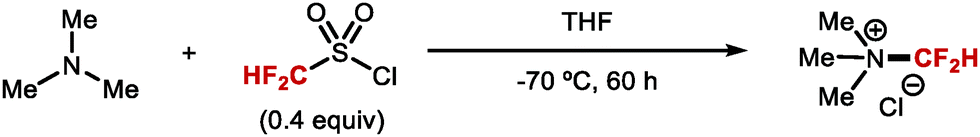
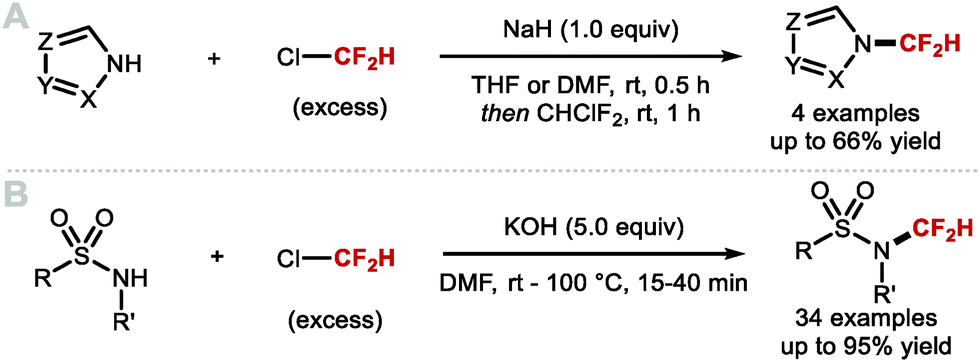
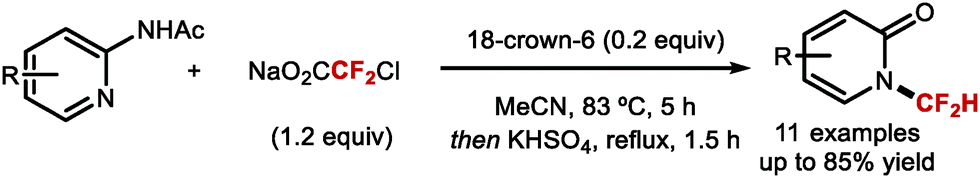
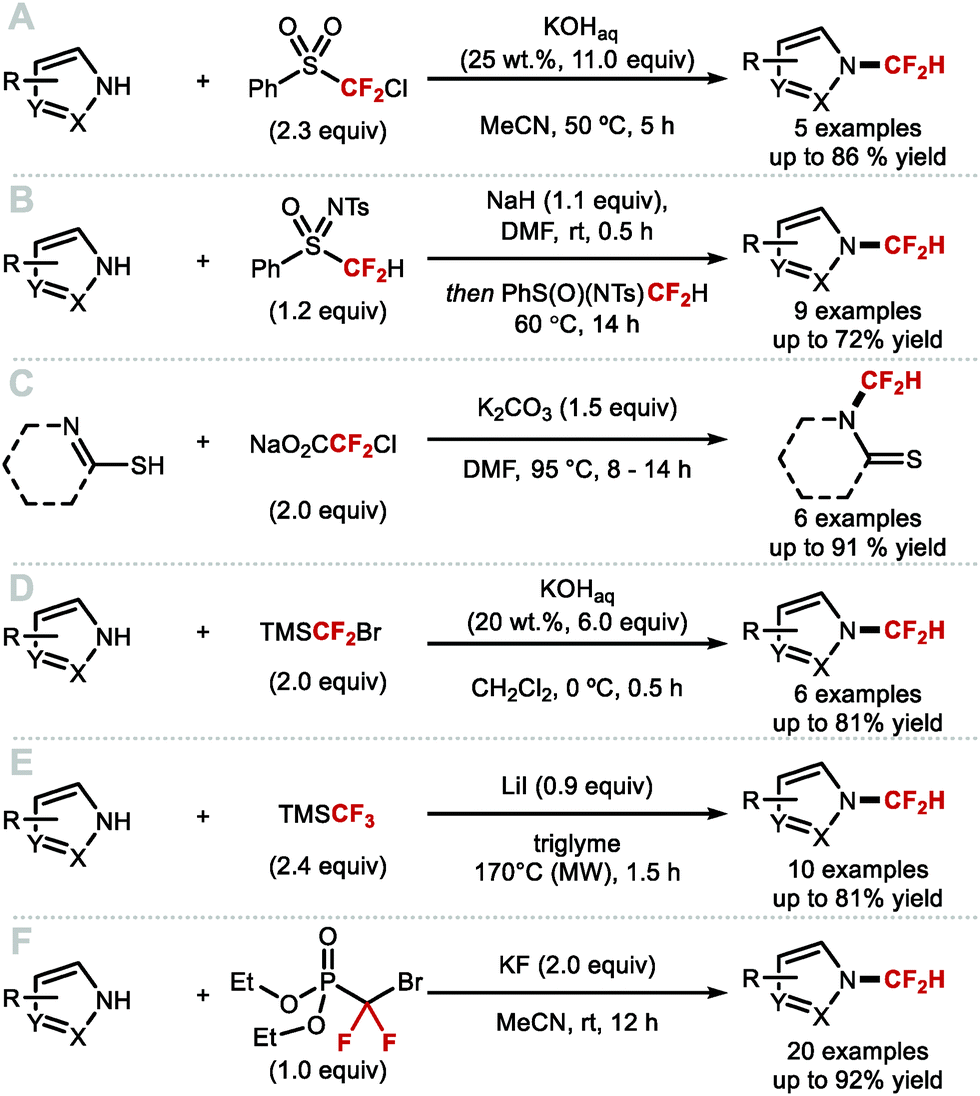
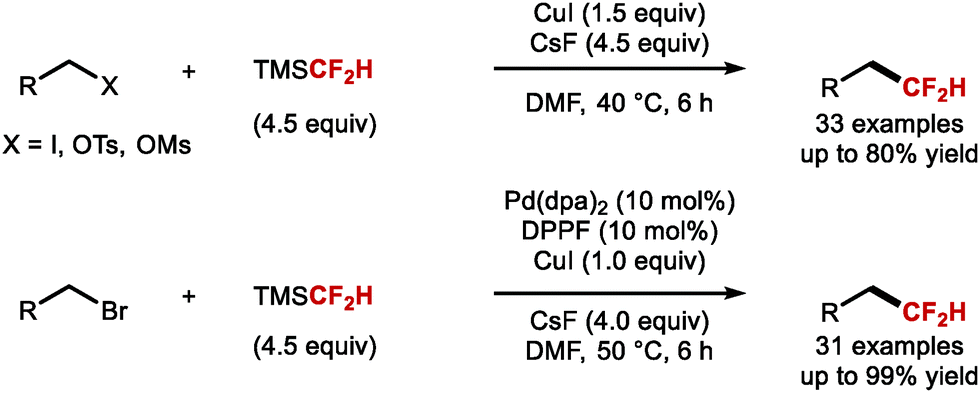DOI:
10.1039/D1CS00360G
(Review Article)
Chem. Soc. Rev., 2021,
50, 8214-8247
Late-stage difluoromethylation: concepts, developments and perspective
Received
13th April 2021
First published on 2nd June 2021
Abstract
This review describes the recent advances made in difluoromethylation processes based on X–CF2H bond formation where X is C(sp), C(sp2), C(sp3), O, N or S, a field of research that has benefited from the invention of multiple difluoromethylation reagents. The last decade has witnessed an upsurge of metal-based methods that can transfer CF2H to C(sp2) sites both in stoichiometric and catalytic mode. Difluoromethylation of C(sp2)–H bond has also been accomplished through Minisci-type radical chemistry, a strategy best applied to heteroaromatics. Examples of electrophilic, nucleophilic, radical and cross-coupling methods have appeared to construct C(sp3)–CF2H bonds, but cases of stereoselective difluoromethylation are still limited. In this sub-field, an exciting departure is the precise site-selective installation of CF2H onto large biomolecules such as proteins. The formation of X–CF2H bond where X is oxygen, nitrogen or sulfur is conventionally achieved upon reaction with ClCF2H; more recently, numerous protocols have achieved X–H insertion with novel non-ozone depleting difluorocarbene reagents. All together, these advances have streamlined access to molecules of pharmaceutical relevance, and generated interest for process chemistry.

Jeroen B. I. Sap
| Dr Jeroen Sap received his MSci degree from Imperial College London, where he developed development of dual-modal (MRI/optical) imaging probes for apoptosis in the group of Prof. Ramón Vilar. He moved to the University of Oxford as a DPhil student under the supervision of Prof. Véronique Gouverneur working on late-stage functionalisation and F-18 radiolabelling in collaboration with Pfizer. He obtained his DPhil in 2020, and is currently an EPSRC Doctoral Prize Fellow in the Gouverneur group. |

Claudio F. Meyer
| Dr Claudio Meyer received his MSc degree from the University of Basel in Switzerland, after working on the synthesis of mTOR inhibitors with Prof. Matthias Wymann in collaboration with PIQUR Therapeutics AG. He then moved to Oxford to work with Prof. Véronique Gouverneur, where he secured his DPhil working on novel 18F-fluoroalkylation and sulfonylation reactions in collaboration with Janssen. |

Natan J. W. Straathof
| Dr Natan Straathof completed in 2009 his MSc degree in chemistry at the University of Leiden to under the supervision of Prof. Hermen Overkleeft and Prof. Gijs van der Marel (Netherlands). He moved to Eindhoven University of Technology for his doctorate under the supervision of Prof. Timothy Noël and Prof. Volker Hessel. He then joined the University of Oxford to work under the supervision of Prof. Véronique Gouverneur on the development of novel 18F PET tracers and automation in collaboration with UCB and Trasis (Belgium). |

Ndidi Iwumene
| Ndidi Iwumene received her MSc degree from Imperial College London, where she worked in the group of Prof. Anthony Barrett on the syntheses of meroterpenoids, using Pd-catalysed decarboxylative allyl migration reactions. She moved to the University of Oxford for her DPhil, where she works on Rh-catalysed hydroacylation reactions under the supervision of Prof. Michael Willis. |

Christopher W. am Ende
| Dr Christopher W. am Ende is an Associate Research Fellow at Pfizer Inc. where he leads a team focused on chemical biology and drug discovery. He obtained his BS in Biochemistry from the University of Delaware, conducting research with Neal J. Zondlo developing lanthanide-binding peptides. During graduate studies, he worked with Peter J. Tonge designing slow, tight binding inhibitors of InhA, the enoyl reductase from M. tuberculosis and then with Kathlyn A. Parker, where he completed the first total synthesis of the natural product bisabosqual A. Chris also serves on the steering committee for the New York Academy of Sciences Chemical Biology Discussion Group, is an adjunct instructor of chemistry at Connecticut College, and has published more than 60 journal articles and patents. |

Andrés A. Trabanco
| Dr Andrés A. Trabanco received his PhD degree from Universidad de Oviedo-Spain in 1999. He then moved to Imperial College London for his postdoctoral studies. He joined the Neuroscience Med Chem Team at Janssen in 2000. He is currently a Scientific Director and has been a chemistry leader and an active team member in several programs within the areas of schizophrenia, depression, anxiety, and cognition/Alzheimer's disease, which have delivered various clinical candidates. He is an inventor of 59 patents/patent applications, and author of over 85 scientific publications. |

Véronique Gouverneur
| Prof. Véronique Gouverneur FRS obtained a PhD in chemistry at the Université Catholique de Louvain (Belgium). In 1992, she moved to a postdoctoral position at the Scripps Research Institute (California, USA). After four years as Maître de Conférence at the University Louis Pasteur in Strasbourg (France), she started her independent research career at the University of Oxford (UK) in the chemistry department and was promoted to Professor in 2008. Since her appointment in Oxford, she holds a tutorial fellowship at Merton College Oxford where she teaches organic chemistry. Her research on fluorine (radio)chemistry has been disseminated in more than 200 publications and awarded numerous prizes and distinctions. |
1. Introduction
It is widely recognised that the introduction of one or more fluorine atoms into molecules can have a significant impact on their physicochemical and biological properties.1,2 A key advantage of fluorinated motifs is their ability to mimic functional groups widespread in biologically active molecules.3 The C–F bond is strong with a bond dissociation energy (BDE) of up to 130 kcal mol−1, and is intermediate in length between C–H and C–O bonds albeit closer to the C–O bond (C–H 1.09 Å, C–F 1.35 Å, C–O 1.43 Å).4,5 The C–F bond is also highly polarised with its stability deriving from an electrostatic Cδ+Fδ− component. As a result, incorporation of fluorine can serve the purpose to improve metabolic robustness as well as modulate cellular membrane permeability.6 Today, the inclusion of one or more fluorine atoms into biologically active compounds has become a common strategy for the design of pharmaceutical drugs and agrochemicals.
Over the past decade, fluorination chemistry has focused on carbon–fluorine (C–F) bond formation,7,8 as well as the introduction of perfluoroalkyl (CnFm) group including numerous studies on trifluoromethylation (CF3).9–17 In this review, we discuss the recent advances made in the field of difluoromethylation, where we formulate the key challenges, solutions, and future directions related to X–CF2H bond formation where X is C(sp3), C(sp2), C(sp), O, N or S. A brief historical overview of key discoveries and developments is provided followed by a discussion of the unique properties of CF2H. Strategies applied to access difluoromethylarenes (ArCF2H) as well as methods leading to C(sp3)–CF2H bond formation are described next. The discussion then focuses on difluorocarbene reagents for the construction of X–CF2H (where X = C, N, O, or S) bonds via X–H insertion, and on strategies to incorporate CF2H onto alkenes and alkynes. Finally, we discuss the application of these technologies in an industrial context.
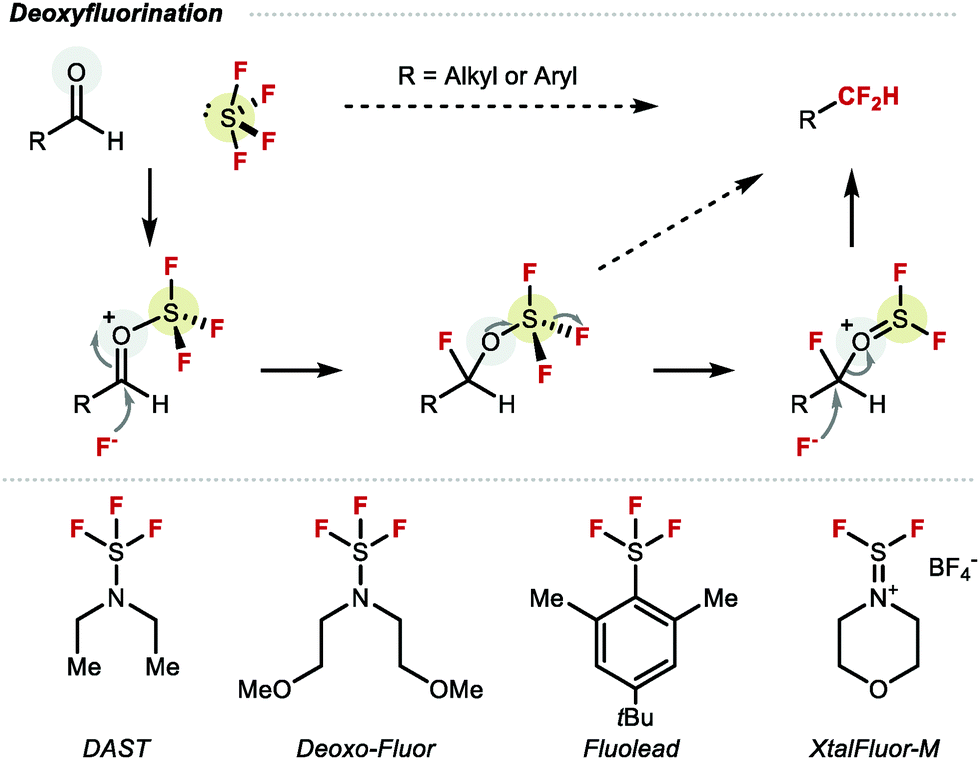
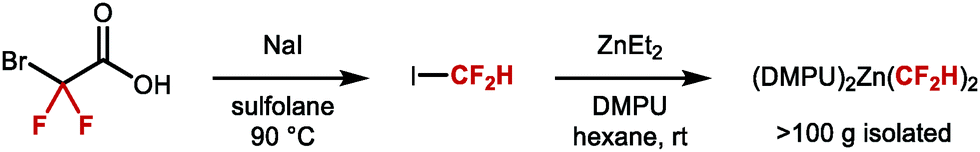
An early example of a molecule featuring CF2H was disclosed by Swarts in the early 20th century, in the form of chlorodifluoromethane (ClCF2H), also known as Freon-22.18 This compound was primarily used as refrigerant, industrial cooling agent, fire repellent as well as a difluoromethylation reagent. However, the realisation that ClCF2H and other chlorofluorocarbons CFCs contribute to ozone depletion led to their phasing out. In 1987, the Montreal protocol agreement imposed a permanent ban of CFCs on a global scale, thereby encouraging the chemistry community to develop alternative perfluoroalkylation reagents, including those required for difluoromethylation.19 For many years, the most widely adopted reaction to construct the CF2H motif was the deoxyfluorination of aldehydes (Scheme 1).20–22 Many reagents were developed for this transformation such as N,N-diethylaminosulfur trifluoride (DAST) or bis(2-methoxyethyl)aminosulfur trifluoride (Deoxo-Fluor®), all derived from gaseous sulfur tetrafluoride (SF4). While these reagents have provided access to a variety of CF2H containing compounds, limitations include scope, scalability, explosivity, and toxicity.23 An important advance was the development of the bench stable crystalline solid XtalFluor-M®.24 Nonetheless, the incompatibility of these reagents with some key functional groups such as unprotected alcohols, carbonyls or carboxylic acids, along with the necessity to pre-install the necessary aldehyde functionality, remains restrictive.25 As a result, new technologies which exploit easy-to-install reactive handles such as halide or boron motifs, or direct C–H difluoromethylation are attractive alternatives, and are discussed in this review. Undoubtedly, these advances have benefited from the invention of a multitude of bespoke difluoromethylation reagents. Approaches towards RCF2H based on carbon–fluorine bond formation or hydrodefluorination are not described in this review.26–30
 |
| | Scheme 1 Deoxyfluorination of the aldehyde functional group. | |
1.1 Properties of the CF2H group
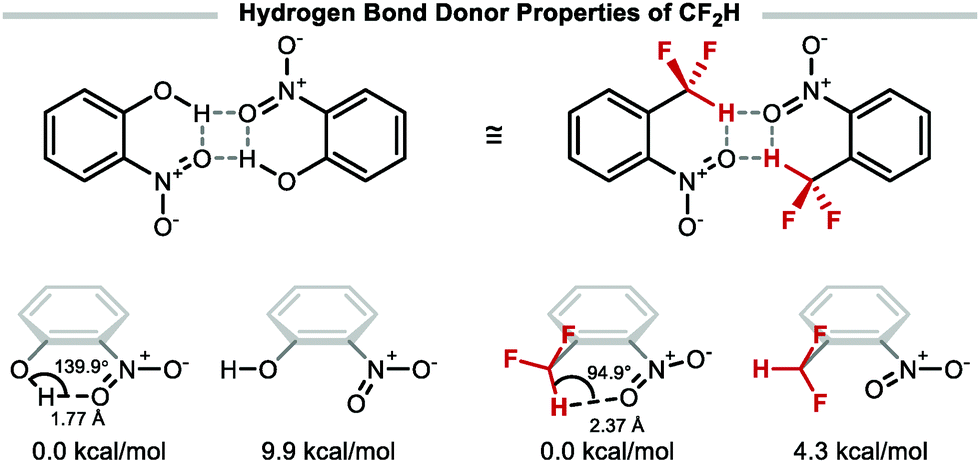
The highly polarised C–H bond of CF2H makes this group a competent hydrogen bond donor, a unique characteristic amongst polyfluorinated motifs.31 The suitability of CF2H as a bioisostere for alcohol, thiol, or amine group, has resulted in its incorporation in numerous bioactive compounds including drugs, herbicides, fungicides, and agrochemicals.2 Recent studies have shown that 1-(difluoromethyl)-2-nitrobenzene can form a dimeric complex similar to the hydrogen bonded dimer of 2-nitrophenol (Fig. 1).32 The conformer enabling intramolecular hydrogen bonding interaction is 4.3 kcal mol−1 lower in energy with respect to the conformer lacking such interaction. Comparatively, the stabilisation gained from intramolecular hydrogen bonding of o-nitrophenol is 9.9 kcal mol−1.
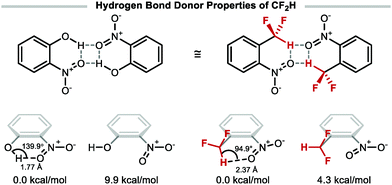 |
| | Fig. 1 Hydrogen bond donor properties of the difluoromethyl group. | |
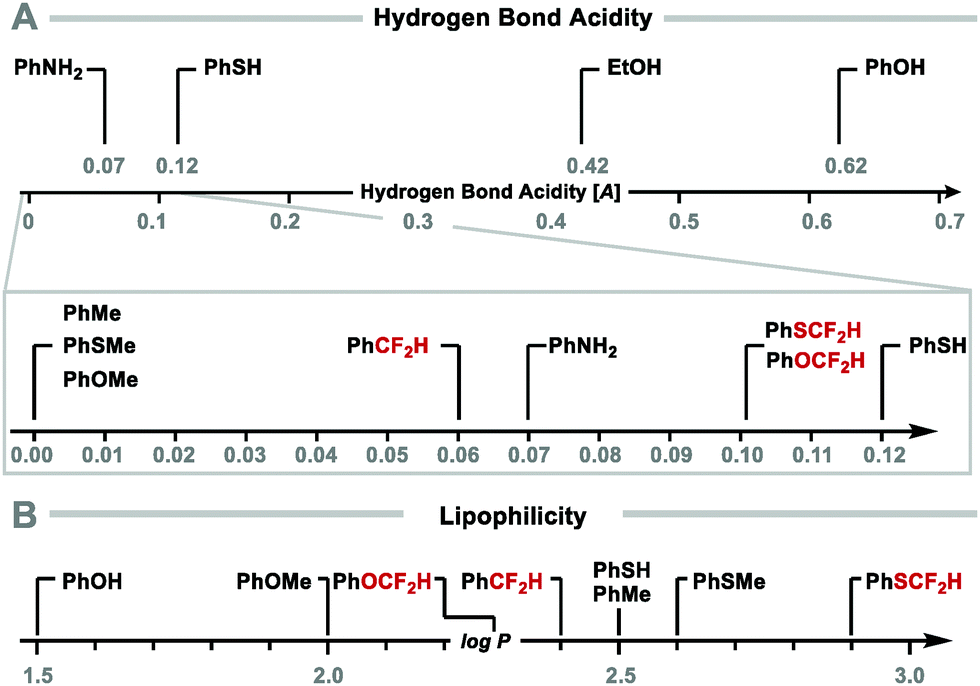
The ability of the difluoromethyl group to form hydrogen bonds has been quantified as [A], a parameter defining hydrogen bond acidity.33,34 The [A] value is derived from Abraham's solute 1H NMR analysis, whereby the chemical shift of the CF2H-proton is measured in both deuterated DMSO and CDCl3. The difference in chemical shift (Δδ = δ(DMSO-d6) − δ(CDCl3)) correlates with a molecule's ability to act as a hydrogen-bond donor, where [A] = 0.0065 + 0.133Δδ. These studies revealed that compounds bearing a CF2H group (A > 0.05) are better hydrogen-bond donors than their methylated analogues (A < 0.01). Interestingly, ArOCF2H and ArSCF2H (A = 0.10) have a similar hydrogen bond acidity to thiophenol (A = 0.12) and aniline (A = 0.07) (Fig. 2A).
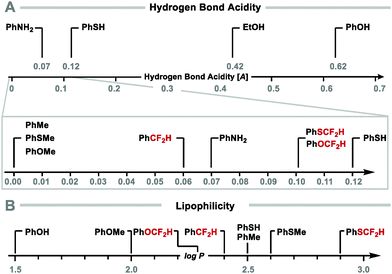 |
| | Fig. 2 (A) Hydrogen bond acidity of difluoromethyl compounds. (B) Lipophilicity of difluoromethyl compounds. | |
CF2H substitution can also modulate lipophilicity with difluoromethylbenzene (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P = 2.4) being more lipophilic than phenol (logP = 1.5). Similarly, difluoromethyl phenyl sulfide (A = 0.10; logP = 2.9) is more lipophilic than the weak hydrogen bond donor thiophenol (A = 0.12; logP = 2.5) (Fig. 2B).34 Studies on the effect of fluorine substitution on ADME properties (absorption, distribution, metabolism, and excretion) in a set of lead-like drug structures containing anisoles informed that ArOCF2H compounds tend to display decreased lipophilicity (>0.5 reduction in logD), yet higher permeability compared to ArOCF3 analogues.35
P = 2.4) being more lipophilic than phenol (logP = 1.5). Similarly, difluoromethyl phenyl sulfide (A = 0.10; logP = 2.9) is more lipophilic than the weak hydrogen bond donor thiophenol (A = 0.12; logP = 2.5) (Fig. 2B).34 Studies on the effect of fluorine substitution on ADME properties (absorption, distribution, metabolism, and excretion) in a set of lead-like drug structures containing anisoles informed that ArOCF2H compounds tend to display decreased lipophilicity (>0.5 reduction in logD), yet higher permeability compared to ArOCF3 analogues.35
Variable fluorine substitution can furthermore influence conformation. Whilst the O–CH3 bond in ArOCH3 generally adopts a coplanar structure, the O–CF3 bond in ArOCF3 preferentially adopts an orthogonal orientation to the aromatic plane. The preference of OCH3 to be coplanar with the phenyl ring stems from the conjugation of the oxygen lone pairs with the aromatic π system. This conformation is ∼3.0 kcal mol−1 lower in energy than the conformer displaying the O–CH3 bond orthogonal to the aromatic ring (Fig. 3). In contrast, ArOCF3 compounds adopt a conformation whereby the C–F bonds are out of plane due to both steric and stereoelectronic effects. This out-of-plane conformation results in anomeric nO–σ*C–F stabilisation which reduces the conjugation between the O-lone pair and the aromatic π-system, thereby eliminating the preference for a coplanar conformation. In this series, ArOCF2H compounds stand out as they have no orientational preference for either conformation (dihedral angle (θ) = 0°–50°). Difluoromethyl ethers can therefore adopt the conformation that enables optimal binding to a target protein.1,35
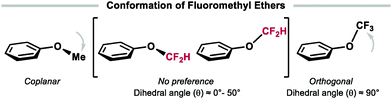 |
| | Fig. 3 Conformational preference of fluorinated ethers. | |
2. C(sp2)–CF2H bond formation for arenes and heteroarenes
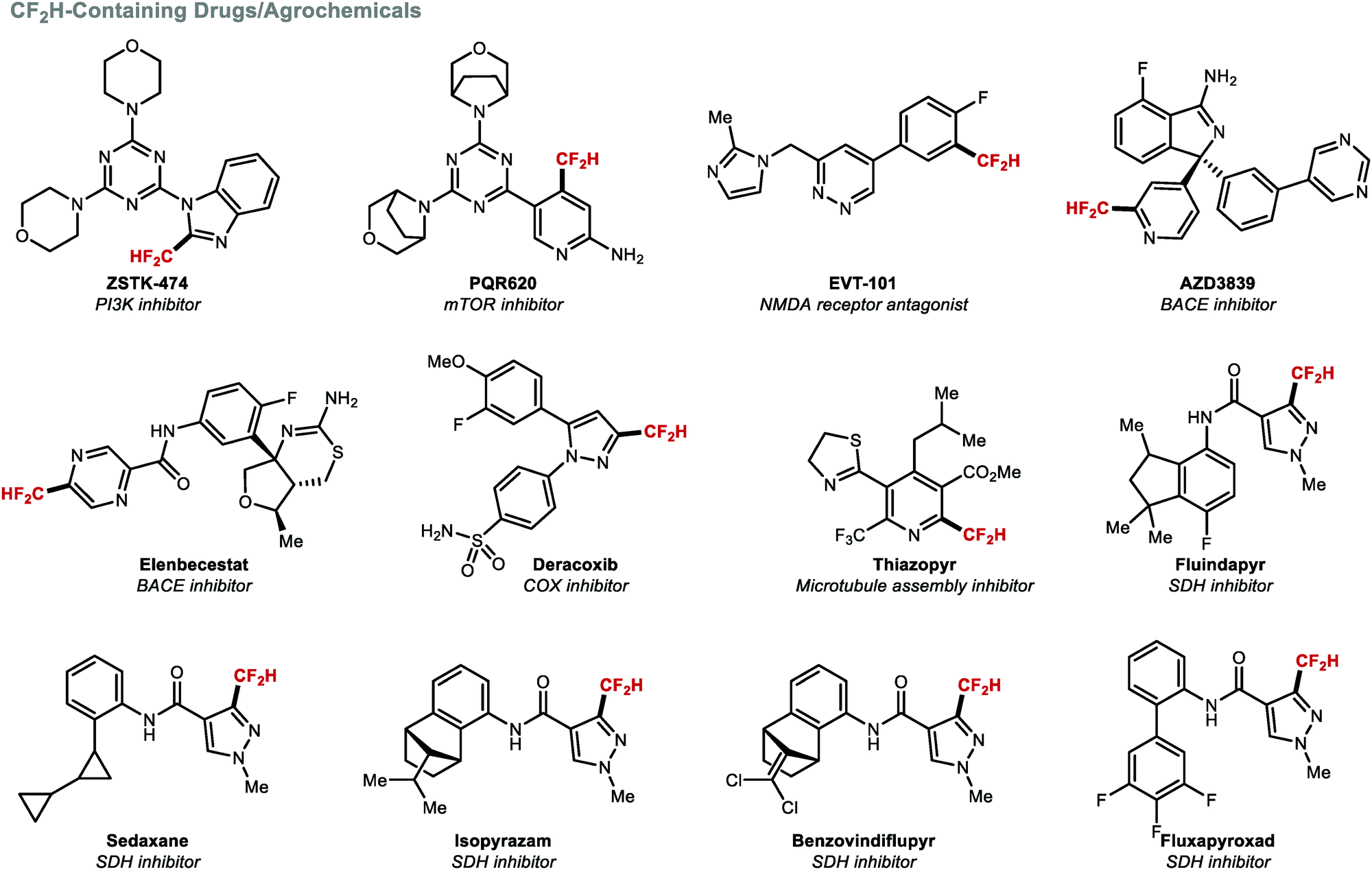
The incorporation of a CF2H group into (hetero)arenes is one of the most investigated difluoromethylation transformations because multiple drug candidates feature such structural motif (Fig. 4). The introduction of the CF2H group can lead to increased potency and/or selectivity. For example, Wymann and co-workers demonstrated that the CF2H group in the mTORC1/2 selective inhibitor PQR620 played a vital role in achieving >1000-fold mTOR selectivity over PI3Kα. Computational modelling studies suggested that the CF2H group forms a beneficial hydrogen bonding interaction with Glu2190 of mTOR.36
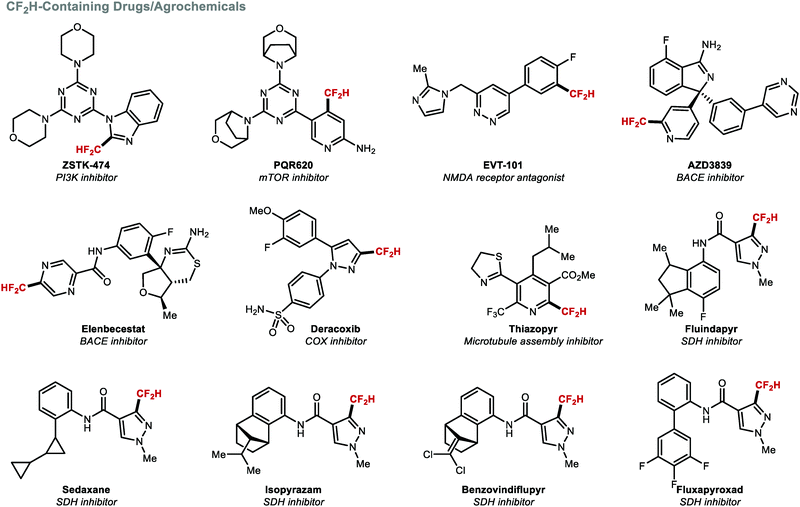 |
| | Fig. 4 Biologically active compounds containing a (hetero)arene–CF2H bond. | |
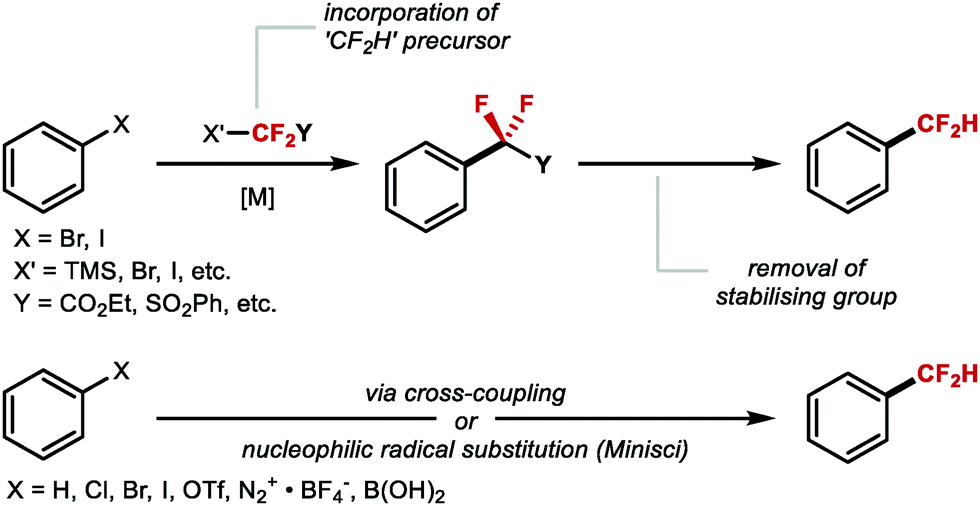
ArCF2H are accessible applying either a stepwise sequence or directly from a CF2H reagent. For the former, different reagents which transfer a CF2Y motif (with Y being a stabilising electron-withdrawing group) can be introduced under transition metal catalysis, more often copper. Reagents featuring the CF2Y motif include BrCF2CO2Et, FSO2CF2CO2H, TMSCF2SO2Ph, BrCF2SO2Ph, BrCF2P(O)(OEt)2, ICF2SO2Ph and TMSCF2CO2Et. After attachment of CF2Y, the stabilising ester, sulfone or phosphonate “Y” group is cleaved to generate CF2H.37–42 Alternatively, the introduction of a CF2H group can be achieved via direct cross-coupling facilitated by a transition metal or through Minisci-type radical chemistry using a suitable difluoromethylation reagent. In the context of late-stage functionalisation (LSF), direct methods are the most attractive (Scheme 2).
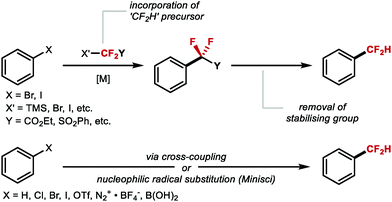 |
| | Scheme 2 Metal-mediated stepwise difluoromethylation reactions. (A) Stepwise route. (B) Direct route. | |
2.1 C(sp2)–CF2H bond formation: cross-coupling
In 1988 and 1990, Burton reported the synthesis of difluoromethyl cadmium and its reactivity with allylic halides and propargylic (pseudo)halides to afford allylic difluoromethyl products and difluoromethyl allenes, respectively.43,44 In 2007, he reported that difluoromethyl copper also permits difluoromethylation of allylic halides, propargylic derivatives and 1-iodoalkynes.45 A cross-coupling mechanism was proposed whereby oxidative addition into the difluoromethyl copper complex affords a CuIII intermediate which underwent reductive elimination to afford the difluoromethylated products. Cross-coupling reactions involving a difluoromethyl group and an aryl electrophile or nucleophile were not investigated in these seminal reports.
2.1.a Copper-mediated C(sp2)–CF2H bond formation.
