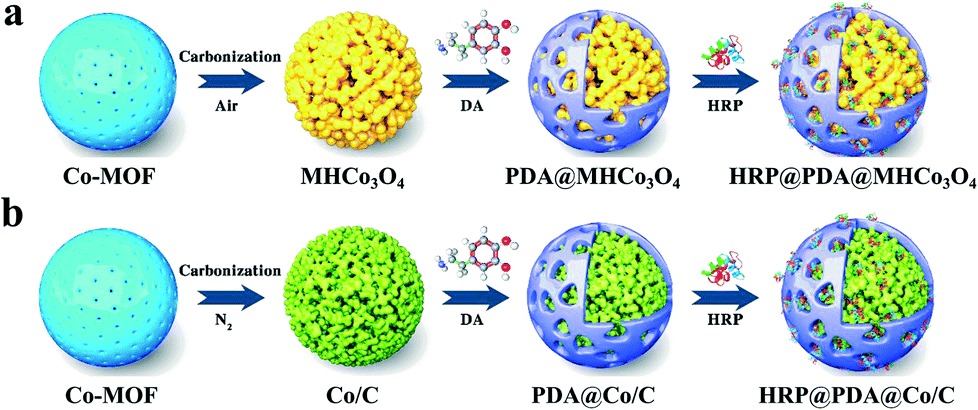
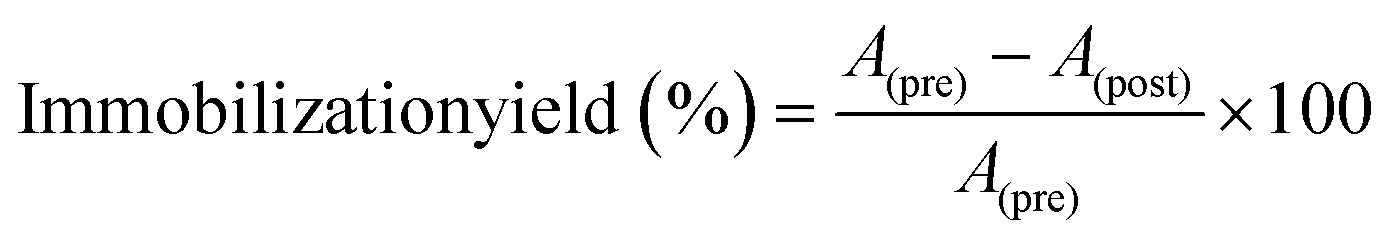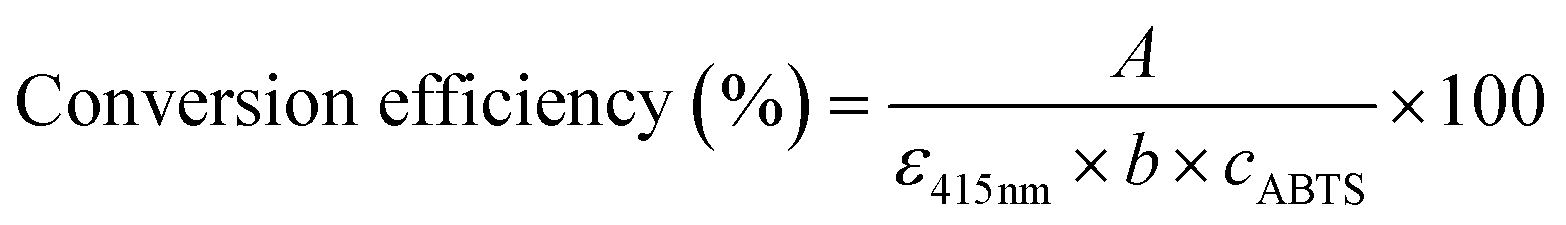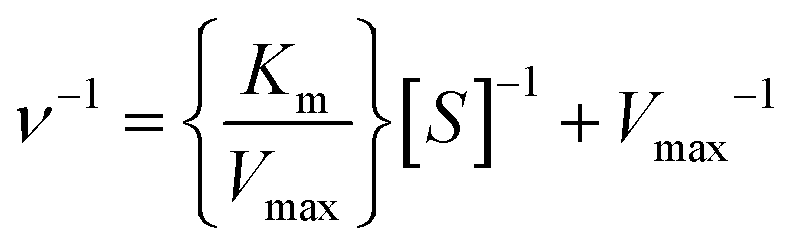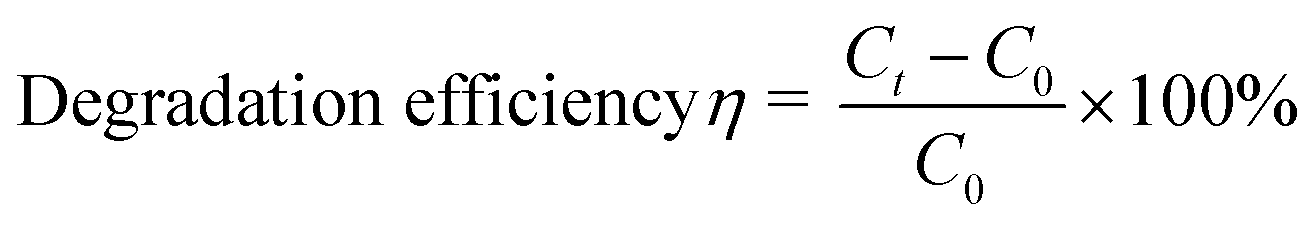Facile preparation of MOF-derived MHCo3O4&Co/C with a hierarchical porous structure for entrapping enzymes: having both high stability and catalytic activity†
Received
3rd August 2021
, Accepted 29th October 2021
First published on 30th October 2021
Abstract
MOF-derived porous materials Co3O4 and Co/C with hierarchical structure (MHCo3O4&Co/C) were prepared by the self-sacrificial template strategy, and the surfaces of such materials were functionally modified with a “polydopamine (PDA)” bionic membrane for entrapping horseradish peroxidase (HRP) in this work. MHCo3O4 was prepared under air atmosphere, and Co/C was prepared under nitrogen atmosphere based on the thermolysis of Co-MOF. HRP@PDA@MHCo3O4 and HRP@PDA@Co/C could retain 96.8% and 93.7% catalytic activity compared to the free HRP, respectively, and exhibited better thermal stability and reusability. After 1 h incubation at 70 °C, HRP@PDA@MHCo3O4 and HRP@PDA@Co/C retained 84.8% and 70.8% of the initial activity compared with less than 20% retained by free HRP. After 10 cycles, HRP@PDA@MHCo3O4 and HRP@PDA@Co/C retained 92.4% and 85.4% of the initial activity. Moreover, HRP@PDA@MHCo3O4&Co/C exhibited extreme catalytic degradation efficiency for 2,4-dichlorophenol in simulated wastewater, and 2 mmol L−1 of 2,4-dichlorophenol was completely degraded within 15 min. The superior properties of HRP@PDA@MHCo3O4&Co/C were researched by kinetics and thermodynamics analyses. The results suggested that the mass transfer resistance of the substrates was lower than that of the bulk solution for the reason that the substrates were originally enriched on the hierarchical porous carrier close to the enzyme. The specificity and binding affinity of the enzymatic reactors to substrates were enhanced by the functionalized “PDA” bionic membrane on the carrier surface, compared with free enzyme in bulk buffer.
