
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Advances in applied supramolecular technologies
George T.
Williams
 a,
Cally J. E.
Haynes
*b,
Mohamed
Fares
c,
Claudia
Caltagirone
*d,
Jennifer R.
Hiscock
*a and
Philip A.
Gale
*ce
a,
Cally J. E.
Haynes
*b,
Mohamed
Fares
c,
Claudia
Caltagirone
*d,
Jennifer R.
Hiscock
*a and
Philip A.
Gale
*ce
aSchool of Physical Sciences, University of Kent, Canterbury, Kent CT2 7NH, UK. E-mail: j.r.hiscock@kent.ac.uk
bDepartment of Chemistry, UCL, London, WC1H 0AJ, UK. E-mail: cally.haynes@ucl.ac.uk
cSchool of Chemistry, The University of Sydney, Sydney, NSW 2006, Australia. E-mail: philip.gale@sydney.edu.au
dDipartimento di Scienze Chimiche e Geologiche, Università degli Studi di Cagliari, S.S. 554 Bivio per Sestu, 09042 Monserrato (CA), Italy. E-mail: ccaltagirone@unica.it
eThe University of Sydney Nano Institute (Sydney Nano), The University of Sydney, NSW 2006, Australia
First published on 13th January 2021
Abstract
Supramolecular chemistry is a comparatively young field that to date has mainly been focused on building a foundation of fundamental understanding. With much progress in this area, researchers are seeking to apply this knowledge to the development of commercially viable products. In this review we seek to outline historical and recent developments within the field of supramolecular chemistry that have made the transition from laboratory to market, and to bring to light those technologies that we believe have commercial potential. In doing so we hope we may illuminate pathways to market for research currently being conducted.
 George T. Williams | George Williams is a postdoctoral research fellow at the University of Kent in the group of Dr Jennifer Hiscock. He completed his undergraduate studies and PhD at the University of Bath under the supervision of Prof. Toby Jenkins. His research primarily focusses around the use of supramolecular chemistry to develop sensors, drug delivery vehicles and stimuli responsive materials. |
 Cally J. E. Haynes | Cally Haynes is a Lecturer in Organic Chemistry and Chemical Biology at UCL. She studied for a PhD in the group of Prof. Phil Gale at the University of Southampton and carried out postdoctoral research in the same group until 2013. Following an editorial position with the Royal Society of Chemistry, she joined the research group of Prof. Jonathan Nitschke at the University of Cambridge as a postdoctoral researcher in 2015 before joining UCL in 2019. Her research interests in supramolecular chemistry include molecular transport and separations, coordination driven self-assembly and the design of functional soft materials. |
 Mohamed Fares | Mohamed Fares is a postdoctoral researcher at the School of Chemistry, University of Sydney (Gale group). After finishing BPharm (Ain Shams University, Egypt) and MPharm (Cairo University, Egypt), he was awarded the prestigious IPRS in 2015 to pursue his PhD at the University of Wollongong (Australia) under supervision of Professor Paul Keller and Professor Phil Gale. Mohamed's research interests include the anion transport supramolecular chemistry and small molecule drug discovery. |
 Claudia Caltagirone | Claudia Caltagirone obtained her PhD in Chemistry under the supervision of Prof. Vito Lippolis at the University of Cagliari (Italy) in 2006. In the same year she became assistant professor in Inorganic Chemistry and then she moved to the University of Southampton (UK) to work as an academic visitor in the group of Prof. Philip A. Gale. Since 2016 she is associate professor in Inorganic Chemistry at the University of Cagliari. Her research mainly focuses on the design of novel supramolecular systems for anion sensing and for the development of new materials. |
 Jennifer R. Hiscock | Jennifer Hiscock is a Reader in Supramolecular Chemistry, Director of Innovation and Enterprise and UKRI Future Leaders Research Fellow in the School of Physical Sciences at the University of Kent (UK). She studied for a PhD in the group of Prof. Phil Gale at the University of Southampton and carried out postdoctoral research in the same group until 2015. Following this she moved to the University of Kent as the Caldin Research Fellow and was appointed Lecturer in Chemistry at this same institution in 2016. Her current research interests focus on the development of Supramolecular Self-associating Amphiphiles (SSAs) as antimicrobial, anticancer and drug adjuvant agents. |
 Philip A. Gale | Phil Gale is Professor and Head of the School (Chemistry), and Associate Dean (International) in the Faculty of Science at the University of Sydney. Phil's research interests focus on the supramolecular chemistry of anionic species and in particular the molecular recognition, sensing and lipid bilayer transport of anions. His research group's contributions to these areas were recognized in 2018 by the International Izatt-Christensen Award in Macrocyclic and Supramolecular Chemistry. |
Introduction
The term supramolecular chemistry was first coined by Jean-Marie Lehn in the 1970s.1 Although this was first use of the term, the study of non-covalent molecular complexation, a field that we now identify as being part of supramolecular chemistry, was already under investigation within the scope of other scientific fields such as enzymology, organic chemistry and inorganic chemistry.2–4 Today, supramolecular chemistry broadly encompasses the study of all types of intermolecular non-covalent bond formation in designed molecular systems, and also incorporates dynamic covalent chemistry.5–7 The body of work undertaken in this field since the late 1960s has twice resulted in the award of the Nobel prize in Chemistry, first in 1987 and again in 2016, for the development of host–guest binding molecules8 and molecular machines respectively.9As a rapidly growing scientific field of study, supramolecular chemistry is now expanding beyond the chemical space, finding applications at the interfaces with many other disciplines, and offering novel solutions to age old problems in biology, physics, engineering and pharmacy.10 This has caused the field of supramolecular chemistry to move towards commercial, real world applications whilst fundamental studies still continue. As a result industry has now begun to embrace this field of chemistry, incorporating aspects of these next generation technologies into a variety of everyday commercial, marketable products.7,11–13
Examples of applied supramolecular innovation include the development of medical diagnostic sensors, enabled through advances in selective host–guest complex formation;7,14 maintenance free materials, created through incorporation of reversible non-covalent moieties within material constructs;15,16 and ion extraction realised through the development of molecular encapsulation technologies.17,18 Herein, we update and expand on a previous review by Kolesnichnko and Anslyn,19 to discuss the application of supramolecular chemistry in the real world. This includes commercial examples of supramolecular chemistry in everyday items, sensors, medicine, materials and extraction technologies. Additionally, we provide insight into recent supramolecular developments that may have the potential to make the transition from laboratory to market.
Supramolecular chemistry in the household
Supramolecular chemistry underpins the workings of many products and, perhaps surprisingly, many of these are simple household items. An example of these items are the cyanoacrylate adhesives, more commonly known as superglue. The late M. Anthony McKervey and his group at Queen's University, Belfast were calixarene chemists who reported (in collaboration with Schwing-Weill and co-workers) a number of ionophoric calix[n]arenes formed by appending additional groups to the lower-rim. For example, p-tert-butylcalix[n]arene where n = 4 or 6 could be reacted with ethyl bromoacetate in dry acetone in the presence of potassium carbonate as base by refluxing for several days to afford the calix[4]arene tetra-ester 1 or calix[6]arene hexa-ester 2 respectively.20 These compounds have high affinities for group 1 metal cations with compound 1 showing Na+ selectivity over other group 1 metal cations whilst compound 2 is selective for larger metal cations. McKervey and co-inventors found that these types of ionophoric calixarenes could be used in super glue as accelerators by sequestering group 1 metal cations. For example, the addition of 0.1% compound 2 to a commercially available cyanoacrylate-based adhesive reduced fixture time from between 20 to 25 minutes to between 3 and 5 minutes, allowing porous surfaces to be effectively bonded together before the glue had diffused away (Fig. 1).21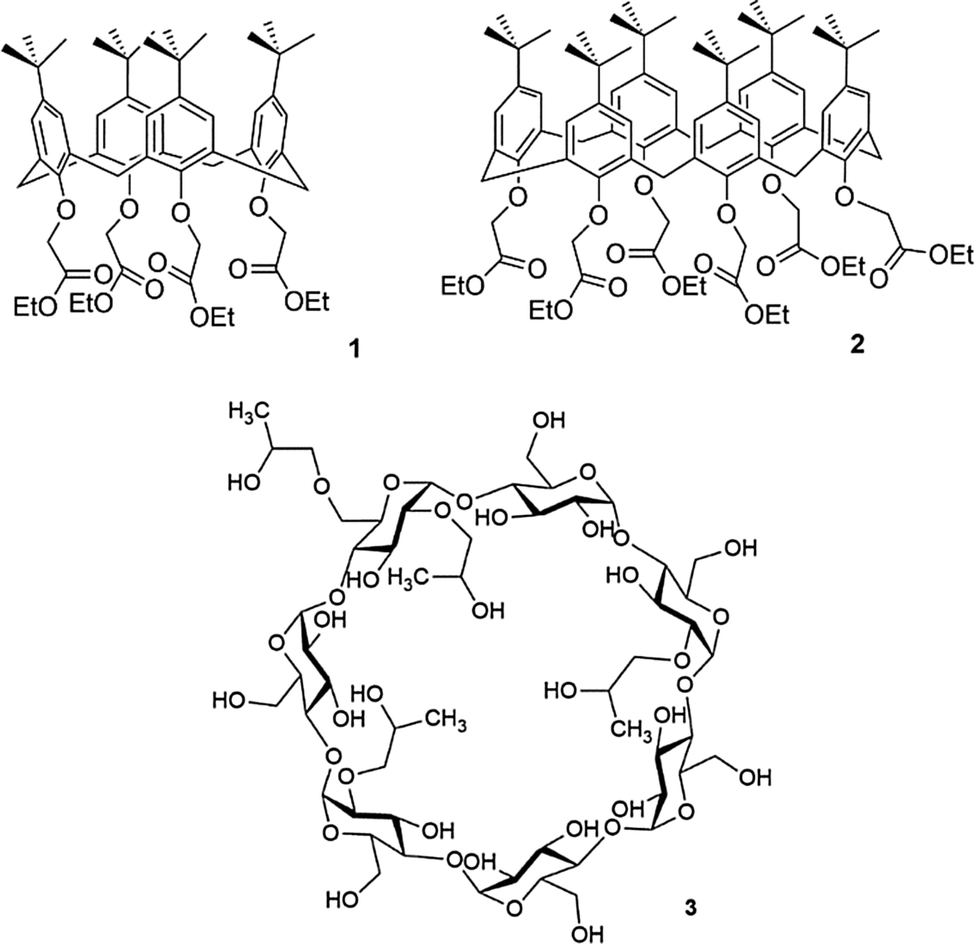 | ||
| Fig. 1 Calix[4]arene tetra-ester (1) and calix[6]arene hexa-ester (2) which have found use in superglue formulation, and hydroxypropyl beta-cyclodextrin (3), an odour trapping molecule found in Febreze. | ||
Another class of supermolecular host used as components of household products are the cyclodextrins (CDs), which are cyclic oligosaccharides that consist of glucose monomers linked by α-1,4-glycosidic bonds.22 These compounds were first isolated by Antoine Villiers in 1891 and are produced from starch by an enzymatic degradation process.22,23 Another household use of supramolecular chemistry is in the product Febreze which is manufactured by Procter and Gamble. Febreze is a spray that eliminates household odours and can be used on fabrics.11 The spray contains hydroxypropyl beta-cyclodextrin (HPBCD) (3), a cyclodextrin derivative with increased solubility. In this case 3 is reputed to complex odour molecules within the cavity of the macrocycle and so suppresses odours. HPBCD as a mixture of compounds with various degrees and patterns of hydroxypropylation and along with other cyclodextrin derivatives has been used as an excipient to improve the water solubility and bioavailability of drugs.24
Supramolecular chemistry in medicine
The three parent cyclodextrins, α (4), β (5) and γ-CDs (6) are formed from 6, 7 and 8 D-glucose units, respectively (Fig. 2).25 Synthetic CD derivatives include the hydrophilic HPBCD, the hydrophobic 2,6-di-O-ethyl-β-CD and the ionizable sulfobutylether-β-CD.26 The approximate dimensions of α, β and γ-CDs are illustrated in Fig. 3.22 Cyclodextrins have a doughnut shaped structure with a hydrophobic cavity and a hydrophilic outer surface (Fig. 4), allowing them to accommodate small hydrophobic drugs or moieties inside the cavity.27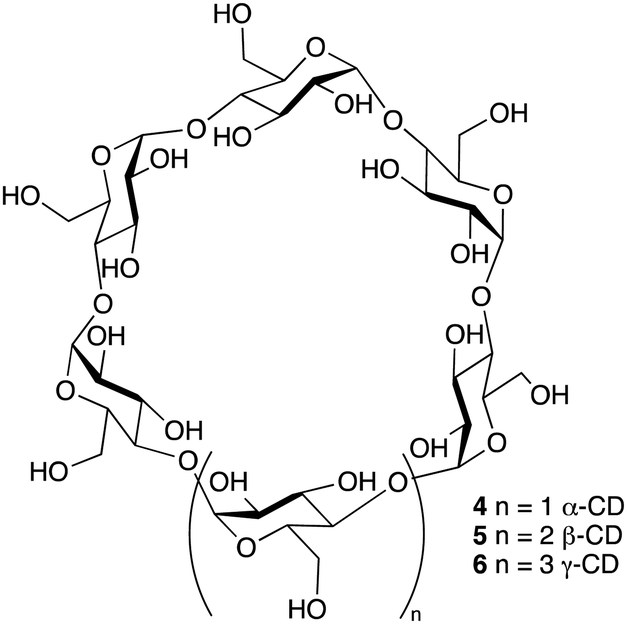 | ||
| Fig. 2 Chemical structure of α, β and γ-cyclodextrin. | ||
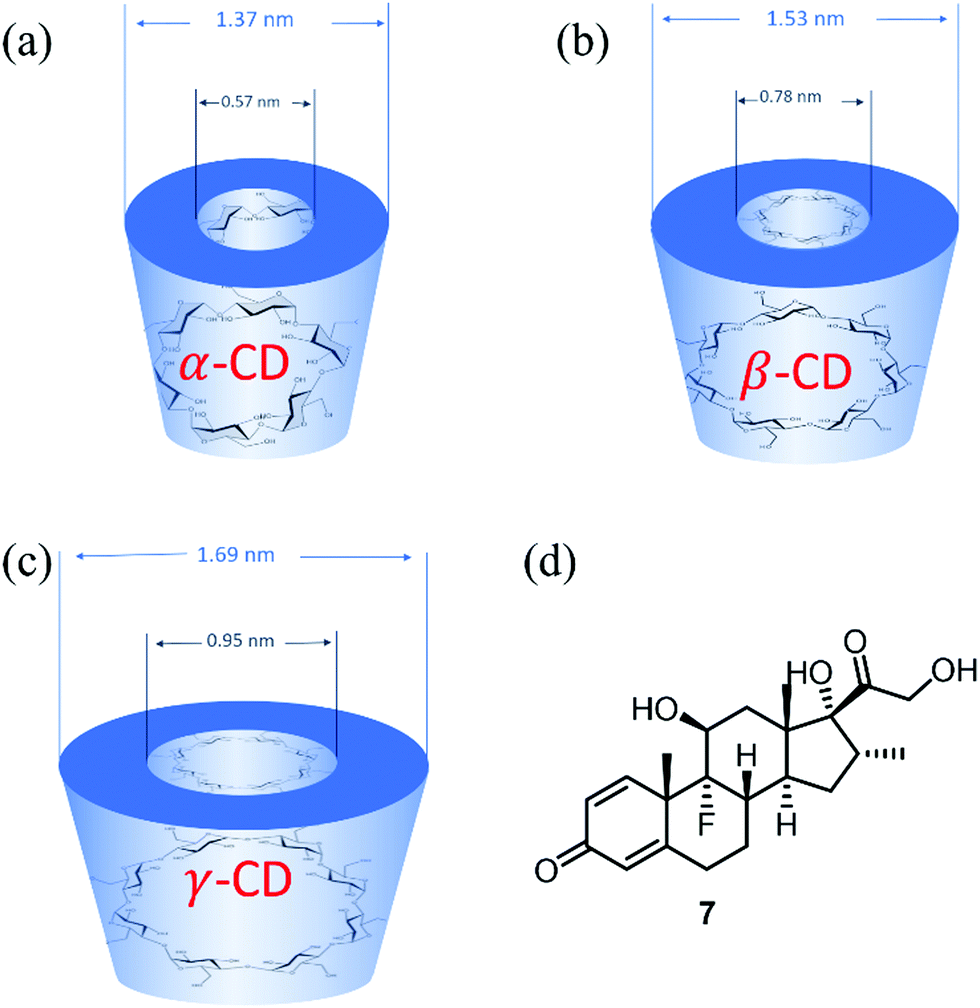 | ||
| Fig. 3 (a–c) Geometric dimensions of α, β and γ-cyclodextrin, (d) structure of dexamethasone, 7. | ||
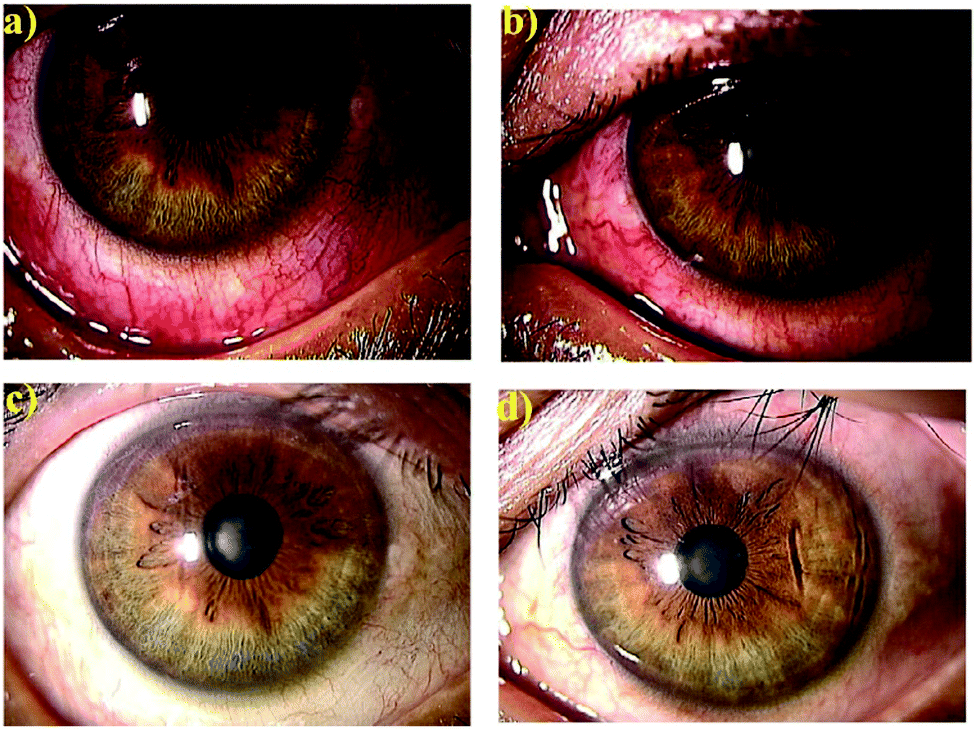 | ||
| Fig. 4 Anteriorocular photographs of a 45-year-old man with anterior scleritis before (a and b) and after 4 weeks treatment with OCS-01 (c and d). Right eye (a and c) and left eye (b and d).35 Reproduced from ref. 35 with permission from Wiley, copyright 2019. | ||
The scalable synthesis of the parent α, β and γ-CDs also makes this scaffold a commercially attractive prospect with a comparatively low price point and ease of purification. The 30 year development in CD related technologies has led to wide applications in pharmaceuticals, drug delivery systems, cosmetics, and the food and chemical industries.26,28–30 Amongst notable applications of CDs are their use in overcoming the limitations of certain pharmaceutical compounds by forming an inclusion complex, enhancing the drug solubility and stability and improving drug permeation and bioavailability.22,26 These inclusion complexes enhance the physicochemical characteristics of the drugs without changing their intrinsic properties. However, the limitations of CDs include the small cavity size of α-CD, the low water solubility of β-CD and low production yields of γ-CD.27
Interestingly, a topical drug delivery system (eye drops) based on dexamethasone 7 (Fig. 3(d)) and γ-CD nanoparticles has been developed by Oculis®.31 This non-invasive system contains 1.5% dexamethasone and γ-CD nanoparticles (OSC-01) and showed a promising reduction of the central macular thickness (CMT) and an increase in visual acuity in diabetic macular oedema (DMO) patients.32In vivo studies in humans showed that OCS-01 achieved a 10 times higher concentration of dexamethasone in the eye tissue after 4 h of application, when compared to Maxidex® (0.1% dexamethasone only eye drops). This was mainly due to the higher ocular absorption and a longer retention of the γ-CD–drug nanoparticles at the site of action.33 OCS-01 completed phase-2 clinical trials (DX-216) for treating inflammation and pain following cataract surgery.34 Of the 144 randomized patients who started this study, 131 completed the trial. Results from DX-211 showed the efficacy of OSC-01 in patients with DME when compared to the vehicle. OCS-01 showed no significant or unexpected ocular adverse effects.34