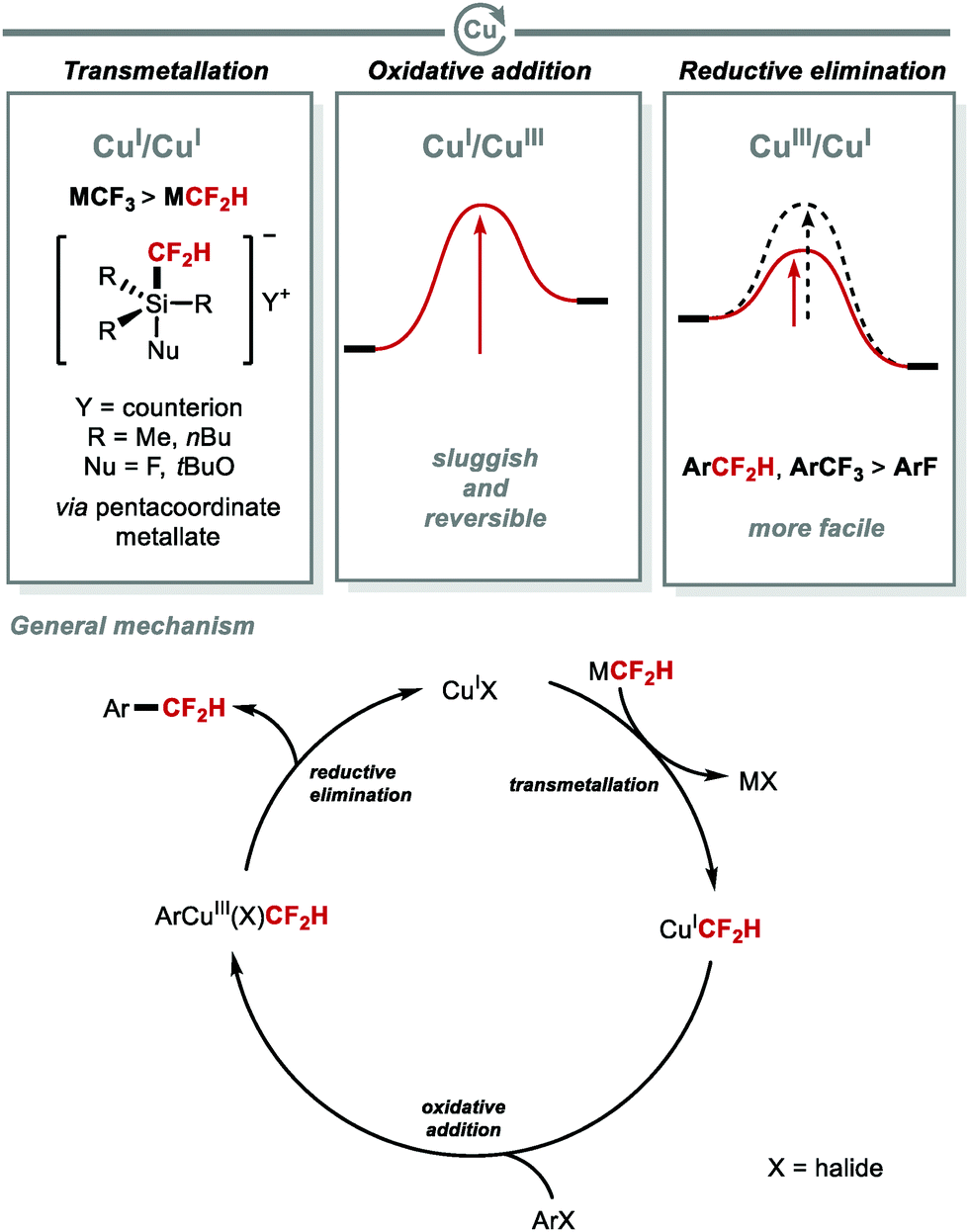
Copper-catalysed C(sp2)–CF2H bond formation is a demanding process (Fig. 5). Unlike thermally stable CuCF3 complexes, the first isolable CuCF2H complex was not reported until 2017, and required stabilisation from an NHC ligand (IPr) to enable its isolation.46 Mechanistically, the high energy barrier associated with oxidative addition to CuI is one of the factors that renders copper cross-coupling reactions involving CF2H challenging. Also, transmetallation with M–CF2H (M = SnR3, SiR3) is less effective than with M–CF3 because CF3 is more electronegative than CF2H. As a result, the formation of the pentacoordinate metallate necessary for transmetallation occurs more easily for M–CF3 than M–CF2H. Despite these challenges, copper has the advantage to undergo facile reductive elimination from high-valent CuIII species. Notably, reductive elimination of Ar–CF2H from CuIII occurs under milder conditions than for Ar–F.
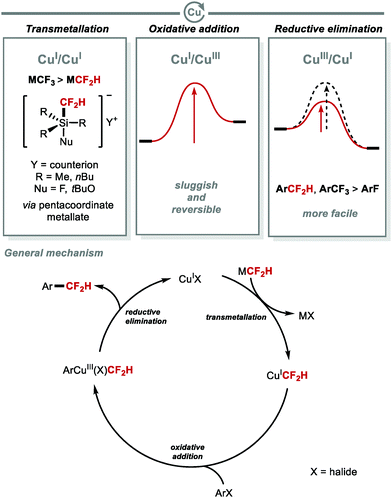 |
| | Fig. 5 Copper-mediated C(sp2)–CF2H bond formation. | |
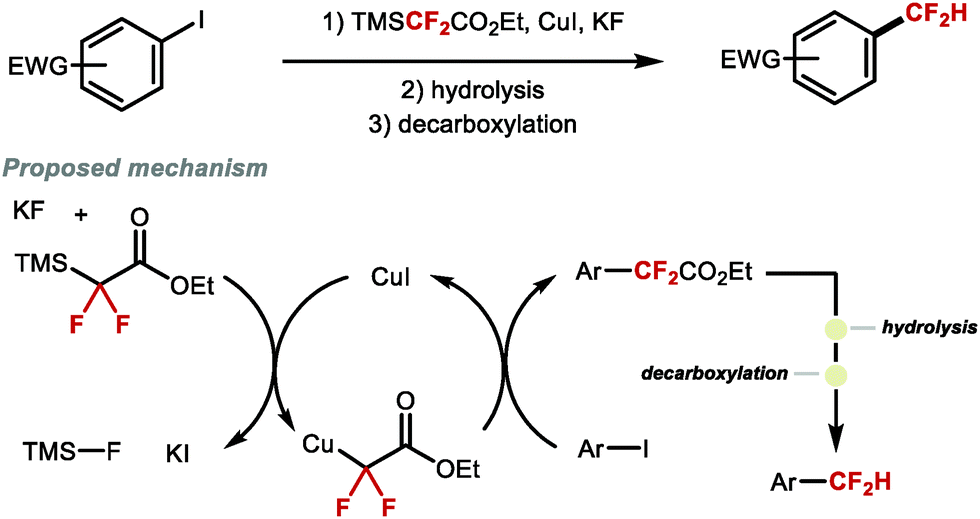
Prior to the development of direct methods for Cu-mediated Ar–CF2H bond formation, Amii and co-workers conceived a stepwise strategy to access difluoromethylated arenes (Scheme 3).37 In this early study, the coupling of aryl iodides with ethyl 2,2-difluoro-2-(trimethylsilyl)acetate gave access to α-aryl-α,α-difluoroacetates. In subsequent steps, the resulting ester was hydrolysed followed by decarboxylation at high temperature to yield the desired difluoromethylarene. The decarboxylation step was successful only for intermediates derived from electron-deficient iodoarenes or iodopyridine. This three-step-one-pot protocol marked the first general route to access ArCF2H from aryl halides.
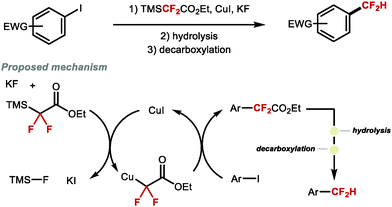 |
| | Scheme 3 Three-step sequence towards difluoromethylarenes from aryl iodides. | |
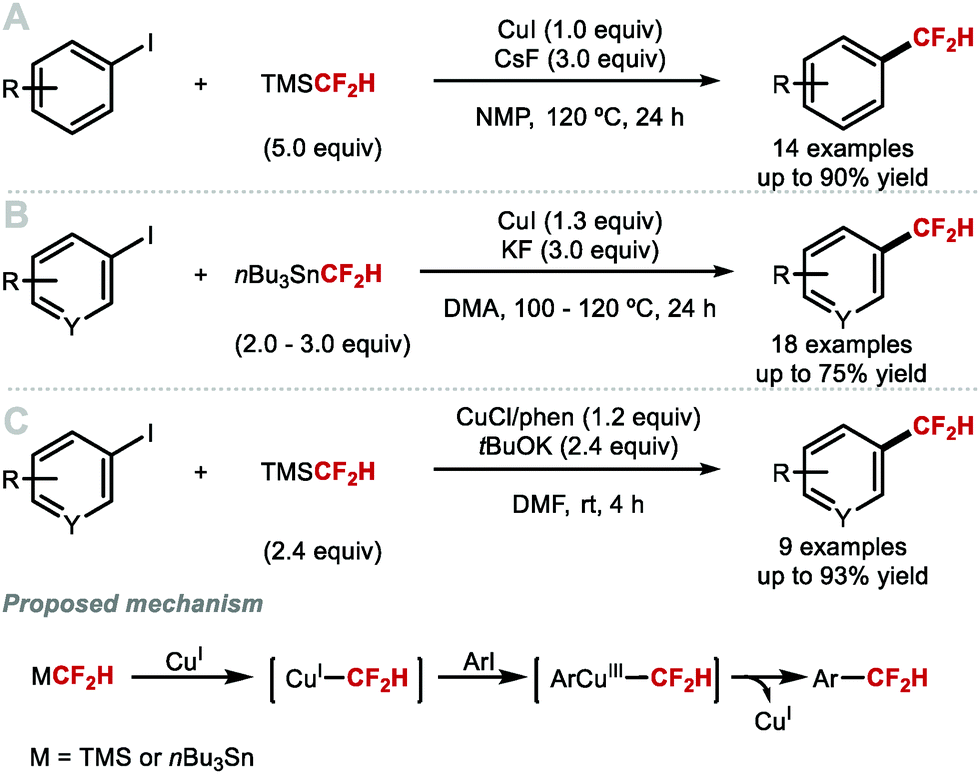
Hartwig and co-workers reported in 2012 the first direct copper-mediated difluoromethylation of aryl iodides using CuI, CsF and commercially available TMSCF2H (Scheme 4A).47 The key discovery of this study was the necessity to use an excess of TMSCF2H (5.0 equiv.) to convert unstable [CuCF2H] into the more stable disubstituted [Cu(CF2H)2]− cuprate complex. Electron-neutral, electron-rich, and sterically hindered aryl iodides readily underwent difluoromethylation in good yields. Electron-deficient aryl iodides did not perform well because competing protodeiodination took place. Ketone and aldehyde functional groups were not tolerated due to competing addition of CF2H onto the carbonyl group. This contribution nevertheless sparked the development of alternative Cu-mediated reactions to facilitate C(sp2)–CF2H bond formation in a direct fashion. In the same year, Prakash and co-workers demonstrated that nBu3SnCF2H can be utilised as a CF2H source to functionalise (hetero)aryl iodides using stoichiometric copper iodide (Scheme 4B).48 In contrast to Hartwig's method, iodoarenes functionalised with electron-withdrawing functional groups were readily difluoromethylated. Notably, whilst Hartwig's method suffered from competing nucleophilic addition to carbonyl groups, Prakash and co-workers did not observe these side-products under their reaction conditions. In 2014, Qing and co-workers reported that modifications of Hartwig's procedure including the use of a more soluble base (tBuOK) and the introduction of phenanthroline ligand, allowed for electron-poor aryl iodides and a selection of heteroaryl iodides to undergo difluoromethylation at ambient temperature (Scheme 4C).49
 |
| | Scheme 4 Copper-mediated difluoromethylation of aryl iodides. Phen = phenanthroline. | |
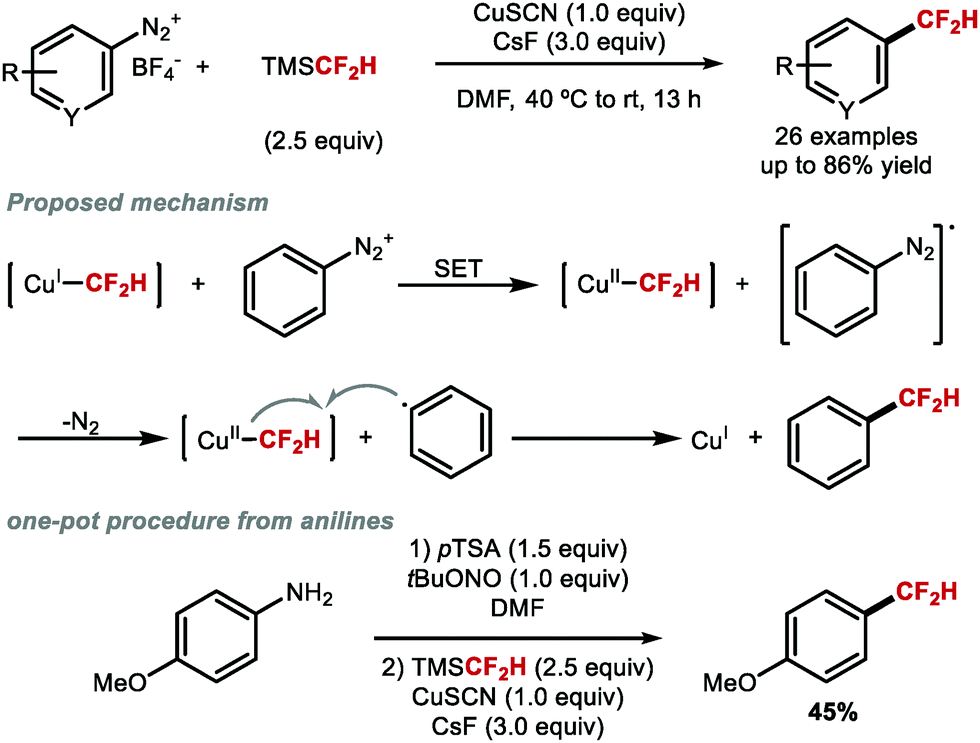
In the same year, Gooßen and co-workers illustrated that difluoromethylation of (hetero)aryl diazonium salts was possible using TMSCF2H and CuSCN (Scheme 5).50 Radical inhibition and trapping experiments suggested that this reaction likely proceeds through a radical pathway involving first transmetallation of the CF2H group at CuI followed by single electron transfer (SET) generating a CuII species and an aryl radical. Outer sphere transfer of CF2H to the aryl radical yielded the desired difluoromethylarene. This scenario is distinct from Hartwig,47 Prakash48 and Qing's49 methods, where transmetallation is proposed to occur at CuI, followed by oxidative addition of an aryl iodide. Gooßen and co-workers also developed a one-pot sequence from aniline precursors via in situ generation of the corresponding diazonium salts.
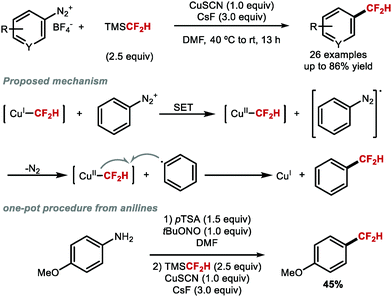 |
| | Scheme 5 Copper-mediated difluoromethylation of aryl diazonium salts. | |
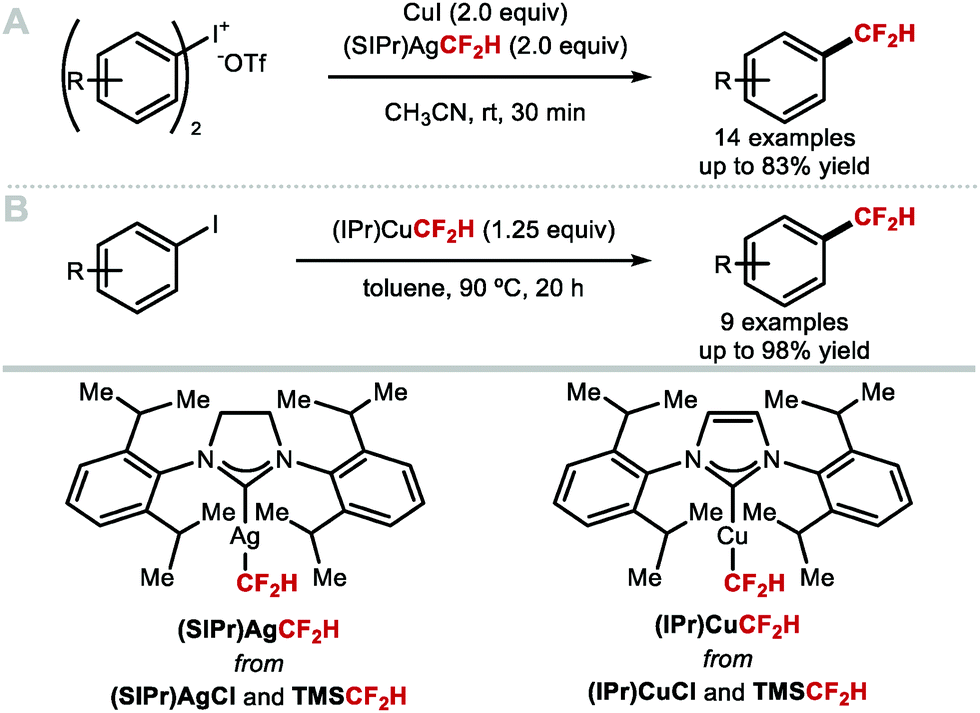
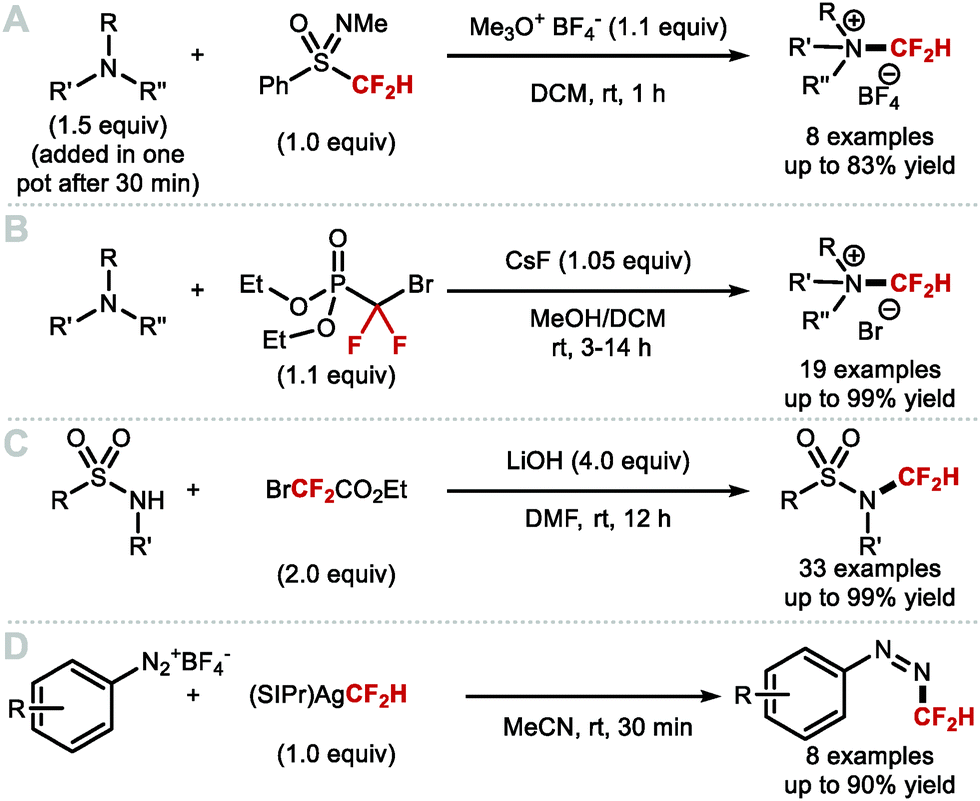
Shen and co-workers investigated alternative approaches to generate [CuCF2H] in situ (Scheme 6A).51 Specifically, the N-heterocyclic carbene (NHC) silver complex [(SIPr)Ag(CF2H)] (SIPr = 1,3-bis(2,6-diisopropylphenyl)imidazolin-2-ylidene) prepared from [(SIPr)AgCl] is air and moisture stable, and allowed in situ generation of LnCuCF2H upon treatment with CuI. This strategy led to rapid formation of difluoromethylarenes from diaryliodonium triflates at room temperature. Substrates bearing electron-donating and electron-withdrawing substituents were tolerated and afforded the desired products in good yields. Inspired by this work, Sanford and co-workers developed a protocol towards an NHC–copper–CF2H complex which alleviated the need for a bimetallic system (Scheme 6B).46 Similarly to [(SIPr)Ag(CF2H)], [(IPr)Cu(CF2H)] was prepared from [(IPr)CuCl] and TMSCF2H, and found sufficiently stable for isolation. Using a stoichiometric amount of [(IPr)Cu(CF2H)], several electron-deficient aryl iodides readily underwent difluoromethylation. Electron-rich substrates were also suitable but led to difluoromethylated products in lower yields.
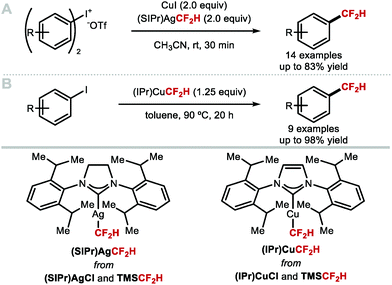 |
| | Scheme 6 (NHC)MCF2H complexes for the difluoromethylation of diaryl iodonium salts and aryl iodides. | |
2.1.b C(sp2)–CF2H bond formation under copper catalysis.
Mikami and co-workers reported the first Cu-catalysed difluoromethylation of aryl iodides using [(DMPU)2Zn(CF2H)2], a reagent developed by Vicic and co-workers (Scheme 7A).52,53 This CF2H reagent is prepared from commercially available difluoroiodomethane and diethylzinc, in the presence of two equivalents of DMPU. Transmetallation from the zinc reagent to the copper catalyst (CuI) at room temperature generates the [Cu(CF2H)2]− complex previously proposed by Hartwig.47 Under these conditions, a variety of electron-deficient aryl iodides afforded the desired difluoromethylated products in moderate to excellent yields.53 Similarly, Sanford's report on the synthesis of [(IPr)Cu(CF2H)], illustrates that the same cuprate complex can be generated in situ using catalytic amounts of [(IPr)CuCl], and TMSCF2H (Scheme 7B).46 Under these conditions, electron-rich substrates performed well, whilst electron-deficient substrates performed better under the stoichiometric conditions described in Scheme 6B.
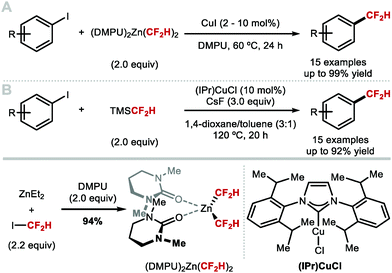 |
| | Scheme 7 Copper-catalysed difluoromethylation of aryl iodides. | |
2.1.c C(sp2)–CF2H bond formation under palladium catalysis.
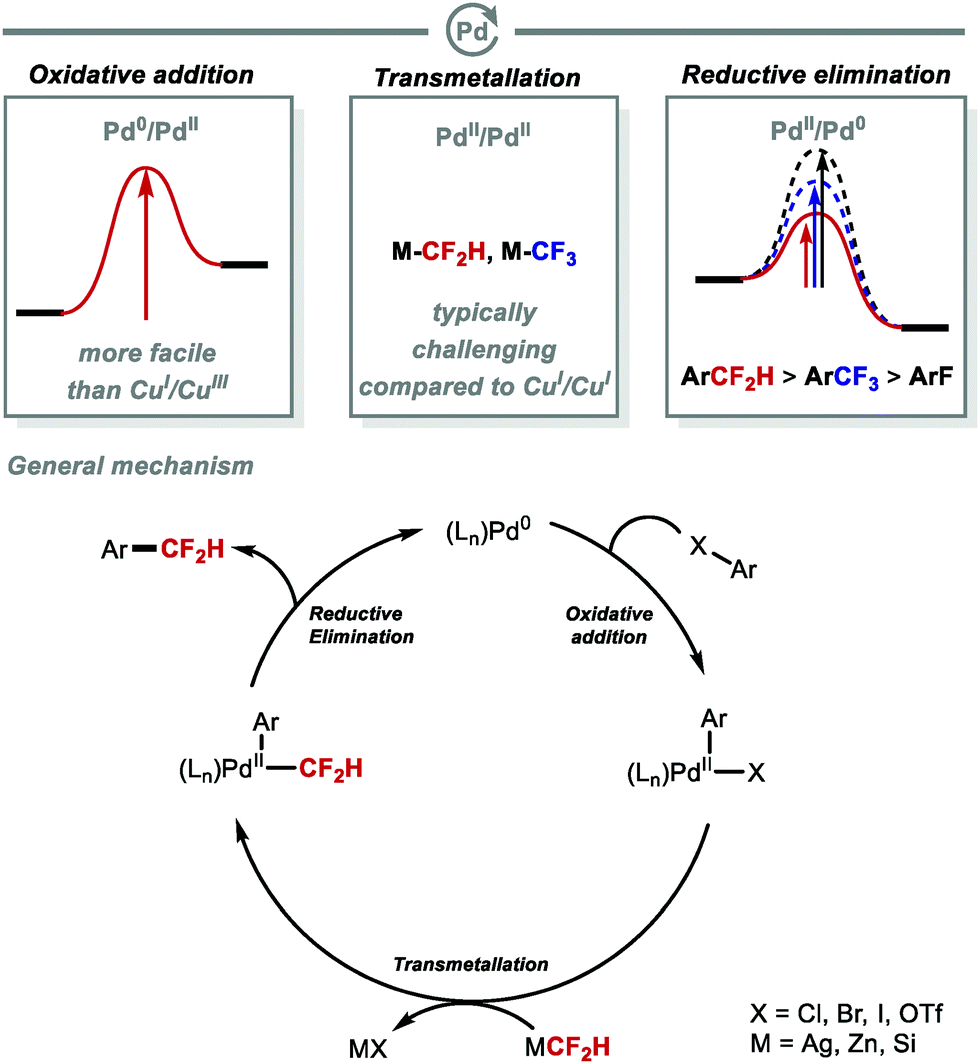
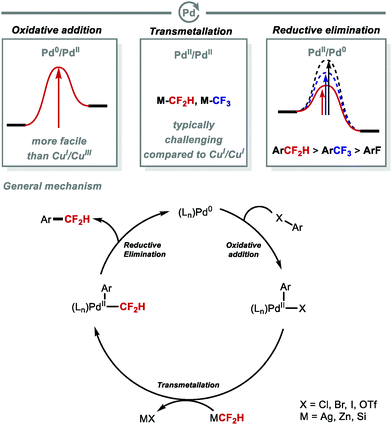
The oxidative addition of aryl halides occurs more readily at Pd0 than CuI. However, a challenge associated with Pd lies in the slower transmetallation to transfer CF2H onto PdIIversus CuI. These challenges are comparable to those associated with transmetallation of CF3 at PdII. Shen and co-workers reported that reductive elimination of Ar–CF2H at PdII is more facile than for Ar–CF3 (Fig. 6).54–57
 |
| | Fig. 6 C(sp2)–CF2H bond formation under palladium catalysis. | |
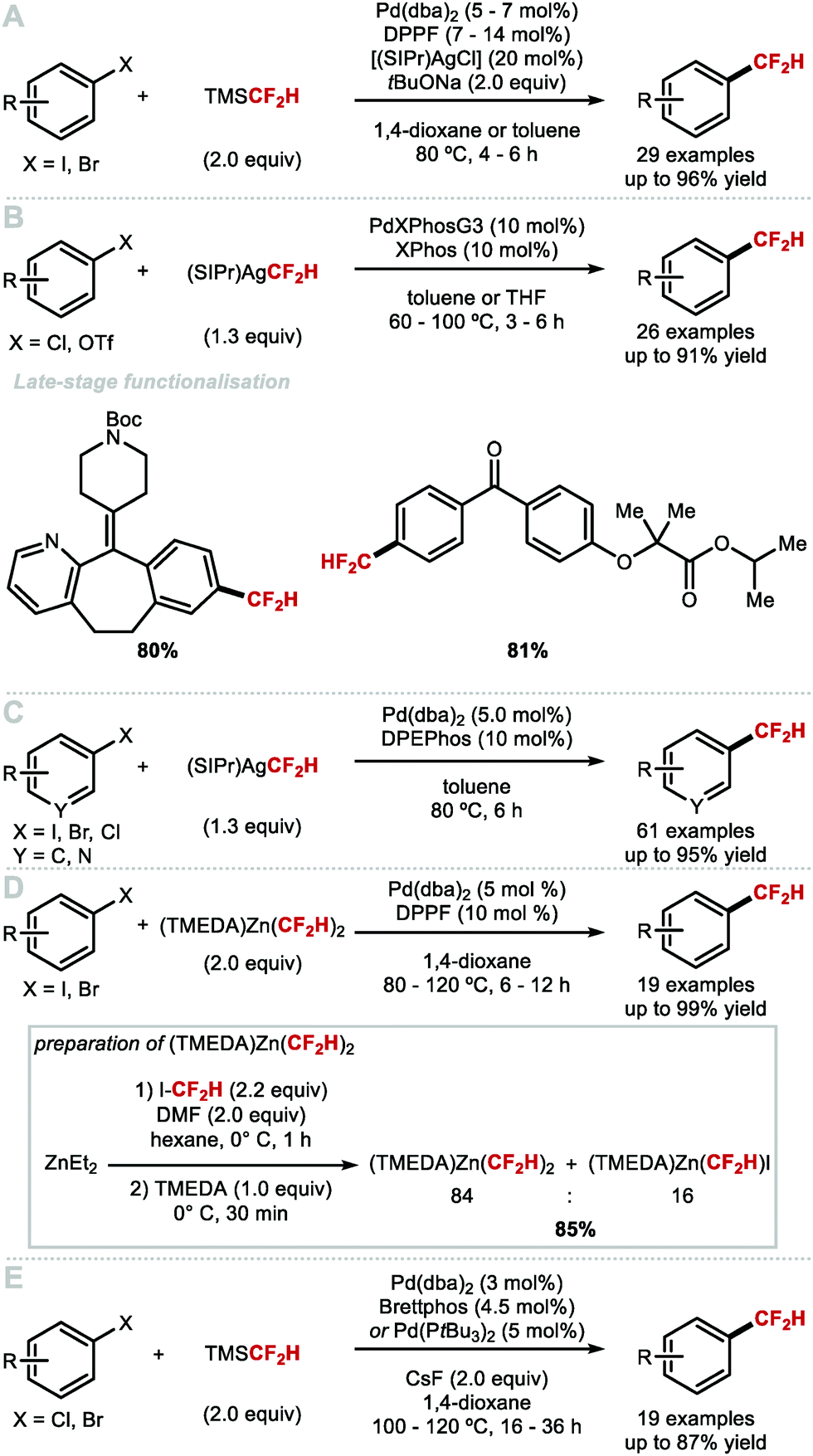
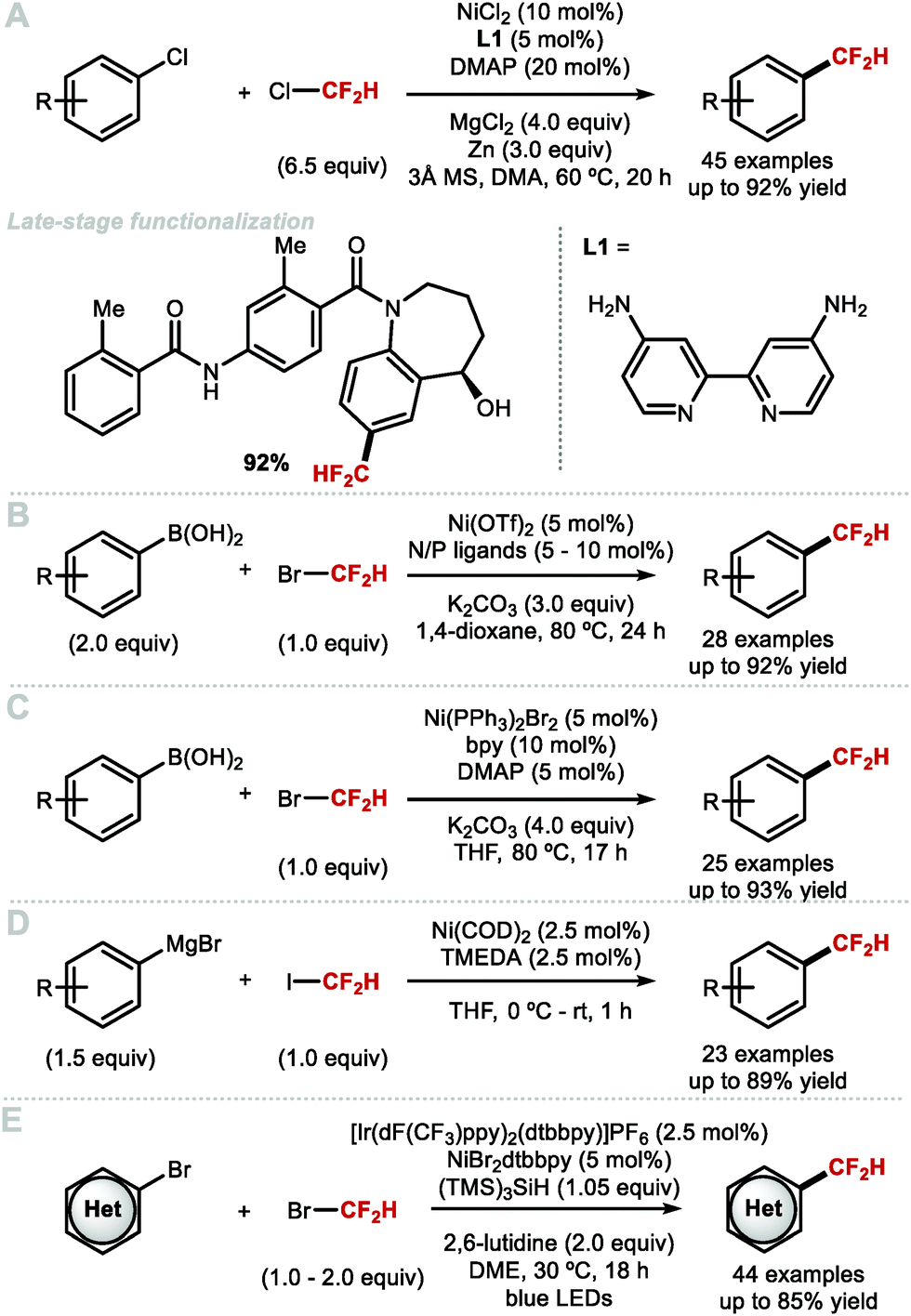
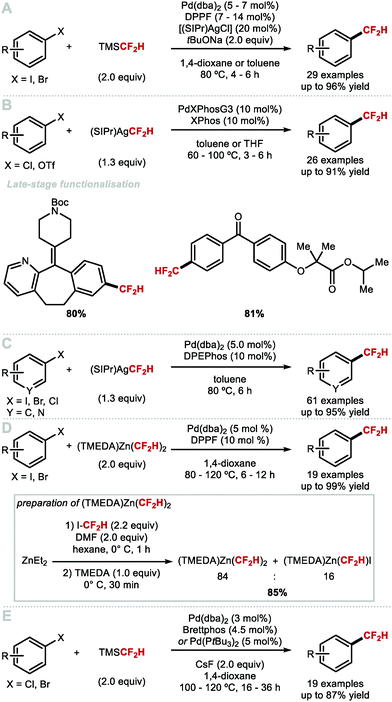
To overcome the challenges of transmetallation for CF2H at PdII, Shen and co-workers developed an effective cooperative Pd/Ag catalytic system for the synthesis of difluoromethylarenes from aryl bromides or aryl iodides in the presence of TMSCF2H (Scheme 8A).57 The data imply that the in situ generated [(SIPr)AgCF2H] complex acts as a transmetallation shuttle in this system. The use of stoichiometric pre-formed [(SIPr)Ag(CF2H)] in combination with the electron-rich and sterically hindered Buchwald catalyst PdXPhosG3 (10 mol%) in the presence of XPhos (10 mol%), allowed for a broader selection of aryl chlorides and triflates to undergo difluoromethylation in high yields (Scheme 8B).58 A selection of functionalised molecules were reacted in high yields, giving medicinal chemists a new tool to access CF2H-containing drug-like molecules in a late-stage fashion. The same authors further illustrated that (hetero)aryl chlorides are amenable to difluoromethylation in the presence of stoichiometric [(SIPr)Ag(CF2H)] and Pd(dba)2 (5 mol%) (Scheme 8C).59 This procedure provides a useful alternative to radical methods which are generally promiscuous with respect to regioselectivity (see Section 2.2). In addition to protocols which use AgI complexes to readily transfer CF2H to Pd, Mikami and co-workers reported a Pd-catalysed Negishi-type cross-coupling of aryl halides with [(TMEDA)Zn(CF2H)2] (Scheme 8D).60 This reagent was readily prepared from difluoroiodomethane and diethylzinc in presence of N,N,N′,N′-tetramethylethylenediamine (TMEDA). Electron-deficient and electron-rich (hetero)aryl halides were suitable substrates for this difluoromethylation protocol, affording the desired products in good to excellent yields. In 2019, Sanford and co-workers reported the Pd-catalysed difluoromethylation of aryl chlorides/bromides with TMSCF2H (Scheme 8E).61 The authors identified optimal catalysts, either Pd(dba)2/BrettPhos or Pd(PtBu3)2, and found that electron-neutral and electron-rich substrates performed well under the optimised reaction conditions. The authors postulated that this reaction operates under a Pd0/PdII catalytic cycle.