Introduction
Enzyme-catalyzed reactions have many advantages, such as excellent catalytic efficiency, high specificity, moderate reaction conditions, and green environmental protection. However, the industrialization application of enzymes is restricted by problems, such as poor stability, high cost and difficulty in recycling from the reaction system.1,2 Loading biological enzyme molecules onto the solid carrier is considered to be one of the most effective ways, which can be reused by solid–liquid separation so as to reduce the cost of using the enzyme.3,4 So far, a large number of solid phase carriers, such as silica,5 anodic alumina,6 TiO2,7,8 chitosan,9 metal nanoparticles10 and graphene oxide11 for entrapping enzymes have been reported, in which nanomaterials with porous structures have been widely taken into consideration.12–15
In recent years, porous nanomaterials with hierarchical structures have attracted much attention due to their unique performance advantages, such as high surface area, excellent thermal and chemical stability,16–18 and they have potential application prospects in the fields of catalysis,19 adsorption,20 supercapacitors,21 energy storage and conversion.22 Various methods have been developed for the preparation of porous nanomaterials.23–25 Among numerous methods, the self-sacrificial template strategy on the basis of calcination of solid precursors is regarded as one of the best prospective methods for preparing porous nanomaterials because of its low cost and good effect. Metal organic frameworks (MOFs) can be taken as solid precursors to prepare porous nanomaterials, and the prepared porous materials not only possess the advantages of the parent MOFs, but also have the excellent characteristics of adjustable pore structure, good thermal stability and chemical stability, which can be an ideal carrier for enzyme immobilization. However, due to the complexity and difficulty in controlling the process of preparing porous materials by thermal decomposition of MOFs and the immobilization of enzymes, these shortcomings greatly limit the large-scale application of MOFs as a template to prepare porous materials. So far, there are merely a few reports on the immobilization of enzymes based on MOF-derived oxide/carbon composites to our best knowledge,26–28 and most of the reported studies on enzymatic reactors have focused on the study of enzymatic properties,27,28 which greatly hindered their application.
In the previous research work, HRP/Cyt c@MHNiO were constructed by introducing HRP and Cyt c into MOF-derived MHNiO, which was synthesized by the self-sacrificial template strategy in our group.29 On the basis of previous research, MOF-derived Co3O4 and Co/C with hierarchical porous structure (MHCo3O4&Co/C) were prepared by the self-sacrificial template strategy, and the surfaces of such materials were functionally modified with a “polydopamine (PDA)” bionic membrane for entrapping horseradish peroxidase (HRP) in this work (shown in Fig. 1). Two types of hierarchical porous Co3O4 (uniformly expressed as MHCo3O4) were prepared under air atmosphere, and three types of hierarchical porous Co/C (uniformly expressed as Co/C) were prepared under nitrogen atmosphere based on the thermolysis of Co-MOF. On the basis of evaluating the carrier by the loading capacity and enzyme activity, the carrier materials with the surface functionalized by the “PDA” bionic membrane were evaluated by the binding affinity of the enzymatic reactors to the substrate. The thermostability and reusability of the enzymatic reactors were investigated. The enzyme-substrate interactions in the enzymatic reactors were researched by kinetic and thermodynamic parameters analysis. The application of the enzymatic reactors in the treatment of wastewater containing 2,4-dichlorophenol was investigated.
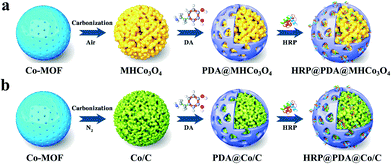 |
| | Fig. 1 Schematic representation for the preparation of HRP@PDA@MHCo3O4 (a) and HRP@PDA@Co/C (b). | |
Results and discussion
Design strategy of enzymatic reactors
Recently, MOFs were employed as sacrificial templates for the preparation of MOF-derived porous materials, and the pore size of such materials can be regulated by adjusting the thermo-decomposed temperature and time.30–33 In this work, Co-MOF has been selected as a model MOF to research the feasibility of MOFs as sacrificial templates for the preparation of hierarchical porous materials. Two types of hierarchical porous Co3O4 (MHCo3O4) were prepared under air atmosphere at different pyrolysis temperatures (500 and 600 °C), and three types of hierarchical porous Co/C were prepared under nitrogen atmosphere at different pyrolysis temperatures (500, 600, and 800 °C) based on the thermolysis of Co-MOF.
A series of screening tests were carried out on the above prepared materials, and such materials were selected as carriers for entrapping HRP by physical adsorption method, respectively, of which the loading capacity and catalytic activity of HRP were evaluated. As shown in Table 1, the average mesoporous diameters of the two types of MHCo3O4 were 13.8 nm and 18.6 nm, respectively, in addition to the original microporous pores. Both the higher enzyme loading capacity and catalytic activity were achieved for HRP by MHCo3O4 with a mesoporous pore size of 13.8 nm. The average mesoporous diameters of the three types of Co/C were 5.7 nm, 6.8 nm and 7.4 nm, respectively, in addition to the original microporous pores. Both the higher enzyme loading capacity and catalytic activity were achieved for HRP by Co/C with a mesoporous pore size of 7.4 nm. To investigate further, the isothermal titration calorimetry (ITC) technique was adopted to screen the ideal carrier, and the isothermal titration calorimetry curves (Fig. S1†) were obtained by on-line measuring the heat flux of the enzyme-catalyzed reaction in the surface-functionalized enzymatic reactors with time. The thermodynamic parameters were obtained by nonlinear least-square fitting method (Table S1†).
Table 1 Carrier screening experimental results and pore features
| Carrier |
Pyrolysis temperature (°C) |
Load amount of enzyme (mg g−1) |
Immobilization yield (%) |
Conversion efficiency of ABTS (%) |
S
BET (m2 g−1) |
V
t (cm3 g−1) |
Microporous and mesoporous size (nm) |
Adsorption average pore diameter (nm) |
| MHCo3O4 |
500 |
139.5 |
70.0 |
93.5 |
26.92 |
0.14 |
1.8, 2.7, 9.4, 18.6 |
13.8 |
| 600 |
119.2 |
59.6 |
90.9 |
21.27 |
0.15 |
1.5, 12.6, 18.5, 25.2 |
18.6 |
| Co/C |
500 |
126.6 |
63.3 |
81.7 |
213.43 |
0.14 |
0.7, 1.2, 5.5, 8.7 |
6.8 |
| 600 |
137.4 |
68.7 |
90.6 |
217.54 |
0.18 |
0.8, 1.3, 5.5, 8.7 |
7.4 |
| 800 |
97.5 |
48.8 |
70.3 |
112.78 |
0.08 |
0.8, 1.2, 3.0, 5.0 |
5.7 |
It can be clearly seen from Table S1† that enzyme-catalyzed reactions obtained from the two types of MHCo3O4 and the three types of Co/C with the surface functionalized by “PDA” bionic membrane were all endothermic reactions. The Ka (binding constant) value of HRP@PDA@MHCo3O4-(500 °C) was greater than that of HRP@PDA@MHCo3O4-(600 °C). Meanwhile, the Ka value of HRP@PDA@Co/C-(600 °C) was greater than that of HRP@PDA@Co/C-(500 °C) and HRP@PDA@Co/C-(800 °C). This indicates that HRP@PDA@MHCo3O4-(500 °C) had a greater binding affinity to substrate than HRP@PDA@MHCo3O4-(600 °C), and HRP@PDA@Co/C-(600 °C) had a greater binding affinity to substrate than HRP@PDA@Co/C-(500 °C) and HRP@PDA@Co/C-(800 °C). Therefore, MHCo3O4 and Co/C with average pore sizes of 13.8 nm and 7.4 nm, respectively, were selected as the carrier for entrapping HRP in this work.
Characterization of carriers and enzymatic reactors
The purple powder Co-MOF precursor, as shown in Fig. S2a,† was prepared with Co2+ as the metal center ion, pyrazine and H3BTC as the organic ligands, and the polymer surfactant PVP as the reaction assistant. The amount of pyrazine was changed during the preparation process to achieve the regulation of the morphology of the Co-MOF. It can be seen from Fig. S2b† that the intensity of the peak in the XRD patterns gradually decreases with the increase of the amount of pyrazine, indicating that the crystallinity of Co-MOF was higher when the dosage of pyrazine was 0.9 mmol. The skeleton structure of Co-MOF can be stabilized to 400 °C under both air and nitrogen atmospheres (Fig. S2c†). When the amount of pyrazine was 0.9 mmol, the morphology of the Co-MOF presented a relatively regular spherical shape (Fig. S2d†). With the increase of pyrazine dosage, the morphology of the material presented a mixture of spicules and spheres (Fig. S2e–h†), which indicated that Co-MOF had a good morphology when the dosage of pyrazine was 0.9 mmol, and could be used as an ideal precursor for subsequent tests.
