
Studies on activation factors for oxidative coupling of methane over lithium-based silicate/germanate catalysts†
Tomohiro
Matsumoto
 ,
Satoshi
Ishikawa
,
Miwa
Saito
,
Wataru
Ueda
and
Teruki
Motohashi
*
,
Satoshi
Ishikawa
,
Miwa
Saito
,
Wataru
Ueda
and
Teruki
Motohashi
*
Department of Materials and Life Chemistry, Kanagawa University, Yokohama 221-8686, Japan. E-mail: t-mot@kanagawa-u.ac.jp
First published on 22nd October 2021
Abstract
Several oxides containing group 14 elements were characterized to investigate catalytic activity factors and the reaction mechanism for oxidative coupling of methane (OCM). Lithium-based silicate Li2SiO3 and germanate Li2GeO3 showed much higher selectivities for C2 hydrocarbons than their Sn-based counterpart Li2SnO3. Our XPS study suggested that strong metal–oxygen bonds in silicates and germanates could be of importance to effectively suppress the deep oxidation of methane at the catalyst surface, resulting in enhanced C2 selectivities. In contrast, weakly bound surface oxygen of Sn-based oxides is subject to direct oxygenation of methane to form COx as by-products, emphasizing the importance of metal–oxygen bonding strength. To achieve high OCM catalytic activity, effective methane activation by strong basicity is also required, and the local environment around oxygen sites could be a key factor to enhance CH4 conversion. Based on the chemical/structural viewpoints, we found that silicate and germanate with higher Li contents, Li4SiO4 and Li4GeO4, are significantly OCM active, comparable to the previously reported catalyst Li2CaSiO4.
1. Introduction
Nowadays, fossil fuels such as coal and crude oil have been mainly used for energy generation and chemical production. There is, however, a concern about the depletion of natural carbon resources, and the development of sustainable alternative processes will be necessary for future industries.1 The effective utilization of methane as a primary component of natural gases is thus noteworthy.2 Nevertheless, methane is the most stable and inactive alkane with no functional groups, limiting its use mostly to simple combustions.3 The oxidative coupling of methane (OCM) is fascinating because this reaction can directly convert methane into ethylene, one of the essential chemicals for manufacturing various chemical products. The OCM process to C2 hydrocarbons proceeds viaeqn (1) and (2), but non-selective reactions, represented by eqn (3) and (4), occur simultaneously:| CH4 + 1/4O2 → 1/2C2H6 + 1/2H2O ΔH = −89 kJ mol−1 | (1) |
| CH4 + 1/2O2 → 1/2C2H4 + H2O ΔH = −141 kJ mol−1 | (2) |
| CH4 + 3/2O2 → CO + 2H2O ΔH = −608 kJ mol−1 | (3) |
| CH4 + 2O2 → CO2 + 2H2O ΔH = −890 kJ mol−1 | (4) |
Since the initial report by Keller and Bhasin in the early 1980s,4 approximately 2300 publications on OCM have been published. Among various catalysts investigated so far, a composite consisting of manganese oxides and sodium tungstates on a silica support (Mn–Na2WO4/SiO2) is known to be one of the most active OCM catalysts.3,5 Kiani and coworkers recently reported experimental evidence for Na-coordinated WO4 surface sites acting as active centers.6–8 However, the chemical/structural complexities of Mn–Na2WO4/SiO2 prevent the establishment of catalytic design concepts towards developing practical OCM catalysts with sufficient activity and durability.
Previous mechanistic studies on OCM9–20 indicated that the formation of C2 hydrocarbons predominantly involves a gas-phase association of methyl radicals (·CH3) generated at surface oxygen sites of the catalyst. Therefore, the methane activation by the catalyst is suggested as a dominating factor of the OCM activity, and it is crucial to trigger the hydrogen abstraction reaction effectively while suppressing side reactions. Several groups have claimed the importance of the electronic state of oxygen species on the catalyst surface.15,21–24 However, there is no concrete view about the role of these oxygen species, and the activation factors of OCM at the catalyst surface are not completely understood.