The OCS-01 eye drops were also investigated for their anti-inflammatory effect in 10 human eyes for six patients (five female) complaining of different ocular inflammatory diseases, including anterior scleritis, posterior scleritis, unidentified uveitis, Vogt–Koyanagi–Harada disease with papillitis and cystoid macular oedema.35 All treated eyes responded to OCS-01 with a rapid resolution of congestion with no patients experiencing impaired visual acuity after the treatment started.35Fig. 4 shows both eyes of a 45-year-old man with anterior scleritis before (Fig. 4(a and b)) and after 4 weeks treatment with OCS-01 (Fig. 4(c and d)). After treatment, ocular congestion almost disappeared, and the pain was reduced significantly and did not recur.35
Poor control of blood glucose levels is implicated with increased morbidity and mortality, especially in hospital intensive care unit (ICU) patients.36,37 Continuous glucose monitoring over a 24 h period is useful when seeking to acquire control of blood sugar and to avoid hypo- and hyper-glycaemia, especially in diabetic patients.38,39 Great efforts have been made in the development of D-glucose sensors based on supramolecular interactions between boronic acid motifs and sugars via hydrogen bonding and ester formation.40–44 Boronic acids are a reliable group for sugar sensing and have showed a high affinity for vicinal diols, catechols and compounds appended with α-hydroxycarboxylate when incorporated into different scaffolds.44 However phenylboronic acid shows significant binding to carbohydrates only above physiological pH, indicating its limited application.41,45 The inclusion of an α-aminomethyl group in the ortho-position of the boronic acid was found to tune the selectivity and improve the binding affinity at neutral pH.46–49 Interestingly, mono or simple boronic acids have been found to bind to saccharides in the order D-fructose > D-galactose > D-mannose > D-glucose.50–52 Changing the binding site to a diboronic acid motif and expanding the structure into more complex systems, capable of binding at multiple sites, perturbed the D-glucose selectivity.51 Finally, incorporating fluorophores such as pyrene and anthracene into the α-aminomethylphenylboronic acid scaffold has yielded hosts that have been used for sensing and cellular imaging.44,52
As a result of these innovations, two companies, namely Eversense (by Senseonics) and Glysure Ltd, have been founded by Tony James and his collaborators, exploiting the application of fluorescent o-aminomethylphenylboronic acids, (8) (Fig. 5).53–55 The Glysure system (9, Fig. 6(a)) and the Eversense (10, Fig. 6(b)) detection chemistry are based on the covalent interaction between the diboronic acids motifs and D-glucose and the formation of cis-1,2-diols or 1,3-diols resulting in the formation of either a five or six membered ring system.54 The acidity of the boron atoms increases when D-glucose binds to the boronic acid groups leading to the formation of weak boron–nitrogen (B^N) bonds. The B^N interaction disrupts electron transfer from the tertiary amines to covalently tethered fluorophores thus leading to fluorescence emission.54 The resulting fluorescence is proportional and quantitative to the D-glucose concentration. Both systems demonstrated a valid and accurate continuous monitoring of blood glucose levels, receiving CE Mark approval in 2015 (Glysure) (Fig. 6(a)) and 2017 (Eversense) (Fig. 6(b)). The Eversense system is derived from the unprecedentedly selective glucose chemosensor (10) developed by James, Sandanayake and Shinkai in 1994, while Glysure system (9) is based on the fluorescent sensor discovered by Arimori, Frimat and James in 2001 as part of a collaboration with Beckman Coulter.51–53,56,57
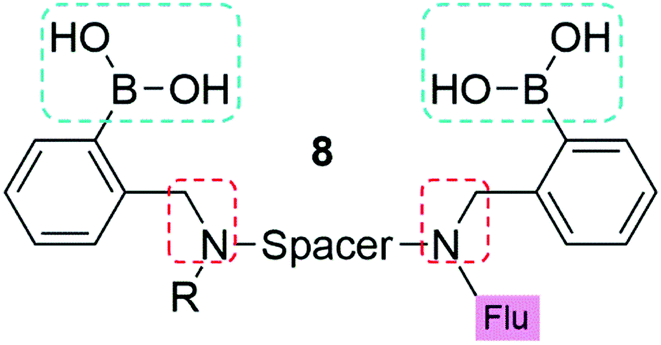 | ||
| Fig. 5 The general structure of the boronic acid based glucose sensors used for continuous glucose monitoring. Blue boxes outline the two boronic acids used to give D-glucose selectivity. Red boxes indicate the ortho α-amino acid to improve saccharide binding affinity at neutral pH. R represents the anchor point to a hydrogel, for example poly(2-hydroxyethylmethacrylate). A six carbon chain is used as the spacer to give glucose selectivity, and may be aliphatic (Glysure LTD) or aromatic (Eversense) in nature. | ||
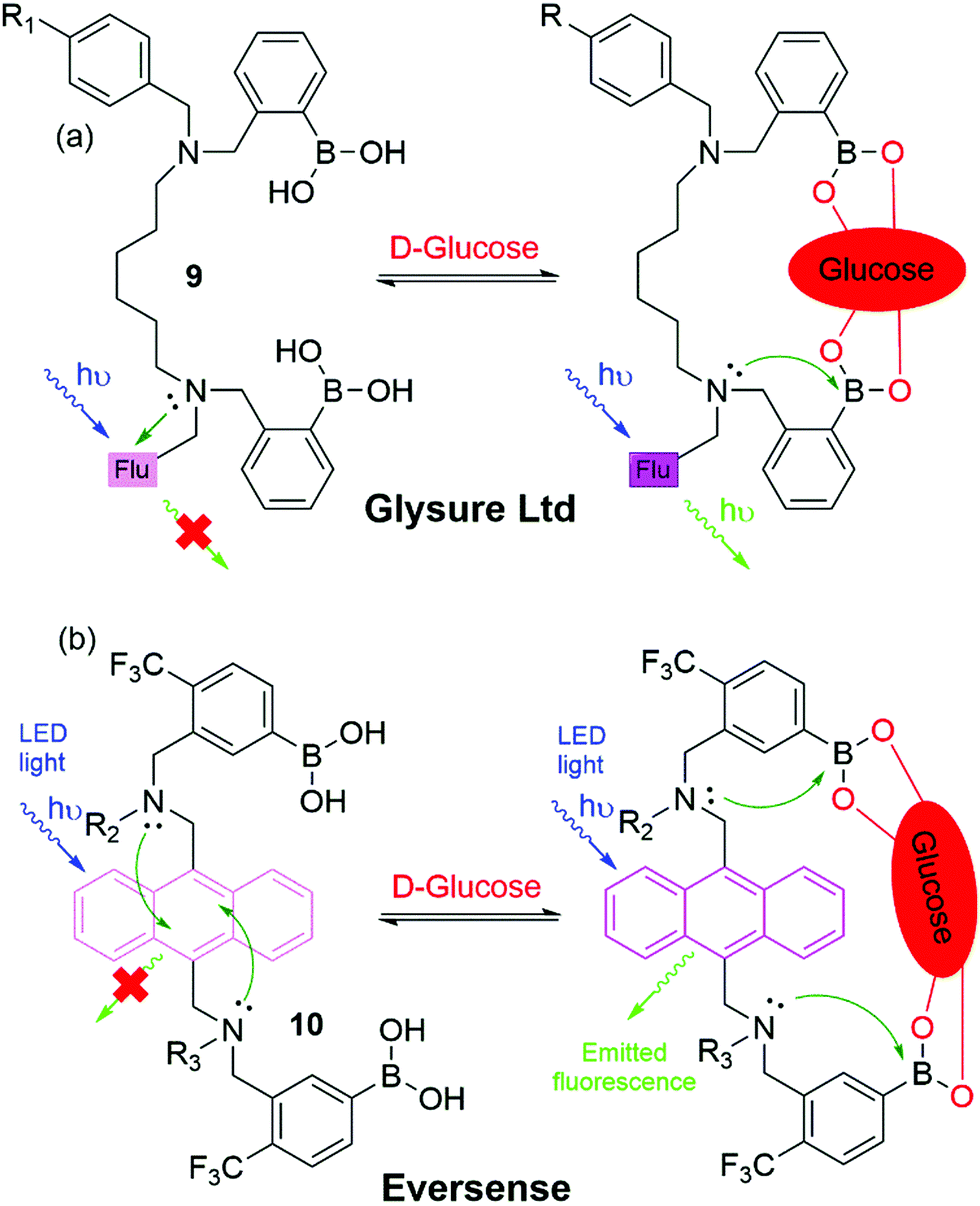 | ||
| Fig. 6 Chemical structure and proposed glucose binding mode of (a) the Glysure Ltd system 9 (R1 = hydrogel) and (b) the Eversense system 10 (R2 = propionic acid side chain, R3 = hydrogel). | ||
Blood glucose monitoring devices incorporating the Glysure and Eversense systems are illustrated in Fig. 7. The Glysure system shown in Fig. 6(a) is immobilized within a hydrogel, while the Eversense system (Fig. 6(b)) is immobilized in a poly(2-hydroxyethylmethacrylate) (pHEMA) hydrogel which is copolymerized with a fluorescent indicator.54 The Glysure sensor device is composed of the immobilized fluorescent glucose indicator cell, (Fig. 7(a)).54 The immobilized hydrogel fills the optical fibre which is surrounded by a dialysis membrane, and consequently fills the optical cell. The microporous membrane, with a pore diameter ∼0.1 μM, prevents blood cells from entering the sensor (Fig. 7(a)).54
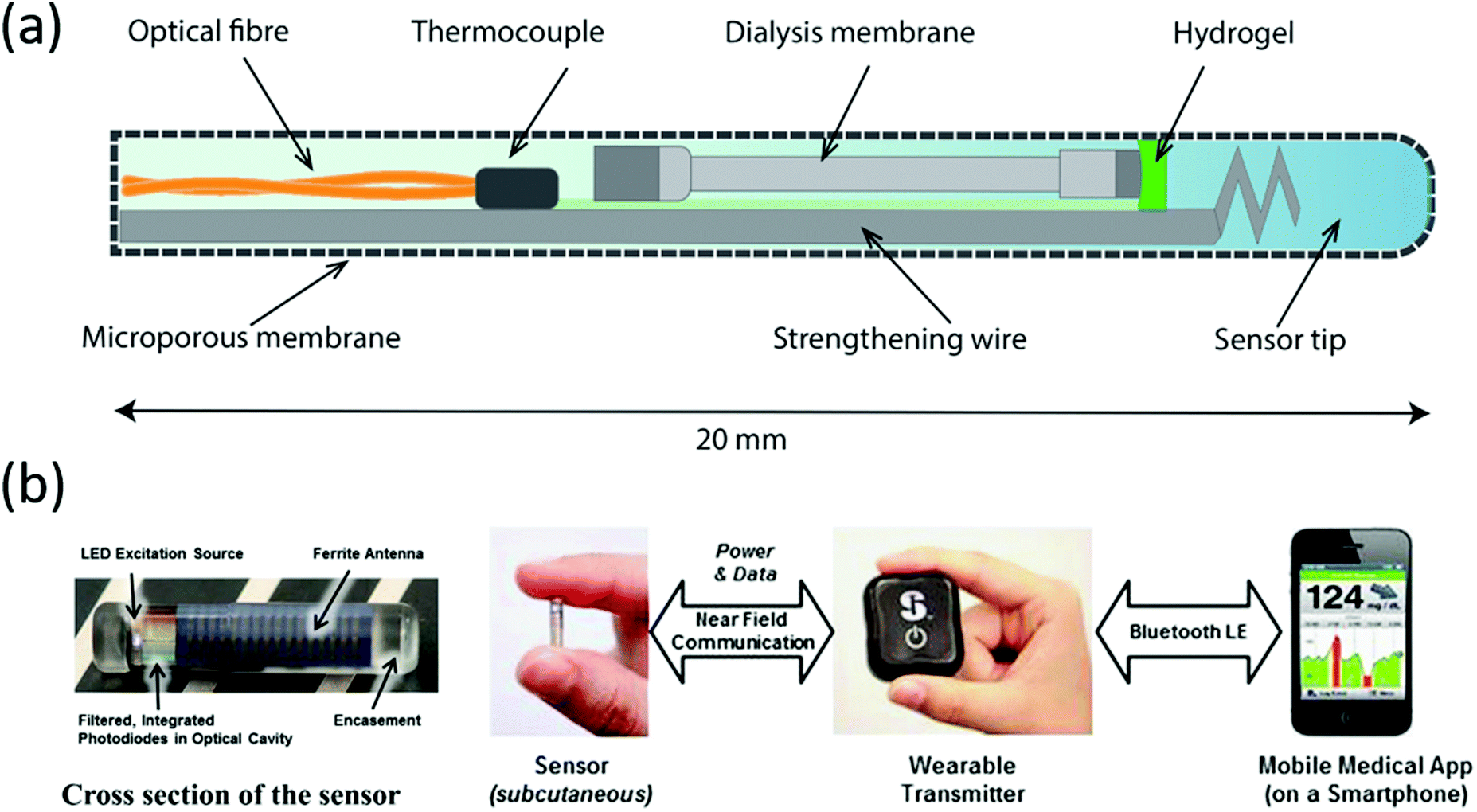 | ||
| Fig. 7 Continuous glucose monitoring system components of (a) Glysure Ltd and (b) Eversense, reproduced from ref. 53 with permission from Elsevier, copyright 2014. | ||
The subcutaneous Eversense sensor monitors glucose concentration in interstitial fluids (Fig. 7(b)).58 This sensor has a diameter of 3.3 mm and a length of 15 mm, which is then enclosed in a rigid and biocompatible polymer capsule (Fig. 7(b)).58 The glucose detection hydrogel is copolymerized onto the capsule surface, while a light emitting diode (LED) provides the excitation source for the fluorescent gel. Two filtered photodiodes measure the fluorescence intensity, while the antenna is responsible for communication with the transmitter.
An externally worn device communicates with the inserted sensor and receives the recorded information. The measurements can also be displayed on a secondary output wirelessly via Bluetooth™ and can be downloaded through a Universal Serial Bus (USB) port (Fig. 7(b)).53
Cucurbiturils (CB) are synthetic barrel-shaped macrocycles obtained from the condensation of formaldehyde and glycolurils.59,60 Although first synthesized more than 100 years ago, cucurbit[6]uril (CB[6]), 11, shown in Fig. 8, was only characterized in the 1980s.59 Over the last two decades, other members of the CB family have been synthesized and purified, including CB[5], CB[7], CB[8], and CB[10] containing 5, 7, 8, or 10 glycoluril subunits, respectively.60,61 CBs display unique physical, chemical and biological properties, primarily owing to their intrinsic ability to undergo complexation with diverse chemical species resulting in the formation of dynamic host–guest complexes. Recently, CBs have gained interest within an increasingly diverse range of applications including nanotechnology, molecular recognition, catalysis, drug delivery, analytical and environmental chemistry and chemical biology.60–64 For example, CB[7] typically binds one guest molecule and can be crosslinked with a variety of motifs, such as adamantane and gold nanoparticles, to prepare supramolecular hydrogels, functional surfaces and nanoparticles with commercial applications.65,66
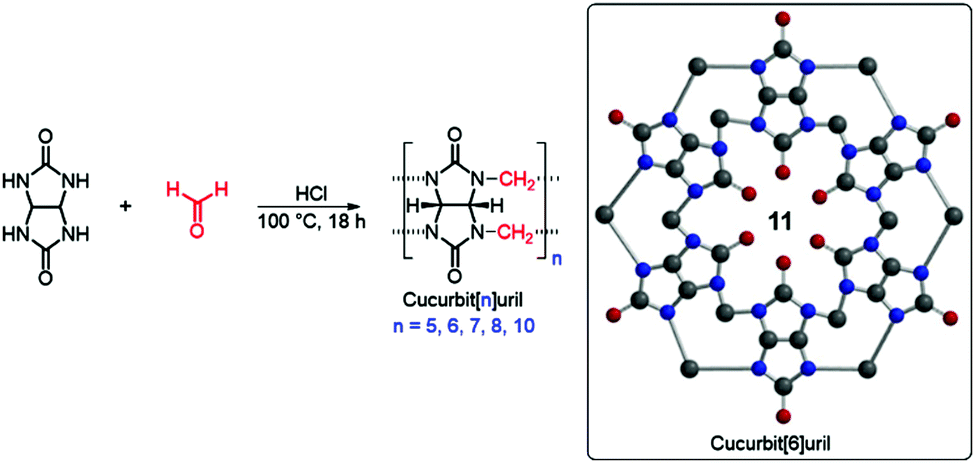 | ||
| Fig. 8 Standard synthesis of CB[n] and 2D chemical structure of CB[6] 11. | ||
A recent study in this area has employed CB[7] as a noncovalent linker motif to enhance the interaction between catechol functionalized chitosan (CAT-CS) and superparamagnetic γ-Fe2O3 nanoparticles.67Fig. 9 shows the synthesis of a magnetic hydrogel nanocomposite composed of CB[7] linked to superparamagnetic γ-Fe2O3 nanoparticles, bound to the CAT-CS polymer backbone. Here the polysaccharide chitosan is post-functionalized with the catechol motif yielding CAT-CS, which was is then complexed with CB[7] via inclusion of the catechol side chains in the cavities of the CB[7] macrocycles. The carbonyl groups of the CB[7] then bind with the γ-Fe2O3 nanoparticles through electrostatic interactions, resulting in the formation of a noncovalent network (Fig. 9). The hybrid supramolecular hydrogel (HSH) displays heat generation through vibrational movement upon exposure to an alternating magnetic field. This offers a promising stimuli-responsive thermo-chemotherapy, which has been investigated for biological activity both in vitro and in vivo.67 Here, in vitro cellular thermal studies conducted using the apoptosis detection kit annexin V-FITC/PI in HeLa cells, showed that when the HSH was exposed to an alternating magnetic field for 12 minutes, cell apoptosis was induced through an increase in temperature.67 Additionally, comparative in vivo intravital fluorescence imaging and infrared thermography studies were performed in tumour-bearing nude mice. In this study, mice were injected with indocyanine green-loaded HSH and, it was shown that in the presence of alternating magnetic pulses tumour size decreased. These effects were shown to be dependent on exposure time, with longer exposures leading to a greater reduction in tumour size (Fig. 10(a–c)). This indicates that the intermittent exposure to magnetic pulses was the major contributing factor to both the therapeutic outcomes and overall mouse fitness, as indicated by comparative difference in mouse body weight. Finally, hematoxylin and eosin (H&E) staining and the immunohistochemical examination of biomarkers for cell proliferation and apoptosis were also performed (Fig. 10(d)). These results showed that in this instance thermo- and chemotherapies were acting synergistically, while the immunohistochemical examinations suggested that the intermittent magnetic irradiation provided an efficient way to suppress tumour growth in vivo.67
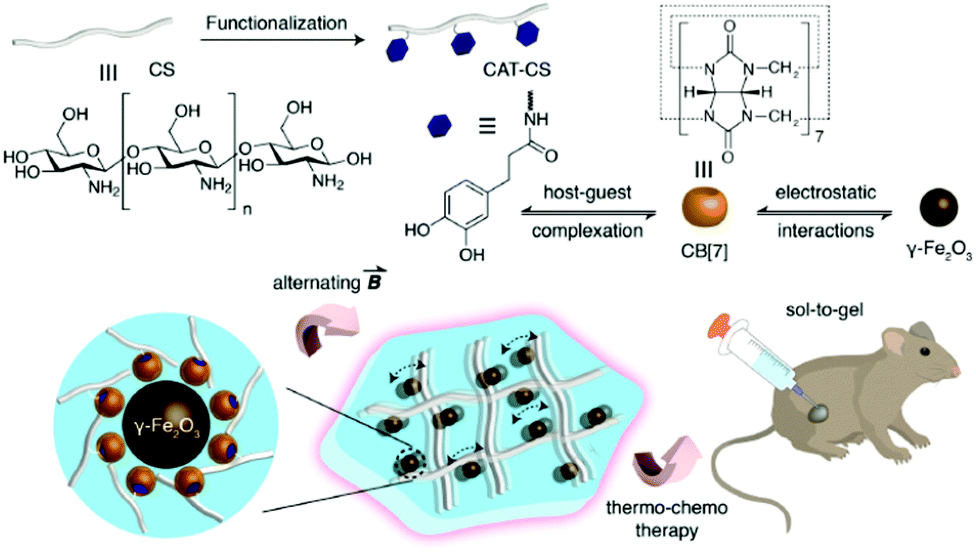 | ||
| Fig. 9 Schematic representation of the hybrid supramolecular hydrogel (HSH) fabrication via CB[7]-mediated electrostatic interactions molecular recognition.67 Reproduced from ref. 67 with permission from Wiley, copyright 2019. | ||
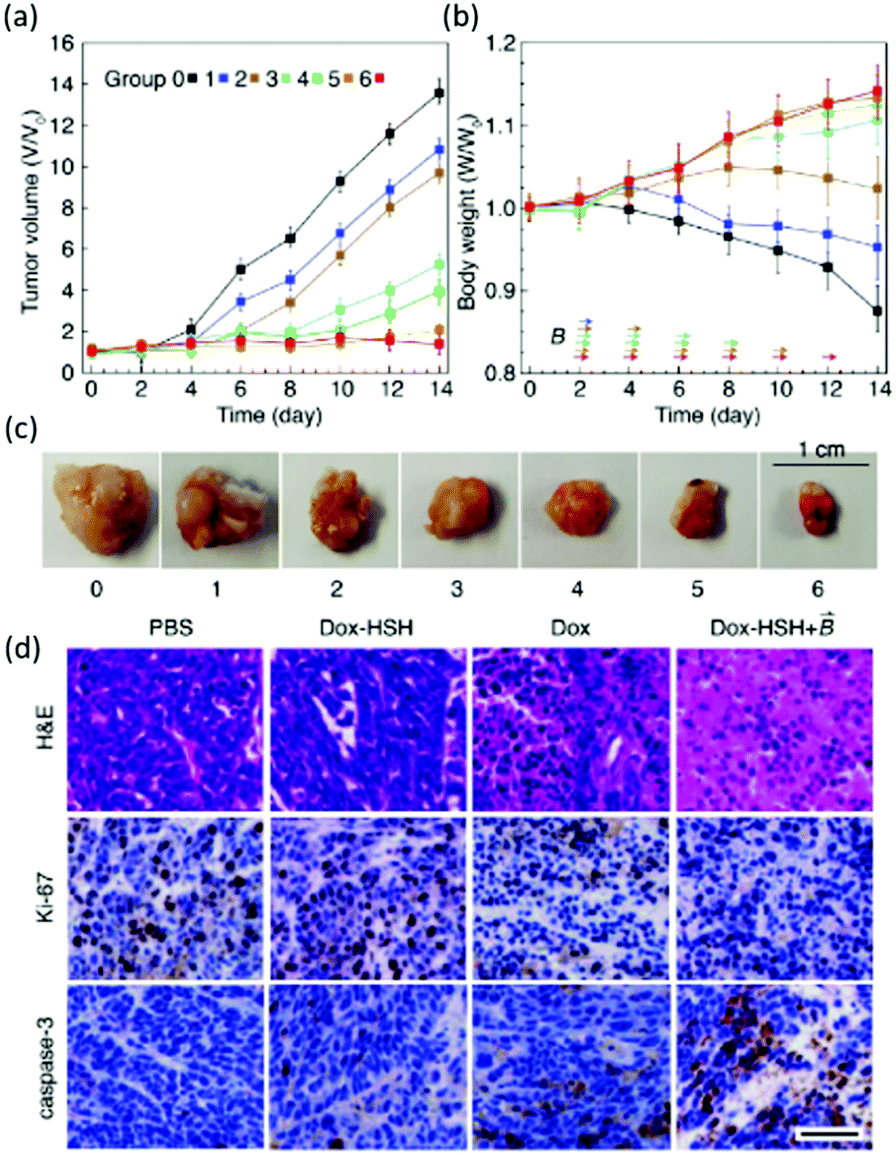 | ||
| Fig. 10 Changes in (a) tumour volume (b) and body weight within 14 days of treatment, arrows indicate the time when the magnetic field was applied. (c) Images of tumors isolated at the end of the experiment. (d) (H&E staining) and immunohistochemical (Ki-67 for cell proliferation and cleaved caspase-3 for apoptosis) analysis was performed on HeLa tumour tissues in mice treated with PBS buffer (control), Dox-loaded HSH (absences of the magnetic field), Dox (presence of the magnetic field), Dox-loaded HSH (presence of the magnetic field), Scale bar is 50 μm.67 Reproduced from ref. 67 with permission from Wiley, copyright 2019. | ||
Supramolecular sensors
Since the pioneering work of Czarnick and de Silva reported in the 1980's and early 1990's, relating to the development of molecular chemosensors,68–74 numerous papers have appeared in the literature on the subject. Quite surprisingly, only a few systems have found application in real life so far. The company Optimedical Inc. commercialised a medical diagnostic analyzer (OPTI LION®) based on three molecular fluorescent chemosensors 12–14, (Fig. 11), selective for Na+, K+, and Ca2+, respectively, designed by de Silva and collaborators. This device can quantify the concentrations of electrolytes contained within blood serum via fluorescence measurements. This system is currently marketed for use within hospital critical care units, ambulances, and even in veterinary settings.75,76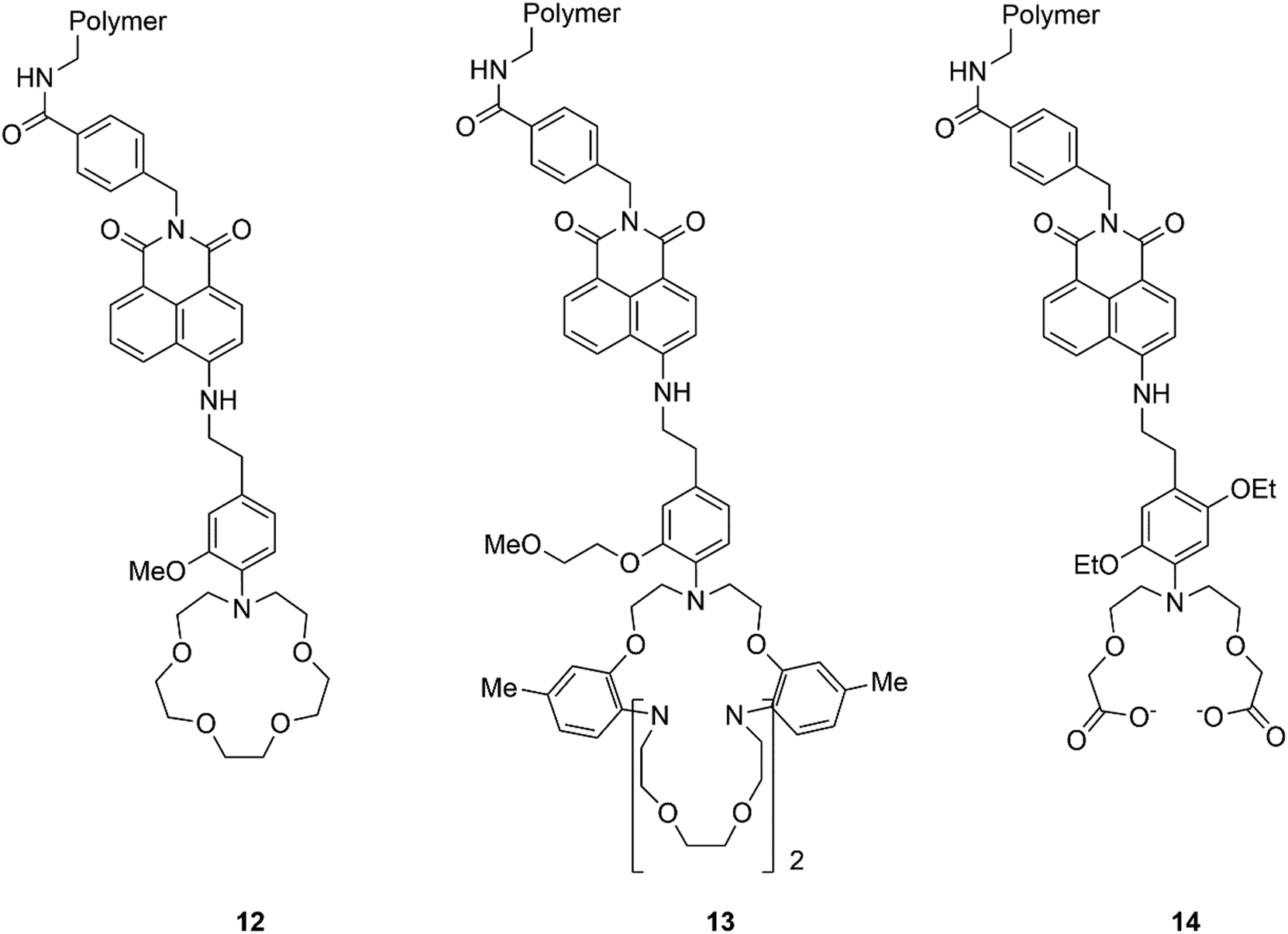 | ||
| Fig. 11 Structure of sensors 12–14. | ||
Additionally, a spin-out company from the University of Bristol, UK, Zylo, founded by Anthony Davis and focused on the development of biomimetic glucose binding molecules was acquired by the global healthcare company Novo Nordisk in 2018. Davis and co-workers are still working in the field, in a new company named Carbometics, and have recently described a highly selective synthetic receptor able to bind glucose in water with a remarkable Ka = 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1, comparable with natural receptor systems.77 This lectin mimic features a highly polar hexaurea core that form hydrogen bonds with the hydroxyl groups within glucose, and a triethylmesitylene moieties that form hydrophobic/CH–π interactions with the CH groups, while the high solubility in water is guaranteed by three peripheral nonacarboxylate groups. This rational design towards binding saccharides with equatorial all equatorial hydroxyls enables this lectin mimic to bind glucose over mannose, a related sugar differing by only a single axial hydroxyl group, with a 100 fold increased binding affinity (Ka = 18600 and 140 M−1 respectively). The system also showed high thermal stability and no toxicity towards HeLa cells. This specificity towards glucose in biological mixtures is crucial in the development technologies to help control diabetes.