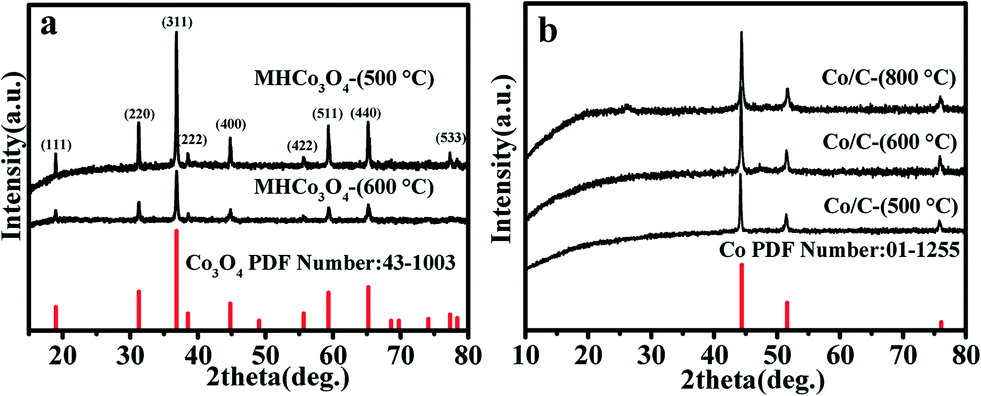
To determine the components and structures of the calcined product, Fig. 2 shows the XRD patterns to research the crystal phases of MHCo3O4 and Co/C. All the diffraction peaks of MHCo3O4 match well with the face-centered cubic Co3O4 (JCPDS 43-1003), indicating the Co-MOF precursor was transformed completely into crystalline Co3O4 under air atmosphere (Fig. 2a). Fig. 2b shows the diffraction characteristic peaks at the diffraction angles of 2θ = 44.37°, 51.59° and 76.08°, which are attributed to the (111), (200) and (220) crystal planes of the Co (JCPDS 01-1255), respectively. The diffraction peaks of the products at different calcining temperatures are in the same position, and the intensity of the diffraction peaks increases as the calcining temperature increases. This indicates that the temperature has little effect on the phase composition of the products, but had an effect on the pyrolysis degree of the Co-MOF precursor. The EDX spectra in Fig. S3† indicate that the MHCo3O4 contain Co and O elements, and the Co/C contain Co and C elements. Combined with the result of the EDX spectra, it can be further proved that the Co-MOF precursor was completely decomposed into Co/C under nitrogen atmosphere. As shown in Fig. S4a and c,† there exists a distinct hysteresis in the N2 adsorption–desorption isotherms of MHCo3O4 and Co/C, indicating the presence of a mesoporous pore. BJH analysis (Fig. S4b and d†) confirmed the presence of hierarchical porosity. Typical morphologies are shown in Fig. S5† and 3. Apparently, the calcined products maintain the similar sizes and shapes of the Co-MOF precursor. The FESEM and FETEM images show that MHCo3O4 is a nanoscale particle accumulation structure formed by homogeneous nanoparticles and a tremendous amount of pores between nanoparticles, while Co/C is a 3D supramolecular nanometer microsphere structure composed of homogeneous nanoparticles and a tremendous amount of pores between nanoparticles (Fig. S5† and 3). After entrapping the enzyme, the morphology and structure of MHCo3O4 and Co/C were almost unaffected.
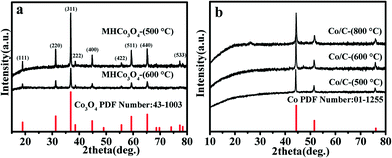 |
| | Fig. 2 (a) XRD patterns of Co3O4 and MHCo3O4, and (b) XRD patterns of Co and Co/C. | |
 |
| | Fig. 3 FETEM images of Co-MOF (a and b), MHCo3O4 (c), HRP@PDA@MHCo3O4 (d), Co/C (e) and HRP@PDA@Co/C (f). | |
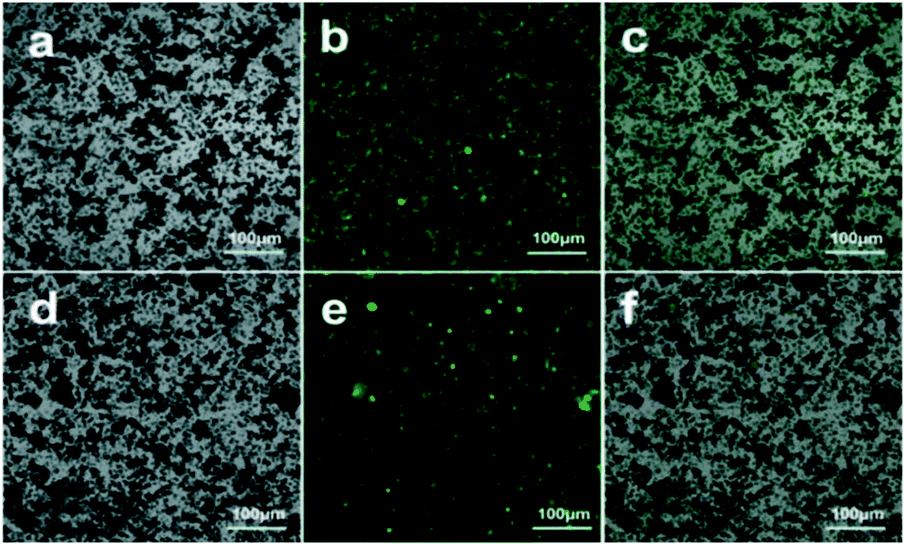
To confirm the successful immobilization of HRP on PDA@MHCo3O4 or PDA@Co/C, HRP was marked with the fluorochrome fluorescein isothiocyanate (FITC). As shown in Fig. 4, many bright green fluorescent spots in the image of FITC-HRP@PDA@MHCo3O4 (b) or FITC-HRP@PDA@Co/C (e) were observed, while PDA@MHCo3O4 or PDA@Co/C itself has no fluorescence (a and d). The overlap of a and b, or d and e can directly verify the successful immobilization of HRP on PDA@MHCo3O4 or PDA@Co/C.
 |
| | Fig. 4 Laser confocal microscopy images of the PDA@MHCo3O4 (a), FITC-HRP@PDA@MHCo3O4 (b and c), PDA@Co/C (d) and FITC-HRP@PDA@Co/C (e and f). | |
The catalytic performance of enzymatic reactors
The mass transfer resistance of the substrate in the enzymatic reactors is usually greater than that of the free enzyme. Meanwhile, the enzyme–carrier interaction often impacts the structure of the enzyme, resulting in the decrease of the enzyme activity. However, it has been reported that the substrate can be enriched around some porous materials (such as ZIFs), to facilitate mass transfer to achieve the higher apparent activity of enzymatic reactors.34–38 In this work, the same amounts of free HRP and enzymatic reactors were adopted for catalyzing the oxidation of ABTS, and the activities of enzymatic reactors were compared with those of free HRP. As shown in Fig. S6† and Table 1, HRP@PDA@MHCo3O4&Co/C can retain 96.8% or 93.7% activity, respectively, while HRP@MHCo3O4&Co/C can retain 93.5% or 90.6% activity compared to the free HRP. The superior activity of the enzymatic reactors was ascribed to the following two aspects: (1) due to the hierarchical porous structure of the carrier, the substrate can be enriched in the micropores around the mesoporous area of the enzymatic reactors, so as to reduce the mass transfer resistance of the substrate and make it close to the active site of the enzyme; (2) through pore size regulation, the mesoporous pore size of the carrier can be matched with the size of the enzyme molecules, so as to avoid the agglomeration of the enzyme molecules. In order to prove the oxidation of ABTS by pure PDA@MHCo3O4 (or PDA@Co/C) material, the blank control experiment of the pure material was added, which indicated that the pure material could not oxidize ABTS to produce ABTS˙+, but the catalytic oxidation of ABTS could be realized by enzymatic reactors. Therefore, the catalytic oxidation reaction of HRP@PDA@ MHCo3O4 (or HRP@PDA@Co/C) to ABTS was not affected by the pure material. Compared with HRP@MHCo3O4 and HRP@Co/C, HRP@PDA@MHCo3O4 and HRP@PDA@Co/C can retain higher catalytic activity. This result verified that PDA can be used as a kind of “adhesive” to bind the enzyme to the carrier material, and promote the preservation of the catalytic activity of the enzymatic reactors.
