA
iCenter for Excellence in Regional Atmospheric Environment, Institute of Urban Environment, Chinese Academy of Sciences, Xiamen 361021, China
First published on 24th July 2021
Abstract
Organic aerosol (OA), a large fraction of fine particles, has a large impact on climate radiative forcing and human health, and the impact depends strongly on size distributions. Here we conducted size-resolved OA measurements using a high-resolution aerosol mass spectrometer at urban and rural sites in the North China Plain (NCP) in summer and winter. Our results showed substantially different size distributions of OA with the diameters peaking at ∼550 nm in summer, and 420 nm and 350 nm at urban and rural sites, respectively, during wintertime. Positive matrix factorization (PMF) of size-resolved high-resolution mass spectra of OA resolved various OA factors at urban and rural sites. In particular, we found that the mass spectra of the same type of secondary OA (SOA) from bulk PMF analysis can be largely different across different sizes. Biomass burning OA (BBOA) and fossil-fuel-related OA (FFOA) showed broad size distributions peaking at 350 nm in winter at the rural site, where primary OA (POA = BBOA + FFOA) dominated OA across different sizes. Comparatively, secondary OA (SOA) in the NCP peaked at ∼400–500 nm during wintertime, and ∼500–650 nm in summer. SOA played an enhanced role during more severely polluted days with peak diameters shifting to larger sizes, while the changes in POA size distributions were small. The size-resolved oxygen-to-carbon (O/C) ratios were also determined and linked with the hygroscopicity parameter of OA (κOA). The results showed that κOA increased substantially with particle size, with higher values in summer in Beijing (0.28 ± 0.021) than those during wintertime (0.17 ± 0.019 and 0.12 ± 0.018). The size-resolved κOA would benefit a better prediction of cloud condensation nuclei than bulk κOA in future studies.
Environmental significanceThe impact of organic aerosol (OA) on climate radiative forcing and human health depends strongly on its size distributions. However, our knowledge of size-resolved OA properties, particularly in a highly polluted environment in the North China Plain is very limited. In this work, we demonstrate large variations in the mass spectra and oxidation state of OA as a function of particle size by analyzing the size-resolved high-resolution mass spectra of OA at urban and rural sites in summer and winter. In particular, we found that the secondary OA factors from previous bulk analysis can be substantially different across different sizes. Positive matrix factorization elucidated the largely different size distributions of primary and secondary OA factors between urban and rural sites. We also demonstrate a clear increase in the hygroscopicity parameter (κ) as the particle size increases. The estimated size-resolved κ of OA has significant implications to reduce uncertainties in predicting the number concentrations of cloud condensation nuclei. |
1 Introduction
Aerosol properties, e.g., acidity, hygroscopicity and mixing state,1–3 and their health and climatic effects depend strongly on particle sizes. Aerosol particle sizes change substantially in different chemical environments due to different sources and atmospheric processes, e.g., new particle formation and growth, photochemical production and aqueous-phase reactions. As a result, size-resolved aerosol compositions including inorganic and organic species have been widely characterized worldwide during the past two decades. Among which, the Aerodyne aerosol mass spectrometer (AMS) is one of the unique instruments in quantitively measuring real-time size-resolved non-refractory submicron aerosol species.4–7 The results found that secondary inorganic aerosol (SIA, e.g., sulfate, nitrate, and ammonium) often peaked in accumulation mode with relatively similar peak diameters, suggesting that they were likely internally mixed.7 Comparatively, the size distributions of organic aerosol (OA) were mostly broader than those of SIA due to the contributions of primary emissions at small sizes. For example, OA showed a large increase in the size of 100–300 nm due to the impact of biomass burning at a rural site.8 A considerable fraction of ultrafine mode (<100 nm) in OA was also found at night in Beijing due to the influences of local primary emissions.9 The OA showed negligible seasonal variations in peak diameter (∼500 nm) in Hong Kong,10 while the peak diameter of OA in winter (∼700 nm) was much larger than that in other seasons (∼500–600 nm) in Beijing likely due to extreme haze episodes during wintertime.11 Such a wide range in size distributions of OA was partly related to the different sources and chemical processing in different environments.The size distributions of OA factors can be derived with various approaches. In early studies, the size distributions of primary OA (POA) and secondary OA (SOA) were estimated based on the tracer m/z-based method.12,13 For example, the size distributions of SOA can be derived from that of m/z 44 (mainly CO2+) that has been identified as an excellent tracer for SOA in previous studies.12,14 The difference between OA and SOA was then defined as POA. In addition, the multiple linear regression (MLR) technique15–17 and 3-vector decomposition and vector-matrix decomposition methods18 were also used to derive the size distributions of OA factors. For example, Ulbrich et al.18 found that the peak diameter of hydrocarbon-like OA (HOA) shifted to a larger size in a case with a large secondary aerosol formation event, implying the mix of HOA-containing particles with secondary species. Sun et al.19 found that SOA showed a significant reduction in accumulation mode, while POA showed an increase below 400 nm during periods with strict emission controls in Beijing. We noticed that most previous studies7 assume constant spectral profiles of OA factors for different sizes, which could introduce additional uncertainties in characterization of sources and processes of OA at different size ranges. Also, the size distributions of OA factors were mostly derived from unit mass resolution (UMR) mass spectra, and the changes in oxidation properties of OA factors as a function of size remain poorly understood. Previous studies also found that the evolution of haze episodes was generally associated with increased oxygen-to-carbon (O/C) ratios and peak diameters at urban sites.19,20 However, our understanding of the evolution of size-resolved OA compositions under different particle matter (PM) levels is still limited.
The water uptake capacity of aerosol as indicated by the hygroscopicity parameter (κ) affects atmospheric radiative forcing, aerosol–cloud interactions and atmospheric multiphase chemistry.2 The κ was widely characterized by using a Hygroscopicity Tandem Differential Mobility Analyzer (HTDMA, denoted as κHTDMA), and can also be estimated by the Zdanovskii–Stokes–Robinson (ZSR) mixing rule (denoted as κchem). While the hygroscopicity of inorganic aerosol is well known, our understanding of κ of OA (κorg) is not complete due to the extremely complex OA compositions and aging processes. As a result, uncertainty remains in the estimation of the hygroscopicity parameter and number concentration of cloud condensation nuclei (NCCN). For example, Zhang et al.22 found that NCCN was overestimated by 45% due to the effect of primary emissions in autumn and winter in Beijing, while an underestimation of NCCN (22%) was observed in an environment with dominant highly oxidized aerosol species at a rural site in the North China Plain (NCP) in summer. These discrepancies partly resulted from the κ calculated from the bulk chemical composition. Indeed, many studies characterized κchem by assuming constant κ values for OA, e.g., 0.1 for SOA, 0 for POA23,24 and 0.3 for water-soluble organic compounds (WSOCs).25,26 However, the OA properties varied as a function of size, e.g., oxidation degree indicated by f44 (fraction of m/z 44 in total OA) and O/C, resulting in size-dependence of κ of OA. Using size-resolved rather bulk chemical composition measurements would improve the estimation of hygroscopicity of OA and help to evaluate the impact of OA on CCN formation. Therefore, a more detailed investigation of size-resolved κ in the NCP in different seasons is essential.
In this study, we demonstrate new results from high resolution spectral analysis of size-resolved OA properties at urban (Beijing) and rural (Gucheng) sites in summer and winter. The mass spectra of OA and the tracer ion contributions as a function of sizes are characterized. The OA factors which reflect the sources of different sizes are identified by positive matrix factorization (PMF), and the size-resolved OA compositions and hygroscopicity parameters are investigated.
2 Experimental methods
2.1 Sampling and instrumentation
The size-resolved OA was measured using an Aerodyne HR-AMS in the NCP in summer and winter seasons. The summer and winter campaigns in Beijing were both conducted at an urban site, i.e., of Institute of Atmospheric Physics from 20 May to 23 June 2018, and from 20 November to 25 December 2018, respectively. A detailed description of the sampling site is given in Xu et al.13 The measurements at Gucheng, a rural site located approximately 120 km southwest of Beijing in Hebei province, were conducted from 10 December 2019 to 13 January 2020. More details on the Gucheng site are given in Kuang et al.27 The setup and operation of the HR-AMS were similar to our previous study.28 Briefly, after passing through a PM2.5 cyclone, ambient aerosol particles with a diameter less than 2.5 μm were drawn into the sampling line, dried by a Nafion dryer, and then sampled using the HR-AMS. The HR-AMS was operated in V-mode by cycling between the mass spectrum and particle time-of-flight (PToF) modes every 10 s, and the time resolution was 3 min.2.2 Data analysis and source apportionment
The HR-AMS data were analyzed by PIKA (v 1.62F http://cires1.colorado.edu/jimenez-group/ToFAMSResources/ToFSoftware/index.html, last access: 24 February 2021). The ionization efficiency (IE) was calibrated following the standard protocols.4 The default relative ionization efficiency (RIE) of organics (1.4) was used in this study. The composition-dependent collection efficiency30 was applied for the quantification of non-refractory submicron aerosol (NR-PM1) species, including organics, sulfate, nitrate, ammonium, and chloride. The size-resolved high-resolution mass spectra of OA were also analyzed to determine the size distributions of fragment ions. Due to the limited mass resolution of V-mode and relatively low signal to noise ratios of PToF mode, only m/z's between 12 and 100 were analyzed. In addition, we found that the mass concentrations of OA below 100 nm detected by AMS in Beijing were generally low,9,11 we thereby limited the size-resolved OA analysis in the range of 100–1000 nm which was further binned into 14 size ranges (Table S1†). The size-resolved elemental ratios of OA including O/C, hydrogen-to-carbon (H/C) and organic mass to organic carbon (OM/OC) ratios were estimated with the “Improved-Ambient (I-A)” method.31 Note that the O/C estimated by the I-A method might have additional uncertainties for OA with f44 < 4%. According to recent laboratory studies,32 primary OA with a low oxidation state shows higher RIE than 1.4 used for the total OA in this study. Therefore, the POA concentrations might be overestimated due to the underestimated RIE, particularly for small particles with the dominance of POA. Consistently, the size-resolved POA/SOA ratios would be overestimated to a certain extent as well and the overestimation would be higher in smaller particle size ranges. Because of the potential overestimation of POA, the O/C and f44 of bulk OA are likely underestimated considering the overall lower values of O/C and f44 for POA than SOA. However, due to the challenges in accurate quantification of RIE of POA in field measurements, we used a universal RIE = 1.4 for both POA and SOA.PMF (PET v 3.05C)33,34 was performed to OA in each size bin to resolve OA factors. Similar to Ulbrich et al.,18 we created the error matrices of size-resolved OA using eqn (1).35,36
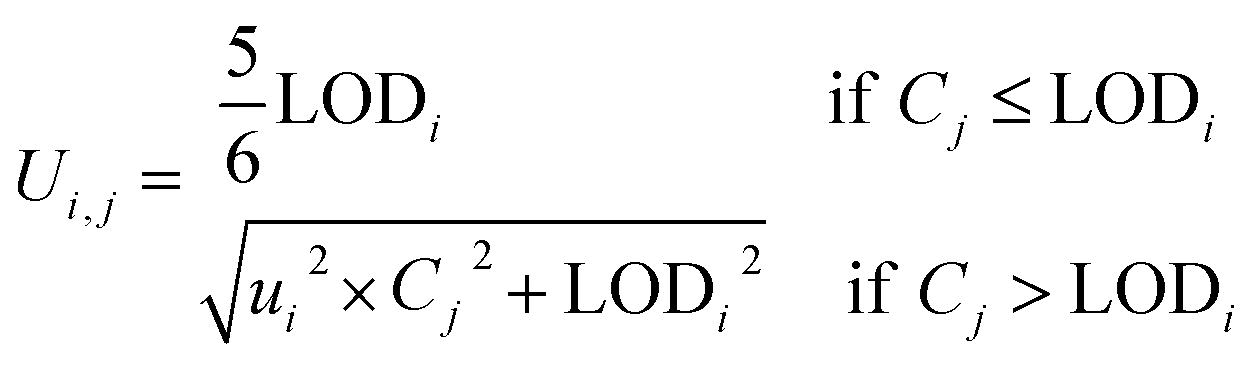 | (1) |
Fig. S2 and S3† show the comparisons of OA composition between size-resolved PMF, size-all PMF, and bulk OA PMF. The fractions of SOA from size-resolved PMF were lower than those from size-all PMF in winter in both Beijing (30.6% vs. 39.9%) and Gucheng (44.8% vs. 54.9%), while they were comparable in Beijing in summer (78.5% vs. 79.2%). However, we noticed that the apportionment of two SOA factors (oxygenated OA (OOA1 and OOA2)) from the size-all and sized-resolved PMF analyses in Beijing summer has some differences. The concentration of OOA1 from size-resolved PMF was overall higher than that from size-all PMF across different sizes (Fig. S3†), while that of OOA2 was lower. We also compared the size-resolved PMF results with those from PMF analysis of bulk OA. The results are overall consistent although the SOA fractions have slight differences in summer and winter seasons. Considering that the mass spectra of OA in different sizes are unlikely the same, we focused on the discussions of size-resolved PMF results unless otherwise stated.