 |
| | Scheme 8 Palladium-catalysed difluoromethylation of aryl (pseudo)halides. | |
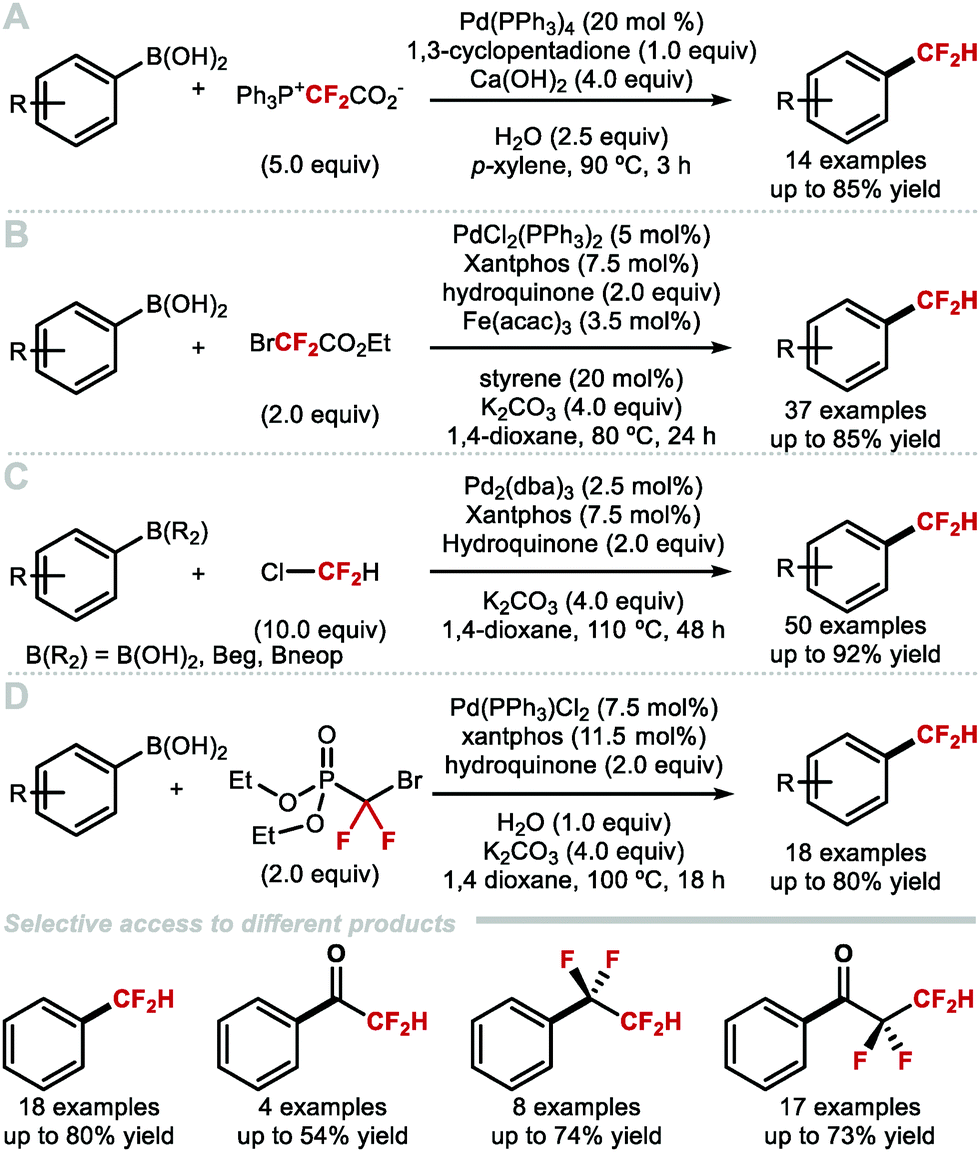
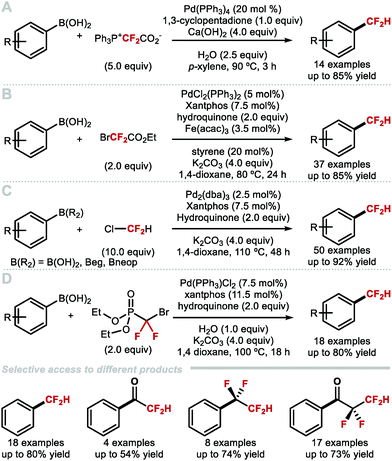
Difluorocarbene reagents can be used to generate MCF2H complexes upon protonation of in situ formed (difluorocarbene)metal ([M = CF2]) complexes. Several groups have exploited this strategy to convert aryl boron reagents to ArCF2H compounds under Pd-catalysis.62–65 Aryl boron reagents are advantageous candidates for difluoromethylation because they are often commercially available or easy to prepare. Furthermore, they are often bench-stable precursors, a desirable feature for cross-coupling reactions.66–68 Xiao and co-workers developed a Pd-catalysed difluoromethylation of aryl boronic acids with PDFA as a difluorocarbene source in 2016 (Scheme 9A).63 In their report the authors suggest that the Pd![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CF2 species could act as a difluorocarbene transfer reagent. Zhang and co-workers developed a Pd-catalysed difluoromethylation of aryl boronic acids using commercially available ethyl bromodifluoroacetate (Scheme 9b).62 This protocol has a broad substrate scope and functional group compatibility. Bioactive molecules such as flavanone-, ezetimibe- and estrone-derived aryl boronic acids all underwent difluoromethylation without the need to protect carbonyl or hydroxyl groups. Preliminary mechanistic studies demonstrated that a PdCF2 intermediate is generated in this reaction. In 2017, Zhang and co-workers reported the palladium-catalysed difluoromethylation of (hetero)arylboronic acids and esters using chlorodifluoromethane as a difluorocarbene source (Scheme 9C).64 The reaction proceeded smoothly for electron-rich and electron-deficient (hetero)arylboronic acids, tolerated a variety of functional groups, and allowed access to various bioactive CF2H-containing analogues. The need for an excess of ozone-depleting chlorodifluoromethane is a limitation of this technology. In 2019, Zhang and Houk developed a controllable Pd-catalysed difluorocarbene transfer reaction which employs aryl boronic acids and BrCF2P(O)(OEt)2 as alternative difluorocarbene precursor (Scheme 9D).65 Alterations of the reaction conditions afforded four distinct types of products, specifically difluoromethylated and tetrafluoroethylated arenes as well as the corresponding fluoroalkylated ketones.
CF2 species could act as a difluorocarbene transfer reagent. Zhang and co-workers developed a Pd-catalysed difluoromethylation of aryl boronic acids using commercially available ethyl bromodifluoroacetate (Scheme 9b).62 This protocol has a broad substrate scope and functional group compatibility. Bioactive molecules such as flavanone-, ezetimibe- and estrone-derived aryl boronic acids all underwent difluoromethylation without the need to protect carbonyl or hydroxyl groups. Preliminary mechanistic studies demonstrated that a PdCF2 intermediate is generated in this reaction. In 2017, Zhang and co-workers reported the palladium-catalysed difluoromethylation of (hetero)arylboronic acids and esters using chlorodifluoromethane as a difluorocarbene source (Scheme 9C).64 The reaction proceeded smoothly for electron-rich and electron-deficient (hetero)arylboronic acids, tolerated a variety of functional groups, and allowed access to various bioactive CF2H-containing analogues. The need for an excess of ozone-depleting chlorodifluoromethane is a limitation of this technology. In 2019, Zhang and Houk developed a controllable Pd-catalysed difluorocarbene transfer reaction which employs aryl boronic acids and BrCF2P(O)(OEt)2 as alternative difluorocarbene precursor (Scheme 9D).65 Alterations of the reaction conditions afforded four distinct types of products, specifically difluoromethylated and tetrafluoroethylated arenes as well as the corresponding fluoroalkylated ketones.
 |
| | Scheme 9 Pd-Catalysed difluoromethylation of aryl boron precursors via a difluorocarbene mechanism. | |
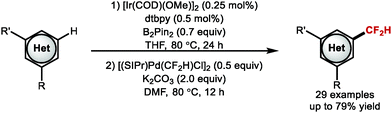
In 2020, Shen and co-workers developed a two-step one-pot C–H borylation–difluoromethylation protocol which allowed site-selective difluoromethylation of a range of (hetero)arenes (Scheme 10).69 The authors suggest a mechanism which involves concurrent ligand transfer that delivers both the CF2H group and NHC ligand from [(SIPr)Ag(CF2H)] to PdII, generating the [(SIPr)Pd(CF2H)Cl] complex in situ. From a medicinal chemist's perspective, this novel methodology represents an attractive strategy as it is suitable for the late-stage site-selective introduction of CF2H onto (hetero)arenes.
 |
| | Scheme 10 Two-step C–H borylation–difluoromethylation of arenes. | |
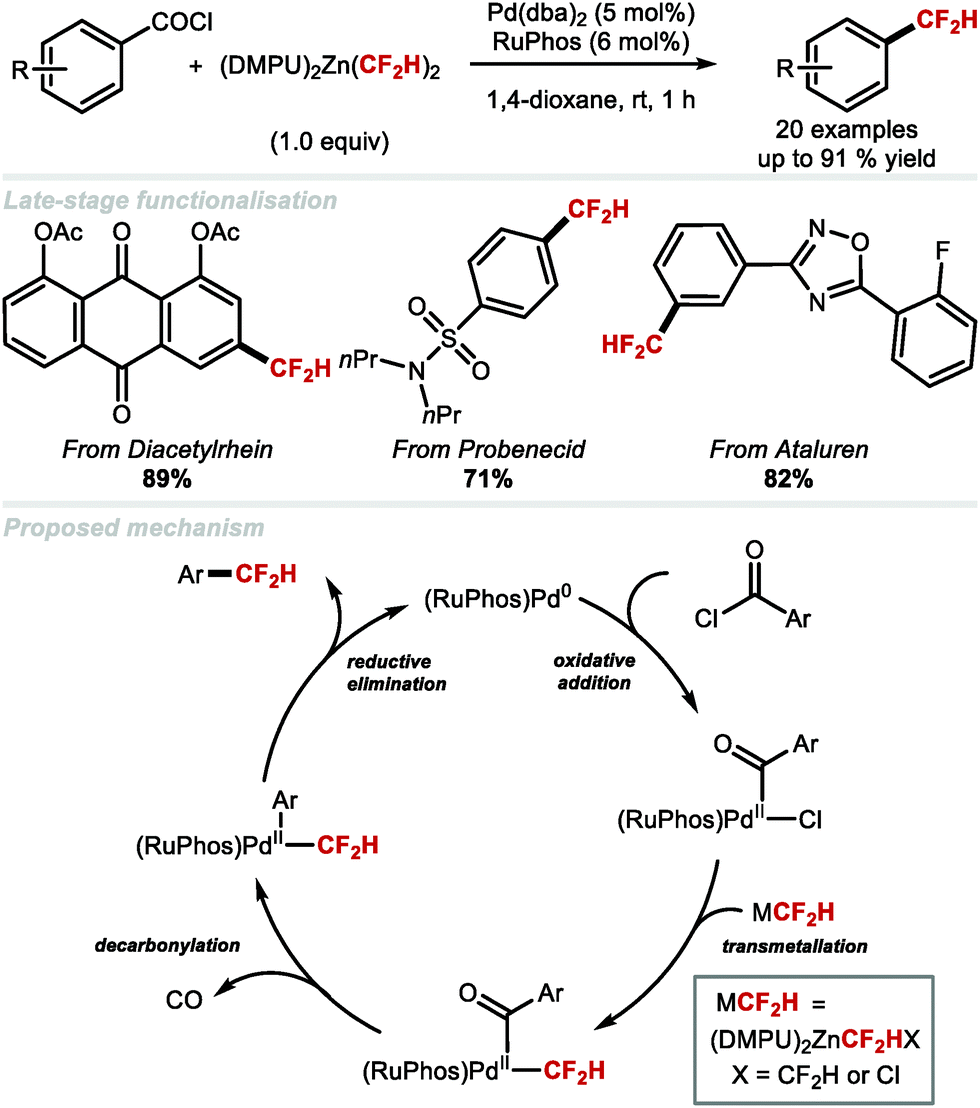
In 2018, the Ritter group reported the catalytic decarbonylative difluoromethylation of aroyl chlorides, a reaction requiring 5 mol% Pd(dba)2, 6 mol% RuPhos and 1 equivalent of [(DMPU)2Zn(CF2H)2] (Scheme 11).70 Electron-deficient, electron-rich and heterocycle-containing benzoyl chlorides all underwent difluoromethylation at ambient conditions. A range of bioactive compounds including diacetylrhein, probenecid and ataluren led to the desired products in good to excellent yields. The proposed catalytic cycle involves oxidative addition of the aroyl chloride to Pd0 which affords [(aroyl)PdII(Cl)], followed by CF2H transmetallation from (DMPU)2Zn(CF2H)2, an event leading to the PdII intermediate undergoing decarbonylation. Reductive elimination furnishes the desired difluoromethylarene with regeneration of the Pd0 catalyst.
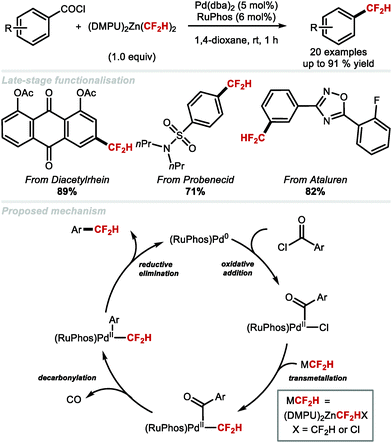 |
| | Scheme 11 Pd-Catalysed decarbonylative difluoromethylation of aroyl chlorides. | |
2.1.d C(sp2)–CF2H bond formation under nickel catalysis.
Nickel's open-shell electronic configurations (i.e. NiI, NiIII) display higher stability than its second and third period counterparts (Pd, Pt) (Fig. 7).71 As a result, the activation of an electrophile by Ni can occur through either a two-electron (oxidative addition) or via a single electron process. In addition, Ni0 has the ability to readily undergo oxidative addition to NiII, NiII transmetallation is facile, as is radical addition converting NiII to NiIII. Furthermore, rapid reductive elimination for Ar–CF2H bond formation from both NiII and NiIII makes nickel an attractive metal for arene functionalisation including difluoromethylation.
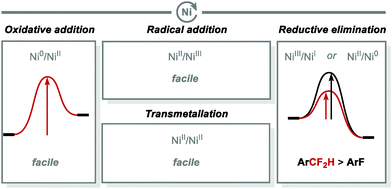 |
| | Fig. 7 C(sp2)–CF2H bond formation under nickel catalysis. | |
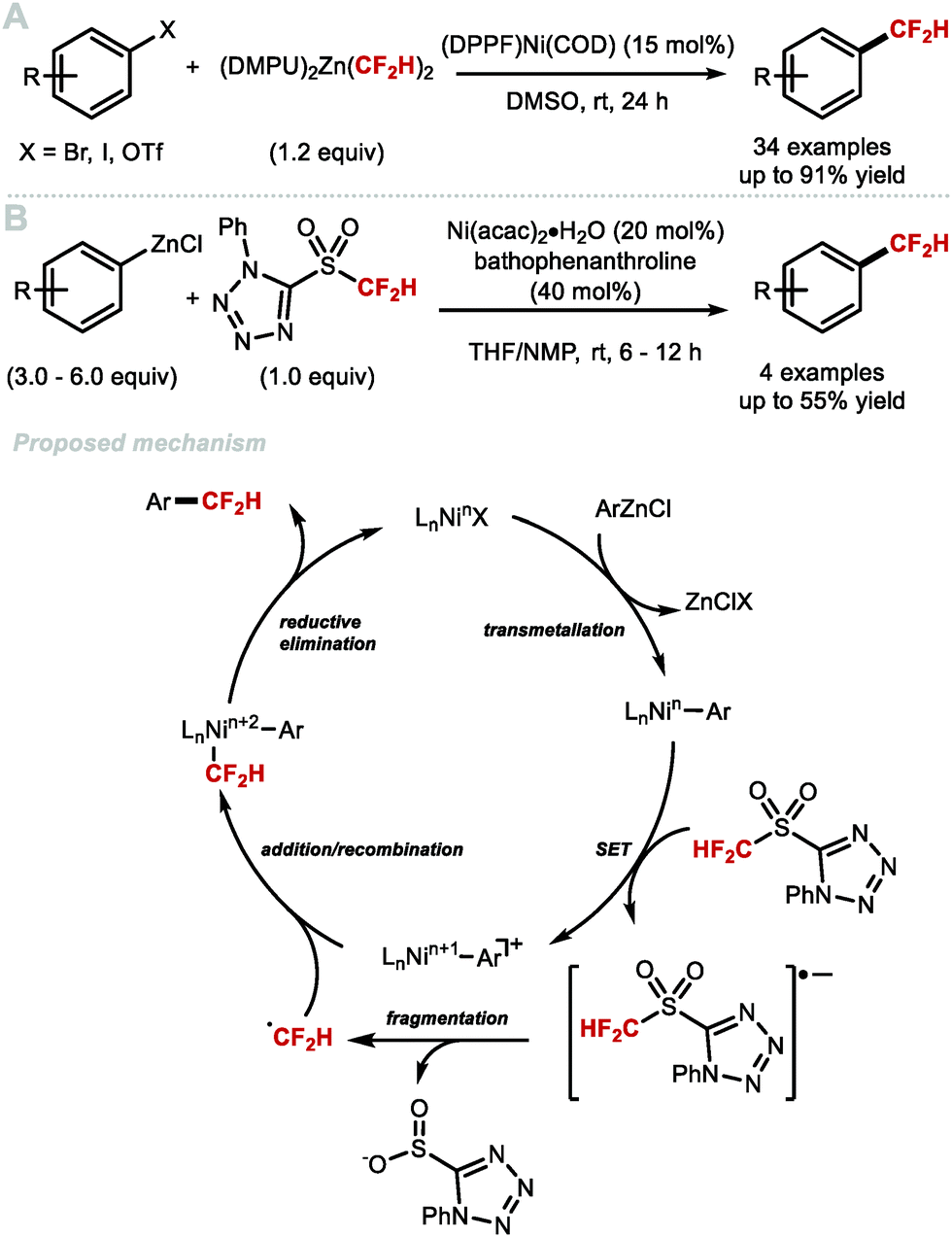
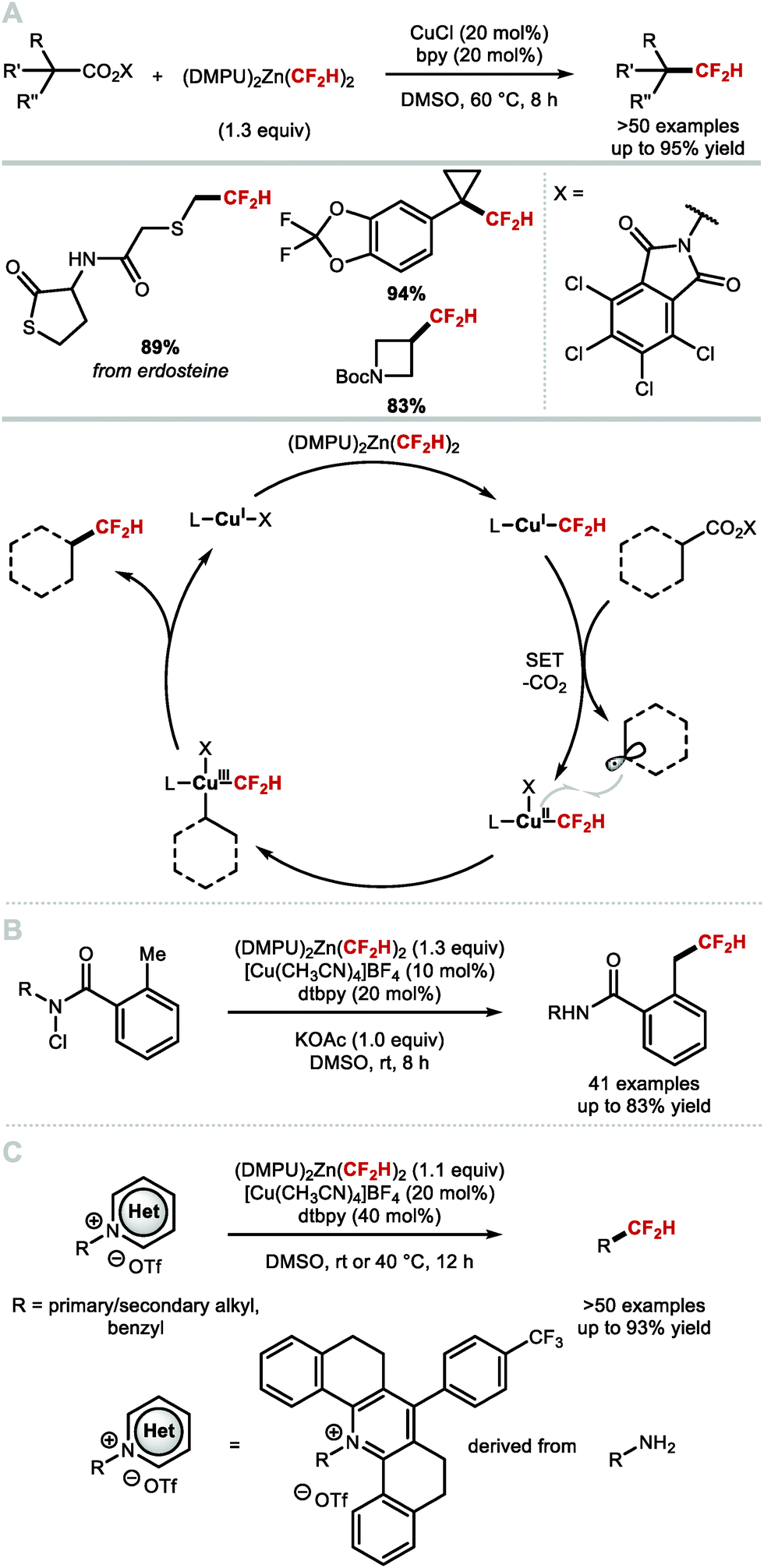
In 2016, Vicic and co-workers reported the nickel-catalysed difluoromethylation of aryl halides and triflates with [(DMPU)2Zn(CF2H)2 (Scheme 12A).52 [(DMPU)2Zn(CF2H)2] was found to be stable for long periods under an inert atmosphere and readily reacts with a variety of electron-deficient arenes in the presence of the nickel pre-catalyst [(DPPF)Ni(COD)]. Electron-rich substrates gave lower yields. The authors postulated that this Negishi-type cross-coupling operates under a traditional Ni0/NiII redox shuttle. Baran and co-workers reported the synthesis of difluoromethylated products using redox active 5-((difluoromethyl)sulfonyl)-1-phenyl-1H-tetrazole combined with Ni(acac)2·H2O and an excess of aryl zinc reagents (Scheme 12B).72 The authors postulate a mechanism whereby the aryl zinc reagent first undergoes transmetallation with nickel, yielding a nickel aryl species. This species can then reduce 5-((difluoromethyl)sulfonyl)-1-phenyl-1H-tetrazole, oxidizing the nickel aryl complex in the process. Fragmentation of the 5-((difluoromethyl)sulfonyl)-1-phenyl-1H-tetrazole radical anion yields the difluoromethyl radical which adds to [LnNin+1Ar]. The resulting [Ar–Nin+2–CF2H] complex undergoes reductive elimination to release the difluoromethylarene with regeneration of LnNinX.
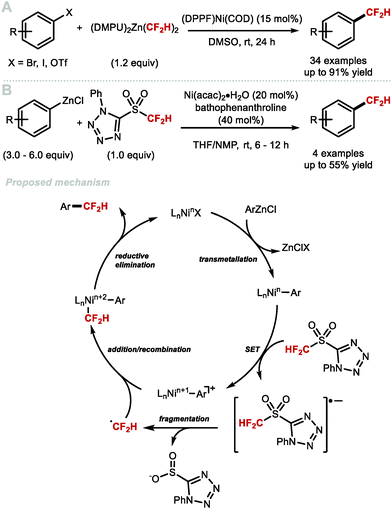 |
| | Scheme 12 Nickel-catalysed difluoromethylation of aryl (pseudo)halides and monoaryl zincates. DPPF = 1,1′-ferrocenediyl-bis(diphenylphosphine), COD = 1,5-cyclooctadiene, acac = acetyl acetonate. | |
Zhang and co-workers modified their Pd0-catalysed difluoromethylation of aryl chlorides with the development of a method that utilizes a NiII catalyst (Scheme 13A).73 The authors suggest a mechanism which commences with the reduction of the NiII catalyst to Ni0 in the presence of Zn0 serving as stoichiometric reductant. Subsequent oxidative addition of the aryl chloride, and reaction with an in situ generated CF2H radical yielded the Ar[NiIII]CF2H intermediate which readily underwent reductive elimination to furnish the difluoromethylarene product. The method is scalable (up to 10 g), and the mild reaction conditions are suitable for LSF of biologically relevant compounds. The requirement of an excess of ozone depleting ClCF2H is a disadvantage of this methodology. In 2018, Wang and Zhang simultaneously reported the nickel-catalysed difluoromethylation of arylboronic acids, with bromodifluoromethane. Wang and co-workers screened different combinations of phosphine and nitrogen ligands in a high-throughput fashion, with various iterations being necessary to optimise the reaction conditions found to be substrate-dependent (Scheme 13B).74 Zhang and co-workers reported that commercially available 2,2′-bipyridine in combination with [Ni(PPh3)2Br2] (5–10 mol%) was sufficient to access various electron-rich and electron-withdrawing difluoromethylarenes in good yields (Scheme 13C).75 Both reports postulate a NiI/NiIII catalytic cycle whereby the initial step involves transmetallation of the aryl boronic acid. Subsequent reaction of [(Ar)NiILn] with bromodifluoromethane via SET, followed by radical recombination, yields the [(Ar)(CF2H)NiIIILnBr] intermediate which reductively eliminated to furnish the desired difluoromethylarene. In the same year, Mikami and co-workers reported that aryl Grignard reagents underwent successful difluoromethylation in the presence of difluoroiodomethane, [Ni(COD)2] (2.5 mol%) and TMEDA (2.5 mol%) (Scheme 13D).76 In the same year, the MacMillan group merged nickel and photoredox catalysis for the difluoromethylation of a broad range of (hetero)aryl halides (Scheme 13E).77 This nickel/photoredox-catalyzed cross-electrophile coupling reaction proceeds at room temperature. A unique mechanistic feature which allows this reaction to proceed under mild conditions is the implementation of a silyl radical mediated bromine atom abstraction from bromodifluoromethane. This mild activation mode allows for the LSF of biologically relevant compounds. Of note is the tolerance of the method to several pharmaceutically-relevant heterocycles such as electron-rich pyrimidines, pyrazoles, and indoles. In particular, the successful difluoromethylation of 1-(3-bromo-1H-pyrazol-1-yl)ethan-1-one is of interest considering that 3-(difluoromethyl)-1-methyl-1H-pyrazole is a motif important to the agrochemical industry.
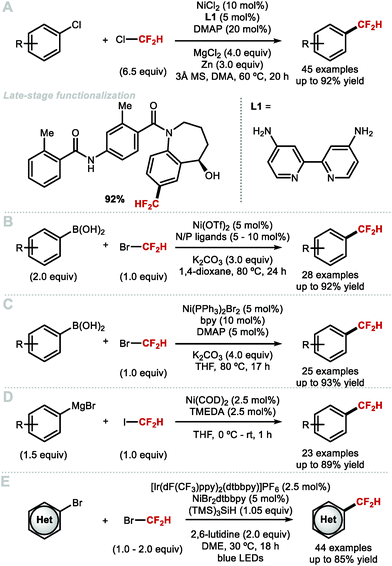 |
| | Scheme 13 Nickel-catalysed difluoromethylation of aryl halides, aryl boronic acids and aryl Grignard reagents. | |
2.1.e C(sp2)–CF2H bond formation under iron catalysis.
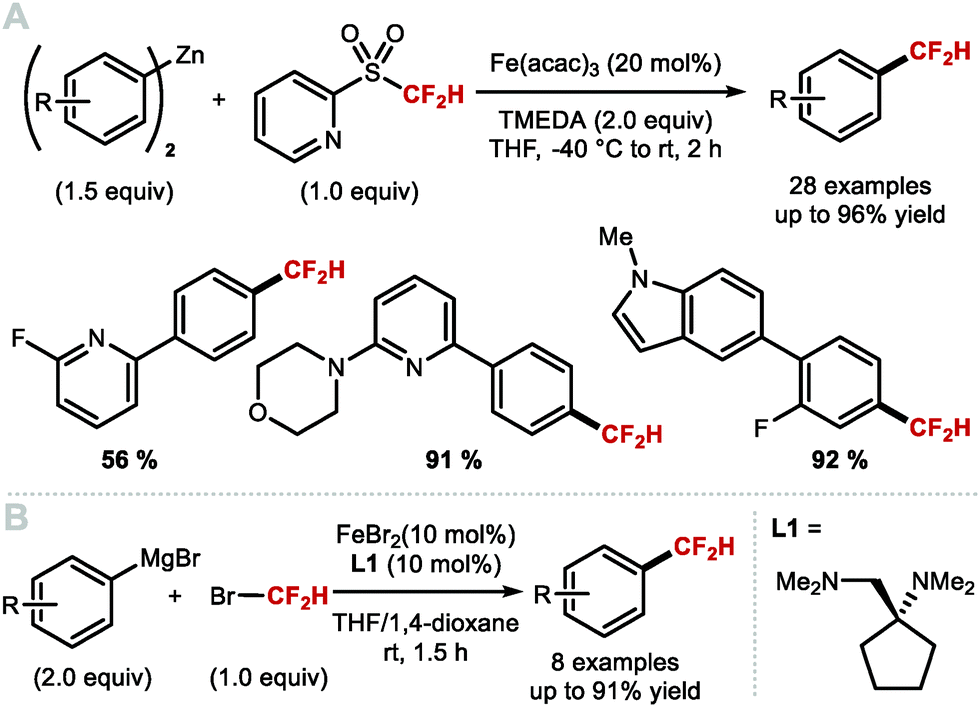
Using widely abundant earth crust iron, Hu and co-workers reported in 2018 a mild and broadly applicable Fe-catalysed cross-coupling protocol for the difluoromethylation of diaryl zinc reagents. This was achieved, using commercially available difluoromethyl 2-pyridyl sulfone (2-PySO2CF2H), TMEDA and Fe(acac)3 (20 mol%) (Scheme 14A).78 This difluoromethylation proceeds under mild reaction conditions and gives access to electron-rich and electron-deficient difluoromethylarenes. In the same year, Zhang and co-workers reported the iron-catalysed difluoromethylation of aryl Grignard reagents with bromodifluoromethane in combination with catalytic FeBr2. The use of the bulky diamine ligand 1-((dimethylamino)methyl)-N,N-dimethylcyclopentan-1-amine (L1) over other commonly used diamine ligands such as N,N,N′,N′-tetramethyl-ethane-1,2-diamine (TMEDA) proved crucial to circumvent the formation of by-products resulting from defluorination (Scheme 14B).79
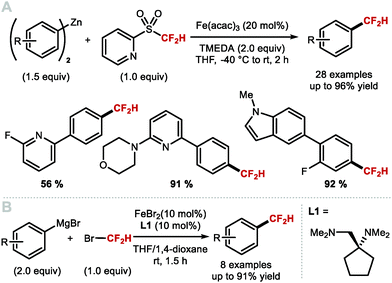 |
| | Scheme 14 Iron-catalysed difluoromethylation of diaryl zinc and aryl Grignard reagents. | |
2.1.f C(sp2)–CF2H bond formation mediated by gold.
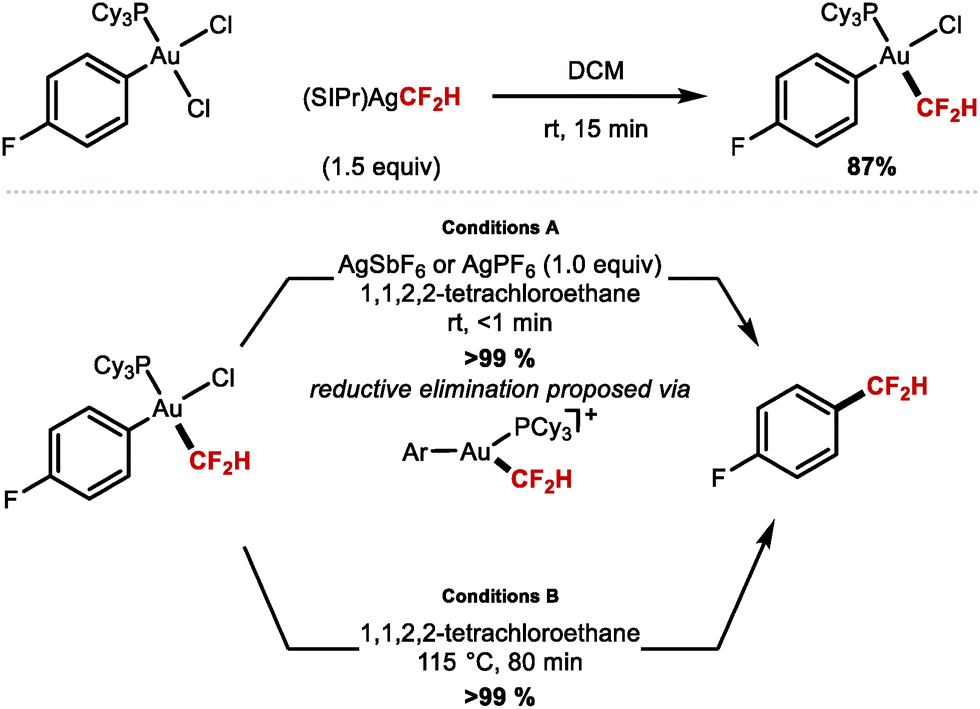
The demand for stable yet reactive MCF2H complexes led Shen and co-workers to prepare the first isolable AuCF2H complex, cis-[Au(PCy3)(4-FC6H4)(CF2H)(Cl)] through transmetallation from [(SIPr)Ag(CF2H)] and cis-[Au(PCy3)(4-FC6H4)(Cl)2] (Scheme 15).80 The reaction of this complex at room temperature with a silver salt such as AgSbF6 or AgPF6, allowed for facile reductive elimination to yield 4-(difluoromethyl)fluorobenzene in very short reaction times (<1 min). In the absence of silver, the reductive elimination proceeded at elevated temperatures (115 °C), and required longer reaction times (80 min). The authors suggest that the cationic intermediate [Au(PCy3)(4-F-C6H4)(CF2H)]+ can form from cis-[Au(PCy3)(4-F-C6H4)(CF2H)(Cl)] in the presence of AgI. This cationic Au complex is required to achieve rapid reductive elimination at room temperature.