Our recent study achieved the discovery of a highly active catalyst Li2CaSiO4 with a maximum C2 yield of 26% (800 °C, CH4/O2 = 2/1) in a single-phase form, which essentially differs from conventional catalysts with chemical and structural complexities.25 It should be noted that the C2 yield of Li2CaSiO4 is comparable to or even higher than that of the known active catalyst Mn–Na2WO4/SiO2 under the same reaction condition (Table S1†). The fact that Li2CaSiO4 exhibits chemical stability even in long-term catalytic tests ensures that any decomposition products are not likely reactive centers, but the excellent OCM activity stems from bulk nature or a specific surface structure of Li2CaSiO4. Indeed, the compound essentially acts as a single phase, facilitating mechanistic studies on OCM catalysts.
Statistical analysis of the past catalytic data suggested that oxides containing strongly basic elements such as lithium tend to be high-performance catalysts.26 However, the basicity alone seems insufficient to enhance both the CH4 conversion and C2 selectivity, suggesting that the coexistence of silicon plays an important role in the catalytic performance of Li2CaSiO4.25 In the present work, fundamental characterization was conducted on several oxides containing group 14 elements, including Li2MO3 (M = Si, Ge, Sn; Fig. S1 of the ESI†), Li4SiO4, and Li4GeO4, to investigate the effects of metal–oxygen bonding strength on the C2 selectivity. Furthermore, based on these findings, possible activation factors and the reaction mechanism are discussed.
Experimental section
The gas composition was set as CH4/O2/N2 = 1.00/0.25/8.75 unless otherwise noted. A blank test resulted in small methane conversion even at 800 °C (Table S8†). CH4 conversion rate XCH4, C2 selectivity SC2, and C2 yield YC2 were calculated by the following formulas. Slight amounts of higher hydrocarbons such as C3H6 and C3H8 were often detected, and these products were taken into account for calculations of YC2 and SC2.
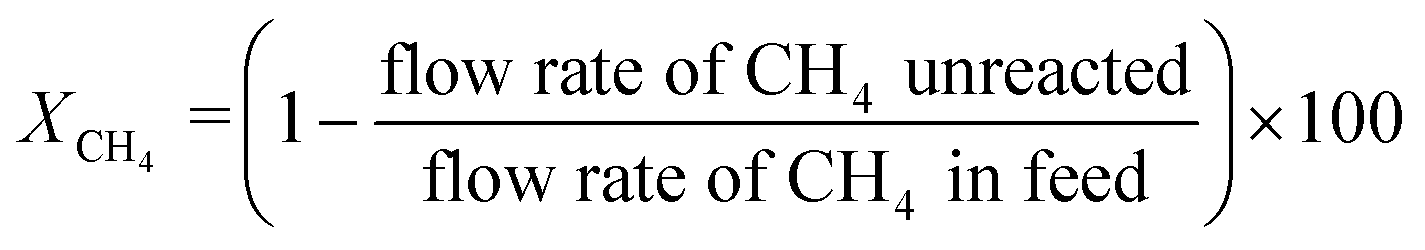

The carbon balance and mass balance were calculated by the following equations. These values sometimes exceeded 100% due to inevitable quantitative errors, especially when the CH4 conversion was small. Alternatively, the emission of CO2 adsorbed on the catalyst surface would overestimate these values.



2. Results and discussion
2.1. Catalytic activity of Li2MO3 (M = Si, Ge, Sn)
The OCM catalytic activity of Li2MO3 (M = Si, Ge, Sn) is presented in Fig. 1. The C2 selectivity and C2 yield reach 52.0% and 12.3%, respectively, for Li2SiO3 at 800 °C. While Li2GeO3 exhibits similar catalytic performance (50.3% and 11.7%), the values for Li2SnO3 fall off to 36.6% and 7.1%. Li2SnO3 is featured with its much lower C2 selectivity, suggesting that the choice of constituent group 14 elements is crucial for the catalyst design. It has been generally recognized that oxides with weak metal–oxygen bonds tend to give active oxygen species that promote deep oxidation reactions.27–29 Therefore, the much lower C2 selectivity of Li2SnO3 may be explained by weakly bound surface oxygen, which preferentially produces CO2 and CO as by-products. The relatively high C2 selectivity of Li2SiO3 and Li2GeO3 is attributed to strong metal–oxygen bonds with Si and Ge that moderately deactivate the surface oxygen to suppress deep oxidation. | ||
Fig. 1 OCM catalytic activity of Li2MO3 (M = Si, Ge, Sn) in a flowing gas mixture of CH4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O2:N2 = 1.0:0.25:8.75. The red curves denote contour lines of the yield. BET surface areas (m2 g−1) are as follows. Li2SiO3: 1.1, Li2GeO3: 0.9, Li2SnO3: 1.2. :O2:N2 = 1.0:0.25:8.75. The red curves denote contour lines of the yield. BET surface areas (m2 g−1) are as follows. Li2SiO3: 1.1, Li2GeO3: 0.9, Li2SnO3: 1.2. | ||
Our previous work revealed that the C2 selectivity of Li2CaSiO4 gets enhanced with smaller specific surface areas.25 Therefore, we attempted to synthesize and characterize Li2MO3 samples with varying their surface areas (Fig. S3 and Table S2†). As seen in Fig. S4,† the samples with smaller surface areas show inferior OCM performance due to the lowered CH4 conversion, in sharp contrast to Li2CaSiO4 for which high values of CH4 conversion are retained irrespective of the reduced surface area. Also noticeably, the C2 selectivity of Li2SnO3 is again smaller than those of the Si and Ge counterparts. It thus turns out that catalysts containing lithium are not necessarily highly OCM active, and the combination of constituent metals is essential.