2.4 Estimation of the hygroscopicity parameter
The hygroscopicity parameter (κchem) of aerosol can be estimated using the ZSR rule and chemical composition data (eqn (2)):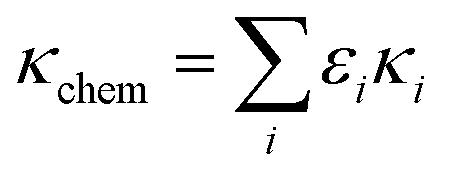 | (2) |
3 Results and discussion
3.1 Characterization of OA size distributions
Fig. 1 presents the average size distributions of OA, CO2+ and C4H9+ during three campaigns. The peak diameter of total OA in summer (∼550 nm) was larger than that in winter (∼420 nm) in Beijing mainly due to the stronger secondary formation and relatively weaker primary emissions in summer. This is further supported by the larger peak diameter of CO2+, a marker ion indicative of oxygenated OA,12 in summer (∼600 nm) than winter (∼480 nm). Comparatively, the peak diameter of OA was ∼350 nm in Gucheng, which was smaller than that in Beijing during wintertime. Previous studies often showed large peak diameters of OA at rural sites than urban areas due to aging processes during the transport.40,41 Such differences between this study and previous study were mainly caused by large emissions of POA, e.g., coal combustion and biomass burning during wintertime in rural areas in the NCP. For example, Sun et al.42 found that POA on average accounted for ∼80% of the total OA at the Gucheng site in winter 2018, which was much higher than that reported in Beijing.9 This is also consistent with much higher C4H9+ signal intensity, a marker ion for primary emissions,14 at the Gucheng site than Beijing.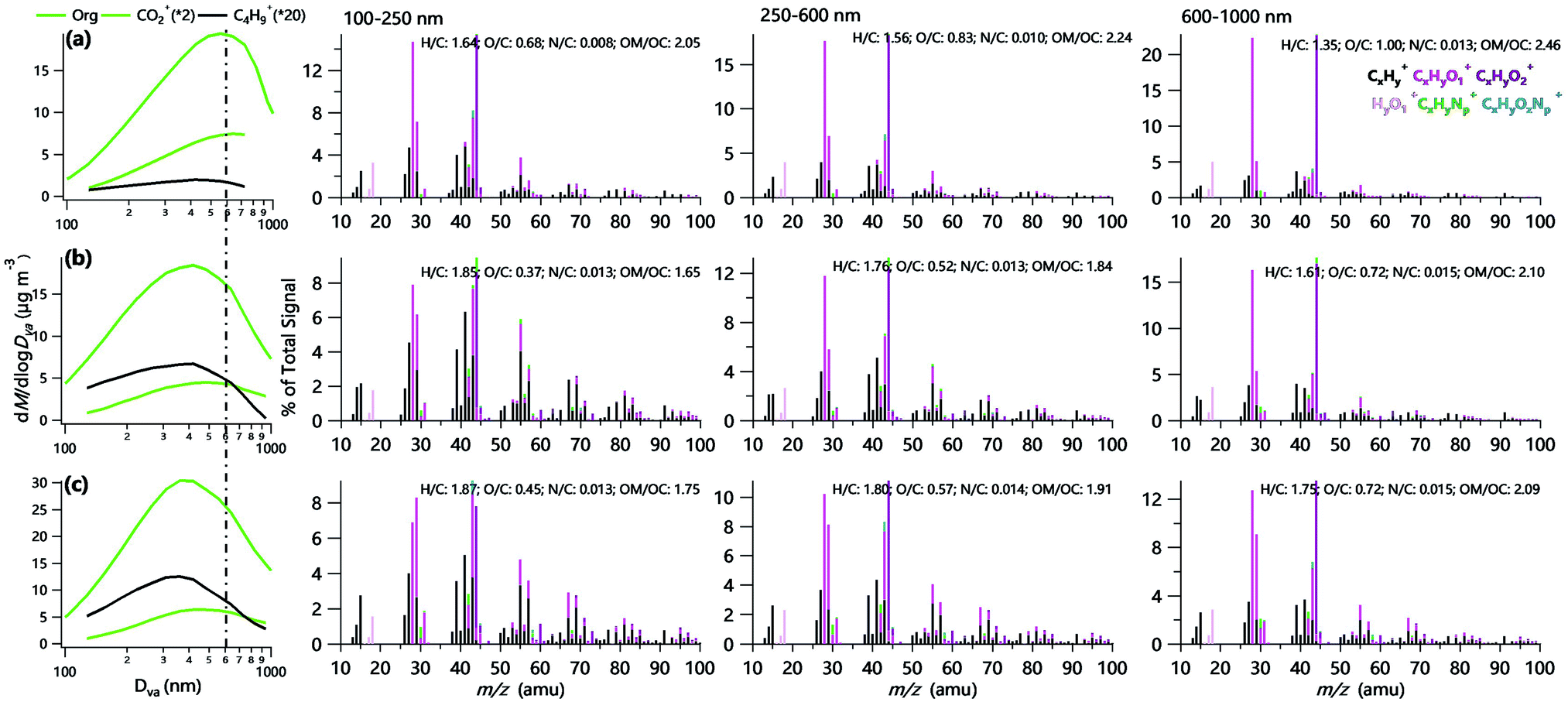 | ||
| Fig. 1 Average size distributions of Org, CO2+ and C4H9+ in (a) summer and (b) winter in Beijing, and (c) winter in Gucheng (left panel). The region on the left side of the dashed line (100–600 nm in this study) generally has a 100% lens transmission efficiency, while it decreases rapidly outside the region.4 The right three panels present average high-resolution mass spectra of OA at 100–250 nm, 250–600 nm and 600–1000 nm, respectively during three campaigns. Also shown are elemental ratios including H/C, O/C, N/C, and OM/OC. | ||
Fig. 1 also presents the average high-resolution mass spectra (HRMS) of OA at three different size ranges, i.e., 100–250 nm, 250–600 nm, and 600–1000 nm. It can be seen that the HRMS of OA showed substantial differences among different size ranges, in particular, OA in winter showed clear changes from primary to secondary OA as the particle size increases. For example, the OA spectrum of 100–250 nm in winter was characterized by the typical hydrocarbon ion series of CnH2n−1+ and CnH2n+1+ (e.g., C3H7+, C4H9+ and C5H11+), highlighting the large influences of primary emissions on small particles. Moreover, the pronounced m/z 60 (mainly C2H4O2+) and 73 (mainly C3H5O2+) in the spectrum were observed, particularly at the rural site, suggesting the influences of biomass burning emissions.43 As the particle size increases, both O/C and f44 showed considerable increases, for instance, O/C increased from 0.68 to 1.0 in summer, and from 0.37–0.45 to 0.72 in winter as particle sizes changed from 100–250 nm to 600–1000 nm. Overall, our results highlight the very different OA properties in different sizes with more oxidized particles in larger sizes.44
Fig. 2 shows the variations of fractions of fragment ions as a function of size. The fCO2+ showed continuous increases while those of C4H7+ and C4H9+ decreased correspondingly with the increase of particle sizes, suggesting that OA becomes more oxidized at larger sizes. Note that marked increases (decreases) in fCO2+ (fC4H7+ and fC4H9+) as the particle size increases were found in winter in Beijing above ∼600 nm, implying the change of sources at large sizes likely due to the influence of regional transport. However, the changes in fC2H3O+ as a function of particle size were relatively small in winter although a clear decrease was observed in summer. These results might indicate that oxidation of freshly oxidized SOA (C2H3O+ as a marker ion) to form more oxidized SOA was more significant in summer than winter at larger sizes. We also noticed small changes in fC2H4O2+ across different sizes, yet it was ubiquitously above 0.3%, a value indicative of biomass burning,44 in winter (∼0.5% in Beijing, and 0.5–1.2% at Gucheng), indicating that biomass burning had influenced OA of all sizes, and BBOA at the rural site was more fresh than the urban site. However, the evolutionary patterns of f44vs. f60 were different between Beijing and Gucheng. While the increase in f44 with increasing particle size was associated with the decrease in f60 at Gucheng site, f60 remained relatively stable for particles of all sizes despite the increase in f44 in Beijing during wintertime. These results might indicate a more significant aging of fresh biomass burning emissions at the rural site while that in Beijing was already aged during the transport. In addition, the difference of f44/f60 between Beijing and Gucheng tended to be slight with increasing particle size, which could be partly attributed to the aging processes. Fig. 2e shows a linear decrease of f55/f57 with increasing particle size, implying the fairly stable ratios of f55/f57 = across different sizes, and they were much higher in summer than winter, and urban than rural sites. For example, f55/f57 varied from 2.3 to 2.6 in summer, and 1.8–1.9 in winter in Beijing, both of which are close to that of cooking OA identified in previous studies at the same site,9,13 demonstrating the impact of cooking emissions on OA in urban areas. However, f55/f57 was much lower at the rural site (1.3–1.5) due to the negligible influence of cooking emissions.29