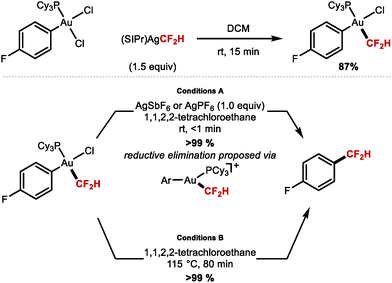 |
| | Scheme 15 Synthesis and reductive elimination from the cis-[Au(PCy3)(4-FC6H4)(CF2H)(Cl)] complex. | |
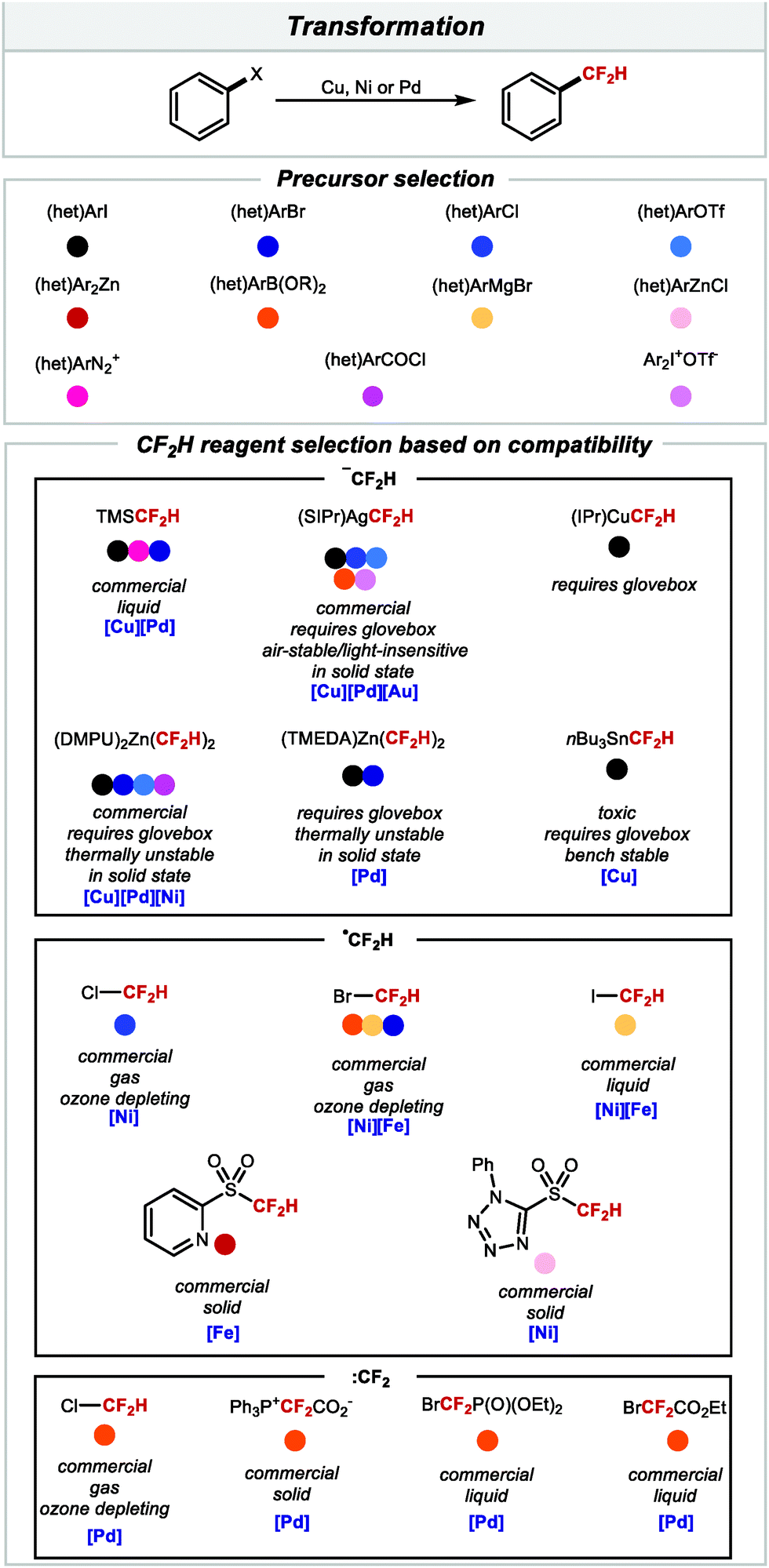
From Section 2.1, it is evident that there is currently an abundance of methods to access difluoromethylarenes in a single step under catalytic conditions from a wide array of accessible precursors. Scheme 16 provides a guide for practitioners of this chemistry, illustrating some key features with respect to the CF2H source and activation manifold employed for each class of substrates. This information may aid selection of the methodology best suited to the problem at hand.
 |
| | Scheme 16 Overview of precursors and difluoromethylating reagents used to access difluoromethyl(hetero)arenes. | |
2.2 C(sp2)–CF2H bond formation via C–H difluoromethylation
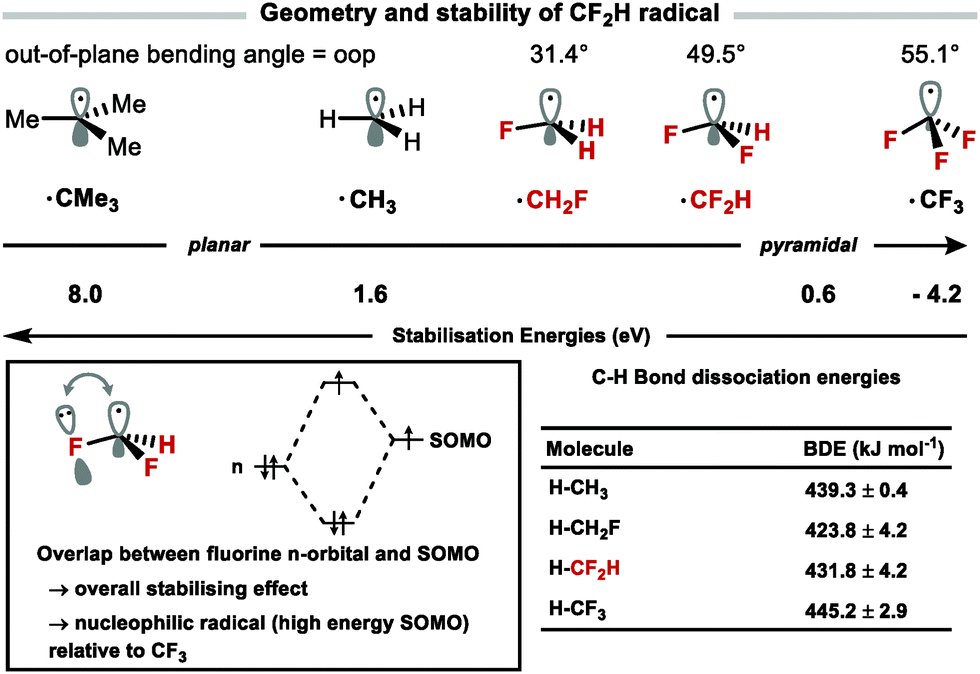
The field of C–H difluoromethylation mostly relies on the generation and reactivity of the difluoromethyl radical although cross-coupling methodologies have appeared. The radical stability of fluoroalkylated radicals varies with fluorine content (CH2F > CF2H > CF3).81 The geometry of fluoroalkyl radicals is also affected by the degree of fluorine substitution, becoming progressively more tetrahedral with increased fluorine substitution as evidenced by the respective out of plane (oop) bending angles, oopCF3 = 55.1°, oopCF2H = 49.5°, oopCH2F = 31.4°.82 This distortion from planarity results in less effective overlap of fluorine lone pairs with the single occupied molecular orbital (SOMO) (Fig. 8). Combined with the lesser electron-withdrawing effect of CF2H versus CF3, the CF2H radical has a comparatively higher energy SOMO and is more nucleophilic than the CF3 radical.83 These properties lead to differences in reactivity and selectivity between CF2H and CF3 radicals for example in Minisci-type chemistry.
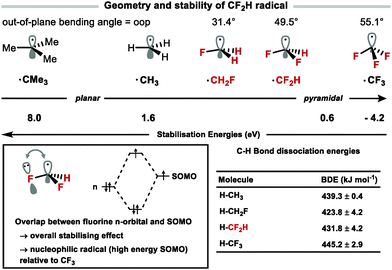 |
| | Fig. 8 Geometry and stability of the CF2H radical versus others. Out-of-plane bending angles calculated computationally (B3LYP/6-31G(d)). | |
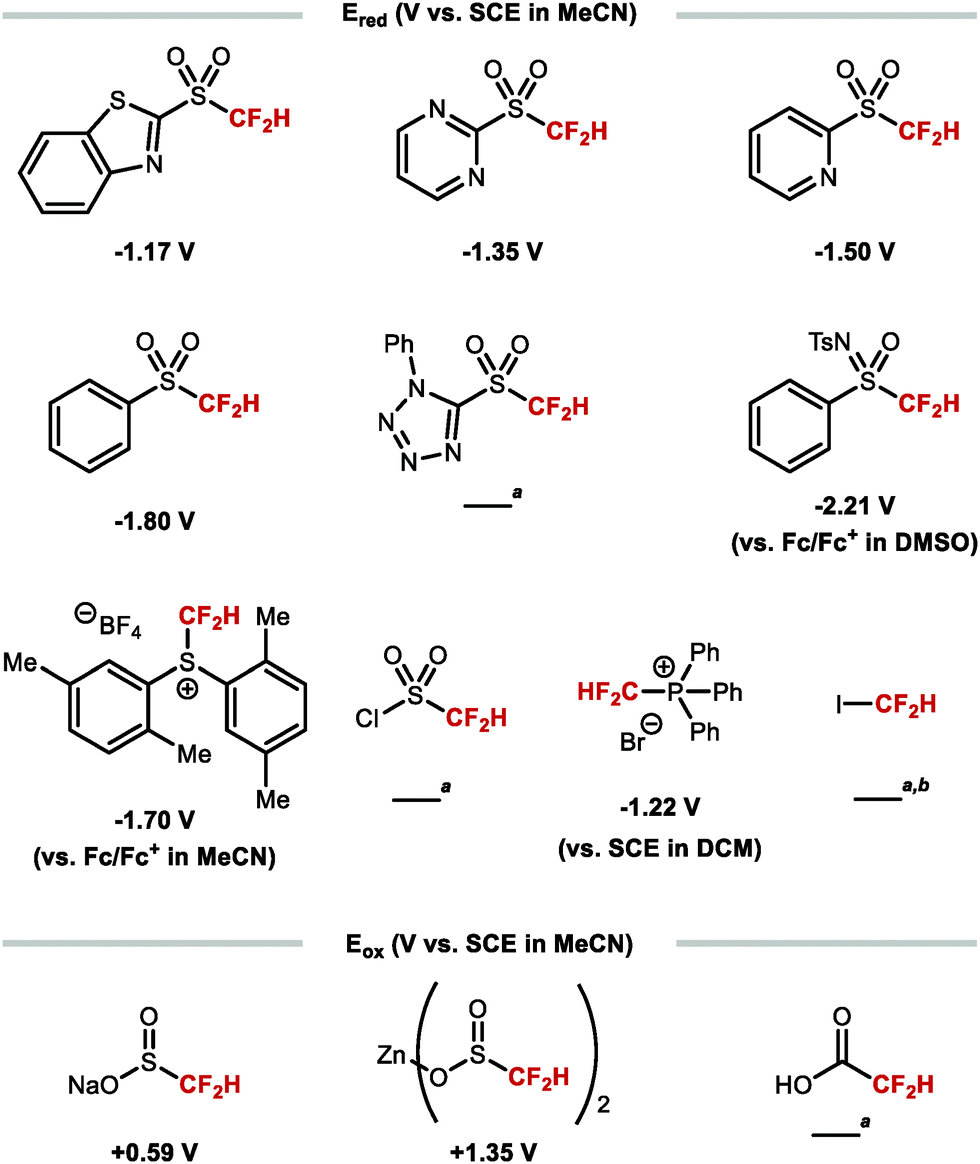
The difluoromethyl radical is often generated by SET from a suitable difluoromethyl radical precursor. The most common reagents and their redox potentials are listed in Fig. 9.84–89
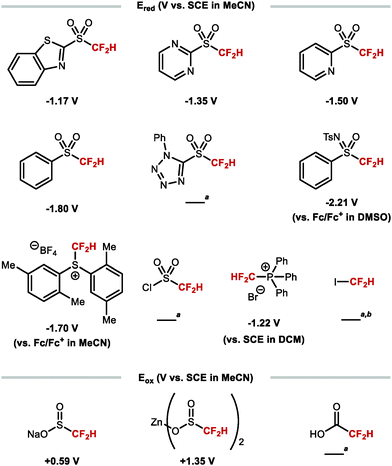 |
| | Fig. 9 Redox properties of various difluoromethyl radical sources. aReduction potential unreported to date. bBDEC–I = 57 ± 6 kcal mol−1. | |
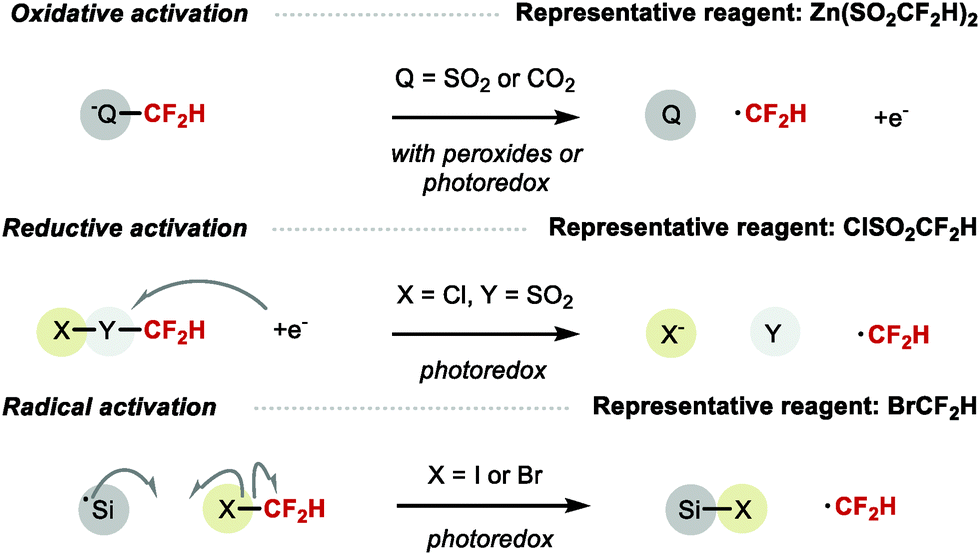
Methods for radical difluoromethylation have proliferated rapidly over the last few years. Several reagents can serve as CF2H radical precursor via a variety of activation processes including single-electron oxidation (e.g. Zn(SO2CF2H)2), single-electron reduction (e.g. ClSO2CF2H), or radical abstraction (e.g. BrCF2H) (Scheme 17).
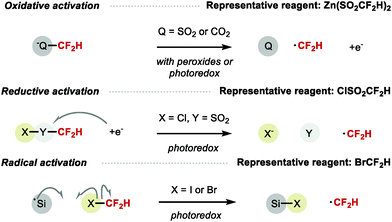 |
| | Scheme 17 Radical CF2H formation. | |
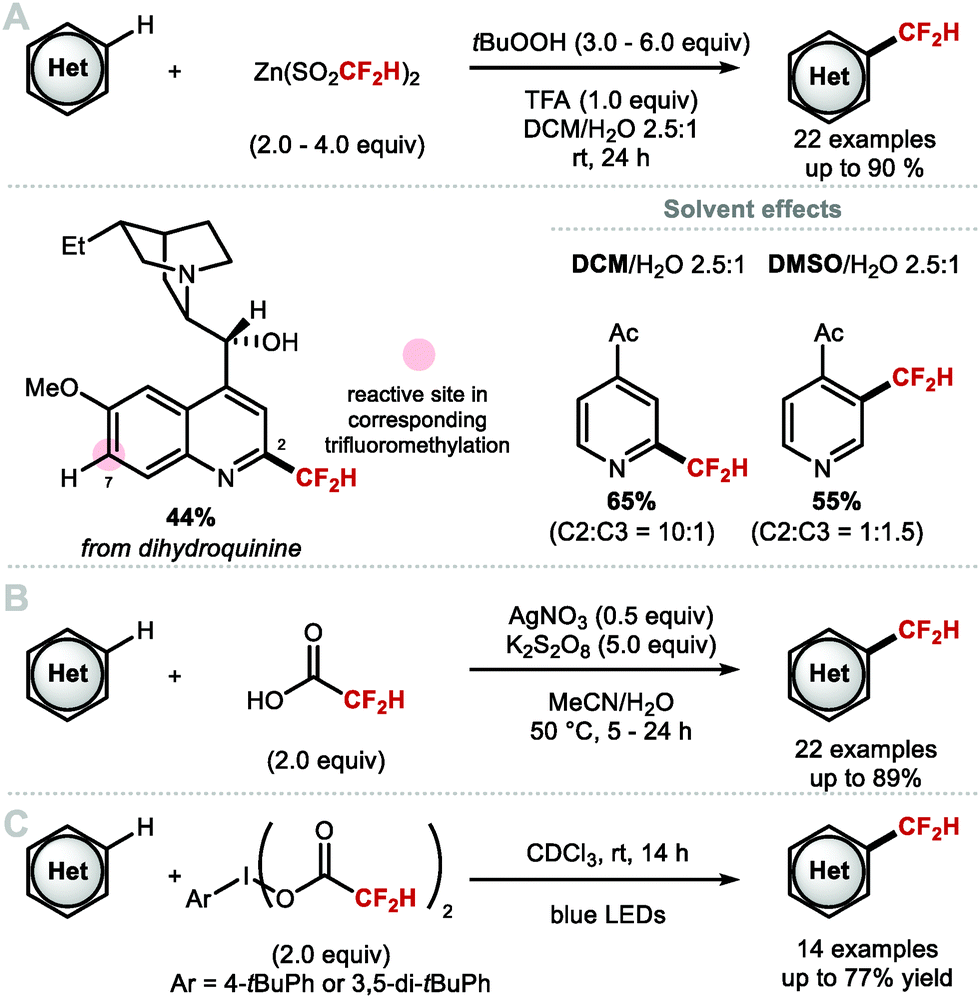
Pioneering work by Chen and co-workers showed that gaseous CF2HI can be used as a source of CF2H radical for the iododifluoromethylation of alkenes.90 This report inspired Baran and co-workers to develop [Zn(SO2CF2H)2] (DFMS), an easy-to-handle bench stable solid CF2H radical precursor.83 With this reagent, N-heterocyclic substrates undergo C–H difluoromethylation in the presence of excess tBuOOH and trifluoroacetic acid (TFA). This report marks the first general C–H difluoromethylation protocol for heteroarenes (Scheme 18A). Their results corroborate the nucleophilic character of ˙CF2H radical which contrasts with the electrophilicity of ˙CF3. This was exemplified with the difluoromethylation of dihydroquinine, which was selective for C2 rather than C7, the latter being the preferred site for CF3 radical addition. The solvent system had a profound effect on selectivity. Using 1-(pyridin-4-yl)ethan-1-one as model substrate, a C2:C3 selectivity superior to 10:1 was obtained in DCM/H2O (2.5:1). When the same reaction was conducted in DMSO/H2O (2.5:1), the C3 regioisomer was formed preferentially (C2:C3, 1:1.5). Christensen, Nielsen and co-workers showed that inexpensive difluoroacetic acid undergoes Minisci-type difluoromethylation with a variety of pyridines and other heteroarenes (Scheme 18B).91 Mono-difluoromethylation versus bis-difluoromethylation was controlled by tuning the reaction temperature, with mono-difluoromethylated compounds obtained at lower temperatures. Maruoka and co-workers developed a hypervalent iodine(III) reagent adorned with difluoroacetoxy ligands. This reagent enables C–H difluoromethylation of heteroarenes upon photolysis under blue light irradiation (Scheme 18C).92
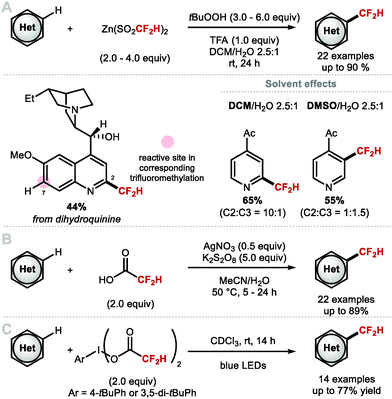 |
| | Scheme 18 Synthesis of heteroaryl-CF2H via radical C–H difluoromethylation. | |
In 2019, Stephenson and co-workers illustrated that the CF2H radical can be generated via photocatalytic activation of pyridinium or quinolinium complexes generated from the corresponding N-oxides and difluoroacetic anhydride (Scheme 19A).93 In 2018, Zhang and Deng developed a visible-light driven metal-free difluoromethylation of coumarins (Scheme 19B).94 The authors generated the difluoromethyl radical from sodium difluoromethanesulfinate activated by Eosin Y, with air as stoichiometric oxidant. More recently, Meng, Li, and co-workers found that the use of the same reagent combined with rose bengal as photocatalyst and green LEDs as light source, enabled difluoromethylation of a broad range of heteroarenes relevant to medicinal chemistry, including pyridines, pyrazines, imidazoles, pyrazoles, indoles and electron-rich arenes (Scheme 19C).95 In 2019, König and co-workers disclosed an alternative activation of DFMS, using an organic semiconductor photocatalyst with applications to C–H difluoromethylation of heteroarenes (Scheme 19D).96 This strategy provides a milder alternative to peroxide activation of DFMS reported by Baran but requires irradiation with blue light. Qing, Chu and co-workers recently reported a silver mediated C–H difluoromethylation of a range of heteroarenes employing TMSCF2H as ˙CF2H source (Scheme 19E).97 The reaction proceeded in high yields for quinoxalin-2(1H)-ones. Other heteroarenes including a benzoxazole derivative also afforded the difluoromethylated product.
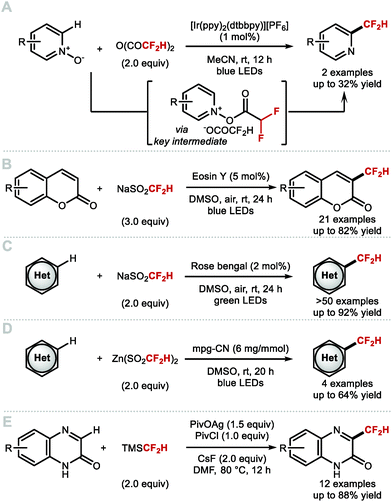 |
| | Scheme 19 Recent radical C–H difluoromethylation methods. | |
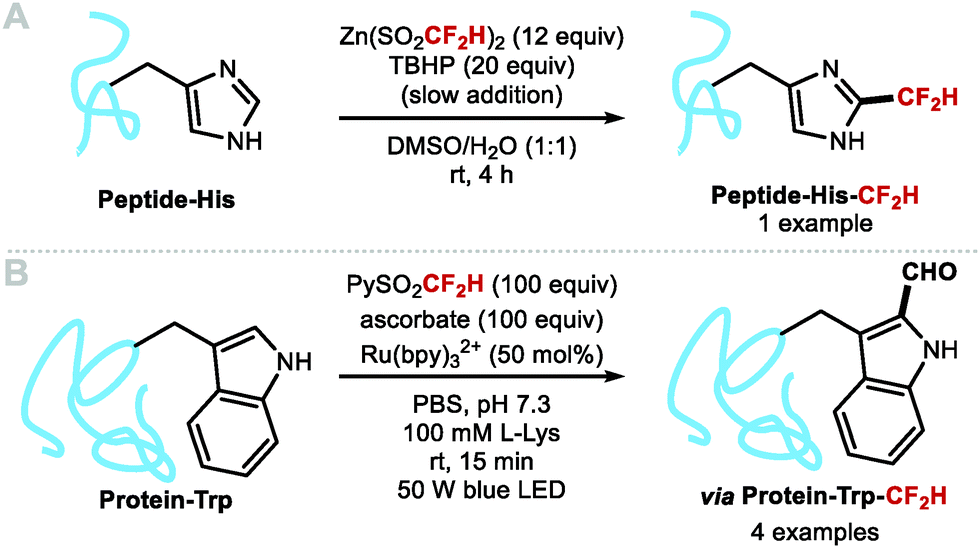
In 2019, Noisier, Gopalakrishnan and co-workers reported a single example of a C–H difluoromethylation of a histidine residue in a pentapeptide (Scheme 20A).98 In 2021, Davis and co-workers demonstrated the site-selective difluoromethylation of tryptophan (Trp) residues in proteins (Scheme 20B).99 A range of proteins undergoing difluoromethylation at the C2 position of Trp, include annexin A5, lysozyme, cationic trypsin and lactalbumin. Once installed, the CF2H spontaneously hydrolysed resulting in net C–H formylation. The HC(O)–Trp residue is a privileged handle for further functionalisation.
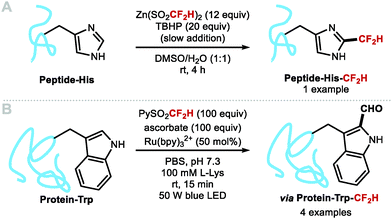 |
| | Scheme 20 C–H difluoromethylation of tryptophan residues in proteins. | |
To overcome the limitations of site-selectivity in C–H difluoromethylation, Qing and co-workers envisioned that a two-electron cross-coupling of heteroarenes with a [CuCF2H] complex could be achieved under oxidative conditions (Scheme 21).100 The authors found that when [CuCF2H] was generated in situ from [CuCN] and TMSCF2H, a variety of heterocycles activated under basic conditions could coordinate CuI. When this CuI complex was further oxidised with 9,10-phenanthrenequinone, subsequent reductive elimination furnished the difluoromethylated product. This report hallmarked the first transition-metal mediated C–H difluoromethylation protocol under oxidative conditions. C–H bonds of oxazoles, thiazoles, imidazoles, 1,3,4-oxadiazoles, benzo[d]oxazoles, benzo[d]thiazoles, benzo[b]thiophenes, pyridines, thiophenes, and thiazolo[5,4-c]pyridines were readily difluoromethylated in good to excellent yields.
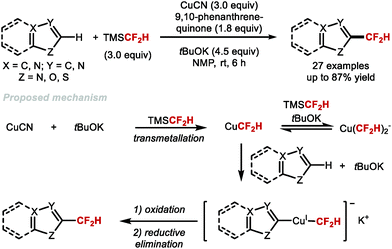 |
| | Scheme 21 Copper-mediated difluoromethylation of acidic heteroaryl C–H bonds. | |
Complementary to the radical C–H difluoromethylation protocol developed by Baran and co-workers,83 Qing's copper mediated oxidative C–H difluoromethylation100 illustrated that substrates bearing multiple reactive sites such as thiazole[5,4-c]pyridine or 5-(quinoxaline-6-yl)oxazole undergo oxidative difluoromethylation at the more acidic C–H azole bond. In contrast, conditions developed by Baran and co-workers enable difluoromethylation at the most electron-deficient carbons adjacent to the nitrogen of the 6-membered heterocyclic ring (Scheme 22).
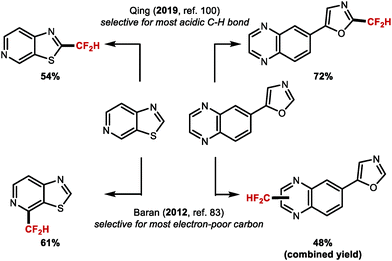 |
| | Scheme 22 Selectivity of radical versus oxidative C–H difluoromethylation. | |
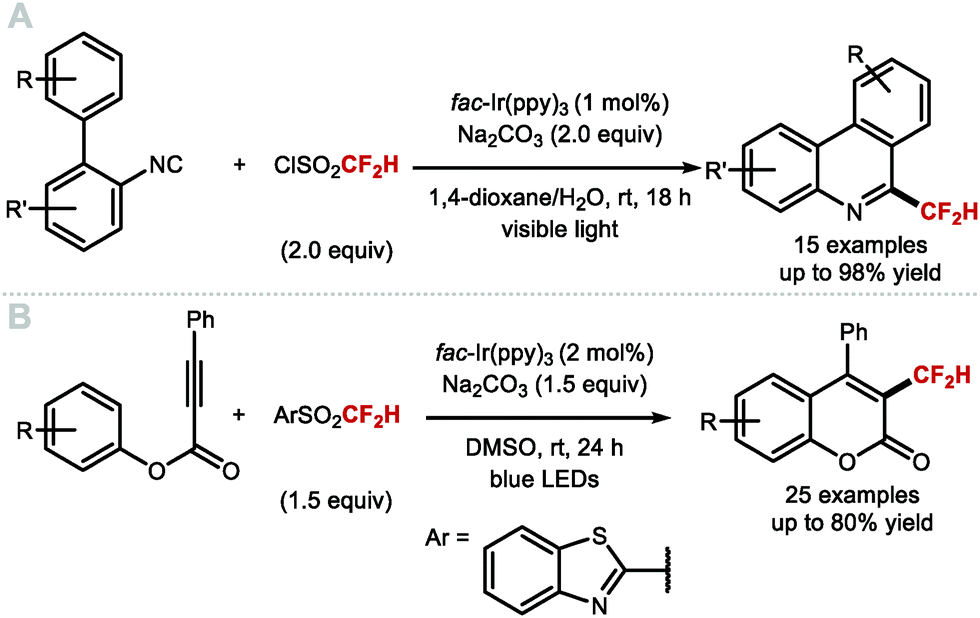
Other than direct methods to install CF2H groups onto heteroaromatics via Minisci-type chemistry, Dolbier and co-workers envisioned that the CF2H radical could react with 2-isocyano-1,1′-biphenyl to generate a series of substituted 6-(difluoromethyl)phenanthridines (Scheme 23A).101 In this transformation, fac-Ir(ppy)3 (1 mol%) activates HCF2SO2Cl to generate the CF2H radical which subsequently adds to a series of biphenyl isocyanides, affording the corresponding difluoromethylated phenathridine products upon oxidation of the intermediate radical species followed by proton loss. Similarly, Fu and co-workers prepared a selection of 3-difluoromethylated coumarins from aryl alkynoates in a process suggested to proceed through a difluoromethylation/cyclisation sequence (Scheme 23B).102
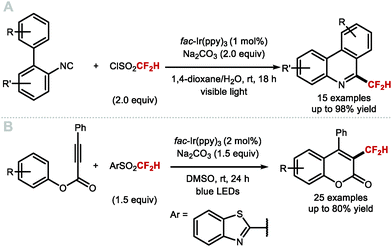 |
| | Scheme 23 Synthesis of heteroaryl–CF2H via radical difluoromethylation–cyclisation. | |
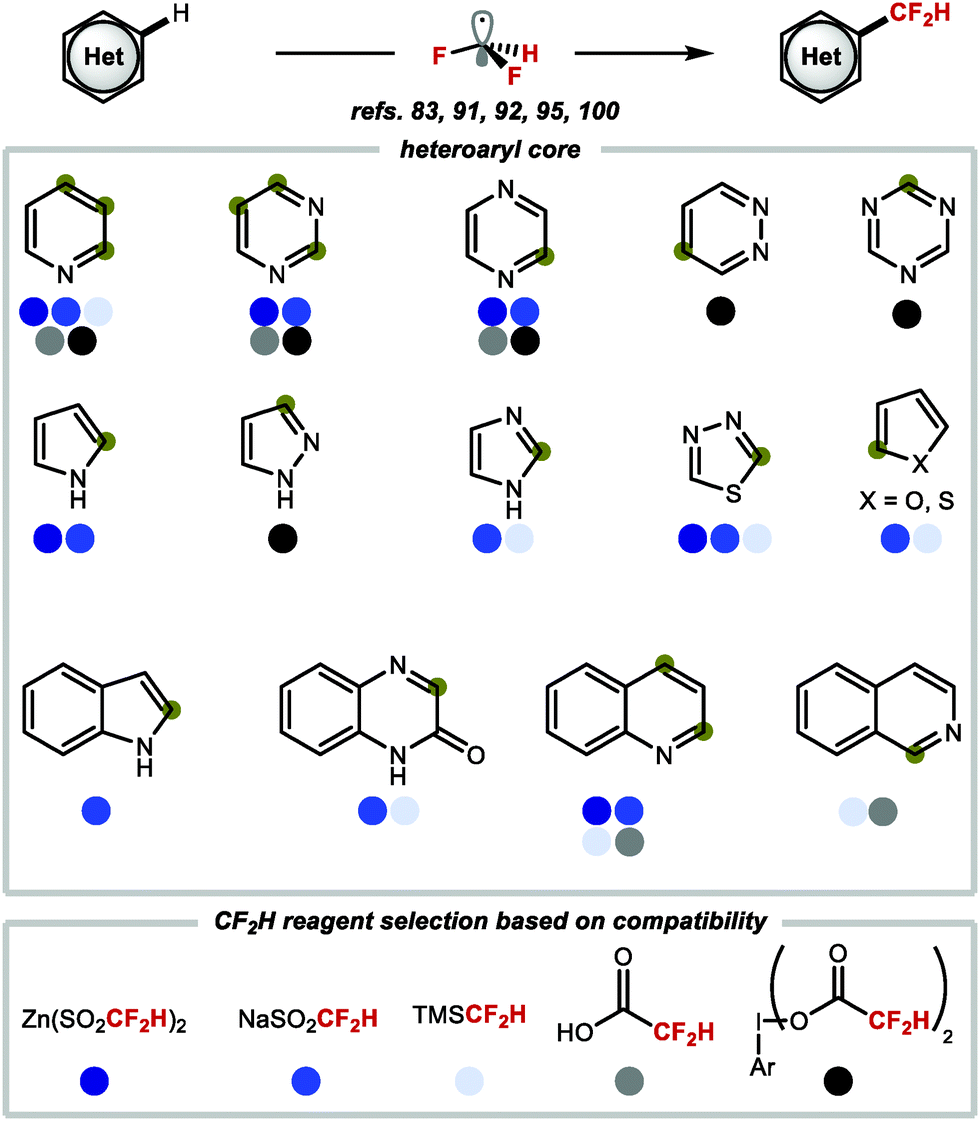
In summary, it is evident that there is an abundance of methods to access a range of heteroarenes substituted with CF2H groups in a single step through C–H difluoromethylation. Scheme 24 provides a selection of heteroarenes that are important to medicinal chemists, and the difluoromethylation reagents that have been used for selective C–H difluoromethylation of functionalised analogues of these heteroarenes.
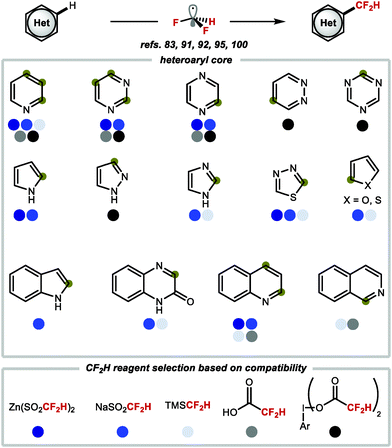 |
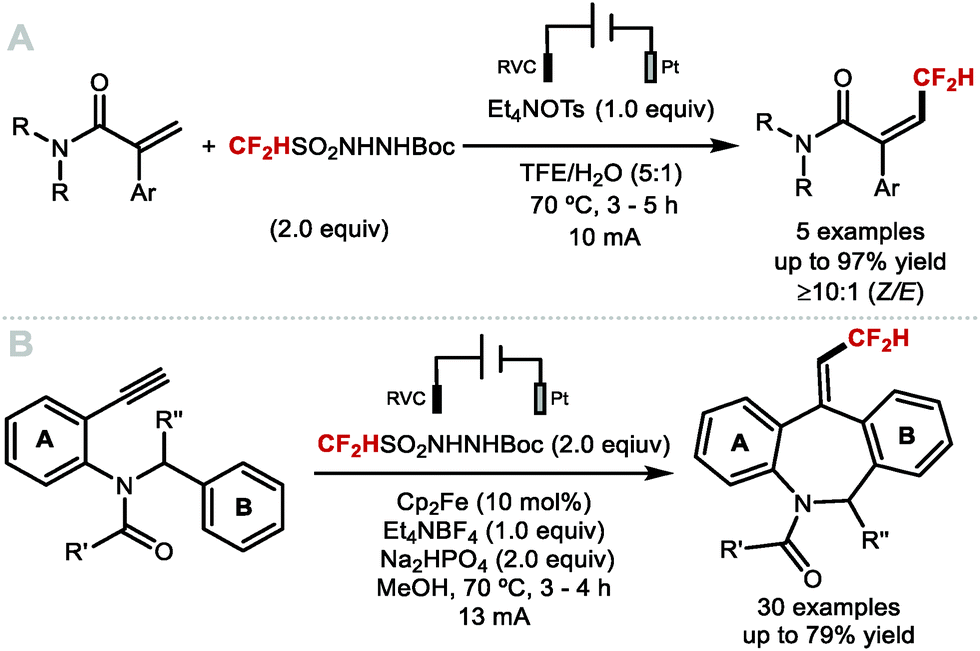
| | Scheme 24 Heterocycles commonly found in medicinal chemistry and suitable reagents for radical C–H difluoromethylation. | |
2.3 C(sp2)–[18F]CF2H bond formation for PET radiochemistry
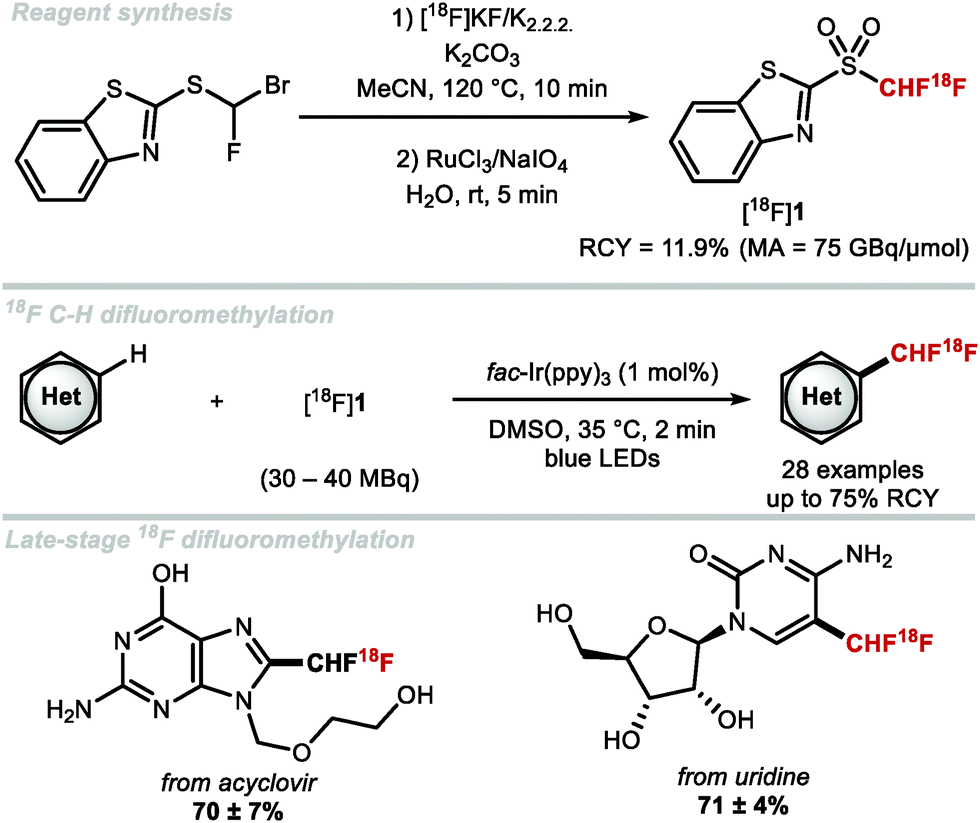
The first radiosynthesis towards [18F]ArCF2H was disclosed by Gouverneur and co-workers in 2013 and accomplished using [18F]F2-derived [18F]selectfluor bis(triflate).103 In 2016, Gouverneur and co-workers disclosed a more broadly applicable method which involved the AgI-mediated halogen exchange reaction of electron-rich (chlorofluoromethyl)arenes and [18F]fluoride.104 In the same year, Ritter and co-workers disclosed an alternative radiosynthesis of [18F]ArCF2H from aryl (pseudo)halides and [18F]fluoride.105 In 2017, Liang and co-workers disclosed a two-step strategy towards [18F]]ArCF2H.106 This was accomplished through nucleophilic radiofluorination of benzyl (pseudo)halides with [18F]fluoride followed by oxidative C–H fluorination with Selectfluor™. In 2019, Gouverneur and co-workers published a novel approach towards [18F]ArCF2H using aryl boronic acids, ethyl bromofluoroacetate and [18F]fluoride.107 The reaction sequence commences with a copper-catalysed cross-coupling with ethyl bromofluoroacetate and an aryl boronic acid. In situ hydrolysis then yielded a wide selection of α-fluoroarylacetic acids. The radioisotope 18F was introduced in the final step applying a Mn-mediated fluorodecarboxylation using Mn(tmp)Cl, iodosylbenzene and [18F]fluoride. Pre-complexation of two equivalents of α-fluoroarylacetic acid with iodosylbenzene to generate the hypervalent iodine complex prior to 18F-fluorination led to increased F-18 incorporation. The application of the methodology was demonstrated with the radiosynthesis of a [18F]CF2H analogue of the COX-II inhibitor ZA140 which was obtained in 15% ± 2% radiochemical yield (RCY). In contrast to the aforementioned reports which disclosed radiosyntheses towards [18F]ArCF2H through halogen exchange and fluorodecarboxylation procedures, Genicot and Luxen disclosed the first 18F-difluoromethylation (Scheme 25).108 For this purpose, 2-[18F]((difluoromethyl)sulfonyl)benzo[d]thiazole was prepared in 11.9% ± 1.4% RCY, and a decay corrected molar activity (MA) of up to 75 GBq μmol−1. This reagent was activated photocatalytically to generate the [18F]CF2H radical that was incorporated into a variety of N-heteroarenes including pharmaceutical drugs. The authors made a first attempt towards a more user-friendly methodology by fully automating the protocol on the ‘AllinOne’ TRASIS module.109 This seminal report marks the first application of an F-18 labelled reagent to access the [18F](het)ArCF2H motif.