2.2. Catalyst surface properties
O 1s XPS spectra were measured for reference samples SiO2, GeO2, and SnO2 to investigate the catalyst surface, because the M–O bonding strength could be one of the key factors of the OCM catalytic activity. As shown in Fig. 2, the O 1s spectra are deconvoluted with multiple Gaussian components. Previous works indicated various types of oxygen species on the catalyst surface.15,21–24,30–35 These oxygen species are discriminable with XPS as lower and higher energy peaks (Table 1). The spectra are deconvoluted into two peaks for SiO2 and GeO2: the lower energy peak is assigned to lattice oxygen (M–O–M) that bridges multiple metal atoms, while the higher energy peak as oxygen without bridging metal atoms, presumably mainly hydroxyl groups (M–OH).30–35 For SnO2, three peaks are observed in which the highest energy peak would be attributed to adsorbed water.31,32 For all the three oxides, the relative intensities of the higher energy peaks decreased upon etching, consistent with the assumption that weakly bound surface oxygen species are preferentially removed by the etching process. It should be noted that the O 1s spectra of SiO2 and GeO2 are similar to each other, whereas that of SnO2 differs significantly. This feature is reminiscent that Sn-based catalysts tend to show poor OCM performance compared to the Si and Ge-counterparts. | ||
| Fig. 2 XPS O 1s spectra of SiO2, GeO2, and SnO2. | ||
| H2O [%] | OH− [%] | O2− [%] | |
|---|---|---|---|
| SiO2 | 88.5 | 11.5 | |
| SiO2 (Ar-etched) | 79.6 | 20.4 | |
| GeO2 | 75.9 | 24.1 | |
| GeO2 (Ar-etched) | 70.2 | 29.8 | |
| SnO2 | 10.2 | 57.8 | 32.0 |
| SnO2 (Ar-etched) | 16.9 | 83.1 |
The XPS analysis reveals a larger M–OH fraction for SiO2 and GeO2 than SnO2. This tendency may be attributed to their different chemical bonding nature. The bandgap widths are theoretically estimated at 5.4, 3.4, and 0.7 eV for SiO2, GeO2, and SnO2, respectively. Electronic excitation beyond the bandgap involves the decreased/increased numbers of electrons in the valence/conduction bands. Since the valence/conduction bands have bonding/antibonding characters, the excitation of valence electrons into the conduction band results in weakened bonding strength between metal and oxygen atoms. Thus, the bonding strength depends on the bandgap width, and oxygen atoms are bound more tightly in SiO2 and GeO2 than in SnO2. Taking into account that M–OH has lower stability than M–O–M, the M–OH species of SnO2 would be too reactive to exist in large quantities. This situation is in sharp contrast to that in SiO2 and GeO2, where the M–OH peak intensity remains large and almost unchanged upon etching. The XPS result supports the above discussion and suggests that the stronger metal–oxygen bond is a dominant factor in suppressing the deep oxidation of methane, i.e., enhancing OCM catalytic activity. We note that the XPS results only represent the chemical properties of the catalyst surface and are not directly related to the catalytic reaction. In situ and operando analyses are indispensable to specify actual species that oxidize methane molecules during the OCM reaction.
