
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The design of responsive luminescent lanthanide probes and sensors
David
Parker
 *ab,
Jack D.
Fradgley
a and
Ka-Leung
Wong
b
*ab,
Jack D.
Fradgley
a and
Ka-Leung
Wong
b
aDepartment of Chemistry, Durham University, South Road, Durham, DH1 3LE, UK. E-mail: david.parker@dur.ac.uk
bDepartment of Chemistry, Hong Kong Baptist University, Kowloon Tong, Hong Kong, China
First published on 1st June 2021
Abstract
The principles of the design of responsive luminescent probes and sensors based on lanthanide emission are summarised, based on a mechanistic understanding of their mode of action. Competing kinetic pathways for deactivation of the excited states that occur are described, highlighting the need to consider each of the salient quenching processes. Such an analysis dictates the choice of both the ligand and its integral sensitising moiety for the particular application. The key aspects of quenching involving electron transfer and vibrational and electronic energy transfer are highlighted and exemplified. Responsive systems for pH, pM, pX and pO2 and selected biochemical analytes are distinguished, according to the nature of the optical signal observed. Signal changes include both simple and ratiometric intensity measurements, emission lifetime variations and the unique features associated with the observation of circularly polarised luminescence (CPL) for chiral systems. A classification of responsive lanthanide probes is introduced. Examples of the operation of probes for reactive oxygen species, citrate, bicarbonate, α1-AGP and pH are used to illustrate reversible and irreversible transformations of the ligand constitution, as well as the reversible changes to the metal primary and secondary coordination sphere that sensitively perturb the ligand field. Finally, systems that function by modulation of dynamic quenching of the ligand or metal excited states are described, including real time observation of endosomal acidification in living cells, rapid urate analysis in serum, accurate temperature assessment in confined compartments and high throughput screening of drug binding to G-protein coupled receptors.
 David Parker | David Parker FRSC FRS was educated in the state sector and read Chemistry at Oxford University (1974–1978), where he studied with John Brown for a DPhil (1980) before taking up a NATO Fellowship with Jean-Marie Lehn in Strasbourg. He was appointed in the autumn of 1981 as a Lecturer in Chemistry at Durham University, rising to Professor in 1992 and served twice as Head of Department. He has gained several national and international awards and prizes for his multidisciplinary research that has embraced the design of a wide variety of sensing systems, the creation of targeted imaging and therapeutic agents and conjugates, as well as major contributions to chiral analysis. In each domain his work has been characterized by incisive mechanistic studies. |
 Jack D. Fradgley | Jack Fradgley graduated in 2017 with a First Class Honours degree in Chemistry from Durham University, working for his final year project with Gareth Williams on luminescent noble metal coordination complexes. He gained his PhD working with David Parker in 2021, working on pH responsive chiral europium complexes. During this period he worked closely with Cisbio Bioassays in France, and was a co-inventor on a patent application in 2020. |
 Ka-Leung Wong | Ka-Leung (Gary) Wong is the Dr Mok Man Hung Endowed Professor of Chemistry and Head of the Department at Hong Kong Baptist University. His undergraduate and graduate training was undertaken at Hong Kong University where he worked with Wing-Tak Wong, followed by post-doctoral periods at City University Hong Kong and Durham University. His research interests span bioinorganic chemistry, the study of responsive luminescent materials as well as numerous applications of lanthanide science and optical spectroscopy. Recent work embraces the development of new probes and drugs for cancer diagnostics and photodynamic therapy. |
1. Introduction
The low molar absorption coefficients of the lanthanide ions, associated with the forbidden nature of f–f transitions, mean that direct excitation of the metal ion is a very inefficient means of stimulating lanthanide luminescence. Accordingly, an indirect pathway is required in which an energy matched sensitising group is incorporated into the metal complex to harvest light efficiently. Ideally, the excitation wavelength should be tuned to the output of common laser or pulsed LED sources (e.g. 337, 355, 365, 405 nm). Careful consideration must be given to the photophysical properties of the sensitising (antenna) groups, notably their molar absorptivities (high ε), internal charge transfer (ICT), singlet and triplet energies and the size of the energy gap that determines the facility of inter-system crossing. Small S1–T1 energy gaps favour fast kISC, which typically needs to be >109 s−1 to compete with kf which is of the order of 3 × 108 s−1. These features determine the choice of the excitation wavelength (λexc), the efficiency of triplet excited state formation and the feasibility of an energetically downhill process that allows intramolecular energy transfer to populate the lanthanide excited state efficiently.1–51.1. Competing mechanistic pathways in sensitised luminescence
A mechanistic scheme can be put forward, (Scheme 1), characterising the competing pathways. The salient excited states have a short lived existence and may follow various radiative or non-radiative decay processes, each defined by a rate constant. Simple energetics determine the need for a downhill energy cascade, such that the sensitising excited state donor must lie close but be more than 10kBT (2050 cm−1 at ambient temperature) higher in energy than the accepting Ln excited state. In this way, the back energy transfer step is not prone to thermal activation.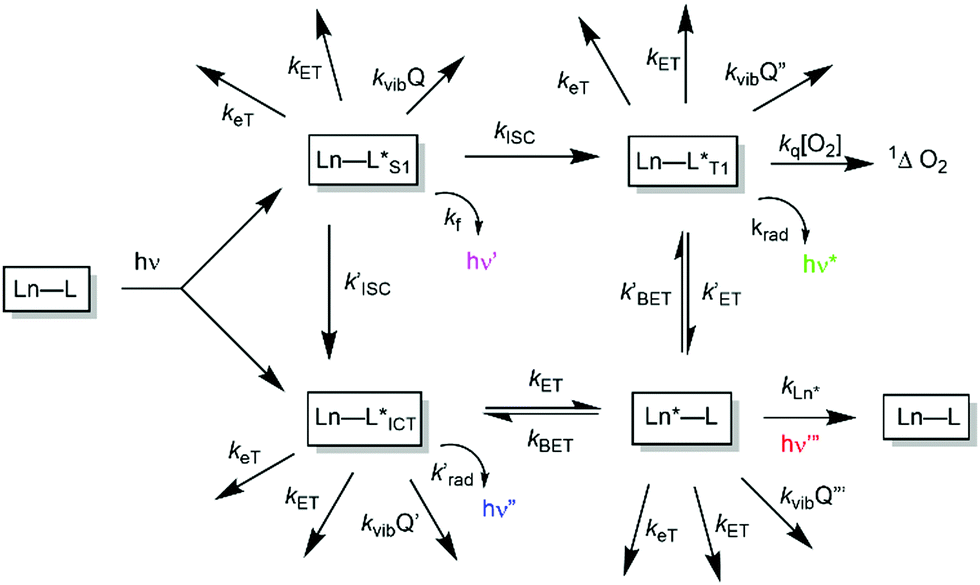 | ||
| Scheme 1 Mechanistic pathways in sensitised lanthanide luminescence, where keT and kET/kBET refer to electron and forward/back energy transfer processes, kISC refers to the rate constant of inter-system crossing, kvib relates to vibrational energy transfer to neighbouring energy matched oscillators and kq is the second order rate constant for collisional quenching of a ligand triplet state (ca. 109 M−1 s−1) by molecular oxygen, forming singlet oxygen. | ||
In such an analysis, there are up to three excited states that can be perturbed to give ‘responsive probes’, for which changes in the observed lanthanide luminescence emission behaviour may occur, potentially modulating the intensity, spectral form, lifetime or circular polarisation of the emitted light.6–9 These excited states have very different lifetimes: the relaxed S1 and ICT states typically exist for no more than a few nanoseconds, the triplet state for up to a microsecond but the lanthanide excited state lifetime falls in the microsecond (e.g. Dy, Yb, Nd, Er) to millisecond (Eu, Tb) range. The longer lifetimes and large pseudo-Stokes’ shifts of sensitised lanthanide luminescence allow the use of time-gated methods of data acquisition for spectroscopy or microscopy, averting the problems associated with light scattering, self-absorption and autofluorescence that may compromise the scope and utility of analyses using short lived fluorophores. A selection of the chromophores that have been explored as sensitisers for lanthanide emission, with their triplet energies, (ET), and absorption maxima (λexc), highlights the predominance of heterocyclic and aromatic systems (Scheme 2).10–26 The Ln ion needs to be as close as possible in space to the chromophore, in order to optimise the efficiency of the ligand to metal energy transfer step, and so is best coordinated to the lanthanide ion by an integral donor, typically a pyridine nitrogen or an anionic oxygen atom. By minimizing the donor–acceptor distance, efficient wave function overlap is created that allows optimal energy transfer to occur by the Dexter electron exchange mechanism.27,28
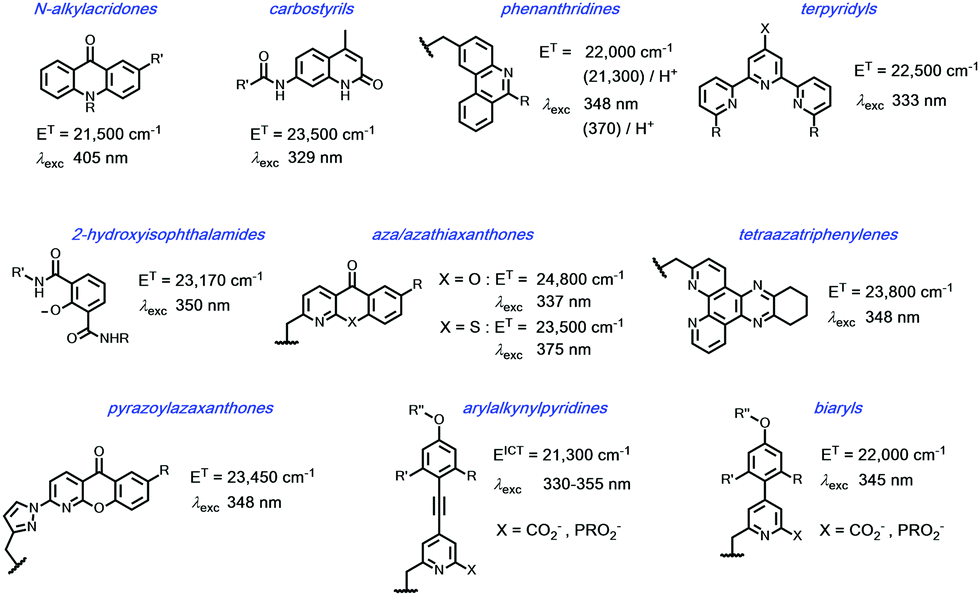 | ||
Scheme 2 Selected sensitising groups with typical triplet or ICT energies and absorption maxima in their lanthanide complexes.10–19 The emissive states of Eu (5D1/5D0) lie at 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 050/17220 cm−1, Tb 5D4 22450 cm−1, Dy 4F9/2 21060 cm−1; Yb 2F5/2 10250 cm−1. 050/17220 cm−1, Tb 5D4 22450 cm−1, Dy 4F9/2 21060 cm−1; Yb 2F5/2 10250 cm−1. | ||
The N-alkyl acridone,10,11 carbostyril antennae12,13 and phenathridinium14 sensitising groups were originally placed more than 5 Å away from the lanthanide centre and often gave modest overall emission quantum yields, owing to the slower rate of the ligand to lanthanide energy transfer step. With more remote antennae, kET from the ligand T1 state may be of the same order as kq[O2], so that a sensitivity to dissolved oxygen concentration may occur. For the acridones and azathiaxanthones,17 the rate of fluorescence, kf is competitive with kISC, so that radiative decay of the excited singlet state competes with triplet formation and fluorescence emission was commonly observed to be more intense than lanthanide luminescence.
In certain cases, europium sensitisation is believed to take place via an efficient intramolecular energy-transfer process that involves a relaxed internal charge transfer (ICT) excited state rather than by a ligand triplet state. In particular, such a situation arises for sensitisers with strongly electron donating groups on the aryl ring, e.g. in arylalkynylpyridines and certain acridones.11,21–25 In the former case with relatively weak donor groups on the aryl ring, the ICT state lies at higher energy and consequently a classical triplet-mediated sensitisation process occurs, with energy transfer via the Dexter exchange mechanism.
Examples of energy transfer directly from the singlet excited state to a proximate Eu ion have also been established, notably when the rate of S1–T1 inter-system crossing is relatively slow.29–32 Such a situation occurs with some coumarin sensitising groups, where the Eu 5D1 excited state may be populated preferentially, decaying within a few microseconds by vibrational relaxation to the longer lived 5D0 Eu emissive state.
In each example of terbium complexes incorporating chromophores with triplet energies of 22000 cm−1 or less, reverse energy transfer, kBET is competitive with the rate of radiative emission from the Ln ion, kLn*, rendering the observed luminescence intensity and emission lifetime sensitive to variations in dissolved oxygen concentration and to changes in temperature. Sensitisation of Eu, Dy and Tb is only possible for four of the shown systems: namely, the azaxanthone, tetraazatriphenylene, pyrazoylazaxanthone and certain terpyridyl series. And, whilst the isophthalamides are excellent sensitisers for Tb3+ and Dy3+ they are not normally used for Eu sensitisation, as keT from the ligand singlet excited state to the ground state Eu3+ ion is fast with respect to kISC.
2. Competitive excited state quenching processes
Considering each of the electron transfer and electronic energy or vibrational energy transfer steps in more detail, the key competitive quenching processes for the ligand or lanthanide excited states can be identified, in the pursuit of effective and efficient responsive probes. In every case, the facility of both intermolecular and intramolecular processes needs to be considered. In this section, we trace the development of the thinking that allows mechanistic understanding of the role of quenching that is a key aspect in how responsive systems should be designed.2.1. Electron transfer quenching, keT
An electron transfer process may quench the singlet or ICT excited state, involving charge transfer to Eu3+ and Yb3+ (but not to Dy3+ or Tb3+), from the high energy ligand excited state. In ligands with electron rich aryl groups, the emission intensity and overall emission quantum yields are reduced but the lanthanide excited state lifetime is not perturbed.A simple example involves electron transfer from the singlet excited state of the sensitising chromophore to a nearby lanthanide ion. Such a process is not relevant for complexes of Dy3+ or Tb3+, as these ions are difficult to reduce (E1/2(red) < −3.5 V), (Table 1). In complexes of Eu3+, Yb3+ and to a lesser extent Sm3+, with octadentate ligands, for example polyamino–carboxylates or carboxamides, the reduction potential occurs in the range −0.8 to −1.2 V for Eu and −1.4 to −2.0 V for the Yb(III) analogues. Thus, while simple phenyl rings and substituted analogues (e.g. X = OMe, CN or CO2Me) can be superb, simple sensitizing groups for Tb3+ in complexes containing this chromophore, the overall quantum yield for Eu emission is much lower. Thus, with the cationic tetra-amide complex, [EuL1]3, ϕem is more than three orders of magnitude lower when X = H, and is even smaller when X = OMe.33 Similar reasoning explains the higher emission quantum yields found for terbium peptide complexes, compared to their Eu analogues, where the metal ion is close in space to electron rich Trp, Tyr or Phe residues.
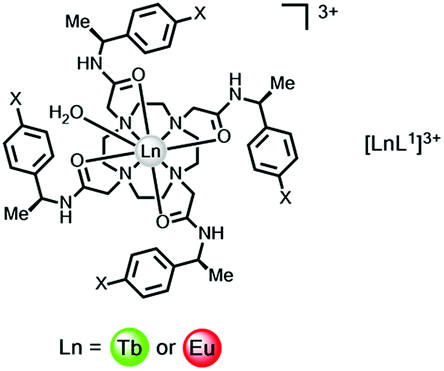 a
a
| X = H | X = CN | X = CO2Me | |
|---|---|---|---|
| a Note the lack of significant variation of the Eu and Tb emission lifetime, as X varies; each complex has one coordinated water.33 | |||
| τ Eu(H2O) | 0.58 | 0.57 | 0.58 |
| τ Tb(D2O) | 1.74 | 1.69 | 1.75 |
| τ Eu(H2O) | 2.44 | 2.27 | 2.44 |
| τ Tb(D2O) | 3.45 | 3.03 | 3.13 |
| ϕ Eu(D2O) | 3 × 10−3 | 0.01 | 0.12 |
| ϕ Tb(D2O) | 0.48 | 0.41 | 0.81 |
The likelihood of electron transfer can be predicted, using the approach first put forward by Weller34 for photo-induced electron transfer processes, eqn (1).
| ΔGET = nF([Eox − Ered] − ES − e2/εr) J mol−1 | (1) |
In the original description, Eox is the oxidation potential of a donor (e.g. a nitrogen lone pair or the HOMO of an electron rich aromatic group), Ered is the reduction potential of the acceptor (e.g. an aryl group or a Ln3+ ion), ES is the singlet excited state energy in eV and e2/εr is an attractive energy term related to formation of a radical ion pair; this term is often less than +0.2 eV. The Weller equation therefore estimates the free energy of activation for the electron transfer step involving a particular excited state that drives the overall process. Application of the equation highlights the facility of photoinduced electron transfer to the Eu3+ centre.
Consider the case in which a simple phenyl group is the hypothetical sensitiser, eqn (2), where ES is 4.46 eV, Eox is +1.76 V and Ered is −1.1 V for a Eu(III) complex.
| ΔGET = F[(2.48 + 1.1) − 4.90 − 0.15] = −1.47F = −142 kJ mol−1 | (2) |
This quenching process is energetically very favourable but it does not occur with the para-carboxymethyl analogue. In this case, the electron-withdrawing CO2Me group raises the oxidation potential of the aryl ring beyond +3.3 V (cf. +2.48 V and +1.76 V for Ph–H and Ph–OMe). Accordingly, the overall quantum yield for sensitised emission in the p-CO2Me substituted Eu complex is much higher, (12%, Table 1).