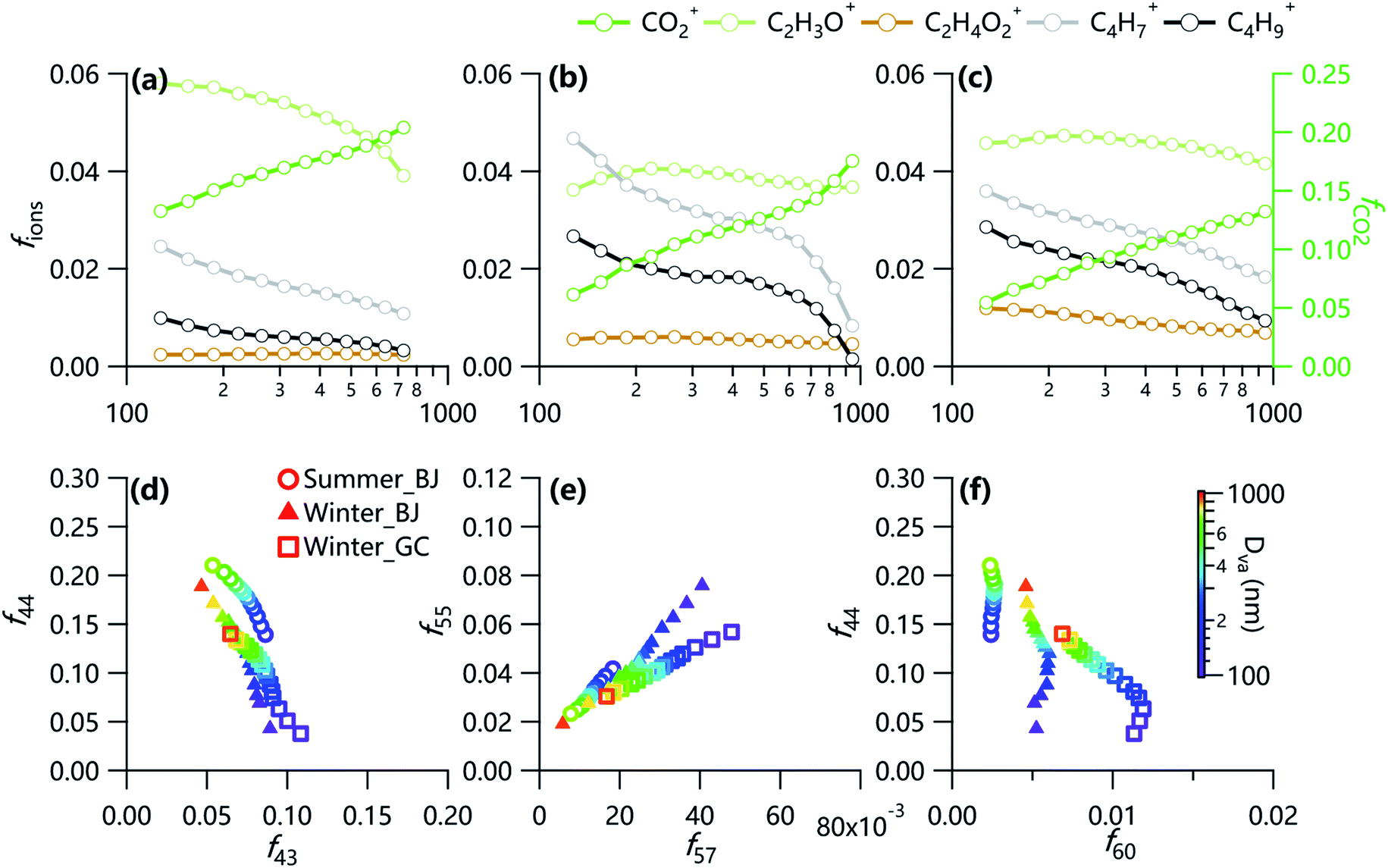 | ||
| Fig. 2 Variations of fractions of CO2+, C2H3O+, C2H4O2+, C4H7+ and C4H9+ as a function of particle sizes in (a) summer and (b) winter in Beijing, and (c) winter in Gucheng. The below panel shows scatter plots of (d) f44vs. f43, (e) f55vs. f57 and (f) f44vs. f60 color coded by aerodynamic diameters during three campaigns. | ||
Fig. 3 shows the diurnal evolution of size-resolved O/C ratios and oxidation states during three campaigns. The O/C ratios and oxidation state of OA showed overall increasing trends across all sizes in the afternoon in both summer and winter, indicating the formation of oxidized SOA through photochemical processing. Another reason was associated with the decreased primary emissions in the day, leading to much higher fractions of SOA in the total OA, and hence a higher oxidation state of bulk OA.9,42,45 Consistently, the lower O/C ratios at nighttime were mainly associated with enhanced primary emissions, e.g., coal combustion and biomass burning. Note that the O/C ratio of OA showed considerable and rapid increases at small sizes (<200 nm) between 9:00 and 10:00 in winter, suggesting an important role of photochemical processing in the formation of SOA at small sizes. This was further supported by decreasing NO3/SO4 (Fig. S4†) as a function of size in winter because NO3 was found to be dominant from photochemical production in winter, while SO4 was more from aqueous-phase processing.46,47 Previous studies found that photochemical aqueous-phase reactions played an important role in SOA formation at Gucheng which might also be one of the reasons for the increases in O/C ratios in early morning.27 Comparatively, O/C ratios above ∼0.6 evolved downwards from ∼500 nm to 200 nm were also observed in summer in Beijing, further demonstrating the photochemical formation of oxidized OA in the day. Also, the O/C ratios in summer were ubiquitously higher than those in winter across different sizes, consistent with higher SOA fractions in OA in summer than winter. We also observed high O/C ratios above 500 nm in early morning in winter at the Gucheng site rather in Beijing. The diurnal cycles and evolution of H/C ratios were on the opposite of O/C ratios. Overall, the changes in size-resolved O/C and H/C ratios elucidate very different OA properties in different seasons and sites.
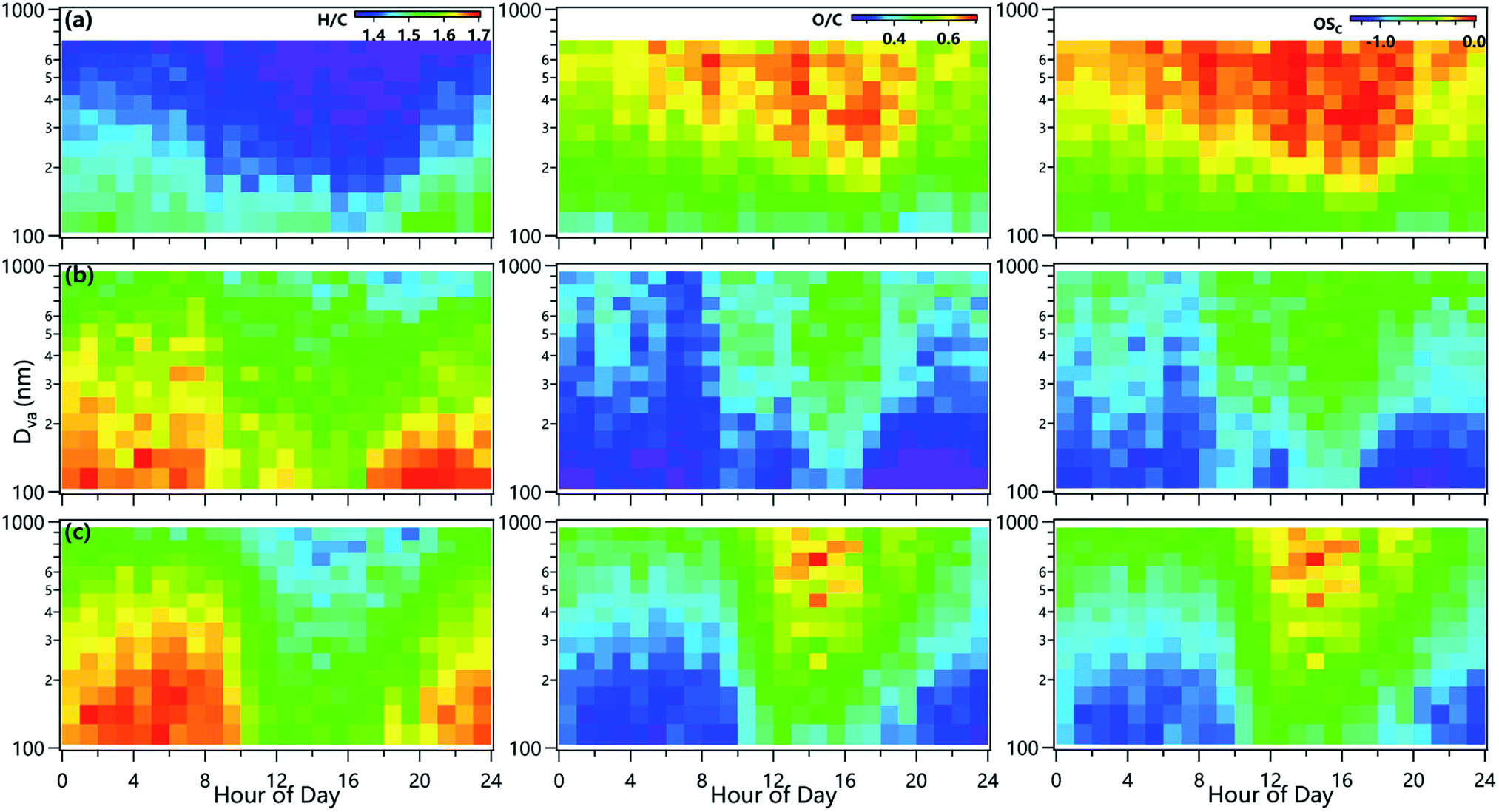 | ||
| Fig. 3 Average diurnal variations of the size distributions of H/C (left), O/C (middle) and oxidation state (right) in (a) summer and (b) winter in Beijing, and (c) winter in Gucheng. | ||
3.2 Size distributions of OA factors
Two factors including a POA and an oxygenated organic aerosol (OOA) were identified below 300 nm, while three factors (POA, OOA1, OOA2) were identified in the range of 300–800 nm in summer in Beijing. Comparatively, three OA factors were resolved during winter across all sizes, including OOA, oxidized POA (OPOA) and POA in Beijing, and OOA, biomass burning OA (BBOA) and fossil-fuel-related OA (FFOA) at Gucheng. The reconstructed size distributions agreed well with those of measured (Fig. S5†). The high-resolution mass spectra of OA factors for each size during three campaigns are shown in Fig. S6–S8.†The mass spectrum of OOA1 in summer was characterized by the relatively high signals of C2H3O+ and CO2+, and the O/C of OOA1 varied from 0.89 to 1.07 across different sizes. As shown in Fig. 4, OOA1 showed an enhancement in the afternoon in summer, indicating that photochemical processing played an important role in the formation of OOA1, consistent with the behaviors of OA (Fig. S9†). This was further supported by the tight correlation between OOA1 and bulk LO-OOA (Fig. S10†), a factor related to photochemical processing48 in summer. Comparatively, OOA2 showed a peak value in the morning, in agreement with the elevated RH in summer in Beijing. Such diurnal variations suggested that OOA2 is likely related to aqueous-phase processing, which was supported by the high CHO+ in the spectrum (Fig. S6†), a marker ion for aqueous-phase processing.49 As shown in Fig. 5, OOA2 peaked at ∼500 nm in summer in Beijing, which was in the range of SOA in previous observations (400–550 nm).16,17,19 We noticed that the fraction of OOA2 decreased from ∼26% at 300 nm to 16% at 800 nm, while OOA1 was elevated from ∼54% to ∼70% with the increase of particle size. One explanation was that the freshly formed OOA1 condensed on preexisting particles more readily than OOA2. Note that the OOA1 also showed a dominant contribution in small size, suggesting the stronger role of photochemical processing in the formation of small particles. This was further supported by the fact that OOA2 cannot be resolved in small sizes. These results indicated the different size distributions of OA formed from photochemical and aqueous-phase processing in summer.