Temperature programmed O2 desorption (O2-TPD) was performed on the Li2MO3 catalysts (M = Si, Ge, Sn) to discuss their different catalytic activity. As shown in Fig. S5,† all the catalysts show large desorption peaks above 200 °C. The onset temperature of O2 desorption increases in the order of Li2SnO3 (200 °C), Li2GeO3 (450 °C), and Li2SiO3 (600 °C). This result indicates that oxygen in Sn-based oxides is bound more loosely than in the Ge- and Si-counterparts, consistent with the systematic trend in bandgap widths of SnO2, GeO2, and SiO2. We thus conclude that the weakly bound oxygen species in Li2SnO3 causes deep methane oxidation, resulting in the reduced C2 selectivity.
For Li2CaSiO4, O 1s XPS spectra were also measured to investigate the electronic state at the catalyst surface (Fig. 3 and Table 2). The O 1s spectra of Li2CaSiO4 are deconvoluted with three components located at EB = 529.9 eV, 532.1 eV, and 533.8 eV. The peaks at 529.9 eV and 533.8 eV would come from oxygen species bound to metal atoms. The peak at 532.1 eV is attributable to carbonates derived from chemisorbed carbon dioxide (carbonate-derived peaks are present in the C 1s region at 288–290 eV).21–24,33–39 The two oxygen species bound to metal atoms are similarly assigned to lattice oxygen and hydroxyl groups, and the latter is dominant for Li2CaSiO4. The result indicates that the metal–oxygen bonding force is strong for this compound enough to suppress the deep oxidation of methane. Besides, Li2CaSiO4 shows a Li 1s peak around 56–57 eV even after a durability test for 50 hours (Fig. S6 and Table S3†), implying the stable presence of Li atoms at the outmost surface. This feature would originate from the robust Si–O covalent framework that prevents surface reconstruction, leading to the high catalytic durability of Li2CaSiO4.
 | ||
| Fig. 3 XPS O 1s spectra of Li2CaSiO4. | ||
| OH− [%] | CO32− [%] | O2− [%] | |
|---|---|---|---|
| Li2CaSiO4 | 61.0 | 36.5 | 2.5 |
| Li2CaSiO4 (Ar-etched) | 43.2 | 38.9 | 17.9 |
2.3. Effect of specific surface area on catalytic activity
For Li2MO3 (M = Si, Ge, Sn), the magnitude of the specific surface area is found to affect the catalytic activity significantly. Although the Li2MO3 catalysts with small surface areas hardly catalyze methane because of their weak basicity, they show much higher CH4 conversion with the increasing surface area. This activity increment is too drastic to attribute only to the increased surface area, suggesting that different oxygen species have been involved. Differences in the specific surface area may change the number of physisorbed oxygen molecules, thereby affecting the constitution of oxygen species at the catalyst surface. Then, the OCM activity of Li2CaSiO4 is compared with those of Li2SiO3 and SiO2, focusing on the specific surface area. As presented in Fig. 4, the Li2CaSiO4 catalyst with a larger surface area shows lower C2 selectivity than the smaller-surface-area catalyst. For the larger-surface-area Li2CaSiO4 catalyst, the surface roughness and the increased defect density promote the adsorption of oxygen molecules, being subject to deep oxidation and lowered C2 selectivity.40 On the other hand, the oxygenation of methane is effectively suppressed for the smaller-surface-area catalyst because of the dominance of strongly bound oxygen species. A similar trend is observed for Li2SiO3 and SiO2 (Fig. S7†). However, the reduced surface area concomitantly leads to extremely low CH4 conversion due to small amounts of basic sites for Li2SiO3 and SiO2 (Fig. S8 and Table S2†). The high CH4 conversion of the large-surface-area SiO2 having weak basicity implies that oxygen molecules adsorbed on the surface are highly reactive to cause deep oxidation of methane. | ||
| Fig. 4 (a) OCM catalytic activity of Li2CaSiO4 with specific surface areas of 0.2 and 0.5 m2 g−1 in a flowing gas mixture of CH4:O2:N2 = 1.0:0.25: 8.75. (b) XRD patterns for the two Li2CaSiO4 catalysts. | ||
Next, OCM activity tests upon varying the catalyst amount were conducted. The specific surface areas were determined by triplicate measurements for each sample to be 1.1 m2 g−1 for Li2SiO3 and 0.2 m2 g−1 for Li2CaSiO4 (Table S4†). For Li2SiO3, the CH4 conversion is enhanced with the increasing catalyst amount from 0.1 g to 0.3 g (Fig. 5). The strongly mass-dependent activity of Li2SiO3 indicates that the catalyst surface allows for the physisorption of many oxygen molecules. Meanwhile, a similar performance for 0.3 g and 1.0 g implies that the supplied oxygen (reactant) has been mostly consumed in these conditions. Irrespective of the catalyst-weight vs. flow-rate (W/F) ratio, the C2 selectivity is enhanced as the temperature increases (Fig. S9†). This behavior is interpreted as a consequence that the surface oxygen desorbs at elevated temperatures. At temperatures below 750 °C, many oxygen molecules adsorb at the catalyst surface such that the deep oxidation of methane is predominant, and the OCM reaction cannot be promoted even though the catalyst amount is optimized.