Intermolecular electron transfer from a halide anion to the singlet excited state of the chromophore occurs may occur readily (I− > Br− > Cl−), especially for cationic complexes that favour ion-pair encounter; the reduction of ligand fluorescence is echoed by the diminution of lanthanide luminescence intensity. The lanthanide emission lifetime is unaffected in each case. For example, with the N-methyl-phenanthridinium complexes, [EuL2]+, Stern–Volmer quenching constants, Ksv (eqn (3)), have been examined for iodide, bromide and chloride quenching, monitoring the modulation of Eu emission intensity. The KSV−1 values for I−, Br− and Cl− quenching were 50, 16, and 5 mM−1 respectively, consistent with the ease of oxidation of the halide anion.14
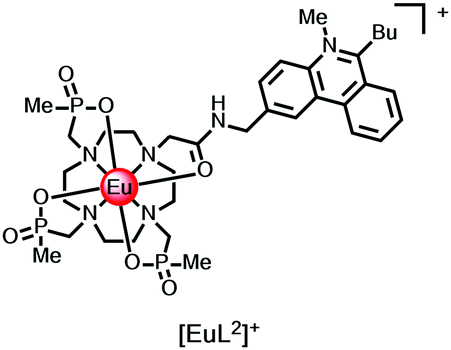
| I0/I = 1 + Ksv[X−] | (3) |
The ligand triplet excited state is always at a lower energy than the singlet and typically has a longer lifetime, lengthening the time window for competitive quenching to occur. For example, aromatic amines and other electron rich aromatics that can form resonance stabilised radical cations,35 quench the T1 state by fast electron transfer, even when kISC is fast with respect to keT for the S1 state from which it derived. Evidently, such aromatic and heterocyclic ring systems are not going to be effective intramolecular sensitisers, because fast electron transfer quenches either the S1 or the T1 excited state. In examples of intermolecular quenching for example, quenching rate constants are of the order of 109–1010 M−1 s−1.
With many electron poor sensitising groups (Scheme 2), exciplex formation may occur between an electron rich species and the ligand triplet excited state. Exciplex formation can be considered to involve partial charge transfer, and the ligand excited state is lowered in energy and broadened as a result. The best examples reported have involved intermolecular quenching by the natural anti-oxidants, urate and ascorbate, and by certain catecholates that each serve to lower the energy of the ligand triplet excited state.36 In doing so, notably for terbium complexes, more efficient, thermally activated back energy transfer, kBET, becomes possible from the higher lying terbium 5D4 state to the transient exciplex, leading to dynamic quenching of the metal excited state and a reduction in its emission lifetime.
Quenching by urate, ascorbate and selected catechols, such as dopamine (ascorbate, Eox = +0.30 V; urate, +0.59 V; catecholates, ca. +0.53 V at pH 7), is more effective for Tb(III) over Eu(III) complexes. Excited state complex formation occurs between the electron-rich reductant and the electron-poor heterocyclic sensitising moiety, as in the Ln complexes [LnL3–5] with tetraazatriphenylene, azaxanthone and pyrazoyl-azaxanthone groups, (Fig. 1).36,37
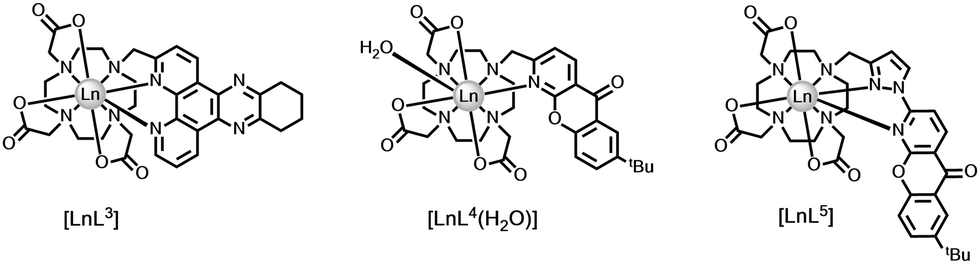 | ||
| Fig. 1 Lanthanide complexes sensitive to dynamic quenching by urate or ascorbate involving exciplex formation; I− quenches by collisional encounter with the ligand triplet.36,37 | ||
The efficiency of quenching did not faithfully follow trends based on consideration of ligand reduction and quencher oxidation potentials, (Table 2), but was sensitive to the steric shielding created by the local ligand environment, notably when the carboxylate donors were replaced by more bulky phosphinate or amide carbonyl donors. Moreover, in the presence of serum albumin, when a non-covalent complex forms with the ligand sensitising moiety, urate quenching was much less efficient and could be completely suppressed with the complexes based on pyrazoyl-azaxanthones, which have the highest association constants with serum albumin.20
| Quencher | Tb/Eu–L3 | Tb/Eu–L4 | Tb/Eu–L5 |
|---|---|---|---|
| Urate | 0.005/0.11 | 0.012/0.28 | 0.03/0.45 |
| Ascorbate | 0.35/2.92 | 0.57/8.90 | 1.39/8.43 |
| Iodide | 2.1/103 | 53/125 | 5.4/72 |
With urate and ascorbate quenching of the lanthanide excited state was reduced as temperature increases, as long-lived exciplex formation is an entropically disfavoured bimolecular process, with a negative entropy of activation. Such a temperature dependence may be contrasted with the less efficient dynamic quenching by iodide (Eox = +0.53 V), where collisional encounter with the ligand triplet is favoured by Coulombic attraction, and the quenching efficiency increased with temperature (vide infra for direct keT to Ln*).
The metal based excited states may be populated directly by laser excitation, for Eu at 397 (7F0–5L6), 457 nm (7F0–5D2) and for Tb at 488 nm (7F6–5D4). Under this mode of excitation, no change was found in the lifetime of the lanthanide excited state nor the metal-based emission intensity, following addition of 0.1 mM urate, 4 mM ascorbate or 100 mM iodide to solutions containing the Eu(III) and Tb(III) complexes of L3–L5. Such observations establish unequivocally that intermolecular quenching of the lanthanide excited state is occurring by a process that involves the excited state of the sensitising moiety.37 Direct quenching of the lanthanide excited state by intermolecular electron transfer with these electron rich species does not occur.
| Lanthanide | [LnL6] k(H2O)/ms−1 | [LnL6] k(D2O)/ms−1 | [LnL3] k(H2O)/ms−1 | [LnL3] k(D2O)/ms−1 |
|---|---|---|---|---|
| Tb | 2.13 | 1.45 | 0.64 | 0.56 |
| Eu | 1.45 | 0.99 | 0.93 | 0.60 |
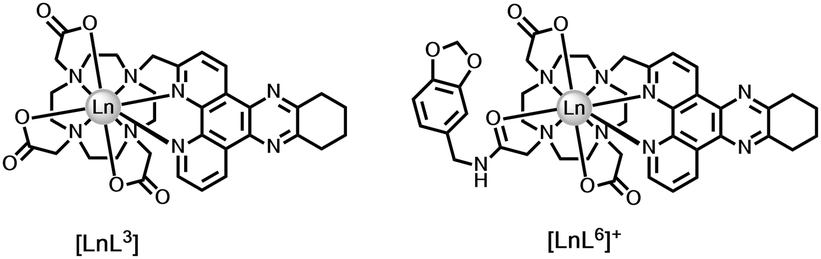
We can extend this thinking to systems with an integral electron-rich moiety. Comparing emission lifetimes for the pair of dpqC complexes, [LnL3] and [LnL6], the Tb and Eu lifetimes are lower for the complex with the trans-catecholamide group (Table 3). Here, intramolecular charge transfer may occur shortening the metal excited state lifetime in water and in D2O, thereby reducing the overall emission quantum yield. The observation by mass spectroscopy of H/D exchange in the dpqC chromophore (Ered = −1.1 V) of [TbL6], but not with [TbL3] following excitation, and the greater quenching of the Tb excited state compared to the Eu analogue, support the hypothesis of intramolecular exciplex formation, with transient radical anion character in the dpqC moiety37.
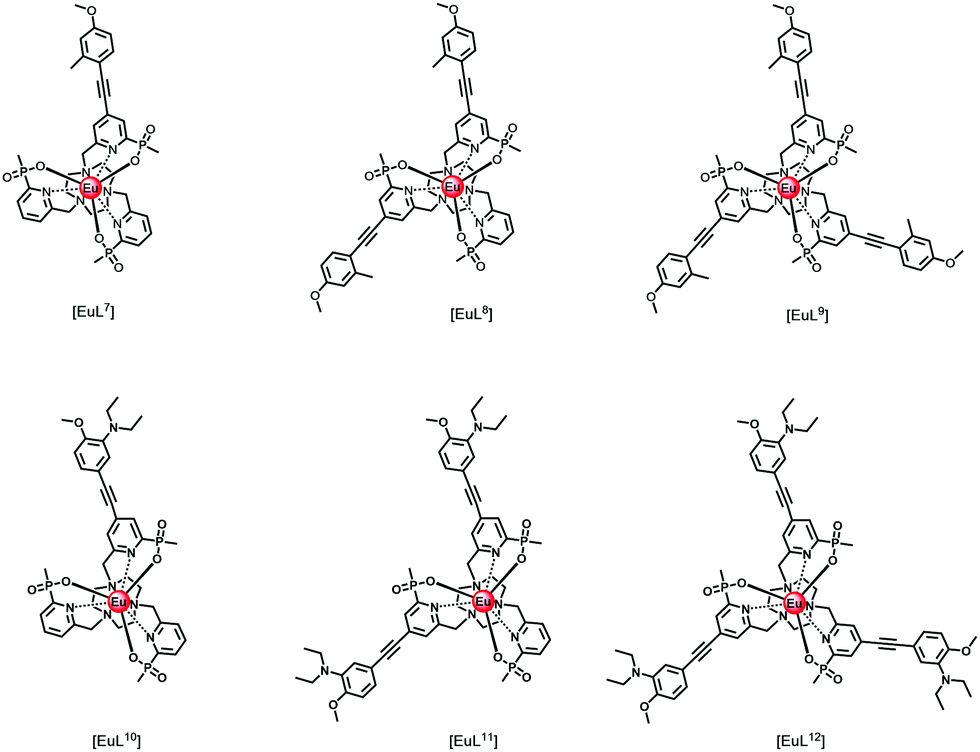
Thinking more generally, quenching of the lanthanide excited state itself may in principle occur by intramolecular electron transfer from a proximate electron rich aryl group or a N lone pair, reducing the excited state lifetime. Consider the following series of complexes, [EuL7–12], (Fig. 2).23,24,38 The set of three Eu complexes, [EuL7–9], containing increasing numbers of electron rich chromophores and can be compared with the set of complexes, [EuL10–12] where the chromophore also possesses a conjugated nitrogen lone pair in the upper aryl ring,38 (Table 4). With [EuL7–9], there is a small decrease in the lifetime of emission as the number of chromophores increases, suggesting that electron transfer from the electron rich sensitising moiety quenches the Eu excited state, to some extent.
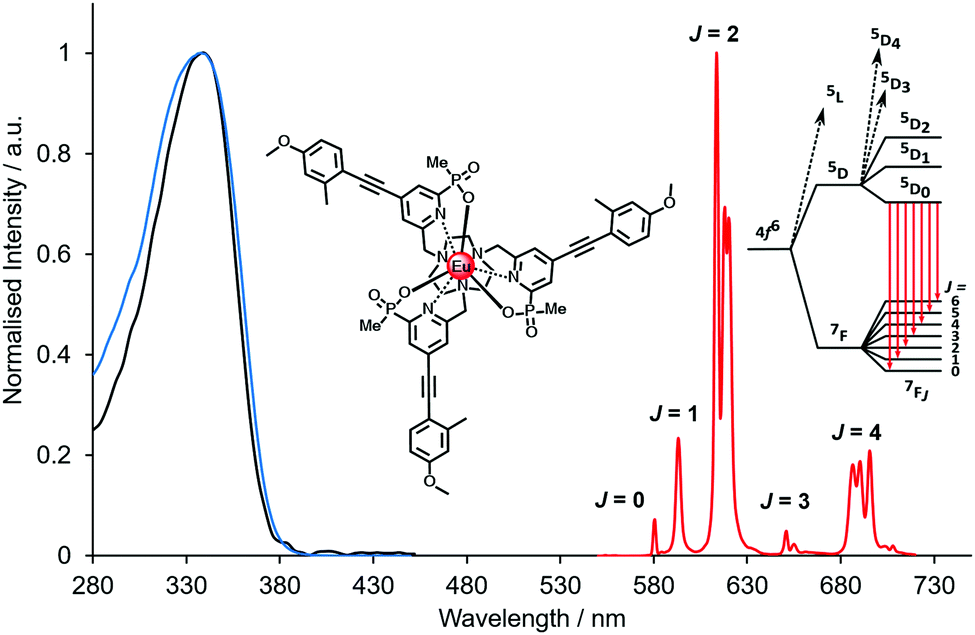 | ||
| Fig. 2 Absorption (blue), excitation and emission spectra of [EuL9], showing the five major transitions in the visible region from the 5D0 Eu excited state (295 K, 0.1 M NaCl). Adapted with permission from the Royal Society of Chemistry.24 | ||
| Complex | τ Eu/msa | Comment |
|---|---|---|
| a 295 K, 0.1 M NaCl, H2O. b pH 8, 0.1 M NaCl, H2O. c pH 4, 0.1 M NaCl, H2O; pKa values are 6.75, 6.30 and 6.21, respectively. | ||
| [EuL7] | 1.39 | Small lifetime decrease with increasing number of electron rich chromophores |
| [EuL8] | 1.27 | |
| [EuL9] | 1.21 | |
| [EuL10] | 0.53b | PeT quenching effect increases with number of N-substituted chromophores |
| [EuL11] | 0.34b | |
| [EuL12] | 0.25b | |
| [EuHL10]+ | 1.16c | N-Protonation suppresses electron transfer quenching |
| [EuHL11]+ | 1.00c | |
| [EuHL12]+ | 0.84c | |
The Weller equation considers the free energy of activation for the electron transfer process and can be applied to assess the feasibility of the quenching of the europium excited state by intramolecular electron transfer from the lowest lying orbital, i.e. the nitrogen lone pair orbitals present on the antenna(e) in [EuL10–12]. The oxidation potential of a typical N,N-dialkylaniline, such as N,N-dimethylaniline, is +0.53 V in acetonitrile compared to +1.2 V for a typical trialkylamine, e.g. triethylamine. The reduction potential of the Eu3+ ion lies in the range −0.8 to −1.2 V, when coordinated by a polar polydentate ligand (estimated as −1.1 V here). The energy of the excited Eu3+ 5D0 state lies at 17220 cm−1 (equivalent to +2.13 eV or 206 kJ mol−1), compared to the Tb3+ 5D4 excited state at 20450 cm−1 (2.54 eV, 245 kJ mol−1) and 10200 cm−1 for 2F5/2 Yb3+ (+1.26 eV, 122 kJ mol−1).
| ΔGET = F[(0.53 + 1.1) − 2.13 − 0.15] = −0.65 F = −63 kJ mol−1 | (4) |
Given these values, the free energy for the electron transfer process from the π-conjugated donor of an N,N-dialkylaniline to the Eu3+ ion is energetically favourable, eqn (4). This analysis assumes that the excited state reduction potential of the europium ion is the same as in the ground state. Moreover, such an electron transfer process to the lanthanide excited state is not favourable for analogues where a methylene group spaces the nitrogen lone pair from the π system.
The Eu excited state is prone to quenching by an intramolecular electron transfer process associated with the amine N lone pair of the diethylamino group. Consider the behaviour of the Eu(III) complexes of ligands L10–L12. The rate of proton transfer to and from nitrogen (typically 1010 to 1011 s−1) occurs much faster than the rate of decay of the excited europium ion (103 s−1) and of the intermediate ligand ICT or triplet excited states. Furthermore, the rate of electron transfer from the unprotonated chromophore to the excited Eu ion is much faster than the rate of energy transfer populating 5D0 Eu* and of its own radiative rate of emission. Thus, during the long lifetime of the Eu 5D0 excited state, deprotonation of the amine group can occur, allowing fast electron transfer to occur from the unprotonated chromophore to the Eu ion, quenching the Eu emission and shortening the ‘time-averaged’ observed lifetime. Such an effect is more likely to occur with increasing numbers of chromophores bearing the amine group, consistent with the observation that the trisubstituted complex, [EuL10], has the shortest observed emission lifetime (4), and the complex with one amine-containing chromophore, [EuL12], has the longest, both at pH 4 and 8. Moreover, in a key control experiment the same pH dependence of emission lifetime was observed for [EuL9–11] following direct excitation of the Eu ion at 397 nm. In this case, it is the Eu excited state that is quenched and not an intermediate ligand excited state.
2.2. Electronic energy transfer quenching, kET
Electronic energy transfer, in principle, may occur at any point along the photochemical pathway, perturbing the ligand-based singlet, ICT and triplet excited states or the lanthanide excited state, (Scheme 1). The process may be inter- or intramolecular in nature, and in each case the relevant excited state lifetime is reduced.39There are two mechanisms that have been suggested to describe energy transfer from a donor to an acceptor. In Dexter transport, the exciton is believed to hop between components in a short-range process that depends on the orbital overlap between the donor and the acceptor. In this case, the symmetry of each component is preserved and triplet to singlet energy transfer is not formally allowed. However, a change in spin symmetry may be possible by incoherent electron exchange when the donor exciton breaks up and reforms on the acceptor. Förster put forward an alternative rationale in which molecular transition dipoles are coupled and can exchange energy. In this case, the energy transfer efficiency, ηET can be described by eqn (5), in which kr and knr are the rates of radiative and non-radiative decay of the donor, r is the donor–acceptor separation, kET is the rate of energy transfer and R0 is the distance for a 50% efficient transfer. In Förster theory, both kET and kr are directly proportional to the oscillator strength of the transition.
| ηET = kET/(kET + kr + knr) = 1/(1 + (r/R0)6) | (5) |
The energy transfer step that leads to population of the emissive lanthanide excited state is of great importance in determining the efficiency of sensitised luminescence, revealed by the value of the overall emission quantum yield. The value of kET is of the order of 108 s−1 for many systems where the chromophore is directly bound to the lanthanide ion, e.g. with isophthalalamide16 and arylalkynylpyridine21–24 sensitisers, where the Dexter exchange mechanism probably predominates, certainly for Eu and Yb complexes. The decay of the intermediate ligand triplet or ICT excited state can be observed to match the grow-in of lanthanide emission, using transient triplet–triplet and X-ray absorption spectroscopy.1,28,40–42 Efficiencies of energy transfer in these cases are believed to be of the order of 90 to 99%.