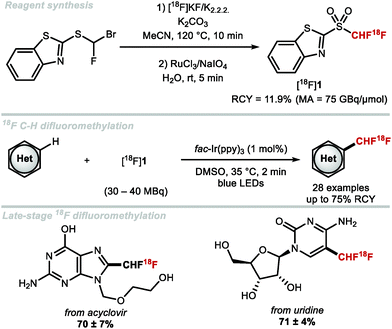 |
| | Scheme 25 Radiosynthesis of [18F]1 and 18F-difluoromethylation of N-heterocycles. | |
3. C(sp3)-Difluoromethylation
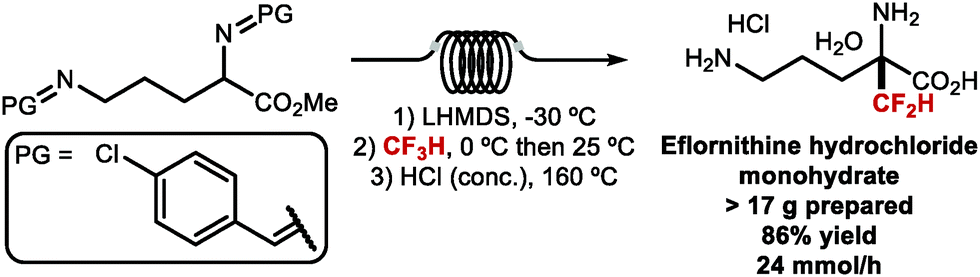
CF2H groups linked to C(sp3) have attracted great interest in medicinal chemistry as a bioisosteric replacement of aliphatic alcohols and thiols.110 Several compounds featuring C(sp3)–CF2H are FDA approved drugs or are currently undergoing clinical trials (Fig. 10). Eflornithine, an essential medicine according to the World Health Organization, contains a CF2H group bound to the α-carbon of the non-proteinogenic amino acid ornithine.111 GDC-0077, a phosphoinositide 3-kinase (PI3K) inhibitor, contains a CF2H bound to an oxazolidinone fragment and is in clinical trials for the treatment of tumours.112 Glecaprevir and Voxilaprevir both featuring an unnatural β-difluoromethylated α-amino acid are marketed macrocyclic protease inhibitors for the treatment of hepatitis C.113,114 In a structure–activity relationship (SAR) study of Voxilaprevir analogues, the CF2H group was predicted to play a beneficial role by improving metabolic stability compared with the ethyl or vinyl groups.115
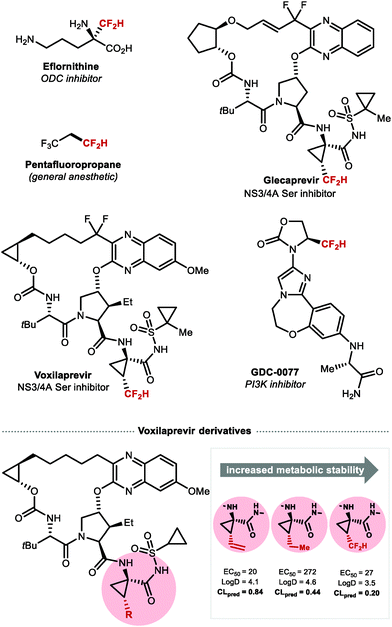 |
| | Fig. 10 Biologically relevant molecules containing C(sp3)-linked difluoromethyl groups. | |
Analogously to traditional routes towards C(sp2)–CF2H, deoxyfluorination of C(sp3)–CHO with sulfur tetrafluoride-derived reagents such as DAST®, Deoxo-Fluor® or XtalFluor® has served as a robust strategy towards C(sp3)–CF2H.116 Both GDC-0077 and Voxilaprevir are prepared by deoxyfluorination of an aldehyde-containing building block. As discussed for the synthesis of difluoromethylarenes, the lack of chemoselectivity for substrates containing reactive functional groups such as alcohols, ketones and carboxylic acids is a limitation of this approach. Therefore, alternative methods to install a CF2H group through nucleophilic, electrophilic, or radical difluoromethylation pathways are of immense benefit, especially in the context of LSF.
3.1 C(sp3)–CF2H bond formation: nucleophilic difluoromethylation
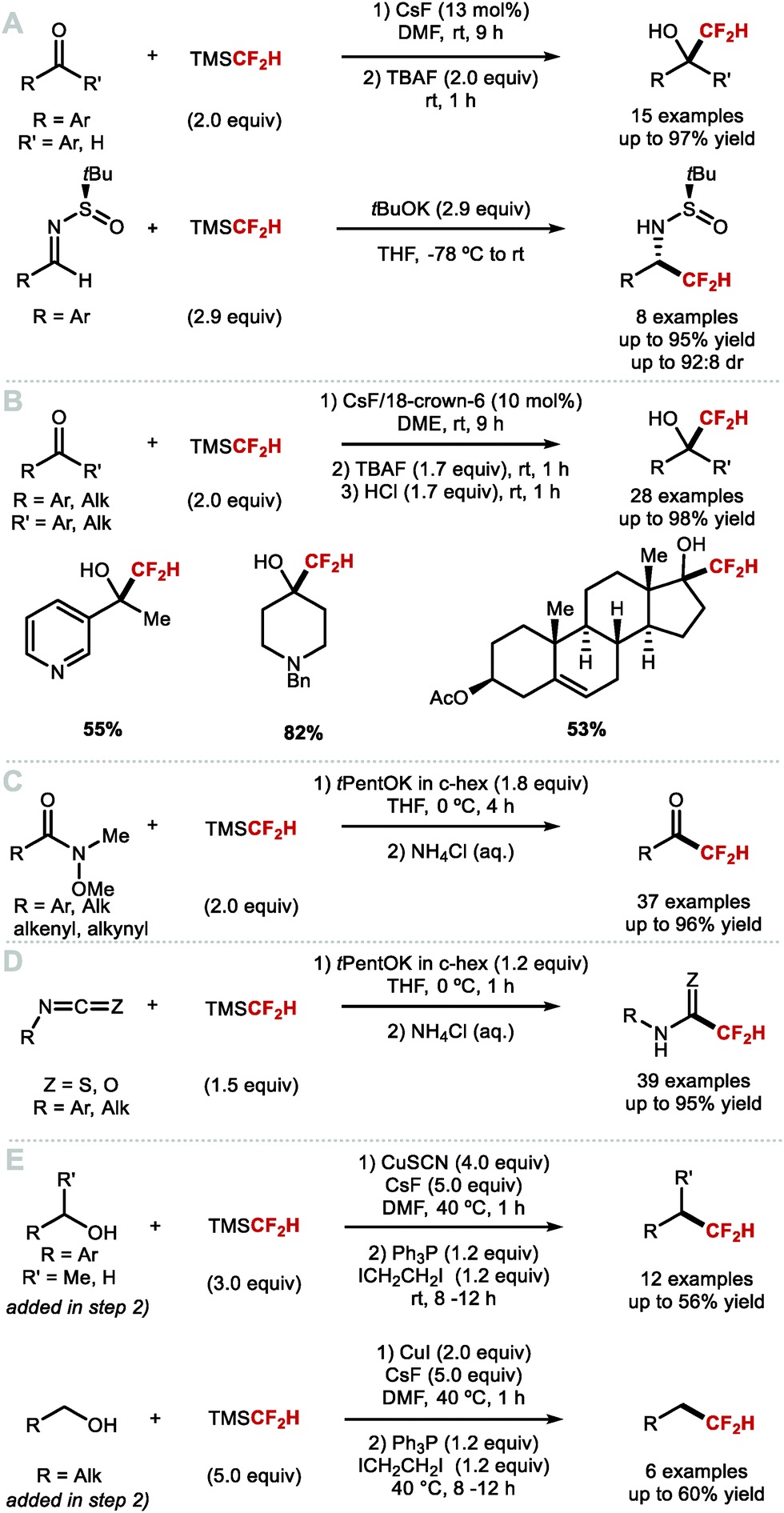
The most common nucleophilic difluoromethylation reagent used for C(sp3)–CF2H bond formation is TMSCF2H. Various difluoromethylation reactions with silane reagents have been reported as early as the 1990s but required harsh conditions.9 In 2011, Hu and co-workers reported a general and mild protocol to access a broad variety of CF2H containing alcohols and sulfonamides (Scheme 26A).117 The use of TMSCF2H as “–CF2H” synthon was demonstrated with nucleophilic addition reactions to aldehydes, ketones and N-tert-butylsulfinyl imines. In the case of aldehydes, a catalytic amount of CsF in DMF was required for activation. TBAF was also added after completion of the reaction to deprotect the in situ formed silylated difluoromethyl carbinol. These reaction conditions were low yielding for less electrophilic substrates such as ketones that afforded the difluoromethylated products in 30–40% yield; this is because DMF competes as electrophile. This limitation was circumvented by the use of tBuOK to serve as a stoichiometric activator of the silane reagent. However, such basic conditions did not allow the difluoromethylation of enolisable ketones. Base activation was also applied to Ellman's N-tert-butylsulfinyl imines. These reactions were generally high yielding and showed good diastereoselectivity. In 2015, He and co-workers disclosed the use of the organic Lewis base phosphazene to activate TMSCF2H.118 The method transformed (hetero)aryl-aldehydes and cinnamaldehyde into the difluoromethyl addition products in up to 99% yield. For enolisable aldehydes and diarylketones, the yields did not exceed 46%. In 2016, Hu and co-workers demonstrated that in situ formation of the pentavalent [(CH3)3Si(CF2H)2]− anionic species enabled difluoromethylation of enolisable ketones (Scheme 26B).119 In this instance, the activation of TMSCF2H was accomplished by employing catalytic amounts of CsF or tBuOK. A crown ether (10 mol%) was required to stabilise the pentavalent silicate anion and to increase the activator's nucleophilicity. This methodology gave access to a broad variety of aliphatic difluoromethyl carbinols in good to excellent yields. In 2019, Pace and co-workers reported the synthesis of difluoromethyl ketones and difluoromethyl(thio)amides (Scheme 26C).120 Specifically, when TMSCF2H was activated by potassium tert-pentoxide (tPentOK), Weinreb amides underwent difluoromethylation leading to difluoromethyl ketones. The same authors reported an efficient difluoromethylation of iso(thio)cyanates leading to difluoromethyl(thio)amides (Scheme 26D).121 For this reaction, TMSCF2H was also activated by tPentOK as the use of this sterically hindered base prevented undesired addition of the base itself onto the isothiocyanates, a side reaction observed with tBuOK. In 2020, Wu and Xiao reported the dehydroxylative difluoromethylation of alcohols.122 Their system involves activation of the alcohol using R3P and ICH2CH2I and requires stoichiometric quantities of preformed [CuCF2H]. Their reaction proceeds under mild conditions and shows good functional group tolerance (Scheme 26E).
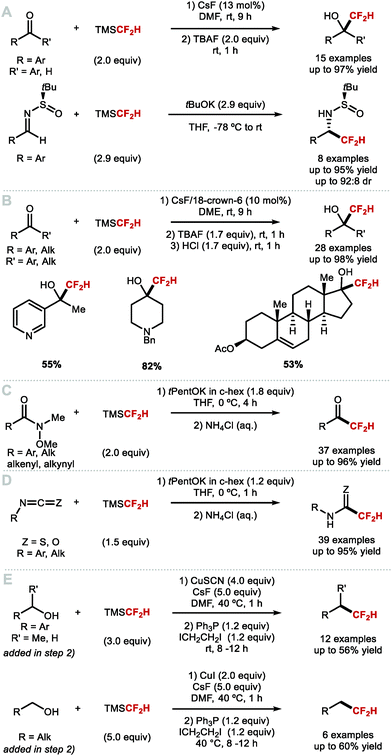 |
| | Scheme 26 Methods of nucleophilic difluoromethylation. | |
3.2 C(sp3)–CF2H bond formation: electrophilic difluoromethylation
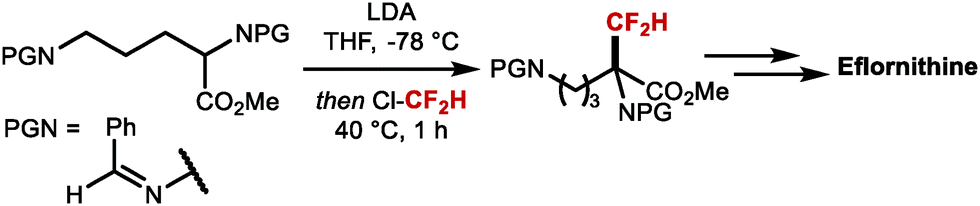
Net electrophilic difluoromethylation at C(sp3)-centres has been well studied. For example, the key step in the synthesis of the ornithine decarboxylase (ODC) inhibitor Eflornithine, a drug developed in the 1970's and brought to market in 1990 to treat sleeping sickness is prepared by α-difluoromethylation with ClCF2H, an ozone-depleting substance (ODS) (Scheme 27).111 This transformation was proposed to proceed via difluorocarbene mechanism.
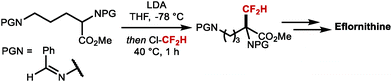 |
| | Scheme 27 Key step in the synthesis of Eflornithine. | |
With the Montreal protocol urging scientists to develop chemistries not employing ozone-depleting fluorine-containing reagents, fluoroform was considered as a suitable alternative. The activation of HCF3 for difluoromethylation of C(sp3) centres was achieved by Mikami and co-workers in 2012 with the difluoromethylation of lithium enolates (Scheme 28A).123 It is well known that the high bond dissociation energy of the C–F bond (117 kcal mol−1) poses a challenge to SN2 reactions of organofluorides. In this case, the authors propose that the high enthalpy of formation for LiF (147 kcal mol−1) allows for nucleophilic substitution with fluoride displacement on fluoroform. The methodology was applied to a variety of ketones, amides and esters, and later to nitriles (Scheme 28B).124 In 2018, Kappe and co-workers extended this methodology to continuous flow conditions for the α-difluoromethylation of esters.125,126 Several substrates were subjected to difluoromethylation including protected amino acids (Scheme 28C).
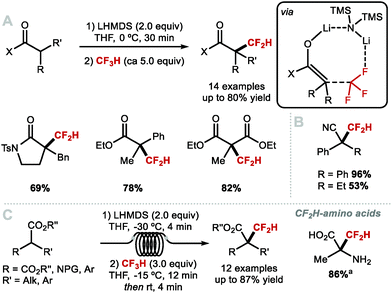 |
| | Scheme 28 Difluoromethylation reactions with fluoroform. aProduct obtained after N-deprotection and ester hydrolysis. | |
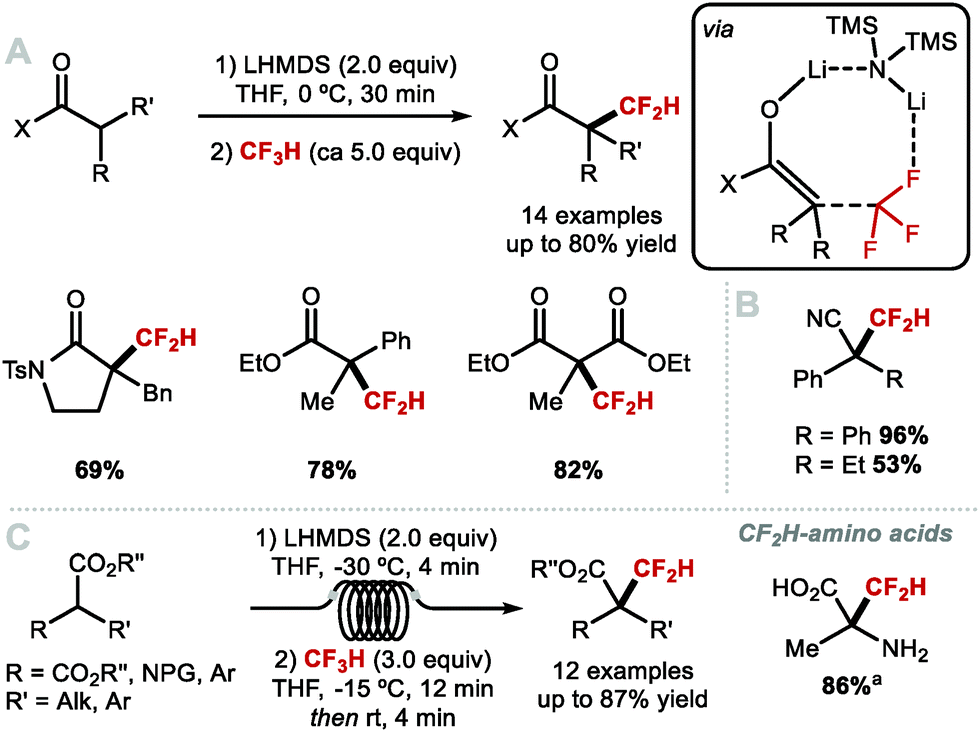
Various difluoromethylations were performed with non-gaseous reagents (Scheme 29). In 2011, Shibata and co-workers reported the electrophilic difluoromethylation of β-ketoesters using the sulfoximine reagent developed by Hu and co-workers (Scheme 29A).127 When comparing the reactivity of this difluoromethyl reagent with the corresponding monofluoromethyl reagent (O-selectivity) and trifluoromethyl reagent (C-selectivity), they noted the formation of a mixture of products resulting from C- and O-difluoromethylation (63:37). In 2018, Shen and co-workers developed a highly C-selective process for β-ketoesters (Scheme 29B); up to 100:0, C- vs. O-alkylation) using a difluoromethyl sulfonium ylide suggested to serve as difluorocarbene source.128 This method allowed the direct α-difluoromethylation of ketene silyl acetals. In 2019, Hu and co-workers demonstrated that commercially available TMSCF2Br was an efficient reagent for the C-difluoromethylation of a range of C(sp3) and C(sp) nucleophiles, such as esters, amides, fluorenes, terminal alkynes, malonates, β-ketoesters and other activated C–H nucleophiles (Scheme 29C).129 For this protocol, the authors also propose a difluorocarbene mechanism.
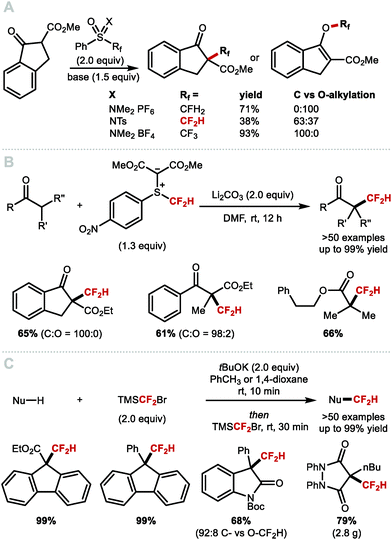 |
| | Scheme 29 Electrophilic difluoromethylations of carbon nucleophiles and difluoromethylation under difluorocarbene conditions. | |
3.3 C(sp3)–CF2H bond formation: radical difluoromethylation
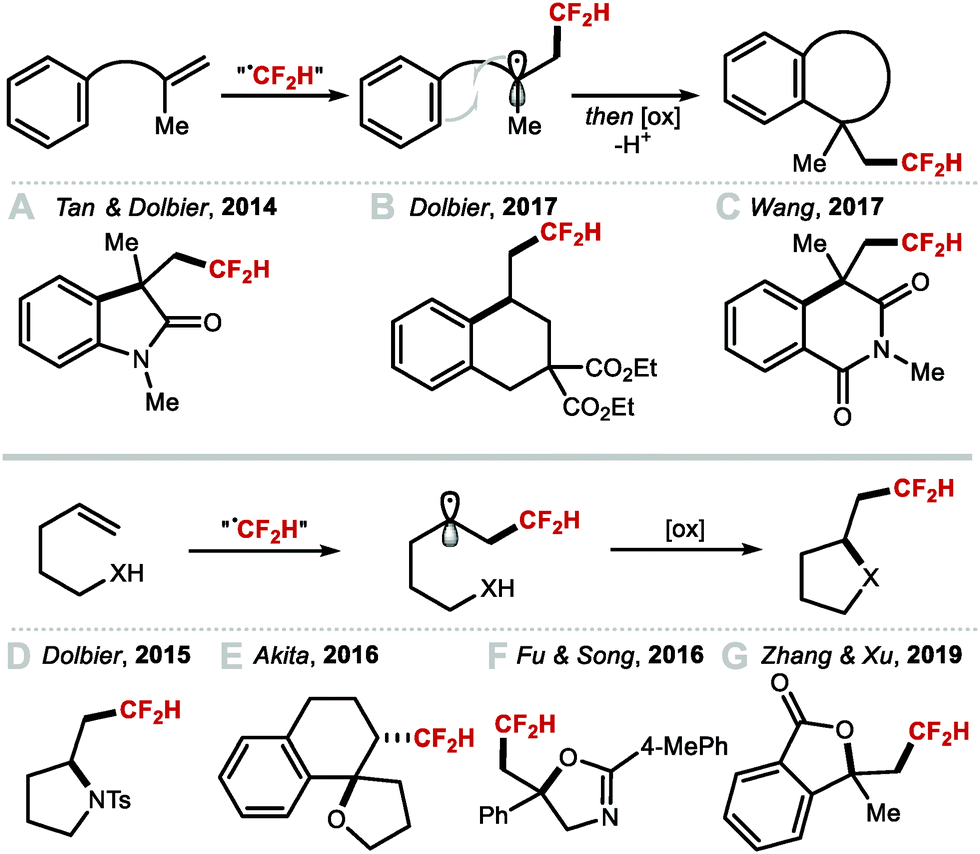
In 2012, Baran and co-workers disclosed the hydrodifluoromethylation of enones with DFMS (Scheme 30A).83 Dolbier and co-workers reported in 2015 a more broadly applicable and higher yielding hydrodifluoromethylation of electron-deficient alkenes by making use of the photocatalyst fac-Ir(ppy)3 to activate difluoromethanesulfonyl chloride with tris(trimethylsilyl)silane ((TMS)3SiH) as hydrogen-atom donor (HAD) (Scheme 30B).130 In the same year, Qing and co-workers expanded the hydrodifluoromethylation to alkenes of different electronic profiles, as well as alkene-containing bioactive molecules (Scheme 30C).131 This method required a non-commercial difluoromethylation reagent and careful handling under inert atmosphere, limiting its applicability to large-scale synthesis. Inspired by Maruoka's report for the direct C–H difluoromethylation of heteroarenes, Gouverneur and co-workers disclosed an operationally simple procedure using difluoroacetic acid, PIDA and THF (Scheme 30D).132,133 In contrast to other hydrodifluoromethylation procedures, this methodology does not require an inert atmosphere and was demonstrated on decagram scale, providing access to medicinally relevant building blocks. Recently, Maestro and Alemán developed a photocatalytic hydrodifluoromethylation of (a)cyclic imines upon activation of DFMS with rhodamine 6G (Scheme 30E).88 In 2020, Davis and Gouverneur reported for the first time a protocol enabling precise incorporation of a CF2H group on a protein (Scheme 30F).134 2-PySO2CF2H was employed to release a CF2H radical capable of reacting at the terminal carbon of a SOMOphilic dehydroalanine (Dha) residue. The reaction is performed at ambient and biocompatible conditions, allowing for the preparation of hydrodifluoromethylated proteins in high yields (up to quantitative) and short reaction times. This seminal work by Davis and Gouverneur sets a benchmark for further developments for the late-stage incorporation of perfluoroalkyl group onto biomolecules.
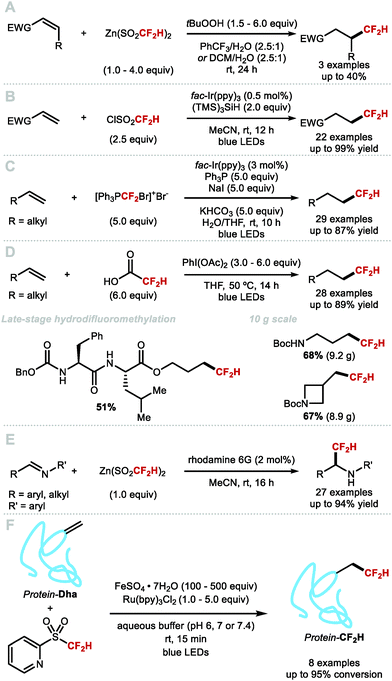 |
| | Scheme 30 Radical hydrodifluoromethylation reactions. | |
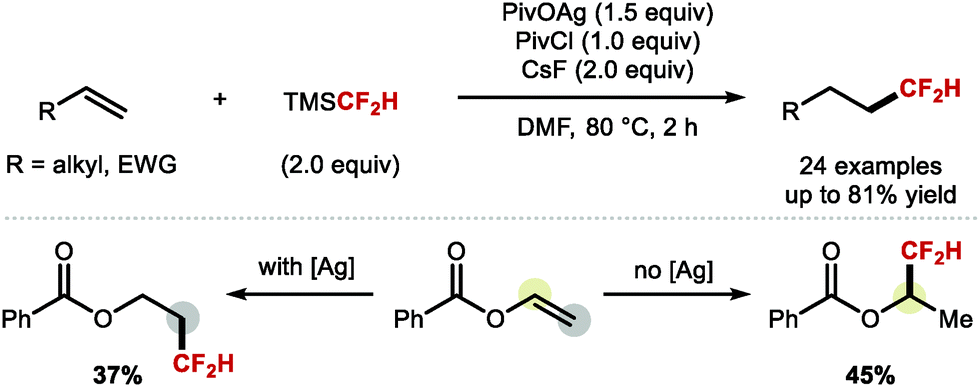
A more recent report from Qing, Chu and co-workers provided a solution to the important problem of regiocontrolled hydrodifluoromethylation of alkenes (Scheme 31).97 The authors disclosed a silver mediated difluoromethylation of a broad range of alkenes using TMSCF2H as the difluoromethyl radical source. The authors reported high regioselectivity towards either the anti-Markovnikov or the Markovnikov product when reacting vinyl benzoate in presence or absence of silver, respectively.
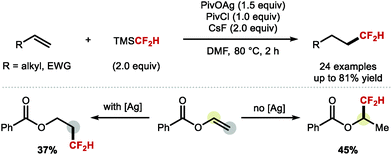 |
| | Scheme 31 Radical hydrodifluoromethylation reactions. | |
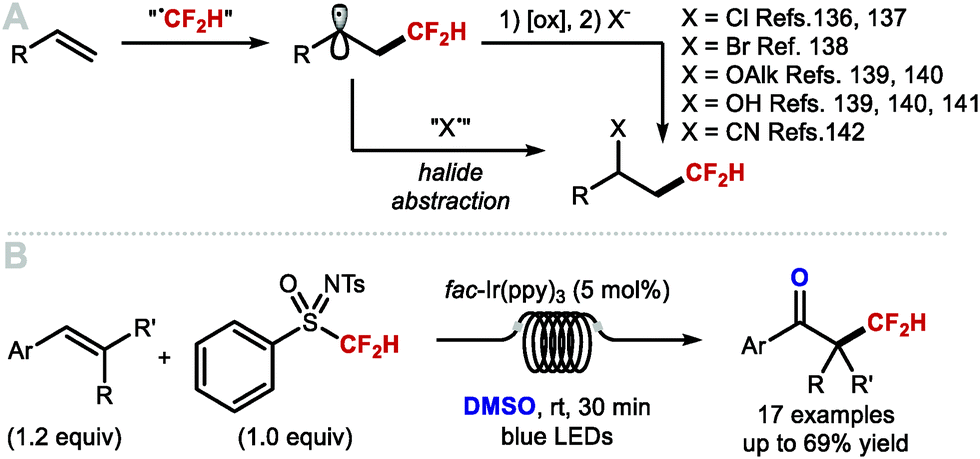
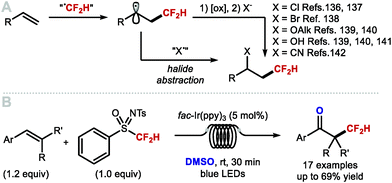
In 2015, Dolbier and co-workers disclosed an atom-economical atom transfer radical addition (ATRA) enabling chlorodifluoromethylation of alkenes with difluoromethyl sulfonyl chloride (Scheme 32A; X = Cl).135,136 In 2016, Qing and co-workers disclosed a related photocatalytic bromodifluoromethylation of alkenes with difluoromethylphosphonium bromide (Scheme 32A; X = Br).137 The method tolerates a broad range of substrates bearing different functional groups. Moreover, a one-pot bromo difluoromethylation–elimination process enabled access to α,β-unsaturated CF2H motifs (see Section 5.1). In the same year, Qing and co-workers expanded their ATRA protocol to the oxydifluoromethylation of styrenes (Scheme 32A; X = OAlk).138 Running the reaction in alcoholic solvents rendered etherified products, whilst the use of water-acetone mixture afforded alcohol-containing products (Scheme 32A, X = OH). Shortly after, Akita and co-workers also disclosed an oxydifluoromethylation of styrenes (Scheme 32A; X = OAlk, OH).139 More recently, Xu and co-workers reported an electrochemical variant to access hydroxydifluoromethylated products from N-arylacrylamides, utilizing CF2HSO2NHNHBoc as a source of CF2H radical (Scheme 32A, X = OH).140 In 2019, Xiao and co-workers disclosed a photoredox catalysed cyanodifluoromethylation of alkenes (Scheme 32A, X = CN).141 In 2019, Koike, Akita and co-workers reported that styrenes can undergo photoredox catalysed keto-difluoromethylation in flow by using a sulfoximine based difluoromethylating reagent and DMSO as oxidant and oxygen source (Scheme 32B).85 Compared to electrophilic methods that mainly focus on the synthesis of CF2H groups bound to quaternary centres, Koike and Akita's method is amenable to the synthesis of CF2H groups bound to secondary and tertiary centres.
 |
| | Scheme 32 Methods for vicinal difluoromethylation–functionalisation. | |
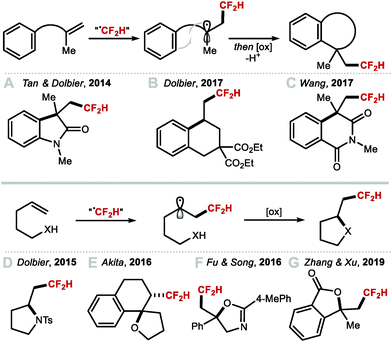
Intramolecular vicinal functionalisations have also been considered. Tan and co-workers reported that the combination of DFMS and ammonium persulfate with catalytic silver nitrate, allowed for a one-step synthesis of 2,2-difluoroethyl oxindoles (Scheme 33A).142,143 In 2017, Dolbier, and co-workers demonstrated that allyl malonates also undergo difluoromethylation–cyclisation to give fluoroalkyl tetralins (Scheme 33B).144 In the same year, Wang and co-workers disclosed the difluoromethylation–cyclisation of N-methacryloyl benzamides affording difluoromethyl-containing isoquinolinediones (Scheme 33C).145 Dolbier and co-workers reported a photoredox difluoromethylation–cyclisation of N- and O-based nucleophiles to form pyrrolidines and γ-lactones (Scheme 33D).146 Activation of difluoromethanesulfonyl chloride (CF2HSO2Cl) by a copper-based photocatalyst rendered the CF2H radical which added to the terminal alkene. The resulting C-centred radical intermediate underwent oxidation and cyclisation, or halide abstraction to form a chlorinated intermediate that readily cyclised under the reaction conditions. In 2016, a similar strategy was developed by Akita and co-workers used for the highly diastereoselective synthesis of difluoromethyl spiroethers from aryl-fused cycloalkenylalkanols (Scheme 33E).147 In the same year, Song and co-workers used amides as nucleophiles in an Ir-catalysed difluoromethylation–cyclisation leading to difluoroethyloxazolines and benzoxazines (Scheme 33F).148 In 2019, Xu and co-workers disclosed the electrochemical difluoromethylation-lactonization of alkenes using CF2HSO2Na in a Pt/Pt cell immersed in a 7:1 MeCN/H2O mixture (Scheme 33G).149 Under these mild acidic conditions (HOAc (3 equiv.)), various carboxydifluoromethylated products were obtained in moderate to good yields (up to 76%).
 |
| | Scheme 33 Difluoromethylation–cyclisation reactions. | |
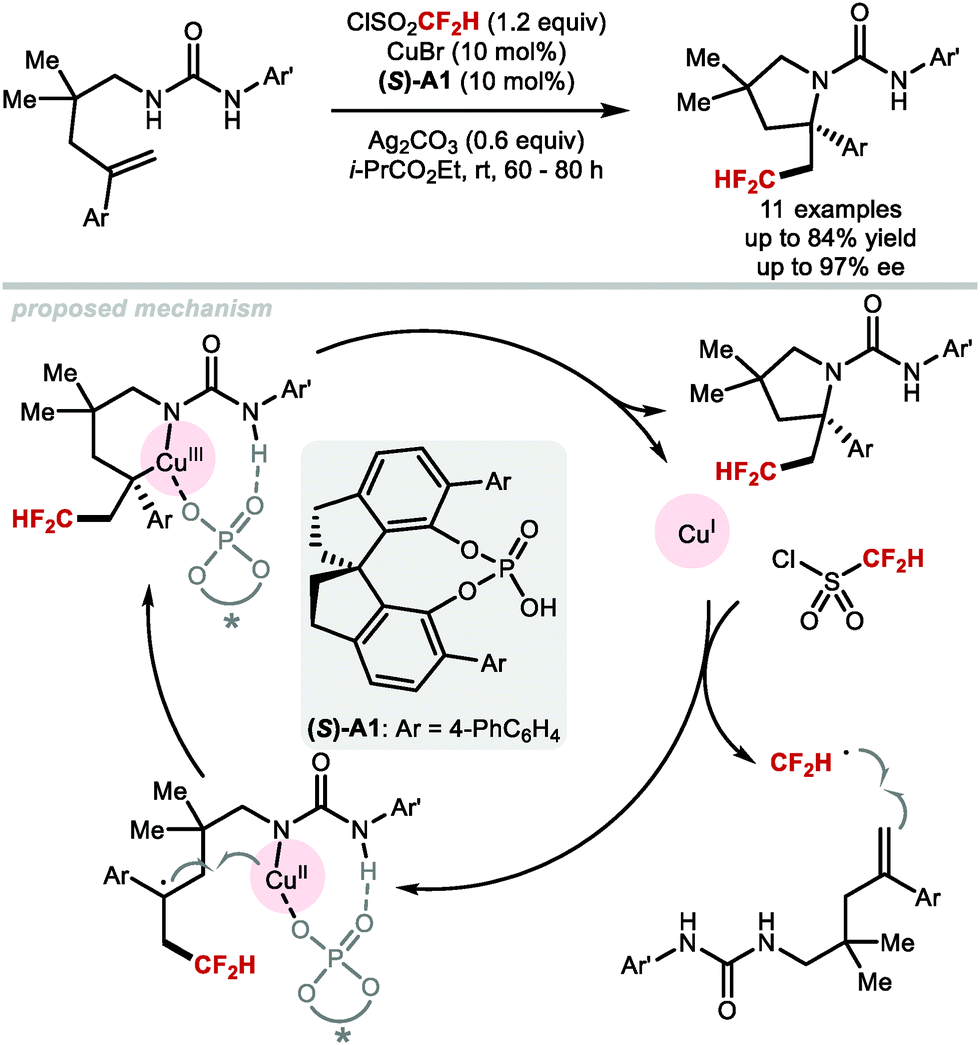
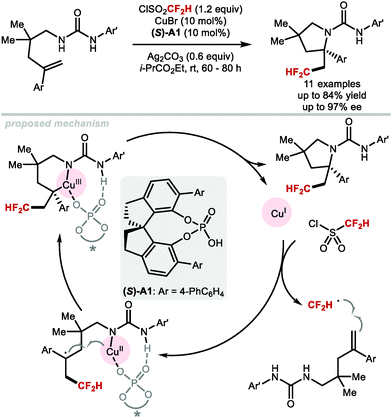
An elegant asymmetric difluoromethylation–cyclisation reaction was reported by Liu and co-workers in 2017 (Scheme 34).150 Urea-containing styrenes underwent difluoromethylation–cyclisation under CuI catalysis in the presence of catalytic amount of chiral phosphoric acid (S)-A1. The products were formed in enantiomeric excesses up to 97%. The CuI catalyst is involved both in the activation of difluoromethanesulfonyl chloride and the enantiodetermining cyclisation of the urea to form enantioenriched pyrrolidines.