 | ||
| Fig. 5 OCM catalytic activity of (a) Li2SiO3 and (b) Li2CaSiO4 with various catalyst amounts in a flowing gas mixture of CH4:O2:N2 = 1.0:0.25:8.75. | ||
In contrast, Li2CaSiO4 with a small surface area shows superb performance regardless of the catalyst amount. Rather, the low-temperature activity of Li2CaSiO4 gets enhanced as the catalyst amount increases. It should be noted that the C2 yield for the 1.0 g catalyst at 700 °C is comparable to that for 0.1 g at 800 °C (Table S5†). Regarding the catalytic activity with the 1.0 g catalyst, both the CH4 and O2 conversions slightly decrease when the feed-gas flow rate increases from 10 mL min−1 to 50 mL min−1, but the OCM performance is not influenced significantly (Tables S6 and S9†). We suggest that the small surface area of the Li2CaSiO4 catalyst effectively suppresses the physisorption of oxygen molecules, enabling us to lower the reaction temperature upon increasing the catalyst amount while preventing the direct oxygenation of methane.
2.4. Activation factors and reaction mechanism
To improve the C2 selectivity, it is now evident that oxygen species at the catalyst surface must be tightly bound to prevent direct oxygenation of methane. Besides, a high degree of basicity is crucial for activating C–H bonds to accelerate the CH4 conversion.22,41–45 The hydroxyl group M–OH observed by XPS would not directly induce the CH4 conversion because of its low electron density and weak basicity. Our previous work revealed a significant difference in the CH4 conversion capability of four oxides having the same elemental combination: Li2CaSiO4, Li2Ca2Si2O7, Li2Ca2Si5O13, and Li2Ca4Si4O13.25 The XPS analysis reveals similar O 1s spectra of these oxides with a predominant M–OH component (Fig. S10†). This result supports our view that the hydroxyl group does not contribute to the activation of C–H bonds.Despite the absence of redox metal species, Li2CaSiO4 efficiently catalyzes partial oxidation of methane. Regarding a possible catalytic mechanism for OCM, Aika et al. reported that the generation of methyl radicals at the catalyst surface could be the initial step, followed by radical coupling in the gas phase.46–48 OCM activity tests with/without Li2CaSiO4 were carried out at various CH4/O2 ratios at 800 °C (Fig. S11 and S12†). The OCM reaction proceeds even under a non-catalytic condition in the absence of oxygen, resulting in the formation of ethane and hydrogen (Fig. S11(a)†). The hydrogen is also detected in the presence of oxygen (Fig. S11(b) and (c) and S12†). This experimental fact suggests that hydrogen atoms are taken from methane to produce methyl radicals. The resultant methyl radicals are coupled in the gas phase to form ethane molecules. Under an anaerobic condition with the Li2CaSiO4 catalyst, the amount of C2 hydrocarbons produced is larger than that in the non-catalytic reaction (Fig. S13†). Furthermore, Li2CaSiO4 shows stable OCM activity over 30 hours at 800 °C (Fig. S14 and S15†) without phase decomposition (Fig. S16 and S17†). These features indicate that the lattice oxygen works as an activator of methane dehydrogenation. Based on this mechanistic view, the surface oxygen would involve the catalytic cycle: the surface oxygen receives hydrogen from methane to produce water molecules, and the gaseous oxygen subsequently fills the oxygen-deficient sites, as reported previously.9–20,46–49
2.5. Materials design for high-performance OCM catalysts
Based on the mechanistic aspect described above, we suggest that high-performance OCM catalysts must contain electropositive elements such as alkaline metals, as well as elements that form robust frameworks with strong metal–oxygen bonds for suppressing the deep oxidation with surface oxygen species. Here, we report that silicate and germanate with higher alkaline metal contents, Li4SiO4 and Li4GeO4 (Fig. S19 and S20†), are also potential OCM catalysts. Their OCM activities are compared with the Li2CaSiO4 catalyst (Fig. 6 and Table S10†). Both Li4SiO4 and Li4GeO4 exhibit significant OCM activity comparable to Li2CaSiO4, reaching C2 yields of 26.2% and 24.7%, respectively, under the stoichiometric CH4/O2 = 2/1 condition at 800 °C. The CH4 consumption rate per unit surface area is much faster for Li4SiO4 and Li4GeO4 than for Li2MO3, demonstrating that the Li-rich silicate/germanate can effectively activate CH4 molecules (Fig. S22†). The C2 hydrocarbons production rate is similarly fast, contributing to their superior C2 selectivity. In addition, Li4SiO4 shows stable OCM performance in a durability test over 30 hours at 800 °C (Fig. S23†) without phase decomposition (Fig. S24†). | ||