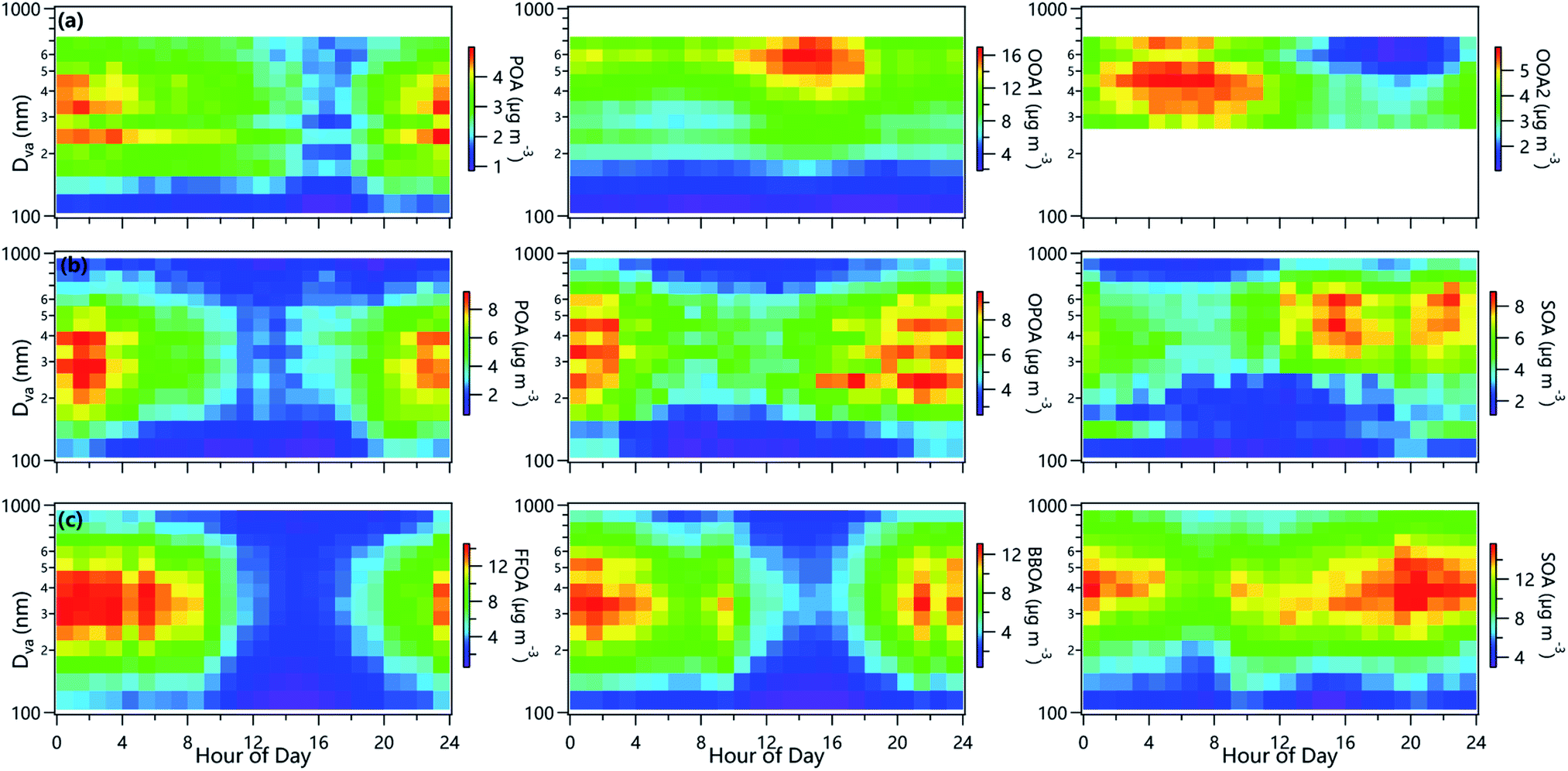 | ||
| Fig. 4 Average diurnal variations of the size distributions of OA factors in (a) summer and (b) winter in Beijing, and (c) winter in Gucheng. | ||
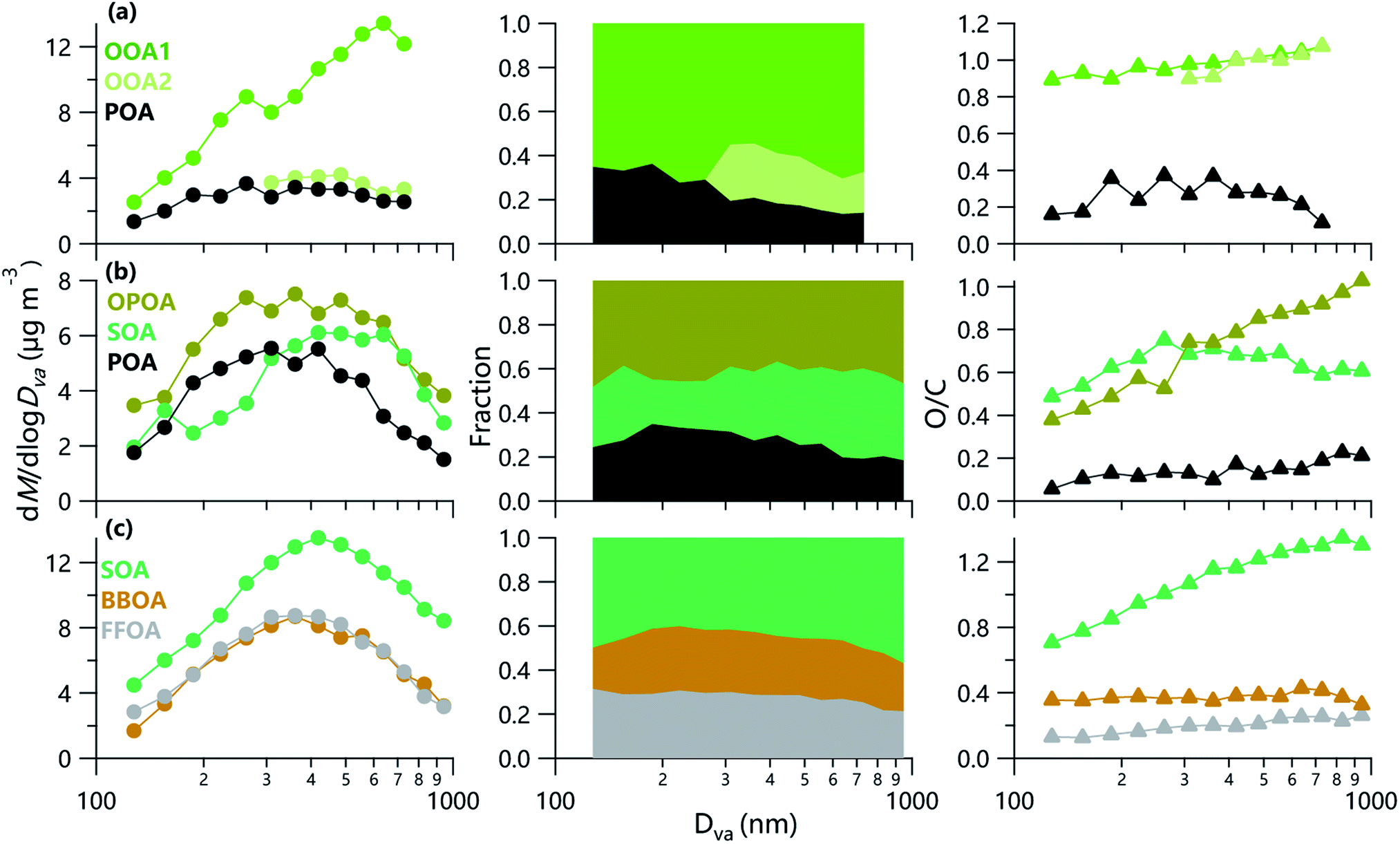 | ||
| Fig. 5 Average size distributions (left), fractional contributions (middle) and O/C (right) ratios of OA factors in (a) summer and (b) winter in Beijing, and (c) winter in Gucheng. | ||
The size-resolved SOA in Gucheng and Beijing during wintertime correlated well with secondary inorganic aerosol (SIA), bulk SOA and CxHyOz+ ions, e.g., C2H3O+ and CO2+ (Fig. S10†). Similar to the elevated SOA fraction as a function of size in summer, SOA also showed a rising trend with the increase of particle size in winter, implying the important role of secondary formation at larger sizes. As indicated in Fig. 5, the O/C of SOA increased from 0.38 to 1.02 and from 0.71 to 1.34 as particle size increased from 100 nm to 1000 nm in Beijing and Gucheng during wintertime, respectively, indicating that aging processes facilitated the formation of large particles, consistent with the fact that large accumulation mode particles are generally more aged than smaller particles.12 Our results highlight that the OA composition varied largely at different sizes which is also supported by the changes in f44/f43, f55/f57 and f44/f60 for OA factors as a function of size (Fig. S11†). The peak diameters of SOA during wintertime (∼400–500 nm) in Beijing and Gucheng were smaller than that in summer in Beijing likely due to weaker secondary formation in winter although some organic acids from aqueous-phase processes during the severe hazes in winter can have large diameters.50 This is consistent with the higher fraction of SOA of the same size and higher O/C (0.68 vs. 0.42–0.54) in summer than that in winter.