000 M−1, comparable with natural receptor systems.77 This lectin mimic features a highly polar hexaurea core that form hydrogen bonds with the hydroxyl groups within glucose, and a triethylmesitylene moieties that form hydrophobic/CH–π interactions with the CH groups, while the high solubility in water is guaranteed by three peripheral nonacarboxylate groups. This rational design towards binding saccharides with equatorial all equatorial hydroxyls enables this lectin mimic to bind glucose over mannose, a related sugar differing by only a single axial hydroxyl group, with a 100 fold increased binding affinity (Ka = 18600 and 140 M−1 respectively). The system also showed high thermal stability and no toxicity towards HeLa cells. This specificity towards glucose in biological mixtures is crucial in the development technologies to help control diabetes.
A selection of other chemosensors that could be implemented for applications in the real world are discussed hereafter. Lippolis and co-workers have described a very simple example of a Hg2+ optode by incorporating receptor 15, into a PVC membrane, (Fig. 12). This system is able to detect Hg2+ in real samples such as an amalgam alloy, hair and well water.78
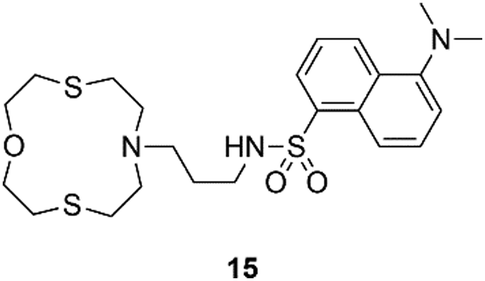 | ||
| Fig. 12 Structure of chemosensor 15. | ||
The sensor is commercially available from Merck.
The recognition of various methamphetamines (MDMA, cocaine, amphetamine, and 3-fluoromethamphetamine) through a synergistic effect of weak interactions (H-bonds, CH–π) was achieved using tetraphosphonate cavitands.79 In particular, pyrene-tetraphosphonate cavitands embedded into using silica nanoparticles have been successfully demonstrated by Dalcanale, Prodi and co-workers to detect ecstasy in water.80 As shown in Fig. 13, the pyrenes in the distal position can form an excimer which is the transducer element of the chemosensor. In the presence of a bulky N-methylammonium guest the excimer formation is perturbed, and the monomer emission can be observed.
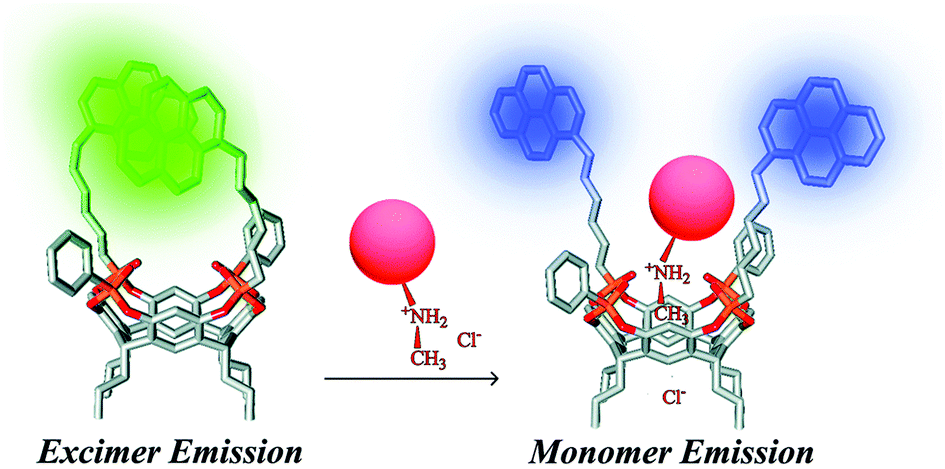 | ||
| Fig. 13 Proposed sensing mode for ecstasy recognition.80 Reproduced from ref. 80 with permission from the Royal Society of Chemistry, copyright 2015. | ||
The same class of compounds, anchored on silicon surfaces, have also been used to recognize sarcosine (recently linked to the occurrence of aggressive prostate cancer forms) from its nonmethylated precursor glycine, in water and urine.81 The selective molecular recognition of sarcosine by a tetraphosphonate cavitand was further implemented to develop an electroluminescent approach.82 Generating luminescence via electrochemistry allows for very low background and high sensitivity, good temporal and spatial resolution, robustness, versatility, and low fabrication cost.83 In a typical electrochemiluminescence experiment in water, the energy to generate the emitting excited state comes from an electrochemical oxidation–reduction process between a dye, classically Ru(bpy)32+, and a co-reactant (typically an amine that can partially deprotonate forming a reactant radical that can reduce the dye to Ru(bpy)3+). At the same time Ru(bpy)32+ can be oxidized at the electrode surface and react with its reduced form generating the emitting excited state Ru(bpy)32+*. In this case, sarcosine acts as the co-reactant able to reduce the ruthenium complex. By functionalising the surface of magnetic beads with the cavitand, the authors were able to form a complex with sarcosine at acidic pH; separate the complex from the matrix by capturing the magnetic beads with a magnetic field; release sarcosine by increasing the pH; and measure its concentration via electrochemiluminescence.82
Anslyn has long been a pioneer within the field of chirality detection, it was almost 20 years ago that a simple, yet very elegant dynamic four-component reversible covalent assembly for secondary alcohol binding was proposed, Fig. 13.84 The reversible self-assembly of pyridine-2-carboxaldehyde, di-(2-pyridylmethyl)amine, Zn(OTf)2 and a secondary alcohol lead to the generic complex 16, Fig. 14, which was easily characterized via1H NMR, and circular dichroism demonstrated the chiral discrimination of secondary alcohols. Such dynamic assemblies are studied towards the development of molecular machines and complex architectures on an atomic scale, with applications in a variety of fields.85
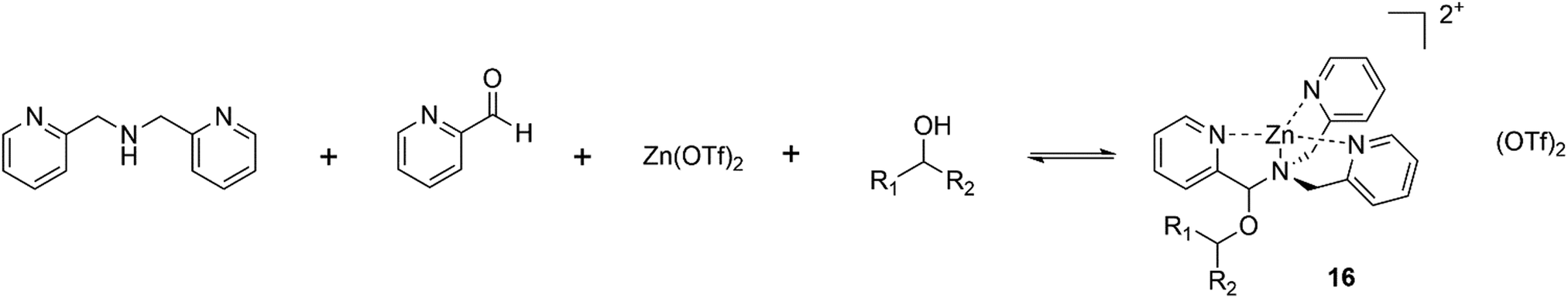 | ||
| Fig. 14 Self-assembly of pyridine-2-carboxaldehyde, di-(2-pyridylmethyl)amine, Zn(OTf)2 and a secondary alcohol to give 8, capable of chiral discrimination through circular dichroism or 1H NMR. | ||
Paolesse and co-workers have shown various examples of sensors based on supramolecular porphyrin aggregates.86 These researchers have recently demonstrated that grafting chiral porphyrin derivative 17 onto ZnO nanoparticles could yield a gas sensor, able to detect and recognize vapours of enantiomeric pairs, Fig. 15(a).87 By layering this material onto quartz microbalances the authors were able to evaluate the total amounts of adsorbed analytes and the relative magnitude of stereoselective interactions with respect to overall interactions when the films were exposed to vapours of (R)- and (S)-limonene, (R)- and (S)-butan-2-ol, and (−)- and (+)-α-pinene enantiomers, displaying a remarkable selectivity for limonene enantiomers.
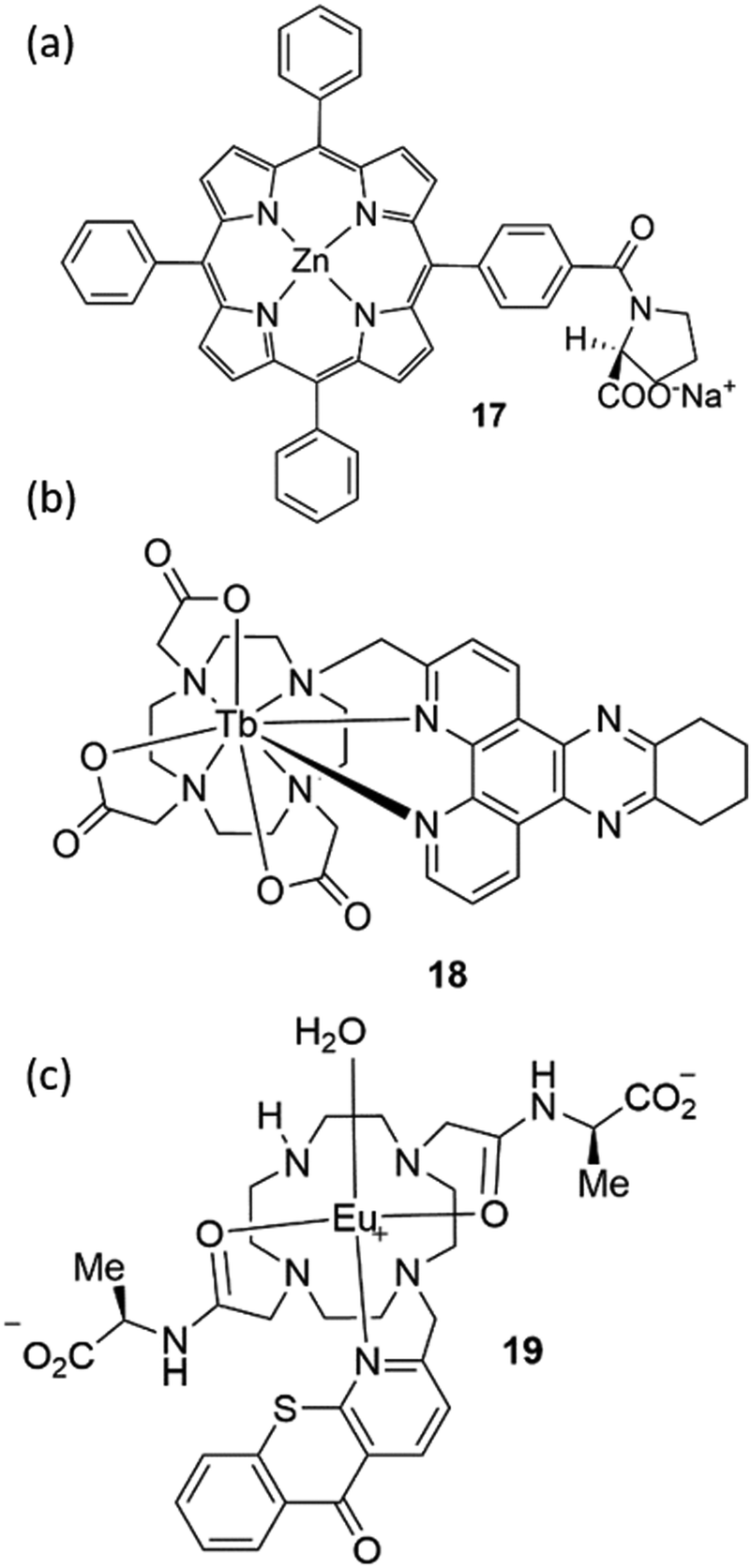 | ||
| Fig. 15 The structure of chemosensors (a) 17, (b) 18 and (c) 19. | ||
Parker and co-workers have extensively studied lanthanide complexes as emissive optical probes to detect key biochemical species such as citrate, lactate and urate.88–90 An assay to measure uric acid in biological fluids was developed by using the luminescent Tb3+ complex 18, Fig. 15(b). By evaluating the Stern–Volmer quenching constants, representing the concentration of analyte needed to reduce the lifetime or emission intensity to 50% of its original value, these authors demonstrated that urate quenching was 50 times more effective than ascorbate.
This result is particularly appealing as the classic clinical assay used for uric acid detection is an enzymatic assay that is subject to interference from ascorbate.91 Parker is the co-founder of FScan Ltd, a University-based company in Durham (UK), which is focused on the development of commercial applications from its luminescent lanthanide chemistry technology platform. In particular they have developed a prostate cancer test using citrate level analysis in semen or prostatic fluid samples. The sensor is based on a luminescent Eu3+ complex (19) able to selectively bind citrate, Fig. 15(c).92
In addition, a great contribution to the field of molecular imaging has been made by Smith and co-workers,93 who have produced an efficient imaging agent for the detection of mammary and prostate tumors. This was developed based on zinc(II)–dipicolylamine (Zn–DPA) unit and a near infrared carbocyanine fluorophore (20), shown in Fig. 16.94 Due to the presence of Zn–DPA, which is well known for its affinity for phosphorylated peptides,95 sensor 20 is able to target dead and dying cells with exposed anionic phosphatidylserine and can selectively accumulate in prostate and mammary tumour cells. Interestingly, the same sensor was successfully used to detect cell death in a brain cryolesion mouse model that replicates certain features of traumatic brain injury.96 Smith and co-workers have also developed squaraine rotaxanes as a novel family of deep-red fluorescent dyes with extremely high brightness and stability.97–100 These dyes exhibit high stability of the encapsulated squaraine which is protected from the attack of water thanks to the interlocked rotaxane structure.
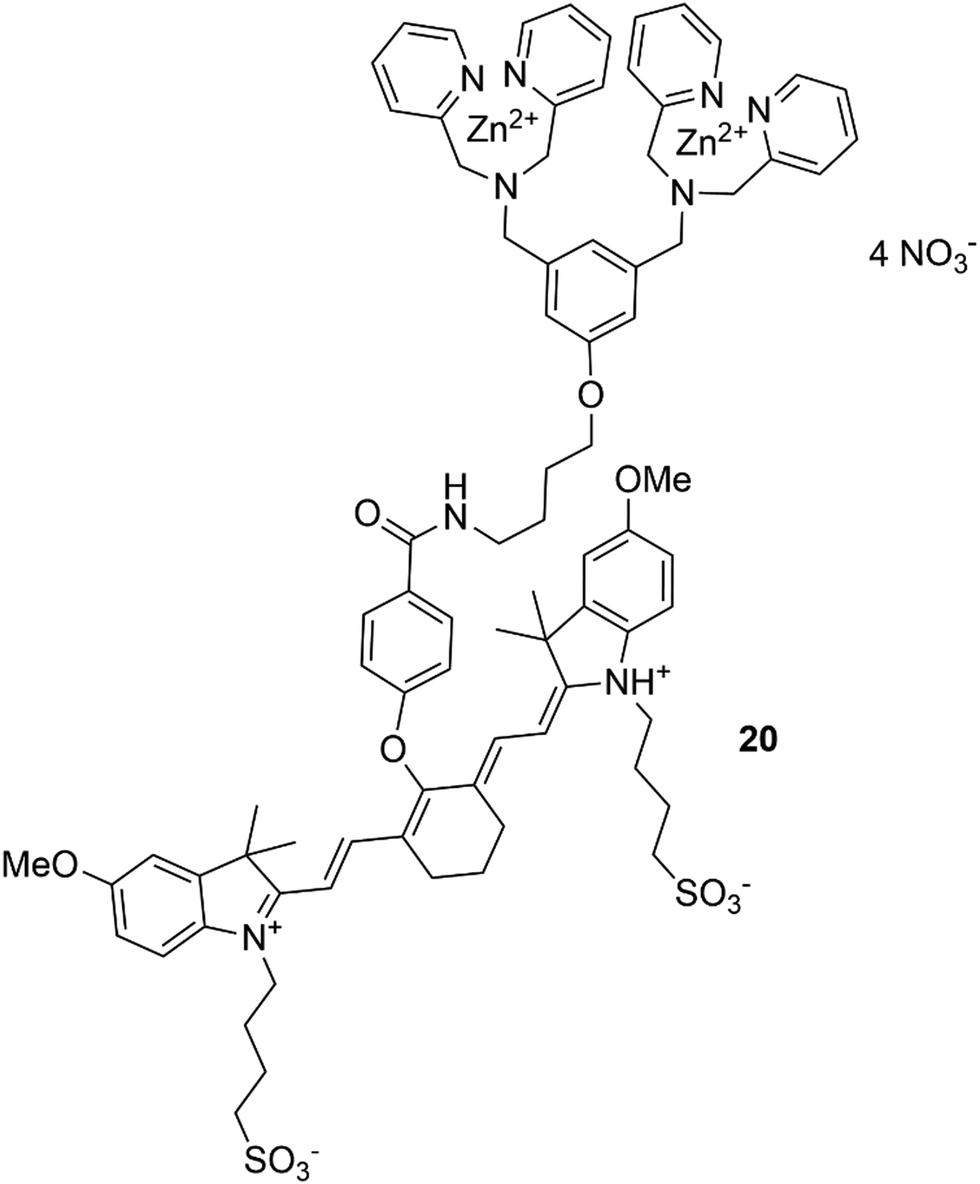 | ||
| Fig. 16 The structure of chemosensor 20. | ||
Vapochromic materials are an interesting class of chemosensors with potential environmental applications. These materials are able to change colour/fluorescence when exposed to vapours of volatile compounds. The first example of a vapochromic chemosensor was reported by Mann and co-workers in the late 1990s and is based on [Pt(arylisocyanide)4][Pt(CN)4] (where aryl-isocyanide = p-CNC6H4CnH2n+1; n = 1, 6, 10, 12, 14) complexes which undergo a bathochromic shift in the NIR region upon exposure to volatile organic compounds (VOCs).101 Since this time two main classes of vapochromic materials have been developed. The first based on coordination complexes or organometallic compounds and the second on organic compounds, as recently reviewed by Huang and co-workers.102 Recent research from this group has led to the development of vapochromic materials, based on nonporous adaptive crystals (NACs) formed from pillar[n]arenes.103
A transformation of the NACs structure occurs upon exposure to vapours of certain guest molecules, resulting in the generation of intrinsic or extrinsic porosity. With the guest molecules removed the NAC structure returns to a nonporous state. For example, NACs of pillar[4]-arene[1]quinone work particularly well in detecting vapours of aliphatic aldehydes of different chain lengths.104 Adsorption of these aldehydes leads to a colour change of this NAC. Crystal structure analysis reveals this colour change is due to alteration of the π–π stacking interactions between the benzoquinone and the 1,4-diethoxybenzene units of the pillar[4]-arene[1]quinone scaffold.105
Additionally, the fluorescence emission properties of an anthracene appended pillar[5]-arene (21) NAC were also found to alter upon exposure to vapours of short chain alkyl ketones such as 2-butanone, 2-pentanone, and 2-hexanone (Fig. 17).106 Within this material, observed fluorescence is attributed to the formation of an excimer between two adjacent anthracene moieties. On adsorption of an appropriate short chain ketone the excimer is disrupted and the fluorescence of the monomeric anthracene only is observed. Further to the use of NACs as vapochromic receptors, their application in chemical purification is outlined later in this review.
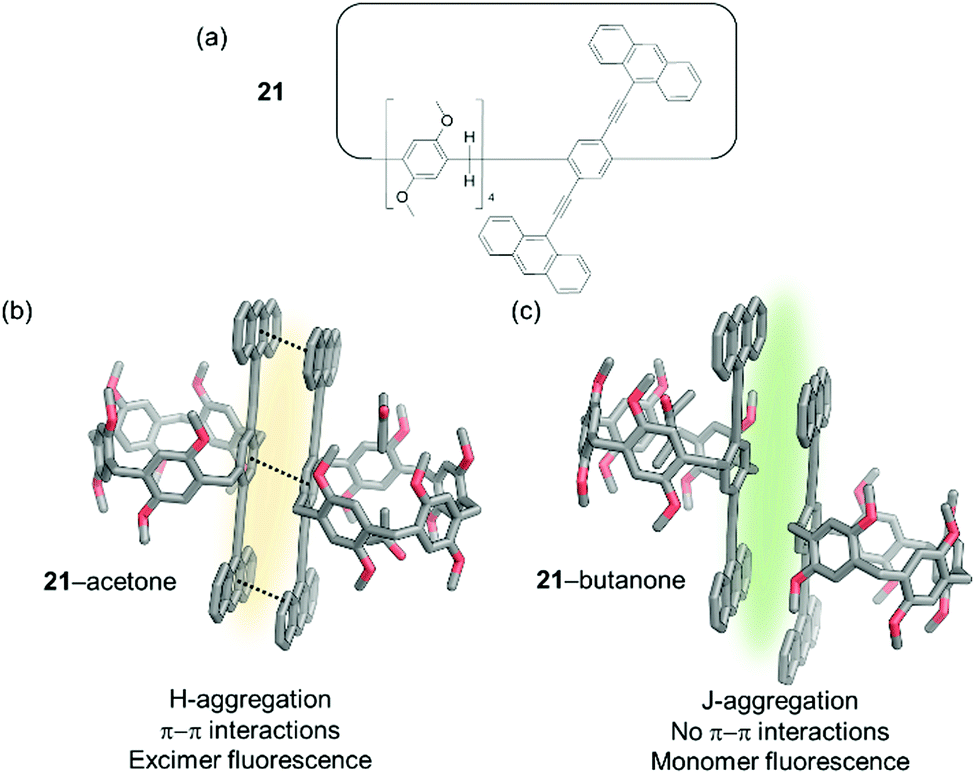 | ||
| Fig. 17 (a) The structure of macrocycle 21; (b) fluorescence emission of H-aggregated 21 caused by excimer formation; (c) fluoresce emission of J-aggregates of 21 caused by monomer emission.106 | ||
Chemical warfare agent sensing and remediation
Chemical warfare agents (CWAs) such as organophosphorus (OP) species and sulfur mustards (HD) are volatile and highly toxic molecules and potentially lethal, uncontrollable weapons when deployed.107 Due to the continued use of these agents in conflict situations and attacks on civilians, there is continued research into methods to remediate these compounds after release and to sense their presence. Such technology would also be of military value to mitigate the risks of producing and storing these species. As a result, significant effort has been put in over recent years to develop sensors able to recognize phosphorous-containing nerve agents. Gale and co-workers have described several examples of ureas/thioureas108–110 or polyureas able to recognize the chemical warfare agent soman (GD) and other organophosphorous species. The recognition event relates to the formation of H-bond interactions between the organophosphonate guests and the urea/thiourea hosts. Some of the proposed systems were able to form organogels and in the presence of the organophosphorous species a disruption of the material could be observed as a sensory output.111–115The first example of fluorescent chemosensors for phosphonate recognition, able to give a turn-on fluorescent response in the presence of hydrolysis products of the nerve gas sarin, isopropyl methylphosphonate (IMP), and methylphosphonate (MP) was reported by Anzenbacher and co-workers.116 Eight fluorescent tripodal sensors with a 1,3,5-triethylbenzene core were dissolved in DMSO and found to give a finger printed response for IMP and MP over other common anions, as demonstrated by linear discrimination analysis (LDA). A chromogenic probe based on BODIPY (22) was developed in Martínez-Máñez group for the detection of DFP (a sarin and soman mimic) and DCNP (a tabun mimic).117 As shown in Fig. 18 this system has three reactive sites; i.e. (i) a nucleophilic phenol group able to undergo phosphorylation with nerve gases, (ii) a carbonyl group as a reactive site for cyanide; and (iii) a triisopropylsilyl (TIPS) protecting group that is known to react with fluoride. The colour change in response to fluoride or cyanide is indicative of the presence of DFP or DCNP respectively. Dramatic and differing colour changes were observed in MeCN upon addition of one equivalent of DFP and DCNP, showing the ability of this system to detect and differentiate these CWA mimics.