The stability of enzymatic reactors
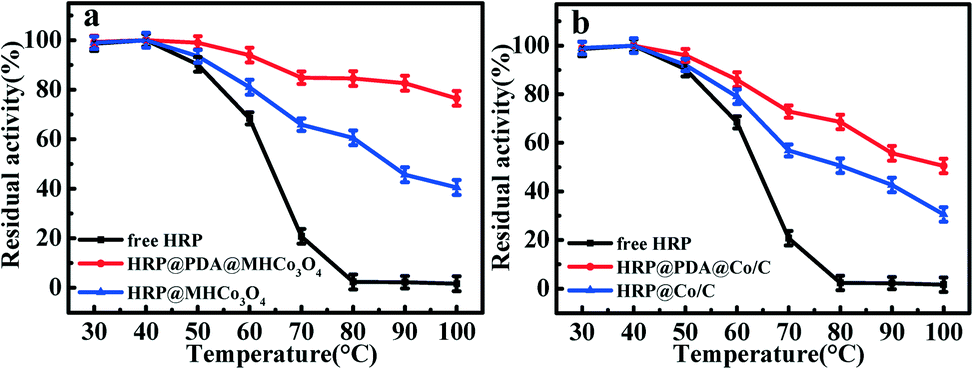
As shown in Fig. 5, the activities of free HRP and enzymatic reactors basically kept unchanged after incubation for 1 h within a range of 30–50 °C, but the superiority in the thermostability of HRP@PDA@MHCo3O4&Co/C was highlighted under higher temperatures. For instance, HRP@PDA@MHCo3O4 and HRP@PDA@Co/C can retain 84.8% and 70.8% activity, respectively, after incubation at 70 °C for 1 h, while free HRP can only retain 18.1% activity. Both HRP@MHCo3O4 and HRP@Co/C can only retain 65.7% and 57.3% activity, respectively, under the same condition. The result confirmed that the thermal stability of HRP@PDA@MHCo3O4&Co/C was remarkably superior to that of the free HRP, which is consistent with the results reported in related literature,39–42 and has been caused by the affinity between the enzyme molecules and the carrier increases after being immobilized in the carrier. The carrier provides a barrier for the free enzyme to resist high temperature, and weakens the influence of high temperature on the conformation of the enzyme reactor. This result further confirmed that the construction of the “PDA” bionic membrane can strengthen the interaction between enzyme and carrier so as to inhibit enzyme shedding, which can result in the excellent thermal stability. In addition, HRP@PDA@MHCo3O4 had a slightly better catalytic effect than HRP@PDA@Co/C at 40–100 °C.
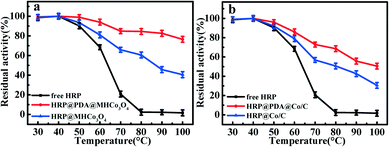 |
| | Fig. 5 (a) Temperature stability of free HRP, HRP@PDA@MHCo3O4 and HRP@MHCo3O4; (b) temperature stability of free HRP, HRP@PDA@Co/C and HRP@Co/C. | |
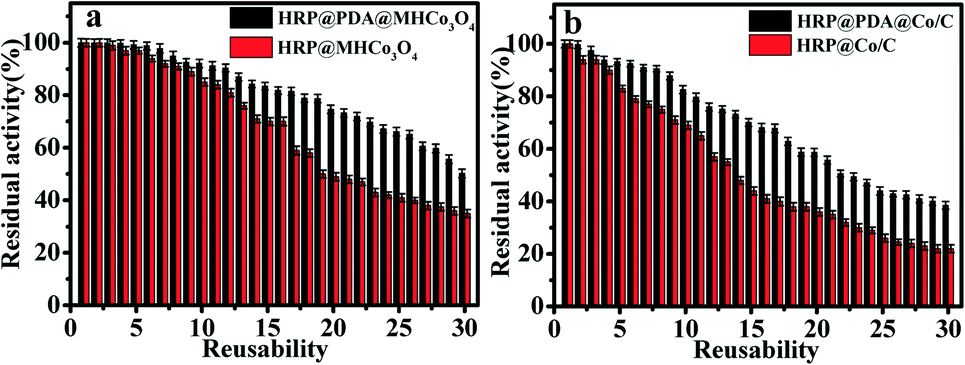
Reusability of enzymatic reactors
As shown in Fig. 6, HRP@PDA@MHCo3O4 and HRP@PDA@ Co/C both show better reusability than HRP@MHCo3O4 and HRP@Co/C. For example, HRP@PDA@MHCo3O4 can retain 92.4% of the initial activity after 10 cycles, while HRP@PDA@Co/C retained 85.4%. The results were superior to those reported in the literature,27,43–49 which was possibly attributed to the functional modification of the carrier surface by the “PDA” bionic membrane (Table 2). The above results confirmed that PDA can be used as a kind of “adhesive” to bind the enzyme tightly to the carrier.
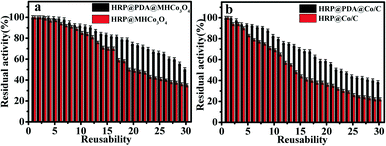 |
| | Fig. 6 Reusability of HRP@PDA@MHCo3O4 (a) and HRP@PDA@Co/C (b). | |
Table 2 Comparison of reusability of various enzymatic reactors
| Enzyme@(material) |
Reusability |
Residual activity (%) |
Ref (year) |
| Laccase@(bimodal mesoporous Zr-MOF) |
10 |
50.0 |
43 2016 |
|
Pseudomonas cepacia lipase@(MCNCs) |
6 |
66.2 |
44 2016 |
| BCL@CMIL-100(Al)-600 |
7 |
53.0 |
27 2017 |
| Lipase@(mesoporous silica) |
7 |
61.3 |
45 2017 |
| Glucoamylase@MOF |
6 |
57.0 |
46 2017 |
| Multi-enzymes@(inorganic nanoreactor) |
6 |
60.0 |
47 2018 |
| Microperoxidase-8@nanoMIL-101 |
5 |
66.0 |
48 2018 |
| Cellulase@cellulose nanofibers |
6 |
85.0 |
49 2019 |
| HRP@PDA@MHCo3O4 |
7 |
97.8 |
This work |
| 10 |
92.4 |
| HRP@PDA@Co/C |
7 |
90.9 |
| 10 |
85.4 |
To further investigate whether the morphology and structure of the materials could be maintained after repeated use, the enzymatic reactors before and after repeated use were used for field-transmission electron microscopy (FETEM) and X-ray powder diffraction (XRD) tests. Fig. S7a and c† shows the FETEM images of the newly prepared enzymatic reactors, and Fig. S7b and d† shows the FETEM images of the enzymatic reactors after repeated use for 30 times, while Fig. S8† shows the XRD patterns of enzymatic reactors before and after repeated use. As shown in Fig. S7 and S8,† the morphology of the materials could be maintained after repeated use, and the XRD diffraction peaks of the materials after repeated use were consistent with those of the newly prepared materials, indicating that the structure of the enzymatic reactors would not be damaged after repeated use and could be stable.