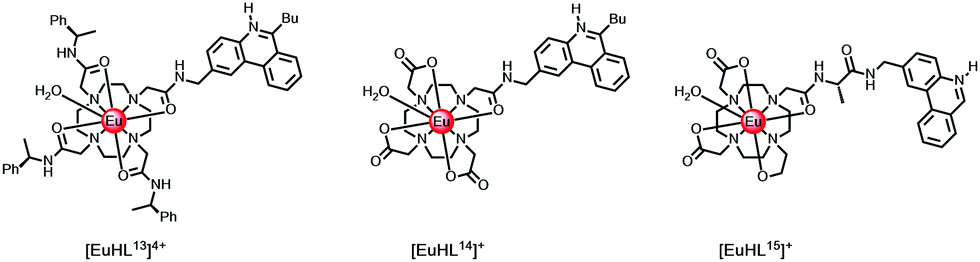
In cases where the sensitiser is located close to the putative R0 value (typically 5.5 Å) for 50% efficient Förster energy transfer, e.g. in [LnHL13]4+ and [EuHL14]+ where the protonated phenanthridinium chromophore is 5.2 Å distant, (Scheme 2), the rates of energy transfer from the ligand triplet to Eu 5D1,0 or Tb 5D4 are around 105 s−1, while kET is 107 s−1 to the Nd 4F3/2 state. Slower rates of energy transfer occur when the chromophore is spaced even further away. Indeed, in a valine spaced phenanthridinium complex, [EuHL15]+, the sensitising heterocyclic moiety is about 8.2 Å from the Eu ion, and the overall Eu luminescence quantum yield is a factor of 20 lower in D2O.43 Similar conclusions concerning the distance dependence of energy transfer efficiency have been made for a series of coumarin sensitised Eu complexes, notwithstanding an energy transfer process that involves the coumarin S1 state.29,44
As noted earlier, when the accepting Ln* is within 10kBT of the triplet energy level, kBET becomes competitive with kLn*, and a photo-equilibrium is set up such that the ligand triplet is much longer lived. Under these circumstances, kq[O2] is comparable to kET and the Ln* lifetime is rendered sensitive to changes in the concentration of dissolved oxygen in solution, and to temperature variations. Oxygen solubilities in different solvents vary by more than two orders of magnitude, so particular care must be taken experimentally when such sensitivity arises. For example, the solubilities of oxygen, expressed as mole fractions at 298 K and 101.3 kPa, are: MeOH (4.2 × 10−4); EtOH (5.7 × 10−4); iPrOH (6.8 × 10−4); 2-BuOH (8.3 × 10−4); H2O (2.3 × 10−5); glycerol (4.8 to 5.5 × 10−6). In contrast, solvent viscosity values (298 K, cP) are independent of the degree of sample aeration: MeOH (0.54); H2O (0.89); EtOH (1.02); iPrOH (2.04); tBuOH (4.02); glycerol (9.3 × 102).45,46 In a small number of cases, where kISC must be relatively slow, evidence has been presented for fast direct energy transfer from the ligand S1 state to the lanthanide 5D1 and 5D0 states.29–31,47,48
The most common type of triplet–triplet energy transfer is associated with collisional quenching of the sensitiser T1 state by triplet oxygen. Other cases may arise when there are two chromophores in proximity in a lanthanide complex. For example, an indirect intramolecular triplet–triplet energy transfer process occurs when the triplet energies of two chromophores are judiciously selected. In [LnL16(H2O)]+ for example, selective excitation of the azaxanthone chromophore (λexc 337 or 355 nm) leads to efficient population of the lanthanide excited state. Intramolecular energy transfer from the azaxanthone triplet to the proximate naphthyl group (ET 20850 cm−1) may then compete with transfer to the Tb 5D4 state (20450 cm−1). The long-lived naphthyl excited state also can sensitise Tb, but now back energy transfer competes, a photo-equilibrium is set up and the naphthyl triplet becomes long lived and is susceptible to quenching by oxygen. Such behaviour occurs in the Tb complex, but not with the Eu analogue.49 In support of this premise, rather than the Tb ion acting as an mediator in an energy relay process, i.e. T1(azaxanthone) to 5D4 Tb to T1(naphthyl), the low-temperature phosphorescence spectrum of [GdL16]+ (77 K) showed emission bands attributed to the azaxanthone triplet (from 400 to 520 nm) alongside a fingerprint emission band from the triplet naphthyl group (475–580 nm), consistent with an intramolecular energy transfer process.
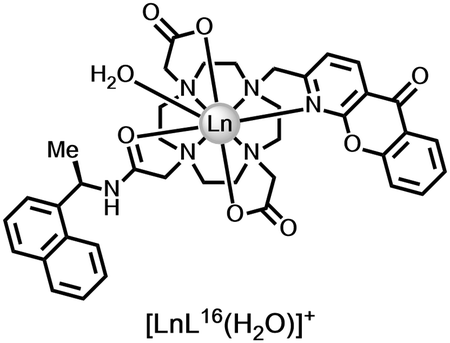
Thus, the ratio of the Tb/Eu lifetimes or the red/green intensity ratio (e.g. 620/545 nm) becomes sensitive to dissolved oxygen variations. By using mixtures of the two complexes in appropriate ratios, bearing in mind their different quantum yields, the red/green intensity ratio can be calibrated to measure [O2] in solution. Similar strategies to assess [O2] have been developed for an innovative pair of heterodinuclear complexes,50 containing both Tb and Eu in different proximity to a ligand naphthyl moiety that also served as the sensitiser, notwithstanding its poor intrinsic ability to sensitise Eu, owing to competitive quenching of the naphthyl S1 state by Eu(III).
2.3. Energy transfer quenching, kET, involving the metal excited state
Electronic energy transfer from the long-lived lanthanide excited state to an energy matched acceptor can be intermolecular or intramolecular in nature. In the more common former case, the long time window for multiple diffusional encounters can give rise to efficient Förster resonance energy transfer (FRET) from Tb* or Eu*, with R0 values around 6 to 8 nm. Energy transfer from small Eu or Tb donor complexes to energy matched acceptors in solution is considerably enhanced by the fact that during its millisecond lifetime, the excited Tb/Eu complex diffuses extensively over a range of about 2 μm. The excited complex encounters a large number of energy acceptors before it decays, e.g. at 1 μM, the average separation between neighbouring acceptors is around 65 nm.39 The overall effect is to average the interactions between the donor and acceptor(s) over all allowed directions and orientations, thereby reflecting the equilibrium behaviour of the system.The accepting near-IR dye adopts the lifetime of the parent donor, and so time-gated observation of its long lived luminescence is possible, allowing ratiometric analysis of its emission intensity relative to the shorter wavelength lanthanide donor.51,52 The energy-matched acceptor must be chosen judiciously, and can be tagged to proteins or antibodies. In examples used commercially, strongly absorbing cyanine dyes are often selected based both on the goodness of their spectral overlap with the donor Eu emission and the wavelength of delayed emission. The acceptor emission band should minimally overlap with the Eu emission profile to permit separate ratiometric analysis of the major 620 and 670 nm emission bands, using optical filters, (Fig. 3).
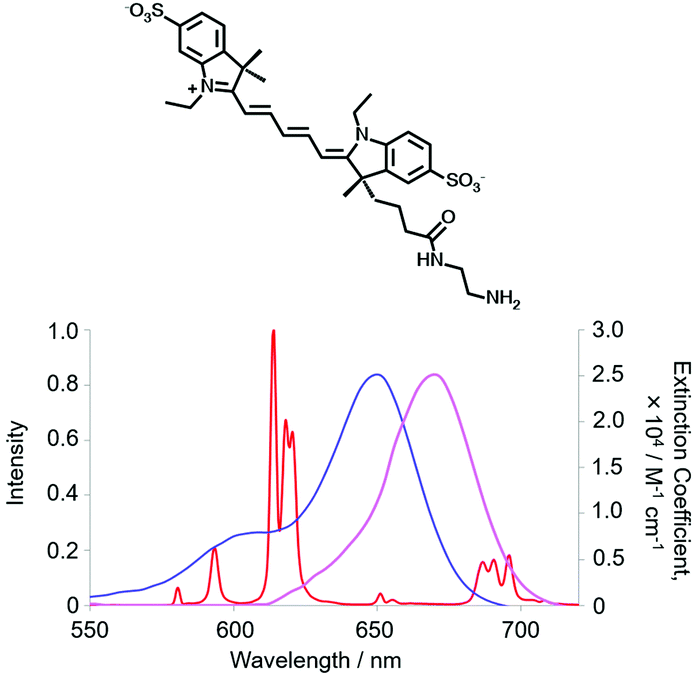 | ||
| Fig. 3 Cyanine dye absorbance (blue), overlapping the Eu spectral output, and dye emission (purple), minimising overlap with [EuL9] emission (red), (MeOH, 295 K). | ||
Typically this FRET process is used to establish the spatial proximity of the donor and acceptor, often tagging the drug receptor site with the lanthanide complex, and the agonist for the receptor with the IR-dye. Such a strategy enables the rapid screening of potential receptor-binding drug candidates (antagonists) in the nanomolar range, using high throughput competition assays in vitro or in cellulo.51–53
In live cell microscopy, FRET has been demonstrated between a cell-impermeable Eu donor and a fluorescent acceptor dye that localised with reasonable but not perfect selectivity in the outer cell membrane. Examination of the long-lived emission from the acceptor dye allowed a much better visualisation of the cell membrane, (Fig. 4), examining the time-gated FRET channel. Energy transfer between the extracellular complex, [EuL17]3− and the dye can only occur over a short range at the membrane interface. The clarity of the time-resolved FRET image defining the cell membrane was far better than that obtained in the absence of the Eu complex, examining cell images observing the short-lived dye fluorescence.54
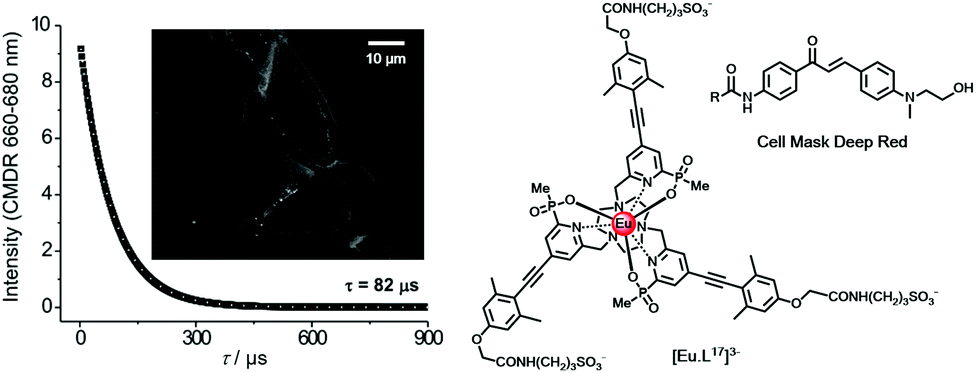 | ||
| Fig. 4 Lifetime decay and time-gated (tg 10 μs; (λexc 365 nm) FRET microscopy image of an NIH-3T3 cell membrane (scale bar 10 μm), observing acceptor dye emission (CMDR) at 660–680 nm following energy transfer from the Eu complex located near the cell membrane. Adapted with permission from ref. 54. Copyright (2016) Wiley-VCH. | ||
Energy transfer may also occur over a long range between ions. The lanthanide ion can act as an acceptor, and in other cases it may be the donor. For example, Tb* to Eu energy transfer occurs in the solid state55,56 and in solution, and can be controlled in well-designed series of heterodinuclear complexes.57 In a dinuclear system with the Tb and Eu ions believed to be separated by 10.6 Å, it has been found that energy transfer from Tb 5D4 to Eu 7F1 was most efficient and was thermally activated. Earlier work had shown that if the transitions are not exactly at resonance, for a Förster transfer mechanism the energy difference must be conserved by phonons. Since phonon relaxation is weak at lower temperatures, the energy transfer efficiency decreases considerably at low temperature. The calculated average R0 distance between the Tb donor and the Eu acceptor held in a sol gel glass was 37 Å, within the usual Förster range.
Numerous examples of energy transfer from a variety of transition metal complexes to rare earth coordination complexes have been reported, including many cases where the near-IR emitting ions Yb, Nd and Er, with their lower lying emissive states, are the acceptors.58–61 Examples of Dexter energy transfer from the Ln* to d-block aqua ions and complexes are also known, but have less often been studied in depth with well-defined complexes. Thus, the 5D0 Eu* is dynamically quenched by the Cu2+ aqua ion in solution (λmax 810 nm; ε 11 M−1 cm−1), whereas 5D4 Tb* is quenched more effectively by the high spin d5 Mn2+ aqua ion (λmax 525 nm; ε 0.018 M−1cm−1), notwithstanding the feeble spectral overlap integral with the broad aqua ion visible absorption bands, associated with the weak oscillator strength of d–d transitions.62,63
2.4. Vibrational energy transfer quenching, kvibQ
Excited state quenching involving vibrational energy transfer is a major cause of non-radiative deactivation for both ligand and lanthanide excited states. It is of particular importance in reducing the lanthanide excited state lifetime, where ligand and solvent XH oscillators, NH > OH ≫ CH, normally cause the largest effect. Lifetimes of Ln* can be compared to deuterated analogues to reveal their relative importance, as νXD values are about 1/1.41 times less than νXH, assuming a harmonic oscillator model, leading to a less efficient Franck–Condon wave function overlap. Such comparisons have allowed good estimations of complex hydration state to be made, comparing rates of decay in water and D2O, for many Eu, Tb, Nd and Yb coordination complexes.64–69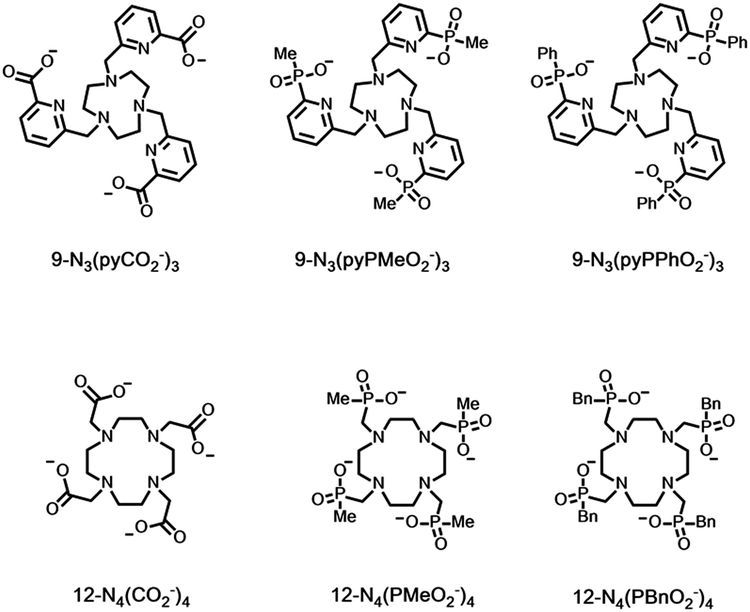
Vibrational energy transfer predominantly occurs by a dipole–dipole mechanism, with an r−6 distance dependence, so that coordinated water molecules with two OH oscillators about 3.1 Å from the Ln* quench very effectively. Effective wave function overlap occurs between 5D0 Eu* and the 3rd or 4th harmonic of νOH, whereas for 5D4 Tb* it is with the fifth vibrational overtone, leading to less efficient Franck–Condon overlap and reduced sensitivity to multi-phonon relaxation by a factor of about 4.
In complexes of the central lanthanides, Ln–O and Ln–N bond lengths are about 2.5 (±0.15) Å, and the distance between a bound OH or NH oscillator (with maximum motion for the lighter H atom) and the Ln ion is around 3 Å. If this value represents 100% efficient vibrational energy transfer, it is possible to calculate the effect of increasing the distance on the energy transfer rate. For OH oscillators that are 3.5–3.6 A away from the Ln centre, the rate corresponds to 25% of the bound water case. Distances of this order correspond to those associated with the ‘second sphere of hydration’, and changes to such second spheres of hydration are mainly determined by local ligand structure, i.e. its hydrophobicity, and the absence or presence of hydrogen bonds involving or close to the ligand donor atoms.69
These effects are apparent on inspection of rates of decay of lanthanide excited state lifetimes and are most often examined for Eu and Tb complexes (Table 5). Quenching rates are smallest in D2O and for Tb compared to Eu, in this set. Values in water are highest for the aqua ion in water and are smallest when no water is coordinated to the Ln*, particularly when the ligand sterically inhibits close diffusional approach of the solvent, e.g. with the relatively hydrophobic tetrabenzylphosphinate Tb complex, where the Tb* lifetime is over 4 ms in water.
| Ligand | k Eu H2O | k Tb H2O | k Eu D2O | k Tb D2O | Ref. |
|---|---|---|---|---|---|
| a Data for perchlorate salts; there is an anion effect for aqua ion lifetimes, as the anion determines the nature and extent of the second hydration sphere. b Ligand is often called DOTA; replacing on average 15 of the 16 ring H atoms by D gave values as follows: kEu H2O = 1.49; kEu D2O = 0.30; kTb H2O = 0.60; kTb D2O = 0.40, consistent with greater quenching of the Eu excited state by C–H oscillators but for Tb the effect is slightly greater for C–D. c The nearest water in the solid state lattice is 5.6 Å from the Ln ion and is 4.25 Å distant in solution, as estimated by NMRD analyses of Gd analogues.68 | |||||
| 9N3(pyCO2−)3 | 0.99 | 0.56 | 0.83 | 0.52 | 70 |
| 9N3(pyPMeO2−)3 | 0.64 | 0.39 | 0.62 | 0.34 | 71 |
| 9N3(pyPPhO2−)3 | 0.74 | 0.47 | 0.65 | 0.38 | 71 |
| (H2O)9a | 8.90 | 2.50 | 0.35 | 0.26 | 72 |
| EDTA(H2O)3 | 2.90 | 0.93 | 0.48 | 0.29 | 73 |
| 12N4(CO2−)4(H2O)b | 1.60 | 0.66 | 0.53 | 0.37 | 68 |
| 12N4(PMeO2−)4 | 0.80 | 0.34 | 0.54 | 0.27 | 68 |
| 12N4(PBnO2−)4c | 0.63 | 0.24 | 0.48 | 0.22 | 68 |
There is a significant of quenching of the Eu 5D0 state by CH oscillators as well, revealed by selective ligand deuteration studies. This quenching effect is even more important for Yb, Nd and Er complexes with their much lower lying emissive excited states matching the overtones of νCH more efficiently than in Eu examples. However, C–D oscillators may have a greater quenching effect than C–H in certain cases, e.g. with Pr(III) complexes.74 Indeed, in general the spectral overlap integrals in multi-phonon relaxation of lanthanide excited states with C–(H/D) overtones need to be assessed carefully for each lanthanide/oscillator combination. Other ligand oscillators also contribute to Ln* deactivation: comparing the values for carboxylate and phosphinate complexes (Table 5) in D2O suggests that higher harmonics of νCO (at 1610 cm−1) quench more effectively than νPO, and a pyridine ring vibration (also around 1600 cm−1) also quenches significantly (top 3 entries Table 5). These effects are most apparent for the Tb complexes, because deactivation of the metal excited state by overlap with low-lying LMCT states, i.e. keT, is not operative.