| Fig. 6 OCM catalytic activity of Li4SiO4 and Li4GeO4 in flowing gas mixtures of (a) CH4:O2:N2 = 1.0:0.25:8.75, and (b) CH4:O2:N2 = 1.0:0.5:8.5. BET surface areas (m2 g−1) are as follows. Li4SiO4: 0.3, Li4GeO4: 0.3, Li2CaSiO4: 0.2. | ||
Interestingly, Li4SiO4 and Li4GeO4 have crystallographic similarities to Li2CaSiO4. As schematically drawn in Fig. S25,† every oxygen site in these oxides is bound to one Si/Ge atom and multiple Li atoms, similar to the Li2CaSiO4 crystal lattice.51 While Li atoms adjacent to each lattice oxygen induce strong basicity and promote methane activation, robust Si–O and Ge–O bonds suppress the deep oxidation of methane, resulting in simultaneously enhanced CH4 conversion and C2 selectivity.22,24 From this structural point of view, the relatively low OCM activity of Li2SiO3 and Li2GeO3 may also be explained. These oxides contain oxygen sites bound to two Si/Ge atoms (in other words, Si–O–Si or Ge–O–Ge bonds; see Table S11†), which would accordingly reduce the basicity at the oxygen site, thereby lowering CH4 conversion. It should be noted that Li2MO3 and MO2 (M = Si and Ge) possibly exhibit high values of CH4 conversion when their surface areas are sufficiently large, but the simultaneous increases in CH4 conversion and C2 selectivity seem impossible because the increased surface area inevitably induces deep methane oxidation by physisorbed oxygen molecules. These results demonstrate that the local environment around oxygen sites plays an important role in the OCM activity, and precise structural control is the key to improving catalytic performance.
As discussed in our previous report on Li2CaSiO4,25 this aspect is valid only when the atomic arrangement at the crystalline surface is intimately related to the bulk structure. We believe that our Li-based silicate/germanate catalysts meet this condition, taking into account the significantly different OCM activity between the Li4MO4 and Li2MO3 systems. In fact, the chemical analysis by X-ray photoelectron spectroscopy evidences similar surface atomic ratios for these systems, implying that their catalytic activity is hardly accounted for in terms of surface Li concentrations (Table S12†).
3. Conclusions
Through systematic characterization of several oxides containing group 14 elements, we demonstrate catalytic activity factors and the reaction mechanism for the oxidative coupling of methane (OCM). Lithium-based silicate Li2SiO3 and germanate Li2GeO3 showed much higher selectivities of C2 hydrocarbons than their Sn-based counterpart Li2SnO3. The XPS study suggested that strong metal–oxygen bonds in silicates and germanates could be of importance to effectively suppress the deep oxidation of methane at the catalyst surface, resulting in enhanced C2 selectivities. In contrast, weakly bound surface oxygen of Sn-based oxides is subject to direct oxygenation of methane to form COx as by-products, emphasizing the importance of metal–oxygen bonding strength. To achieve high OCM catalytic activity, the basicity of the catalyst is another primary factor for promoting methane activation, which is likely related to the local environment around oxygen sites.Based on the chemical/structural viewpoints in the present study, we found that silicate and germanate with higher Li contents, Li4SiO4 and Li4GeO4, are significantly OCM active, comparable to the potential catalyst Li2CaSiO4. The significant OCM activity of Li4SiO4 and Li4GeO4 is believed to originate from the crystallographic similarities to Li2CaSiO4, where every oxygen site is bound to one Si/Ge atom and multiple Li atoms, resulting in simultaneously enhanced CH4 conversion and C2 selectivity.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Prof. Ryo Maezono and Prof. Kenta Hongo of Japan Advanced Institute of Science and Technology for their help in DFT calculations. This work was supported in part by JSPS Grant-in-Aid for Science Research JP20H02827 (B) and JP19H04707 (Innovative Area “Mixed Anion”).References
- M. Zhao, S. Ke, H. Wu, W. Xia and H. Wan, Ind. Eng. Chem. Res., 2019, 58, 22847 CrossRef CAS.