Enzyme-substrate interaction in the enzymatic reactors
As shown in Table 3, the Km values of HRP@PDA@MHCo3O4&Co/C were lower than that of the free HRP and HRP@MHCo3O4&Co/C. This indicated that the affinity of the enzyme to substrate was improved in the enzymatic reactors, and the functional modification on the surface of the carrier material increased the affinity of the enzymatic reactors to the substrate. The Vmax values of HRP@MHCo3O4 and HRP@PDA@MHCo3O4 were increased compared to the free HRP, from 0.5355 mmol L−1 s−1 of HRP to 1.4005 mmol L−1 s−1 and 1.6792 mmol L−1 s−1 of HRP@MHCo3O4 and HRP@PDA@MHCo3O4, respectively. The increase of the Vmax values may be due to the accumulation of substrates in the micropores and the embedding of the enzymes into the adjacent mesopores, thus reducing the diffusion distance of substrates to the enzymes in HRP@MHCo3O4 and HRP@PDA@MHCo3O4. However, the Vmax values of HRP@Co/C and HRP@PDA@Co/C were decreased compared to the free HRP. We speculate that this is because the enzyme molecules need a turnover space in the catalytic process. Although the pore size of Co/C can just accommodate the entry of HRP, it cannot provide enough turnover space, thus leading to the decrease of the Vmax value. To justify the Vmax values of the enzymatic reactors, we carried out an FTIR study on the carrier and enzymatic reactors, respectively. Fig. S9† shows that the peak position of PDA@MHCo3O4 before and after loading enzymes is the same, while the peak position of PDA@Co/C before and after loading enzymes is slightly different, which may be the result of the conformational change of the enzyme molecule during the immobilization process caused by the aperture limit effect of the carrier. Basically, the decrease of Vmax could be attributed to a modification of the protein conformation due to the immobilization process.50–52 Therefore, the above speculation has been further confirmed. The kcat/Km values of the HRP@PDA@MHCo3O4&Co/C were higher than the free HRP, revealing that equal amounts of enzyme could transform ABTS rapidly when it was embedded in PDA@MHCo3O4&Co/C. In addition, the kcat/Km values of the HRP@PDA@MHCo3O4 and HRP@PDA@Co/C were higher than that of the corresponding HRP@MHCo3O4 and HRP@Co/C, which indicated that the enzymatic reactors on the surface of the carrier material functionally modified with “PDA” bionic membrane improved the specificity of the substrate.
Table 3 Enzymatic kinetic parameters of free HRP and enzymatic reactors
| Enzyme |
V
max (mmol L−1 s−1) |
K
m (mmol L−1) |
k
cat (s−1) |
k
cat/Km (L mmol−1 s−1) |
| Free HRP |
0.5355 |
0.4093 |
21.4200 |
52.3333 |
| HRP@MHCo3O4 |
1.4005 |
0.1280 |
56.0200 |
437.6563 |
| HRP@PDA@MHCo3O4 |
1.6792 |
0.0936 |
67.1600 |
717.5214 |
| HRP@Co/C |
0.1416 |
0.1007 |
5.6640 |
56.2463 |
| HRP@PDA@Co/C |
0.1564 |
0.0567 |
6.2560 |
110.3351 |
As can be seen from Table 4, all of the ΔG values were negative, in which the negative ΔG values of the enzymatic reactors were greater than that of free HRP. This indicates that the spontaneity of the HRP@PDA@MHCo3O4&Co/C enzyme-catalyzed reaction is significantly enhanced. The positive ΔH values indicated that all of the above enzyme-catalyzed reactions were endothermic processes. However, the required energy decreased dramatically, as for HRP@PDA@MHCo3O4&Co/C, which was mainly owing to the hierarchical porous structure and the functionalized modification of the carrier surface. This resulted in the mass transfer resistance of the substrates being lower than that in bulk buffer. The measured binding site number (n) has aroused extensive concern. For free HRP, the n value of 1.23 proved the substrate was combined with enzyme as 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. However, as to HRP@PDA@MHCo3O4&Co/C, the n values significantly increased, indicating the substrate was concentrated on PDA@MHCo3O4&Co/C. This further verified the promoting effect of the “PDA” biomimetic membrane functionalized modification on the enzymatic catalytic reaction. TheΔS value of the reaction can reveal the structural changes of the reactants. The ΔS values were decreased dramatically by using HRP@PDA@MHCo3O4&Co/C compared to using the free HRP. The decreased ΔS values verified that the rigidity of the enzyme was enhanced after they were loaded onto the carrier. According to the contribution of ΔH and TΔS to ΔG, the oxidation of ABTS is an entropy-driven process. The Ka values of HRP@PDA@MHCo3O4 and HRP@PDA@Co/C were superior to that of the free HRP, indicating the functional modification on the surface of the carrier material increased the affinity of the enzymatic reactors to substrate, which was in accordance with the conclusion obtained by the kinetic parameters.
:1. However, as to HRP@PDA@MHCo3O4&Co/C, the n values significantly increased, indicating the substrate was concentrated on PDA@MHCo3O4&Co/C. This further verified the promoting effect of the “PDA” biomimetic membrane functionalized modification on the enzymatic catalytic reaction. TheΔS value of the reaction can reveal the structural changes of the reactants. The ΔS values were decreased dramatically by using HRP@PDA@MHCo3O4&Co/C compared to using the free HRP. The decreased ΔS values verified that the rigidity of the enzyme was enhanced after they were loaded onto the carrier. According to the contribution of ΔH and TΔS to ΔG, the oxidation of ABTS is an entropy-driven process. The Ka values of HRP@PDA@MHCo3O4 and HRP@PDA@Co/C were superior to that of the free HRP, indicating the functional modification on the surface of the carrier material increased the affinity of the enzymatic reactors to substrate, which was in accordance with the conclusion obtained by the kinetic parameters.
Table 4 Thermodynamic parameters from independent site modela
| Enzyme |
K
a × 10−3 (M−1) |
ΔH (kJ mol−1) |
ΔS (J mol−1 K−1) |
n
|
TΔS (kJ mol−1) |
ΔG (kJ mol−1) |
|
Measured in phosphate buffer (pH = 5.0) at 298 K, corrected for dilution response.
|
| Free HRP |
0.75 ± 0.01 |
1088.0 ± 0.01 |
3703.0 ± 0.2 |
1.23 ± 0.1 |
1104.0 ± 0.1 |
−16.41 ± 0.04 |
| HRP@MHCo3O4 |
0.78 ± 0.01 |
999.1 ± 0.02 |
3359.7 ± 0.1 |
1.39 ± 0.1 |
1016.0 ± 0.2 |
−16.86 ± 0.06 |
| HRP@PDA@MHCo3O4 |
1.24 ± 0.01 |
97.8 ± 0.01 |
387.3 ± 0.1 |
6.23 ± 0.1 |
115.5 ± 0.2 |
−17.66 ± 0.06 |
| HRP@Co/C |
0.97 ± 0.02 |
988.0 ± 0.02 |
3372.7 ± 0.2 |
1.28 ± 0.1 |
1017.0 ± 0.2 |
−17.06 ± 0.05 |
| HRP@PDA@Co/C |
1.01 ± 0.02 |
37.3 ± 0.02 |
182.5 ± 0.2 |
5.79 ± 0.1 |
54.4 ± 0.2 |
−17.14 ± 0.05 |
Degradation of contaminant in wastewater
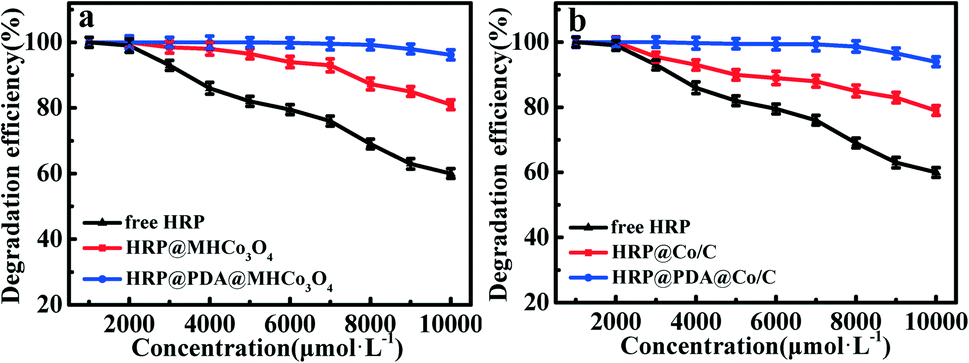
A majority of phenolic compounds, particularly chlorophenol, a highly toxic compound, are very difficult to be degraded.53–55 In this work, HRP@PDA@MHCo3O4&Co/C were adopted for enzymatic degradation of 2,4-dichlorophenol in simulated wastewater (Fig. 7 and S10†). As shown in Fig. 7a, compared with HRP@MHCo3O4, HRP@PDA@MHCo3O4 shows a higher catalytic degradation efficiency of 2,4-dichlorophenol in simulated wastewater. The 2,4-dichlorophenol at 2 mmol L−1 could be completely degraded in 15 min. In addition, the degradation efficiency of 2,4-dichlorophenol at concentrations up to 10 mmol L−1 could reach up to 96.5%. Compared with HRP@Co/C, HRP@PDA@Co/C shows a higher catalytic degradation efficiency of 2,4-dichlorophenol in simulated wastewater (Fig. 7b). The 2,4-dichlorophenol (2 mmol L−1) could be completely degraded in 15 min. Nearly 95% of 2,4-dichlorophenol was degraded when the concentration of 2,4-dichlorophenol was increased to 10 mmol L−1. This result was superior to the degradation effect reported in the literature (Table 5).56–62 In order to prove the catalytic degradation of 2,4-dichlorophenol in simulated wastewater by pure carrier materials, blank control experiments were added. The results showed that the pure carrier materials could not catalytically degrade 2,4-dichlorophenol in the simulated wastewater, but it could be achieved by loading HRP, which proved that the catalytic degradation of 2,4-dichlorophenol in the enzymatic reactors was not affected by the pure carrier materials.