The steric shielding of the lanthanide excited state from solvent diffusional encounter is therefore an effective means of maximising the emission lifetime. Ligand design should bear in mind this key factor; the nature and steric demand of the ligand donors needs to be chosen thoughtfully, (Fig. 5).
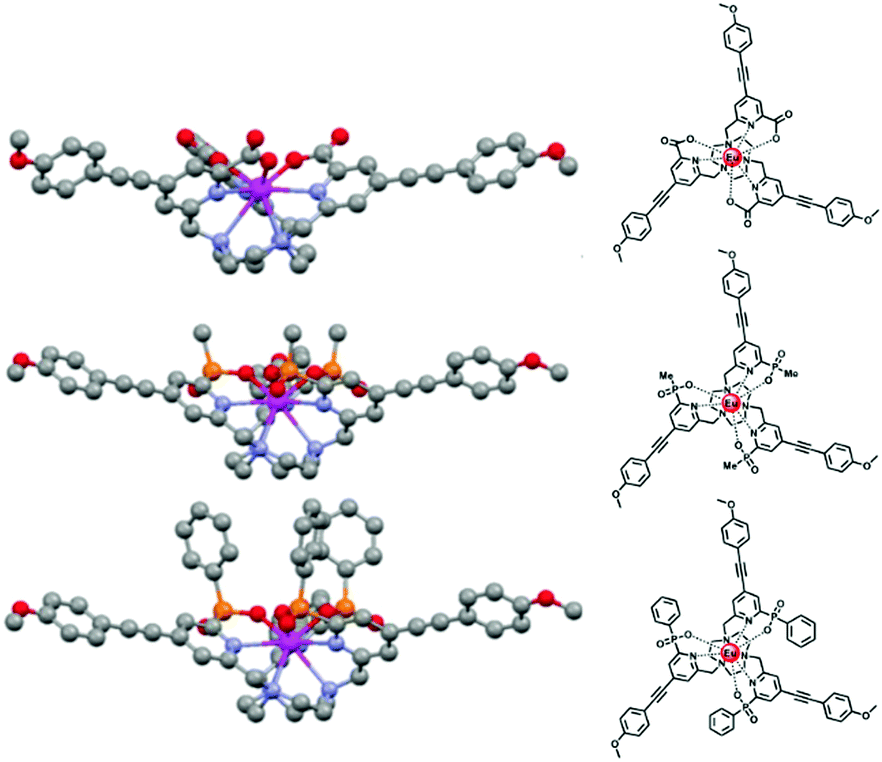 | ||
| Fig. 5 Increased steric shielding of the Ln centre from vibrational quenching as the ligand carboxylate is replaced by P–Me and P–Ph phosphinate groups, increases the europium emission lifetime; (upper to lower: τEu (MeOH) = 0.99, 1.18 and 1.30 ms). Adapted with permission from ref. 23. Copyright (2014) Wiley-VCH.23 | ||
Clearly, the sensitivity of the Ln* to deactivation by vibrational energy transfer offers great scope for creativity in designing responsive probes, based on modulation of the degree of vibrational quenching. For lanthanide complexes of heptadentate ligands, one or two solvent molecules are coordinated. The coordinated solvent can be displaced reversibly by anions, leading to large changes in the Ln* lifetime and in spectral form as the local ligand field changes (see below). Such reversible binding can involve either intermolecular or intramolecular anion ligation. A representative selection of ternary anion lanthanide adducts, (Scheme 3), reveals the opportunities to distinguish chelating oxyanions such as citrate, lactate and bicarbonate from monodentate anions, such as the wide range of phosphorus(V)–oxyanions.75–77
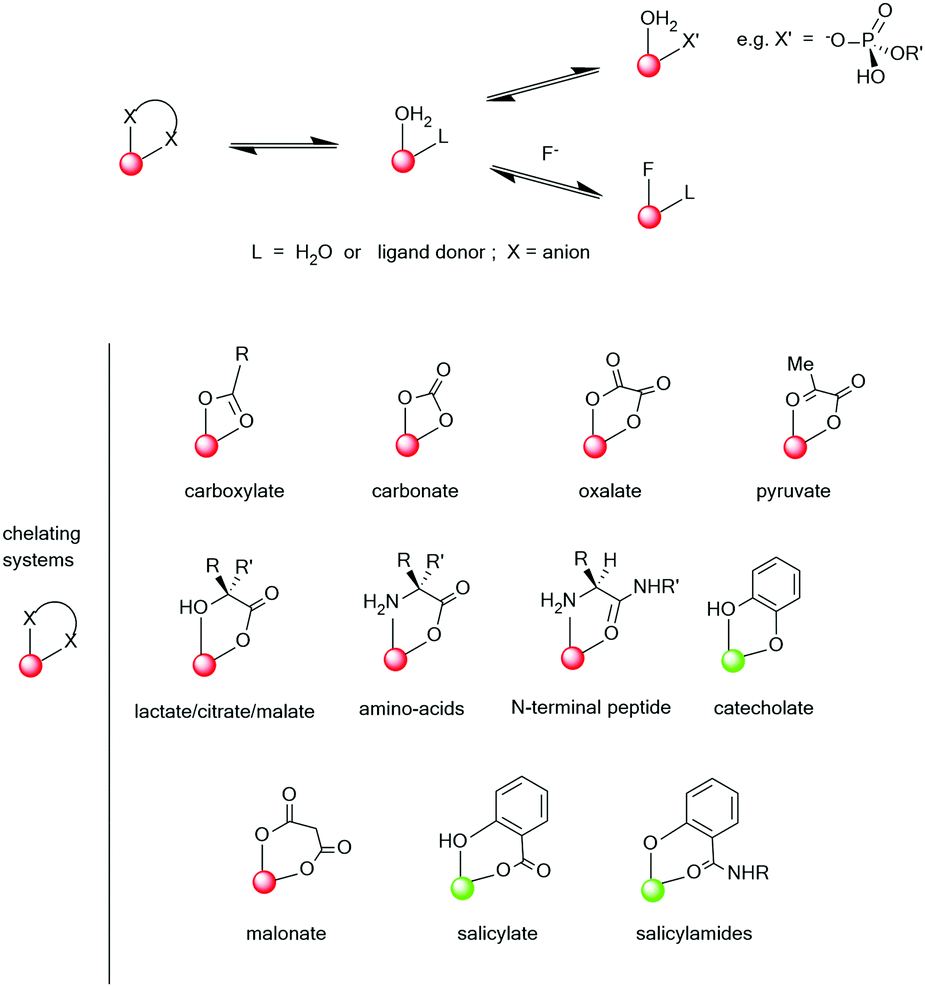 | ||
| Scheme 3 Typical ternary anion adducts formed by lanthanide aqua complexes (red indicates Eu and green Tb, as the preferred ion for stronger emission). Adapted with permission from the Royal Society of Chemistry.75 | ||
3. Classification of responsive systems by the signal observed
Four major types of responsive system can be distinguished, based on the nature of the signal being observed, offering a simple classification of responsive probes.3.1. Emission intensity variation with no change in spectral form
In this case, the lanthanide emission varies in overall intensity, but the spectral form does not change. Examples include the perturbation of the ligand S1 excited state by fast electron transfer, the altering of the S1 and T1 energy by reversible coordination to a structurally modified sensitiser, e.g. by protonation or metal ion binding without changing the lanthanide ligand field, and the quenching of Ln* by charge or electron transfer; each example is echoed by Ln* emission intensity diminution.Issues of calibration often limit the utility of these systems, as the observed emission remains a function of complex concentration. Sometimes excitation spectroscopy can be used to probe the reversible modification of the absorption spectral profile of the sensitising group, where it occurs. In other cases, keT, kf and kISC may be of the same magnitude, so that ligand fluorescence and lanthanide luminescence both occur and their relative intensities can be compared.14
3.2. Ratiometric analysis of emission intensity
In this set, the relative intensity of separate emission bands for one ion may change, or the ratio of intensities of two different lanthanide complexes, ideally of a common ligand, may vary. In the former case, Eu(III) complexes are pre-eminent.78 The absence of degeneracy of the 5D0 emissive state simplifies the spectrum compared to all other luminescent lanthanides. Moreover, the ΔJ = 2 and 4 bands are electric dipole allowed and hypersensitive to perturbation of the Eu ligand field, (Fig. 2), while the ΔJ = 1 transition is magnetic dipole allowed and less often varies in total intensity. Hence, the ΔJ = 2/ΔJ = 1 intensity ratio is often monitored and is independent of complex concentration. Thus, when the coordination environment and the local ligand field of the lanthanide ion are changed, these ratios can be monitored to report the perturbation faithfully. The old dogma that lanthanide complexes have a spectral signature that is determined by the nature of the ion is a very crude first approximation and is rather misleading. Complexes of Yb(III) also possess a near-IR emission fingerprint that varies greatly with ligand field despite the broad spectral form, associated with multiple transitions from the 2F5/2 excited state. A few reports are emerging of responsive near-IR probes that take advantage of this behaviour.79,80For two complexes of or within the same ligand that have a common sensitising group, the relative intensity of the major emission bands from the two Ln* can be measured simultaneously, and the ratio is independent of both probe concentration and excitation source fluctuation. The red/green intensity ratio for Eu and Tb complexes is often used to report changes in temperature, pH, pO2 and pX.50,81–86
3.3. Lanthanide emission lifetime variation
Each luminescent lanthanide complex possesses an emission lifetime that is characteristic of the local ligand structure and the associated ligand field, governed by the primary and the second sphere coordination environment. Measurements of emission lifetime changes, therefore, can be very useful in monitoring both reversible and irreversible transformations to the structure and the physical and chemical environment of probe complexes.A careful recent analysis concluded that ligand field splitting in lanthanide complexes is a complex function of a variety of factors.87 The primary determinants of the lanthanide ligand field are the constitution, configuration and the dynamic speciation of the coordination complex. With kinetically labile systems the ligand field will vary with time, depending upon the rate of ligand exchange. Such exchange dynamics need to be considered carefully, with respect to the timescale of the observed emission lifetime. Additional factors can also be very important: the molecular polarisability of the ligand and its donor atoms; the degree and type of any polyhedral geometric distortion, that may change local symmetry; the presence and extent of metal and ligand solvent dipolar interactions; the occurrence of directed hydrogen bonding effects and the nature and degree of supramolecular order in the system, e.g. complex aggregation and non-covalent association. Each of these factors may be critical in defining the ligand field experienced by the metal; their relative importance varies for a given lanthanide/ligand combination.
Clearly, by working with well defined lanthanide coordination complexes of known speciation and kinetic stability, there is great scope for probe design based on perturbation of the ground state ligand field, hand in hand with consideration of the relative importance of dynamic, often temperature dependent, processes that may perturb the lanthanide excited state and alter its lifetime, i.e. through variations in the relative magnitude of keT, kET and kvibQ (Scheme 1).
3.4. Circularly polarised luminescence: perturbation of chiral systems
Faraday appreciated in the 19th century that ‘polarised light is a most subtle and delicate investigator of molecular condition’. How right he was! A circularly polarised luminescence (CPL) spectrum contains much more information than the total emission spectrum, as it gives a sign and intensity for each transition, allowing spectral resolution of many overlapping multiplets, (Fig. 6). Uniquely, it can give information about the local chiral environment.88–101 Lanthanide complexes act as pure spherical emitters, avoiding the issues associated with anisotropy that may complicate other CPL active systems.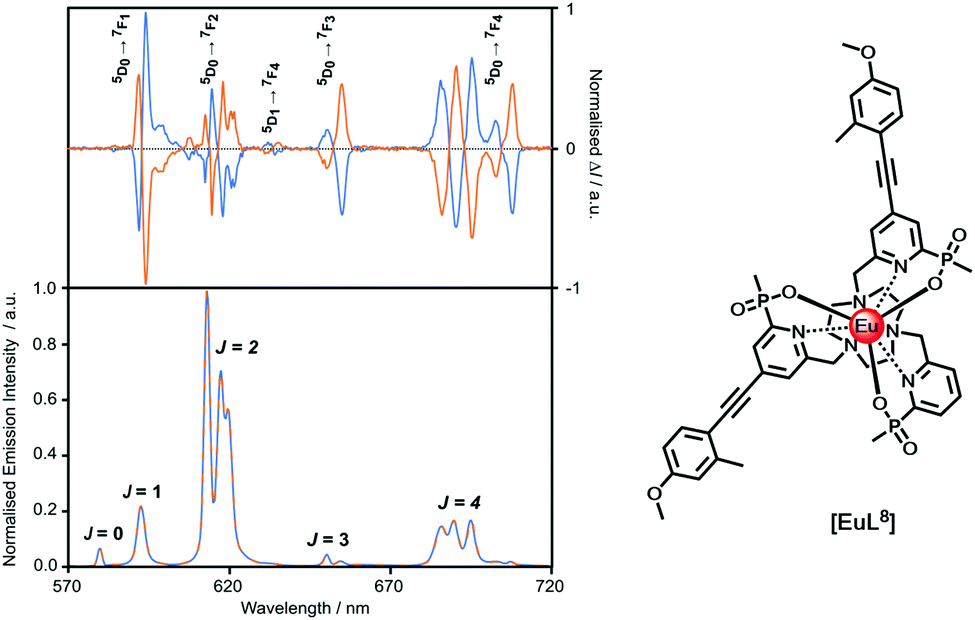 | ||
| Fig. 6 Comparison of the CPL and total emission spectra for the SSS-Δ (blue) and RRR-Λ enantiomers of [EuL8] (295 K, H2O), revealing the greater resolution afforded by CPL spectroscopy. | ||
By definition, a lanthanide coordination complex is chiral if it lacks an improper axis of rotation. The vast majority of the coordination complexes discussed herein are synthesised as racemic mixtures of the Δ and Λ (or P and M) enantiomers. In many cases, enantiomer inter-conversion rates are fast at ambient temperature, but for an increasing number of well-defined macrocyclic complexes the enantiomers have been resolved by chiral HPLC or can be formed stereoselectively.
There are two main classes of CPL chiral probes.88 The first is based on dynamically racemic complexes, where reversible binding with a chiral species changes the constitution of the complex by ligand displacement or non-covalent association (e.g. with an anion or protein). Binding ‘switches on’ an induced CPL signal that fingerprints the structure of the bound species. Examples have been described signalling the concentration of α-acid glycoprotein in serum for example, and of drugs that can compete with protein binding to the Eu complex.95–100 The second class uses enantiopure (or scalemic) probe complexes, where reversible binding with a chiral or an achiral species, modulates the CPL signal. In the limit, inversion of configuration (dynamic helicity inversion) may occur, and was observed when a cationic SSS–Δ–LndpqC triamide complex bound reversibly to drug site II in serum albumin.101
4. Modes of action of responsive luminescent probes
A large number of responsive luminescent lanthanide probes have emerged in the period between 1996 and 2021. A selection is discussed here, and these have been chosen for didactic and pragmatic reasons to reflect the work of the authors, in order to exemplify and rationalise their different modes of action. Necessarily it involves the selection of some more classical examples together with recent ones, so that the development of the different families can be traced and exemplified.They are arranged here in four categories, giving rise to an alternative classification. The first two involve reversible or irreversible changes to the constitution of the sensitiser in which the latter is better considered as a set of activity based probes, as they lack chemical reversibility. The second pair relate either to changes in the constitution or configuration of the metal centre or to modulation of excited state dynamic quenching, i.e. they involve the perturbation of the ligand ICT/T1 states or the lanthanide excited state itself. In each case, probe design must: tune the affinity of the probe to the levels of the analyte found in the sample or environment; assess the impact of potential interferents in the analysis; ensure accurate calibration of the real system; be aware of the response time of the fluctuation monitored, with respect to the time it takes for data acquisition.