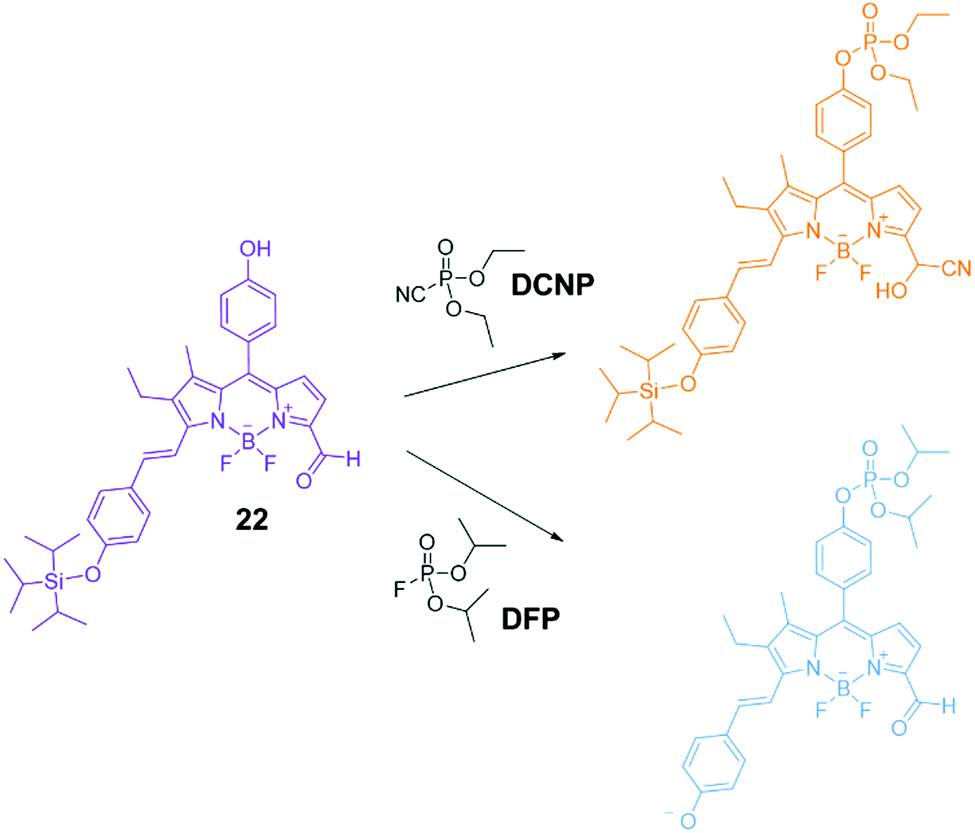 | ||
| Fig. 18 Proposed mechanism of the chromogenic response of sensor 22 in the presence of DCNP and DFP.117 Reproduced from ref. 117 with permission from the Royal Society of Chemistry, copyright 2016. | ||
Although OP species eventually degrade via hydrolysis, this is a slow process at neutral pH. To avoid the use of excess base or other reagents,107 catalysis underpinned by molecular recognition strategies has been investigated as a route to promote the hydrolysis of bound OPs to safe by-products. However, investigating strategies to remediate or sense live CWAs is extremely challenging due to the acute toxicity of these species. While significant progress has been made using simulants of lower toxicity and greater ease of handling, their behaviour invariably differs from live agents.107 Here we will discuss recent reports that show progress in the remediation of live CWAs which demonstrate the potential for application in the field.
The application of supramolecular chemistry towards this purpose has focussed on strategies to immobilise the agent and subsequently trigger its breakdown, with recent progress made in systems which can achieve both of these elements. While organogels formed from gelators that can act as destructive agents to trigger the decomposition of the CWA are known, it is also possible to immobilise reactive decontaminants within an organogel matrix to form a multicomponent material from simple precursors.114,118 In recent work funded by the Defence Science and Technology Laboratories (Dstl), Holder, Blight and co-workers examined the immobilisation of a catalytic metal organic framework (MOF) inside a polymer sponge network.119 This work combined the previously reported ability of styrenic poly high internal phase emulsions (pHIPEs) to swell and trap a range of OP and mustard series nerve agents120 with the known propensity of zirconium MOFs to function as heterogeneous catalysts for the hydrolysis of OPs.121 The authors prepared a composite material consisting of a styrene or vinyl based pHIPEs and zirconium MOF-808 at a loading of 25 wt% relative to the monomer weight. MOF-808 was chosen due to its activity in catalysing OP hydrolysis, and because the linker is commercially available making this MOF suitable for cost-effective scale-up. Phosphorus NMR studies using the CWA simulant dimethyl p-nitrophenylphosphate (DMNP) indicated that OP hydrolysis was significantly accelerated in the presence of the composite material in 0.45 M NEM buffer and THF/H2O/D2O (2:1:1), and performed better than experiments in which the catalytic MOF was added as a powder, due to the better dispersion of the catalyst. Minimal hydrolysis was observed in the absence of the MOF. Following on from this, hydrolysis studies using the live agent VX demonstrated that the composite materials could trigger hydrolysis in THF/water solution, with half-lives of 1 hour. Furthermore, neat VX could also be degraded in the absence of any solvent; with a total MOF-808 catalyst loading of 0.18% relative to VX, near full hydrolysis was observed after 2 weeks under ambient humidity. Over the same period without catalysis, the hydrolysis of VX would be expected to reach 50% completion only. The authors noted that potentially 4.5 kg of the composite would be able to absorb, immobilise and degrade the entire contents of a standard barrel (208 L) of VX.
Supramolecular separations and hydrometallurgy
Supramolecular and coordination chemistry have been fruitfully applied in the extractive metallurgy of metals such as copper, nickel, cobalt and zinc for a number of years.122 Solvent extraction using phenolic oxime ligands, extensively studied by Tasker,123 has accounted for up to ca. 25% of global copper production.124 IBC Advanced Technologies, Inc. (IBC),125 co-founded by Izatt, Bradshaw and Christensen in 1988, have pioneered the use of so-called ‘Molecular Recognition Technology’ (MRT) – macrocycles such as BOB Calix (a calixarene-crown ether) – for the selective removal or exchange of metal ions. This type of technology has been used in applications such as the removal of caesium from nuclear waste at the Savannah River Site126 and its potential in green chemistry approaches to metal recovery from electronic waste has been recently highlighted.127One area of recent progress has been in the application of supramolecular hosts for the isolation of precious metals such as gold.128 Key advances in this field have targeted lowering both the costs and the environmental footprint of precious metal mining projects, avoiding the use of highly toxic and environmentally damaging reagents such as cyanide. Another target is the capacity to recycle gold from Waste Electrical and Electronic Equipment (WEEE), such as mobile phones and computers, which represents an ‘urban mine’ for gold that could reduce the requirement for environmentally damaging mining processes.129,130 Approaches to this challenge have been recently reviewed by Love,129 who highlighted that a typical waste printed circuit board contains up to 60 different chemical elements,131 thus demonstrating the need for selectivity in precious metal extraction.
Notable progress in supramolecular approaches to gold extraction has been made based on the work of Stoddart et al., who first reported the capacity of α-cyclodextrin to recognise and isolate gold in the form of alkali metal haloaurate salts (produced from gold bearing raw materials through etching followed by neutralisation) in 2013.132 The underlying principle of this work is the spontaneous precipitation of an extended {[K(OH2)6][AuBr4]⊂(α-cyclodextrin)2}n chain superstructure from water. Metallic gold can then be isolated from the α·Br precipitate by reduction with sodium metabisulfite (Na2S2O5), while the α-cyclodextrin can be recovered for re-use by recrystallisation. This extraction strategy inspired the establishment of Cycladex in 2014,133 a company whose mission is to reduce operating costs and toxic waste by-products in commercial precious metal extraction. As a result of these efforts a patented method by which precious metals can be recovered has been developed,134 and the company has reportedly demonstrated this in a pilot plant at the ton level.135
The success of Cycladex has also inspired studies into gold precipitation using other macrocyclic hosts. Tao et al. reported that cucurbit[8]uril (CB[8]) macrocycles can imprison [AuCl4]− anions through co-precipitation in honeycomb-like frameworks.136 Cucurbiturils (CB[n]s) are known to form supramolecular frameworks through precipitation/crystallisation, which arise due to interactions between the positively charged outer surface of the CB[n] with either the negatively charged opening portals of adjacent CB[n]s or with structure directing agents, such as anions. In this case, Tao et al. observed that [AuCl4]− anions were trapped between macrocycles via outer surface interactions yielding an extended CB[8] framework (Fig. 19). The direct combination of CB[8] and HAuCl4 in aqueous HCl or HNO3 yielded framework F1, in which six [AuCl4]− anions were contained in hexagonal cavities defined by six CB[8] macrocycles. This process was largely unaffected by the presence of other metal chlorides including ZnCl2, CdCl2, NiCl2, CoCl2, MnCl2, FeCl3 and CuCl2 and H2PtCl6 if the precipitation was performed from hot solutions; however, some co-crystallisation of [PtCl6]2− occurred if the samples were left overnight. Frameworks F2 and F3, formed in the absence of [AuCl4]− anions, could take up [AuCl4]− to yield framework F1. Meanwhile, gold could be isolated from framework F1 through reduction with hydrazine, producing a mixture of framework F2 and metallic gold. Thus, a route was developed by which gold can be isolated from aqueous solutions of [AuCl4]− with the regeneration of a host framework which could be re-used.
 | ||
| Fig. 19 (a) Crystal structures showing the relationship of the central CB[8] molecule with eight neighbouring macrocycles in F1 and F2, reported by Tao et al.; (b–d) representative outer surface interactions between the central CB[8] molecule with neighbouring macrocycles; (e) the arrangement of ten [AuCl4]− anions around the central CB[8] macrocycle; (f–h) representative outer surface interactions between the central CB[8] macrocycle and ten surrounding [AuCl4]− anions.136 Reproduced from ref. 136 with permission from the Royal Society of Chemistry, copyright 2019. | ||
Following on from this work, the groups of Tao137 and Stoddart138 have independently examined the ability of smaller cucurbit[n]uril (CB[n = 5–7]) macrocycles to precipitate [AuCl4]− and [AuBr4]− anions from acidic aqueous solution. The smaller CB[n] macrocycles are easier to synthesise and isolate than CB[8], hence a recovery process using these homologues is attractive on economic grounds. Tao et al. discovered that CB[n = 5–7]s can each bind to and co-precipitate [AuCl4]− anions, with a range of coordination modes and stoichiometries observed via single crystal X-ray diffraction. A common feature was the interaction of the [AuCl4]− anions with the exterior surface of the CB[n], including ion–dipole interactions between [AuCl4]− anions and the portal carbonyl groups of the macrocycle and [Au–Cl⋯H–C] hydrogen-bonding interactions between [AuCl4]− anions and methylene or methine hydrogen atoms (Fig. 20), as also observed in the distinct structures obtained by Stoddart et al. from CB[6] and both [AuCl4]− and [AuBr4]− anions.
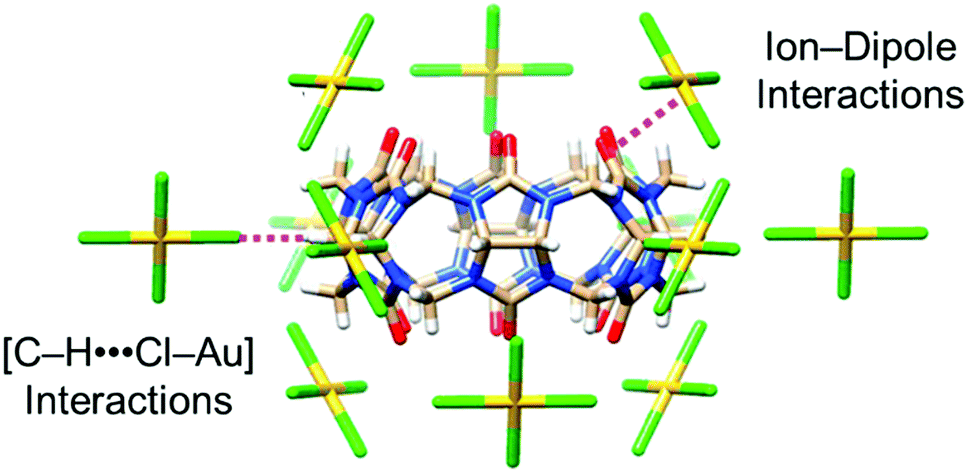 | ||
| Fig. 20 Stick representation of the X-ray crystal structure of (CB[6]·[AuCl4]−), reported by Tao and co-workers.137 The CB[6] macrocycle is surrounded by 12 [AuCl4]− anions, which form interactions with the outer surface of the macrocycle. Reproduced from ref. 137 with permission from the American Chemical Society, copyright 2020. | ||
Interestingly, CB[7] was observed to form [Au(OH2)4]3+⊂CB[7] inclusion complexes, leading Tao et al. to suggest that CB[7] has the most suitable cavity dimensions to encapsulate this hydrated cation. Additionally, Tao observed that CB[6] exhibited better performance in gold extraction experiments than the other CB[n] homologues, while an optimised protocol from Stoddart138 achieved an impressive gold recovery efficiency of 99.2% based on the co-precipitation of CB[6] and HAuCl4. This extraction efficiency is greater than the 78% achieved with α-cyclodextrin.139 The extraction was selective for gold over Cu, Zn and Ag (Stoddart) and Cu, Cd, Ni, and Zn (Tao). Both groups observed that reduction with hydrazine hydrate could yield gold metal and allow recovery of the macrocycle for re-use.
Love et al. have focussed on strategies to isolate gold from WEEE waste through solvent extraction. They reported a series of simple amides 23–25 (Fig. 21(a)) able to extract [AuCl4]− from acidic single-metal and simulated WEEE solutions into toluene and other hydrophobic solvents.140,141 Insight from ESI-MS, density functional theory and molecular dynamics studies involving 23 indicated that phase transfer occurred through the assembly of clusters of [AuCl4]− with one protonated and one neutral amide, yielding overall charge neutral structures stabilised by a combination of hydrogen bonding and columbic attraction (Fig. 21(b)).140 The overall charge neutrality of the clusters means that ion pairing is not required for transport into the organic phase to occur. Primary amide 23 demonstrated remarkable selectivity for extracting gold from mixed-metal systems representative of mixtures obtained by acidic leaching of printed circuit boards, such as those found in mobile phones. Once the organic solvent phase containing these clusters had been isolated, the addition of a clean, aqueous solution resulted in the back transfer of the [AuCl4]− into the new aqueous phase. This allowed the gold to be easily isolated and the ligand to be re-used for further extractions. Ligands 24 and 25 were found to extract [AuCl4]− more efficiently than 23 from single metal solutions; however, their efficiency in extracting gold from mixed metal solutions was greatly reduced. The authors found that the presence of other metals such as Fe(III) and Sn(IV) caused the formation of a gold-rich third phase in extractions using 24 and 25. The presence of Fe(III) and Sn(IV) in the third phase in addition to [AuCl4]− complicated the clean isolation of gold species. This finding demonstrates the complications that can arise when increasing the complexity of model experiments to better represent real-world applications.
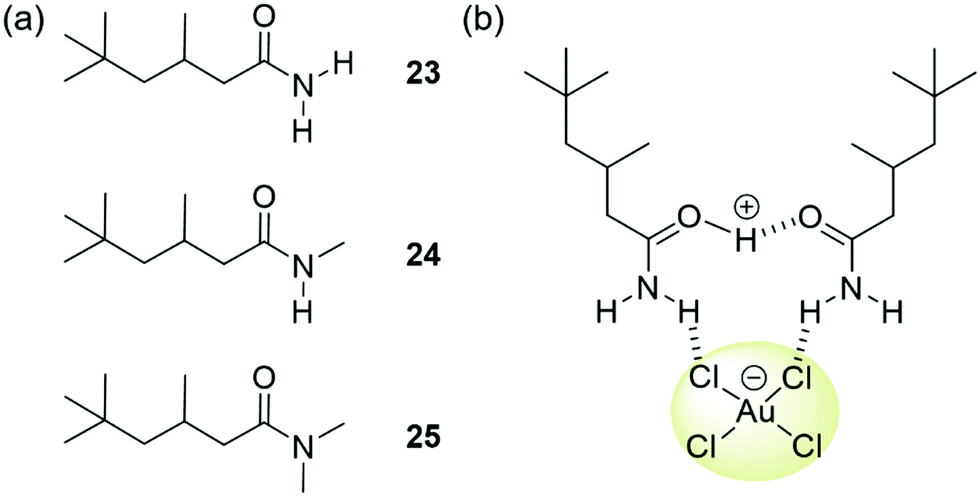 | ||
| Fig. 21 (a) The structure of ligands 23–25, reported by Love; (b) extraction of [AuCl4]− is thought to occur via the formation of clusters of one protonated and one neutral ligand. | ||
Environmental remediation
Oil spills are hugely damaging environmental disasters that have a severe, adverse effect on freshwater and marine ecosystems.142 Effective clean-up strategies can help to mitigate the damage caused by oil spills by dispersing or removing the oil from the aqueous environment. The use of organogelators in this application has been reviewed by Sureshan and Vibhute.143 Another established strategy for the remediation of light oil spills is the addition of elasticity agents, such as Elastol,144 to the oil. Elastol is a powdered formulation of a long-chain hydrophobic polymer which dissolves in the oil and modifies its mechanical properties through gelatinisation/solidification. The effect is to limit the dispersion of the oil and to increase the ease of physically recovering it from the surface of the water.Targeting a similar remediation strategy, there is interest in developing supramolecular phase-selective organogelators (PSOGs) that can form organogels from biphasic oil/water mixtures and thus trap the oil.145 In a recent review, Okesola and Smith highlighted that supramolecular gelators with a plausible chance of application in this context must be based on low cost, non-toxic components and must gel rapidly in high salt conditions without the need for a carrier solvent, heating or agitation.146 The gels formed must be stable at low temperatures and to shear forces to aid with physical recovery. The possibility of recovering the oil via distillation is also economically attractive and could facilitate re-use of the gelator.
While a number of patents exist describing the application of supramolecular gelators such as dibenzylidene-D-sorbitol147 and 12-hydroxystearic acid148 in this context, developing PSOG systems that meet all of the above requirements remains an ongoing challenge. In recent work, Zhang, Wang, Song and co-workers reported gelators 26–28 (Fig. 22(a)) via a 2-step synthesis.149 While all three gelators formed stable gels in aromatic and hydrocarbon solvents via the heating–cooling method, and 26 and 28 could gel fuel oils such as kerosene and diesel with low critical gelation concentrations (<0.1% w/v), only gelator 26 could form room temperature, biphasic gels with fuel oils when applied as a powder (i.e. in the absence of a carrier solvent). A combination of FT-IR and 1H NMR analysis indicated the self-assembly of 26–28 was driven by the synergistic formation of hydrogen bonding (amide and hydroxyl groups), π–π stacking (aromatic units) and van der Waals forces (alkyl chains) as shown in Fig. 22(b). SEM, PXRD and FT-IR of xerogels and pristine powders indicated that unsaturation (26) or branching (27) in the chains of the gelators lead to looser packing in the solid state, yielding porous structures with lipophilic surfaces. The authors concluded that these structures were more accessible to oil solvents and that the van der Waals interactions between chains were weaker when the packing was looser and more easily disrupted by oil. Hence, only gelator 26 was able to dissolve to form a gel without a carrier solvent or heating.
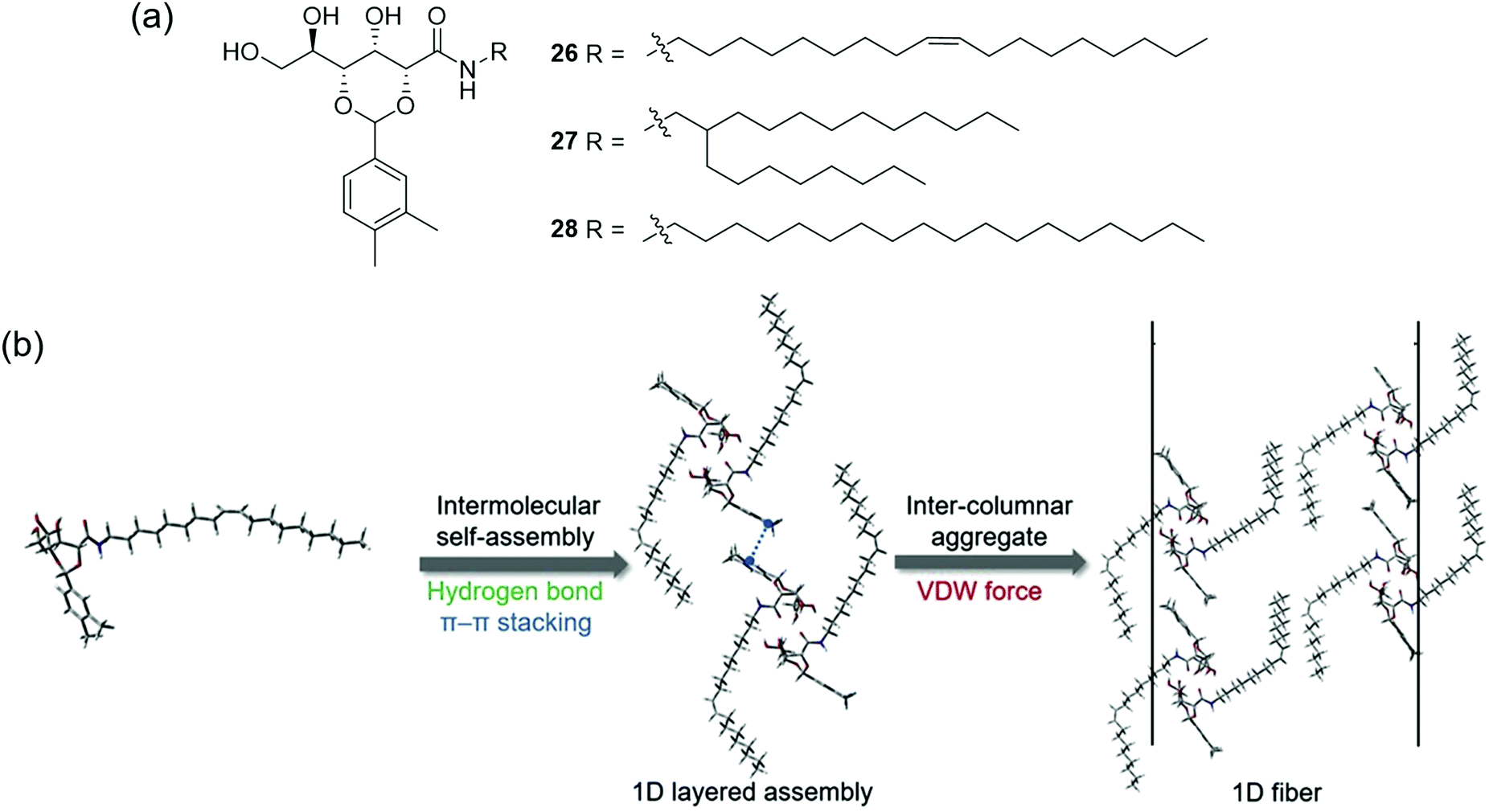 | ||
| Fig. 22 (a) The structure of gelators 26–28; (b) Gelation of hydrophobic solvents occurs via the formation of intermolecular hydrogen bonds, π–π stacking and van der Waals interactions between lipophilic chains.149 Adapted from ref. 149 with permission from Elsevier, copyright 2020. | ||
Powdered gelator 26 was also found to gel a range of fuel oils including gasoline, diesel and seven types of crude oil in biphasic seawater mixtures without the need for a carrier solvent, heating or agitation. The gels formed at biphasic critical gelation concentrations between 2–7% w/v, and were stable to acidic and basic media. Light oils could be gelled within 1 minute (5% w/v), although heavy crude oils required a longer gelation time (up to 7 minutes at 10% w/v). Lighter oils could still be gelled at the lower temperature of 5 °C, while heavier oils solidified. Rheometry measurements indicated good mechanical properties and strength which could mean that the gels are suitable for simple salvage in a remediation process. This gelator was also found to be non-toxic in zebrafish, and the crude oil recovery could be scaled up to recover 35 mL of crude oil in an attempt to better simulate a large scale, real-life recovery.
Another environmental problem that supramolecular chemistry has been applied to is the removal of organic micropollutants from water. These pollutants, such as pharmaceuticals and organic dyes, have detrimental effects both to the health of ecosystems, and to human health.150 In response to this, Alsbaiee and co-workers have developed mesoporous polymers by crosslinking β-CD with tetrafluoroterephthalonitrile, yielding a material capable of forming host–guest complexes with a range of common organic pollutants including plastic components, pharmaceuticals, pesticides and aromatic carcinogens. Not only does this material offer adsorption rates of 15–200 times greater than leading alternatives, it can also be regenerated through mild washing processes with no loss in performance.151 Such technology would be well suited to continuous flow water treatment.
Anion extraction from nuclear waste
In 2013 a team of scientists lead by Bruce Moyer at Oak Ridge National Labs in the U.S., highlighted the potential for supramolecular anion recognition to be applied in the separation of sulfate from nuclear waste.152 The presence of sulfate causes numerous problems in the handling and long-term storage of nuclear waste, particularly in converting the waste into a form that is readily stored. However, the authors noted that that no technology to meet this need had yet been identified and concluded that there was great potential for molecular-recognition approaches to provide a solution to this problem. They noted that successful new technologies must be “competitive against a field of alternatives and therefore must fulfil a given industrial need with the least cost”.Despite the relative ease of engineering anion receptors that bind strongly to sulfate (particularly in organic solvents), its high hydration energy means that extracting sulfate from aqueous solution is challenging. Another key consideration for nuclear waste treatment is the ability to recognise and extract sulfate anions in the presence of high concentrations of nitrate anions, which are less hydrophilic and thereby an easier target to extract into a hydrophobic environment.
In 2018, Williams and Moyer et al. reported the simple di(imino)guanidinium receptor 29-Cl shown in Fig. 23(a).18 The synthesis of this receptor was relatively straightforward (68.6% over 3 steps). In liquid–liquid anion extraction experiments using 1,2-DCE as a receiving phase, 29 was found to efficiently extract sulfate from an aqueous phase and outperformed the commercial anion extractant Aliquat 336 (A336) in both extraction efficiency and selectivity. Receptor 29 displayed a remarkable selectivity for sulfate over chloride, even when chloride was present in 100× excess. When the extraction was performed with the solvent Isopar L (a commonly used solvent in industrial extractions), the selectivity factor (SF) for sulfate over chloride was as high as 4300; for context, the authors noted that SF values >1 are rare in the literature.