The mass spectra of POA across different sizes showed similar features of primary emissions in both summer and winter, which were dominated by CnH2n−1+ and CnH2n+1+ (e.g., C3H7+, C4H8+, C4H9+), and the size-resolved H/C ratios were in the range of those of POA in the NCP.7,11,51 Also, POA was generally well correlated with species typically from primary emissions, e.g., BC, NOx and CO. The peak diameter of POA (∼300 nm) in summer in Beijing was slightly larger than that in previous studies (∼200 nm).16–18 Comparatively, the POA size in winter was even larger than that in summer peaking at ∼350 nm in winter in Beijing. One reason was due to the influences of additional primary emissions, e.g., biomass burning and fossil fuel combustion. The OPOA identified in winter in Beijing showed a relatively high fraction of CxHy+ and H/C (1.52–1.75), and also a relatively high correlation with primary species or fragment ions, e.g., C2H4O2+, C3H5O2+, C8H5+, bulk FFOA, bulk OPOA, BC and Chl (Fig. S10†). The broad size distributions of OPOA indicated that OPOA here may not represent only fresh POA. For example, OPOA was affected by the aging process and/or coated by other species during transport, which was supported by the moderate O/C (0.49–0.75) of OPOA. Different from POA that deceased from 35% at 200 nm to 19% at 1000 nm in winter in Beijing, the fraction of OPOA remained relatively constant across different sizes likely related to aging processes at larger size, which was supported by the elevated O/C as a function of size. Comparatively, the f55/f57 in POA in summer was around 2 below 300 nm, while it showed an obvious decrease above 300 nm likely due to the variations of POA compounds, for instance, f55/f57 in COA is greater than 2, while that in BBOA and FFOA is less than 2.52
BBOA identified in Gucheng in each size bin showed pronounced signals of C2H4O2+ and C3H5O2+, two marker ions for biomass burning44,53 and tight correlations with bulk BBOA, C2H4O2+ and C3H5O2+. BBOA showed much similar size distributions with FFOA in Gucheng, which peaked at ∼350 nm. This peak diameter is slightly larger than that in Mexico City (∼300 nm)18 and Fresno (200 nm)16 likely due to the differences in fuels and aerosols mixing state. The O/C of BBOA across different sizes was comparable to those reported in previous campaigns,7 but slightly higher than those of primary emissions (0.28–0.35)52 even at small size (100 nm). Such results might suggest that BBOA at 100 nm in winter in Gucheng already showed the characteristics of oxidation. Also, the O/C of FFOA showed a remarkable increase from 0.13 to 0.26, indicating more aged FFOA in larger size. Indeed, the f44/f60 in both FFOA and BBOA during wintertime in Gucheng showed clear enhancements as a function of size (Fig. S11†), likely due to the fact that BBOA is mixed with FFOA, yet cannot be completely separated.
3.3 OA size distributions under different PM levels
Fig. 6 shows the variations of size distributions of OA factors at three different PM levels (the total mass concentrations of NR-PM1 species). A summary of frequencies of three different PM levels is shown in Table S2.† Similar to previous studies,13,20 the peak diameters of SOA factors shifted to larger sizes with elevated PM levels during three campaigns. This was partly related to the hygroscopic growth of aerosol particles at high RH due to ubiquitously increased PM levels as a function of RH. In addition, the aging processes during the transport in the atmosphere is another reason for the increased size at high PM levels because highly polluted periods were often found to be largely influenced by regional transport.54,55 As shown in Fig. S12,† the differences of OA loading between relatively clean days and moderately polluted conditions (35 μg m−3 < NR-PM1 < 70 μg m−3) peaked at ∼300 nm, which was smaller than that between relatively cleaner days and seriously (NR-PM1 > 70 μg m−3) polluted conditions (∼480 nm), highlighting that particles with relatively small sizes play an important role in the formation of moderate pollution compared to severe pollution. Comparatively, the increase in OA loading in both moderately and seriously polluted conditions in summer was attributed to particles with a peak diameter of ∼630 nm, highlighting the role of secondary formation during polluted periods in summer. It is interesting to note that the contribution of OOA1 to the increase in OA loading above 300 nm showed an obvious enhancement as a function of size in polluted conditions, yet not in OOA2 in summer in Beijing. Results here indicate that the photochemical processing played an important role in larger size particle in processes of pollution formation in summer in Beijing.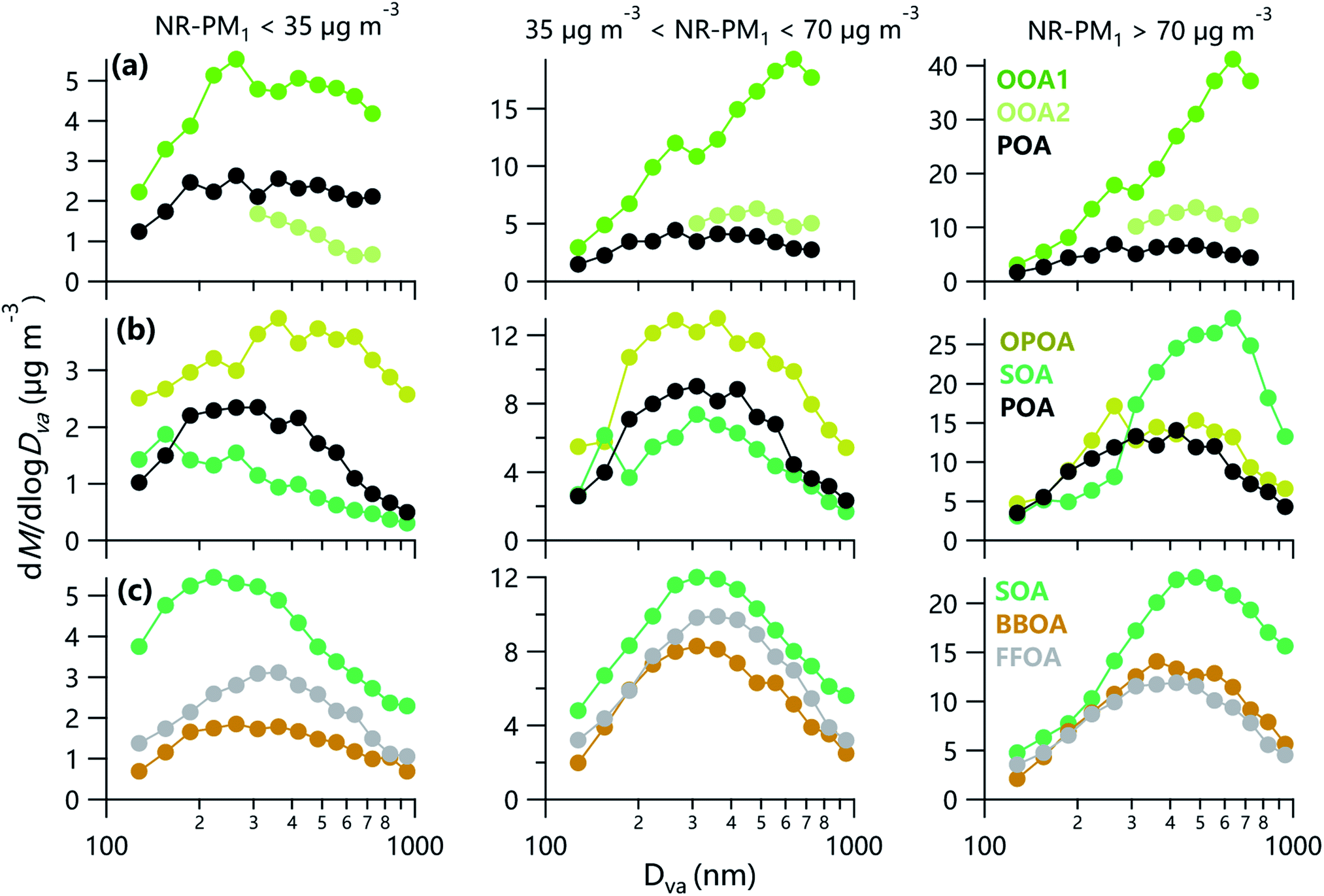 | ||
| Fig. 6 Average size distributions of OA factors at different pollution levels: NR-PM1 < 35 μg m−3 (left), 35 μg m−3 < NR-PM1 < 70 μg m−3 (middle) and NR-PM1 > 70 μg m−3 (right) in (a) summer and (b) winter in Beijing, and (c) winter in Gucheng. | ||
As indicated in Fig. 6, the peak diameters of POA in summer showed negligible changes with elevated PM levels, while POA shifted from ∼300 nm on relatively clean days to ∼400 nm on polluted days during wintertime in Beijing. These results are likely due to the variations of POA compositions with the increase of PM levels in winter in Beijing. For example, the elevated contribution of BBOA with a relatively larger size and decreased fraction of COA (a factor showing a negligible signal above 300 nm) with the elevated PM was found in winter in Beijing.9 In addition, BBOA and FFOA in Gucheng also showed increased peak diameters with the elevated PM, which were mainly due to the aging of BBOA and FFOA in polluted conditions. The peak diameter of OOA1 shifted from 550 nm on relatively clean days to ∼700 nm on moderately polluted days, consistent with the behaviors of OOA2 which shifted from 300 nm to 500 nm. Nevertheless, OOA1 and OOA2 showed negligible changes in peak diameters from moderately to severely polluted days in summer, suggesting similar atmospheric processes from moderate to severe pollution in summer in Beijing. Similar OA factor contributions in moderate and severe pollution (Fig. S13†) further support our conclusion above. These behaviors were possible because regional transport was the major factor driving the formation of polluted days with NR-PM1 > 35 μg m−3 in summer. In contrast, the peak diameter of SOA gradually increased from 300 nm on moderately polluted days to ∼650 nm (∼400 nm) on highly polluted days in Beijing (Gucheng) during wintertime, indicating the change of driving factor from moderate to severe pollution in winter in the NCP. For example, Sun et al.56 focused on four polluted episodes during 10–14 January 2013 and found the four main factors (e.g. stagnant meteorological conditions, coal combustion, secondary production, and regional transport) driving the evolution of pollution in Beijing during wintertime. Such conclusions were further supported by the altered OA compositions in moderately and seriously polluted conditions in winter (Fig. S13†). In fact, despite the dramatic changes of chemical compositions with the elevated size in moderately and seriously polluted conditions, comparable OA compositions were found below 300 nm. Results here suggest that the OA compositions remain relatively constant in small size with the increase of PM levels.