4.1. Reversible change of ligand constitution: Iem
N-Alkylphenanthridinium sensitisers undergo reversible pseudo-base formation following attack by hydroxide, (Scheme 4). A unique example with [EuL18]+ offers a means of measuring pH in the range 11 to 13, and is signalled by the ‘switching off’ of Eu luminescence, with intensity dropping by two orders of magnitude.14 Parallel information can be gleaned, examining changes in sensitiser fluorescence intensity, as kf and kISC are competitive and excitation spectroscopy may be used to track the change.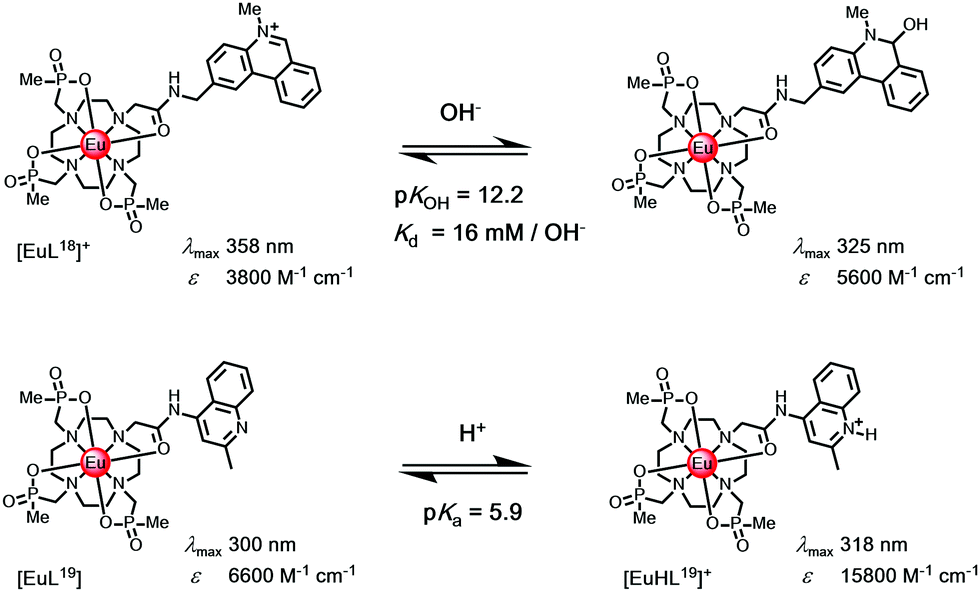 | ||
| Scheme 4 (upper) Reversible attack by hydroxide modifies the sensitiser constitution, switching off Eu emission; (lower) N-protonation reduces keT with a three-fold Eu intensity enhancement associated with suppression of photoinduced electron transfer.14 | ||
Much more commonly, protonation of the sensitiser modifies its S1 and T1 energies, and is associated with a large reduction in keT.102 These cases where either sensitiser protonation, e.g. with [EuL19], or diamagnetic metal ion binding (e.g. K+, Zn2+, Mg2+) occur are common and lead to suppression of photoinduced electron transfer. Typically, they lead to intensity enhancements of a factor of three or so, when exciting at the isosbestic wavelength. Thus, examples have been reported of the detection of millimolar potassium in aqueous samples of a Eu complex with a remote phenanthridine sensitiser,103 of millimolar magnesium levels in calf serum with a pyridyl arylalkynyl Eu complex104 and of zinc at the micromolar level with a terbium complex.105 In each case, the lanthanide emission lifetime is not changed.
4.2. Irreversible change of ligand constitution: Iem and τLn*
Most conventional sensors achieve selectivity for a target analyte using reversible and selective binding, based on design concepts arising from an understanding of the principles of ionic and molecular recognition. In contrast, activity based sensing (ABS) harnesses the inherent chemical reactivity of the analyte to enable specific and sensitive detection with spatial and temporal resolution. In this approach, the selectivity comes from the ability of the analyte to carry out a chemical transformation, resulting in an emission intensity change. This approach most commonly makes use of an irreversible reaction, and the luminescence signal either increases with time (‘switch-on’ systems), or diminishes with time; the former systems are favoured.106–108The majority of the reported ABS systems described involve irreversible transformation of the sensitiser constitution, leading to Iem modulation, and have used shorter-lived fluorophores. However, a series of lanthanide probes also operate in this manner, and in some cases the lanthanide emission lifetime may be modulated if the transformed functional group leads to changes in the extent of Ln* quenching by electron transfer. Occasionally, chemical transformation of the sensitiser leads to significant changes in the S1 and T1 energy levels. For example, if the absorption spectrum is modified, excitation spectroscopy can be used to track the extent of change, provided that the product remains reasonably luminescent. Alternatively, when the energy of the triplet is changed lanthanide emission might be switched off; or more usefully, when the T1 level is raised the energy transfer step leading to population of Ln* is more efficient, leading to intensity enhancement.
Examples have included: the switching on of Tb and Eu emission by the analyte-triggered formation of a sensitizing antenna from a non-sensitising caged precursor;109 the reaction of an aryl azide with H2S to give a p-amino aryl ketone with a strong ICT band;110 the cyclisation of a 1,2 diaminoaryl derivative with nitric oxide to give a 1,2,3-triazole,111 the selective oxidative cleavage of an O-benzylboronate ether with peroxynitrite112 to reveal a chelating 8-hydroxyquinoline, in this latter case switching off Eu emission, (Scheme 5).113,114
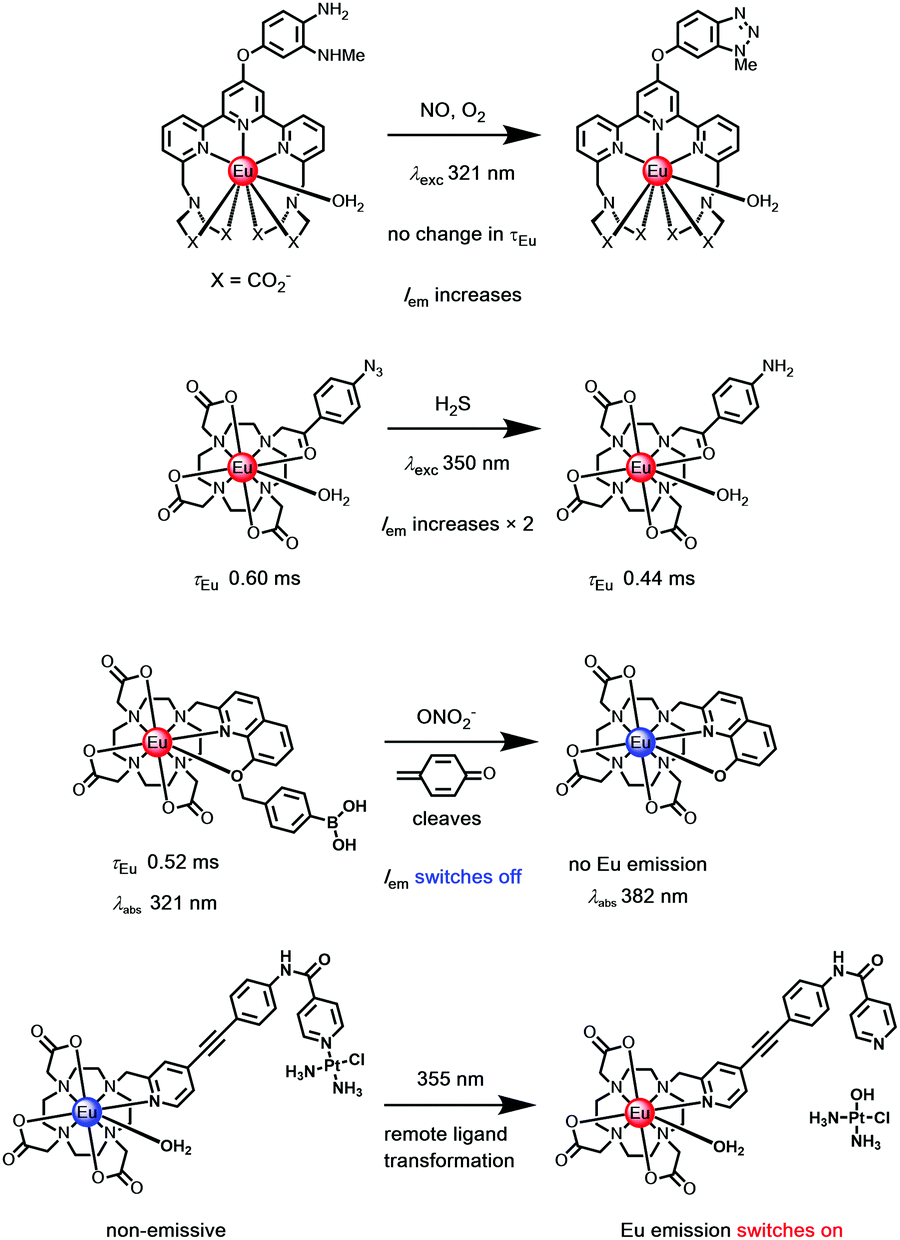 | ||
| Scheme 5 Examples of irreversible transformation with ‘activity based’ Eu(III) probes.109–115 | ||
The irreversible transformation of the sensitising ligand may occur while the complex is in its excited state. The strength of certain lanthanide–ligand bonds, e.g. Ln–Npy, may weaken following excitation, for example, triggering a remote dissociative process (Scheme 5), as in a unique Pt–pyridine coordination complex linked to the pyridylalkynylaryl sensitiser, where emission switches on after irradiation.115
4.3. Reversible change of metal primary coordination sphere: Iratio and τLn*
Changes to the number and nature of the ligand or solvent donors coordinating to the lanthanide ion alter the magnitude of the ground state ligand field splitting, leading to changes in emission spectral form and lifetime. In certain cases, the reversible coordination of a new donor occurs that has a different quenching effect. In the simplest case, this is the solvent, and the largest effect is seen when water is involved, as it modifies kvib greatly. Two simple examples illustrate this concept, involving reversible intramolecular coordination. First, N-protonation of a 5-ring chelate sulphonamide leads to cleavage of the Ln–N bond and replacement by a water molecule;116,117 second, the dissociation of coordinated pyridyl and tertiary N atoms may occur when an added metal ion chelates preferentially, leading to solvent coordination to the lanthanide ion, (Scheme 6).118,119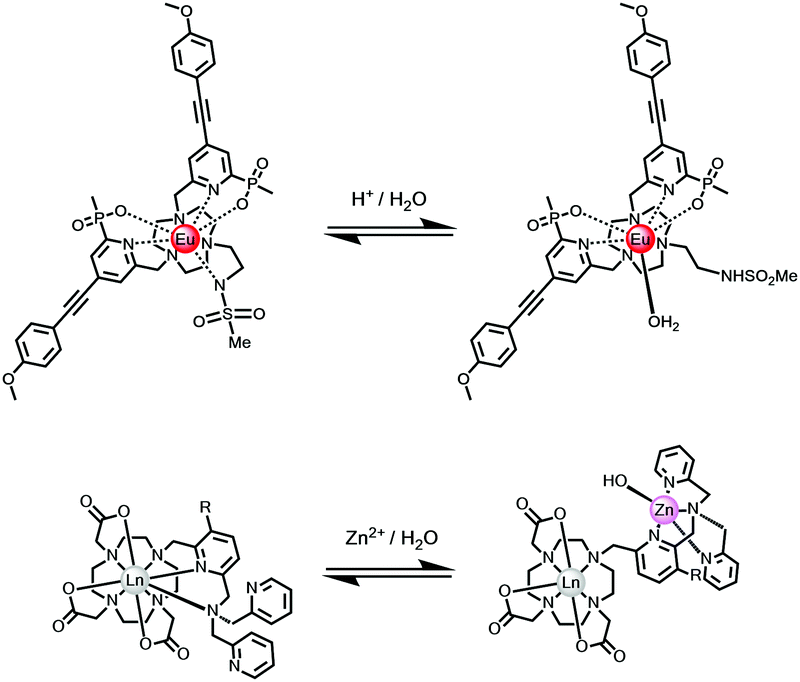 | ||
| Scheme 6 The speciation at Eu in each complex changes reversibly following addition of acid (upper) or metal ion (lower), with dissociation of a nitrogen donor(s) and replacement by water.116–119 | ||
The large class of intermolecular examples involving reversible N and O donor coordination by an added anion, discussed above (Scheme 3), permit anion sensing and signalling, vide infra. Other examples of a reversible change in speciation at the lanthanide centre may occur in the excited state, and have been observed in certain cases, notably involving protein association where reversible ligation of a glutamate side chain residue occurs with alpha-acid glycoprotein, for example.97
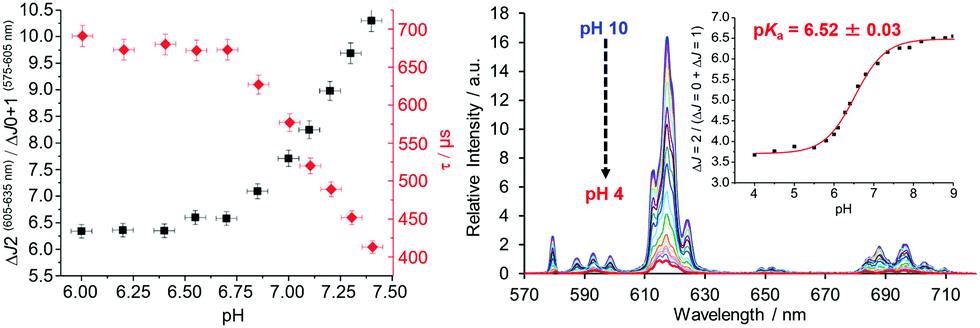 | ||
| Fig. 7 (right) Variation of the Eu emission spectrum with pH for the sulphonamide complex shown in Scheme 6 (0.1 M NaCl, 295 K); (left) emission band intensity ratio and lifetime dependence with pH in the endoplasmic reticulum of live mouse skin fibroblasts, where the pKa is 7.15 (±0.05), from a calibration undertaken in situ. Adapted with permission from the Royal Society of Chemistry.116 | ||
A similar approach involving an integral sulfonamide moiety has been used to assess lysosomal pH in living cells, using the Tb and Eu complexes of a common chiral ligand with a remote azaxanthone sensitising group that localised quickly to the cellular lysosomes.117 The Tb/Eu emission intensity ratio (Iem 545/620 nm; λexc 355 nm) or Eu CPL emission dissymmetry factor were used to monitor the local pH and the system was calibrated in situ (pKa = 5.80 (0.10)), using a classical fluorescent pH probe.
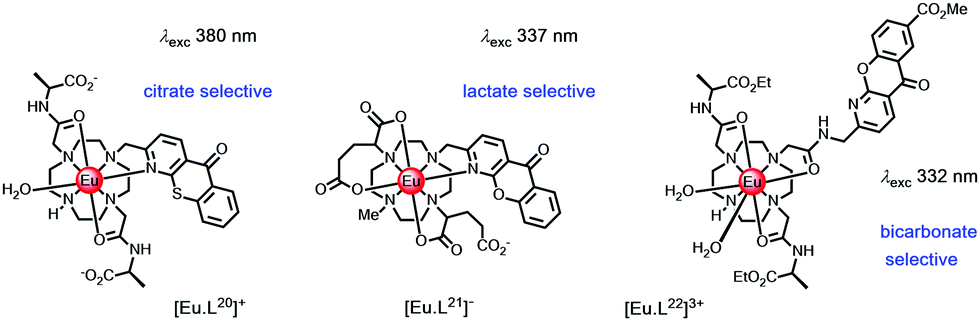 | ||
| Scheme 7 Complexes designed to bind citrate, lactate and bicarbonate selectively.120–123 | ||
The bicarbonate anion is an important target in probe development because it plays a central role in a wide range of chemical and biochemical processes. These include intracellular pH homeostasis, kidney function and sperm maturation. Low levels of bicarbonate in patients with chronic kidney disease have been suggested to be a reliable indicator of metabolic acidosis. For patients in intensive care, bicarbonate levels been flagged as a reliable means of signalling metabolic acidosis. In clinical studies, values below 22 mM have been linked to increased rates of mortality.
The Tb and Eu complexes of the azaxanthone triamide complex, [EuL22]3+ have been used to measure bicarbonate in human serum. In addition, these complexes are trafficked to cellular mitochondria and have permitted analyses of intracellular bicarbonate levels.104 Reversible binding of this anion is characterized by a large increase in the ΔJ = 2 Eu emission band, following carbonate chelation, (Fig. 8).123 Binding could be followed either by examining Eu emission intensity ratios, Eu/Tb ratios or by monitoring changes in the Eu CPL gem values, where one of the ΔJ = 1 transitions inverted sign.122,124
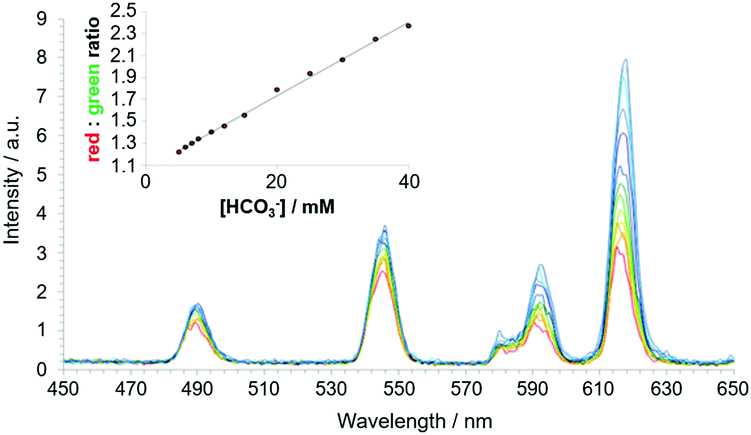 | ||
| Fig. 8 Time-gated spectral data showing the variation of emission for [EuL22]3+/[TbL22]3+ following addition of bicarbonate (λexc 365 nm, 10 μs time gate, pH 7.4, 295 K, 0.1 M NaCl, 0.4 mm human serum albumin, 2.3 mM lactate, 0.13 mM citrate, 1 mM HPO42−, 1 mM ATP). The inset shows the red/green ratio change with increasing bicarbonate concentration. Adapted with permission from ref. 122. Copyright (2012) Wiley-VCH.122 | ||
Bicarbonate binding at the metal centre is competitive with non-covalent protein binding. The complex forms a 1:1 complex with albumin with an affinity in the same range (logK ca. 3.50) as the binding constant for bicarbonate. With the Eu complex, protein binding gave rise to quenching of emission that was greater than with Tb analogues. Presumably, electron-rich aromatic groups in the protein (e.g., Tyr, Trp) take part in a quenching charge-transfer interaction with the azaxanthone triplet excited state. Thus, the rate of quenching competes with the intramolecular energy transfer process that is more efficient for Tb than Eu. In the Tb complexes, the energy gap between the azaxanthone triplet at 23500 cm−1 and the Tb 5D4 excited state (20450 cm−1) is smaller compared to the Eu 5D1 and 5D0 excited states (19200 and 17220 cm−1). In media containing protein, e.g., serum or in cellulo, an increase in the concentration of bicarbonate causes de-quenching of lanthanide emission and is signalled faithfully either by a ratiometric change in Eu emission-band intensities, or by measuring the Eu/Tb intensity ratio, with bands selected using optical filters.