 |
| | Scheme 34 Catalytic asymmetric difluoromethylation–cyclisation. | |
3.4 C(sp3)–CF2H bond formation: cross-coupling
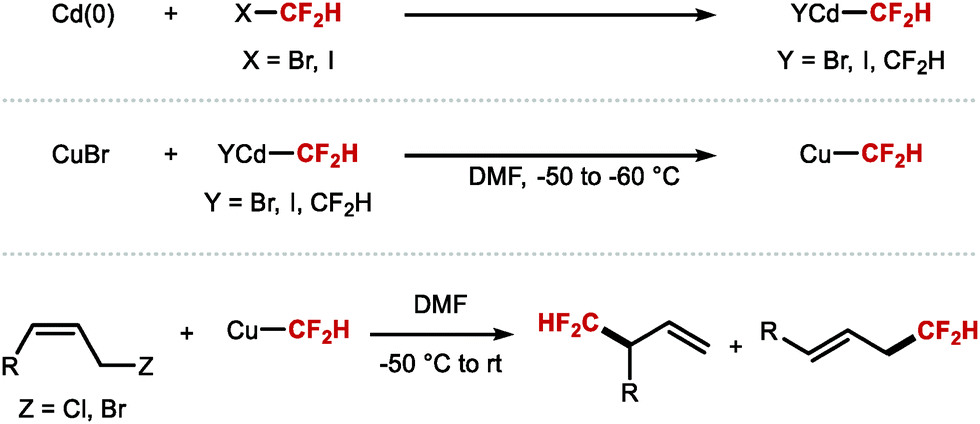
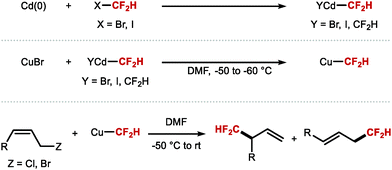
As eluded to in the introduction of Section 2.1, the first reports disclosing the use of transition metal–CF2H complexes for C(sp3)–CF2H bond formation date back to 1988 when Burton demonstrated that a difluoromethyl cadmium reagent was prepared by metal insertion into difluoroiodomethane (Scheme 35).43,44 This cadmium reagent was used for the difluoromethylation of allylic halides and propargylic (pseudo)halides to afford allylic difluoromethyl products and difluoromethyl allenes, respectively. In 2007, the same authors demonstrated the transmetallation of CdCF2H species to CuI, as well as the use of both CdCF2H and CuCF2H for the difluoromethylation of allylic halides, propargylic derivatives and 1-iodoalkynes.45 The authors found that CuCF2H is less stable than CdCF2H, and decomposes readily at room temperature. Moreover, the copper species allows for higher level of regiocontrol in allylic difluoromethylation.
 |
| | Scheme 35 Pioneering studies on the synthesis and reactivity of [CdCF2H] and [CuCF2H] complexes for C(sp3)–CF2H bond formation. | |
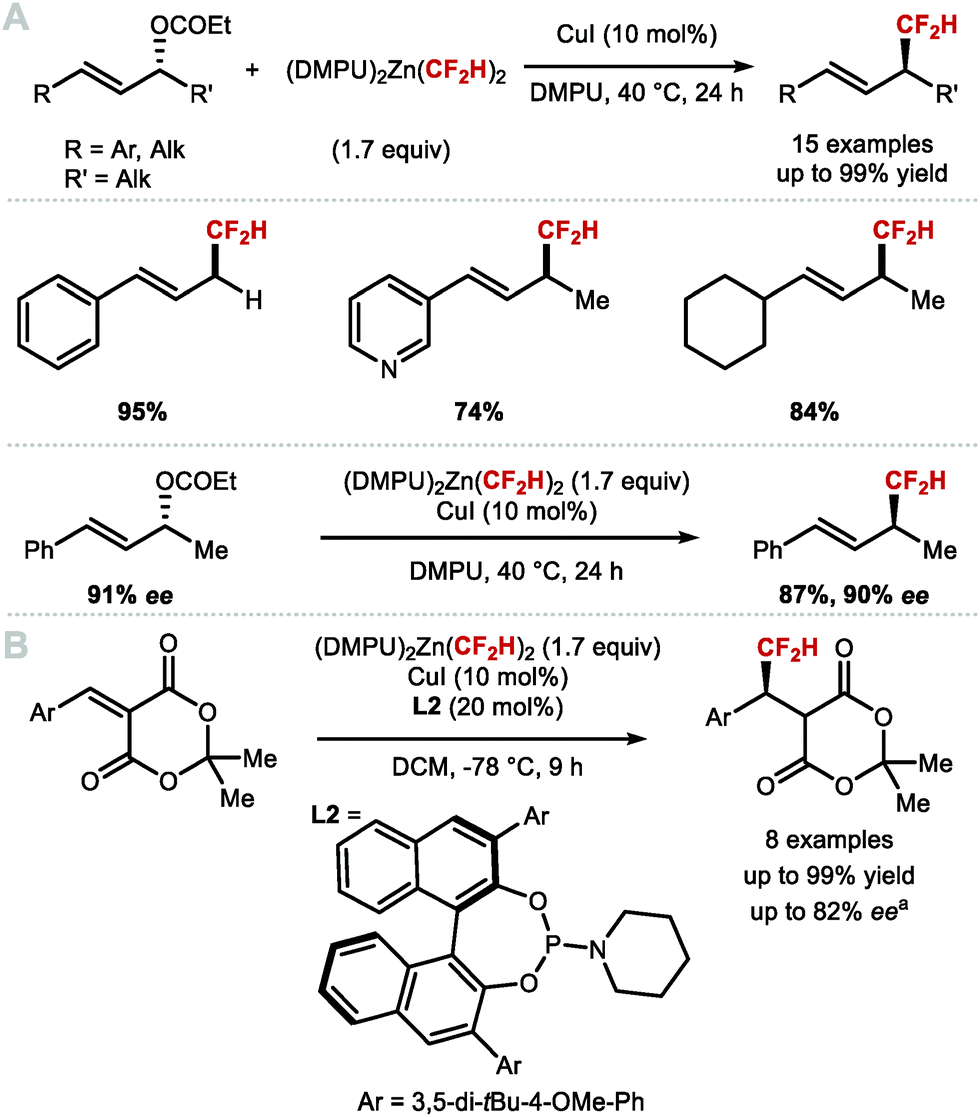
These findings inspired Mikami and co-workers to re-investigate allylic difluoromethylation. They reported that regioselective difluoromethylation of allylic carbonates was feasible using nucleophilic [(DMPU)2Zn(CF2H)2] in the presence of catalytic CuI (Scheme 36A).151 In 2019, the same authors disclosed an asymmetric variant employing CuI and the chiral phosphoramidite ligand L2 in-substoichiometric amount (Scheme 36B).152
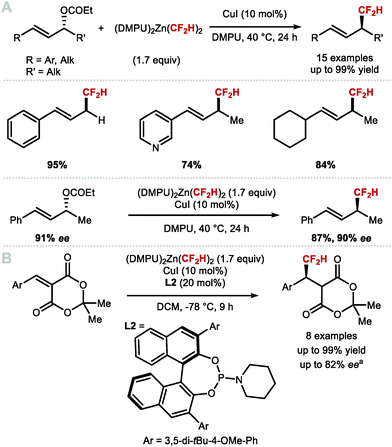 |
| | Scheme 36 C(sp3)–CF2H metal mediated or catalysed bond formation. aEnantiomeric excess measured on a derivative of the product shown. | |
In 2019, Liu and co-workers applied a Cu-catalysed C(sp3)-difluoromethylation to alkyl radicals generated from redox active esters (Scheme 37A).153 [(DMPU)2Zn(CF2H)2] was used for transmetallation onto CuI to form a LnCuICF2H species. This species participates into a single-electron reduction of the redox-active esters. After decarboxylation, the alkyl radical recombines with LnCuIICF2H species to afford LnCuIII(CF2H)(alkyl) that undergoes reductive elimination to afford the difluoromethyl alkane with concomitant regeneration of the CuI catalyst. The applicability of this methodology was demonstrated on primary, secondary and tertiary carboxylic acids, as well as biologically relevant molecules. In the same year, the same authors disclosed a benzylic C–H difluoromethylation via H-atom abstraction by amidyl radicals formed from N-chlorocarboxamides (Scheme 37B).154 The method was demonstrated on several examples (up to 91% yield), and allowed for the difluoromethylation of primary and secondary C(sp3)–H bonds; however, only substrates bearing a benzylic C–H bond were sufficiently activated to furnish the desired product. More recently, Liu and co-workers reported a two-step deaminative difluoromethylation (Scheme 37C).155 Various pyridinium salts derived from amines underwent difluoromethylation under Cu-catalysis (>50 examples, up to 93% yield).
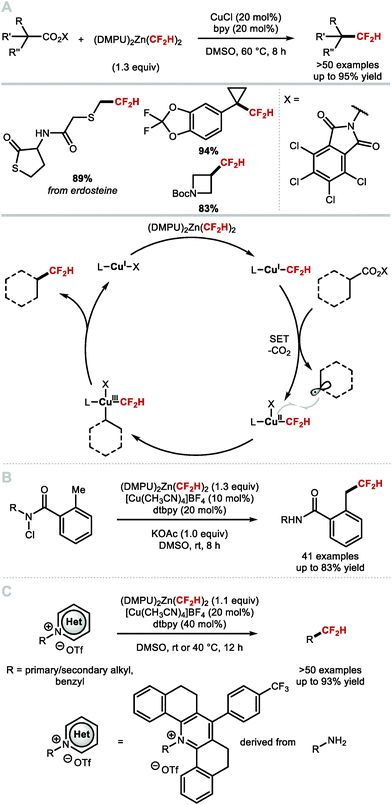 |
| | Scheme 37 Decarboxylative difluoromethylation, C–H difluoromethylation and deaminative difluoromethylation of C(sp3) centres. | |
In 2021, Shen and co-workers described the direct difluoromethylation of unactivated alkyl bromides, iodides, tosylates and mesylates (Scheme 38).156 The authors illustrated that alkyl iodides, tosylates, and mesylates readily reacted with TMSCF2H and CsF as activator under copper catalysis. Alkyl bromides required a combination of stoichiometric CuI and catalytic amounts of Pd(dba)2. This method is complementary to existing hydrodifluoromethylation technologies applied to access similar products from unactivated alkenes.
 |
| | Scheme 38 Difluoromethylation of alkyl(pseudo)halides. | |
4. (O/S/N)–Difluoromethylation
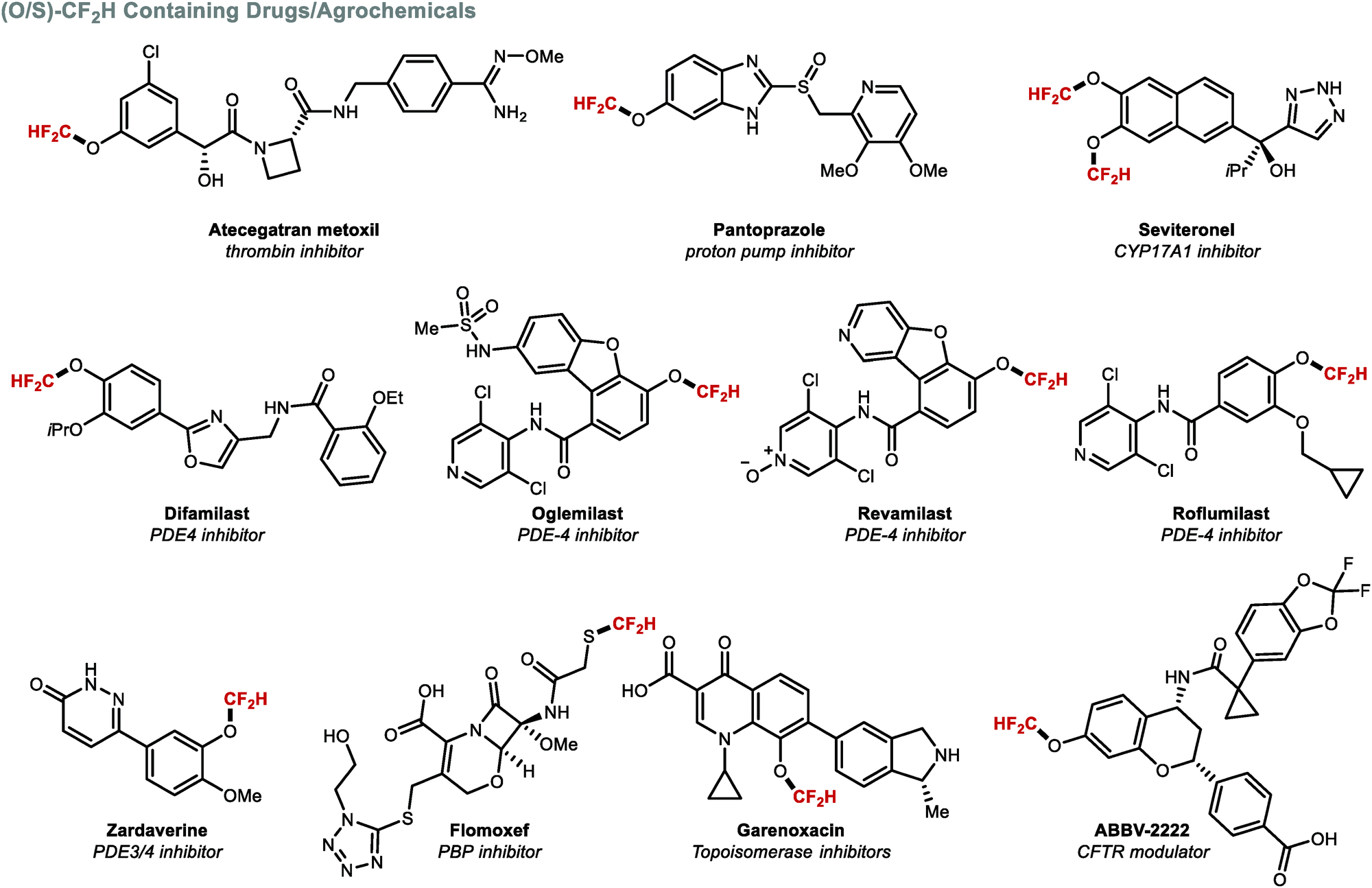
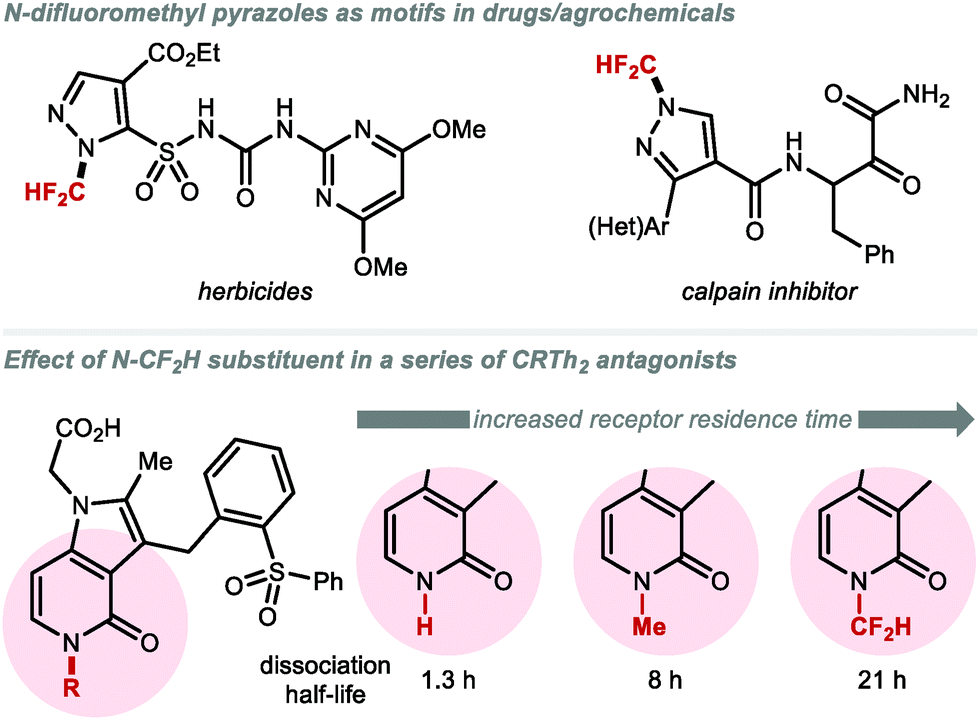
Apart from their different conformational preference (Fig. 3), (per)fluoroalkoxy (RfO) groups vary in a number of additional parameters such as Hammett constants σm and σp.157,158 While the OCF3 group has σm and σp values of 0.38 and 0.35 respectively, the OCF2H group exhibits weaker electron-withdrawing effects (σm = 0.31 and σp = 0.18). A further decrease is observed for OCH2F (σm = 0.20 and σp = 0.02). These trends have direct implications on physicochemical properties. For example, lipophilicity and metabolic stability tend to increase with fluorine substitution. Due to its intermediary status within the RfO family, the OCF2H group has become a prevalent motif amongst pharmaceuticals and agrochemicals, with several FDA approved drugs bearing this motif (Fig. 11). Among these, a prominent example is the block-buster drug Pantoprazole®, a marketed proton-pump inhibitor used in the treatment of gastroesophageal reflux disease (GERD).159
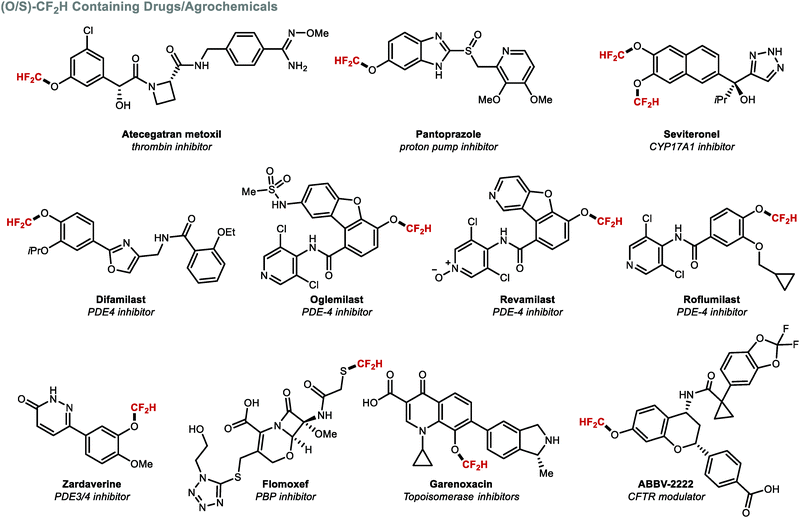 |
| | Fig. 11 Biomolecules containing the (O/S)CF2H motif. | |
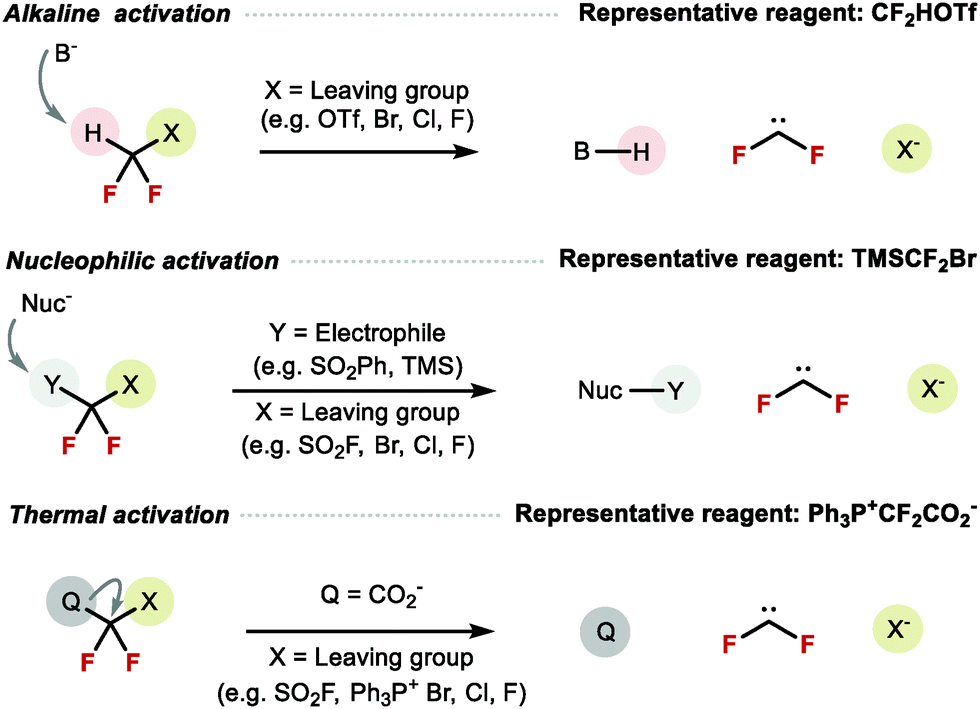
In the past decade, with the growing number of RfO containing bioactive molecules in drug discovery pipelines, the demand for novel methods to construct X–CF2H (X = O, S, N) both on aromatic and aliphatic backbones increased. The most common strategy for their synthesis is difluorocarbene insertion into the X–H bond. Alternative methods include decarboxylative fluorination,103 electrophilic difluorination,160 and more recently difluoromethoxylation161 and difluoromethylthiolation.162 This review focuses on X–CF2H bond disconnection, which relies on the availability of difluorocarbene reagents. Various modes of activation of these reagents are known including the use of base, nucleophile, as well as thermal activation (Scheme 39).
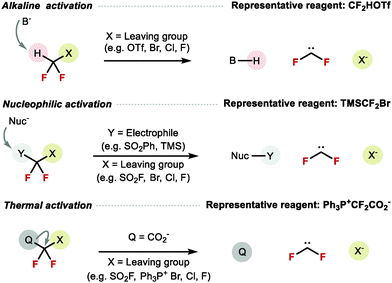 |
| | Scheme 39 Modes of activation to generate difluorocarbene. | |
4.1 O–/S–difluoromethylation
4.1.a Difluoromethylation of (thio)phenols applying difluorocarbene chemistry.
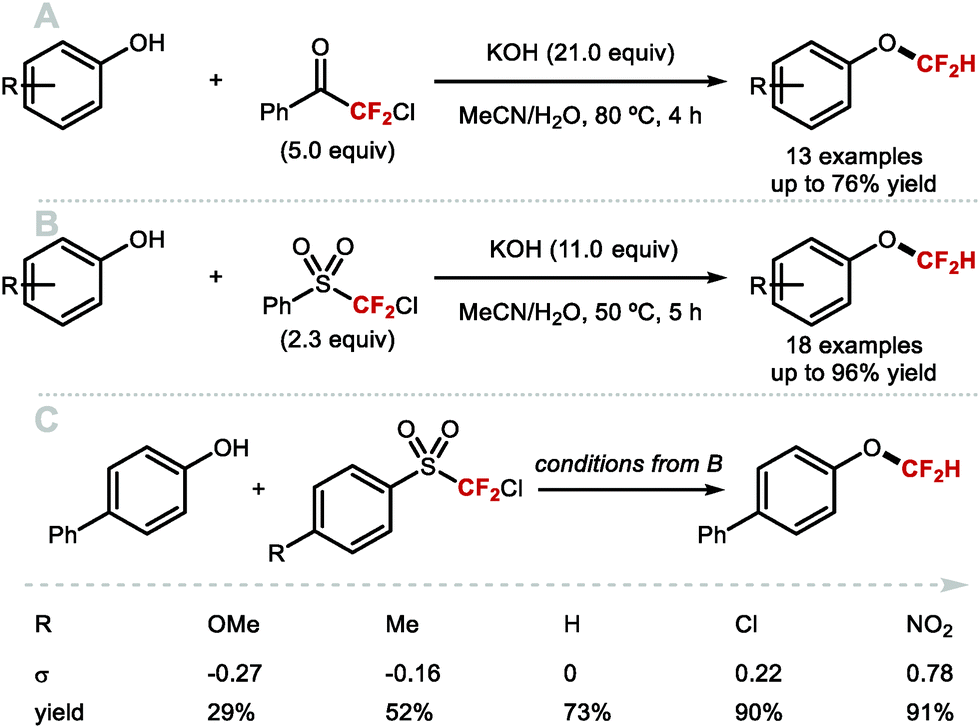
As early as 1960, Haszeldine reported sodium chlorodifluoroacetate (ClCF2CO2Na) as a difluorocarbene reagent.163 Since then, various difluorocarbene reagents have been developed. Many of these hazardous gaseous reagents are limited in scope and require harsh reaction conditions for their activation. These characteristics hamper wider application in modern organofluorine chemistry. As a result, many research groups have developed a new generation of non-ozone depleting difluorocarbene reagents which can be activated under mild conditions and exhibit broader functional group tolerance. In 2006, Hu and co-workers invented the new difluorocarbene reagent 2-chloro-2,2-difluoroacetophenone, which was prepared without the need to use ozone-depleting chemicals (Scheme 40A).164 This reagent was successfully employed for the difluoromethylation of phenol derivatives, offering moderate yields of up to 76%. One year later, the same authors reported the synthesis of chlorodifluoromethyl phenyl sulfone, another non-ODS-based difluorocarbene reagent, which provided access to N–CF2H (further detail in section 4.2) as well as O-difluoromethylated phenols with different ring electronics (Scheme 40B).165 As an extension of their work, Hu and co-workers published an additional report in 2011 on the effect of aromatic ring substituents on the reactivity of these difluorocarbene reagents (Scheme 40C).166 Substituent effects were more pronounced for the sulfone-based reagents. Extensive screening revealed that p-chlorophenyl chlorodifluoromethyl sulfone and p-nitrophenyl chlorodifluoromethyl sulfone were the most efficient for transferring difluorocarbene to phenols.
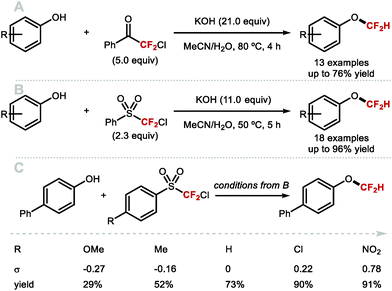 |
| | Scheme 40 Non-ODS based difluorocarbene reactions. | |
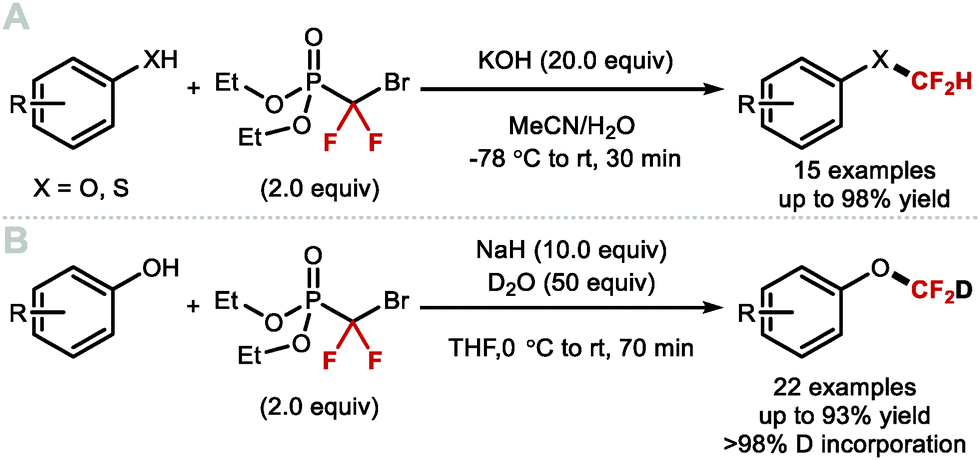
In 2009, Zafrani and Segall, described the difluorocarbene reactivity of BrCF2P(O)(OEt)2 on both phenols (9 examples, up to 96% yield) and thiophenols (6 examples, up to 98% yield) (Scheme 41A).167 The difluoromethylated products were obtained in the presence of 20 equivalents of KOH in a solvent mixture of MeCN/H2O within 30 minutes. Wu and Zou further extended this methodology to the preparation of ArOCF2D (Scheme 41B).168 The authors found that their protocol was broad in scope, tolerating electron-rich and electron-deficient arenes, as well as heterocyclic substrates (22 examples, up to 93% yield) with excellent deuterium incorporation (>98%D). The protocol was scaled up to 30 g, with no compromise on yield (90%) or deuterium incorporation (99%D). A general method to access ArOCF2D presents a valuable addition to the medicinal chemist toolbox for application in drug discovery programs. It is indeed well established that substitution of hydrogen for deuterium can lead to improvement of the pharmacokinetics, pharmacodynamics and overall metabolic stability of a drug molecule.
 |
| | Scheme 41 Difluoromethylation and deuterodifluoromethylation of (thio)phenols with diethyl bromodifluoromethylphosphonate. | |
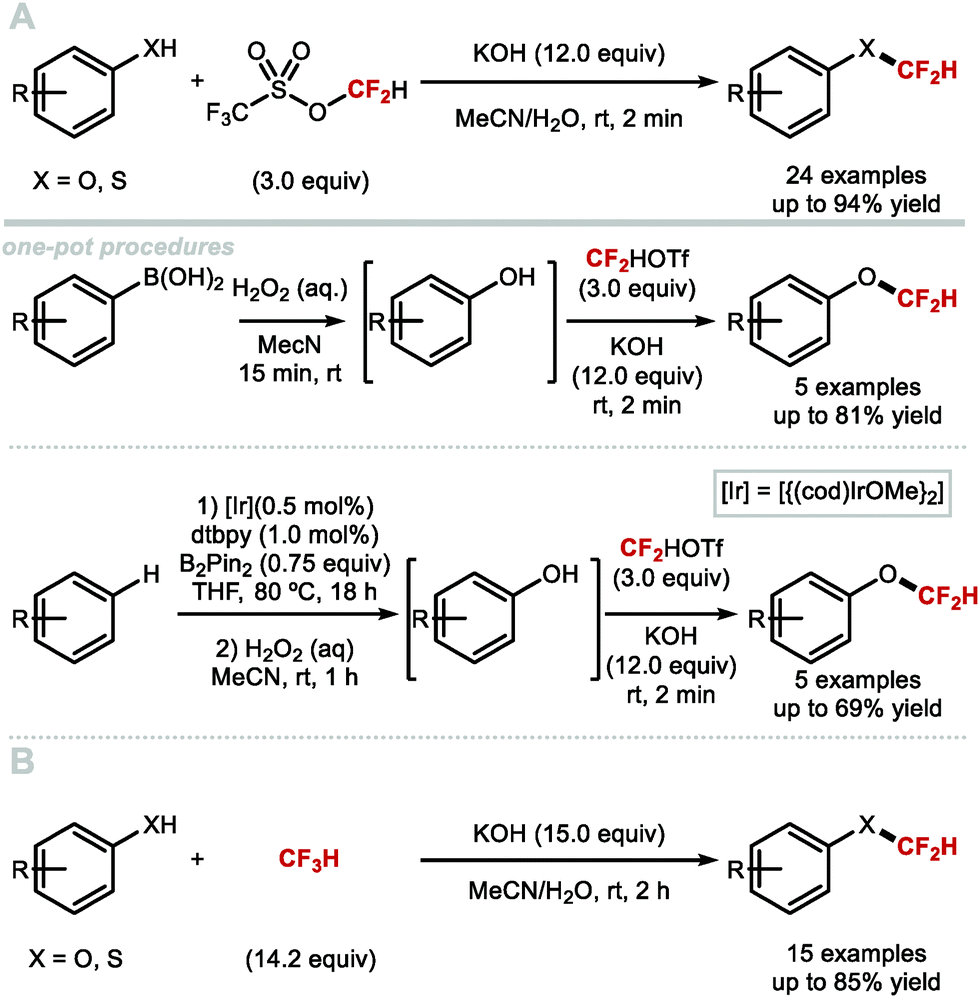
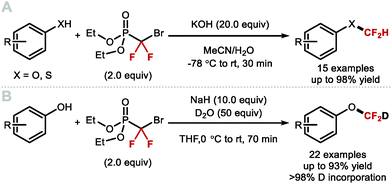
In 2013, the Hartwig group described the synthesis difluoromethyl triflate and its application as a difluorocarbene reagent for the difluoromethylation of phenols and thiophenols (Scheme 42A).169 This commercially available non-gaseous and non-ODS reagent allows difluoromethyl ethers and sulfides to be prepared within a few minutes at ambient conditions under aqueous basic conditions. The broad substrate scope and short reaction times make this method amenable to one-pot sequences involving in situ generation of phenols from either aryl boronic acids or simple arenes. For electron-rich phenols that led to side-products, difluoromethyl triflate was replaced by difluoromethyl nonaflate, a modification increasing conversions towards the desired difluoromethyl ether products. Leroux and co-workers expanded the original substrate scope reported by Hartwig to N-containing heteroaromatics.170 They also prepared a series of OCF2H analogues of imidacloprid and thiacloprid, two blockbuster insecticides. Dolbier and co-workers reported that fluoroform is suitable for the difluoromethylation of phenols (15 examples, up to 88% yield) and thiophenols (4 examples, up to 76% yield). A large excess of fluoroform was however required to obtain satisfactory yields (Scheme 42B).171
 |
| | Scheme 42 (Difluoromethyl)triflate as a mild and reactive difluorocarbene reagent. | |
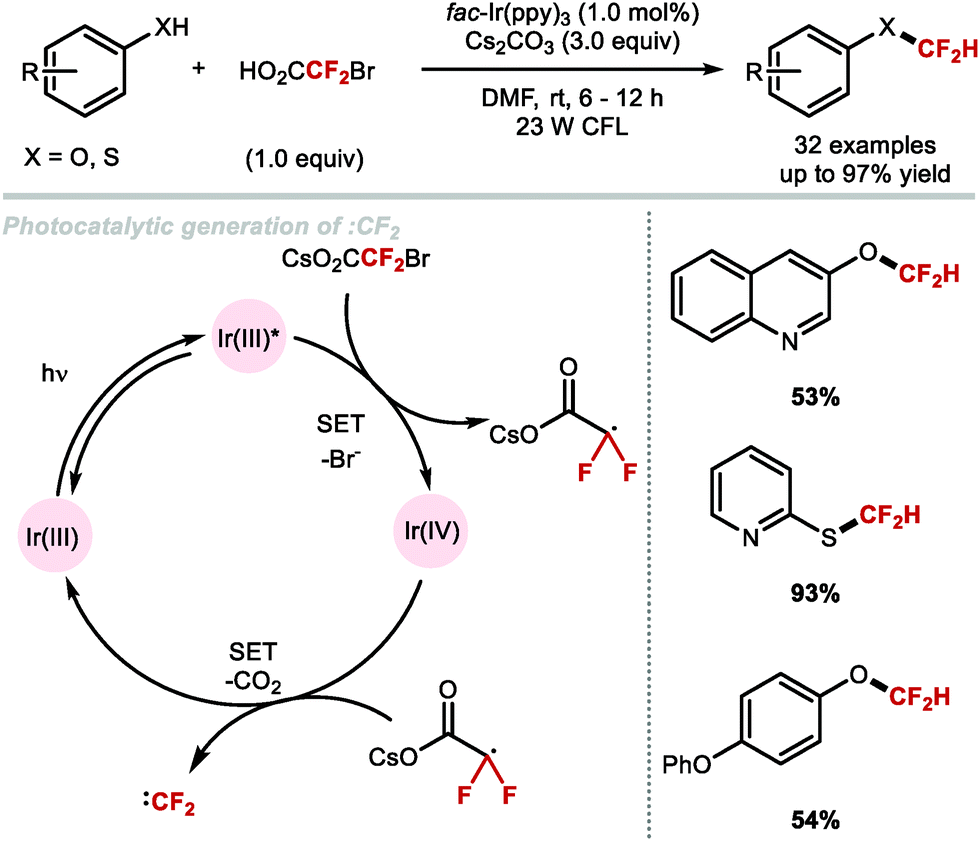
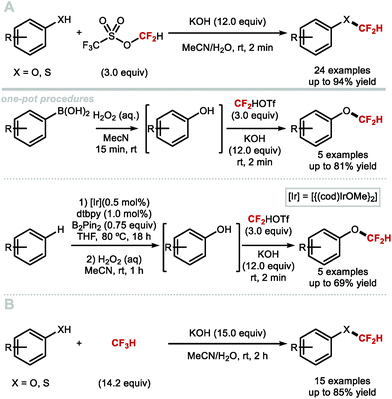
In 2017, Fu and co-workers disclosed the first example of difluorocarbene generation under photoredox catalysis (Scheme 43).172 Mechanistically, the cesium salt of bromodifluoroacetic acid (BrCF2CO2Cs) is proposed to quench the excited state of the Ir(III)* photocatalyst (PC*). The resulting oxidised Ir(IV) species was then returned to its native oxidation state through SET, thereby generating the difluorocarbene species. Reaction with a selection of phenolates and thiophenolates afforded the resulting difluoromethylated products in excellent yields.