3.4 Size-resolved hygroscopicity of OA
Fig. 7 shows the variations of estimated κchem and κorg as a function of particle size. Both κchem and κorg during three campaigns showed increasing trends with the increase of particle size mainly due to the elevated contributions of SIA and SOA, consistent with the results from previous studies.2 The κorg varied from 0.03 to 0.18 at Gucheng in winter, which was comparable to that (0.08) observed in winter of 2018 at the same site.57 Note that the κorg at Gucheng was lower than that in Beijing during wintertime at the same size, which was attributed to the higher contributions of POA. As indicated in Fig. S2,† the fraction of FFOA, a hydrophobic factor,58 at Gucheng was higher than that in Beijing (32.7% vs. 19.6%), although the fraction of bulk SOA in Beijing was lower than that in Gucheng (41.9% vs. 48.6%). The other cause of such differences is the different functional groups during two campaigns,59 which was supported by the differences in f44/f43, f55/f57 and f44/f60 as shown in Fig. 2. Results here indicate that the OA is more hygroscopic in urban than that at the rural site during wintertime in the NCP at the same size.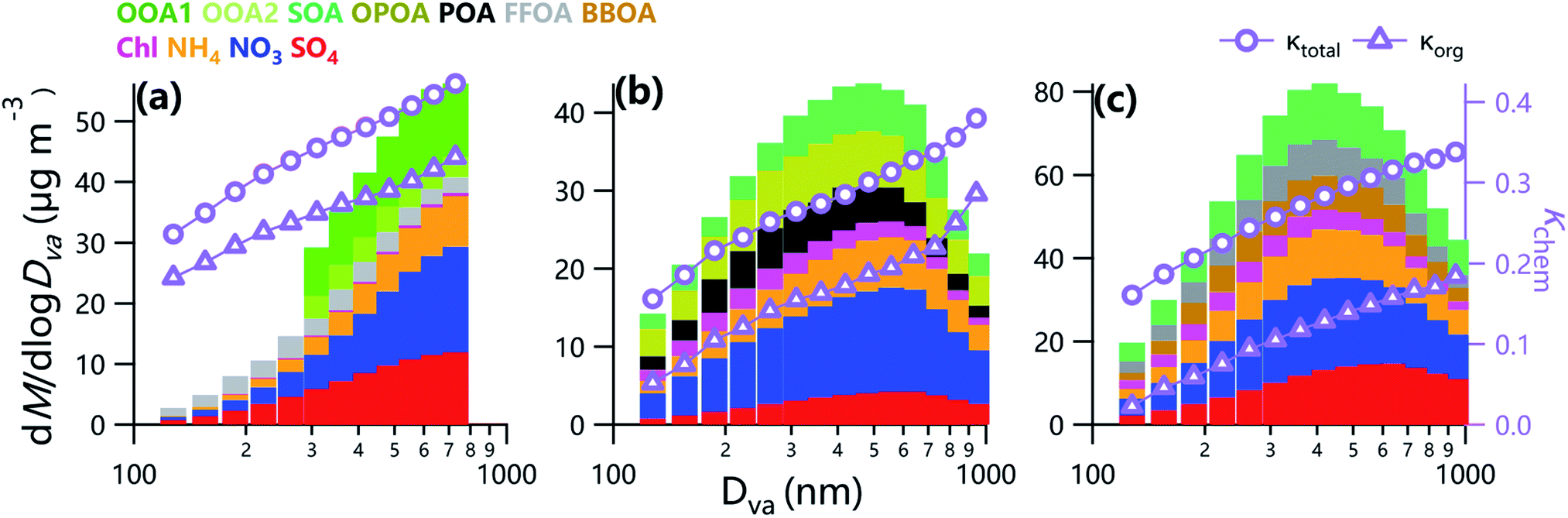 | ||
| Fig. 7 Average size distributions of κchem and κorg, and the contributions of NR-PM1 species (SO4, NO3, NH4, Chl, and OA factors) in (a) summer and (b) winter in Beijing, and (c) winter in Gucheng. | ||
As shown in Fig. 7, the κorg in summer was higher than that in winter in the NCP at the same size due to the dominant contribution of SOA, implying enhanced ability of water uptake in summer in the NCP. These results are consistent with those from HTDMA measurements in Beijing,21 but contradict with previous results from off-line measurements which showed higher hygroscopicity of OA in winter in Beijing.26 Such differences can be partly attributed to the differences in water-soluble particulate matter and total particles in two seasons. For example, previous studies found that the fraction of water-soluble OA (69%) in summer was higher than that in winter (47%) in Beijing.60
While the κchem in summer in Beijing (0.24–0.37) was overall comparable to that observed in summer of 2009,25 the values above 400 nm were higher than that in 2009. In addition to the variations of precursors and meteorological conditions in different environments, such differences of hygroscopicity between the two studies at large size can be partly attributed to the constant κorg of 0.3 used in summer of 2009. It is recommended that future studies of hygroscopicity should consider the size distributions of the hygroscopicity parameter.
4 Conclusions
We analyzed the size-resolved high-resolution mass spectra of OA that were measured at an urban and rural site in summer and winter in the NCP. Our results showed large variations in mass spectra and oxidation state of OA as a function of particle size. For example, the O/C ratio increased with the increase of particle size at both sites, suggesting more oxidized OA at large sizes. In particular, the O/C below 200 nm in the afternoon in winter showed an obvious enhancement suggesting the role of photochemical processing in the formation of small particles. PMF analysis of size-resolved HRMS of OA identified various POA and SOA factors in Beijing and Gucheng. We found that the differences in the properties of POA and SOA factors at different sizes can be substantial. For example, POA species (e.g. OPOA, FFOA) showed elevated O/C ratios as a function of size highlight the aging of POA at a large size. These results highlight the variations of physical and chemical properties of OA factors at different sizes. SOA peaked at 400–500 nm during wintertime, which was smaller than that in summer due to the weaker secondary formation in winter. Comparatively, the peak diameters of BBOA and FFOA in winter in Gucheng were comparable (∼350 nm), which were larger than that of POA related to the traffic and cooking emissions. Significant changes of the variations in OA compositions as a function of size were found at different levels of pollution during wintertime in both urban and rural sites indicating the different roles of POA and SOA in haze formation in the NCP. We further estimated κOA using the size-resolved oxidation state of OA, and the results showed clear increases in κOA as a function of size with higher values in summer than winter. Therefore, the size-resolved κOA will help reduce the uncertainties in closure studies of CCN that often assume a constant κ value for OA.Data availability
The data in this study are available from the authors upon request (E-mail: sunyele@mail.iap.ac.cn).Author contributions
YS and WeX designed the research. WeX, CC, YQ, CX, NM and WaX conducted the measurements. WeX, CC and YC analyzed the data. CC, YC, PF, ZW, PL, JZ and NLN reviewed and commented on the paper. WeX and YS wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge Donna T. Sueper at the Aerodyne Research Inc. for helpful discussions. This work was supported by the National Natural Science Foundation of China (92044301 and 41975170).References
- R. L. Craig, P. K. Peterson, L. Nandy, Z. Lei, M. A. Hossain, S. Camarena, R. A. Dodson, R. D. Cook, C. S. Dutcher and A. P. Ault, Direct Determination of Aerosol pH: Size-Resolved Measurements of Submicrometer and Supermicrometer Aqueous Particles, Anal. Chem., 2018, 90, 11232–11239 CrossRef CAS PubMed
.