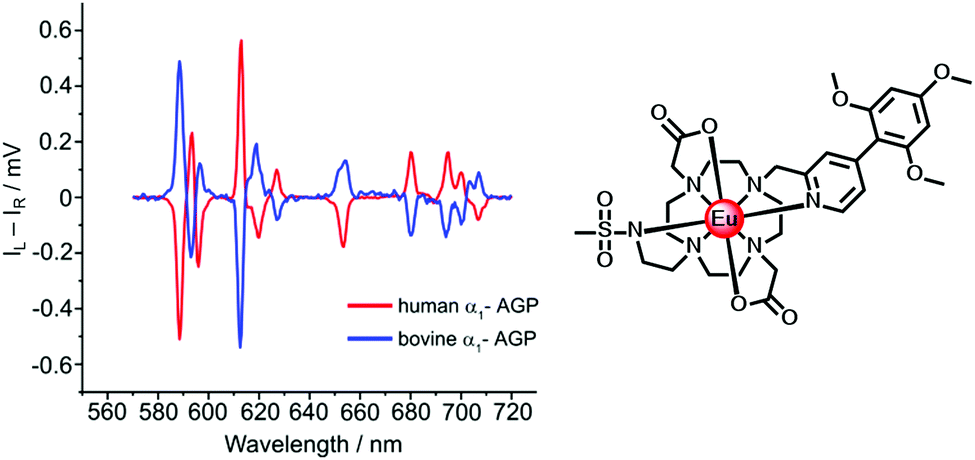 | ||
| Fig. 9 Induced CPL spectra of the Eu complex when bound to human (Δ) and bovine (Λ) α1-AGP, under conditions when the sulfonamide arm is coordinated (λexc 310 nm, 295 K). Adapted with permission from the Royal Society of Chemistry.96 | ||
A second example of probe helicity inversion occurs following binding of ADP and ATP to the dinuclear Zn/Eu complex, formed in situ when zinc is added and displaces two of the ligand N donors, (Scheme 6, above).95 This Eu complex is dynamically racemic, with enantiomer interconversion occurring by cooperative arm rotation and independent macrocyclic ring inversion. On binding ADP and ATP, the induced CPL signature showed opposite helicities for the anion complexes, (Fig. 10).
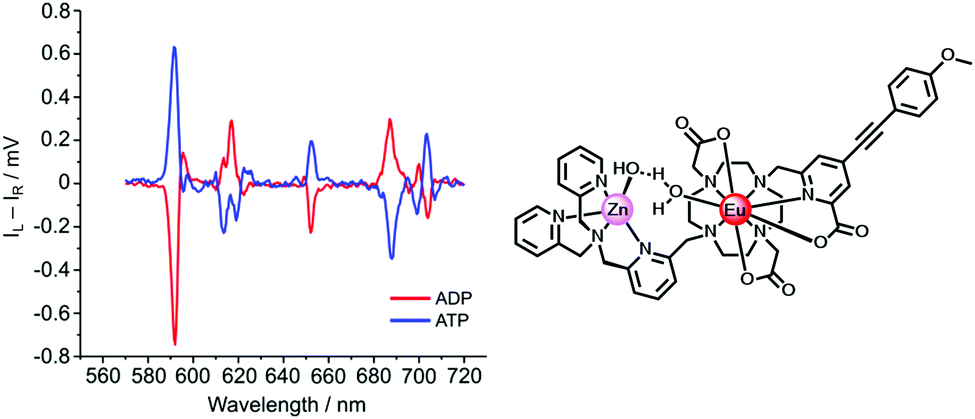 | ||
| Fig. 10 Induced CPL spectra signalling the binding of ADP and ATP (λexc 335 nm, pH 7.4, 5 μM complex, 50 μM Zn2+, 100 μM ADP or ATP) in adducts of opposite helicity. Adapted with permission from ref. 95. Copyright (2018) Wiley-VCH. | ||
The total Eu emission spectra were identical for the ADP and ATP adducts. The CPL differences allowed the relative amounts of ADT and ATP in mixtures to be estimated, based on analysis of the CPL emission dissymmetry value, gem (592 nm).
In the ternary adducts with AMP, ADP and ATP, the terminal phosphate can bridge the Zn and Eu ions. The Λ form is the more stable than the Δ isomer in the ADP (and AMP) adduct by 23 kJ mol−1, whereas the Δ form is energetically more stable than the Λ isomer in the ATP adduct by 32 kJ mol−1, (Fig. 11). In the latter case, favourable directed H-bonding occurs between the furanose OH groups and the ATP α-phosphate (and to the β-phosphate in the ADP adduct) and to the carbonyl oxygen of an acetate group.95
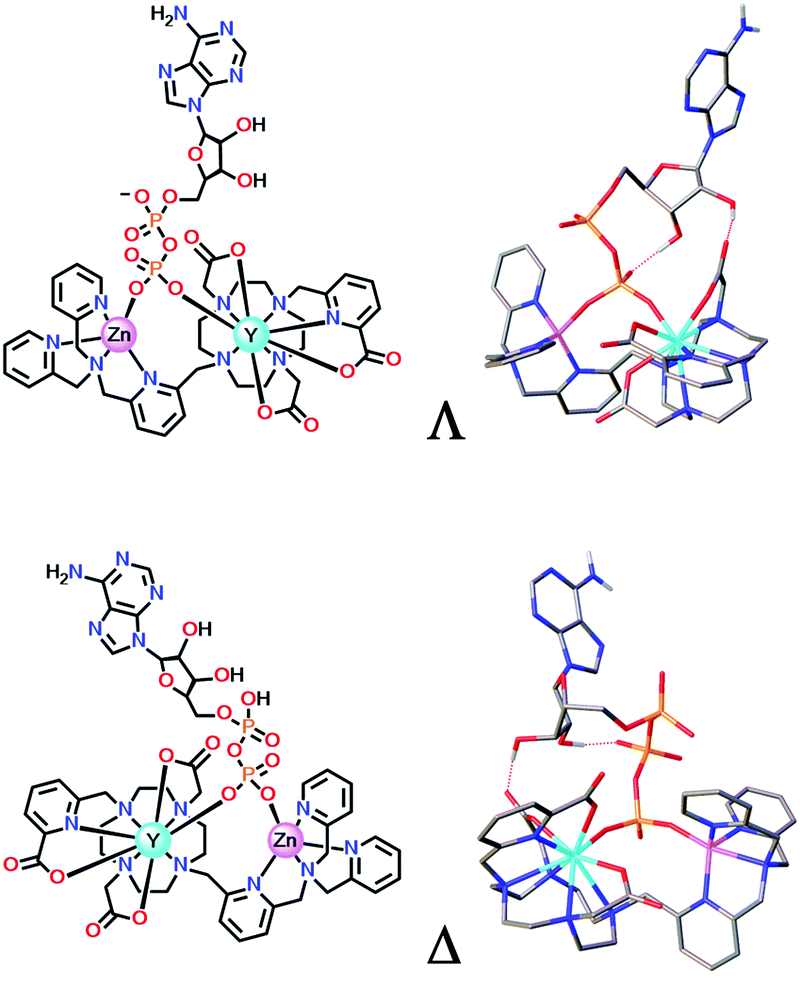 | ||
| Fig. 11 DFT optimised model geometries for the favoured ternary adducts with ADP (top) and ATP (lower) showing different arrangements of the exocyclic ring substituents (Λ/Δ); Y was used as a surrogate for Eu. Adapted with permission from ref. 95. Copyright (2018) Wiley-VCH. | ||
4.4. Dynamic quenching involving perturbation of ligand T1 and Ln excited states
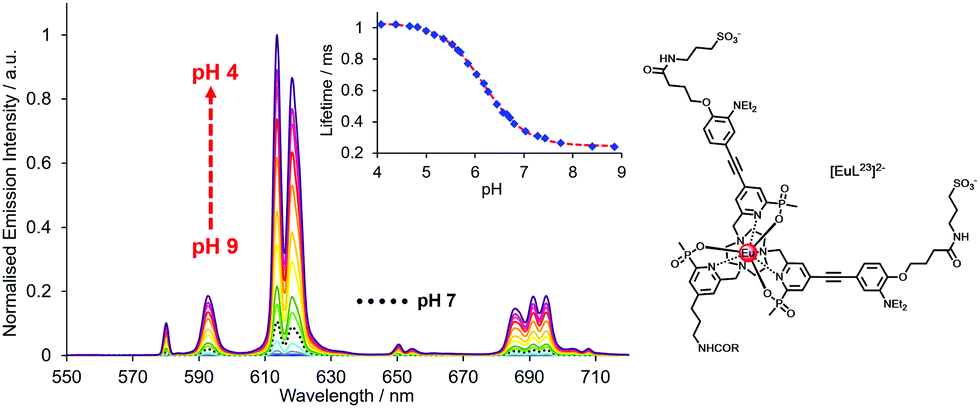 | ||
| Fig. 12 pH Variation of europium emission of [EuL23]2− (λexc 332 nm, 295 K, 0.1 M NaCl); showing the variations of the europium emission intensity and lifetime (λexc 332 nm, λem 613 nm) with a pKa value of 6.18(03). Reproduced with permission from the Royal Society of Chemistry.125 | ||
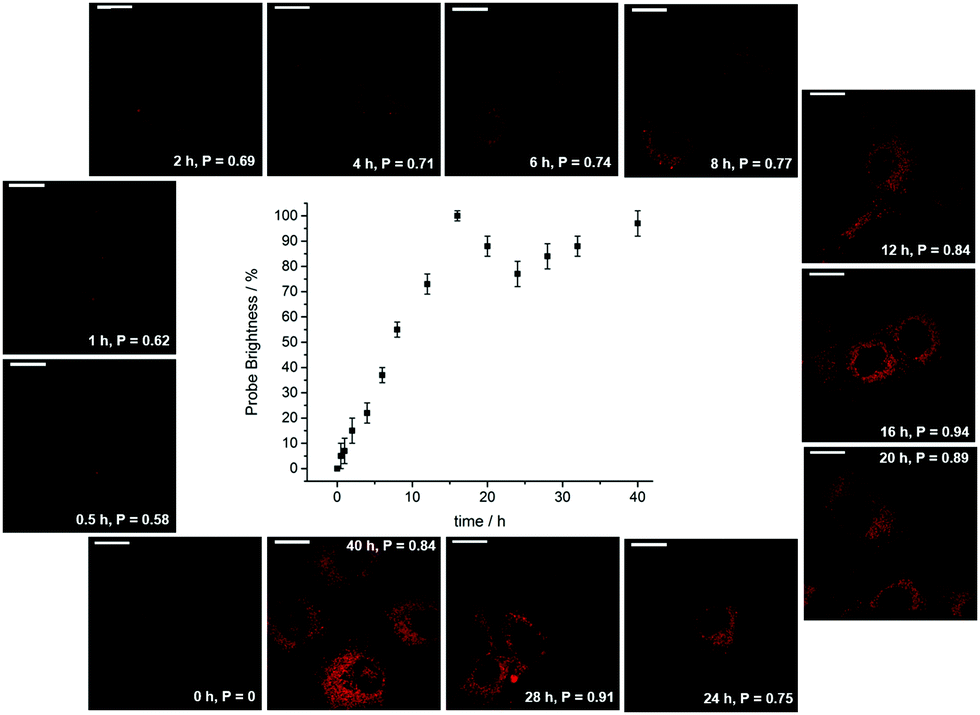 | ||
| Fig. 13 Confocal microscopy images (λexc 355 nm) revealing the time dependence of endosomal/lysosomal localisation with the protonated form of [EuL12], showing the order of magnitude increase in probe brightness as the organelles acidify over the 18 h cell cycle. Adapted with permission from ref. 38. Copyright (2021) Wiley-VCH. | ||
During the ageing process of endosomes, the pH reduces with time until eventually they evolve into lysosomes. The cytosolic pH of healthy cells is 7.2, while the pH of endosomes is typically from 6.5 to 5.5; in mature lysosomes the resting pH is typically around 4.5. Receptor internalisation and endosomal uptake can be monitored when the species that is internalised is tagged with a pH sensitive emissive probe, (Fig. 13). This approach has been described to follow the agonist-induced internalisation of the glucagon-like peptide-1 receptor (GLP-1R): a 5-fold increase in europium emission intensity was observed 1 h after agonist-induced internalisation of the receptor labelled with [EuL23]2−, (Scheme 8).125 The GLP-1R receptor is a target for anti-diabetic drugs because it is involved in the metabolic pathway for insulin production.
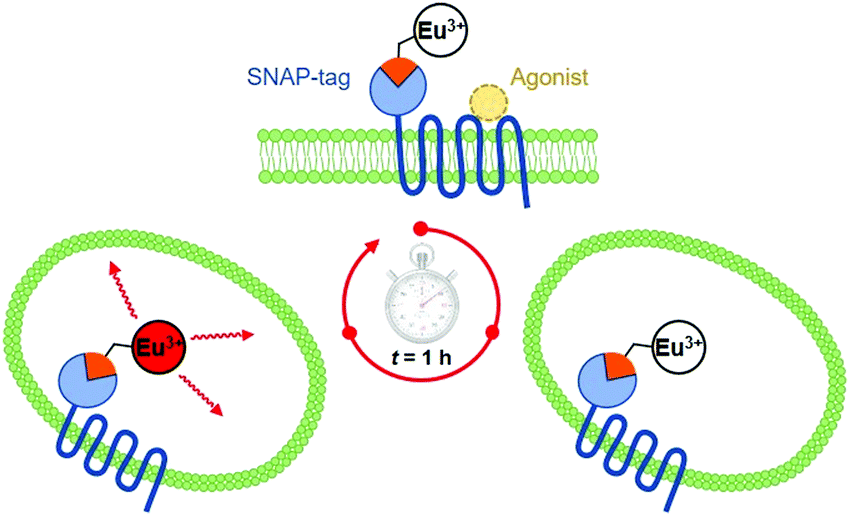 | ||
| Scheme 8 Cartoon depicting receptor mediated internalisation of the Eu labelled GLP-1R receptor, switching on Eu emission following endosomal acidification. Reproduced with permission from the Royal Society of Chemistry.125 | ||
A different approach to pH monitoring, observing changes in Tb/Eu emission intensity ratios, involves a 4-hydroxypyridine derivative of a substituted terpyridyl ligand (λexc 321 nm, pKa 5.8). In this case, both the Eu and Tb complexes are emissive in the protonated form, but in the deprotonated state Eu luminescence is quenched by internal charge transfer from the phenolate group, (Fig. 14). Such an effect probably occurs by dynamic quenching of the Eu excited state (i.e. not via quenching of the ligand S1 state by Eu3+) without affecting the emission intensity or lifetime in the Tb analogue. No Eu lifetime variations were reported in this work to resolve this issue, nor was the pH dependence of the Eu lifetime assessed after direct excitation of Eu at 396 nm, that could have distinguished these possibilities.
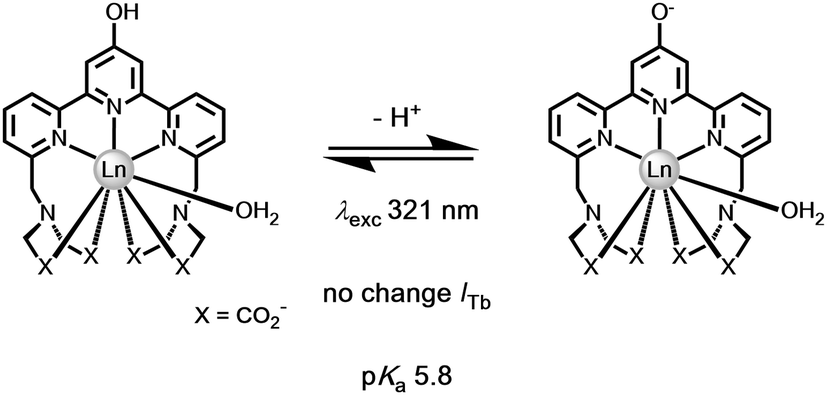 | ||
| Fig. 14 Deprotonation of the 4-OH pyridine switches off Eu but not Tb luminescence.126 | ||
In both of these examples, the Eu excited state is prone to quenching by an intramolecular electron transfer process associated with the ligand amine or phenoxide lone pair. The rate of proton transfer to and from the N or O atom is >1010 s−1, much faster than the slow rate of decay of the excited europium ion (103 s−1) or the lifetime of the intermediate ligand based excited states. Furthermore, the rate of electron transfer from the unprotonated chromophore to the excited Eu ion is faster than the rate of energy transfer populating it or indeed of its own radiative rate of emission. Thus, during the millisecond lifetime of the Eu 5D0 excited state, as the pH rises loss of a proton from the protonated amine or phenol group occurs, allowing fast electron transfer to the Eu ion, quenching its emission and shortening the 'time-averaged' observed lifetime. An alternative viewpoint is to say that there is a potential energy surface crossing of the 5D0 Eu excited state with the internal ligand to metal charge transfer state (LMCT) that involves the conjugated heteroatom and the extended π system.127
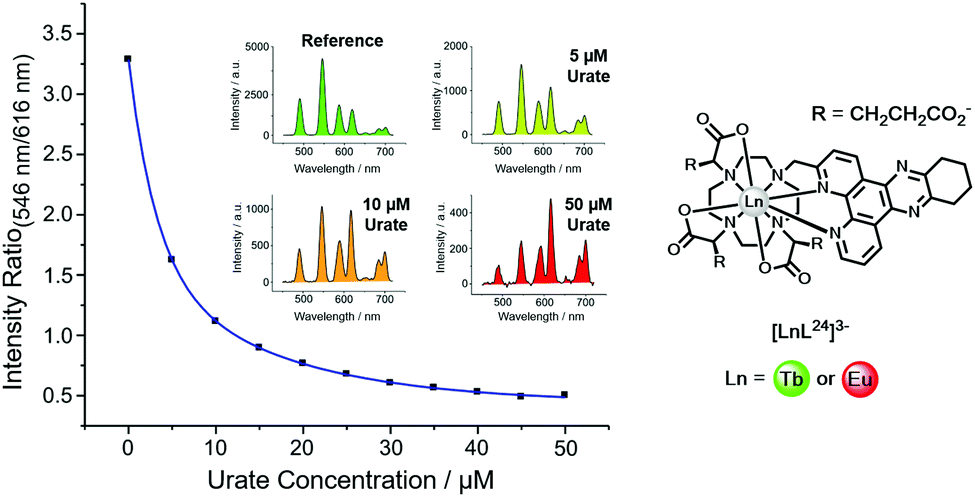 | ||
| Fig. 15 Variation of the Tb (546 nm) to Eu (616 nm) intensity ratio (λexc, 348 nm, pH 7.4, 295 K) as a function of urate concentration using [LnL24]3−. Reproduced with permission from the Royal Society of Chemistry.128 | ||
Uric acid is formed in the liver as the final breakdown product in the metabolism of adenine and guanine. Levels of uric acid in the body are set by the balance between the rate of urinary elimination and its rate of formation. Hyperuricaemia is a condition that is commonly associated with gout and also indicates an increased breakdown of cell nuclei or the onset of kidney disease. Patients on chemotherapy for cancer, such as myeloma, lymphoma or leukaemia typically are often hyperuricaemic, and their uric acid levels are monitored carefully to avoid the risk of kidney damage.