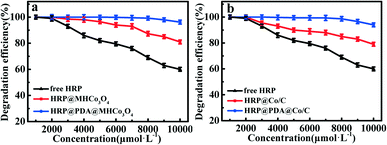 |
| | Fig. 7 (a) Degradation of 2,4-dichlorophenol by free HRP, HRP@PDA@MHCo3O4 and HRP@MHCo3O4. (b) Degradation of 2,4-dichlorophenol by free HRP, HRP@PDA@Co/C and HRP@Co/C. | |
Table 5 Comparison of the degradation efficiency of various catalyst materials to organic toxins reported previously with that in this work
| Catalyst material |
Toxicant |
Real water sample |
Degradation time |
Degradation efficiency (%) |
Ref (year) |
| Planar falling film reactor |
2,4-Dichlorophenol |
Aqueous solutions |
1.5 h |
73.0 |
56 2018 |
| TiO2@CuFe2O4 |
2,4-Dichlorophenoxyacetic acid |
Wastewater |
1 h |
97.2 |
57 2018 |
| POM@MOFs |
Phenol |
Aqueous solutions |
1 h |
97.0 |
58 2018 |
| TiO2 electrode Ru–IrO2 electrode |
2,4-Dichlorophenol |
HF/HNO3/H2O mixed solution |
1 h |
94.3 |
59 2018 |
| 83.9 |
| BioMnOx |
Phenol |
Aqueous solutions |
40 min |
100.0 |
60 2018 |
| MgO@Ag/TiO2 |
Phenol |
Aqueous solutions |
2 h |
95.0 |
61 2019 |
| Z-Scheme LaCoO3/g-C3N4 |
Phenol |
Aqueous solutions |
5 h |
85.0 |
62 2019 |
| HRP@PDA@MHCo3O4 |
2,4-Dichlorophenol |
SimulatedWastewater |
15 min |
100.0 |
This work |
| HRP@PDA@Co/C |
2,4-Dichlorophenol |
SimulatedWastewater |
15 min |
100.0 |
However, we found an interesting phenomenon that free HRP exhibited comparatively lower degradation efficiency than the enzymatic reactors containing the same amount of enzyme under equal condition (Table 6). The improved catalytic activity of the enzymatic reactors for degrading 2,4-dichlorophenol may be ascribed to the following two aspects: (1) although the degradation reaction was performed in simulated wastewater, the free HRP embedded in the pores can withstand the adverse conditions. Thus, the free HRP lost more activity than the enzymatic reactors in simulated wastewater; (2) the “PDA” bionic membrane was used to enhance the hydrothermal stability of the carrier, so that the enzymatic reactors had the advantage of high enzyme loading capacity and retained the catalytic activity of the enzyme to the greatest extent.
Table 6 The degradation of 2,4-dichlorophenol by free HRP and enzymatic reactors
| Enzyme |
Real water sample |
Enzyme concentration (μmol L−1) |
2,4-Dichlorophenol concentration (mmol L−1) |
Degradation efficiency (%) |
| Free HRP |
Simulated wastewater |
2.0 |
6.0 |
80.2 ± 0.02 |
| 9.0 |
62.5 ± 0.01 |
| HRP@PDA@MHCo3O4 |
Simulated wastewater |
2.0 |
6.0 |
99.8 ± 0.01 |
| 9.0 |
98.0 ± 0.01 |
| HRP@PDA@Co/C |
Simulated wastewater |
2.0 |
6.0 |
99.6 ± 0.01 |
| 9.0 |
96.6 ± 0.01 |
Conclusion
MOF-derived Co3O4&Co/C with hierarchical porous structure (MHCo3O4 and Co/C) were prepared in air and nitrogen atmospheres by the self-sacrificial template strategy, and the surfaces of such materials were functionally modified with the “PDA” bionic membrane for entrapping horseradish peroxidase (HRP) for the first time here. Those enzymatic reactors exhibited outstanding catalytic activity, excellent thermostability, and potential reusability mainly based on three factors: (1) the hierarchical pores give room for the enzymes and substrates separately, which is within the neighborhood of each other to make it easy for substrates to enter the active sites of the enzymes; (2) the rigid framework of PDA@MHCo3O4&Co/C possesses a shielding effect on the embedded enzyme to protect it from the influence of heat and other inactivation factors; (3) the functional modification of the carrier surface by the “PDA” bionic membrane retains the enzyme activity to the maximum extent. The reaction thermodynamics and kinetics of HRP@PDA@MHCo3O4&Co/C have been investigated systematically. The results showed that the specificity and binding affinity of the enzymatic reactors to substrates were strengthened compared with that of the free enzymes.
Experimental
Preparation of MHCo3O4&Co/C
The preparation procedure for MHCo3O4&Co/C includes two main steps: the Co-MOF prepared by a solvothermal procedure was used as a precursor, and then the MHCo3O4 and Co/C were synthesized by self-sacrificial template strategy, which is based on the thermolysis of Co-MOF in air and nitrogen atmospheres, respectively. The detailed preparation processes of MHCo3O4 and Co/C were described in the ESI.†
Preparation of PDA@MHCo3O4&Co/C
Amounts of 0.241 g of MHCo3O4 (or 0.200 g of Co/C) and 0.0095 g of DA were dispersed in 50 mL of ultra-pure water, and then 0.102 g of HMTA was added. The mixture was swirled for 30 s and incubated for 3 h at 90 °C and then cooled naturally. Subsequently, the resulted PDA@MHCo3O4 and PDA@Co/C were dried under vacuum at 60 °C for 12 h.
Preparation of enzymatic reactors
An amount of 5 mg of PDA@MHCo3O4 or PDA@Co/C was dispersed in 0.1 mol L−1 phosphate buffer (PBS), and 100 μL of HRP (0.25 mmol L−1) was added. After full adsorption, the solid phase was collected by centrifugation, and washed three times by PBS (0.1 mol L−1), then dried under vacuum at 30 °C for 12 h. The immobilization yield of HRP was measured through absorbance before and after immobilization at the characteristic absorption peak of HRP at 403 nm.| | 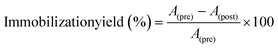 | (1) |
Determination of enzymatic activity
To investigate the enzymatic activity of HRP, the peroxidation of ABTS was taken as the model reaction (Fig. 8).63 The measurements of the absorbance of the ABTS cation radicals (ABTS˙+) at 415 nm were carried out by applying 5 mg of HRP@PDA@MHCo3O4 or HRP@PDA@Co/C in PBS (0.1 mol L−1) containing 30 μL of ABTS (0.01 mol L−1) and 30 μL of H2O2 (0.1 mol L−1) for 5 min, and the mixtures were then centrifuged for 2 min. The conversion efficiency of ABTS was calculated by the following formula.| | 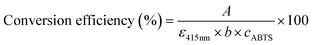 | (2) |
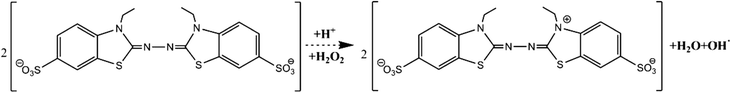 |
| | Fig. 8 Peroxidation of ABTS to ABTS˙+ catalyzed by HRP. | |
Investigation of thermal stability and reusability
The thermal stability was investigated and represented as residual activity by incubating the same amount of the enzymatic reactors and free enzymes at different temperatures (30–100 °C) for 1 h, and compared with that before incubation.