- Y. Kuang, W. Xu, J. Tao, N. Ma, C. Zhao and M. Shao, A Review on Laboratory Studies and Field Measurements of Atmospheric Organic Aerosol Hygroscopicity and Its Parameterization Based on Oxidation Levels, Curr. Pollut. Rep., 2020, 6, 410–424 CrossRef CAS
- N. Riemer, A. P. Ault, M. West, R. L. Craig and J. H. Curtis, Aerosol Mixing State: Measurements, Modeling, and Impacts, Rev. Geophys., 2019, 57, 187–249 CrossRef
- J. T. Jayne, D. C. Leard, X. Zhang, P. Davidovits, K. A. Smith, C. E. Kolb and D. R. Worsnop, Development of an aerosol mass spectrometer for size and composition analysis of submicron particles, Aerosol Sci. Technol., 2000, 33, 49–70 CrossRef CAS
- Q. Zhang, J. L. Jimenez, M. R. Canagaratna, J. D. Allan, H. Coe, I. Ulbrich, M. R. Alfarra, A. Takami, A. M. Middlebrook, Y. L. Sun, K. Dzepina, E. Dunlea, K. Docherty, P. F. DeCarlo, D. Salcedo, T. Onasch, J. T. Jayne, T. Miyoshi, A. Shimono, S. Hatakeyama, N. Takegawa, Y. Kondo, J. Schneider, F. Drewnick, S. Weimer, K. Demerjian, P. Williams, K. Bower, R. Bahreini, L. Cottrell, R. J. Griffin, J. Rautiainen, J. Y. Sun, Y. M. Zhang and D. R. Worsnop, Ubiquity and dominance of oxygenated species in organic aerosols in anthropogenically-influenced northern hemisphere mid-latitudes, Geophys. Res. Lett., 2007, 34, L13801 Search PubMed
- M. R. Canagaratna, J. T. Jayne, J. L. Jimenez, J. D. Allan, M. R. Alfarra, Q. Zhang, T. B. Onasch, F. Drewnick, H. Coe, A. Middlebrook, A. Delia, L. R. Williams, A. M. Trimborn, M. J. Northway, P. F. DeCarlo, C. E. Kolb, P. Davidovits and D. R. Worsnop, Chemical and microphysical characterization of ambient aerosols with the aerodyne aerosol mass spectrometer, Mass Spectrom. Rev., 2007, 26, 185–222 CrossRef CAS PubMed
- W. Zhou, W. Xu, H. Kim, Q. Zhang, P. Fu, D. R. Worsnop and Y. Sun, A review of aerosol chemistry in Asia: insights from aerosol mass spectrometer measurements, Environ. Sci.: Processes Impacts, 2020, 22, 1616–1653 RSC
- X. F. Huang, L. Y. He, M. Hu, M. R. Canagaratna, J. H. Kroll, N. L. Ng, Y. H. Zhang, Y. Lin, L. Xue, T. L. Sun, X. G. Liu, M. Shao, J. T. Jayne and D. R. Worsnop, Characterization of submicron aerosols at a rural site in Pearl River Delta of China using an Aerodyne High-Resolution Aerosol Mass Spectrometer, Atmos. Chem. Phys., 2011, 11, 1865–1877 CrossRef CAS
- W. Xu, Y. Sun, Q. Wang, J. Zhao, J. Wang, X. Ge, C. Xie, W. Zhou, W. Du, J. Li, P. Fu, Z. Wang, D. R. Worsnop and H. Coe, Changes in Aerosol Chemistry From 2014 to 2016 in Winter in Beijing: Insights From High-Resolution Aerosol Mass Spectrometry, J. Geophys. Res.: Atmos., 2019, 124, 1132–1147 CAS
- Y. J. Li, B. P. Lee, L. Su, J. C. H. Fung and C. K. Chan, Seasonal characteristics of fine particulate matter (PM) based on high-resolution time-of-flight aerosol mass spectrometric (HR-ToF-AMS) measurements at the HKUST Supersite in Hong Kong, Atmos. Chem. Phys., 2015, 15, 37–53 CrossRef
- W. Hu, M. Hu, W. W. Hu, J. Zheng, C. Chen, Y. Wu and S. Guo, Seasonal variations in high time-resolved chemical compositions, sources, and evolution of atmospheric submicron aerosols in the megacity Beijing, Atmos. Chem. Phys., 2017, 17, 9979–10000 CrossRef CAS
- Q. Zhang, D. R. Worsnop, M. R. Canagaratna and J. L. Jimenez, Hydrocarbon-like and oxygenated organic aerosols in Pittsburgh: Insights into sources and processes of organic aerosols, Atmos. Chem. Phys., 2005, 5, 3289–3311 CrossRef CAS
- W. Q. Xu, Y. L. Sun, C. Chen, W. Du, T. T. Han, Q. Q. Wang, P. Q. Fu, Z. F. Wang, X. J. Zhao, L. B. Zhou, D. S. Ji, P. C. Wang and D. R. Worsnop, Aerosol composition, oxidation properties, and sources in Beijing: results from the 2014 Asia-Pacific Economic Cooperation summit study, Atmos. Chem. Phys., 2015, 15, 13681–13698 CrossRef CAS
- Q. Zhang, M. R. Alfarra, D. R. Worsnop, J. D. Allan, H. Coe, M. R. Canagaratna and J. L. Jimenez, Deconvolution and quantification of hydrocarbon-like and oxygenated organic aerosols based on aerosol mass spectrometry, Environ. Sci. Technol., 2005, 39, 4938–4952 CrossRef CAS PubMed
- Y. L. Sun, Q. Zhang, J. J. Schwab, T. Yang, N. L. Ng and K. L. Demerjian, Factor analysis of combined organic and inorganic aerosol mass spectra from high resolution aerosol mass spectrometer measurements, Atmos. Chem. Phys., 2012, 12, 8537–8551 CrossRef CAS
- X. Ge, A. Setyan, Y. Sun and Q. Zhang, Primary and secondary organic aerosols in Fresno, California during wintertime: Results from high resolution aerosol mass spectrometry, J. Geophys. Res., 2012, 117, D19301 Search PubMed
- J. Z. Xu, J. S. Shi, Q. Zhang, X. L. Ge, F. Canonaco, A. S. H. Prevot, M. Vonwiller, S. Szidat, J. M. Ge, J. M. Ma, Y. Q. An, S. C. Kang and D. H. Qin, Wintertime organic and inorganic aerosols in Lanzhou, China: sources, processes, and comparison with the results during summer, Atmos. Chem. Phys., 2016, 16, 14937–14957 CrossRef CAS
- I. M. Ulbrich, M. R. Canagaratna, M. J. Cubison, Q. Zhang, N. L. Ng, A. C. Aiken and J. L. Jimenez, Three-dimensional factorization of size-resolved organic aerosol mass spectra from Mexico City, Atmos. Meas. Tech., 2012, 5, 195–224 CrossRef CAS
- Y. Sun, Z. Wang, O. Wild, W. Xu, C. Chen, P. Fu, W. Du, L. Zhou, Q. Zhang, T. Han, Q. Wang, X. Pan, H. Zheng, J. Li, X. Guo, J. Liu and D. R. Worsnop, “APEC Blue”: Secondary Aerosol Reductions from Emission Controls in Beijing, Sci. Rep., 2016, 6, 20668 CrossRef CAS PubMed
- S. Guo, M. Hu, M. L. Zamora, J. Peng, D. Shang, J. Zheng, Z. Du, Z. Wu, M. Shao, L. Zeng, M. J. Molina and R. Zhang, Elucidating severe urban haze formation in China, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 17373–17378 CrossRef CAS PubMed
- X. Fan, J. Liu, F. Zhang, L. Chen, D. Collins, W. Xu, X. Jin, J. Ren, Y. Wang, H. Wu, S. Li, Y. Sun and Z. Li, Contrasting size-resolved hygroscopicity of fine particles derived by HTDMA and HR-ToF-AMS measurements between summer and winter in Beijing: the impacts of aerosol aging and local emissions, Atmos. Chem. Phys., 2020, 20, 915–929 CrossRef CAS
- F. Zhang, Y. Y. Wang, J. F. Peng, J. Y. Ren, D. Collins, R. Y. Zhang, Y. L. Sun, X. Yang and Z. Q. Li, Uncertainty in Predicting CCN Activity of Aged and Primary Aerosols, J. Geophys. Res.: Atmos., 2017, 122, 11723–11736 Search PubMed
- Z. J. Wu, J. Zheng, D. J. Shang, Z. F. Du, Y. S. Wu, L. M. Zeng, A. Wiedensohler and M. Hu, Particle hygroscopicity and its link to chemical composition in the urban atmosphere of Beijing, China, during summertime, Atmos. Chem. Phys., 2016, 16, 1123–1138 CrossRef CAS
- Y. Wang, Z. Li, Y. Zhang, W. Du, F. Zhang, H. Tan, H. Xu, T. Fan, X. Jin, X. Fan, Z. Dong, Q. Wang and Y. Sun, Characterization of aerosol hygroscopicity, mixing state, and CCN activity at a suburban site in the central North China Plain, Atmos. Chem. Phys., 2018, 18, 11739–11752 CrossRef CAS
- H. J. Liu, C. S. Zhao, B. Nekat, N. Ma, A. Wiedensohler, D. van Pinxteren, G. Spindler, K. Mueller and H. Herrmann, Aerosol hygroscopicity derived from size-segregated chemical composition and its parameterization in the North China Plain, Atmos. Chem. Phys., 2014, 14, 2525–2539 CrossRef
- P. Zhao, X. Du, J. Su, J. Ding and Q. Dong, Aerosol hygroscopicity based on size-resolved chemical compositions in Beijing, Sci. Total Environ., 2020, 716, 137074 CrossRef CAS PubMed
- Y. Kuang, Y. He, W. Xu, B. Yuan, G. Zhang, Z. Ma, C. Wu, C. Wang, S. Wang, S. Zhang, J. Tao, N. Ma, H. Su, Y. Cheng, M. Shao and Y. Sun, Photochemical Aqueous-Phase Reactions Induce Rapid Daytime Formation of Oxygenated Organic Aerosol on the North China Plain, Environ. Sci. Technol., 2020, 54, 3849–3860 CrossRef CAS PubMed
- W. Xu, C. Chen, Y. Qiu, Y. Li, Z. Zhang, E. Karnezi, S. N. Pandis, C. Xie, Z. Li, J. Sun, N. Ma, W. Xu, P. Fu, Z. Wang, J. Zhu, D. R. Worsnop, N. L. Ng and Y. Sun, Organic aerosol volatility and viscosity in the North China Plain: contrast between summer and winter, Atmos. Chem. Phys., 2021, 21, 5463–5476 CrossRef CAS
- W. Xu, C. Chen, Y. Qiu, Y. Li, Z. Zhang, E. Karnezi, S. N. Pandis, C. Xie, Z. Li, J. Sun, N. Ma, W. Xu, P. Fu, Z. Wang, J. Zhu, D. R. Worsnop, N. L. Ng and Y. Sun, Organic aerosol volatility and viscosity in North China Plain: Contrast between summer and winter, Atmos. Chem. Phys. Discuss., 2020, 2020, 1–22 Search PubMed
- A. M. Middlebrook, R. Bahreini, J. L. Jimenez and M. R. Canagaratna, Evaluation of Composition-Dependent Collection Efficiencies for the Aerodyne Aerosol Mass Spectrometer using Field Data, Aerosol Sci. Technol., 2012, 46, 258–271 CrossRef CAS
- M. R. Canagaratna, J. L. Jimenez, J. H. Kroll, Q. Chen, S. H. Kessler, P. Massoli, L. Hildebrandt Ruiz, E. Fortner, L. R. Williams, K. R. Wilson, J. D. Surratt, N. M. Donahue, J. T. Jayne and D. R. Worsnop, Elemental ratio measurements of organic compounds using aerosol mass spectrometry: characterization, improved calibration, and implications, Atmos. Chem. Phys., 2015, 15, 253–272 CrossRef