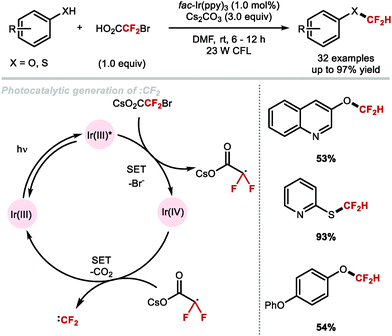 |
| | Scheme 43 Difluorocarbene generation under photoredox catalysis for the difluoromethylation of (thio)phenols. | |
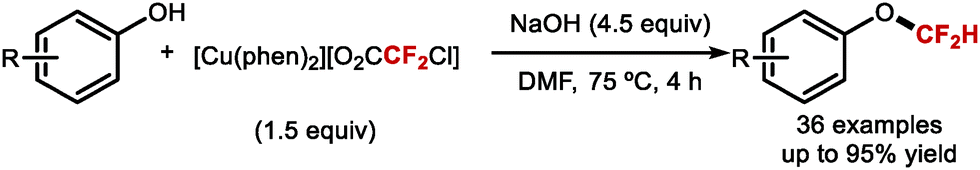
Building on the studies of Zhang62,64,65 and Xiao63 who had investigated the reactivity of [LnPd = CF2] complexes, Weng designed [Cu(phen)2][O2CCF2Cl], a stable copper complex readily prepared from CuCl and ClCF2CO2H (Scheme 44).173 Under aqueous basic conditions and heat, [Cu(phen)2][O2CCF2Cl] readily releases difluorocarbene and converts phenols to the corresponding difluoromethoxyarenes.
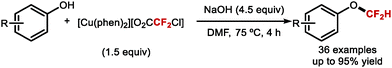 |
| | Scheme 44 Difluorocarbene release from the [Cu(phen)2][O2CCF2Cl] complex. | |
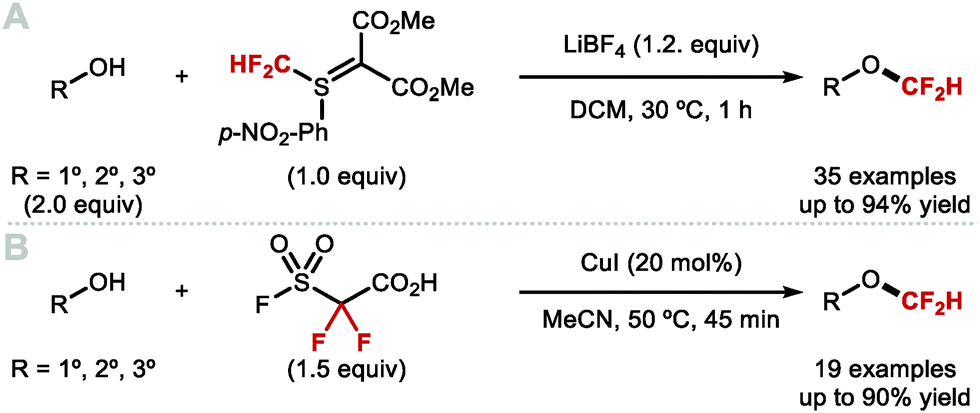
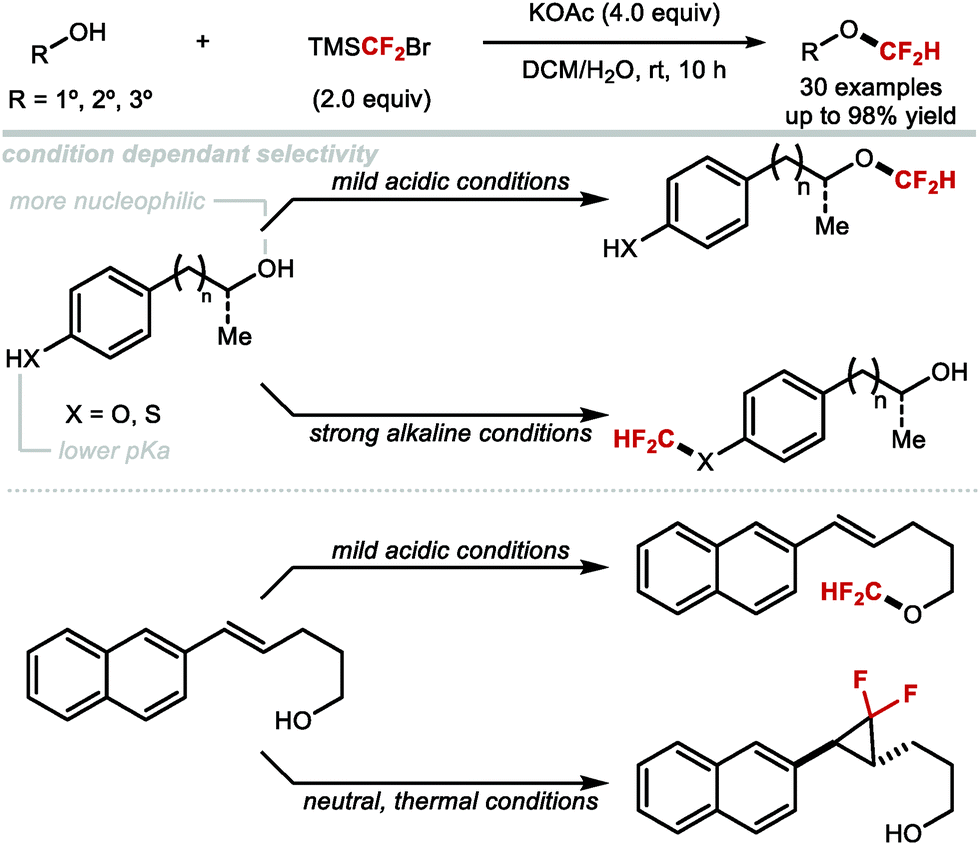
4.1.b Difluoromethylation of aliphatic alcohols/thiols.
The syntheses of aryldifluoromethyl ethers have focused on difluorocarbene chemistry under basic conditions. Applying similar conditions to the difluoromethylation of alkyl alcohols was found less effective, an observation consistent with the higher pKa of alkyl alcohols (pKa ∼ 16) compared to phenols (pKa ∼ 10). Notably, alkyl alcohols can react with difluorocarbene without prior deprotonation.174 These insertion reactions required mild reaction conditions to avoid competing pathways triggered by additives such as bases. Nonetheless, Hine and Tanabe,175 and later Mizukado176 reported that the difluoromethylation of aliphatic alcohols was possible with ClCF2H under basic conditions (Scheme 45). Similarly, Burton177 and Hu178 showed that the difluoromethylation of aliphatic alcohols was possible with BrCF2P(O)OEt2 and TMSCF2Br under basic conditions, albeit in low conversions. A variety of early reports which operate under neutral conditions, have proven more effective. As early as 1965, Mitsch and Robertson179 showed that the photolysis of difluorodiazirine could generate difluorocarbene and permit the difluoromethylation of a limited selection of alcohols. In 1995, Miethchen and co-workers reported the O-difluoromethylation of monosaccharides mediated by trifluoromethylzinc bromide,180 and in 2005, Mizukado and co-workers described the use of hexafluoropropene oxide as a difluorocarbene reagent capable of reacting with aliphatic alcohols.181 However, all of these methods either exhibit poor reactivity or lack generality in scope.
 |
| | Scheme 45 Sub-optimal difluoromethylation of aliphatic alcohols under basic and neutral difluorocarbene conditions. | |
More recently, several developments within this area have addressed the above-mentioned limitations which has led to more general protocols for the difluoromethylation of aliphatic alcohols. In 2016, the Shen group discovered that difluoromethyl-(4-nitrophenyl)-bis-(carbomethoxy)-methylide sulfonium ylide is ideal for the difluoromethylation of alkyl alcohols (Scheme 46A).182 This reagent in conjunction with a Lewis acid activator (LiBF4) resulted in the synthesis of a series of alkyl difluoromethylethers in good yields. Mechanistically, this reaction is different from classical difluorocarbene based reactions, as shown to proceed via nucleophilic substitution. Inspired by Weng's work, Mykhailiuk and co-workers reported the activation of FSO2CF2CO2H with CuI for the synthesis of structurally diverse difluoromethyl ethers from polyfunctional alcohols (Scheme 46B).183 This methodology astutely exploits copper catalysis to mediate the transfer of difluorocarbene. The conditions are mild and produce the desired products in moderate to high yields. The method was scalable and tolerated various functional groups such as carbamates and esters. However, only primary and secondary alcohols displayed useful reactivity. Significantly lower yields were indeed obtained for tertiary alcohols.
 |
| | Scheme 46 Difluoromethylation of aliphatic alcohols under difluorocarbene conditions. | |
In 2017, Hu showed that activation of TMSCF2Br with NaOH, KOAc or KHF2 allowed for the difluoromethylation of a variety of aliphatic alcohols (primary, secondary and tertiary) (Scheme 47).174 For tertiary alkyl difluoromethylethers, excess TMSCF2Br was required to obtain high yields. The authors noted that the higher nucleophilicity of aliphatic alcohols compared to phenols, permitted difluoromethylation under mild acidic conditions. In contrast, phenols required basic conditions with difluoromethylation proceeding via phenolate anions. Furthermore, chemoselective difluoromethylation was elegantly achieved by tuning the reaction conditions. Under weakly acidic conditions, employing KHF2 as an activator, difluorocarbene selectively inserted into the aliphatic O–H bond of 4-(4-hydroxypentyl)phenol in good yield (80%). Conversely, when KOH was used, difluoromethylation occurred at the phenol oxygen in good yield (71%). Similarly, in the case of (4-mercaptophenyl)methanol, difluorocarbene inserted into the O–H bond when the reaction was performed under mild acidic conditions. When aqueous NaOH was used, difluoromethylation occurred at the thiol. With (E)-5-(naphthalen-2-yl)pent-4-en-1-ol, a two-phase system consisting of DCM and water facilitated the insertion of difluorocarbene into the O–H bond at 0 °C under mild acidic conditions. In contrast, under homogeneous conditions (toluene at high temperature), the difluorocyclopronated product was formed exclusively in the presence of nBu4NBr as activator.
 |
| | Scheme 47 TMSCF2Br as a multi-purpose difluorocarbene reagent for the chemoselective difluoromethylation of aliphatic alcohols. | |
In 2019, Zhang and co-workers reported that S-(difluoro-methyl)sulfonium salt is suitable for the facile difluoromethylation of aliphatic alcohols (Scheme 48).184 The optimised reaction conditions involved the use of NaOAc (5.0 equivalents) and nBu4NBF4 (20 mol%) as initiator, in a solvent mixture of CH2Br2 and H2O at room temperature. Similar to the seminal reports from Hu and Shen, a wide array of functional groups including ester, nitro, methoxy and boronic ester were tolerated in the alcohol substrate. The method showed high selectivity for the difluoromethylation at the aliphatic OH site in the presence of functional groups such as phenol, carbamate, alkyne, alkene, or N-heterocycles. Furthermore, difluoromethylation of aliphatic thiols was possible by omitting the initiator (nBu4NBF4), changing the counterion of their difluorocarbene reagent from PF6− to BF4−, the base from NaOAc to KOH (2.4 equiv.), and the solvent to MeCN.
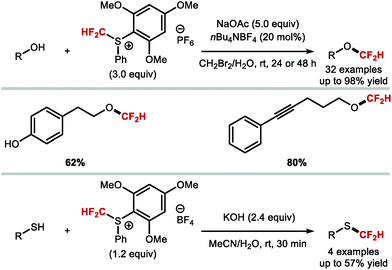 |
| | Scheme 48
S-(Difluoromethyl)sulfonium salts as effective difluoromethylating reagents of aliphatic alcohols and thiols. | |
4.1.c Difluoromethylation of thiols under radical conditions.
One-electron chemistry has been shown to proceed with selectivity complementary to two-electron pathways. Baran and co-workers were the first to successfully difluoromethylate with DFMS, a series of heteroaromatic thiols including 2-mercaptobenzothiazole, 2-mercaptobenzoxazole and 2-mercapto-1-methylbenzimidazole in moderate yields (Scheme 49A).83 In 2017, Yi and co-workers developed an alternative silver-catalysed difluoromethylation process (Scheme 49B).185 The reaction employs NaSO2CF2H as source of CF2H radical, K2S2O8 as oxidant and AgNO3 (10 mol%). Products were formed in good yields, showing good functional group tolerance including groups that would be reactive towards difluorocarbene. The Studer group recently presented a facile difluoromethylation of various thiols using (difluoromethyl)triphenylphosphonium bromide (Scheme 49C).86 Mechanistic studies revealed that an SRN1-type mechanism is at play. The authors illustrate the power of this radical process with the selective difluoromethylation of thiol with no competing reaction taking place at phenol or aniline. Aliphatic thiols were not reactive.
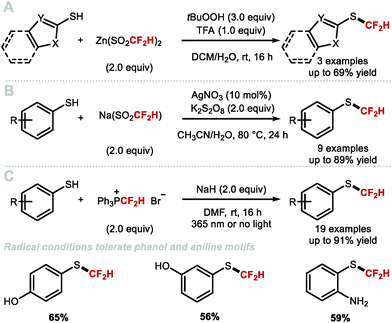 |
| | Scheme 49 Difluoromethylation of aromatic and heteroaromatic thiophenols under radical conditions. | |
4.2
N-Difluoromethylation
N-Heteroaromatic scaffolds such as imidazoles and benzimidazoles, are prevalent structural motifs in medicinal chemistry.186 Methods for the selective insertion of a CF2H group into a N–H bond have therefore a myriad of applications ranging from medicinal to agricultural chemistry. In this context, N-difluoromethylated pyrazoles were investigated in SAR studies of calpain inhibitors and in herbicide research (Fig. 12). Recently, Andrés and co-workers were able to modulate receptor residence times in a family of pyridone-containing CRTh2 antagonists by varying the substituent at the pyridine nitrogen.187 Their study showed that N-difluoromethyl 2-pyridones had a significantly higher dissociation half-life than the corresponding non-substituted or N-methylated pyridones.
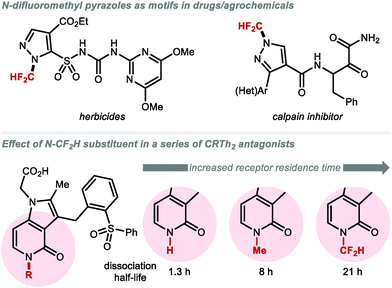 |
| | Fig. 12 Application of N–CF2H compounds. | |
Sundermeyer was amongst the first to report the difluoromethylation of an N-nucleophile as early as 1985, by reaction of CF2HSO2Cl with trimethylammonium chloride (Scheme 50).188
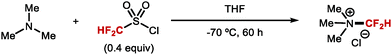 |
| | Scheme 50 Sundermeyer's observation in the reaction of trimethylamine with difluorocarbene. | |
In the years that followed, more general strategies were reported for N-difluoromethylation, but mostly employed gaseous ODS such as ClCF2H. In 1998, Lyga et al. described the difluoromethylation of five-membered NH-heterocycles using sodium hydride as a base followed by treatment with excess ClCF2H (Scheme 51A).189 In 2002, Petko et al. expanded the difluoromethylation of nitrogen nucleophiles to various sulfonamides using ClCF2H under strong alkaline conditions (Scheme 51B).190 Since then, further difluoromethylation protocols for additional classes of N-nucleophiles have emerged.
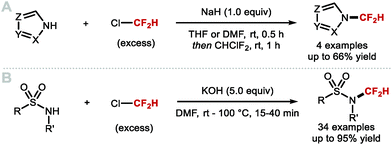 |
| | Scheme 51
N-Difluoromethylation reactions with ClCF2H. | |
In 2006 Ando et al. reported the difluoromethylation of 2-acetamidopyridines (Scheme 52).191 The use of catalytic amounts of 18-crown-6 in combination with sodium chlorodifluoroacetate (SCDA) allowed for chemoselective difluoromethylation at the pyridine nitrogen. Treatment with KHSO4 under reflux gave the corresponding N-difluoromethyl-2-pyridones in good yields.
 |
| | Scheme 52 Synthesis of N-difluoromethyl-2-pyridones with SCDA. | |
In 2007, Hu and co-workers developed chlorodifluoromethyl phenyl sulfone as a novel non-ODS difluorocarbene source.165 The reagent was activated under aqueous basic conditions for the N-difluoromethylation of NH-heterocycles including imidazoles, benzimidazoles and benzotriazoles (Scheme 53A). The same group reported the difluoromethylation of similar substrates in non-aqueous conditions using instead N-tosyl-S-difluoromethyl-S-phenylsulfoximine (Scheme 53B).192 This sulfoximine-based reagent is a crystalline solid prepared via a copper(II)-catalysed nitrene transfer. The requirement of mild temperatures and a small excess of the sulfoximine are advantageous characteristics of this methodology. However, the need to react imidazole substrates with a strong base (sodium hydride) prior to addition of the difluoromethylation reagent hampered functional group tolerance. In 2013, a more cost-effective strategy for difluoromethylation of various nucleophiles emerged making use of SCDA in the presence of K2CO3 at 95 °C (Scheme 53C).193 Under these conditions, SCDA readily undergoes decarboxylation generating the difluorocarbene required for capture by nitrogen nucleophiles. A variety of nitrogenous heteroaromatic thiols showed good reactivity leading to difluoromethylated products in high yields, albeit in some cases with poor chemoselectivity. Overall, despite the low cost of SCDA, the method is blighted by the requirement of high temperatures and strong alkaline conditions. Simultaneously, Hu and co-workers reported the Freon-free synthesis of TMSCF2Br, a powerful difluorocarbene reagent.178 (Benz)imidazoles, benzotriazole and tetrazoles underwent N-difluoromethylation under concentrated aqueous basic conditions (Scheme 53D). Shortly after, Prakash et al. showed that similar (benz)imidazoles can be difluoromethylated in the presence of TMSCF3 and LiI in less than 2 hours in good to excellent yields (Scheme 53E).194 The reaction was performed at high temperatures (170 °C) using microwave irradiation or conventional heating. Electron-donating and electron-withdrawing substituents were tolerated, albeit with no control over regioselectivity or chemoselectivity. The methodology was extended to the difluoromethylation of biologically active molecules such as theophylline and 8-(1H-benzoimidazol-2-yl)-quinoline (8-BQ). In 2018, He and co-workers demonstrated that diethyl bromodifluoromethylphosphonate can be activated under mild conditions to afford N-difluoromethylated (benz)imidazoles and pyrazoles in high yields (Scheme 53F).195
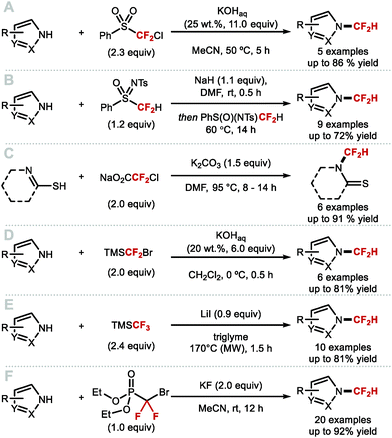 |
| | Scheme 53 Difluoromethylation of N-heterocycles. | |
Following the pioneering discoveries of Sundermeyer,188 Prakash and co-workers reported in 2011 that in situ formed N,N-dimethyl-S-difluoromethyl-S-phenylsulfoxinium tetrafluoroborate enabled efficient difluoromethylation of various tertiary amines (Scheme 54A).196 Zafrani and co-workers further reported the difluoromethylation of tertiary amines employing BrCF2P(O)(OEt)2 activated by CsF (Scheme 54B).197 The authors proposed that BrCF2P(O)(OEt)2 reacts with fluoride liberating the difluorocarbene intermediate. Nucleophilic attack by the amine followed by protonation affords α-difluoromethylated quaternary ammonium salts. Notably, hydroxyl, alkenyl, alkynyl and ester groups were all tolerated under these mild conditions. In 2018, Jana and co-workers disclosed alternative conditions for the difluoromethylation of N-tosyl protected anilines (Scheme 54C).198 Using an aqueous solution of LiOH in DMF to generate difluorocarbene from BrCF2CO2Et at room temperature, a broad range of N-difluoromethylated products were accessible. Neither electronic or steric perturbation compromised reactivity. In 2015, Shen et al. extended the use of [(NHC)Ag(CF2H)] complex to successfully difluoromethylate various aryldiazonium salts at nitrogen (Scheme 54D).51 The reaction afforded difluoromethyl diazene compounds in good to excellent yields. Good functional group tolerance was observed and reactions with aryldiazonium salts bearing electron-donating or electron-withdrawing groups all resulted in high yields. This N-difluoromethylation is unique because it does not proceed through a difluorocarbene mechanism.
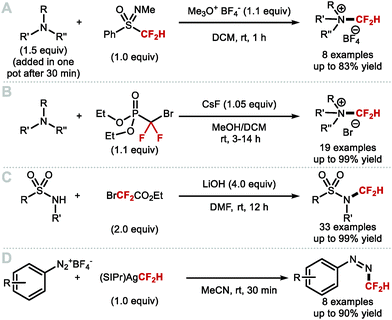 |
| | Scheme 54 Difluoromethylation of other nitrogen-based substrates. | |
The Tang group reported a unique transformation leading to N-difluoromethylated thioureas from azoles (Scheme 55A).199 To successfully prepare these products, the authors reacted elemental sulfur (S8), HOCH2SO2Na, and BrCF2CO2Et in DMA at 100 °C. The reaction was successful on a range of triazoles, imidazoles and benzimidazoles with the products obtained in satisfactory yields. Efforts from the Weng group led to a novel method for the synthesis of 3-difluoromethylbenzoxazole-2-thiones from 2-aminophenols (Scheme 55B).200 This alternative one-pot sequence employed S8 and ClCF2CO2Na under basic conditions (tBuONa) in DMF at 70 °C. The use of molecular sieves improved reaction yields.
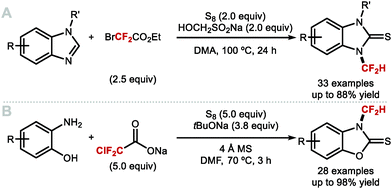 |
| | Scheme 55 Synthesis of N-difluoromethyl-thioureas and N-difluoromethyl benzoxazole-2-thiones. | |
5. Difluoromethylation of alkenes and alkynes
The difluoromethylation of alkenes has only recently been reported. Various difluoromethylation protocols developed for the conversion of aryl halides to difluoromethylarenes are also suitable for the difluoromethylation of vinyl halides.47,48 Furthermore, difluoromethylated alkenes can be accessed through a variety of radical-based methodologies featuring either photochemical or electrochemical activation. In contrast, difluoromethylated alkynes are prepared using difluorocarbene chemistry.
5.1 Difluoromethylation of alkenes
Qing and co-workers described the bromodifluoromethylation of alkenes, with the ATRA products undergoing in situ elimination upon addition of DBU (Scheme 56).137 This two-step one-pot indirect protocol yielded a small selection of difluoromethylated alkenes (4 examples, up to 83%) with good E/Z selectivity (up to 97:3).
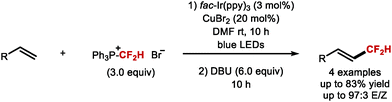 |
| | Scheme 56 Two-step one-pot difluoromethylation of alkenes. | |
In 2019, Zhao and Loh reported the photoredox catalysed difluoromethylation of enamides using difluoromethyltriphenylphosphonium bromide under Ir-photocatalysis (Scheme 57).201 A wide selection of enamides were readily difluoromethylated in good yields under the optimised reaction conditions, and in most cases, with complete E/Z stereoselectivity.
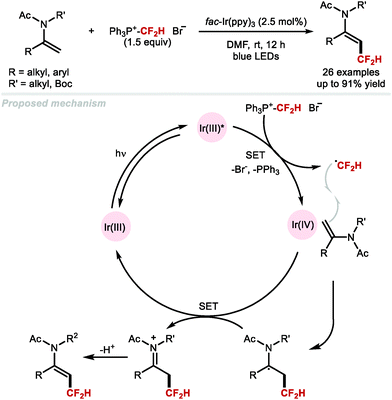 |
| | Scheme 57 Stereoselective difluoromethylation of enamides. | |
With electrochemistry re-emerging as a green strategy for organic synthesis, an electrochemical difluoromethylation approach has recently been considered by Xu and co-workers.202 Electrochemical C–H difluoromethylation of acrylamides was accomplished with CF2HSO2NHNHBoc as precursor of CF2H radical and Et4NOTs as electrolyte (Scheme 58A).140 The use of a reticulated vitreous carbon (RVC)/Pt electrode and applying 10 mA of current at 70 °C in a TFE/H2O mixture (5:1) afforded β-difluoromethylated acrylamides in good yields (5 examples, up to 97%) and good Z/E selectivity (≥10:1). In 2018, Xu and co-workers reported that ferrocene (Cp2Fe) is a highly efficient mediator for the electrochemical activation of CF2HSO2NHNHBoc for the release of CF2H radical (Scheme 58B).203 This protocol provided access to difluoromethylated dibenzazepines upon CF2H radical addition to an alkyne followed by 7-membered ring-forming homolytic aromatic substitution.
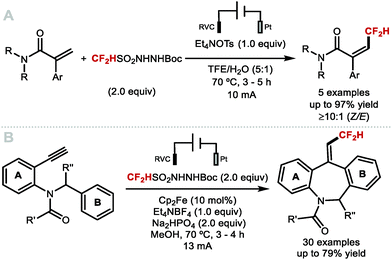 |
| | Scheme 58 Electrochemical methods for the difluoromethylation of alkenes and alkynes. | |
A recent report from Qing, Chu and co-workers demonstrated that styrenes, cinnamic acids and vinyl trifluoroborate salts efficiently underwent Ag-promoted radical difluoromethylation with TMSCF2H (Scheme 59).97 This method affords difluoromethyl alkenes in high yields with no requirement for light irradiation or electrochemical activation.
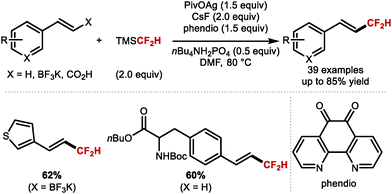 |
| | Scheme 59 Silver-mediated radical difluoromethylation using TMSCF2H. Phendio = 1,10-phenanthroline-5,6-dione. | |
5.2 Difluoromethylation of alkynes
A seminal report by Hu and co-workers in 2009 illustrated the use of N-tosyl-S-difluoromethyl-S-phenylsulfoximine for the difluoromethylation of alkynes (Scheme 60).192 While the methodology is limited to electron-rich alkynes, the protocol represents a valuable alternative to previously known Freon-based approaches. The authors proposed a difluorocarbene mechanism with the first equivalent of lithium acetylide acting as a base to deprotonate N-tosyl-S-difluoromethyl-S-phenylsulfoximine, thereby generating a difluorocarbene species which can react with a second lithium acetylide equivalent. The resulting anion is then quenched resulting in the difluoromethylated product.
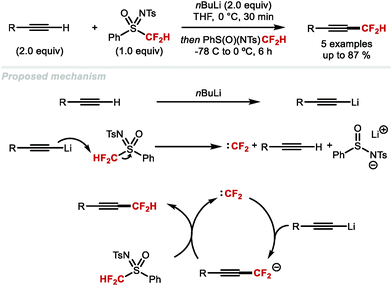 |
| | Scheme 60 Difluoromethylation of lithium acetylides. | |
In 2015 and 2016, Shibata and Mikami reported the difluoromethylation of aryl and heteroaryl-acetylenes under a fluoroform atmosphere in the presence of tBuOK or LHMDS (Scheme 61).204,205 In Shibata's report, further derivatisation of the difluoromethylated alkyne products to difluoromethylated isoxazoles and triazoles through 1,3-cycloaddition and click reactions was demonstrated (Scheme 61A).
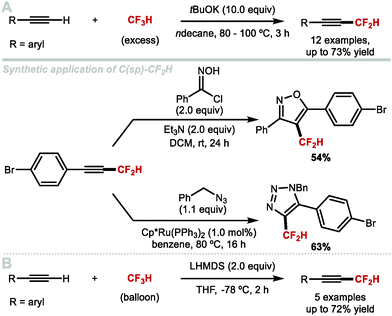 |
| | Scheme 61 Difluoromethylation of alkynes with fluoroform. | |
In 2019, Hu and co-workers reported the use of commercially available TMSCF2Br as a difluoromethylation reagent for a selection of electron-rich and electron-neutral alkynes under ambient conditions (Scheme 62).129 Heterocycles such as dibenzothiophene and benzofuran are compatible under the optimised reaction conditions. The authors suggested that a difluorocarbene mechanism is operating.
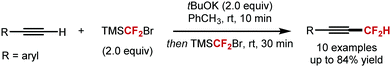 |
| | Scheme 62 Difluoromethylation of alkynes with TMSCF2Br. | |
In 2020, Zhang and co-workers extended the palladium difluorocarbene ([Pd = CF2]) chemistry (described in Section 2.1.c) to the difluoromethylation of alkynes (Scheme 63).206 The protocol employed chlorodifluoromethane as the difluorocarbene precursor, and featured good functional group tolerance, broad substrate scope, and was applied to the synthesis of complex drug molecules. One limitation of this method is the use of chlorodifluoromethane, an ODS.
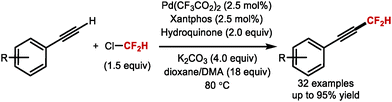 |
| | Scheme 63 Difluoromethylation of alkynes via [Pd = CF2]. | |
6. Industrial state of play
The toolbox of reactions that facilitate access to difluoromethylated compounds is expanding at a rapid pace. Process scientists are continuously considering these reactions to adapt them to large-scale synthesis. For many years, the large scale difluoromethylation of phenols relied on the use of chlorodifluoromethane. The ozone-depleting properties of this reagent have prompted scientists to utilise more environmentally friendly reagents. In 2019, Greszler illustrated that diethyl bromodifluoromethyl-phosphonate is a suitable difluorocarbene reagent to access ABBV-2222, a CFTR corrector for the treatment of cystic fibrosis (Scheme 64).207 Despite the exothermic nature of the difluorocarbene reaction, ABBV-2222 was prepared on 130 g scale.
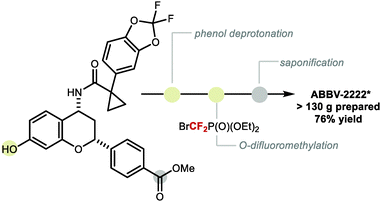 |
| | Scheme 64 Large-scale late-stage difluoromethylation with diethyl bromodifluoromethylphosphonate. *Structure of ABBV-2222 shown in Fig. 11. | |
Scientists from Innocrin Pharmaceuticals recently disclosed that bis-difluoromethylation of a relevant intermediate towards the synthesis of seviteronel is feasible on multikilogram scale using a modified literature procedure (Scheme 65).208 The authors demonstrated that a slight excess of SCDA in combination with excess potassium carbonate in a mixture of DMF/H2O afforded the desired building block in 57% yield on a multikilogram scale.
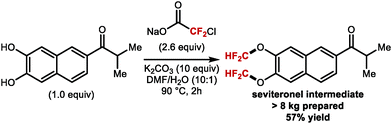 |
| | Scheme 65 Large-scale bis-difluoromethylation of an intermediate towards seviteronel. | |
In 2018, Kappe and co-workers optimised their continuous flow α-difluoromethylation for a telescoped continuous synthesis of Eflornithine (Scheme 66).209 Eflornithine hydrochloride monohydrate was produced in more than 17 grams with a throughput of 24 mmol h−1.
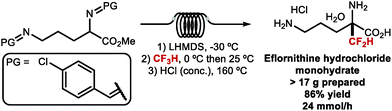 |
| | Scheme 66 Continuous synthesis of Eflornithine hydrochloride monohydrate via electrophilic difluoromethylation with fluoroform. | |
While the above examples illustrate that progress has been made to scale up difluoromethylation protocols, many large-scale syntheses of C(sp2)–CF2H compounds still rely on either deoxyfluorination or a building block approach. The reason may be the limited number of studies focused on the large-scale production of difluoromethylation reagents. Notably, Pfizer recently developed a scalable synthesis of [(DMPU)2Zn(CF2H)2] (Scheme 67).210 One of the key challenges to overcome was the high price of the starting material (CF2HI). To combat this problem, the synthesis of CF2HI from BrF2CCO2H was optimised. With CF2HI in hand, [(DMPU)2Zn(CF2H)2] was prepared from ZnEt2 in quantities superior to 100 g.
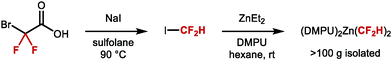 |
| | Scheme 67 Continuous synthesis of [(DMPU)2Zn(CF2H)2]. | |
During the review of this manuscript, a metal-free C–H difluoromethylation process for azines involving phosphonium salt formation followed by sp2–sp3 phosphorus ligand-coupling was reported by Paton, McNally and co-workers. Various pyridines, drug-like fragments, and pharmaceuticals were readily converted into difluoromethyl analogues. No pre-installed functional groups or directing groups are required, and for a range of sterically and electronically distinct pyridines, difluoromethylation occurs selectively at the 4-position.211
7. Conclusion and future outlook
Polyfluoroalkylation is an ever-expanding field of research within organic chemistry. This is also the case for difluoromethylation, where a community of chemists have developed numerous synthetic strategies for the successful incorporation of CF2H onto diverse classes of molecules. Efforts to facilitate the construction of C(sp2)–CF2H bonds have largely focused on cross-coupling and free radical methods. The state of play in cross-coupling has advanced tremendously over the last decade, and to date, nucleophilic, electrophilic, and radical difluoromethylation reagents as well as difluorocarbene precursors are viable cross-coupling partners for a wide range of arenes. While the development of cross-coupling methodologies has primarily focused on the functionalisation of arenes, technologies which harness the innate selectivity of the nucleophilic CF2H radical have been applied to electrophilic heteroarenes. Further developments within the area of radical C–H difluoromethylation that go beyond functionalisation at innately reactive sites would be highly desirable. Progress towards this end has been made by Baran and co-workers who illustrated the effect of solvents on reversing regioselectivity of pyridines.83 Furthermore, Qing and co-workers have shown that under oxidative conditions, C–H difluoromethylation of heteroaromatics can occur at the most acidic C–H bond instead of the most electron-deficient carbon.100 C(sp2)–CF2H bond formation through cross-coupling has also matured to a point where more earth-abundant metals are employed. This is a welcome development, and so is the use of renewable non-ODS CF2H source. Future research directions should focus on difluoromethylation that make use of inexpensive reagents, feedstock chemicals and operationally simple procedures. Furthermore, there are still gaps in the area of C(sp2)–CF2H bond formation such as meta-selective C–H difluoromethylation of (hetero)arenes or decarboxylative difluoromethylation of aryl carboxylic acids. Furthermore, a versatile 18F-difluoromethylation strategy would be highly desirable as current methods are applicable only to starting materials that require multistep synthesis, or display innate selectivity imposed by the use of [18F]CF2H radical.108 The field of C(sp3)–CF2H bond formation has been addressed from various angles. Nucleophilic difluoromethylation is generally achieved with TMSCF2H, whilst electrophilic difluoromethylation often proceeds via a difluorocarbene mechanism. Recently, various radical methods have allowed for the construction of C(sp3)–CF2H bonds in presence of unprotected functional groups, thus allowing for late-stage difluoromethylation. Future developments should aim at filling gaps such as a general method for Markovnikov difluoromethylation of alkenes or undirected C(sp3)–H difluoromethylation. In comparison to asymmetric trifluoromethylation, the field of stereoselective difluoromethylation remains underdeveloped. Specifically, general enantioselective difluoromethylation methodologies which furnish quaternary centres substituted with a CF2H group would be of value to the pharmaceutical industry. Evidently, the use of difluorocarbene reagents has had a tremendous impact in the construction of X–CF2H bonds. Current challenges are accessibility to X–CF2H fragments with high selectivity in presence of multiple reactive functional groups. Finally, the focus of all existing methods has been on small molecules. Today, it is not unthinkable that new methods which allow for incorporation of the CF2H group on biologically relevant systems, such as peptides, proteins and oligonucleotides, are within reach. A small fluorine-containing motif like the CF2H group could be useful for probing biological activity or mechanistic studies by 19F NMR. Seminal work in this direction has recently been reported by Davis, Gouverneur and co-workers, who developed site-selective photocatalytic hydrodifluoromethylation and difluoromethylation of dehydroalanine and tryptophan residues in proteins, respectively.99,134
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We acknowledge the Engineering and Physical Sciences Research Council (EP/N509711/1 and EP/T517811/1) and Pfizer (J. B. I. S.), the European Union's Horizon 2020 Research and innovation program under Marie Skłodowska-Curie Grant Agreement 721902 and Janssen (C. F. M.). The EPSRC Centre for Doctoral Training in Synthesis for Biology and Medicine (EP/L015838/1) generously supported by AstraZeneca, Diamond Light Source, Defence Science and Technology Laboratory, Evotec, GlaxoSmithKline, Janssen, Novartis, Pfizer, Syngenta, Takeda, UCB, and Vertex is also acknowledged (N. I.).