The reusability of the enzymatic reactors was investigated by removal from the reaction medium at the end of each cycle, and adding a fresh substrate solution mixture for the next cycle, where the conversion efficiency of ABTS was represented as 100%.
Measurements of the enzymatic kinetic and thermodynamic parameters
The enzymatic kinetics and thermodynamic parameters were measured and calculated using the methods recently reported by our research group.64 The detailed experimental procedures were described in the ESI.† The kinetic parameters, such as Michaelis constant (Km), catalytic turnover frequency (kcat), and catalytic specificity constant (kcat/Km), were calculated according to the regression equations of the initial reaction rates in the presence of varying concentrations of substrate.| | 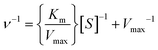 | (3) |
The thermodynamic parameters, such as enthalpy change (ΔH), binding site number (n), equilibrium constant (Ka), entropy change (ΔS) and Gibbs free energy (ΔG), were obtained by the nonlinear least-square fitting method (Fig. S11†).
The application of HRP@PDA@MHCo3O4&Co/C
The degradation of 2,4-dichlorophenol was performed in simulated wastewater65 with an overall volume of 3 mL including enzymatic reactors containing HRP (2.0 μmol L−1) and 2,4-dichlorophenol with the concentrations ranging from 1 mmol L−1 to 10 mmol L−1. The specific experimental procedures were provided in the ESI.† The degradation efficiency (η) was calculated as follows:| | 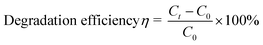 | (5) |
Author contributions
The manuscript was written through contributions of all authors. Y. J. and C. Z. conceived and designed the study. H. P. and Y. L. performed the experiments. X. G. and C. Q. did the microthermal study. X. G., Q. Z., M. H. and S. L. wrote and revised the paper. All authors read and approved the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was sponsored by the National Natural Science Foundation of China (21873061) to Y. Jiang, the National Natural Science Foundation of China (22003037), the Key Laboratory Project of Education Department of Shaanxi Province (20JS043), the Natural Science Foundation Project (19SKY003 and 20SKY005) of Shangluo University to X. Gao, the National Natural Science Foundation of China (22173056), and the Scientific Research Team Project (20SCX01) of Shangluo University to C. Qiao.
Notes and references
- V. Stepankova, S. Bidmanova, T. Koudelakova, Z. Prokop, R. Chaloupkova and J. Damborsky, ACS Catal., 2013, 3, 2823–2836 CrossRef CAS.
- S. Cantone, V. Ferrario, L. Corici, C. Ebert, D. Fattor, P. Spizzo and L. Gardossi, Chem. Soc. Rev., 2013, 42, 6262–6276 RSC.
- Q. Yang, B. Wang, Z. Zhang, D. S. Lou, J. Tan and L. C. Zhu, RSC Adv., 2017, 7, 38028–38036 RSC.
- R. A. Sheldon and S. van Pelt, Chem. Soc. Rev., 2013, 42, 6223–6235 RSC.
- Y. Q. Lin, W. H. Jin, Y. Qiu and G. Y. Zhang, Int. J. Biol. Macromol., 2019, 134, 1156–1169 CrossRef CAS.
- W. Chen, B. Jin, Y. L. Hu, Y. Lu and X. H. Xia, Small, 2012, 8, 1001–1005 CrossRef CAS.
- J. W. Cui, S. He, S. Dai, L. Y. Liu, A. Zhao, L. Lu, P. Yang, J. Chen and N. Huang, Chem. Eng. J., 2021, 424, 130392–130407 CrossRef CAS.
- H. P. Cheng, M. C. Hu, Q. G. Zhai, S. N. Li and Y. C. Jiang, Chem. Eng. J., 2018, 347, 703–710 CrossRef CAS.
- D. Q. Wang and W. F. Jiang, Int. J. Biol. Macromol., 2019, 126, 1125–1132 CrossRef CAS.
- Y. G. Zhou, R. M. Mohamadi, M. Poudineh, L. Kermanshah, S. Ahmed, T. S. Safaei, J. Stojcic, R. K. Nam, E. H. Sargent and S. O. Kelley, Small, 2016, 12, 727–732 CrossRef CAS PubMed.
- W. Zhuang, X. B. Quan, Z. F. Wang, W. F. Zhou, P. P. Yang, L. Ge, B. V. Hernandez, J. L. Wu, M. Li, J. Zhou, C. J. Zhu and H. J. Ying, Chem. Eng. J., 2020, 394, 125038–125050 CrossRef CAS.
- X. L. Wu, M. Hou and J. Ge, Catal. Sci. Technol., 2015, 5, 5077–5085 RSC.
- W. Tang, T. H. Ma, L. Zhou, G. Y. Wang, X. L. Wang, H. J. Ying, C. Chen and P. Wang, Catal. Sci. Technol., 2019, 9, 6015–6026 RSC.
- M. Sharifi, M. J. Sohrabi, S. H. Hosseinali, A. Hasan, P. H. Kani, A. J. Talaei, A. Y. Karim, N. M. Q. Nanakali, A. Salihi, F. M. Aziz, B. Yan, R. H. Khan, A. A. Saboury and M. Falahati, Int. J. Biol. Macromol., 2020, 143, 665–676 CrossRef CAS PubMed.
- C. Zhang, X. R. Wang, M. Hou, X. Y. Li, X. L. Wu and J. Ge, ACS Appl. Mater. Interfaces, 2017, 9, 13831–13836 CrossRef CAS.
- C. Yu, X. P. Li, Z. Y. Liu, X. J. Yang, Y. Huang, J. Lin, J. Zhang and C. C. Tang, Mater. Res. Bull., 2016, 83, 609–614 CrossRef CAS.
- Y. Chang, Z. P. Wei, X. Chang, G. L. Ma, L. L. Meng, T. T. Liu, L. Yang, Y. M. Guo and X. M. Ma, Chem. Eng. J., 2021, 421, 127816–127828 CrossRef CAS.
- H. X. Yang, Y. Xie, M. M. Zhu, Y. M. Liu, Z. K. Wang, M. H. Xu and S. L. Lin, Dalton Trans., 2019, 48, 9205–9213 RSC.
- X. H. Lin, S. B. Wang, W. G. Tu, Z. B. Hu, Z. X. Ding, Y. D. Hou, R. Xu and W. X. Dai, Catal. Sci. Technol., 2019, 9, 731–738 RSC.
- Q. Wang, L. X. Zhang, Y. K. Guo, M. Shen, M. Wang, B. Li and J. L. Shi, Chem. Eng. J., 2020, 396, 125347–125359 CrossRef CAS.
- Y. Han, S. Zhang, N. Shen, D. J. Li and X. F. Li, Mater. Lett., 2017, 188, 1–4 CrossRef CAS.
- P. P. Zhao, C. Deng, Z. Y. Zhao, P. Lu, S. He and Y. Z. Wang, Chem. Eng. J., 2021, 420, 129942–129954 CrossRef CAS.
- K. J. An, S. G. Kwon, M. Park, H. B. Na, S. Baik, J. H. Yu, D. Kim, J. S. Son, Y. W. Kim, I. C. Song, W. K. Moon, H. M. Park and T. Hyeon, Nano Lett., 2008, 8, 4252–4258 CrossRef CAS PubMed.
- S. L. Xiong and H. C. Zeng, Angew. Chem., Int. Ed., 2012, 51, 949–952 CrossRef CAS.
- J. Chen, S. M. He, B. Huang, L. Y. Zhang, Z. Q. Qiao, J. Wang, G. C. Yang, H. Huang and Q. L. Hao, Appl. Surf. Sci., 2018, 457, 508–515 CrossRef CAS.