In the lanthanide uric acid assay, the Tb and Eu complexes of the same ligand are used. Any non-specific effects that could change emission intensity or lifetime occur to the same degree for each complex. Examples of such effects include light scattering caused by particulates, sample-to-sample variations of protein levels, and surface adhesion. Indeed, any static quenching process operating by deactivation of the singlet excited state of the sensitizer occurs to the same extent for each complex. These effects are intrinsically corrected because the lanthanide emission intensity ratio is taken.
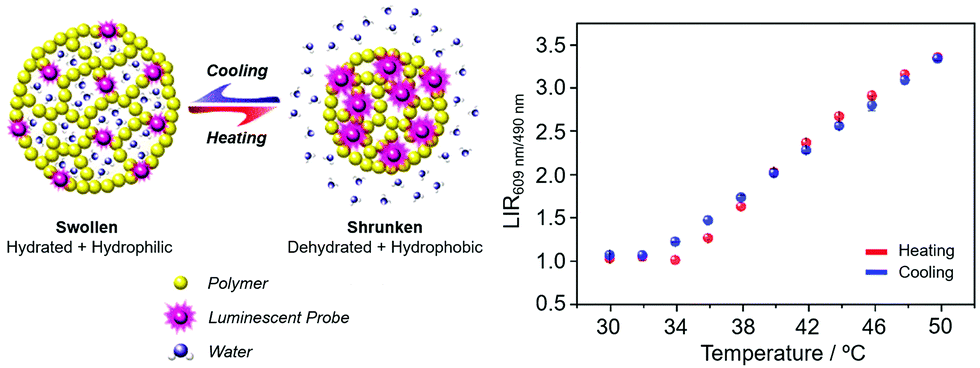 | ||
| Fig. 16 (left) Principle of a nanoparticle thermometer based on changes in the host polymer matrix hydrophilicity; (right) performance of the polymer nanoparticles covalently linked to a Eu/Tb dipicolinate complex in 1:1 ratio (λexc 275 nm). Adapted with permission from the Royal Society of Chemistry and the Centre National de Recherche Scientifique.129 | ||
Ytterbium complexes intrinsically show even greater sensitivity to vibrational quenching by proximate OH oscillators leading to a sensitivity of the Yb emission lifetime to both temperature and medium viscosity, as the frequency of the quenching vibrational oscillator is perturbed.130 It can be recalled here that Förster energy transfer from Tb to Eu has an inherent temperature dependence. In the Förster transfer mechanism, if the transitions are not exactly at resonance, the energy difference must be conserved by phonons. Since phonon relaxation is ineffective at low temperature, the energy transfer efficiency may decrease significantly, in this case below 200 K. Accordingly, chelates of europium and terbium with β-diketonate ligands have been examined in gel glasses and in polydimethylsiloxane matrices. The Tb/Eu emission intensity ratios vary with temperature over the range 200 to 300 K, and 150 to 250 K respectively, suggesting future uses in low T thermometry.131,132
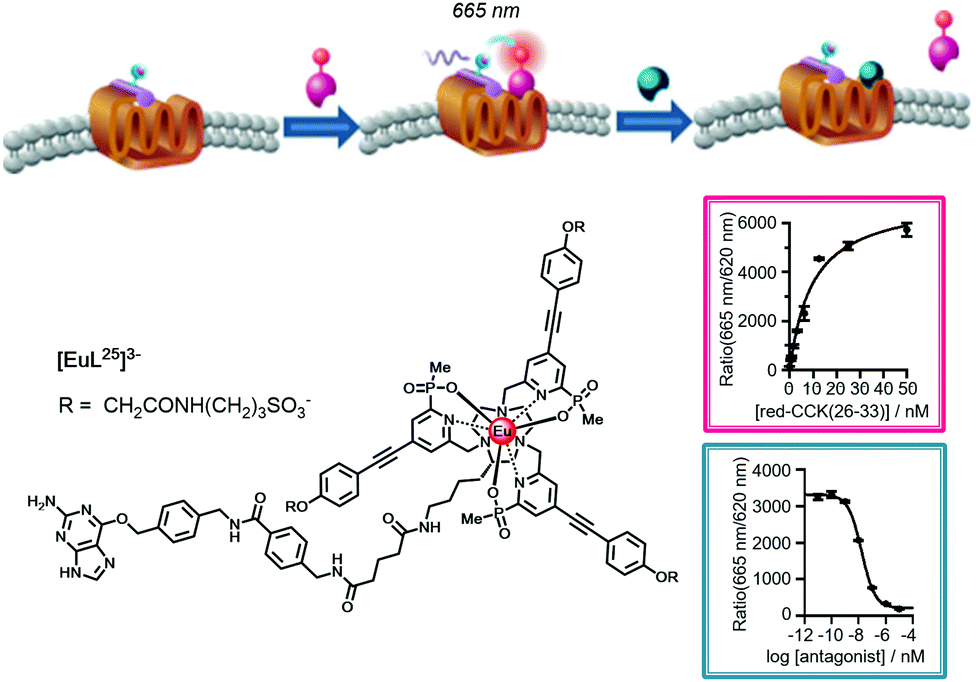 | ||
| Fig. 17 Cartoon depicting the consecutive binding of an IR-dye labelled agonist acceptor to the Eu-SNAP-tagged CCK-2 cell surface receptor, quenching the Eu emission and accompanied by a FRET signal at 665 nm (Kd = 8 nM), followed by competitive displacement of the agonist by an added antagonist (Ki = 6 nM), restoring the europium emission at 620 nm. Adapted with permission from ref. 136. Copyright (2014) Wiley-VCH. | ||
5. Conclusions
In the sensitised emission of lanthanide complexes, there are three excited states that can be perturbed giving rise to ‘responsive probes’. The luminescence changes that may occur modulate emission intensity, spectral form, lifetime and circular polarisation.6–9 The longer lifetimes and large pseudo-Stokes’ shifts of sensitised lanthanide luminescence have allowed time-gated methods of data acquisition to be used for spectroscopy and microscopy, averting the problems associated with light scattering, auto-fluorescence and self-absorption that compromise the scope and utility of analyses using short lived fluorophores.The ligand that coordinates the lanthanide ion has to be chosen carefully, such that the complex has sufficient kinetic and thermodynamic stability to be used without fear of premature metal ion dissociation or unwanted ligand exchange under the low concentration assay conditions. Moreover, the ligand should be designed thoughtfully to minimise vibrational deactivation of the lanthanide excited state by energy-matched oscillators that include OH, NH, CH, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and aromatic ring vibrations, in order to enhance the emission quantum yield and optimise the emission lifetime. Consideration should also given to steric shielding of the lanthanide ion, for which the use of phosphinate rather than carboxylate groups offers much scope in design and enhances the metal excited state lifetime.
O and aromatic ring vibrations, in order to enhance the emission quantum yield and optimise the emission lifetime. Consideration should also given to steric shielding of the lanthanide ion, for which the use of phosphinate rather than carboxylate groups offers much scope in design and enhances the metal excited state lifetime.
The sensitising moiety can be selected from a wide range of aromatic and heterocyclic systems, for which the energies of their singlet, triplet and internal charge transfer states can be easily established. The nature of the sensitising moiety chosen depends very strictly upon the lanthanide ion to be used, notably its ability to quench the ligand singlet excited state, e.g. for Eu, Yb and Sm(III) complexes where the overall free energy change can be estimated based on redox potentials. The ground or excited state of the sensitising moiety can be perturbed, leading to modulation of emission behaviour. Such a perturbation may be reversible, leading to new sensor designs, e.g. for urate analysis with electron poor sensitising groups where exciplex formation occurs with the ligand triplet. This behaviour leads to variation of both the lifetime and the emission intensity and occurs to differing extents for Tb and Eu complexes of the same ligand, allowing ratiometric analysis. In cases where back energy transfer to/from the ligand triplet is engineered, oxygen and temperature sensitivity results, notably for complexes of Tb owing to its high excited state energy.
In a second class of system, the sensitiser is irreversibly transformed by reaction with a target analyte, changing the singlet and triplet excited state energies to permit the creation of ‘activity based probes’ that can be used to signal the presence of a low concentration analyte. This is best done for systems that are designed to ‘switch-on’ or greatly increase the optical signal. Two further classes of responsive probe involve either changes in the speciation or configuration of the metal centre, or are based on modulation of excited state dynamic quenching, i.e. they involve perturbation of the ligand ICT/T1 states or the lanthanide excited state itself. In each case, the design of a new probe must consider four key operational aspects: the tuning of probe affinity to the analyte concentration range in the sample or environment; the assessment of the impact of interfering species in the analysis, notably electron rich species that may quench the ligand or metal excited state by electron transfer; ensuring the accurate calibration of the system in situ; being aware of the overall response time of the optical change that is monitored, taking account of the time that it takes for the data to be acquired with respect to the time-dependence of the variation that is of interest!
Reversible change to the primary and secondary coordination sphere of the lanthanide ion perturbs the ligand field, in an exquisitely sensitive manner that dismisses the old dogma that the optical fingerprint of lanthanide emission is just a function of the metal ion. Anion binding in water to the metal ion complexes of heptadentate ligands (lower coordination number systems tend to be limited by complex instability) occurs reversibly and can be inter- or intramolecular in nature. A large number of sensors have been created that operate directly or in diluted samples of complex biological fluids e.g. urine, serum, semen, allowing the concentration of key anions like lactate, citrate, bicarbonate as well as pH to be measured in vitro. Using spectral imaging in microscopy, related probes have been used to monitor changes in pH and pX in living cells, suggesting a future growth in their use for live cell assays.
In this context, the recent development of circular polarised luminescence microscopy offers promise for further development. Already, a series of responsive CPL probes has emerged based on Eu and Tb complexes, notably involving protein binding. The CPL fingerprint uniquely includes the sign of the optical transition, greatly enhancing spectral resolution and also allowing the configuration of the emissive species to be identified. CPL Probes where the circularly polarised emission switches on following analyte binding to a dynamically racemic complex are of particular interest in this regard, and have been used to report ATP/ADP ratios and the concentration of α1-AGP in serum.95,96 There is great scope for new chiral probe development, as cheaper CPL instrumentation for spectroscopy and microscopy is developed.91–93,137
Efficient systems that function by modulation of the dynamic quenching of the metal excited state have been described, including real time observation of acidification that has allowed the monitoring of receptor internalisation into endosomal compartments and lysosomes. Many cases have been exploited where the metal excited state is quenched by energy transfer to a proximate acceptor, such as a near-IR dye that adopts the longer lifetime of the lanthanide donor. In these FRET processes using terbium and europium complexes that have high intrinsic brightness, the high throughput screening of drug binding to G-protein coupled receptors has been developed, as just one example of a plethora of sensitive biochemical analyses involving these versatile luminescent lanthanide coordination complexes.
Looking to the future, there is always space for creative design that may transcend the classifications offered here. In recent work, for example, heterodinuclear Nd/Yb and Eu/Tb DOTA based complexes have been appended to a zinc-binding peptide.138,139 Metal ion binding regulates the ligand structure and changes the distance between an integral sensitising moiety (a coumarin for the Tb/Eu pair and an anthryl group for the near-IR emitting Nd/Yb system) and the lanthanide ions. As a result, the Nd/Yb and Eu/Tb emission intensity ratios change as a function of zinc concentration, allowing calibration. Future design allows free rein for the creative spirit of the chemist!
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This review is dedicated to the creative chemists who took the key steps in the development of the responsive luminescent systems described here; their names are given below. M’illumino d’immenso.References
- B. Alpha, R. Ballardini, V. Balzani, J.-M. Lehn, S. Perathoner and N. Sabbatini, Photochem. Photobiol., 1990, 52, 299–306 Search PubMed.
- M. H. V. Werts, Sci. Prog., 2005, 88, 101–131 Search PubMed.
- S. Pandya, J. Yu and D. Parker, Dalton Trans., 2006, 2757–2766 Search PubMed.
- J.-C. G. Bünzli, Coord. Chem. Rev., 2015, 293, 19–47 Search PubMed.
- E. Mathieu, A. Sipos, E. Demeyere, D. Phipps, D. Sakaveli and K. E. Borbas, Chem. Commun., 2018, 54, 10021–10035 Search PubMed.
- A. B. Aletti, D. M. Gillen and T. Gunnlaugsson, Coord. Chem. Rev., 2018, 354, 98–120 Search PubMed.
- D. Parker, Coord. Chem. Rev., 2000, 205, 109–131 Search PubMed.
- C. P. Montgomery, B. S. Murray, E. J. New, R. Pal and D. Parker, Acc. Chem. Res., 2009, 42, 925–937 Search PubMed.
- S. Shuvaev, M. Starck and D. Parker, Chem. – Eur. J., 2017, 23, 9974–9989 Search PubMed.
- A. Dadabhoy, S. Faulkner and P. G. Sammes, J. Chem. Soc., Perkin Trans. 2, 2000, 2359–2361 Search PubMed.
- Y. Bretonniere, M. J. Cann, D. Parker and R. Slater, Org. Biomol. Chem., 2004, 2, 1624–1632 Search PubMed.
- D. Kovacs, X. Lu, L. S. Mezaros, M. Ott, J. Andres and K. E. Borbas, J. Am. Chem. Soc., 2017, 139, 5756–5767 Search PubMed.
- M. Xiao and P. R. Selvin, J. Am. Chem. Soc., 2001, 123, 7067–7073 Search PubMed.
- J. A. G. Williams, P. K. Senanayake and D. Parker, J. Chem. Soc., Perkin Trans. 2, 1998, 2129–2139 Search PubMed.
- H. Takalo, V.-M. Mukkala, L. Merio, J. C. Rodriguez-Ubis, R. Sedano, O. Juanes and E. Brunet, Helv. Chim. Acta, 1997, 80, 372–387 Search PubMed.
- S. Petoud, S. M. Cohen, J.-C. G. Bünzli and K. N. Raymond, J. Am. Chem. Soc., 2003, 125, 13324–13325 Search PubMed.
- P. A. Atkinson, K. S. Findlay, F. Kielar, R. Pal, R. A. Poole, H. Puschmann, S. L. Richardson, P. A. Stenson, A. L. Thompson, J. Yu and D. Parker, Org. Biomol. Chem., 2006, 4, 1707–1722 Search PubMed.
- G. Bobba, J.-C. Frias and D. Parker, Chem. Commun., 2002, 890–891 Search PubMed.
- R. A. Poole, G. Bobba, M. J. Cann, J.-C. Frias, R. D. Peacock and D. Parker, Org. Biomol. Chem., 2005, 3, 1013–1024 Search PubMed.
- C. P. Montgomery, D. Parker and L. Lamarque, Chem. Commun., 2007, 3841–3843 Search PubMed.
- A. D’Aleo, A. Picot, A. Beeby, J. A. G. Williams, B. Le Guennic, C. Andraud and O. Maury, Inorg. Chem., 2008, 47, 10258–10268 Search PubMed.
- J. W. Walton, A. Bourdolle, S. J. Butler, M. Soulie, M. Delbianco, B. K. McMahon, R. Pal, H. Puschmann, J. M. Zwier, L. Lamarque, O. Maury, C. Andraud and D. Parker, Chem. Commun., 2013, 49, 1600–1602 Search PubMed.
- M. Soulié, F. Latzko, E. Bourrier, V. Placide, S. J. Butler, R. Pal, J. W. Walton, P. L. Baldeck, B. Le Guennic, C. Andraud, J. M. Zwier, L. Lamarque, D. Parker and O. Maury, Chem. – Eur. J., 2014, 20, 8636–8646 Search PubMed.
- S. J. Butler, L. Lamarque, R. Pal and D. Parker, Chem. Sci., 2014, 5, 1750–1756 Search PubMed.
- S. J. Butler, M. Delbianco, L. Lamarque, B. K. McMahon, E. R. Neil, R. Pal, D. Parker, J. W. Walton and J. M. Zwier, Dalton Trans., 2015, 44, 4791–4803 Search PubMed.
- V. Placide, A. T. Bui, A. Grichine, A. Duperray, D. Pitrat, C. Andraud and O. Maury, Dalton Trans., 2015, 44, 4918–4924 Search PubMed.
- E. G. Moore, A. P. S. Samuel and K. N. Raymond, Acc. Chem. Res., 2009, 42, 542–552 Search PubMed.
- A. Beeby, S. Faulkner, D. Parker and J. A. G. Williams, J. Chem. Soc., Perkin Trans. 2, 2001, 1268–1273 Search PubMed.
- J. Andres and A.-S. Chauvin, Phys. Chem. Chem. Phys., 2013, 15, 15981–15994 Search PubMed.
- M. Kleinerman, J. Chem. Phys., 1969, 51, 2370–2381 Search PubMed.