References
- K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 Search PubMed.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 Search PubMed.
- N. A. Meanwell, J. Med. Chem., 2018, 61, 5822–5880 Search PubMed.
- D. O’hagan, Chem. Soc. Rev., 2008, 37, 308–319 Search PubMed.
- L. Pauling, H. D. Springall and K. J. Palmer, J. Am. Chem. Soc., 1939, 61, 927–937 Search PubMed.
- E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315–8359 Search PubMed.
- J. M. Brown and V. Gouverneur, Angew. Chem., Int. Ed., 2009, 48, 8610–8614 Search PubMed.
- M. G. Campbell and T. Ritter, Chem. Rec., 2014, 14, 482–491 Search PubMed.
- G. K. S. Prakash and A. K. Yudin, Chem. Rev., 1997, 97, 757–786 Search PubMed.
- R. J. Lundgren and M. Stradiotto, Angew. Chem., Int. Ed., 2010, 49, 9322–9324 Search PubMed.
- O. A. Tomashenko and V. V. Grushin, Chem. Rev., 2011, 111, 4475–4521 Search PubMed.
- X. F. Wu, H. Neumann and M. Beller, Chem. – Asian J., 2012, 7, 1744–1754 Search PubMed.
- A. Studer, Angew. Chem., Int. Ed., 2012, 51, 8950–8958 Search PubMed.
- H. Liu, Z. Gu and X. Jiang, Adv. Synth. Catal., 2013, 355, 617–626 Search PubMed.
- C. Zhang, Adv. Synth. Catal., 2014, 356, 2895–2906 Search PubMed.
- A. Hafner, N. Jung and S. Bräse, Synthesis, 2014, 1440–1447 Search PubMed.
- S. Barata-Vallejo, S. M. Bonesi and A. Postigo, RSC Adv., 2015, 5, 62498–62518 Search PubMed.
- H. S. Booth and E. M. Bixby, Ind. Eng. Chem., 1932, 24, 637–641 Search PubMed.
- A. McCulloch, P. M. Midgley and A. A. Lindley, Atmos. Environ., 2006, 40, 936–942 Search PubMed.
- W. J. Middleton, J. Org. Chem., 1975, 40, 574–578 Search PubMed.
- G. S. Lal, G. P. Fez, R. J. Pesaresi, F. M. Prozonic and H. Cheng, J. Org. Chem., 1999, 64, 7048–7054 Search PubMed.
- T. Umemoto, R. P. Singh, Y. Xu and N. Saito, J. Am. Chem. Soc., 2010, 132, 18199–18205 Search PubMed.
- R. Szpera, N. Kovalenko, K. Natarajan, N. Paillard and B. Linclau, Beilstein J. Org. Chem., 2017, 13, 2883–2887 Search PubMed.
- F. Beaulieu, L. P. Beauregard, G. Courchesne, M. Couturier, F. Laflamme and A. L’Heureux, Org. Lett., 2009, 11, 5050–5053 Search PubMed.
- G. W. Rewcastle, S. A. Gamage, J. U. Flanagan, R. Frederick, W. A. Denny, B. C. Baguley, P. Kestell, R. Singh, J. D. Kendall, E. S. Marshall, C. L. Lill, W. J. Lee, S. Kolekar, C. M. Buchanan, S. M. F. Jamieson and P. R. Shepherd, J. Med. Chem., 2011, 54, 7105–7126 Search PubMed.
- D. E. Yerien, S. Barata-Vallejo and A. Postigo, Chem. – Eur.
J., 2017, 23, 14676–14701 Search PubMed.
- N. Levi, D. Amir, E. Gorshonov and Y. Zafrani, Synthesis, 2019, 4549–4567 Search PubMed.
- R. Szpera, D. F. J. Moseley, L. B. Smith, A. J. Sterling and V. Gouverneur, Angew. Chem., Int. Ed., 2019, 58, 14824–14848 Search PubMed.
- J. B. I. Sap, N. J. W. Straathof, T. Knauber, C. F. Meyer, M. Médebielle, L. Buglioni, C. Genicot, A. A. Trabanco, T. Noël, C. W. Ende and V. Gouverneur, J. Am. Chem. Soc., 2020, 142, 9181–9187 Search PubMed.
- Y.-J. Yu, F.-L. Zhang, T.-Y. Peng, C.-L. Wang, J. Cheng, C. Chen, K. N. Houk and Y.-F. Wang, Science, 2021, 1240, 1232–1240 Search PubMed.
- Y. Zafrani, D. Yeffet, G. Sod-Moriah, A. Berliner, D. Amir, D. Marciano, E. Gershonov and S. Saphier, J. Med. Chem., 2017, 60, 797–804 Search PubMed.
- C. D. Sessler, M. Rahm, S. Becker, J. M. Goldberg, F. Wang and S. J. Lippard, J. Am. Chem. Soc., 2017, 139, 9325–9332 Search PubMed.
- M. H. Abraham, R. J. Abraham, J. Byrne and L. Griffiths, J. Org. Chem., 2006, 71, 3389–3394 Search PubMed.
- Y. Zafrani, G. Sod-Moriah, D. Yeffet, A. Berliner, D. Amir, D. Marciano, S. Elias, S. Katalan, N. Ashkenazi, M. Madmon, E. Gershonov and S. Saphier, J. Med. Chem., 2019, 62, 5628–5637 Search PubMed.
- L. Xing, D. C. Blakemore, A. Narayanan, R. Unwalla, F. Lovering, R. A. Denny, H. Zhou and M. E. Bunnage, ChemMedChem, 2015, 10, 715–726 Search PubMed.
- D. Rageot, T. Bohnacker, A. Melone, J. B. Langlois, C. Borsari, P. Hillmann, A. M. Sele, F. Beaufils, M. Zvelebil, P. Hebeisen, W. Löscher, J. Burke, D. Fabbro and M. P. Wymann, J. Med. Chem., 2018, 61, 10084–10105 Search PubMed.
- K. Fujikawa, Y. Fujioka, A. Kobayashi and H. Amii, Org. Lett., 2011, 13, 5560–5563 Search PubMed.
- K. Fujikawa, A. Kobayashi and H. Amii, Synthesis, 2012, 3015–3018 Search PubMed.
- S. Ge, W. Chaładaj and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 4149–4152 Search PubMed.
- M.-C. Belhomme, T. Poisson and X. Pannecoucke, J. Org. Chem., 2014, 79, 7205–7211 Search PubMed.
- X. Sun and S. Yu, Org. Lett., 2014, 16, 2938–2941 Search PubMed.
- J. Jung, E. Kim, Y. You and E. J. Cho, Adv. Synth. Catal., 2014, 356, 2741–2748 Search PubMed.
- G. A. Hartgraves and D. J. Burton, J. Fluor. Chem., 1988, 39, 425–430 Search PubMed.
- D. J. Burton and G. A. Hartgraves, J. Fluor. Chem., 1990, 49, 155–158 Search PubMed.
- D. J. Burton and G. A. Hartgraves, J. Fluor. Chem., 2007, 128, 1198–1215 Search PubMed.
- J. R. Bour, S. K. Kariofillis and M. S. Sanford, Organometallics, 2017, 36, 1220–1223 Search PubMed.
- P. S. Fier and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 5524–5527 Search PubMed.
- G. K. S. Prakash, S. K. Ganesh, J.-P. Jones, A. Kulkarni, K. Masood, J. K. Swabeck and G. A. Olah, Angew. Chem., 2012, 124, 12256–12260 Search PubMed.
- X. L. Jiang, Z. H. Chen, X. H. Xu and F. L. Qing, Org. Chem. Front., 2014, 1, 774–776 Search PubMed.
- C. Matheis, K. Jouvin and L. J. Goossen, Org. Lett., 2014, 16, 5984–5987 Search PubMed.
- Y. Gu, D. Chang, X. Leng, Y. Gu and Q. Shen, Organometallics, 2015, 34, 3065–3071 Search PubMed.
- L. Xu and D. A. Vicic, J. Am. Chem. Soc., 2016, 138, 2536–2539 Search PubMed.
- H. Serizawa, K. Ishii, K. Aikawa and K. Mikami, Org. Lett., 2016, 18, 3686–3689 Search PubMed.
- D. A. Culkin and J. F. Hartwig, Organometallics, 2004, 23, 3398–3416 Search PubMed.
- V. Grushin and W. Marshall, J. Am. Chem. Soc., 2006, 12644–12645 Search PubMed.
- V. Grushin and W. Marshall, J. Am. Chem. Soc., 2006, 128, 4632–4641 Search PubMed.
- Y. Gu, X. Leng and Q. Shen, Nat. Commun., 2014, 5, 1–7 Search PubMed.
- C. Lu, H. Lu, J. Wu, H. C. Shen, T. Hu, Y. Gu and Q. Shen, J. Org. Chem., 2018, 83, 1077–1083 Search PubMed.
- C. Lu, Y. Gu, J. Wu, Y. Gu and Q. Shen, Chem. Sci., 2017, 8, 4848–4852 Search PubMed.
- K. Aikawa, Y. Nakamura, Y. Yokota, W. Toya and K. Mikami, Chem. – Eur. J., 2015, 21, 96–100 Search PubMed.
- D. M. Ferguson, C. A. Malapit, J. R. Bour and M. S. Sanford, J. Org. Chem., 2019, 84, 3735–3740 Search PubMed.
- Z. Feng, Q. Q. Min and X. Zhang, Org. Lett., 2016, 18, 44–47 Search PubMed.
- X. Y. Deng, J. H. Lin and J. C. Xiao, Org. Lett., 2016, 18, 4384–4387 Search PubMed.
- Z. Feng, Q. Q. Min, X. P. Fu, L. An and X. Zhang, Nat. Chem., 2017, 9, 918–923 Search PubMed.
- X. P. Fu, X. S. Xue, X. Y. Zhang, Y. L. Xiao, S. Zhang, Y. L. Guo, X. Leng, K. N. Houk and X. Zhang, Nat. Chem., 2019, 11, 948–956 Search PubMed.
- T. C. Wilson, G. McSweeney, S. Preshlock, S. Verhoog, M. Tredwell, T. Cailly and V. Gouverneur, Chem. Commun., 2016, 52, 13277–13280 Search PubMed.
- J. W. B. Fyfe and A. J. B. Watson, Chem, 2017, 3, 31–55 Search PubMed.
- T. C. Wilson, T. Cailly and V. Gouverneur, Chem. Soc. Rev., 2018, 47, 6990–7005 Search PubMed.
- H. Zhao, S. Herbert, T. Kinzel, W. Zhang and Q. Shen, J. Org. Chem., 2020, 85, 3596–3604 Search PubMed.
- F. Pan, G. B. Boursalian and T. Ritter, Angew. Chem., Int. Ed., 2018, 57, 16871–16876 Search PubMed.
- J. B. Diccianni and T. Diao, Trends Chem., 2019, 1, 830–844 Search PubMed.
- R. R. Merchant, J. T. Edwards, T. Qin, M. M. Kruszyk, C. Bi, G. Che, D. H. Bao, W. Qiao, L. Sun, M. R. Collins, O. O. Fadeyi, G. M. Gallego, J. J. Mousseau, P. Nuhant and P. S. Baran, Science, 2018, 360, 75–80 Search PubMed.
- C. Xu, W. H. Guo, X. He, Y. L. Guo, X. Y. Zhang and X. Zhang, Nat. Commun., 2018, 9, 1–10 Search PubMed.
- J. Sheng, H. Q. Ni, K. J. Bian, Y. Li, Y. N. Wang and X. S. Wang, Org. Chem. Front., 2018, 5, 606–610 Search PubMed.
- X. P. Fu, Y. L. Xiao and X. Zhang, Chinese J. Chem., 2018, 36, 143–146 Search PubMed.
- H. Motohashi and K. Mikami, Org. Lett., 2018, 20, 5340–5343 Search PubMed.
- V. Bacauanu, S. Cardinal, M. Yamauchi, M. Kondo, D. F. Fernández, R. Remy and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2018, 57, 12543–12548 Search PubMed.
- W. Miao, Y. Zhao, C. Ni, B. Gao, W. Zhang and J. Hu, J. Am. Chem. Soc., 2018, 140, 880–883 Search PubMed.
- L. An, Y. L. Xiao, S. Zhang and X. Zhang, Angew. Chem., Int. Ed., 2018, 57, 6921–6925 Search PubMed.
- S. Liu, K. Kang, S. Liu, D. Wang, P. Wei, Y. Lan and Q. Shen, Organometallics, 2018, 37, 3901–3908 Search PubMed.
- C. Ni and J. Hu, Chem. Soc. Rev., 2016, 45, 5441–5454 Search PubMed.
-
H. Zipse, Radical stability – A theoretical perspective, 2006 Search PubMed.
- Y. Fujiwara, J. A. Dixon, R. A. Rodriguez, R. D. Baxter, D. D. Dixon, M. R. Collins, D. G. Blackmond and P. S. Baran, J. Am. Chem. Soc., 2012, 134, 1494–1497 Search PubMed.
- J. Rong, L. Deng, P. Tan, C. Ni, Y. Gu and J. Hu, Angew. Chem., Int. Ed., 2016, 55, 2743–2747 Search PubMed.
- Y. Nakayama, G. Ando, M. Abe, T. Koike and M. Akita, ACS Catal., 2019, 9, 6555–6563 Search PubMed.
- N. B. Heine and A. Studer, Org. Lett., 2017, 19, 4150–4153 Search PubMed.
- N. Noto, T. Koike and M. Akita, Chem. Sci., 2017, 8, 6375–6379 Search PubMed.
- A. F. Garrido-Castro, A. Gini, M. C. Maestro and J. Alemán, Chem. Commun., 2020, 56, 3769–3772 Search PubMed.
- Z. Zou, W. Zhang, Y. Wang, L. Kong, G. Karotsis, Y. Wang and Y. Pan, Org. Lett., 2019, 21, 1857–1862 Search PubMed.
- P. Cao, J. X. Duan and Q. Y. Chen, J. Chem. Soc., Chem. Commun., 1994, 204, 737–738 Search PubMed.
- T. T. Tung, S. B. Christensen and J. Nielsen, Chem. – Eur. J., 2017, 23, 18125–18128 Search PubMed.
- R. Sakamoto, H. Kashiwagi and K. Maruoka, Org. Lett., 2017, 19, 5126–5129 Search PubMed.
- A. C. Sun, E. J. McClain, J. W. Beatty and C. R. J. Stephenson, Org. Lett., 2018, 20, 3487–3490 Search PubMed.
- P. Dai, X. Yu, P. Teng, W. H. Zhang and C. Deng, Org. Lett., 2018, 20, 6901–6905 Search PubMed.
- W. Zhang, X. X. Xiang, J. Chen, C. Yang, Y. L. Pan, J. P. Cheng, Q. Meng and X. Li, Nat. Commun., 2020, 11, 1–10 Search PubMed.
- I. Ghosh, J. Khamrai, A. Savateev, N. Shlapakov, M. Antonietti and B. König, Science, 2019, 365, 360–366 Search PubMed.
- J. Yang, S. Zhu, F. Wang, F. L. Qing and L. Chu, Angew. Chem., Int. Ed., 2020, 4300–4306 Search PubMed.
- A. F. M. Noisier, M. J. Johansson, L. Knerr, M. A. Hayes, W. J. Drury, E. Valeur, L. R. Malins and R. Gopalakrishnan, Angew. Chem., Int. Ed., 2019, 58, 19096–19102 Search PubMed.
- M. Imiołek, P. G. Isenegger, W. L. Ng, A. Khan, V. Gouverneur and B. G. Davis, ACS Cent. Sci., 2021, 7, 145–155 Search PubMed.
- S. Q. Zhu, Y. L. Liu, H. Li, X. H. Xu and F. L. Qing, J. Am. Chem. Soc., 2018, 140, 11613–11617 Search PubMed.
- Z. Zhang, X. Tang and W. R. Dolbier, Org. Lett., 2015, 17, 4401–4403 Search PubMed.
- M. Zhu, W. Fu, Z. Wang, C. Xu and B. Ji, Org. Biomol. Chem., 2017, 15, 9057–9060 Search PubMed.
- S. Mizuta, I. S. R. Stenhagen, M. O’Duill, J. Wolstenhulme, A. K. Kirjavainen, S. J. Forsback, M. Tredwell, G. Sandford, P. R. Moore, M. Huiban, S. K. Luthra, J. Passchier, O. Solin and V. Gouverneur, Org. Lett., 2013, 15, 2648–2651 Search PubMed.
- S. Verhoog, L. Pfeifer, T. Khotavivattana, S. Calderwood, T. L. Collier, K. Wheelhouse, M. Tredwell and V. Gouverneur, Synlett, 2016, 25–28 Search PubMed.
- H. Shi, A. Braun, L. Wang, S. H. Liang, N. Vasdev and T. Ritter, Angew. Chem., Int. Ed., 2016, 55, 10786–10790 Search PubMed.
- G. Yuan, F. Wang, N. A. Stephenson, L. Wang, B. H. Rotstein, N. Vasdev, P. Tang and S. H. Liang, Chem. Commun., 2017, 53, 126–129 Search PubMed.
- J. B. I. Sap, T. C. Wilson, C. W. Kee, N. J. W. Straathof, C. W. Am Ende, P. Mukherjee, L. Zhang, C. Genicot and V. Gouverneur, Chem. Sci., 2019, 10, 3237–3241 Search PubMed.
- L. Trump, A. Lemos, B. Lallemand, P. Pasau, J. Mercier, C. Lemaire, A. Luxen and C. Genicot, Angew. Chem., Int. Ed., 2019, 58, 13149–13154 Search PubMed.
- L. Trump, A. Lemos, J. Jacq, P. Pasau, B. Lallemand, J. Mercier, C. Genicot, A. Luxen and C. Lemaire, Org. Process Res. Dev., 2020, 24, 734–744 Search PubMed.
- N. Malquin, K. Rahgoshay, N. Lensen, G. Chaume, E. Miclet and T. Brigaud, Chem. Commun., 2019, 55, 12487–12490 Search PubMed.
- P. Bey, J.-P. Vevert, V. Van Dorsselaer and M. Kolb, J. Org. Chem., 1979, 44, 2732–2742 Search PubMed.
- C. Han, S. M. Kelly, T. Cravillion, S. J. Savage, T. Nguyen and F. Gosselin, Tetrahedron, 2019, 75, 4351–4357 Search PubMed.
- R. D. Cink, K. A. Lukin, R. D. Bishop, G. Zhao, M. J. Pelc, T. B. Towne, B. D. Gates, M. M. Ravn, D. R. Hill, C. Ding, S. C. Cullen, J. Mei, M. R. Leanna, J. Henle, J. G. Napolitano, N. K. Nere, S. Chen, A. Sheikh and J. M. Kallemeyn, Org. Process Res. Dev., 2020, 24, 183–200 Search PubMed.
- M. Bourlière, S. C. Gordon, S. L. Flamm, C. L. Cooper, A. Ramji, M. Tong, N. Ravendhran, J. M. Vierling, T. T. Tran, S. Pianko, M. B. Bansal, V. de Lédinghen, R. H. Hyland, L. M. Stamm, H. Dvory-Sobol, E. Svarovskaia, J. Zhang, K. C. Huang, G. M. Subramanian, D. M. Brainard, J. G. McHutchison, E. C. Verna, P. Buggisch, C. S. Landis, Z. H. Younes, M. P. Curry, S. I. Strasser, E. R. Schiff, K. R. Reddy, M. P. Manns, K. V. Kowdley and S. Zeuzem, N. Engl. J. Med., 2017, 376, 2134–2146 Search PubMed.
- J. G. Taylor, S. Zipfel, K. Ramey, R. Vivian, A. Schrier, K. K. Karki, A. Katana, D. Kato, T. Kobayashi, R. Martinez, M. Sangi, D. Siegel, C. V. Tran, Z. Y. Yang, J. Zablocki, C. Y. Yang, Y. Wang, K. Wang, K. Chan, O. Barauskas, G. Cheng, D. Jin, B. E. Schultz, T. Appleby, A. G. Villaseñor and J. O. Link, Bioorg. Med. Chem. Lett., 2019, 29, 2428–2436 Search PubMed.
- P. A. Messina, K. C. Mange and W. J. Middleton, J. Fluor. Chem., 1989, 42, 137–143 Search PubMed.
- Y. Zhao, W. Huang, J. Zheng and J. Hu, Org. Lett., 2011, 13, 5342–5345 Search PubMed.
- G. F. Du, Y. Wang, C. Z. Gu, B. Dai and L. He, RSC Adv., 2015, 5, 35421–35424 Search PubMed.
- D. Chen, C. Ni, Y. Zhao, X. Cai, X. Li, P. Xiao and J. Hu, Angew. Chem., Int. Ed., 2016, 55, 12632–12636 Search PubMed.
- M. Miele, A. Citarella, N. Micale, W. Holzer and V. Pace, Org. Lett., 2019, 21, 8261–8265 Search PubMed.
- M. Miele, R. D’Orsi, V. Sridharan, W. Holzer and V. Pace, Chem. Commun., 2019, 55, 12960–12963 Search PubMed.
- W. Zhang, J. H. Lin, W. Wu, Y. C. Cao and J. C. Xiao, Chinese J. Chem., 2020, 38, 169–172 Search PubMed.
- T. Iida, R. Hashimoto, K. Aikawa, S. Ito and K. Mikami, Angew. Chem., Int. Ed., 2012, 51, 9535–9538 Search PubMed.
- K. Aikawa, K. Maruyama, K. Honda and K. Mikami, Org. Lett., 2015, 17, 4882–4885 Search PubMed.
- B. Gutmann, P. Hanselmann, M. Bersier, D. Roberge and C. O. Kappe, J. Flow Chem., 2017, 7, 46–51 Search PubMed.
- M. Köckinger, T. Ciaglia, M. Bersier, P. Hanselmann, B. Gutmann and C. O. Kappe, Green Chem., 2018, 20, 108–112 Search PubMed.
- Y. Nomura, E. Tokunaga and N. Shibata, Angew. Chem., 2011, 123, 1925–1929 Search PubMed.
- J. Zhu, H. Zheng, X. S. Xue, Y. Xiao, Y. Liu and Q. Shen, Chinese J. Chem., 2018, 36, 1069–1074 Search PubMed.
- Q. Xie, Z. Zhu, L. Li, C. Ni and J. Hu, Angew. Chem., Int. Ed., 2019, 58, 6405–6410 Search PubMed.
- X. J. Tang, Z. Zhang and W. R. Dolbier, Chem. – Eur. J., 2015, 21, 18961–18965 Search PubMed.
- Q. Y. Lin, X. H. Xu, K. Zhang and F. L. Qing, Angew. Chem., Int. Ed., 2016, 55, 1479–1483 Search PubMed.
- C. F. Meyer, S. M. Hell, A. Misale, A. A. Trabanco and V. Gouverneur, Angew. Chem., Int. Ed., 2019, 58, 8829–8833 Search PubMed.
- C. F. Meyer, S. M. Hell, J. B. I. Sap, A. Misale, A. Peschiulli, D. Oehlrich, A. A. Trabanco and V. Gouverneur, Tetrahedron, 2019, 130679 Search PubMed.
- B. Josephson, C. Fehl, P. G. Isenegger, S. Nadal, T. H. Wright, A. W. J. Poh, B. J. Bower, A. M. Giltrap, L. Chen, C. Batchelor-McAuley, G. Roper, O. Arisa, J. B. I. Sap, A. Kawamura, A. J. Baldwin, S. Mohammed, R. G. Compton, V. Gouverneur and B. G. Davis, Nature, 2020, 585, 530–537 Search PubMed.
- X. J. Tang and W. R. Dolbier, Angew. Chem., Int. Ed., 2015, 54, 4246–4249 Search PubMed.
- C. S. Thomoson, X. J. Tang and W. R. Dolbier, J. Org. Chem., 2015, 80, 1264–1268 Search PubMed.
- Q. Y. Lin, Y. Ran, X. H. Xu and F. L. Qing, Org. Lett., 2016, 18, 2419–2422 Search PubMed.
- Y. Ran, Q. Y. Lin, X. H. Xu and F. L. Qing, J. Org. Chem., 2016, 81, 7001–7007 Search PubMed.
- Y. Arai, R. Tomita, G. Ando, T. Koike and M. Akita, Chem. – Eur. J., 2016, 22, 1262–1265 Search PubMed.
- H. H. Xu, J. Song and H. C. Xu, ChemSusChem, 2019, 12, 3060–3063 Search PubMed.
- M. Zhang, J. H. Lin and J. C. Xiao, Angew. Chem., Int. Ed., 2019, 58, 6079–6083 Search PubMed.
- J. Liu, S. Zhuang, Q. Gui, X. Chen, Z. Yang and Z. Tan, Eur. J. Org. Chem., 2014, 3196–3202 Search PubMed.
- X. J. Tang, C. S. Thomoson and W. R. Dolbier, Org. Lett., 2014, 16, 4594–4597 Search PubMed.
- Z. Zhang, H. Martinez and W. R. Dolbier, J. Org. Chem., 2017, 82, 2589–2598 Search PubMed.
- G. Zou and X. Wang, Org. Biomol. Chem., 2017, 15, 8748–8754 Search PubMed.
- Z. Zhang, X. Tang, C. S. Thomoson and W. R. Dolbier, Org. Lett., 2015, 17, 3528–3531 Search PubMed.
- N. Noto, T. Koike and M. Akita, J. Org. Chem., 2016, 81, 7064–7071 Search PubMed.
- W. Fu, X. Han, M. Zhu, C. Xu, Z. Wang, B. Ji, X. Q. Hao and M. P. Song, Chem. Commun., 2016, 52, 13413–13416 Search PubMed.
- S. Zhang, L. Li, J. Zhang, J. Zhang, M. Xue and K. Xu, Chem. Sci., 2019, 10, 3181–3185 Search PubMed.
- J. S. Lin, F. L. Wang, X. Y. Dong, W. W. He, Y. Yuan, S. Chen and X. Y. Liu, Nat. Commun., 2017, 8, 1–11 Search PubMed.
- K. Aikawa, K. Ishii, Y. Endo and K. Mikami, J. Fluor. Chem., 2017, 203, 122–129 Search PubMed.
- Y. Endo, K. Ishii and K. Mikami, Tetrahedron, 2019, 75, 4099–4103 Search PubMed.
- X. Zeng, W. Yan, S. B. Zacate, T. H. Chao, X. Sun, Z. Cao, K. G. E. Bradford, M. Paeth, S. B. Tyndall, K. Yang, T. C. Kuo, M. J. Cheng and W. Liu, J. Am. Chem. Soc., 2019, 141, 11398–11403 Search PubMed.
- X. Zeng, W. Yan, M. Paeth, S. B. Zacate, P. H. Hong, Y. Wang, D. Yang, K. Yang, T. Yan, C. Song, Z. Cao, M. J. Cheng and W. Liu, J. Am. Chem. Soc., 2019, 141, 19941–19949 Search PubMed.
- X. Zeng, W. Yan, S. B. Zacate, A. Cai, Y. Wang, D. Yang, K. Yang and W. Liu, Angew. Chem., Int. Ed., 2020, 16398–16403 Search PubMed.
- H. Zhao, C. Lu, S. Herbert, W. Zhang and Q. Shen, J. Org. Chem., 2021, 86, 2854–2865 Search PubMed.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 Search PubMed.
- M. A. McClinton and D. A. McClinton, Tetrahedron, 1992, 48, 6555–6666 Search PubMed.
- A. Dabrowski, B. Štabuc and L. Lazebnik, Prz. Gastroenterol., 2018, 13, 6–15 Search PubMed.
- N. Yoneda and T. Fukuhara, Chem. Lett., 2001, 222–223 Search PubMed.
- J. W. Lee, W. Zheng, C. A. Morales-Rivera, P. Liu and M. Y. Ngai, Chem. Sci., 2019, 10, 3217–3222 Search PubMed.
- X. Xiao, Z. T. Zheng, T. Li, J. L. Zheng, T. Tao, L. M. Chen, J. Y. Gu, X. Yao, J. H. Lin and J. C. Xiao, Synthesis, 2020, 197–207 Search PubMed.
- J. M. Birchall, G. W. Cross and R. N. Haszeldine, Proc. Chem. Soc., 1960, 81 Search PubMed.
- L. Zhang, J. Zheng and J. Hu, J. Org. Chem., 2006, 71, 9845–9848 Search PubMed.
- J. Zheng, Y. Li, L. Zhang, J. Hu, G. J. Meuzelaar and H. J. Federsel, Chem. Commun., 2007, 5149–5151 Search PubMed.
- F. Wang, L. Zhang, J. Zheng and J. Hu, J. Fluor. Chem., 2011, 132, 521–528 Search PubMed.
- Y. Zafrani, G. Sod-Moriah and Y. Segall, Tetrahedron, 2009, 65, 5278–5283 Search PubMed.
- Y. Geng, M. Zhu, A. Liang, C. Niu, J. Li, D. Zou, Y. Wu and Y. Wu, Org. Biomol. Chem., 2018, 16, 1807–1811 Search PubMed.
- P. S. Fier and J. F. Hartwig, Angew. Chem., Int. Ed., 2013, 52, 2092–2095 Search PubMed.
- G. Landelle, E. Schmitt, A. Panossian, J. P. Vors, S. Pazenok, P. Jeschke, O. Gutbrod and F. R. Leroux, J. Fluor. Chem., 2017, 203, 155–165 Search PubMed.
- C. S. Thomoson and W. R. Dolbier, J. Org. Chem., 2013, 78, 8904–8908 Search PubMed.
- J. Yang, M. Jiang, Y. Jin, H. Yang and H. Fu, Org. Lett., 2017, 19, 2758–2761 Search PubMed.
- X. Lin, C. Hou, H. Li and Z. Weng, Chem. – Eur. J., 2016, 22, 2075–2084 Search PubMed.
- Q. Xie, C. Ni, R. Zhang, L. Li, J. Rong and J. Hu, Angew. Chem., Int. Ed., 2017, 56, 3206–3210 Search PubMed.
- J. Hine and K. Tanabe, J. Am. Chem. Soc., 1957, 79, 2654–2655 Search PubMed.
- J. Mizukado, Y. Matsukawa, H. D. Quan, M. Tamura and A. Sekiya, J. Fluor. Chem., 2006, 127, 400–404 Search PubMed.
- R. M. Flynn and D. J. Burton, J. Fluor. Chem., 2011, 132, 815–828 Search PubMed.
- L. Li, F. Wang, C. Ni and J. Hu, Angew. Chem., 2013, 125, 12616–12620 Search PubMed.
- R. A. Mitsch and J. E. Robertson, J. Heterocycl. Chem., 1965, 2, 152–156 Search PubMed.
- R. Miethchen, M. Hein, D. Naumann and W. Tyrra, Liebigs Ann., 1995, 1717–1719 Search PubMed.
- J. Mizukado, Y. Matsukawa, H. D. Quan, M. Tamura and A. Sekiya, J. Fluor. Chem., 2005, 126, 365–369 Search PubMed.
- J. Zhu, Y. Liu and Q. Shen, Angew. Chem., Int. Ed., 2016, 55, 9050–9054 Search PubMed.
- K. Levchenko, O.
P. Datsenko, O. Serhiichuk, A. Tolmachev, V. O. Iaroshenko and P. K. Mykhailiuk, J. Org. Chem., 2016, 81, 5803–5813 Search PubMed.
- G. K. Liu, X. Li, W. B. Qin, X. S. Peng, H. N. C. Wong, L. Zhang and X. Zhang, Chem. Commun., 2019, 55, 7446–7449 Search PubMed.
- J. J. Ma, Q. R. Liu, G. P. Lu and W. B. Yi, J. Fluor. Chem., 2017, 193, 113–117 Search PubMed.
-
M. Gaba and C. Mohan, Development of drugs based on imidazole and benzimidazole bioactive heterocycles: Recent advances and future directions, Springer US, 2016, vol. 25 Search PubMed.
- M. Andrés, M. A. Buil, M. Calbet, O. Casado, J. Castro, P. R. Eastwood, P. Eichhorn, M. Ferrer, P. Forns, I. Moreno, S. Petit and R. S. Roberts, Bioorg. Med. Chem. Lett., 2014, 24, 5111–5117 Search PubMed.
- U. Rheude and W. Sundermeyer, Chem. Ber., 1985, 118, 2208–2219 Search PubMed.
- J. W. Lyga and R. M. Patera, J. Fluor. Chem., 1998, 92, 141–145 Search PubMed.
- K. I. Petko, A. A. Tolmachev and L. M. Yagupol'skii, Russ. J. Org. Chem., 2002, 38, 1030–1034 Search PubMed.
- M. Ando, T. Wada and N. Sato, Org. Lett., 2006, 8, 3805–3808 Search PubMed.
- W. Zhang, F. Wang and J. Hu, Org. Lett., 2009, 11, 2109–2112 Search PubMed.
- V. P. Mehta and M. F. Greaney, Org. Lett., 2013, 15, 5036–5039 Search PubMed.
- G. K. Surya Prakash, S. Krishnamoorthy, S. K. Ganesh, A. Kulkarni, R. Haiges and G. A. Olah, Org. Lett., 2014, 16, 54–57 Search PubMed.
- T. Mao, L. Zhao, Y. Huang, Y. G. Lou, Q. Yao, X. F. Li and C. Y. He, Tetrahedron Lett., 2018, 59, 2752–2754 Search PubMed.
- G. K. S. Prakash, Z. Zhang, F. Wang, C. Ni and G. A. Olah, J. Fluor. Chem., 2011, 132, 792–798 Search PubMed.
- Y. Zafrani, D. Amir, L. Yehezkel, M. Madmon, S. Saphier, N. Karton-Lifshin and E. Gershonov, J. Org. Chem., 2016, 81, 9180–9187 Search PubMed.
- A. Polley, G. Bairy, P. Das and R. Jana, Adv. Synth. Catal., 2018, 360, 4161–4167 Search PubMed.
- J. C. Deng, Y. C. Gao, Z. Zhu, L. Xu, Z. D. Li and R. Y. Tang, Org. Lett., 2019, 21, 545–548 Search PubMed.
- Z. Li, J. Dong, Z. Yuan, D. Y. Yang and Z. Weng, Org. Lett., 2018, 20, 6407–6410 Search PubMed.
- T. H. Zhu, Z. Y. Zhang, J. Y. Tao, K. Zhao and T. P. Loh, Org. Lett., 2019, 21, 6155–6159 Search PubMed.
- E. J. Horn, B. R. Rosen and P. S. Baran, ACS Cent. Sci., 2016, 2, 302–308 Search PubMed.
- P. Xiong, H. H. Xu, J. Song and H. C. Xu, J. Am. Chem. Soc., 2018, 140, 2460–2464 Search PubMed.
- S. Okusu, E. Tokunaga and N. Shibata, Org. Lett., 2015, 17, 3802–3805 Search PubMed.
- K. Aikawa, K. Maruyama, J. Nitta, R. Hashimoto and K. Mikami, Org. Lett., 2016, 18, 3354–3357 Search PubMed.
- X.-Y. Zhang, X.-P. Fu, S. Zhang and X. Zhang, CCS Chem., 2020, 2, 293–304 Search PubMed.
- S. N. Greszler, B. Shelat and E. A. Voight, Org. Lett., 2019, 21, 5725–5727 Search PubMed.
-
M. Sharp, D. D. Wirth, W. J. Hoekstra, S. W. Rafferty and M. F. Bindl, Process for preparation of naphthyl(triazolyl)(isopropyl)methanol as anticancer compound, WO2019113312A1, 2019.
- M. Köckinger, C. A. Hone, B. Gutmann, P. Hanselmann, M. Bersier, A. Torvisco and C. O. Kappe, Org. Process Res. Dev., 2018, 22, 1553–1563 Search PubMed.
- S. Monfette, Y. Q. Fang, M. M. Bio, A. R. Brown, I. T. Crouch, J. N. Desrosiers, S. Duan, J. M. Hawkins, C. M. Hayward, N. Peperni and J. P. Rainville, Org. Process Res. Dev., 2020, 24, 1077–1083 Search PubMed.
- X. Zhang, K. G. Nottingham, C. Patel, J. V. Alegre-Requena, J. N. Levy, R. S. Paton and A. McNally, Nature, 2021 DOI:10.1038/s41586-021-03567-3.
Footnote |
| † These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Claudio F.
Meyer†
a,
Claudio F.
Meyer†