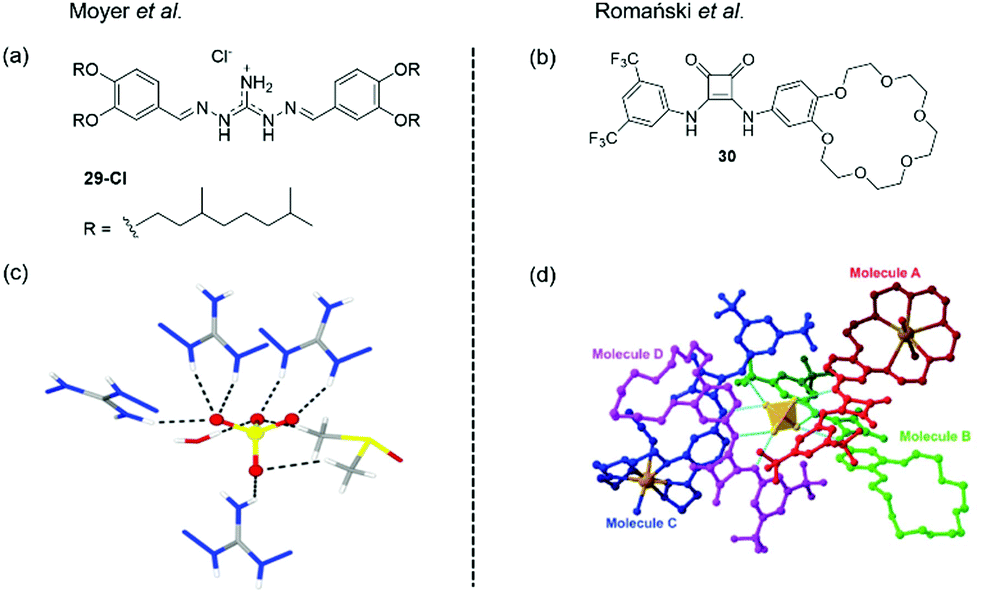 | ||
| Fig. 23 (a) The structure of receptor 29, reported by Moyer and (c) X-ray crystal structure of the complex of a simple analogue of DIG with a sulfate anions, showing the dual-coordination by four receptors, one water molecule and one DMSO molecule. Adapted from ref. 17 with permission from The Royal Society of Chemistry; (b) the structure of ion pair receptor 30, reported by Romański et al. and (d) X-ray crystal structure of the complex of 30 with Na2SO4, showing the coordination of the sulfate anion by 4 receptors, forming a charge neutral complex via the inclusion of 2× Na+ ions in the benzocrown cavities.17,18 Adapted from ref. 18 with permission from the Royal Society of Chemistry, copyright 2018. | ||
Further studies designed to better understand the cause of this selectivity were conducted. The isopar L solutions of 29-Cl were found to contain a high water content which increased linearly with the receptor concentration. Here small angle X-ray scattering data implied the formation of nanoscale colloidal structures thought to be reverse micelles in the organic phase.
Based on previous studies which suggested that iminoguanidiniums can interact with partially hydrated sulfate anions,153–155 the authors speculated that sulfate-water clusters could be extracted by 29-Cl. They reasoned that the sulfate anion cannot achieve coordinative saturation (12 hydrogen bonds) from 29-Cl receptors alone on steric grounds, and hence concluded that the sulfate must also be coordinated by associated water molecules. Correspondingly, the X-ray crystal structure of a simpler analogue of 29 complexed with sulfate revealed that the anion was coordinated by four receptors and, one water molecule and one DMSO molecule (Fig. 23(c)). The ability of 29-Cl to extract sulfate without total de-solvation could explain the remarkable selectivity observed.
Recent work from Romański et al. reported the squaramide-based ion pair receptor 30 (Fig. 23(b)), able to extract K2SO4 from water into organic solvents. While many sulfate extraction systems have been based on charged anion receptors that produce charge-neutral, organic-soluble complexes with sulfate, this can require the use of bulky counterions. In this case the need for bulky counterions is avoided, since the neutral receptor can cooperatively bind to both anions and cations to achieve charge balance. Receptor 30 was found to interact with a range of monovalent anions forming 1:1 complexes, and the addition of one equivalent of Na+ or K+ lead to an enhancement in anion binding strength in all cases, with the greatest effect observed for K+ due to preferential binding to the benzo-18-crown-6 unit. However, titrations with divalent SO42− revealed that more complex binding stoichiometries were forming. In particular, titrations with TBA2SO4 carried out in the presence of K+ produced a two-step binding isotherm that could not be fitted to a simple binding model. Careful analysis of the 1H NMR titration data obtained in CD3CN solution indicated that a 4:1 (receptor:anion) complex forms under these conditions, and DOSY-NMR and DLS confirmed the formation of large supramolecular assemblies with hydrodynamic diameter dH = 28 nm. The X-ray crystal structure of 30 with Na2SO4 is shown in Fig. 23(d), and demonstrates the formation of a 4:1 (receptor:anion) complex, in which the sulfate anion is encapsulated by four ligands (Fig. 23(d)). The overall complex remains charge neutral as two Na+ cations are bound within the benzocrown cavities.
This difference in binding behaviour towards monovalent and divalent anions prompted the authors to investigate the ability of 30 to differentiate these species under extraction conditions. Atomic emission spectroscopy and ion chromatography were used to quantify the extraction efficiency of 30 towards K2SO4 from water to chloroform in the presence of different competing salts. An aqueous mixture of KNO3 and K2SO4 was extracted with a solution of 30 in chloroform; the concentration of sulfates in the aqueous phase was reduced by up to 74%, whilst a drop of just 7.2% was observed for nitrate. The authors proposed that the greater organic-solubility of the 4:1 (receptor:anion) complexes formed with divalent anions compared to the 1:1 complexes formed with monovalent anions underpinned the extraction selectivity.
Recently, Jolliffe and co-workers have developed the lipophilic, macrocyclic squaramide receptor 31 shown in Fig. 24(a), which is capable of extracting sulfate from water into a hydrophobic solvent phase, and also of transporting sulfate across a bulk chloroform membrane in U-tube experiments.156 The authors suggest that the advantage of using a transport rather than extraction process for sulfate removal is that the extractant – in this case the organic macrocycle – can be recovered for re-use. Macrocycle 31 was found to bind tetrabutylammonium sulfate strongly in chloroform-d saturated with water (Ka > 104 M−1 determined by 1H NMR titration) and extract TBA2SO4 from water into a chloroform-d solution. Despite also forming strong complexes with nitrate anions (Ka > 104 M−1 in chloroform-d), 31 could selectively extract sulfate from water to chloroform-d in the presence of TBANO3. The identity of the extracted macrocycle-anion complex was determined based on distinct 1H NMR signatures. However, the lipophilic tetrabutylammonium counterion was required for successful extraction, and no anion extraction was observed when only sodium salts were used. Washing a chloroform-d solution of 31·SO42− with aqueous Ba(NO3)2 lead to the precipitation of BaSO4 and generation of 31·NO3−via anion metathesis. Based on this regeneration of the sulfate-free species, the authors designed a U-tube experiment to investigate sulfate transport across a bulk chloroform membrane (Fig. 24(b)). The sulfate concentrations of the aqueous phases were monitored over time using a modified BaSO4 gravimetric analysis coupled with ICP-MS. In the absence of an ion source in the organic phase, no transport was observed; however, when five equivalents of TBANO3 was dissolved in the organic phase, sulfate transport was enabled. A 2-fold increase in sulfate transport was obtained by adding BaCl2 to the receiving phase, which resulted in BaSO4 precipitation, removing sulfate from solution and driving the transport of further equivalents of the anion.
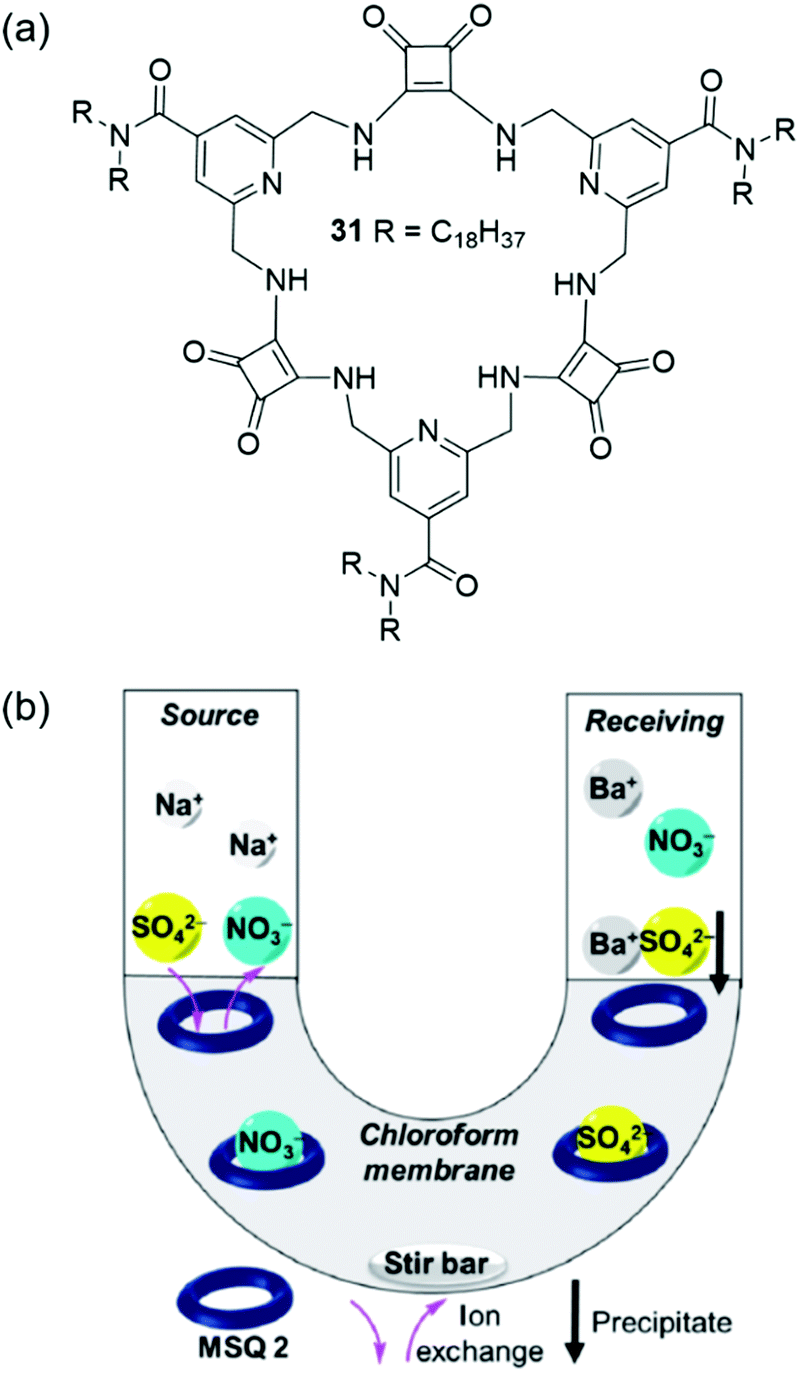 | ||
| Fig. 24 (a) The structures of macrocyclic squaramide receptor 31; (b) the transport of sulfate across a bulk hydrophobic solvent phase.156 Reproduced from ref. 156 with permission from the Royal Society of Chemistry, copyright 2020. | ||
Nonporous adaptive crystals in separations
The separation and refinement of chemical species is an essential industrial process, with a large environmental and economic impact. Indeed, distillation currently accounts for between 10–15% of global energy consumption.157 In 2016 Sholl and Lively outlined ‘seven chemical separations to change the world’, offering adsorption and separation as a pathway to reducing the environmental impact.158 To be effective separation agents, adsorptive materials require high surface areas. Many such materials have been reported, including but not limited to, MOFs,159 covalent organic frameworks (COFs)160 and zeolites.161–163 Whilst these classes of materials represent steps forward within this field, large macromolecular structures lead to poor solubility, and the presence of reversible bonds often limit stability. These factors preclude the use of these materials in solution-based separation processes. Arguably the step-change within this area can be attributed to work by Huang and co-workers who have pioneered the development of NACs, utilizing supramolecular assembly to overcome these limitations.103 As previously discussed, NACs are nonporous in their initial crystalline state however, upon the introduction of guest species a reversible crystal structure transformation occurs, generating porosity in situ. Within the scope of this review we seek to bring to the reader's attention NAC separation systems with commercially relevant applications however, for a more inclusive review of this area we refer the reader to a mini review published by Yang, another of this fields forefathers.164One of the first examples we wish to highlight from this area is that of Cooper, Huang and co-workers, who have demonstrated the ability of NACs constructed of pillar[5]arene 32 (EtP5) and pillar[6]arene 33 (EtP6), Fig. 25, to separate styrene and ethylbenzene,165 one of Sholl and Lively's seven targeted chemical separations.158 Styrene is used in the production of a variety of polymeric materials (e.g. synthetic rubbers, plastics and resins), and is produced through the dehydrogenation of ethylbenzene. Due to inefficiencies in this process, steam produced during the synthetic procedure often contains unreacted ethylbenzene, which must then be removed. Owing to the similar boiling points of styrene and ethylbenzene (146 and 136 °C respectively),166 conventional distillation is not practical, and as a result separating these two compounds requires complex and energy intensive processes.167 The authors instead utilize the EtP5 and EtP6 NACs, taking advantage of the small differences in the geometry of styrene and ethylbenzene (Fig. 26(a–c)), the presence of which initiates crystallization processes resulting in the formation of porous materials with differing stabilities. The crystalline material containing styrene is far more stable than that of ethylbenzene, meaning that the solid crystalline material containing the styrene prevails, and can easily be separated. Starting from a 50:50 mixture of ethylbenzene and styrene, this process has been shown to produce styrene with >99% purity. Further to this, as the crystallization of EtP6 is directed by the presence of styrene, it is not necessary to start with crystalline EtP6, and as such it can be recycled through heating with no loss of styrene capturing ability, Fig. 26.
 | ||
| Fig. 25 The general structure of pillar[n]arenes. | ||
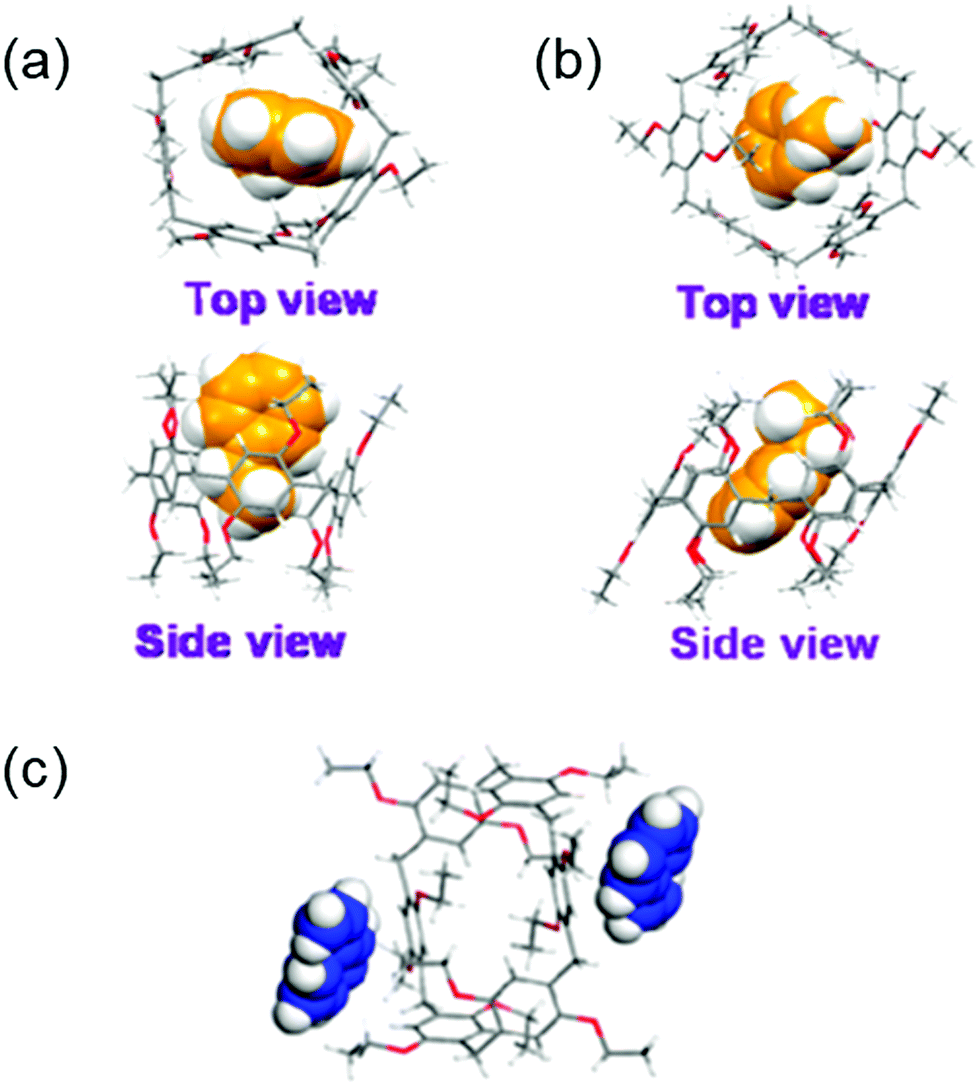 | ||
| Fig. 26 Single crystal structures of (a) ethylbenzene complexed with EtP5, (b) ethylbenzene complexed with EtP6, (c) styrene complexed with EtP6 in 2D channels between pillararenes.165 Reproduced from ref. 165 with permission from the American Chemical Society, copyright 2017. | ||
para-Xylene is another commercially important compound used in the production of a variety of polymers such as polyethylene terephthalate (PET) and polyester.158 However, the synthesis of this compound results in a mixture of ortho-, meta- and para-xylene, which again are difficult to separate using conventional distillation procedures. Here again the NAC EtP6 material was found to be effective in the separation of para-xylene from a mixture with its other isomers, achieving a purity of 90% and providing a less energy intensive route towards another industrially relevant starting material.168 In the interest of developing separation methodologies with even lower energy demands, Khasab and co-workers have further developed this work. These authors utilized a water soluble pillar[5]arene derivate to perform separations of xylene isomers using a liquid–liquid extraction methodology, achieving a selectivity of over 88%.169
Haloalkanes and haloalkenes are another class of chemicals with great industrial significance as high value feedstocks within the petrochemical and pharmaceutical industries.170 Traditionally, these compounds are synthesized through direct halogenation. Due to selectivity issues, this produces reaction mixtures that require complex purification.171 To this end, Yang and co-workers report a new class of NACs, desolvated perethylated leaning pillar[6]arenes (EtLP6). This tilted version of the EtP6 scaffold offers enhanced guest binding ability as well as good cavity adaptability, Fig. 27.172 Through a similar solid vapour adsorption method as previously reported,168 EtLP6 is able to separate the 1-bromo and 2-bromo isomers of bromopropane, bromobutane and bromopentane with 89.6%, 93.8% and 96.3% purity respectively.173 Additionally, the separation of 1-chlorobutane from 2-chlorobutane and vice versa has since been achieved using EtP5 and EtP6 respectively.170 As with EtP6, when freed of guests the EtLP6 material may also be reused, and shows no loss of separation ability after being recycled five times.
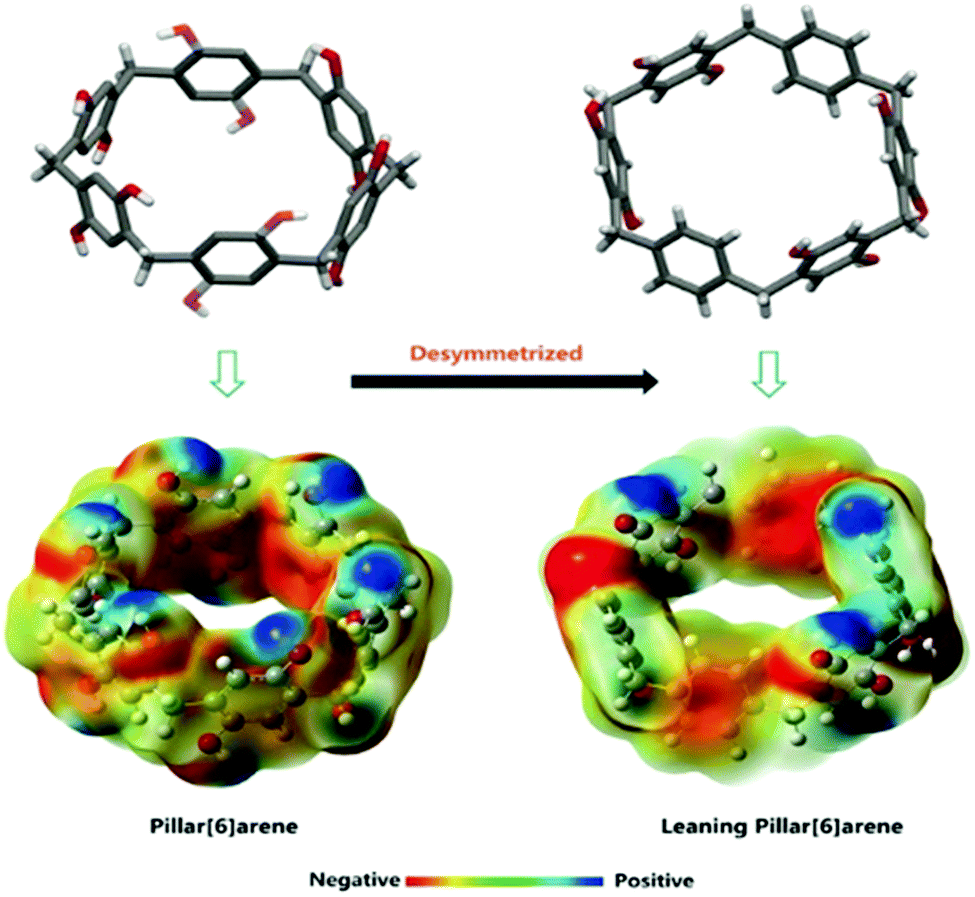 | ||
| Fig. 27 The structures and potential energy maps of traditional and leaning pillar[6]arenes.172 Reproduced from ref. 172 with permission from Wiley, copyright 2018. | ||
Moving beyond pillar[n]arenes, Huang and co-workers have also utilized CB[6], referred to by the authors as (Q[6]), in the formation of NACs, Fig. 28(a–c).174 Toluene and pyridine are both important solvents, and are also used in polymer production and for catalysis. The industrial preparation of toluene is often contaminated with pyridine impurities which, similarly to the previous examples, are problematic to remove through distillation.175 The authors found that pyridine was capable of forming a host:guest complex with CB[6], whilst toluene was not. It is as a result of this selective complexation event that this material has been shown to completely remove the pyridine impurities separate from crude toluene, Fig. 28(d).