- G. A. Hebbink, S. I. Klink, L. Grave, P. G. B. O. Alink and F. C. J. M. van Veggel, ChemPhysChem, 2002, 3, 1014–1018 Search PubMed.
- J. R. G. Thorne, J. M. Rey, R. G. Denning, S. E. Watkins, M. Etchells, M. Green and V. Christou, J. Phys. Chem. A, 2002, 106, 4014–4021 Search PubMed.
- R. S. Dickins, J. A. K. Howard, C. L. Maupin, J. M. Moloney, D. Parker, J. P. Riehl, G. Siligardi and J. A. G. Williams, Chem. – Eur. J., 1999, 5, 1095–1105 Search PubMed.
- A. Weller, Pure Appl. Chem., 1968, 16, 115–124 Search PubMed.
- G. Porcal, S. G. Bertolotti, C. M. Previtali and M. V. Encinas, Phys. Chem. Chem. Phys., 2003, 5, 4123–4128 Search PubMed.
- F. Kielar, C. P. Montgomery, E. J. New, D. Parker, R. A. Poole, S. L. Richardson and P. A. Stenson, Org. Biomol. Chem., 2007, 5, 2975–2982 Search PubMed.
- G.-L. Law, D. Parker, S. L. Richardson and K.-L. Wong, Dalton Trans., 2009, 8481–8484 Search PubMed.
- M. Starck, J. D. Fradgley, R. Pal, J. M. Zwier, L. Lamarque and D. Parker, Chem. – Eur. J., 2021, 27, 766–777 Search PubMed.
- L. Stryer, D. D. Thomas and C. F. Meares, Annu. Rev. Biophys. Bioeng., 1982, 11, 203–222 Search PubMed.
- J. A. G. Williams and D. Parker, J. Chem. Soc., Perkin Trans. 2, 1996, 1581–1586 Search PubMed.
- A. Beeby, D. Parker and J. A. G. Williams, J. Chem. Soc., Perkin Trans. 2, 1996, 1565–1579 Search PubMed.
- M. W. Mara, D. S. Tatum, A.-M. March, G. Doumy, E. G. Moore and K. N. Raymond, J. Am. Chem. Soc., 2019, 141, 11071–11081 Search PubMed.
- I. M. Clarkson, A. Beeby, J. I. Bruce, L. J. G. Govenlock, C. E. Mathieu, M. P. Lowe, D. Parker and K. Senanayake, New J. Chem., 2000, 24, 377–386 Search PubMed.
- J. Andres and A.-S. Chauvin, Eur. J. Inorg. Chem., 2010, 2700–2713 Search PubMed.
- E. R. H. Walter, J. A. G. Williams and D. Parker, Chem. Commun., 2017, 53, 13344–13347 Search PubMed.
- A. T. Bui, A. Grichine, A. Duperray, P. Lidon, F. Riobe, C. Andraud and O. Maury, J. Am. Chem. Soc., 2017, 139, 7693–7696 Search PubMed ; correction: J. Am. Chem. Soc., 2018, 140, 8048.
- E. Kasprzycka, V. A. Trush, V. M. Amirkhanov, L. Jerzykiewicz, O. L. Malta, J. Legendziewicz and P. Gawryszewska, Chem. – Eur. J., 2017, 23, 1318–1330 Search PubMed.
- C. Yang, L.-M. Fu, Y. Wang, J.-P. Zhang, W.-T. Wong, X.-C. Ai, Y.-F. Qiao, B.-S. Zou and L.-L. Gui, Angew. Chem., Int. Ed., 2004, 43, 5010–5013 Search PubMed.
- G. L. Law, R. Pal, L. O. Palsson, D. Parker and K. L. Wong, Chem. Commun., 2009, 7321–7323 Search PubMed.
- T. J. Sorensen, A. M. Kenwright and S. Faulkner, Chem. Sci., 2015, 6, 2054–2059 Search PubMed.
- J. M. Zwier, H. Bazin, L. Lamarque and G. Mathis, Inorg. Chem., 2014, 53, 1854–1866 Search PubMed.
- P. Scholler, J. M. Zwier, E. Trinquet, P. Rondard, J.-P. Pin and L. Prõz, Prog. Mol. Biol. Transl. Sci., 2013, 113, 275–312 Search PubMed.
- M. Delbianco, V. Sadovnikova, E. Bourrier, L. Lamarque, G. Mathis, D. Parker and J. M. Zwier, Angew. Chem., Int. Ed., 2014, 53, 10718–10722 Search PubMed.
- M. Starck, R. Pal and D. Parker, Chem. – Eur. J., 2016, 22, 570–580 Search PubMed.
- T. Kim Anh, T. Ngoc, P. Thu Nga, V. T. Bitch, P. Long and W. Strek, J. Lumin., 1988, 39, 215–221 Search PubMed.
- H. Wang, D. Zhao, Y. Cui, Y. Yang and G. Qian, J. Solid State Chem., 2017, 246, 341–345 Search PubMed.
- G. Bao, K.-L. Wong, D. Jin and P. A. Tanner, Light: Sci. Appl., 2018, 7, 96 Search PubMed.
- T. Lazarides, D. Sykes, S. Faulkner, A. Barbieri and M. D. Ward, Chem. – Eur. J., 2008, 14, 9389–9399 Search PubMed.
- M. D. Ward, Coord. Chem. Rev., 2010, 254, 2634–2642 Search PubMed.
- A. Beeby, R. S. Dickins, S. Fitzgerald, L. J. Govenlock, C. L. Maupin, D. Parker, J. P. Riehl, G. Siligardi and J. A. G. Williams, Chem. Commun., 2000, 1183–1184 Search PubMed.
- R. S. Dickins, J. A. K. Howard, J. M. Moloney, D. Parker and R. D. Peacock, Angew. Chem., Int. Ed. Engl., 1997, 36, 521–523 Search PubMed.
- B. C. Barja, A. Remorino, M. J. Roberti and P. F. Aramendia, J. Argentin. Chem. Soc., 2005, 93, 81–96 Search PubMed.
- D. T. Cronce and W. DeW Horrocks, Biochemistry, 1992, 31, 7963–7969 Search PubMed.
- J. L. Kropp and M. W. Windsor, J. Chem. Phys., 1966, 45, 761 Search PubMed.
- W. R. Dawson, J. L. Kropp and M. W. Windsor, J. Chem. Phys., 1966, 45, 2410–2418 Search PubMed.
- W. D. W. Horrocks and D. R. Sudnick, J. Am. Chem. Soc., 1979, 101, 334–340 Search PubMed.
- R. S. Dickins, D. Parker, A. S. de Sousa and J. A. G. Williams, Chem. Commun., 1996, 697–698 Search PubMed.
- A. Beeby, I. M. Clarkson, R. S. Dickins, S. Faulkner, D. Parker, L. Royle, A. S. de Sousa, J. A. G. Williams and M. Woods, J. Chem. Soc., Perkin Trans. 2, 1999, 493–503 Search PubMed.
- S. Aime, M. Botta, D. Parker and J. A. G. Williams, J. Chem. Soc., Dalton Trans., 1996, 17–23 Search PubMed.
- G. Nocton, A. Nonat, C. Gateau and M. Mazzanti, Helv. Chim. Acta, 2009, 92, 2257–2273 Search PubMed.
- J. W. Walton, R. Carr, N. H. Evans, A. M. Funk, A. M. Kenwright, D. Parker, D. S. Yufit, M. Botta, S. De Pinto and K.-L. Wong, Inorg. Chem., 2012, 51, 8042–8056 Search PubMed.
- G. Stein and G. Wurzberg, J. Chem. Phys., 1975, 62, 208–213 Search PubMed.
- C. C. Bryden and C. N. Reilley, Anal. Chem., 1982, 54, 610–615 Search PubMed.
- C. Doffek, N. Alzakhem, C. Bischof, J. Wahsner, T. Guden-Silber, J. Lugger, C. Platas-Iglesias and M. D. Seitz, J. Am. Chem. Soc., 2012, 134, 16413–16423 Search PubMed.
- S. J. Butler and D. Parker, Chem. Soc. Rev., 2013, 42, 1652–1666 Search PubMed.
- M. C. Heffern, L. M. Matosziuk and T. J. Meade, Chem. Rev., 2014, 114, 4496–4539 Search PubMed.
- S. E. Bodman and S. J. Butler, Chem. Sci., 2021, 12, 2716–2734 Search PubMed.
- K. Binnemans, Coord. Chem. Rev., 2015, 295, 1–45 Search PubMed.
- S. Shuvaev and D. Parker, Dalton Trans., 2019, 48, 4471–4473 Search PubMed.
- Y. Ning, Y.-W. Liu, Y.-S. Meng and J.-L. Zhang, Inorg. Chem., 2018, 57, 1332–1341 Search PubMed.
- R. A. Poole, F. Kielar, S. L. Richardson, P. A. Stenson and D. Parker, Chem. Commun., 2006, 4084–4086 Search PubMed.
- M. S. Tremblay, M. Halim and D. Sames, J. Am. Chem. Soc., 2007, 129, 7570–7577 Search PubMed.
- D. G. Smith, B. K. McMahon, R. Pal and D. Parker, Chem. Commun., 2012, 48, 8520–8523 Search PubMed.
- G.-L. Law, R. Pal, L. O. Palsson, D. Parker and K.-L. Wong, Chem. Commun., 2009, 7321–7323 Search PubMed.
- D. Parker and J. A. G. Williams, Chem. Commun., 1998, 245–246 Search PubMed.
- J. A. Sobrinho, G. A. B. Junior, I. O. Mazali and F. A. Sigoli, New J. Chem., 2020, 8068–8075 Search PubMed.
- D. Parker, E. A. Suturina, I. Kuprov and N. F. Chilton, Acc. Chem. Res., 2020, 53, 1520–1534 Search PubMed.
- R. Carr, N. H. Evans and D. Parker, Chem. Soc. Rev., 2012, 41, 7673–7686 Search PubMed.
- J. L. Lunkley, N. M. Nguyen, K. M. Tuminaro, D. Margittai and G. Muller, Inorganics, 2018, 6, 87–113 Search PubMed.
- Y. Kitagawa, M. Tsurui and Y. Hasegawa, ACS Omega, 2020, 5, 3786–3791 Search PubMed.
- L. E. MacKenzie, L. O. Palsson, D. Parker, A. Beeby and R. Pal, Nat. Commun., 2020, 11, 1676 Search PubMed.
- L. E. MacKenzie and R. Pal, Nat. Rev. Chem., 2021, 5, 109–124 Search PubMed.
- A. T. Frawley, M. Starck, R. Pal and D. Parker, Chem. Sci., 2018, 9, 1042–1049 Search PubMed.
- L. Di Bari and F. Zinna, Chirality, 2015, 27, 1–13 Search PubMed.
- S. Shuvaev, M. A. Fox and D. Parker, Angew. Chem., Int. Ed., 2018, 57, 7488–7492 Search PubMed.
- S. Shuvaev, E. A. Suturina, K. Mason and D. Parker, Chem. Sci., 2018, 9, 2996–3003 Search PubMed.
- R. Carr, L. Di Bari, S. Lo Piano, D. Parker, R. D. Peacock and J. M. Sanderson, Dalton Trans., 2012, 41, 13154–13158 Search PubMed.
- R. Carr, R. Puckrin, B. K. McMahon, R. Pal, D. Parker and L. O. Palsson, Methods Appl. Fluoresc., 2014, 2, 024007 Search PubMed.
- L. Jennings, R. S. Waters, R. Pal and D. Parker, ChemMedChem, 2017, 12, 271–281 Search PubMed.
- S. Shuvaev, R. Pal and D. Parker, Chem. Commun., 2017, 53, 6724–6727 Search PubMed.
- C. P. Montgomery, E. J. New, D. Parker and R. D. Peacock, Chem. Commun., 2008, 4261–4263 Search PubMed.
- T. Gunnlaugsson, D. A. MacDonaill and D. Parker, J. Am. Chem. Soc., 2001, 123, 12866–12876 Search PubMed.
- E. A. Weitz and V. C. Pierre, Chem. Commun., 2010, 47, 541–543 Search PubMed.
- E. R. H. Walter, D. Parker and J. A. G. Williams, Chem. – Eur. J., 2018, 24, 7724–7733 Search PubMed.
- O. Reany, T. Gunnlaugsson and D. Parker, J. Chem. Soc., Perkin Trans. 2, 2000, 1819–1831 Search PubMed.
- J. Ohata, K. J. Bruemmer and C. J. Chang, Acc. Chem. Res., 2019, 52, 2841–2848 Search PubMed.
- A. T. Aron, K. M. Ramos-Torres, J. A. Cotruvo and C. J. Chang, Acc. Chem. Res., 2015, 48, 2434–2442 Search PubMed.
- Y. Yang, Q. Zhao, W. Feng and F. Li, Chem. Rev., 2013, 113, 192–270 Search PubMed.
- E. Pershagen, J. Nordholm and K. E. Borbas, J. Am. Chem. Soc., 2012, 134, 9832–9835 Search PubMed.
- M. Tropiano and S. Faulkner, Chem. Commun., 2014, 50, 4696–4698 Search PubMed.
- Z. Dai, L. Tian, B. Song, Z. Ye, X. Liu and J. Yuan, Anal. Chem., 2014, 86, 11883–11889 Search PubMed.
- M. Liu, Z. Ye, G. Wang and J. Yuan, Talanta, 2012, 99, 951–958 Search PubMed.
- C. Breen, R. Pal, M. R. J. Elsegood, S. J. Teat, F. Iza, K. Wende, B. R. Buckley and S. J. Butler, Chem. Sci., 2020, 11, 3164–3170 Search PubMed.
- C. Szijjarto, E. Pershagen, N. O. Ilchenko and K. E. Borbas, Chem. – Eur. J., 2013, 19, 3099–3109 Search PubMed.
- H. Li, R. Lan, C.-F. Chan, L. Jiang, L. Dai, D. W. J. Kwong, M. H.-W. Lam and K.-L. Wong, Chem. Commun., 2015, 51, 14022–14025 Search PubMed.
- B. K. McMahon, R. Pal and D. Parker, Chem. Commun., 2013, 49, 5363–5366 Search PubMed.
- D. G. Smith, B. K. McMahon, R. Pal and D. Parker, Chem. Commun., 2012, 48, 8520–8523 Search PubMed.
- S. J. A. Pope and R. H. Laye, Dalton Trans., 2006, 3108–3113 Search PubMed.
- A. C. Harnden, A. S. Batsanov and D. Parker, Chem. – Eur. J., 2019, 25, 6212–6225 Search PubMed.
- R. Pal, D. Parker and L. C. Costello, Org. Biomol. Chem., 2009, 7, 1525–1528 Search PubMed.
- R. S. Dickins, J. A. K. Howard, A. S. Batsanov, M. Botta, J. I. Bruce, C. S. Love, R. D. Peacock, H. Puschmann and D. Parker, J. Am. Chem. Soc., 2002, 124, 12697–12705 Search PubMed.
- D. G. Smith, R. Pal and D. Parker, Chem. – Eur. J., 2012, 18, 11604–11613 Search PubMed.
- Y. Bretonniere, M. J. Cann, D. Parker and R. Slater, Org. Biomol. Chem., 2004, 2, 1624–1632 Search PubMed.
- J. I. Bruce, R. S. Dickins, T. Gunnlaugsson, S. Lopinski, M. P. Lowe, R. D. Peacock, J. J. B. Perry, S. Aime, M. Botta and D. Parker, J. Am. Chem. Soc., 2000, 122, 9674–9684 Search PubMed.
- J. D. Fradgley, M. Starck, M. Laget, E. Bourrier, E. Dupuis, L. Lamarque, J. M. Zwier and D. Parker, Chem. Commun., 2021 10.1039/D1CC01029H.
- M. Liu, Z. Ye, C. Xin and J. Yuan, Anal. Chim. Acta, 2013, 761, 149–156 Search PubMed.
- M. T. Berry, P. S. May and H. Xu, J. Phys. Chem., 1996, 100, 9216–9222 Search PubMed.
- R. A. Poole, F. Kielar, S. L. Richardson, P. A. Stenson and D. Parker, Chem. Commun., 2006, 4084–4086 Search PubMed.
- J. A. Sobrinho, G. A. B. Junior, I. O. Mazali and F. A. Sigoli, New J. Chem., 2020, 44, 8068–8075 Search PubMed.
- Y. Ning, Y.-W. Liu, Y.-S. Meng and J.-L. Zhang, Inorg. Chem., 2018, 57, 1332–1341 Search PubMed.
- Y. Liu, G. Qian, Z. Wang and M. Wang, Appl. Phys. Lett., 2005, 86, 071907 Search PubMed.
- D. A. Galico, I. O. Mazali and F. A. Sigoli, New J. Chem., 2018, 42, 18451–18459 Search PubMed.
- S. Blair, R. Kataky and D. Parker, New J. Chem., 2002, 26, 530–535 Search PubMed.
- Y. Amao, I. Okura and T. Miyashita, Bull. Chem. Soc. Jpn., 2000, 73, 2663–2668 Search PubMed.
- J. Yu, l Sun, H. Peng and M. I. J. Stich, J. Mater. Chem., 2018, 20, 6975–6981 Search PubMed.
- M. Delbianco, V. Sadovnikova, E. Bourrier, G. Mathis, L. Lamarque, J. M. Zwier and D. Parker, Angew. Chem., Int. Ed., 2014, 126, 10894–10898 Search PubMed.
- A. T. Frawley, R. Pal and D. Parker, Chem. Commun., 2016, 52, 13349–13352 Search PubMed.
- C. Cepeda, L. Raibaut, G. Fremy, S. V. Eliseeva, A. Romieu, J. Pécaut, D. Boturyn, S. Petoud and O. Sénèque, Chem. – Eur. J., 2020, 59, 13476–13483 Search PubMed.
- M. Isaac, L. Raibaut, C. Cepeda, A. Roux, D. Boturyn, S. V. Eliseeva, S. Petoud and O. Sénèque, Chem. – Eur. J., 2017, 23, 10992–10996 Search PubMed.
| This journal is © The Royal Society of Chemistry 2021 |