- W. B. Wei, S. Y. Dong, G. Q. Huang, Q. Xie and T. L. Huang, Sens. Actuators, B, 2018, 260, 189–197 CrossRef CAS.
- L. H. Liu, Y. H. Shih, W. L. Liu, C. H. Lin and H. Y. Huang, ChemSusChem, 2017, 10, 1364–1369 CrossRef CAS.
- S. Y. Dong, L. Peng, W. B. Wei and T. L. Huang, ACS Appl. Mater. Interfaces, 2018, 10, 14665–14672 CrossRef CAS PubMed.
- X. Gao, Y. Ding, Y. D. Sheng, M. C. Hu, Q. G. Zhai, S. N. Li, Y. C. Jiang and Y. Chen, ChemCatChem, 2019, 11, 2828–2836 CrossRef CAS.
- M. L. Yue, C. Y. Yu, H. H. Duan, B. L. Yang, X. X. Meng and Z. X. Li, Chem. – Eur. J., 2018, 24, 16160–16169 CrossRef CAS PubMed.
- K. Y. Zou and Z. X. Li, Chem. – Eur. J., 2018, 24, 6506–6518 CrossRef CAS.
- B. N. Bhadra, A. Vinu, C. Serre and S. H. Jhung, Mater. Today, 2019, 25, 88–111 CrossRef CAS.
- M. Zhong, L. Kong, N. Li, Y. Y. Liu, J. Zhu and X. H. Bu, Coord. Chem. Rev., 2019, 388, 172–201 CrossRef CAS.
- C. Hu, Y. X. Bai, M. Hou, Y. S. Wang, L. C. Wang, X. Cao, C. W. Chan, H. Sun, W. B. Li, J. Ge and K. N. Ren, Sci. Adv., 2020, 6, eaax5785 CrossRef CAS PubMed.
- X. L. Wu, H. Yue, Y. Y. Zhang, X. Y. Gao, X. Y. Li, L. X. Wang, Y. F. Cao, M. Hou, H. X. An, H. Gu, L. Zhang, S. Li, W. Lou, J. Y. Ma, H. Lin, Y. N. Fu, H. K. Gu, W. Y. Lou, W. Wei, R. N. Zare and J. Ge, Nat. Commun., 2019, 10, 5165–5172 CrossRef PubMed.
- X. L. Wu, J. Ge, C. Yang, M. Hou and Z. Liu, Chem. Commun., 2015, 51, 13408–13411 RSC.
- F. J. Lyu, Y. F. Zhang, R. N. Zare, J. Ge and Z. Liu, Nano Lett., 2014, 14(10), 5761–5765 CrossRef CAS PubMed.
- Y. F. Cao and J. Ge, Chin. J. Catal., 2021, 42, 1625–1633 CrossRef CAS.
- M. L. Verma, R. Rajkhowa, X. Wang, C. J. Barrow and M. Puri, Bioresour. Technol., 2013, 145, 302–306 CrossRef CAS PubMed.
- V. Venezia, F. Sannino, A. Costantini, B. Silvestri, S. Cimino and V. Califano, Microporous Mesoporous Mater., 2020, 302, 110203 CrossRef CAS.
- S. R. Sanchez, M. A. Villena, E. G. Romero, H. L. Ramirez and A. B. Perez, Food Bioprocess Technol., 2014, 7, 1381–1392 CrossRef.
- N. D. Tran and K. J. Balkus, Perspect. Catal., 2011, 1, 956–968 Search PubMed.
- S. L. Pang, Y. W. Wu, X. Q. Zhang, B. N. Li, J. Ouyang and M. Y. Ding, Process Biochem., 2016, 51, 229–239 CrossRef CAS.
- S. L. Cao, Y. M. Huang, X. H. Li, P. Xu, H. Wu, N. Li, W. Y. Lou and M. H. Zong, Sci. Rep., 2016, 6, 20420–20431 CrossRef CAS.
- J. Gao, Y. Wang, Y. J. Du, L. Y. Zhou, Y. He, L. Ma, L. Y. Yin, W. X. Kong and Y. J. Jiang, Chem. Eng. J., 2017, 317, 175–186 CrossRef CAS.
- S. S. Nadar and V. K. Rathod, Int. J. Biol. Macromol., 2017, 95, 511–519 CrossRef CAS.
- W. H. Kong, D. Wu, N. Hu, N. Li, C. J. Dai, X. F. Chen, Y. R. Suo, G. L. Li and Y. N. Wu, Biosens. Bioelectron., 2018, 99, 653–659 CrossRef CAS PubMed.
- E. Gkaniatsou, C. Sicard, R. Ricoux, L. Benahmed, F. Bourdreux, Q. Zhang, C. Serre, J. P. Mahy and N. Steunou, Angew. Chem., Int. Ed., 2018, 57, 16141–16146 CrossRef CAS PubMed.
- M. A. Yassin, A. A. M. Gad, A. F. Ghanem and M. H. A. Rehim, Carbohydr. Polym., 2019, 205, 255–260 CrossRef CAS PubMed.
- Z. Gao and I. Zharov, Chem. Mater., 2014, 26, 2030–2037 CrossRef CAS.
- J. K. Gao, Z. J. Zhang, Y. J. Jiang, Y. Chen and S. F. Gao, Molecules, 2017, 22, 1597–1608 CrossRef.
- Y. Jiang, W. Sun, L. Zhou, L. Ma, Y. He and J. Gao, Appl. Biochem. Biotechnol., 2016, 179, 1155–1169 CrossRef CAS.
- A. Karci, Chemosphere, 2014, 99, 1–18 CrossRef CAS PubMed.
- X. Y. Ji, Z. G. Su, M. F. Xu, G. H. Ma and S. P. Zhang, ACS Sustainable Chem. Eng., 2016, 4, 3634–3640 CrossRef CAS.
- L. Qian, P. Liu, S. Shao, M. J. Wang, X. Zhan and S. X. Gao, Chem. Eng. J., 2019, 360, 54–63 CrossRef CAS.
- K. H. H. Aziz, H. Miessner, S. Mueller, A. Mahyar, D. Kalass, D. Moeller, I. Khorshid and M. A. M. Rashid, J. Hazard. Mater., 2018, 343, 107–115 CrossRef PubMed.
- M. Golshan, B. Kakavandi, M. Ahmadi and M. Azizi, J. Hazard. Mater., 2018, 359, 325–337 CrossRef CAS.
- X. H. Zhong, Y. Lu, F. Luo, Y. W. Liu, X. H. Li and S. X. Liu, Chem. – Eur. J., 2018, 24, 3045–3051 CrossRef CAS PubMed.
- J. Luo, Y. B. Wang, D. Cao, K. Xiao, T. Guo and X. Zhao, Chem. Eng. J., 2018, 343, 69–77 CrossRef CAS.
- N. Tian, X. K. Tian, Y. L. Nie, C. Yang, Z. X. Zhou and Y. Li, Chem. Eng. J., 2018, 352, 469–476 CrossRef CAS.
- T. Scott, H. L. Zhao, W. Deng, X. H. Feng and Y. Li, Chemosphere, 2019, 216, 1–8 CrossRef CAS.
- Z. H. Jin, R. S. Hu, H. Y. Wang, J. N. Hu and T. Ren, Appl. Surf. Sci., 2019, 491, 432–442 CrossRef CAS.
- F. N. Alliny, M. C. R. Ana and F. S. P. Denise, Langmuir, 2007, 23, 1981–1987 CrossRef PubMed.
- X. Gao, Q. G. Zhai, M. C. Hu, S. N. Li and Y. C. Jiang, Catal. Sci. Technol., 2021, 11, 2446–2455 RSC.
- S. V. Mohan, K. K. Prasad, N. C. Rao and P. N. Sarma, Chemosphere, 2005, 58, 1097–1105 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cy01393a |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *ab,
Huibin
Pan
c,
Chengfang
Qiao
a,
Yongliang
Liu
a,
Chunsheng
Zhou
*a,
Quanguo
Zhai
*ab,
Huibin
Pan
c,
Chengfang
Qiao
a,
Yongliang
Liu
a,
Chunsheng
Zhou
*a,
Quanguo
Zhai