- J. H. Lunsford, Catal. Today, 2000, 63, 165 CrossRef CAS.
- E. V. Kondratenko, T. Peppel, D. Seeburg, V. A. Kondratenko, N. Kalevaru, A. Martin and S. Wohlrab, Catal. Sci. Technol., 2017, 7, 366 RSC.
- G. E. Keller and M. M. Bhasin, J. Catal., 1982, 73, 9 CrossRef CAS.
- S. Arndt, T. Otremba, U. Simon, M. Yildiz, H. Schubert and R. Schomäcker, Appl. Catal., A, 2012, 425, 53 CrossRef.
- D. Kiani, S. Sourav, J. Baltrusaitis and I. E. Wachs, ACS Catal., 2019, 9(7), 5912 CrossRef.
- D. Kiani, S. Sourav, W. Taifan, M. Calatayud, F. Tielens, I. E. Wachs and J. Baltrusaitis, ACS Catal., 2020, 10(8), 4580 CrossRef CAS.
- D. Kiani, S. Sourav, I. E. Wachs and J. Baltrusaitis, Catal. Sci. Technol., 2020, 10, 3334 RSC.
- D. J. Driscoll, W. Martir, J. Wang and J. H. Lunsford, J. Am. Chem. Soc., 1985, 107, 58 CrossRef CAS.
- D. J. Driscoll, K. D. Campbell and J. H. Lunsford, Adv. Catal., 1987, 35, 139 CAS.
- K. D. Campbell and J. H. Lunsford, J. Phys. Chem., 1988, 92, 5792 CrossRef CAS.
- Y. Tong, M. P. Rosynek and J. H. Lunsford, J. Phys. Chem., 1989, 93, 2896 CrossRef CAS.
- Y. Feng, J. Niiranen and D. Cutman, J. Phys. Chem., 1991, 95, 6558 CrossRef CAS.
- Y. Feng, J. Niiranen and D. Cutman, J. Phys. Chem., 1991, 95, 6564 CrossRef CAS.
- Y. Gambo, A. Jalila, S. Triwahyono and A. Abdulrasheed, J. Ind. Eng. Chem., 2018, 59, 218 CrossRef CAS.
- L. Luo, X. Tang, W. Wang, Y. Wang, S. Sun, F. Qi and W. Huang, Sci. Rep., 2013, 3, 1625 CrossRef PubMed.
- L. Luo, R. You, Y. Liu, J. Yang, Y. Zhu, W. Wen, Y. Pan, F. Qi and W. Huang, ACS Catal., 2019, 9, 2514 CrossRef CAS.
- A. M. Maitra, Appl. Catal., A, 1993, 104, 11 CrossRef CAS.
- Z. Zhang, X. E. Verykios and M. Baerns, Catal. Rev.: Sci. Eng., 1994, 36, 507 CrossRef CAS.
- M. C. Alvarez-Galvan, N. Mota, M. Ojeda, S. Rojas, R. M. Navarro and J. L. G. Fierro, Catal. Today, 2011, 171, 15 CrossRef CAS.
- V. J. Ferreira, P. Tavares, J. L. Figueiredo and J. L. Faria, Ind. Eng. Chem. Res., 2012, 51, 10535 CrossRef CAS.
- J. Xu, L. Peng, X. Fang, Z. Fu, W. Liu, X. Xu, H. Peng, R. Zheng and X. Wang, Appl. Catal., A, 2018, 552, 117 CrossRef CAS.