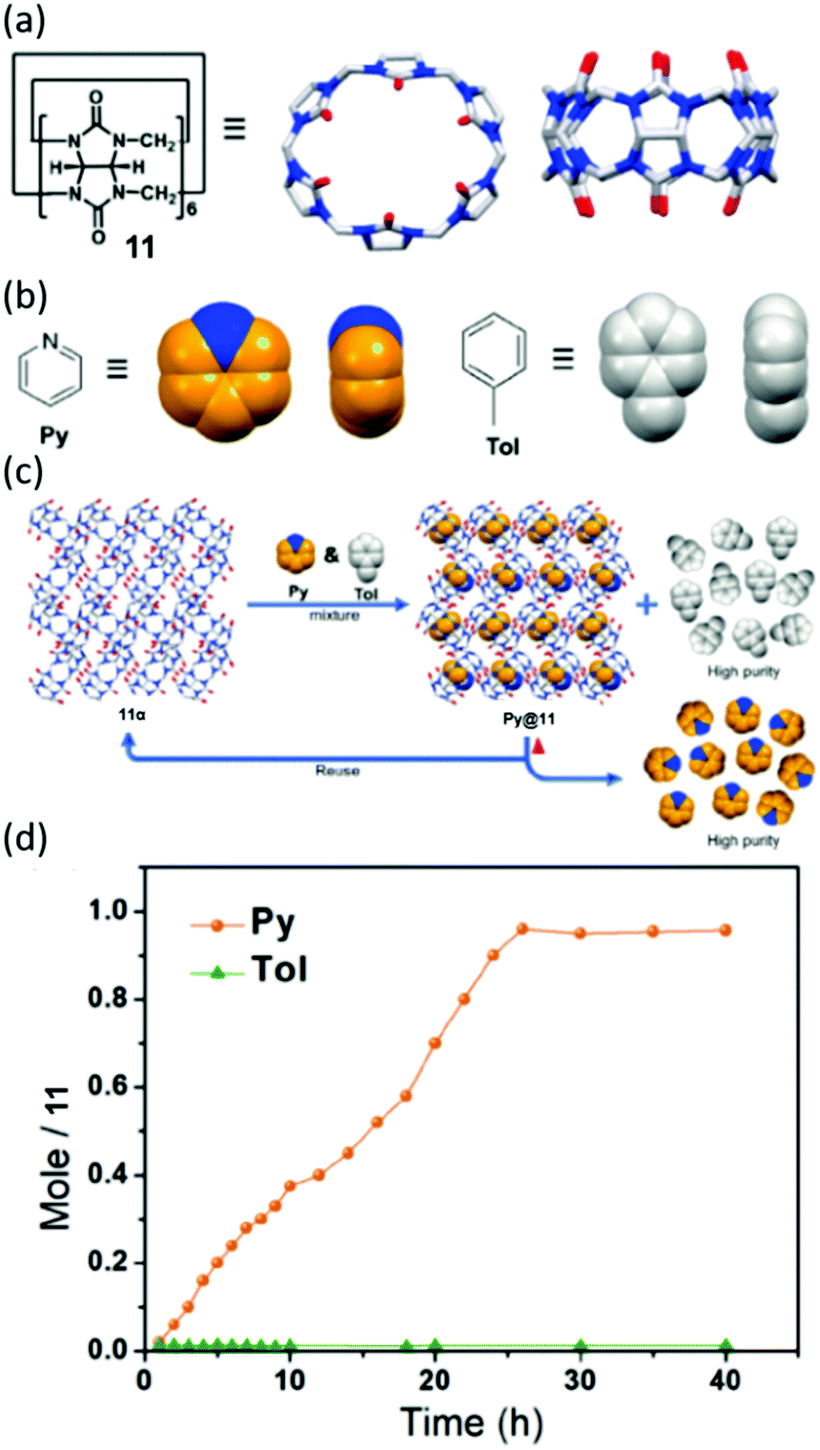 | ||
| Fig. 28 Chemical structures and cartoon representations of (a) 11 and (b) pyridine and toluene. (c) General scheme showing adsorption and separation of pyridine and toluene by NACs of 11. (d) Time dependent solid–vapour sorption profile for single component pyridine and toluene vapours, reproduced with permission.174 Reproduced from ref. 174 with permission from Wiley, copyright 2020. | ||
Supramolecular materials as healable coatings
Supramolecular materials are constructed using non-covalent forces such as hydrogen bonding, electrostatics and hydrophobic/hydrophilic interactions.176 The reversibility and dynamic nature of these interactions results in the production of materials with unique properties,177 capable of responding to temperature,178 mechanical stress,179 pH180 and the presence of small molecules.5,181,182 These materials are often produced through the combination of traditional covalent polymers with functionalities that are capable of binding through supramolecular forces. True supramolecular polymers are those in which the monomers themselves are not polymeric in nature, distinguishing them from supramolecular polymeric hybrid materials.183 They were defined by Sijbesma as “polymeric arrays of monomeric units that are brought together by reversible and highly directional secondary interactions, resulting in polymeric properties in dilute and concentrated solutions, as well as in the bulk. The monomeric units of the supramolecular polymers themselves do not possess a repetition of chemical fragments.”184The reversible nature of supramolecular bonds enables the production of self-healing materials. The term self-healing as used herein, describes materials that, much like a living organism, can repair themselves without any outside intervention. Broadly speaking self-healing materials can be classified into two types: those with the intrinsic ability to heal,185 and composite materials containing encapsulated healing agents for triggerable release.186–188 In the context of this review, the supramolecular materials discussed herein are those with the inherent ability to self-heal. This potential for commercialisation has been realised due to ‘maintenance free’ properties, leading to the development of these systems as coatings, adhesives and synthetic rubbers.
Coatings are applied to surfaces to protect them from the external environment; however, these coatings often need to be re-applied due to everyday ‘wear and tear’ degrading surface integrity. Here, supramolecular self-healing materials, developed as protective coatings, offer increased longevity when compared to traditional alternatives such as paints, resins and plastics.189 Such traditional materials require manual intervention to remove defects such as scratches, particularly important when considering high value goods. These limitations are not experienced by next-generation supramolecular chemistry inspired alternatives.
Coatings such of this type have already been used in the automotive industry. One example is polyrotaxane paints that have been in used in the car manufacturing industry, specifically by Nissan (since 2005),13 to prevent the need for scratch repair on vehicles. The same manufacturer has also expanded the applications of this technology, producing a self-healing smartphone case, reported to be able to fix minor abrasions in under an hour. Working with mobile operator NTT DoCoMo, and with Advanced Softmaterials Inc., a University of Tokyo based company, Nissan also aims to incorporate this class of materials directly into the production of the next generation of smartphones.13
Rotaxanes are a class of interlocked molecules, featuring the general structure shown in Fig. 29.190 Polyrotaxanes, a sub-class of the rotaxane family, feature a single molecular axle that passes through multiple macrocycles. They have been extensively studied by 2016 Nobel Prize winning chemists Stoddart and Sauvage.9,191 The polyrotaxane macrocycles are free to move along the molecular axle due to the lack of covalent bonding between the two, and it is this property that gives rise to their potential commercial material applications.192 Due to the popularity of these systems, multiple comprehensive reviews have been published on the synthesis, properties and potential applications of these compounds.192–194
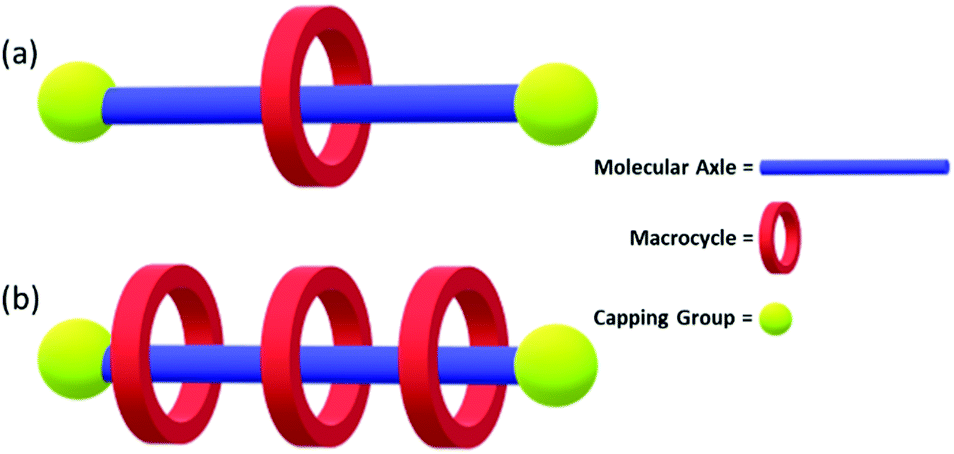 | ||
| Fig. 29 (a) Rotaxane molecule featuring a molecular axle threaded through a macrocycle held in place by a capping group. (b) A polyrotaxane featuring multiple macrocycles. | ||
An example highlighting the application of polyrotaxanes in the production of supramolecular self-healing materials is provided by Harada and co-workers, Fig. 30. This work shows how supramolecular rotaxane systems can be combined with dynamic covalent bonds195 to produce materials that can self-heal through both physical (supramolecular) and chemical (dynamic covalent chemistry) mechanisms. Here, the supramolecular ‘mechanical bond’ allows the physical movement of cyclodextrin macrocycles along their axle to compensate for external stress, while the dynamic covalent bonds can break and reform at new positions, healing the material to a greater degree of strength.
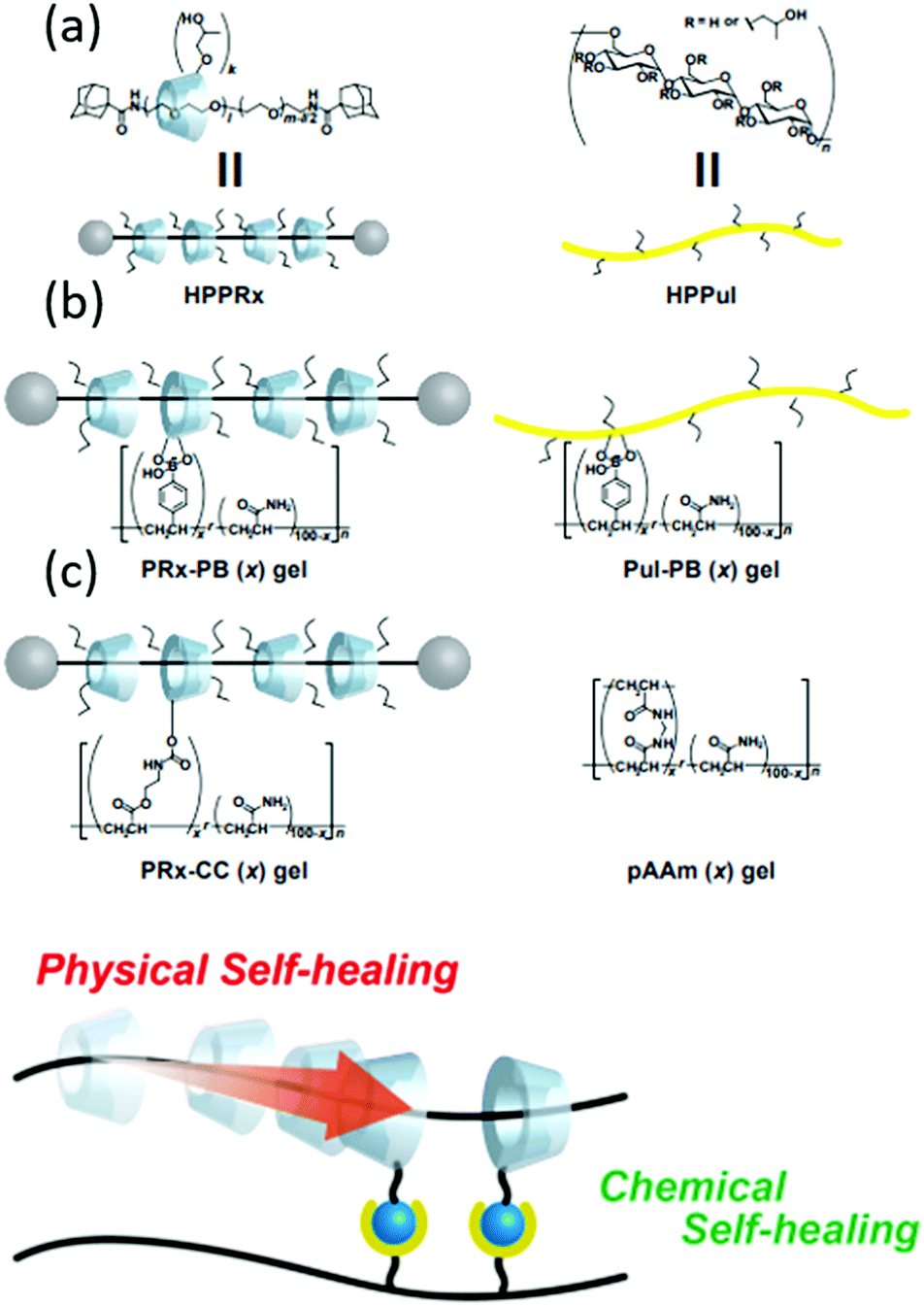 | ||
| Fig. 30 (a) Schematic representation of cyclodextrin/polyacrylamide polyrotaxane system used by Harada et al. (b) The chemical structures of: the polyrotaxane–boronic acid hybrid gel (PRx-PB), the boronic acid containing polyacrylamide–cyclodextrin gel (PuI-PB), covalently bound cyclodextrin–polyacrylamide gel (PRx-CC) and polyacrylamide gels (pAAm). (c) The combination of dynamic chemical and physical bonds that allow efficient self-healing.15 Reproduced from ref. 15 with permission from Cell Press, copyright 2016. | ||
This combination of dynamic covalent and supramolecular chemistry was initially utilised in the development of supramolecular gels, giving rise to systems that when physically split in two, could be re-joined to recover 83% of the original sample strength in only five minutes.15 The recovery of material strength in this instance is attributed to the dynamic nature of the boronic acid–diol bond, a feature that has seen their use in an ever-increasing number of functional materials.6,196 As exemplified by Harada, material self-healing is a common attribute in wet materials such as hydrogels. However, further development of this dynamic covalent-supramolecular system has allowed for a step-change toward also achieving this self-healing functionality within dry materials. This has classically been problematic, due to the decreased diffusivity inherent in systems that are not associated with a solvent. The addition of a commercially available photoinitiator (in this case Irgacure 2959), and subsequent exposure of the mixture to UV light, enabled the polyrotaxane–boronate system to become further crosslinked, resulting in the production of a scratch healable coating that can be applied to hard surfaces.
Here glass slides were coated with three variations of these materials PRx-PB, PuI-PB and PRx-CC materials. An incision was then made through the coating, to expose the glass underneath, and the self-healing ability of the coating was then observed, Fig. 31(a). The dual supramolecular-dynamic covalent physical–chemical healing properties of PRx-PB coating offered full material recovery within 30 minutes. The coatings lacking either supramolecular (PuI-PB) or dynamic covalent (PuI-CC) properties did demonstrate self-healing properties however, these offered a much lower performance over the same 30 minute time frame, Fig. 31(b).
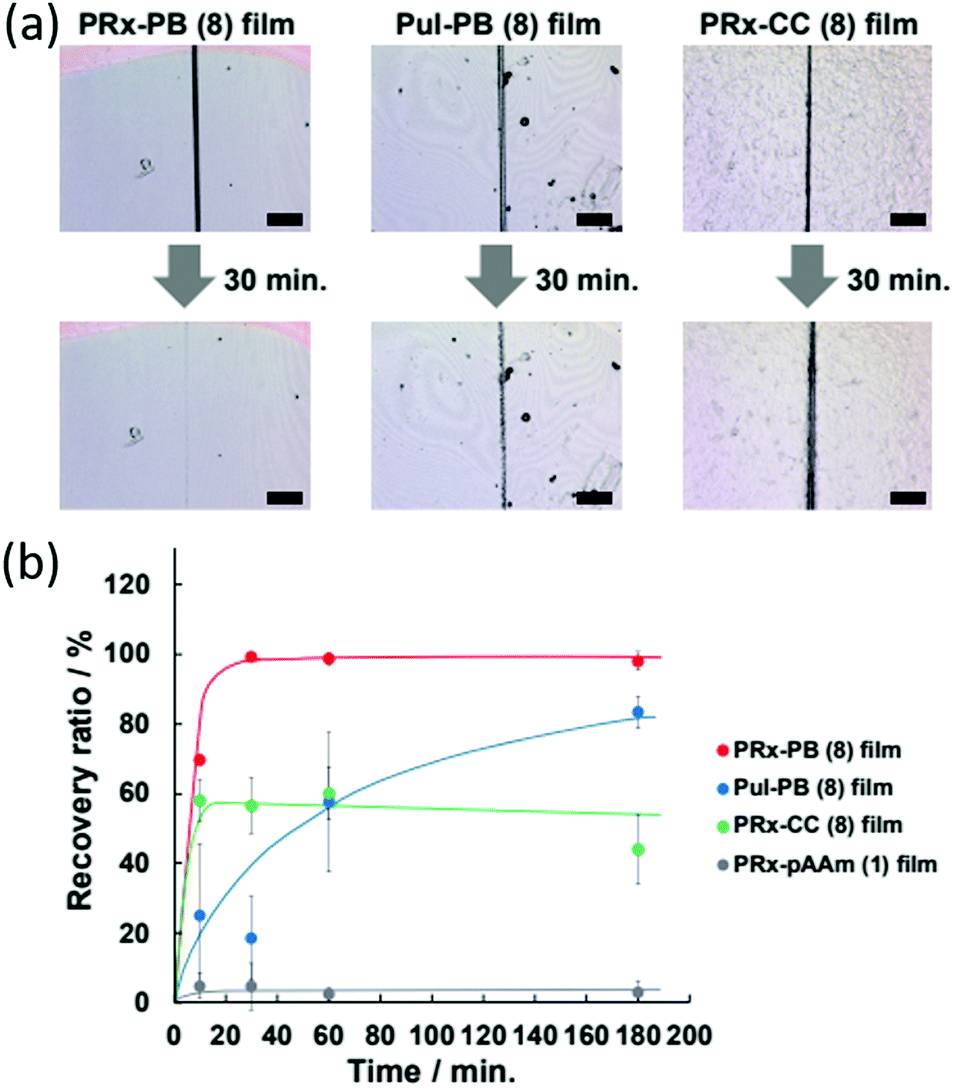 | ||
| Fig. 31 (a) The self-healing abilities of films containing rotaxanes crosslinked with dynamic boronic acid bonds (PRx-PB), films offering dynamic boronic acid bonds without the additional physical recovery due to rotaxane movement (PuI-PB), and films containing rotaxanes covalently bound to the polyacrylamide backbone (PRx-CC). (b) The recovery ratio versus time of each of the previously described materials, as well as polyrotaxanes with neither dynamic nor covalent crosslinkers (PRx-pAAm).15 Reproduced from ref. 15 with permission from Cell Press, copyright 2016. | ||
Self-healing rubbers
Rubbers are a ‘dry’ polymer with numerous applications worldwide from automobile fixtures and fittings to pencil erasers, and have an estimated global market value of 31.3 billion dollars (2019), which is predicted to increase.197 However, these materials suffer from poor longevity, lack of re-processability, and have a heavy environmental impact. Here, we showcase the efforts of supramolecular chemists to develop rubbers that are self-healing, and therefore exhibit longer useful lifetimes. Some of the described supramolecular rubbers can be recycled, resulting in the production of materials that act as a greener alternative to traditional systems. Practical applications of such supramolecular rubbers would be varied; it is easy to imagine that such technology would allow for the development of self-healing tyres with the potential to reduce automotive accidents, and decrease reducing upkeep costs.198As previously mentioned, materials that exhibit self-healing properties in dry conditions are more challenging to develop. The lack of co-solvents makes this process problematic as the production of supramolecular rubbers often involves the combination of non-covalent, reversible (often polar) and covalent (often non-polar) crosslinkers.199 Weitz et al. report the use of randomly branched polymers, containing motifs capable of acting as both reversible supramolecular and covalent crosslinkers (Fig. 32), generating a self-healing hybrid elastomer that exhibited a fracture energy of 13500 J m−2, which is comparable to natural rubber.16
 | ||
| Fig. 32 Schematic representation of the hybrid elastomer produced by Weitz et al. Reproduced with permission.16 Reproduced from ref. 16 with permission from Wiley, copyright 2017. | ||
To develop this material towards commercial applications, Weitz et al. reacted diamine and acrylic acid monomeric units, to generate randomly branched polymers consisting of mono-substituted, di-substituted and tri-substituted amides. The unreacted, pre-polymer, monomeric mixture is a viscous liquid, which can be poured/injected into a mould. On heating the mixture above 150 °C, condensation reactions occur between the amine and carboxyl groups contained within the monomeric units, resulting in covalent crosslinks, which secure a three-dimensional network that can maintain the shape of a mould. The ease by which this material can be formed into specific shapes has advantages when considering material manufacture for a variety of potential applications. The self-healing properties of this class of materials arises from the presence of the secondary amide groups, produced from the reactions of these monomeric units. Here the amide groups act as both hydrogen bond donors and acceptors, further stabilising the structure of the material through the formation of multiple hydrogen bonds (Fig. 32).
Within this material small deformations can be self-healed though the reformation of reversible amide–amide hydrogen bond however, larger deformations that result in the breaking of covalent bonds yield non-reversible material damage.200 In this combination approach, only the reversible hydrogen bonds can reform, therefore the material only recovers 30% of its mechanical strength when stretched beyond the limit of the covalent crosslinks.
Butyl rubber is a commonly used synthetic substitute for natural rubber and is produced from the reaction of isobutylene and isoprene. The inclusion of alkyl halides into butyl rubber has been found to increase material crosslinking efficacy, when incorporating agents such as sulfur and epoxides.201 In line with material developments in this area, Heinrich et al. have developed a simple methodology for the conversion of the widely used bromobutyl material into a self-healing rubber. Here, as shown in Fig. 33, the introduction of butylimidazole groups (34) allowed the material to crosslink with ionic bonds, rather than covalent. The incorporation of reversible supramolecular bonds, resulted in a material capable of self-healing.198
 | ||
| Fig. 33 The introduction of butylimidazole groups into bromobutyl rubber, yielding groups able to crosslink rubbers through ionic bonds. | ||
The introduction of ionic interactions into the covalent bromobutyl rubber network negates the need for further additives such as sulphur or peroxides, yielding a material with an elastic modulus superior to that of conventional rubbers.201
The tensile strength of this butylimidazole incorporated bromobutyl rubber was found to be greater than of the conventional sulphur crosslinked bromobutyl rubber (9 and 8 MPa respectively). In addition, when comparing the stretchability of both materials, the supramolecular system again out-performed the conventional sulfur system (1000% and ≈800% stretchability respectively). As can be seen in Fig. 34, the reversible nature of the ionic interactions within this material is able to facilitate molecular rearrangement, allowing separate pieces of the material to be re-joined. Further alteration of the materials molecular structure (varying the n-alkyl chain length) has led to the identification of a distinct correlation between alkyl chain mobility and self-healing properties. Extending this approach, Böhme et al. have demonstrated that the mechanical properties of these materials may also be tuned through an alteration of chain length.202
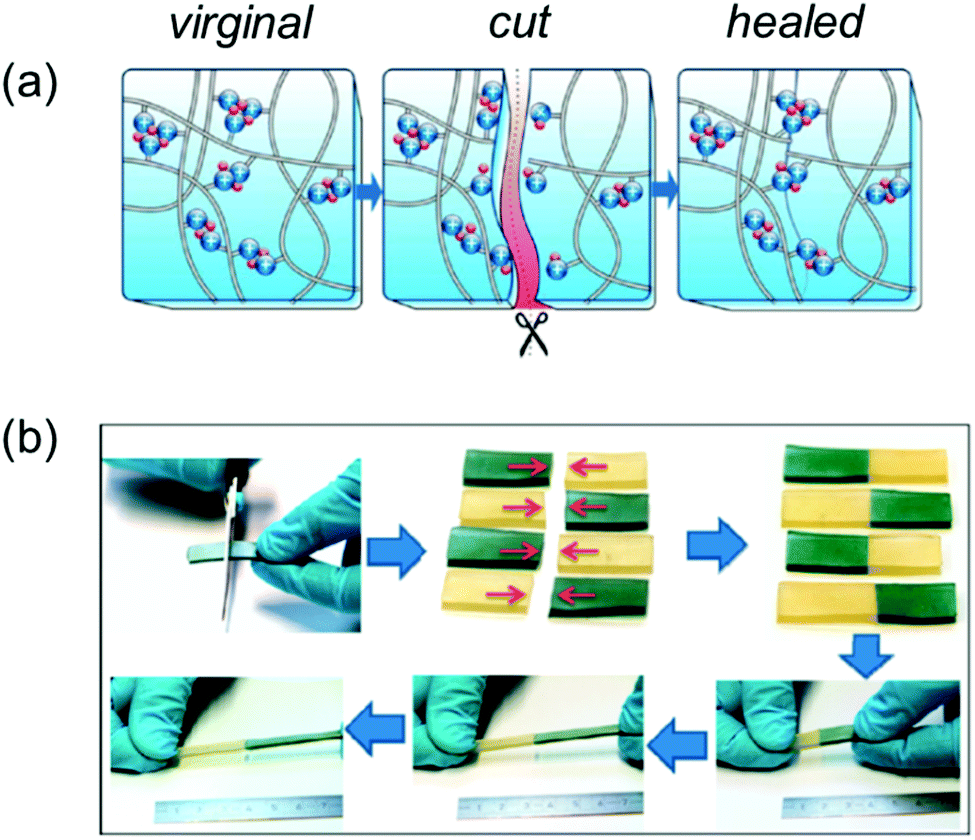 | ||
| Fig. 34 (a) The movement of charge clusters allowing reformation of the material. (b) The self-healing nature of the material, demonstrated by the use of coloured dye.198 Reproduced from ref. 198 with permission from the American Chemical Society, copyright 2015. | ||
The re-processability of covalently crosslinked rubbers is limited, due to the complex and energy intensive processes involved.203 In an effort to overcome this limitation, Böhme et al. introduced complimentary hydrogen bond donating and accepting moieties into the molecular structure of the butyl rubber material, resulting in the incorporation of secondary supramolecular binding modes, in addition to the ionic interactions previously discussed, 35 (Fig. 35(a)).204 This incorporation of complimentary hydrogen bond donating and accepting functionalities allows the material to be re-processed through the implementation of a heating–cooling process annealing process, to allow the reformation of the hydrogen bonding network, which allows the material to be reused (Fig. 35(b)).204 This innovation represents a plausible route towards the development of green alternatives for covalently crosslinked rubbers, which as thermosetting materials cannot be softened or remoulded for future uses.203
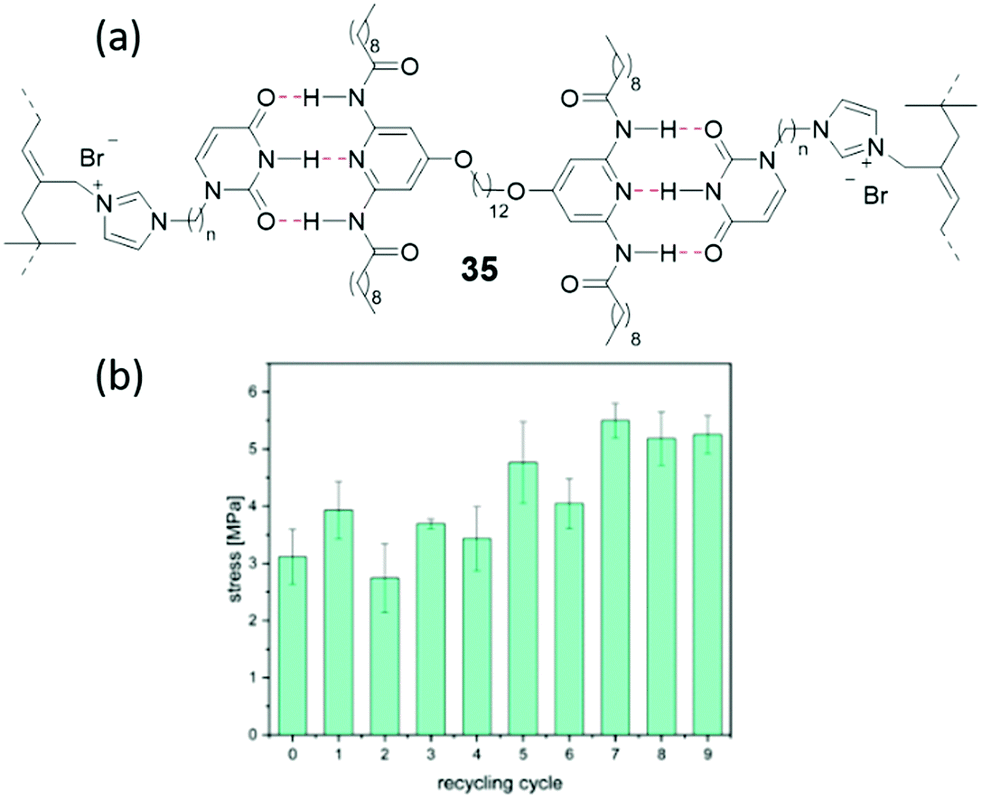 | ||
| Fig. 35 (a) Ionically modified butyl rubber doped with uracil (35) to give hydrogen bonding interactions; red dotted lines indicate hydrogen bonding. (b) Stress at break values after 9 reprocessing cycles.204 Reproduced from ref. 204 with permission from the Royal Society of Chemistry, copyright 2020. | ||
Supramolecular chemistry towards the development of adhesives for use in humid environments
The application of supramolecular chemistry in the development of traditional adhesives has already been discussed, however recent advances in material development have also led to the creation of a ‘velcro’-like material, capable of reversible adhesion underwater.205 Such materials have a wide variety of uses in moist environments, such as the repair of leaks in piping, the fixing of ordnance and beacons to surfaces, as well as in a surgical setting.206 However, creating materials capable of bonding underwater is challenging, due to the interference of water at the interface between the adhesive and adhered surfaces. The presence of water molecules at the interface inhibits adhesive–surface contact, and thus prevents efficient fastening.207Previous attempts to develop such materials have often looked to nature for inspiration. Mussels, barnacles and other sea dwelling species produce natural adhesives that can bind strongly despite the aqueous environments.206,208 This property arises from the increase prevalence of catechol containing 3,4-dihydroxyl-L-phenylalanine (DOPA) residues within the protein adhesives that these aquatic species secrete. This has led to the production of a number of catechol containing biomimetic underwater adhesive polymers.209,210 These adhesive materials have been explored in great detail and have been extensively reviewed.211–213 A limiting factor of the catechol containing adhesives produced to date is their inability to act as reversible bonding agents. Further to this, curing agents must be added to complete the adhesion processes,214 which can be problematic for certain applications. Kim et al. took a purely synthetic route in their approach to the development of a ‘supramolecular velcro’ adhesive. The formation of host–guest supramolecular complexes in aqueous solutions is accompanied by expulsion of water molecules from hydrophobic host cavities.215 One such host–guest pairing, uses CB[7] as the host and aminomethylferrocene as the guest, complexation between the host and guest results in a Ka ≈ 1012 M−1.216 Here, the host CB[7] was bound to one surface to be adhered, and aminomethylferrocene was bound to the second, Fig. 36. When the two surfaces are brought together, complexation occurs between the host and guest, driven entropically by the expulsion of water from the hydrophobic cavity.