- W. Xu, A. Lambe, P. Silva, W. W. Hu, T. Onasch, L. Williams, P. Croteau, X. Zhang, L. Renbaum-Wolff, E. Fortner, J. L. Jimenez, J. Jayne, D. Worsnop and M. Canagaratna, Laboratory evaluation of species-dependent relative ionization efficiencies in the Aerodyne Aerosol Mass Spectrometer, Aerosol Sci. Technol., 2018, 52, 626–641 CrossRef CAS
- I. M. Ulbrich, M. R. Canagaratna, Q. Zhang, D. R. Worsnop and J. L. Jimenez, Interpretation of organic components from Positive Matrix Factorization of aerosol mass spectrometric data, Atmos. Chem. Phys., 2009, 9, 2891–2918 CrossRef CAS
- P. Paatero and U. Tapper, Positive matrix factorization: A non-negative factor model with optimal utilization of error estimates of data values, Environmetrics, 1994, 5, 111–126 CrossRef
- A. V. Polissar, P. K. Hopke, W. C. Malm and J. F. Sisler, Atmospheric Aerosol over Alaska: 2. Elemental Composition and Sources, J. Geophys. Res., 1998, 103, 19045–19057 CrossRef CAS
- J. E. Petit, O. Favez, J. Sciare, F. Canonaco, P. Croteau, G. Močnik, J. Jayne, D. Worsnop and E. Leoz-Garziandia, Submicron aerosol source apportionment of wintertime pollution in Paris, France by double positive matrix factorization (PMF2) using an aerosol chemical speciation monitor (ACSM) and a multi-wavelength Aethalometer, Atmos. Chem. Phys., 2014, 14, 13773–13787 CrossRef
- P. S. K. Liu, R. Deng, K. A. Smith, L. R. Williams, J. T. Jayne, M. R. Canagaratna, K. Moore, T. B. Onasch, D. R. Worsnop and T. Deshler, Transmission efficiency of an aerodynamic focusing lens system: Comparison of model calculations and laboratory measurements for the Aerodyne Aerosol Mass Spectrometer, Aerosol Sci. Technol., 2007, 41, 721–733 CrossRef CAS
- M. D. Petters and S. M. Kreidenweis, A single parameter representation of hygroscopic growth and cloud condensation nucleus activity, Atmos. Chem. Phys., 2007, 7, 1961–1971 CrossRef CAS
- F. Mei, A. Setyan, Q. Zhang and J. Wang, CCN activity of organic aerosols observed downwind of urban emissions during CARES, Atmos. Chem. Phys., 2013, 13, 12155–12169 CrossRef
- M. R. Alfarra, H. Coe, J. D. Allan, K. N. Bower, H. Boudries, M. R. Canagaratna, J. L. Jimenez, J. T. Jayne, A. Garforth, S.-M. Li and D. R. Worsnop, Characterization of urban and regional organic aerosols in the Lower Fraser Valley using two Aerodyne Aerosol Mass Spectrometers, Atmos. Environ., 2004, 38, 5745–5758 CrossRef CAS
- S. Zhou, S. Collier, J. Xu, F. Mei, J. Wang, Y.-N. Lee, A. J. Sedlacek III, S. R. Springston, Y. Sun and Q. Zhang, Influences of upwind emission sources and atmospheric processing on aerosol chemistry and properties at a rural location in the Northeastern U.S, J. Geophys. Res.: Atmos., 2016, 121, 6049–6065 Search PubMed
- Y. Sun, Y. He, Y. Kuang, W. Xu, S. Song, N. Ma, J. Tao, P. Cheng, C. Wu, H. Su, Y. Cheng, C. Xie, C. Chen, L. Lei, Y. Qiu, P. Fu, P. Croteau and D. R. Worsnop, Chemical Differences Between PM1 and PM2.5 in Highly Polluted Environment and Implications in Air Pollution Studies, Geophys. Res. Lett., 2020, 47, e2019GL086288 CAS
- C. Mohr, J. A. Huffman, M. J. Cubison, A. C. Aiken, K. S. Docherty, J. R. Kimmel, I. M. Ulbrich, M. Hannigan and J. L. Jimenez, Characterization of primary organic aerosol emissions from meat cooking, trash burning, and motor vehicles with High-Resolution Aerosol Mass Spectrometry and comparison with ambient and chamber observations, Environ. Sci. Technol., 2009, 43, 2443–2449 CrossRef CAS PubMed
- M. J. Cubison, A. M. Ortega, P. L. Hayes, D. K. Farmer, D. Day, M. J. Lechner, W. H. Brune, E. Apel, G. S. Diskin, J. A. Fisher, H. E. Fuelberg, A. Hecobian, D. J. Knapp, T. Mikoviny, D. Riemer, G. W. Sachse, W. Sessions, R. J. Weber, A. J. Weinheimer, A. Wisthaler and J. L. Jimenez, Effects of aging on organic aerosol from open biomass burning smoke in aircraft and laboratory studies, Atmos. Chem. Phys., 2011, 11, 12049–12064 CrossRef
- Y. Sun, W. Du, P. Fu, Q. Wang, J. Li, X. Ge, Q. Zhang, C. Zhu, L. Ren, W. Xu, J. Zhao, T. Han, D. R. Worsnop and Z. Wang, Primary and secondary aerosols in Beijing in winter: sources, variations and processes, Atmos. Chem. Phys., 2016, 16, 8309–8329 CrossRef CAS
- Y. L. Sun, Z. F. Wang, P. Q. Fu, T. Yang, Q. Jiang, H. B. Dong, J. Li and J. J. Jia, Aerosol composition, sources and processes during wintertime in Beijing, China, Atmos. Chem. Phys., 2013, 13, 4577–4592 CrossRef
- X. Chen, H. Wang, K. Lu, C. Li, T. Zhai, Z. Tan, X. Ma, X. Yang, Y. Liu, S. Chen, H. Dong, X. Li, Z. Wu, M. Hu, L. Zeng and Y. Zhang, Field Determination of Nitrate Formation Pathway in Winter Beijing, Environ. Sci. Technol., 2020, 54, 9243–9253 CrossRef CAS PubMed
- W. Xu, T. Han, W. Du, Q. Wang, C. Chen, J. Zhao, Y. Zhang, J. Li, P. Fu, Z. Wang, D. R. Worsnop and Y. Sun, Effects of Aqueous-Phase and Photochemical Processing on Secondary Organic Aerosol Formation and Evolution in Beijing, China, Environ. Sci. Technol., 2017, 51, 762–770 CrossRef CAS PubMed
- J. Zhao, Y. Qiu, W. Zhou, W. Xu, J. Wang, Y. Zhang, L. Li, C. Xie, Q. Wang, W. Du, D. Worsnop, L. Zhou, X. ge, P. Fu, J. Li, Z. Wang, N. Donahue and Y. Sun, Organic Aerosol Processing During Winter Severe Haze Episodes in Beijing, J. Geophys. Res.: Atmos., 2019, 124, 10248–10263 CAS
- S. Guo, M. Hu, Z. B. Wang, J. Slanina and Y. L. Zhao, Size-resolved aerosol water-soluble ionic compositions in the summer of Beijing: implication of regional secondary formation, Atmos. Chem. Phys., 2010, 10, 947–959 CrossRef CAS
- W. Hu, M. Hu, W. Hu, J. L. Jimenez, B. Yuan, W. Chen, M. Wang, Y. Wu, C. Chen, Z. Wang, J. Peng, L. Zeng and M. Shao, Chemical composition, sources, and aging process of submicron aerosols in Beijing: Contrast between summer and winter, J. Geophys. Res.: Atmos., 2016, 121, 1955–1977 CAS
- W. Xu, Y. He, Y. Qiu, C. Chen, C. Xie, L. Lei, Z. Li, J. Sun, J. Li, P. Fu, Z. Wang, D. R. Worsnop and Y. Sun, Mass spectral characterization of primary emissions and implications in source apportionment of organic aerosol, Atmos. Meas. Tech., 2020, 13, 3205–3219 CrossRef
- A. C. Aiken, D. Salcedo, M. J. Cubison, J. A. Huffman, P. F. DeCarlo, I. M. Ulbrich, K. S. Docherty, D. Sueper, J. R. Kimmel, D. R. Worsnop, A. Trimborn, M. Northway, E. A. Stone, J. J. Schauer, R. M. Volkamer, E. Fortner, B. de Foy, J. Wang, A. Laskin, V. Shutthanandan, J. Zheng, R. Zhang, J. Gaffney, N. A. Marley, G. Paredes-Miranda, W. P. Arnott, L. T. Molina, G. Sosa and J. L. Jimenez, Mexico City aerosol analysis during MILAGRO using high resolution aerosol mass spectrometry at the urban supersite (T0) - Part 1: Fine particle composition and organic source apportionment, Atmos. Chem. Phys., 2009, 9, 6633–6653 CrossRef CAS
- R. Timmermans, R. Kranenburg, A. Manders, C. Hendriks, A. Segers, E. Dammers, Q. Zhang, L. Wang, Z. Liu, L. Zeng, H. D. van der Gon and M. Schaap, Source apportionment of PM2.5 across China using LOTOS-EUROS, Atmos. Environ., 2017, 164, 370–386 CrossRef CAS
- H. Du, J. Li, X. Chen, Z. Wang, Y. Sun, P. Fu, J. Li, J. Gao and Y. Wei, Modeling of aerosol property evolution during winter haze episodes over a megacity cluster in northern China: roles of regional transport and heterogeneous reactions of SO2, Atmos. Chem. Phys., 2019, 19, 9351–9370 CrossRef CAS
- Y. Sun, J. Qi, Z. Wang, P. Fu, J. Li, T. Yang and Y. Yi, Investigation of the sources and evolution processes of severe haze pollution in Beijing in January 2013, J. Geophys. Res.: Atmos., 2014, 119, 4380–4398 Search PubMed
- Y. Kuang, Y. He, W. Xu, P. Zhao, Y. Cheng, G. Zhao, J. Tao, N. Ma, H. Su and Y. Zhang, Distinct diurnal variation in organic aerosol hygroscopicity and its relationship with oxygenated organic aerosol, Atmos. Chem. Phys., 2020, 20, 865–880 CrossRef CAS
- M. Kanakidou, J. H. Seinfeld, S. N. Pandis, I. Barnes, F. J. Dentener, M. C. Facchini, R. V. Dingenen, B. Ervens, A. Nenes, C. J. Nielsen, E. Swietlicki, J. P. Putaud, Y. Balkanski, S. Fuzzi, J. Horth, G. K. Moortgat, R. Winterhalter, C. E. L. Myhre, K. Tsigaridis, E. Vignati, E. G. Stephanou and J. Wilson, Organic aerosol and global climate modelling: a review, Atmos. Chem. Phys., 2005, 5, 1053–1123 CrossRef CAS
- S. R. Suda, M. D. Petters, G. K. Yeh, C. Strollo, A. Matsunaga, A. Faulhaber, P. J. Ziemann, A. J. Prenni, C. M. Carrico, R. C. Sullivan and S. M. Kreidenweis, Influence of Functional Groups on Organic Aerosol Cloud Condensation Nucleus Activity, Environ. Sci. Technol., 2014, 48, 10182–10190 CrossRef CAS PubMed
- Y. Qiu, W. Xu, L. Jia, Y. He and Y. Sun, Molecular composition and sources of water-soluble organic aerosol in summer in Beijing, Chemosphere, 2020, 255, 126850 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ea00025j |
| ‡ Now at: State Key Joint Laboratory of Environmental Simulation and Pollution Control, College of Environmental Sciences and Engineering, Peking University, Beijing 100871, China. |
| This journal is © The Royal Society of Chemistry 2021 |