- P. Huang, Y. Zhao, J. Zhang, Y. Zhu and Y. Sun, Nanoscale, 2013, 5, 10844 RSC.
- Y. Hou, W. Han, W. Xia and H. Wan, ACS Catal., 2015, 5, 1663 CrossRef CAS.
- T. Matsumoto, M. Saito, S. Ishikawa, K. Fujii, M. Yashima, W. Ueda and T. Motohashi, ChemCatChem, 2020, 12, 1968 CrossRef CAS.
- U. Zavyalova, M. Holena, R. Schlçgl and M. Baerns, ChemCatChem, 2011, 3, 1935 CrossRef CAS.
- H. Chang, E. Bjørgum, O. Mihai, J. Yang, H. L. Lein, T. Grande, S. Raaen, Y. Zhu, A. Holmen and D. Chen, ACS Catal., 2020, 10(6), 3707 CrossRef CAS.
- R. K. Grasselli, Top. Catal., 2002, 21, 79 CrossRef CAS.
- Y. Moro-oka, Y. Morikawa and A. Ozaki, J. Catal., 1967, 7, 23 CrossRef CAS.
- A. Hashemi and A. Bahari, Appl. Phys. A: Mater. Sci. Process., 2017, 123, 535 CrossRef.
- H. Jadhav, S. Suryawanshi, M. A. More and S. Sinha, Appl. Surf. Sci., 2017, 419, 764 CrossRef CAS.
- G. Gaggiotti, A. Galdikas, S. Kačiulis, G. Mattogno and A. Setkus, J. Appl. Phys., 1994, 76, 4467 CrossRef CAS.
- G. M. Bancroft, H. W. Nesbitt, R. Ho, D. M. Shaw, J. S. Tse and M. C. Biesinger, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 075405 CrossRef.
- K. Prabhakaran and T. Ogino, Surf. Sci., 1995, 325, 263 CrossRef CAS.
- L. Peng, J. Xu, X. Fang, W. Liu, X. Xu, L. Liu, Z. Li, H. Peng, R. Zheng and X. Wang, Eur. J. Inorg. Chem., 2018, 17, 1787 CrossRef.
- J. Xu, Y. Zhang, X. Xu, X. Fang, R. Xi, Y. Liu, R. Zheng and X. Wang, ACS Catal., 2019, 9, 4030 CrossRef CAS.
- J. Xu, Y. Zhang, Y. Liu, X. Fang, X. Xu, W. Liu, R. Zheng and X. Wang, Eur. J. Inorg. Chem., 2019, 2, 183 CrossRef.
- C. S. Gopinath, S. G. Hegde, A. V. Ramaswamy and S. M. Mahapatra, Mater. Res. Bull., 2002, 37, 1323 CrossRef CAS.
- X. D. Peng, D. A. Richards and P. C. Stair, J. Catal., 1990, 121, 99 CrossRef CAS.
- E. Iwamatsu and K. Aika, J. Catal., 1989, 117, 416 CrossRef CAS.
- L. Peng, J. Xu, X. Fang, W. Liu, X. Xu, L. Liu, Z. Li, H. Peng, R. Zheng and X. Wang, Eur. J. Inorg. Chem., 2018, 17, 1787 CrossRef.
- T. W. Elkins, S. J. Roberts and H. E. Hagelin-Weaver, Appl. Catal., A, 2016, 528, 175 CrossRef CAS.
- A. A. Davydov, M. L. Shepotko and A. A. Budneva, Catal. Today, 1995, 24, 225 CrossRef CAS.
- V. R. Choudhary and V. H. Rane, J. Catal., 1991, 130, 411 CrossRef CAS.
- K. Tanaka and A. Ozaki, J. Catal., 1967, 8, 1 CrossRef CAS.
- E. Iwamatsu and K. Aika, J. Catal., 1989, 117, 416 CrossRef CAS.
- K. Aika, N. Fujimoto, M. Kobayashi and E. Iwamatsu, J. Catal., 1991, 127, 1 CrossRef CAS.
- E. Iwamatsu, T. Moriyama, N. Takasaki and K. Aika, J. Catal., 1988, 113, 25 CrossRef CAS.
- V. I. Lomonosov and M. Yu. Sinev, Kinet. Catal., 2016, 57, 647 CrossRef CAS.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. I. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr., 2005, 220, 567 CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cy01641e |
| This journal is © The Royal Society of Chemistry 2022 |