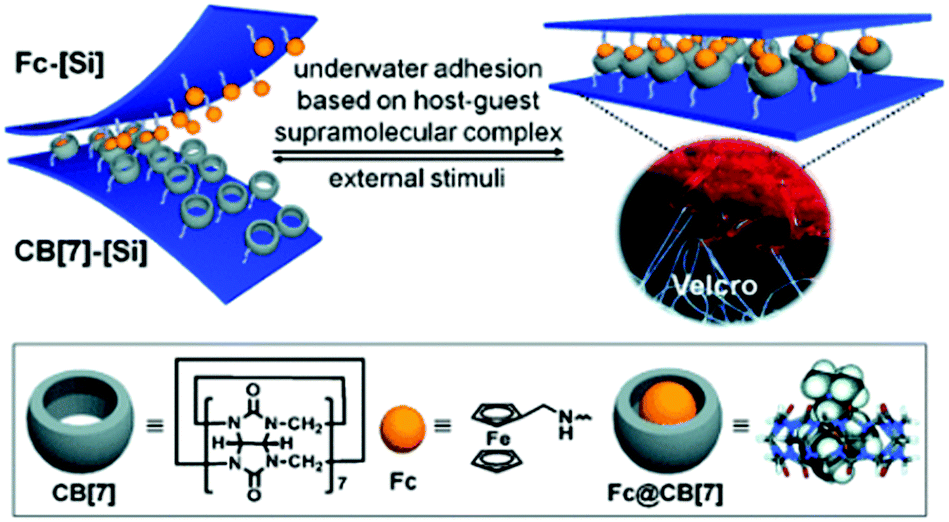 | ||
| Fig. 36 A schematic representation of the CB[7]/aminomethylferrocene ‘velcro’, with a light microscope image of traditional hook and loop Velcro.205 Reproduced from ref. 205 with permission from Wiley, copyright 2013. | ||
The strength of this supramolecular adhesive was found to be greater than that of commercial alternatives (3M™ Velcro, 3M™ double sided tape and 3M™ double sided foam tape), when tested both in air and underwater. Similarly, when compared to previously reported mollusc inspired adhesive polymers, this material displayed similar adhesive strengths, without the need for curing agents (mussel derived adhesion strength: 1.5 MPa217 and 1.2 MPa;218 CB[7] adhesion strength: 1.12 MPa).207
It was possible to tune the strength of adhesion for the CB[7]/aminomethylferrocene ‘velcro’ by varying the density of the host and guest molecules present on the separate surfaces. Much like conventional velcro, this adhesion of the CB[7]/aminomethylferrocene ‘velcro’ was reversible when broken mechanically, owing to its self-healing ability obtained through reversible complexation events. The adhesive bond of the material was also chemically switchable, thanks to the redox-active nature of the aminomethylferrocene. On immersion of the aminomethylferrocene surface in a 10 mM solution of NaClO, the oxidation of the aminomethylferrocene turned off the adhesion, as the oxidised species no longer acts as an effective guest.219 The guest surface was found to be reactivated through exposure to ascorbic acid (20 mM) however, this only resulted in 44% of the original adhesive strength.205
Adhesives, both supramolecular and traditional, are often polymeric in nature. These include resins, bioadhesive gels and all previously discussed systems. In contrast, low molecular weight monomeric units as adhesives have received limited interest in the field, with few systems having been reported.220 One example of such a system was produced by Dong and co-workers, who developed a crown-ether containing, low molecular weight supramolecular adhesive, 36, capable of tough and stable adhesion of a range of materials in seawater over periods of twelve months, Fig. 37.221 This novel material was able to bind to both hydrophobic (PTFE and Teflon) and hydrophilic materials (steel and glass). This is thought to be due to a combination of the large range of supramolecular interactions that it can form, including hydrogen bonds, electrostatic forces, π–π stacking, hydrophobic interactions and metal complexation. Here, similar adhesive strengths (1.94 MPa compared to 2.02 MPa) could be achieved with almost ten times less material (1.25 mg cm−2 compared to 11.6 mg cm−2). Furthermore, to prepare a 20 cm2 area of this adhesive by hand required an hour however, it was found that this same area could also be prepared through electrospinning in just one minute. The versatility of this material has been shown through real life testing in which it has been used as a patch (Fig. 38) to repair breakages in glass flasks, plastic centrifuge tubes and water pipes.
 | ||
| Fig. 37 Structure of small molecular weight supramolecular adhesive developed by Dong et al. | ||
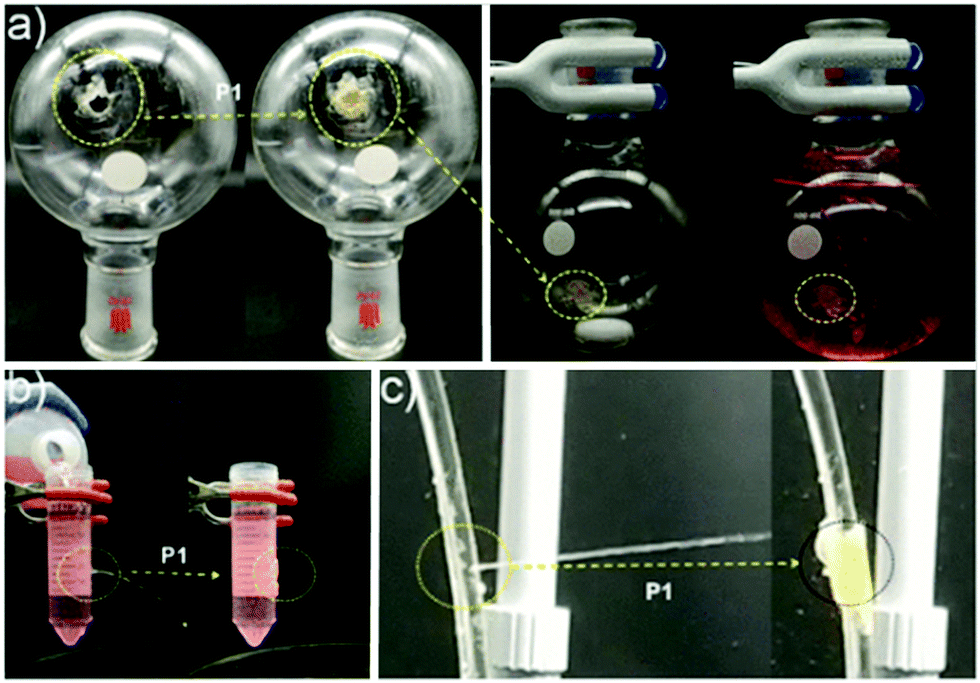 | ||
| Fig. 38 Compound 22 fixing and making watertight (a) a broken glass round bottom flask, (b) a centrifuge tube and (c) a water pipe.221 Reproduced from ref. 221 with permission from the American Chemical Society, copyright 2020. | ||
Supramolecular chemistry in battery materials
As previously discussed, polyrotaxane based materials offer unique physical properties. The ‘sliding’ of the macrocycles along a molecular axle allows them to efficiently dissipate energy. This has sparked investigation into their use as binder materials in lithium ion batteries. The use of silicon particles as an anode material in these batteries shows great promise, however during charge–discharge cycles the silicon undergoes large volume changes, resulting in particle destruction.222 Elastic materials are thus added to prevent this effect, traditionally this material is polyacrylic acid. Choi and co-workers have shown that with the addition of 5% w/w polyrotaxane to the binder material, the elasticity is greatly increased.223 The inclusion abilities of this supramolecular motif has yielded a material capable of increasing the lifetime of these high capacity batteries to commercially viable levels.Conclusions
The varied and unique properties that the application of supramolecular chemistry can bring to products is only now being realised. Huge leaps in host guest chemistries are paving the way towards more accurate real time glucose sensors, previously dangerous chemical warfare agent disposal is being made safe through the use of gels, and soon a punctured tyre may be capable of simply repairing itself. This is by no means an exhaustive list of the expansive research occurring within supramolecular chemistry with great potential for commercialisation, and work continues in many groups that will yield even more impressive materials, sensors and other applications-based chemistries. We hope that by highlighting a range of areas in which supramolecular chemistry has moved out of the lab and into the real world, we may inspire others to make this transition with their own scientific endeavours.Conflicts of interest
There are no conflicts to declare.Acknowledgements
GTW would like to thank the University of Kent GCDC for funding his postdoctoral fellowship. JRH would like to thank the UKRI for her Future Leaders Fellowship funding (MR/T020415/1). MF and PAG acknowledge and pay respect to the Gadigal people of the Eora Nation, the traditional owners of the land on which we research, teach and collaborate at the University of Sydney. MF thanks the University of Sydney for funding. PAG thanks the Australian Research Council (DP180100612, DP200100453 and LE210100144) and the University of Sydney for funding. CJEH thanks University College London for financial support.Notes and references
- J. M. Lehn, Acc. Chem. Res., 1978, 11, 49–57 CrossRef CAS.
- C. R. Patrick and G. S. Prosser, Nature, 1960, 187, 1021 CrossRef CAS.
- D. M. G. Lawrey and H. McConnell, J. Am. Chem. Soc., 1952, 74, 6175–6177 CrossRef CAS.
- W. G. Levine and J. Peisach, Nature, 1965, 207, 406–407 CrossRef CAS.
- E. V. Lampard, A. C. Sedgwick, T. Sombuttan, G. T. Williams, B. Wannalerse, A. T. A. Jenkins, S. D. Bull and T. D. James, ChemistryOpen, 2018, 7, 266–268 CrossRef CAS.
- G. T. Williams, A. C. Sedgwick, S. Sen, L. Gwynne, J. E. Gardiner, J. T. Brewster, J. R. Hiscock, T. D. James, A. T. A. Jenkins and J. L. Sessler, Chem. Commun., 2020, 56, 5516–5519 RSC.
- B. C. Crane, N. P. Barwell, P. Gopal, M. Gopichand, T. Higgs, T. D. James, C. M. Jones, A. Mackenzie, K. P. Mulavisala and W. Paterson, J. Diabetes Sci. Technol., 2015, 9, 751–761 CrossRef.
- https://www.nobelprize.org/prizes/chemistry/1987/press-release/, (accessed 28/07/20).
- https://www.nobelprize.org/prizes/chemistry/2016/summary/%3E, (accessed 27/05/20).
- J. M. Lehn, Chem. Soc. Rev., 2017, 46, 2378–2379 RSC.
- T. Trinh, J. P. Cappel, P. A. Geis, M. L. McCarty, D. Pilosof and S. S. Zwerdling, US Pat., US5714137, 1998 Search PubMed.
- J. K. Tusa and H. R. He, J. Mater. Chem., 2005, 15, 2640–2647 RSC.
- https://europe.nissannews.com/en-GB/releases/release-87812-world-s-first-self-healing-iphone-case, (accessed 30/06/20).
- A. P. de Silva and K. R. A. S. Sandanayake, J. Chem. Soc., 1989, 1183–1185 CAS.
- M. Nakahata, S. Mori, Y. Takashima, H. Yamaguchi and A. Harada, Chem, 2016, 1, 766–775 CAS.
- J. R. Wu, L. H. Cai and D. A. Weitz, Adv. Mater., 2017, 29, 8 Search PubMed.
- D. Jagleniec, L. Dobrzycki, M. Karbarz and J. Romanski, Chem. Sci., 2019, 10, 9542–9547 RSC.
- N. J. Williams, C. A. Seipp, K. A. Garrabrant, R. Custelcean, E. Holguin, J. K. Keum, R. J. Ellis and B. A. Moyer, Chem. Commun., 2018, 54, 10048–10051 RSC.
- I. V. Kolesnichenko and E. V. Anslyn, Chem. Soc. Rev., 2017, 46, 2385–2390 RSC.
- F. Arnaudneu, E. M. Collins, M. Deasy, G. Ferguson, S. J. Harris, B. Kaitner, A. J. Lough, M. A. McKervey, E. Marques, B. L. Ruhl, M. J. Schwingweill and E. M. Seward, J. Am. Chem. Soc., 1989, 111, 8681–8691 CrossRef CAS.
- M. A. McKervey, D. P. Melody, J. Woods and J. M. Rooney, Eur. Pat., EP0151527A1, 1984 Search PubMed.
- J. Szejtli, Chem. Rev., 1998, 98, 1743–1754 CrossRef CAS.
- G. Crini, Chem. Rev., 2014, 114, 10940–10975 CrossRef CAS.
- A. Haimhoffer, A. Rusznyak, K. Reti-Nagy, G. Vasvari, J. Varadi, M. Vecsernyes, I. Bacskay, P. Feher, Z. Ujhelyi and F. Fenyvesi, Sci. Pharm., 2019, 87, 21 CrossRef.
- M. E. Davis and M. E. Brewster, Nat. Rev. Drug Discovery, 2004, 3, 1023–1035 CrossRef CAS.
- S. B. Carneiro, F. Í. Costa Duarte, L. Heimfarth, J. d. S. Siqueira Quintans, L. J. Quintans-Júnior, V. F. d. Veiga Júnior and Á. A. Neves de Lima, Int. J. Mol. Sci., 2019, 20, 642 CrossRef CAS.
- C. Muankaew and T. Loftsson, Basic Clin. Pharmacol. Toxicol., 2018, 122, 46–55 CrossRef CAS.
- O. Adeoye and H. Cabral-Marques, Int. J. Pharm., 2017, 531, 521–531 CrossRef CAS.
- J. R. Lakkakula and R. W. Macedo Krause, Nanomedicine, 2014, 9, 877–894 CrossRef CAS.
- T. Loftsson, J. Inclusion Phenom. Macrocyclic Chem., 2002, 44, 63–67 CrossRef CAS.
- P. Jansook, G. C. Ritthidej, H. Ueda, E. Stefánsson and T. Loftsson, J. Pharm. Pharm. Sci., 2010, 13, 336–350 CAS.
- A. Ohira, K. Hara, G. Jóhannesson, M. Tanito, G. M. Ásgrímsdóttir, S. H. Lund, T. Loftsson and E. Stefánsson, Acta Ophthalmol., 2015, 93, 610–615 CrossRef CAS.
- G. Johannesson, M. D. Moya-Ortega, G. M. Asgrimsdottir, S. H. Lund, M. Thorsteinsdottir, T. Loftsson and E. Stefansson, Acta Ophthalmol., 2014, 92, 550–556 CrossRef CAS.
- S. A. Oculis, 2020, https://www.globenewswire.com/news-release/2020/02/10/1982307/0/en/Oculis-Announces-Positive-OCS-01-Phase-2-Data-in-Patients-with-Diabetic-Macular-Edema-DME.html, (accessed 29/10/2020).
- K. Imamachi, E. Stefansson, A. Ohira and M. Tanito, Acta Ophthalmol., 2019, 97, 824–827 CrossRef.
- J. S. Krinsley, M. Egi, A. Kiss, A. N. Devendra, P. Schuetz, P. M. Maurer, M. J. Schultz, R. T. van Hooijdonk, M. Kiyoshi, I. M. Mackenzie, D. Annane, P. Stow, S. A. Nasraway, S. Holewinski, U. Holzinger, J. C. Preiser, J. L. Vincent and R. Bellomo, Crit. Care, 2013, 17, R37 CrossRef.
- Y. Mamo, F. Bekele, T. Nigussie and A. Zewudie, BMC Endocr. Disord., 2019, 19, 91 CrossRef.
- D. C. Klonoff, D. Ahn and A. Drincic, Diabetes Res. Clin. Pract., 2017, 133, 178–192 CrossRef CAS.
- A. Wood, D. O'Neal, J. Furler and E. I. Ekinci, Intern. Med. J., 2018, 48, 499–508 CrossRef.
- J. P. Lorand and J. O. Edwards, J. Org. Chem., 1959, 24, 769–774 CrossRef CAS.
- G. Wulff, Pure Appl. Chem., 1982, 54, 2093–2102 Search PubMed.
- H. Fang, G. Kaur and B. Wang, J. Fluoresc., 2004, 14, 481–489 CrossRef CAS.
- Z. Guo, I. Shin and J. Yoon, Chem. Commun., 2012, 48, 5956–5967 RSC.
- X. Sun, B. M. Chapin, P. Metola, B. Collins, B. Wang, T. D. James and E. V. Anslyn, Nat. Chem., 2019, 11, 768–778 CrossRef CAS.
- O. Hirata, Y. Kubo, M. Takeuchi and S. Shinkai, Tetrahedron, 2004, 60, 11211–11218 CrossRef CAS.
- W. Yang, S. Gao, X. Gao, V. V. R. Karnati, W. Ni, B. Wang, W. B. Hooks, J. Carson and B. Weston, Bioorg. Med. Chem. Lett., 2002, 12, 2175–2177 CrossRef CAS.
- L. Zhu, S. H. Shabbir, M. Gray, V. M. Lynch, S. Sorey and E. V. Anslyn, J. Am. Chem. Soc., 2006, 128, 1222–1232 CrossRef CAS.
- T. D. James, K. R. A. S. Sandanayake and S. Shinkai, J. Chem. Soc., Chem. Commun., 1994, 4, 477–478 RSC.
- W. Ni, G. Kaur, G. Springsteen, B. Wang and S. Franzen, Bioorg. Chem., 2004, 32, 571–581 CrossRef CAS.
- S. Arimori, M. L. Bell, C. S. Oh and T. D. James, Org. Lett., 2002, 4, 4249–4251 CrossRef CAS.
- S. Arimori, M. L. Bell, C. S. Oh, K. A. Frimat and T. D. James, Chem. Commun., 2001, 1836–1837 RSC.
- S. Arimori, M. L. Bell, C. S. Oh, K. A. Frimat and T. D. James, J. Chem. Soc., Perkin Trans. 1, 2002, 803–808 RSC.
- M. Mortellaro and A. DeHennis, Biosens. Bioelectron., 2014, 61, 227–231 CrossRef CAS.
- B. C. Crane, N. P. Barwell, P. Gopal, M. Gopichand, T. Higgs, T. D. James, C. M. Jones, A. Mackenzie, K. P. Mulavisala and W. Paterson, J. Diabetes Sci. Technol., 2015, 9, 751–761 CrossRef.
- T. D. James, 2019, https://chemistrycommunity.nature.com/posts/53695-measure-for-measure-fluorescent-boronic-acid-sensors-for-continuous-monitoring-of-glucose-based-on-ortho-aminomethylphenylboronic-acids-an-ongoing-shakespearian-drama, (accessed 29/10/2020).
- T. D. James, K. R. A. S. Sandanayake and S. Shinkai, Angew. Chem., Int. Ed. Engl., 1994, 33, 2207–2209 CrossRef.
- T. D. James, K. S. Sandanayake, R. Iguchi and S. Shinkai, J. Am. Chem. Soc., 1995, 117, 8982–8987 CrossRef CAS.
- A. E. Colvin and H. Jiang, J. Biomed. Mater. Res., Part A, 2013, 101, 1274–1282 CrossRef.
- W. Freeman, W. Mock and N. Shih, J. Am. Chem. Soc., 1981, 103, 7367–7368 CrossRef CAS.
- S. J. Barrow, S. Kasera, M. J. Rowland, J. del Barrio and O. A. Scherman, Chem. Rev., 2015, 115, 12320–12406 CrossRef CAS.
- R. S. Cicolani, L. R. R. Souza, G. B. de Santana Dias, J. M. R. Gonçalves, I. d. S. Abrahao, V. M. Silva and G. J. F. Demets, J. Inclusion Phenom. Macrocyclic Chem., 2020 DOI:10.1007/s10847-020-00999-8.
- G. Stephenson, R. M. Parker, Y. Lan, Z. Yu, O. A. Scherman and C. Abell, Chem. Commun., 2014, 50, 7048–7051 RSC.
- D. Das, K. I. Assaf and W. M. Nau, Front. Chem., 2019, 7, 619 CrossRef CAS.
- S. K. Konda, R. Maliki, S. McGrath, B. S. Parker, T. Robinson, A. Spurling, A. Cheong, P. Lock, P. J. Pigram and D. R. Phillips, ACS Med. Chem. Lett., 2017, 8, 538–542 CrossRef CAS.
- J. Kim, I.-S. Jung, S.-Y. Kim, E. Lee, J.-K. Kang, S. Sakamoto, K. Yamaguchi and K. Kim, J. Am. Chem. Soc., 2000, 122, 540–541 CrossRef CAS.
- X. M. Chen, Y. Chen, Q. Yu, B. H. Gu and Y. Liu, Angew. Chem., Int. Ed., 2018, 57, 12519–12523 CrossRef CAS.
- H. Qiao, J. Jia, W. Chen, B. Di, O. A. Scherman and C. Hu, Adv. Healthcare Mater., 2019, 8, 1801458 Search PubMed.
- R. A. Bissell and A. P. De Silva, J. Chem. Soc., Chem. Commun., 1991, 1148–1150 RSC.
- A. J. Bryan, A. P. de Silva, S. A. De Silva, R. A. D. D. Rupasinghe and K. R. A. S. Sandanayake, Biosensors, 1989, 4, 169–179 CrossRef CAS.
- M. Y. Chae and A. W. Czarnik, J. Am. Chem. Soc., 1992, 114, 9704–9705 CrossRef CAS.
- A. P. De Silva and H. Q. N. Gunaratne, J. Chem. Soc., Chem. Commun., 1990, 186–188 RSC.
- P. A. De Silva, N. H. Q. Gunaratne and C. P. McCoy, Nature, 1993, 364, 42–44 CrossRef.
- M. E. Huston, E. U. Akkaya and A. W. Czarnik, J. Am. Chem. Soc., 1989, 111, 8735–8737 CrossRef CAS.
- J. Yoon and A. W. Czarnik, J. Am. Chem. Soc., 1992, 114, 5874–5875 CrossRef CAS.
- A. P. De Silva, T. P. Vance, M. E. S. West and G. D. Wright, Org. Biomol. Chem., 2008, 6, 2468–2481 RSC.
- J. K. Tusa and H. He, J. Mater. Chem., 2005, 15, 2640–2647 RSC.
- R. A. Tromans, T. S. Carter, L. Chabanne, M. P. Crump, H. Li, J. V. Matlock, M. G. Orchard and A. P. Davis, Nat. Chem., 2019, 11, 52–56 CrossRef CAS.
- M. Shamsipur, M. Hosseini, K. Alizadeh, N. Alizadeh, A. Yari, C. Caltagirone and V. Lippolis, Anal. Chim. Acta, 2005, 533, 17–24 CrossRef CAS.
- E. Biavardi, S. Federici, C. Tudisco, D. Menozzi, C. Massera, A. Sottini, G. G. Condorelli, P. Bergese and E. Dalcanale, Angew. Chem., Int. Ed., 2014, 53, 9183–9188 CrossRef CAS.
- D. Masseroni, E. Biavardi, D. Genovese, E. Rampazzo, L. Prodi and E. Dalcanale, Chem. Commun., 2015, 51, 12799–12802 RSC.
- E. Biavardi, C. Tudisco, F. Maffei, A. Motta, C. Massera, G. G. Condorelli and E. Dalcanale, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 2263–2268 CrossRef CAS.
- G. Valenti, E. Rampazzo, E. Biavardi, E. Villani, G. Fracasso, M. Marcaccio, F. Bertani, D. Ramarli, E. Dalcanale, F. Paolucci and L. Prodi, Faraday Discuss., 2015, 185, 299–309 RSC.
- G. Valenti, E. Rampazzo, S. Kesarkar, D. Genovese, A. Fiorani, A. Zanut, F. Palomba, M. Marcaccio, F. Paolucci and L. Prodi, Coord. Chem. Rev., 2018, 367, 65–81 CrossRef CAS.
- L. You, J. S. Berman and E. V. Anslyn, Nat. Chem., 2011, 3, 943–948 CrossRef CAS.
- C. Di Natale, D. Monti and R. Paolesse, Mater. Today, 2010, 13, 46–52 CrossRef CAS.
- S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081–10206 CrossRef CAS.
- M. Stefanelli, G. Magna, F. Zurlo, F. M. Caso, E. Di Bartolomeo, S. Antonaroli, M. Venanzi, R. Paolesse, C. Di Natale and D. Monti, ACS Appl. Mater. Interfaces, 2019, 11, 12077–12087 CrossRef CAS.
- C. P. Montgomery, B. S. Murray, E. J. New, R. Pal and D. Parker, Acc. Chem. Res., 2009, 42, 925–937 CrossRef CAS.
- D. Parker, Chem. Soc. Rev., 2004, 33, 156–165 RSC.
- D. Parker, R. S. Dickins, H. Puschmann, C. Crossland and J. A. K. Howard, Chem. Rev., 2002, 102, 1977–2010 CrossRef CAS.
- R. A. Poole, F. Kielar, S. L. Richardson, P. A. Stenson and D. Parker, Chem. Commun., 2006, 4084–4086 RSC.
- B. D. Smith, Beilstein J. Org. Chem., 2015, 11, 2540–2548 CrossRef CAS.
- R. Pal, D. Parker and L. C. Costello, Org. Biomol. Chem., 2009, 7, 1525–1528 RSC.
- B. A. Smith, W. J. Akers, W. M. Leevy, A. J. Lampkins, S. Xiao, W. Wolter, M. A. Suckow, S. Achilefu and B. D. Smith, J. Am. Chem. Soc., 2010, 132, 67–69 CrossRef CAS.
- A. Ojida, Y. Mito-Oka, M. A. Inoue and I. Hamachi, J. Am. Chem. Soc., 2002, 124, 6256–6258 CrossRef CAS.
- B. A. Smith, B. W. Xie, E. R. Van Beek, I. Que, V. Blankevoort, S. Xiao, E. L. Cole, M. Hoehn, E. L. Kaijzel, C. W. G. M. Löwik and B. D. Smith, ACS Chem. Neurosci., 2012, 3, 530–537 CrossRef CAS.
- E. Arunkumar, C. C. Forbes, B. C. Noll and B. D. Smith, J. Am. Chem. Soc., 2005, 127, 3288–3289 CrossRef CAS.
- E. Arunkumar, N. Fu and B. D. Smith, Chem. – Eur. J., 2006, 12, 4684–4690 CrossRef CAS.
- J. M. Dempsey, C. Zhai, H. H. McGarraugh, C. L. Schreiber, S. E. Stoffel, A. Johnson and B. D. Smith, Chem. Commun., 2019, 55, 12793–12796 RSC.
- J. J. Gassensmith, J. M. Baumes and B. D. Smith, Chem. Commun., 2009, 6329–6338 RSC.
- C. A. Daws, C. L. Exstrom, J. R. Sowa and K. R. Mann, Chem. Mater., 1997, 9, 363–368 CrossRef CAS.
- E. R. Li, K. C. Jie, M. Liu, X. R. Sheng, W. J. Zhu and F. H. Huang, Chem. Soc. Rev., 2020, 49, 1517–1544 RSC.
- K. C. Jie, Y. J. Zhou, E. R. Li and F. H. Huang, Acc. Chem. Res., 2018, 51, 2064–2072 CrossRef CAS.
- E. R. Li, K. C. Jie, Y. J. Zhou, R. Zhao, B. Zhang, Q. Wang, J. Y. Liu and F. H. Huang, ACS Appl. Mater. Interfaces, 2018, 10, 23147–23153 CrossRef CAS.
- E. R. Li, K. C. Jie, Y. J. Zhou, R. Zhao and F. H. Huang, J. Am. Chem. Soc., 2018, 140, 15070–15079 CrossRef CAS.
- Q. Li, H. T. Z. Zhu and F. H. Huang, J. Am. Chem. Soc., 2019, 141, 13290–13294 CrossRef CAS.
- Y. J. Jang, K. Kim, O. G. Tsay, D. A. Atwood and D. G. Churchill, Chem. Rev., 2015, 115, PR1–PR76 CrossRef.
- A. Barba-Bon, A. M. Costero, M. Parra, S. Gil, R. Martínez-Máñez, F. Sancenõn, P. A. Gale and J. R. Hiscock, Chem. – Eur. J., 2013, 19, 1586–1590 CrossRef CAS.
- J. R. Hiscock, N. J. Wells, J. A. Ede, P. A. Gale and M. R. Sambrook, Org. Biomol. Chem., 2016, 14, 9560–9567 RSC.
- M. R. Sambrook, J. R. Hiscock, A. Cook, A. C. Green, I. Holden, J. C. Vincent and P. A. Gale, Chem. Commun., 2012, 48, 5605–5607 RSC.
- J. R. Hiscock, I. L. Kirby, J. Herniman, G. John Langley, A. J. Clark and P. A. Gale, RSC Adv., 2014, 4, 45517–45521 RSC.
- J. R. Hiscock, F. Piana, M. R. Sambrook, N. J. Wells, A. J. Clark, J. C. Vincent, N. Busschaert, R. C. D. Brown and P. A. Gale, Chem. Commun., 2013, 49, 9119–9121 RSC.
- J. R. Hiscock, M. R. Sambrook, J. A. Ede, N. J. Wells and P. A. Gale, J. Mater. Chem. A, 2015, 3, 1230–1234 RSC.
- J. R. Hiscock, M. R. Sambrook, N. J. Wells and P. A. Gale, Chem. Sci., 2015, 6, 5680–5684 RSC.
- F. Piana, M. Facciotti, G. Pileio, J. R. Hiscock, W. Van Rossom, R. C. D. Brown and P. A. Gale, RSC Adv., 2015, 5, 12287–12292 RSC.
- N. A. Esipenko, P. Koutnik, T. Minami, L. Mosca, V. M. Lynch, G. V. Zyryanov and P. Anzenbacher, Chem. Sci., 2013, 4, 3617–3623 RSC.
- A. Barba-Bon, A. M. Costero, S. Gil, R. Martínez-Máñez and F. Sancenón, Org. Biomol. Chem., 2014, 12, 8745–8751 RSC.
- J. R. Hiscock, G. P. Bustone and E. R. Clark, ChemistryOpen, 2017, 6, 497–500 CrossRef CAS.
- Y. Kalinovskyy, A. J. Wright, J. R. Hiscock, T. D. Watts, R. L. Williams, N. J. Cooper, M. J. Main, S. J. Holder and B. A. Blight, ACS Appl. Mater. Interfaces, 2020, 12, 8634–8641 CrossRef CAS.
- A. J. Wright, M. J. Main, N. J. Cooper, B. A. Blight and S. J. Holder, ACS Appl. Mater. Interfaces, 2017, 9, 31335–31339 CrossRef CAS.
- Y. Kalinovskyy, N. J. Cooper, M. J. Main, S. J. Holder and B. A. Blight, Dalton Trans., 2017, 46, 15704–15709 RSC.
- A. M. Wilson, P. J. Bailey, P. A. Tasker, J. R. Turkington, R. A. Grant and J. B. Love, Chem. Soc. Rev., 2014, 43, 123–134 RSC.
- J. R. Turkington, P. J. Bailey, J. B. Love, A. M. Wilson and P. A. Tasker, Chem. Commun., 2013, 49, 1891–1899 RSC.
- G. A. Kordosky, presented in part at the 18th International Solvent Extraction Conference, Cape Town, South Africa, 2002.
- IBC Advanced Technologies, Inc., http://www.ibcmrt.com/, (accessed 29/10/2020).
- D. D. Walker, M. A. Norato, S. G. Campbell, M. L. Crowder, S. D. Fink, F. F. Fondeur, M. W. Geeting, G. F. Kessinger and R. A. Pierce, Sep. Sci. Technol., 2005, 40, 297–309 CrossRef CAS.
- R. M. Izatt, Chem. Eng. Prog., 2015, 111, 48–54 Search PubMed.
- J. J. M. Nelson and E. J. Schelter, Inorg. Chem., 2019, 58, 979–990 CrossRef CAS.
- M. D. Rao, K. K. Singh, C. A. Morrison and J. B. Love, RSC Adv., 2020, 10, 4300–4309 RSC.
- C. Hagelüken and C. W. Corti, Gold Bull., 2010, 43, 209–220 CrossRef.
- A. Greenfield and T. E. Graedel, Resour., Conserv. Recycl., 2013, 74, 1–7 CrossRef.
- Z. Liu, M. Frasconi, J. Lei, Z. J. Brown, Z. Zhu, D. Cao, J. Iehl, G. Liu, A. C. Fahrenbach, Y. Y. Botros, O. K. Farha, J. T. Hupp, C. A. Mirkin and J. Fraser Stoddart, Nat. Commun., 2013, 4, 1855 CrossRef.
- cycladex, http://cycladex.com/, (accessed 29/10/2020).
- R. B. Pettman and D. F. Arnolds, US Pat., WO2017158561A1, 2017 Search PubMed.
- FS ventures, https://www.fs-ventures.co.uk/investee-companies/investee-companies/cycladex-ltd/, accessed 29/10/2020.
- L. X. Chen, M. Liu, Y. Q. Zhang, Q. J. Zhu, J. X. Liu, B. X. Zhu and Z. Tao, Chem. Commun., 2019, 55, 14271–14274 RSC.
- R.-L. Lin, Y.-P. Dong, M. Tang, Z. Liu, Z. Tao and J.-X. Liu, Inorg. Chem., 2020, 59, 3850–3855 CrossRef CAS.
- H. Wu, L. O. Jones, Y. Wang, D. Shen, Z. Liu, L. Zhang, K. Cai, Y. Jiao, C. L. Stern, G. C. Schatz and J. F. Stoddart, ACS Appl. Mater. Interfaces, 2020, 12, 38768–38777 CrossRef CAS.
- Z. Liu, A. Samanta, J. Lei, J. Sun, Y. Wang and J. F. Stoddart, J. Am. Chem. Soc., 2016, 138, 11643–11653 CrossRef CAS.
- E. D. Doidge, I. Carson, P. A. Tasker, R. J. Ellis, C. A. Morrison and J. B. Love, Angew. Chem., Int. Ed., 2016, 55, 12436–12439 CrossRef CAS.
- E. D. Doidge, L. M. M. Kinsman, Y. Ji, I. Carson, A. J. Duffy, I. A. Kordas, E. Shao, P. A. Tasker, B. T. Ngwenya, C. A. Morrison and J. B. Love, ACS Sustainable Chem. Eng., 2019, 7, 15019–15029 CrossRef CAS.
- J. E. Tamis, R. H. Jongbloed, C. C. Karman, W. Koops and A. J. Murk, Integr. Environ. Assess. Manage., 2012, 8, 231–241 CrossRef CAS.
- A. M. Vibhute and K. M. Sureshan, ChemSusChem, 2020, 13, 5343–5360 CrossRef CAS.
- D. P. O’Mara, A. F. Hadermann and J. C. Trippe, US Pat., US4720397A, 1988 Search PubMed.
- B. Doshi, M. Sillanpää and S. Kalliola, Water Res., 2018, 135, 262–277 CrossRef CAS.
- B. O. Okesola and D. K. Smith, Chem. Soc. Rev., 2016, 45, 4226–4251 RSC.
- T. Kobayashi, Y. Kawashima, M. Yoshimura, M. Sugiura, T. Nobe and S. Fujimoto, US Pat., 4502975, 1985 Search PubMed.
- R. G. Weiss and A. M. V. Mallya, Worldwide Pat., WO2012047251A1, 2012 Search PubMed.
- B. Zhang, S. Chen, H. Luo, B. Zhang, F. Wang and J. Song, J. Hazard. Mater., 2020, 384, 121460 CrossRef CAS.
- R. P. Schwarzenbach, B. I. Escher, K. Fenner, T. B. Hofstetter, C. A. Johnson, U. von Gunten and B. Wehrli, Science, 2006, 313, 1072–1077 CrossRef CAS.
- A. Alsbaiee, B. J. Smith, L. L. Xiao, Y. H. Ling, D. E. Helbling and W. R. Dichtel, Nature, 2016, 529, 190–194 CrossRef CAS.
- B. A. Moyer, R. Custelcean, B. P. Hay, J. L. Sessler, K. Bowman-James, V. W. Day and S.-O. Kang, Inorg. Chem., 2013, 52, 3473–3490 CrossRef CAS.
- R. Custelcean, N. J. Williams and C. A. Seipp, Angew. Chem., Int. Ed., 2015, 54, 10525–10529 CrossRef CAS.
- R. Custelcean, N. J. Williams, C. A. Seipp, A. S. Ivanov and V. S. Bryantsev, Chem. – Eur. J., 2016, 22, 1997–2003 CrossRef CAS.
- C. A. Seipp, N. J. Williams, V. S. Bryantsev, R. Custelcean and B. A. Moyer, RSC Adv., 2015, 5, 107266–107269 RSC.
- L. Qin, S. J. N. Vervuurt, R. B. P. Elmes, S. N. Berry, N. Proschogo and K. A. Jolliffe, Chem. Sci., 2020, 11, 201–207 RSC.
- J. L. Humphrey, Separation process technology, McGraw-Hill, New York, London, 1997 Search PubMed.
- D. S. Sholl and R. P. Lively, Nature, 2016, 533, 316 CrossRef.
- X. Zhao, Y. Wang, D.-S. Li, X. Bu and P. Feng, Adv. Mater., 2018, 30, 1705189 CrossRef.
- Z. F. Wang, S. N. Zhang, Y. Chen, Z. J. Zhang and S. Q. Ma, Chem. Soc. Rev., 2020, 49, 708–735 RSC.
- M. Mofarahi and F. Gholipour, Microporous Mesoporous Mater., 2014, 200, 1–10 CrossRef CAS.
- E. J. Garcia, J. Perez-Pellitero, G. D. Pirngruber, C. Jallut, M. Palomino, F. Rey and S. Valencia, Ind. Eng. Chem. Res., 2014, 53, 9860–9874 CrossRef CAS.
- M. Yu, R. D. Noble and J. L. Falconer, Acc. Chem. Res., 2011, 44, 1196–1206 CrossRef CAS.
- J. R. Wu and Y. W. Yang, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202006999.
- K. C. Jie, M. Liu, Y. J. Zhou, M. A. Little, S. Bonakala, S. Y. Chong, A. Stephenson, L. J. Chen, F. H. Huang and A. I. Cooper, J. Am. Chem. Soc., 2017, 139, 2908–2911 CrossRef CAS.
- X. G. Li, C. T. Cui, H. Li and X. Gao, Sep. Purif. Technol., 2019, 228, 10 Search PubMed.
- H. M. V. Tassel, US Pat., US3398063A, 1966 Search PubMed.
- K. C. Jie, M. Liu, Y. J. Zhou, M. A. Little, A. Pulido, S. Y. Chong, A. Stephenson, A. R. Hughes, F. Sakakibara, T. Ogoshi, F. Blanc, G. M. Day, F. H. Huang and A. I. Cooper, J. Am. Chem. Soc., 2018, 140, 6921–6930 CrossRef CAS.
- G. Zhang, B. Moosa, A. Chen and N. M. Khashab, ChemPlusChem, 2020, 85, 1244–1248 CrossRef CAS.
- Y. J. Zhou, K. C. Jie, R. Zhao, E. R. Li and F. H. Huang, J. Am. Chem. Soc., 2020, 142, 6957–6961 CrossRef CAS.
- D. Cantillo and C. O. Kappe, React. Chem. Eng., 2017, 2, 7–19 RSC.
- J. R. Wu, A. U. Mu, B. Li, C. Y. Wang, L. Fang and Y. W. Yang, Angew. Chem., Int. Ed., 2018, 57, 9853–9858 CrossRef CAS.
- J. R. Wu, B. Li and Y. W. Yang, Angew. Chem., Int. Ed., 2020, 59, 2251–2255 CrossRef CAS.
- Q. Li, K. C. Jie and F. H. Huang, Angew. Chem., Int. Ed., 2020, 59, 5355–5358 CrossRef CAS.
- Y. Li, W. S. Li, L. Zhong and C. J. Xu, Ind. Eng. Chem. Res., 2017, 56, 11894–11902 CrossRef CAS.
- D. B. Amabilino, D. K. Smith and J. W. Steed, Chem. Soc. Rev., 2017, 46, 2404–2420 RSC.
- X. Z. Yan, F. Wang, B. Zheng and F. H. Huang, Chem. Soc. Rev., 2012, 41, 6042–6065 RSC.
- V. R. de la Rosa, P. Woisel and R. Hoogenboom, Mater. Today, 2016, 19, 44–55 CrossRef CAS.
- A. Lavrenova, D. W. R. Balkenende, Y. Sagara, S. Schrettl, Y. C. Simon and C. Weder, J. Am. Chem. Soc., 2017, 139, 4302–4305 CrossRef CAS.
- B. Y. Xia, B. Zheng, C. Y. Han, S. Y. Dong, M. M. Zhang, B. J. Hu, Y. H. Yu and F. H. Huang, Polym. Chem., 2013, 4, 2019–2024 RSC.
- I. Yoshimura, Y. Miyahara, N. Kasagi, H. Yamane, A. Ojida and I. Hamachi, J. Am. Chem. Soc., 2004, 126, 12204–12205 CrossRef CAS.
- M. Ikeda, K. Fukuda, T. Tanida, T. Yoshii and I. Hamachi, Chem. Commun., 2012, 48, 2716–2718 RSC.
- L. L. Yang, X. X. Tan, Z. Q. Wang and X. Zhang, Chem. Rev., 2015, 115, 7196–7239 CrossRef CAS.
- L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071–4098 CrossRef CAS.
- D. G. Bekas, K. Tsirka, D. Baltzis and A. S. Paipetis, Composites, Part B, 2016, 87, 92–119 CrossRef CAS.
- N. J. Kanu, E. Gupta, U. K. Vates and G. K. Singh, Composites, Part A, 2019, 121, 474–486 CrossRef CAS.
- V. V. Kumar, G. Balaganesan, J. K. Y. Lee, R. E. Neisiany, S. Surendran and S. Ramakrishna, Polymers, 2019, 11, 19 CrossRef.
- Y. J. Wang, D. T. Pham and C. Q. Ji, Cogent Eng., 2015, 2, 28 Search PubMed.
- F. Behzad, N. Navid and D. Amir, J. Manuf. Mater. Process., 2019, 3, 28 Search PubMed.
- F. M. Raymo and J. F. Stoddart, Chem. Rev., 1999, 99, 1643–1663 CrossRef CAS.
- J. F. Stoddart, Angew. Chem., Int. Ed., 2017, 56, 11094–11125 CrossRef CAS.
- L. Fang, M. A. Olson, D. Benitez, E. Tkatchouk, W. A. Goddard and J. F. Stoddart, Chem. Soc. Rev., 2010, 39, 17–29 RSC.
- T. Takata, Polym. J., 2006, 38, 1–20 CrossRef CAS.
- A. W. Heard and S. M. Goldur, ACS Cent. Sci., 2020, 6, 117–128 CrossRef CAS.
- Y. H. Jin, C. Yu, R. J. Denman and W. Zhang, Chem. Soc. Rev., 2013, 42, 6634–6654 RSC.
- G. M. Peters, X. D. Chi, C. Brockman and J. L. Sessler, Chem. Commun., 2018, 54, 5407–5409 RSC.
- https://www.globenewswire.com/news-release/2020/02/12/1983950/0/en/Industrial-Rubber-Market-to-Garner-44-6-Billion-by-2026-Allied-Market-Research.html, (accessed 05/06/2020).
- A. Das, A. Sallat, F. Bohme, M. Suckow, D. Basu, S. Wiessner, K. W. Stockelhuber, B. Voit and G. Heinrich, ACS Appl. Mater. Interfaces, 2015, 7, 20623–20630 CrossRef CAS.
- Z. L. Yu, F. Tantakitti, T. Yu, L. C. Palmer, G. C. Schatz and S. I. Stupp, Science, 2016, 351, 497–502 CrossRef CAS.
- X. H. Zhao, Soft Matter, 2014, 10, 672–687 RSC.
- J. Kruzelak, R. Sykora and I. Hudec, Chem. Pap., 2016, 70, 1533–1555 CAS.
- M. Suckow, A. Mordvinkin, M. Roy, N. K. Singha, G. Heinrich, B. Voit, K. Saalwachter and F. Bohme, Macromolecules, 2018, 51, 468–479 CrossRef CAS.
- L. Landini, S. G. de Araujo, A. B. Lugao and H. Wiebeck, Eur. Polym. J., 2007, 43, 2725–2731 CrossRef CAS.
- S. Stein, A. Mordvinkin, B. Voit, H. Komber, K. Saalwachter and F. Bohme, Polym. Chem., 2020, 11, 1188–1197 RSC.
- Y. Ahn, Y. Jang, N. Selvapalam, G. Yun and K. Kim, Angew. Chem., Int. Ed., 2013, 52, 3140–3144 CrossRef CAS.
- R. J. Stewart, T. C. Ransom and V. Hlady, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 757–771 CrossRef CAS.
- H. Shao and R. J. Stewart, Adv. Mater., 2010, 22, 729 CrossRef CAS.
- R. Santos, G. da Costa, C. Franco, P. Gomes-Alves, P. Flammang and A. V. Coelho, Mar. Biotechnol., 2009, 11, 686–698 CrossRef CAS.
- M. A. North, C. A. Del Grosso and J. J. Wilker, ACS Appl. Mater. Interfaces, 2017, 9, 7866–7872 CrossRef CAS.
- X. Y. Sha, C. X. Zhang, M. W. Qi, L. H. Zheng, B. K. Cai, F. Chen, Y. L. Wang and Y. F. Zhou, Macromol. Rapid Commun., 2020, 41, 2000055 CrossRef CAS.
- W. Zhang, R. X. Wang, Z. M. Sun, X. W. Zhu, Q. Zhao, T. F. Zhang, A. Cholewinski, F. Yang, B. X. Zhao, R. Pinnaratip, P. K. Forooshani and B. P. Lee, Chem. Soc. Rev., 2020, 49, 433–464 RSC.
- A. H. Hofman, I. A. van Hees, J. Yang and M. Kamperman, Adv. Mater., 2018, 30, 38 CrossRef.
- Q. Guo, J. S. Chen, J. L. Wang, H. B. Zeng and J. Yu, Nanoscale, 2020, 12, 1307–1324 RSC.
- M. H. Lauren and J. J. Wilker, J. Mater. Sci., 2007, 42, 8934–8942 CrossRef.
- Q. H. Wang, Y. L. Zhong, D. P. Miller, X. X. Lu, Q. Tang, Z. L. Lu, E. Zurek, R. Liu and B. Gong, J. Am. Chem. Soc., 2020, 142, 2915–2924 CrossRef CAS.
- K. Kim, N. Selvapalam, Y. H. Ko, K. M. Park, D. Kim and J. Kim, Chem. Soc. Rev., 2007, 36, 267–279 RSC.
- G. Westwood, T. N. Horton and J. J. Wilker, Macromolecules, 2007, 40, 3960–3964 CrossRef CAS.
- M. E. Yu and T. J. Deming, Macromolecules, 1998, 31, 4739–4745 CrossRef CAS.
- D. Sobransingh and A. E. Kaifer, Org. Lett., 2006, 8, 3247–3250 CrossRef CAS.
- J. Courtois, I. Baroudi, N. Nouvel, E. Degrandi, S. Pensec, G. Ducouret, C. Chaneac, L. Bouteiller and C. Creton, Adv. Funct. Mater., 2010, 20, 1803–1811 CrossRef CAS.
- X. Li, Y. Deng, J. L. Lai, G. Zhao and S. Y. Dong, J. Am. Chem. Soc., 2020, 142, 5371–5379 CrossRef CAS.
- I. Kovalenko, B. Zdyrko, A. Magasinski, B. Hertzberg, Z. Milicev, R. Burtovyy, I. Luzinov and G. Yushin, Science, 2011, 334, 75–79 CrossRef CAS.
- S. Choi, T. W. Kwon, A. Coskun and J. W. Choi, Science, 2017, 357, 279–283 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |