
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransition metal-catalyzed alkene isomerization as an enabling technology in tandem, sequential and domino processes
Daniele
Fiorito†
 ,
Simone
Scaringi†
and
Clément
Mazet
*
,
Simone
Scaringi†
and
Clément
Mazet
*
Organic Chemistry Department, University of Geneva, 30 quai Ernest Ansermet, Geneva 1211, Switzerland. E-mail: clement.mazet@unige.ch
First published on 9th December 2020
Abstract
One-pot reactions elaborated around transition metal-catalyzed isomerization of alkenes not only offer the inherent advantages of atom-, step- and redox-economy but also enable the preparation of value-added products that would be difficult to access by conventional methods. In this Review, we cover seminal and recent examples of tandem, sequential and domino processes, which incorporate the most attractive features of olefin isomerization.
 Daniele Fiorito | Daniele Fiorito studied Chemistry at the University of Milano (Italy) where he received his MSc in 2015 working under the supervision of Prof. Cesare Gennari. He completed his PhD in chemistry at the University of Geneva (Switzerland) under the direction of Prof. Clément Mazet working on the development of several transition metal-catalyzed selective reactions (2019). He was recently awarded a Swiss National Science Foundation early postdoc mobility fellowship to pursue postdoctoral studies in the group of Prof. Varinder K. Aggarwal at the University of Bristol (UK). |
 Simone Scaringi | Simone Scaringi was born in Milano (Italy) in 1995. He received his BSc (2017) and MSc (2019) from the University of Pavia (Italy) under the supervision of Prof. Stefano Protti. During his MSc studies, he spent 3 months in the group of Prof. Géraldine Masson at the Institut de Chimie des Substances Naturelles (Gif-sur-Yvette, France). In September 2019, he moved to the University of Geneva (Switzerland) to pursue his doctoral studies under the direction of Prof. Clément Mazet. He is currently working on transition metal-catalyzed selective isomerizations of olefins for remote functionalizations. |
 Clément Mazet | Clément Mazet earned his MSc from the University of Strasbourg (France) where he was introduced to research in organometallic chemistry by the late John A Osborn. He received his PhD from the same institution in 2002 working with Lutz H. Gade. After postdoctoral stays with Andreas Pfaltz (University of Basel, Switzerland) and Eric N. Jacobsen (Harvard University, USA), he began his independent career at the University of Geneva (Switzerland). His interests include mechanistic and synthetic chemistry with emphasis on all aspects of selective catalysis. He received the Zasshikai Lectureship Award from the University of Tokyo (2012) and the Werner Prize from the Swiss Chemical Society (2013). |
1. Introduction
A countless number of catalytic reactions are based on the ability of transition metals to isomerize or migrate carbon–carbon double bonds.1 The scope of applications of these processes spans across multiple areas such as food, fragrance, materials, medicinal chemistry, pharmaceutical and petrochemical. A strategy based on C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond isomerization provides the distinct advantages to (i) control the position and geometry of the unsaturation, (ii) install new functionalities at a different position of the starting alkene if it is judiciously combined with a second chemical transformation. The latter reduces the number of steps, functional group manipulations and protection/deprotection events.2
C bond isomerization provides the distinct advantages to (i) control the position and geometry of the unsaturation, (ii) install new functionalities at a different position of the starting alkene if it is judiciously combined with a second chemical transformation. The latter reduces the number of steps, functional group manipulations and protection/deprotection events.2
The economic and environmental benefits associated with the development of multistep ‘one-pot’ catalytic reactions has raised strong interest in academia and industry.3 The field emulates from Nature's ability to separate different reactive sites within an enzyme, or different enzymes within a cell to accomplish a series of orthogonal transformations in a given sequence without the need to isolate reactive intermediates.4 In this context, numerous research groups have associated olefin isomerization with (at least) one additional chemical transformation using transition metal catalysts, organocatalysts, enzymatic catalysts or any combination thereof.5 In such ventures, a major task for the synthetic chemist is to meet the requirement of temporal resolution and compatibility of all reaction components to a single set of experimental conditions. Importantly, the elaboration of multicatalytic systems is not achieved at the expense of molecular complexity; rather it provides access to novel chemical space by exploiting the reactivity of a transiently generated (sensitive) functional group.
This Review aims at providing the reader with an overview of the most significant advances in the field of ‘one-pot’ transformations articulated around transition metal-catalyzed alkene isomerization. To highlight the inherent benefits of a ‘single-vessel’ approach, our contribution is organized according to the nature of the function transiently generated after the first catalytic transformation. It is further sub-divided according to the nature of the coupled catalytic process at play. For sake of clarity, we follow the generally accepted classification set by Fogg and dos Santos in 2004 and discuss examples of tandem catalysis, sequential catalysis as well as domino reactions (Fig. 1).6Tandem catalysis encompasses one-pot reactions, which occur via two consecutive mechanistically distinct and non-interfering processes.7 Within this category, one can further distinguish auto-tandem catalysis which employs a single catalyst from assisted-tandem catalysis in which the initial catalyst is modified by an external intervention at a given time point to initiate the second transformation.8 A final subset is orthogonal tandem catalysis, which operates with two catalysts present at the beginning of the reaction.9 By contrast, sequential catalysis is based on the use of several distinct precatalysts added sequentially, in the same flask, at different time points of the reaction in order to trigger a series of successive transformations. Finally, domino reactions based on alkene isomerization are one-pot processes in which the migrating alkene can be either trapped intermolecularly with an external reagent or engaged intramolecularly in a skeletal rearrangement.10 Independently of any categorization, the identification of an appropriate thermodynamic driving force for olefin isomerization is an important strategic aspect for the development of one-pot processes relying on this elementary transformation.1
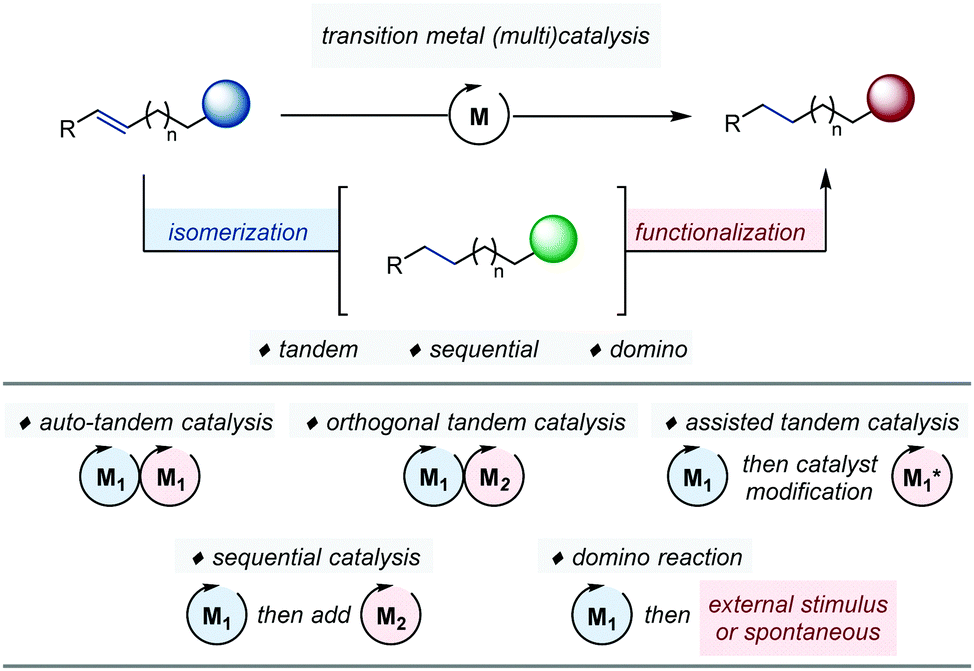 | ||
| Fig. 1 Transition metal-catalyzed alkene isomerization in tandem, sequential and domino processes. | ||
In the present digest, we arbitrarily decided to place our focus on systems which either rely on a single metal precursor or which combine two distinct transition metal catalysts.11 Arguably two of the most common associations falling within this categorization are isomerization/metathesis and isomerization/hydroformylation processes. While the former has been summarized recently and will not be covered in the present conspectus,12 an update of the most significant contributions published since it has been last reviewed in 2015 is presented in Section 2.1 for the latter.13 Of important note, a vast body of the literature covers catalytic isomerizations of alkenes terminated by a functionalization event which operate through a single catalytic cycle (i.e. isomerizing hydrofunctionalization). For this very reason, these systems cannot be defined as tandem, domino or sequential processes and, therefore, fall beyond the scope of this Review.11a,14,15
2. Relocated alkenes as transient intermediates
The formation of thermodynamically more stable alkenes is a commonly employed tactic in olefin isomerization. This can be achieved either by formation of a more substituted double bond or by conjugating the migrating alkene with a neighboring unsaturated functional group (carbonyls, arenes, alkenes…). In the following sections, emphasis has been placed on one-pot processes where a transiently relocated alkene is engaged in a subsequent transformation by means of an auto-tandem, a domino or a sequential approach. The net result is the functionalization of a previously unactivated C(sp3), which does not rely on a directing group strategy.2.1 Tandem processes
As part of a study on Ru-catalyzed addition of aldehydes to conjugated 1,3-dienes to access β,γ-unsaturated ketones, the Ryu group disclosed an isolated example of an auto-tandem isomerization/addition process starting from a non-conjugated 1,5-diene (Fig. 2).16 Based on preliminary investigations, the authors proposed that a coordinatively unsaturated [Ru–H] in situ generated from [RuHCl(CO)(PPh3)3] (Ru1) is likely to chemoselectively react with the terminal alkene and reconjugate the system via successive migratory insertion/β-H elimination sequences (i.e. 2-C chain walking via 1,2-H shift).17 Subsequent formation of a Ru–π-allyl intermediate (A2) by reinsertion of the Ru-hydride across the conjugated diene A1 is supported by NMR spectroscopy.18 This intermediate was proposed to react with aldehydes via a Zimmerman–Traxler 6-membered transition state that minimizes detrimental steric interactions. The resulting alkoxy-ruthenium A3 was proposed to deliver the targeted carbonyl compound (3) upon β-H elimination.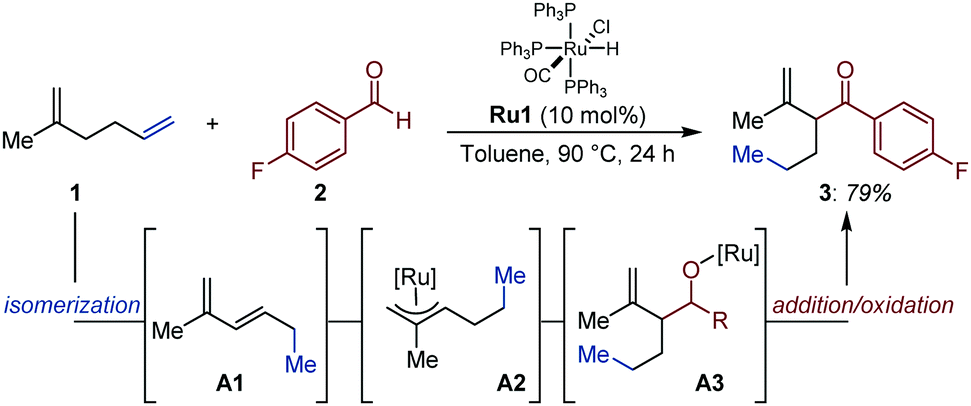 | ||
| Fig. 2 Ru-Catalyzed auto-tandem isomerization/aldehyde addition. | ||
A remote and seemingly unactivated C(sp3) can become the site for C–C bond formation by exploiting its transient electrophilic character. Gooßen and co-workers developed an auto-tandem system in which a Rh catalyst isomerizes ω-unsaturated esters (4) into conjugated esters (B).19 The in situ generated Michael acceptors, in turn, undergo a Rh-catalyzed conjugate addition of C- or N-centered nucleophiles (Fig. 3).20 The authors showed that the rhodium catalyst initially establishes a dynamic equilibrium of positional and geometrical alkene isomers and, once the CC bond is conjugated with the ester group, it is continuously removed from the equilibrium by a rhodium-catalyzed 1,4-addition of either tetraarylborates, primary or secondary amines. The combination of [Rh(acac)(cod)] and a bulky bidentate phosphite (1.5 mol% each) is uniquely effective at catalyzing each individual event. In the case of N-nucleophiles, even though the aza-Michael addition proceeds thermally, the rhodium catalyst led to markedly improved yields.
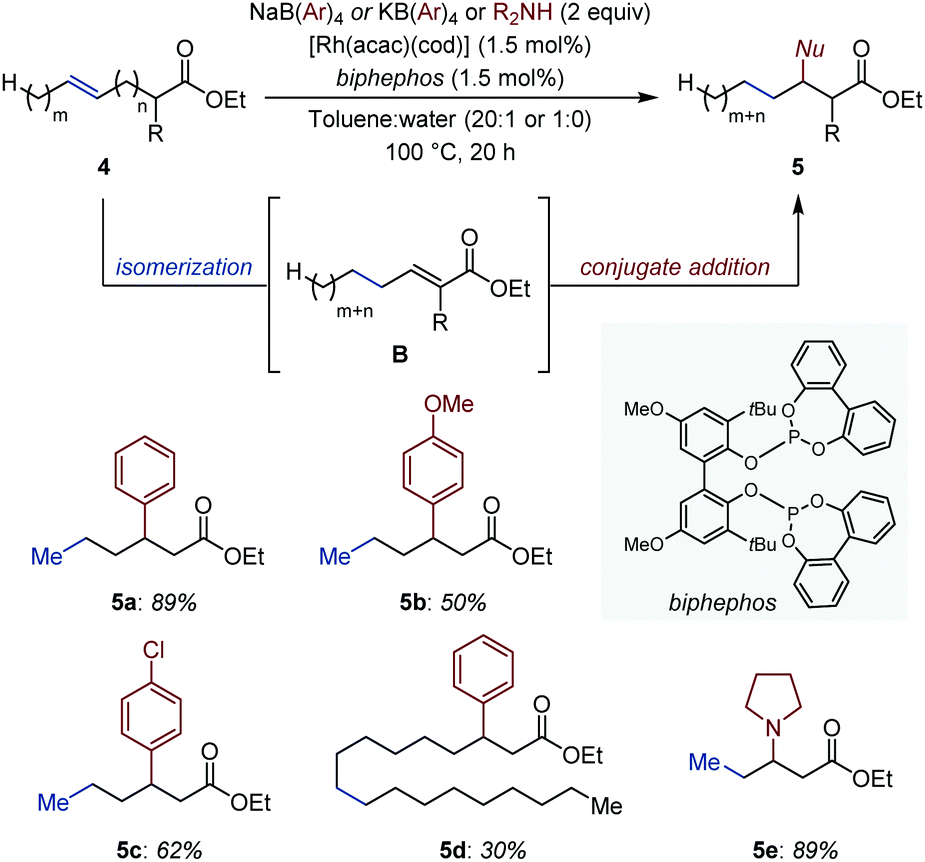 | ||
| Fig. 3 Rh-Catalyzed auto-tandem isomerization/conjugate addition of ω-unsaturated esters. | ||
The conversion of biobased fatty acids to hydrocarbons is a highly sought-after goal to provide alternative sources of raw materials to the petrochemical industry. In this context, the catalytic decarboxylation of mixtures of fatty acids has been identified as a particularly attractive strategy.21 In 2014, Doll and co-workers developed an auto-tandem isomerization/decarboxylation, which gives access to mixtures of alkenes from unsaturated fatty acids using [Ru3(CO)12] as precatalyst and reached promising turn-over frequencies.22 The advantage of this system over existing protocols is that the catalyst dynamically isomerizes the alkene towards the terminal carbonyl moiety and subsequently decarboxylates the most reactive α,β- or β,γ-unsaturated acid derivatives (C1 and C2). Moreover, it maintains efficiency at slightly lower temperatures and it offers a better mass-balance compared to reagent-assisted decarboxylation methods. This protocol is also scalable to multi-gram quantity (Fig. 4).
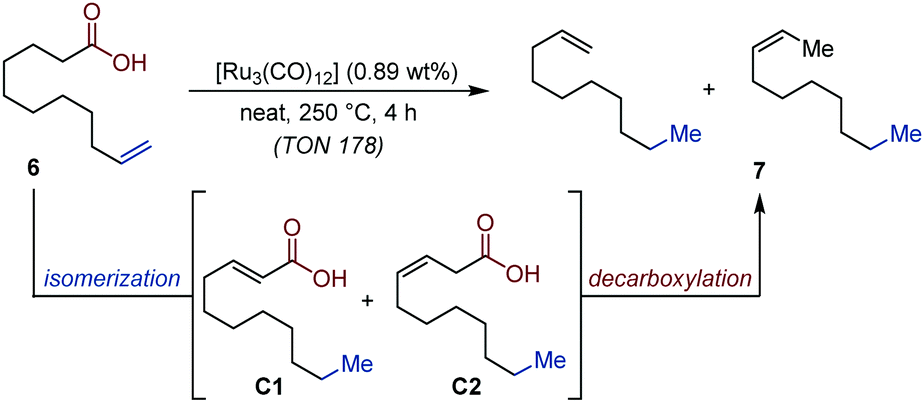 | ||
| Fig. 4 Ru-Catalyzed auto-tandem isomerization/decarboxylation of 10-undecenoic acid into decenes. | ||
Formation of a styrenyl intermediate by conjugative isomerization of a terminal alkene to a distant arene is a robust strategy to relocate an alkene with high positional fidelity. Liu, Chen and co-workers developed an auto-tandem Pd-catalyzed isomerization/Wacker oxidation of terminal alkenes (8) using tert-butyl hydrogen peroxide as oxidant, which operates at room temperature within short reaction times (Fig. 5).23
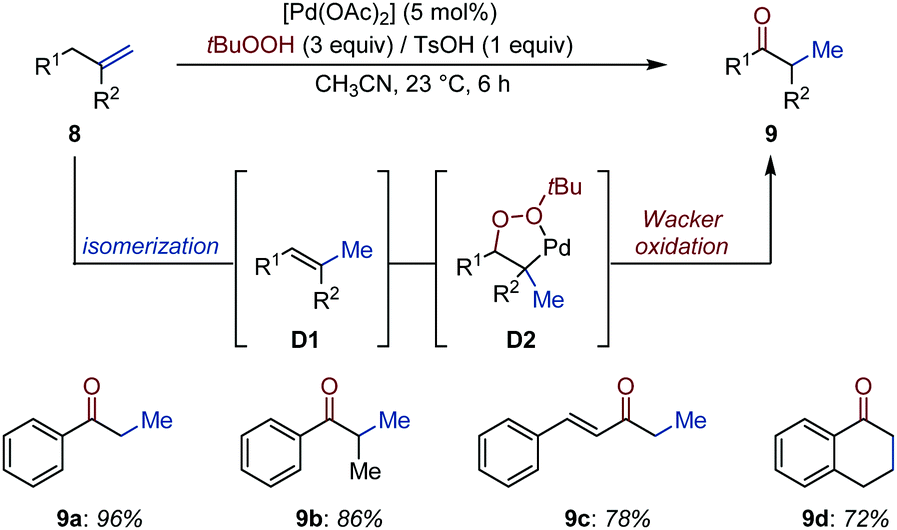 | ||
| Fig. 5 Pd-Catalyzed auto-tandem isomerization/Wacker oxidation of terminal alkenes. | ||
The isomerization catalyst – obtained by addition of 1 equivalent of TsOH to [Pd(OAc)2] (5 mol%) – converts allyl(hetero)aryls into the thermodynamically more stable vinyl(hetero)arenes (D1) by migrating the alkene over 1C atom.24 The in situ generated alkene was proposed to produce a peroxypalladacycle (D2), which degrades into the desired carbonyl compound via heterolytic O–O bond cleavage.25 The method was applied to a dozen of examples and the products were usually isolated in high to very high yield.
Stoltz and co-workers showed that synthetically useful carboxylic acids are accessible by an assisted-tandem 1C isomerization/oxidation of terminal olefins using Grubbs second-generation (Ru2) as transition metal precursor (Fig. 6).26 Their strategy relies on the formation of thermodynamically more stable internal olefins followed by oxidative cleavage of the repositioned unsaturation. Using Nishida's protocol (vide infra), the initial Ru–alkylidene complex Ru2 is converted in situ into a [Ru–H] isomerization catalyst Ru3.27 By a simple solvent switch once the CC bond migration is complete, the metal hydride is converted in situ with NaIO4 into an ill-defined ruthenium–oxo complex Ru4 (presumably resembling [RuO4]) for the subsequent oxidation of the vinyl intermediate into carboxylic acids. The overall transformation is a formal two-carbon scission, which provides expedient access to enantioenriched tri- and tetrasubstituted α-amino acids. Among other examples, the utility of the method was further established with the synthesis of (R)-α-ethyl-ornithine (12) in an elegant sequence that combines the Ru-catalyzed tandem isomerization/oxidation with a Curtius rearrangement followed by a simple hydrolysis.
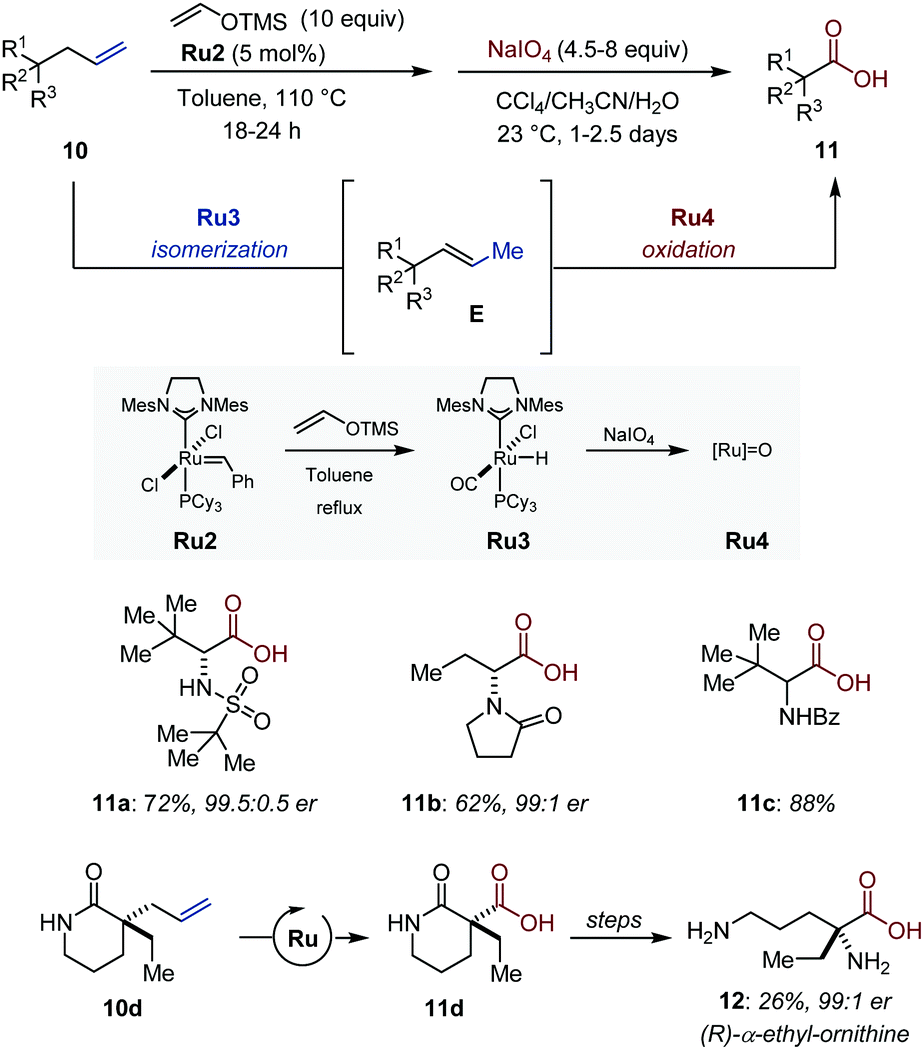 | ||
| Fig. 6 Ru-Catalyzed assisted-tandem isomerization/oxidation of terminal olefins. | ||
Metal-catalyzed isomerization/hydroformylation processes have been the subject of numerous investigations.13 These systems typically rely on orthogonal-tandem catalysis, where an alkene isomerization catalyst produces statistical mixtures of internal and terminal olefins. A second catalyst reacts with the thermodynamically less stable terminal alkenes, and constantly shifts the equilibrium towards what appears to be the contra-thermodynamic product. Recently, Vorholt showed that excellent linear/branched selectivity (14, rr 10.1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) could be achieved with methyl oleate 13 by associating the Pd-catalyst employed by Gooßen for orthogonal-tandem isomerization/metathesis with the well-established [(biphephos)Rh] hydroformylation catalyst (Fig. 7A).28,29 Key to the success of this tandem process is the ability of the Rh-catalyst to react exclusively with terminal olefins under the reaction conditions.30
:1) could be achieved with methyl oleate 13 by associating the Pd-catalyst employed by Gooßen for orthogonal-tandem isomerization/metathesis with the well-established [(biphephos)Rh] hydroformylation catalyst (Fig. 7A).28,29 Key to the success of this tandem process is the ability of the Rh-catalyst to react exclusively with terminal olefins under the reaction conditions.30
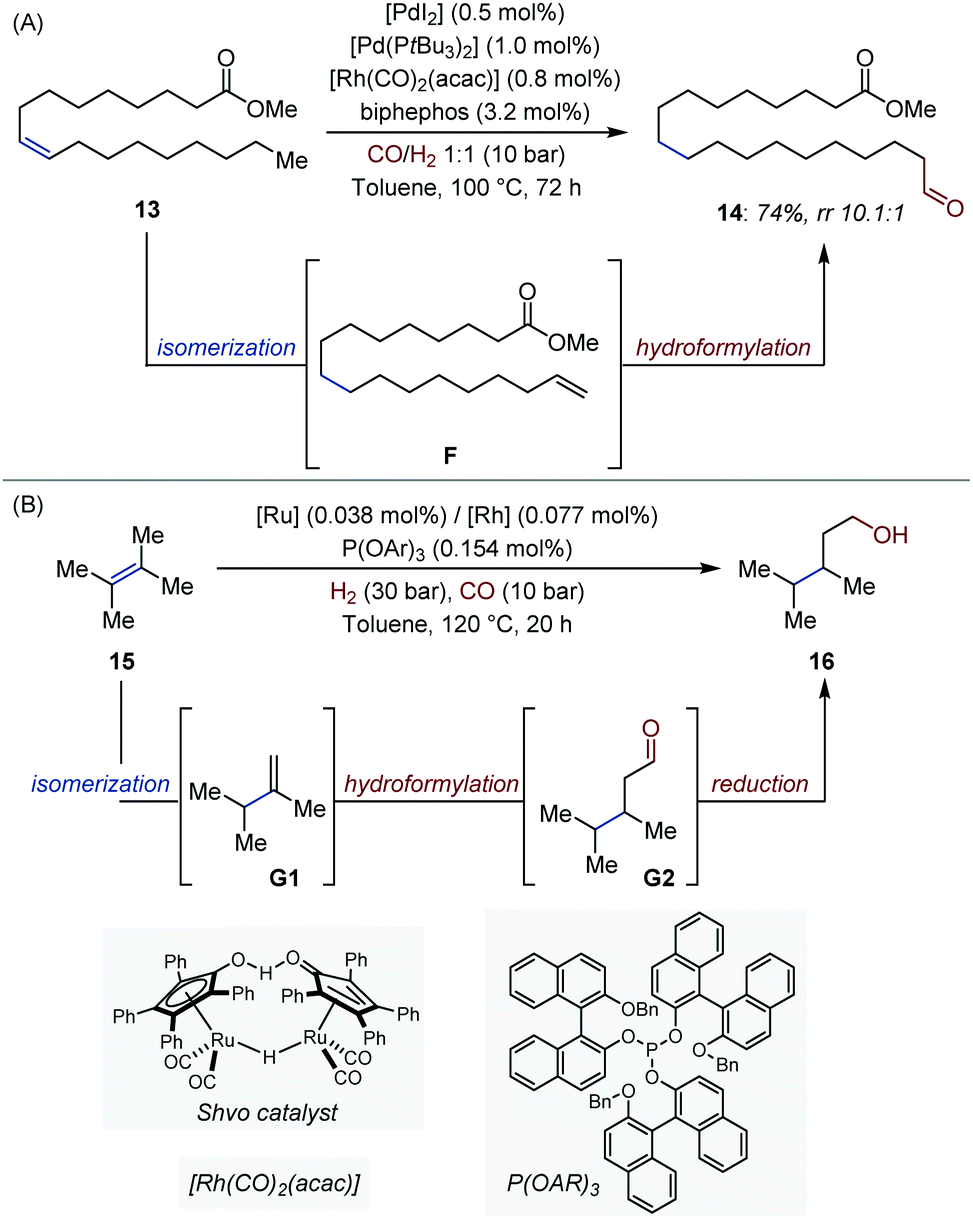 | ||
| Fig. 7 (A) Orthogonal-tandem isomerization/hydroformylation of methyl oleate. (B) Orthogonal-tandem isomerization/hydroformylation of 2,3-dimethylbut-2-ene. | ||
In 2018, the Beller and Pereira groups extended a [Ru/Rh] bimetallic system developed by Nozaki and co-workers five years earlier to tetrasubstituted olefins, a particularly challenging sub-class of alkenes (Fig. 7B).31,32 The system consists of a succession of three non-interfering catalytic cycles (isomerization, hydroformylation, reduction). Similar cooperative effects as in the original report were observed between the Rh and Ru catalysts. High yield, chemo- and regio-selectivity were obtained in many cases, including for stereochemically complex steroid derivatives.
2.2 Domino processes
Upon isomerization, an alkene can be strategically positioned to engage in a subsequent pericyclic transformation. In this context, diastereomerically pure annulenes (18) and cycloheptenones (19) were prepared through an expedient Ir-catalyzed isomerization/Cope rearrangement by the Marek group, starting from readily accessible dialkenyl cyclopropanes or alkenyl silylated cyclopropanols (17) (Fig. 8).33 While the isomerization was found to proceed at room temperature, the pericyclic rearrangement was thermally triggered by heating up the system at 85 °C. These domino procedures appeared to be highly functional group tolerant and afforded densely functionalized bicyclic compounds in moderate to good yield. The authors proposed that the high level of diastereoselectivity obtained originated from excellent control of the double-bond geometry during Ir-catalyzed chain walking, a process sustainable over up to 10 carbon atoms (18b).34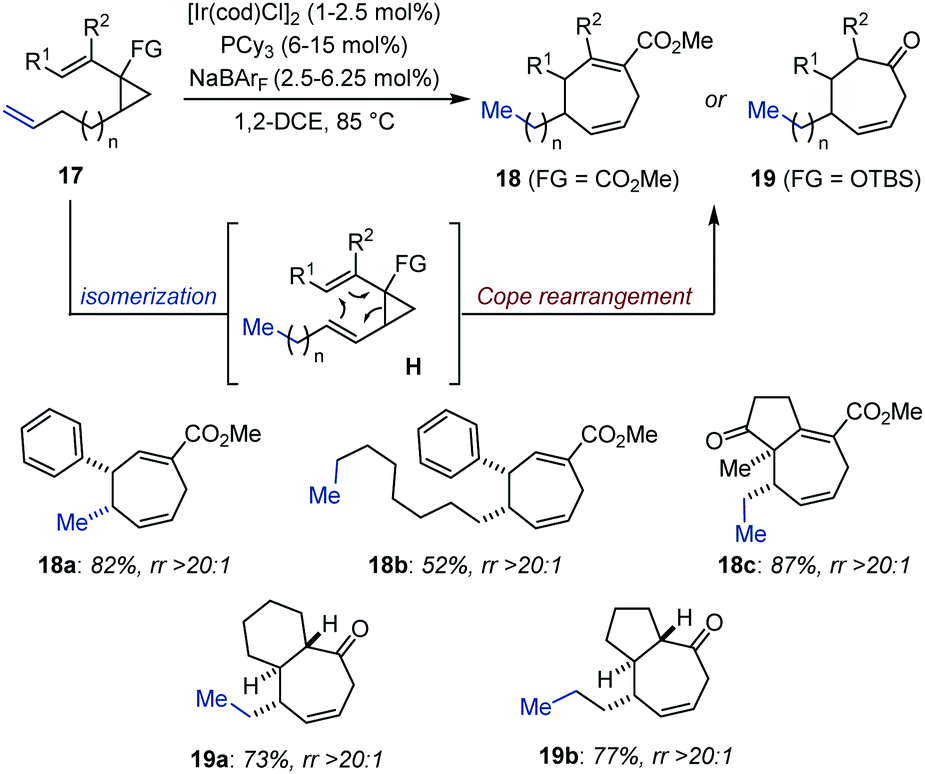 | ||
| Fig. 8 Ir-Catalyzed domino olefin migration/Cope rearrangement. | ||
2.3 Sequential processes
A series of remote functionalizations based on sequential multimetallic catalytic reactions has been reported by Mazet and co-workers.35 The reactions are triggered by hydrometalation of α,β-unsaturated esters using [Pd–H] or [Ru–H] isomerization catalysts and are driven by the formation of thermodynamically more stable (hetero)vinyl arenes (Fig. 9). Intermediates such as I were generated with high levels of stereoselectivity and were engaged in a subsequent functionalization reaction in the same reaction vessel. Several [Pd/Cu] catalytic systems were developed. Specifically, an isomerization/enantioselective α-borylation, an isomerization/β-borylation and an isomerization/enantioselective α-amination provided access to multifunctional compounds with excellent levels of regio- and/or enantiocontrol. The authors showed that the combination of existing protocols is not necessarily trivial, in particular in the context of enantioselective catalysis.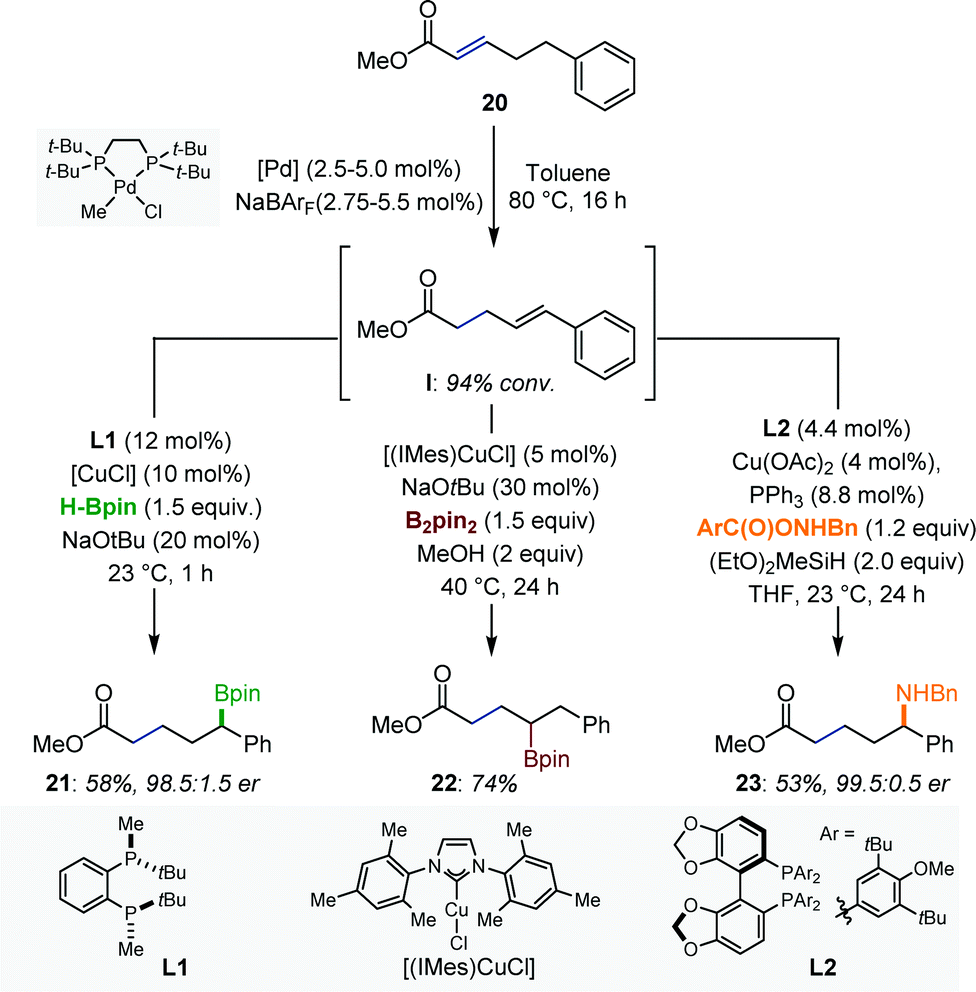 | ||
| Fig. 9 Remote functionalization of α,β-unsaturated esters by sequential multimetallic catalysis. | ||
Finally, difunctionalization can occur simultaneously at the benzylic and homobenzylic positions by combining the deconjugative isomerization with Sharpless asymmetric dihydroxylation (SAD) using either [Pd/Os] or [Ru/Os] couples. Starting from a simple α,β-unsaturated ester, the isomerization/dihydroxylation sequence followed by in situ lactonization delivered a naturally occurring γ-butyrolactone in enantiopure form.
3. Enol ethers as transient intermediates
Enol ethers are synthetically useful entities but their inherent reactivity poses several challenges for their preparation and isolation.36 Catalytic approaches, which consist in generating enol ethers from stable precursors prior to engaging them in situ in a subsequent catalytic reaction, are therefore attractive.3.1 Tandem processes
In 1998, Crivello and Rajaraman reported a Co-catalyzed auto-tandem isomerization/polymerization of allyl ethers (Fig. 10).37 They found that in presence of a silane the precatalyst [Co2(CO)8] disproportionates into [HCo(CO)4] and [R3SiCo(CO)4]. Upon loss of carbon monoxide, the former readily produces [HCo(CO)3], which catalyzes the isomerization of allyl ethers into a mixture of cis- and trans-1-propenyl ethers. These were subsequently polymerized by the silyl-cobalt complex [R3SiCo(CO)4] at room temperature. Multifunctional allylic ethers were also polymerized by this method to produce cross-linked insoluble matrices. Finally, because polymerization was inhibited by Lewis bases, a cationic polymerization mechanism was proposed to be operating.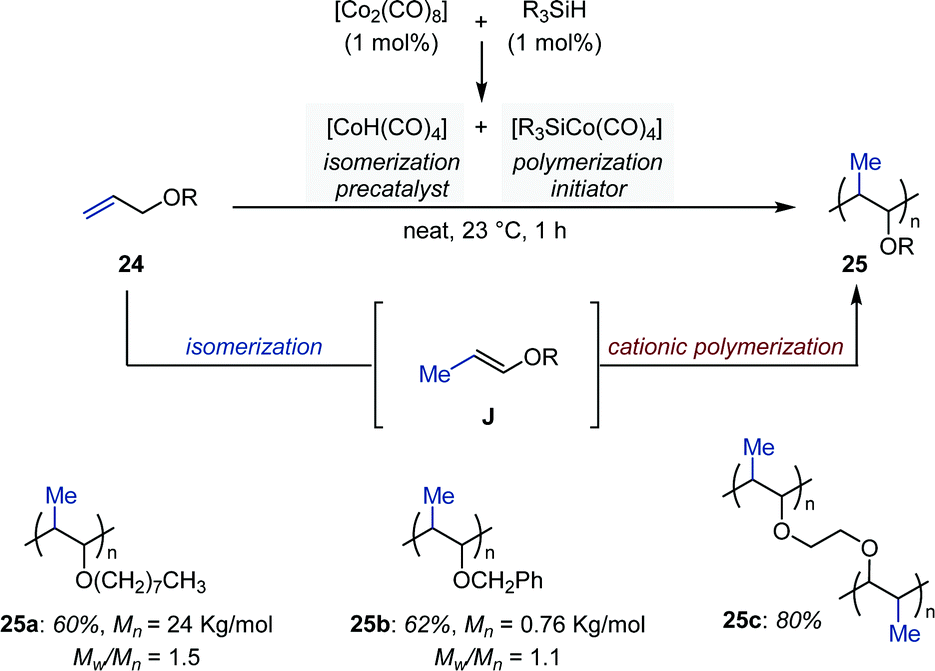 | ||
| Fig. 10 Co-Catalyzed auto-tandem isomerization/polymerization of allyl ethers. | ||
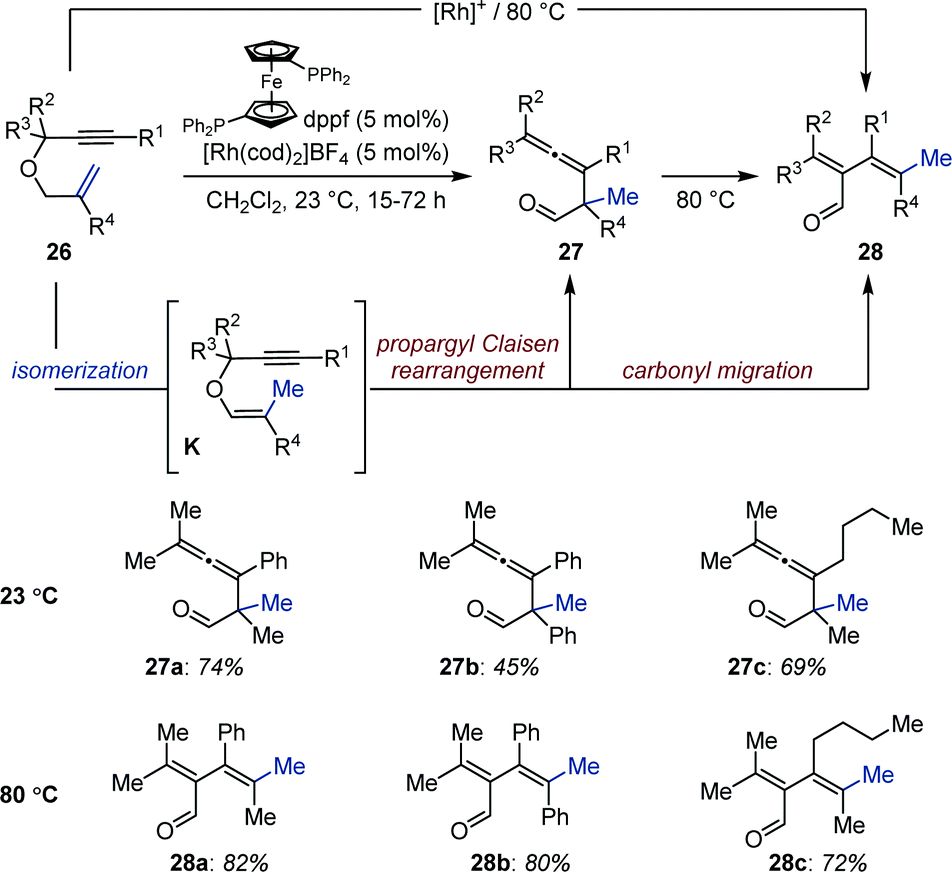
By exploiting the propensity of enol ethers to undergo intramolecular rearrangements when brought in close proximity of another reactive unsaturation, Tanaka and co-workers identified a cationic bisphosphine–rhodium complex ([(ddpf)Rh(cod)]BF4) for the isomerization/propargyl Claisen rearrangement of allyl propargyl ethers (26) to allenes (27) (Fig. 11).38 The reaction proceeds via enol ethers K generated by a Rh-catalyzed 1-carbon olefin isomerization. The system operates at room temperature and the products are isolated in good yield after reduction using sodium borohydride in methanol. The authors found that carbonyl migration occurred when the reactions were conducted at 80 °C to afford the corresponding dienals in high yield (28). However when primary and secondary propargyl ethers were investigated under these conditions the system afforded allenes (27) resulting from a propargyl-Claisen rearrangement. A sequence consisting in aldehyde C–H activation, hydrorhodation of the allene, carbonyl migration and reductive elimination was proposed to account for the formation of dienals 28. The intermediacy of enol ethers K and aldehydes 27 was confirmed by monitoring the reaction by 1H NMR spectroscopy.
 | ||
| Fig. 11 Thermally-controlled Rh-catalyzed isomerization/propargyl Claisen rearrangement and isomerization/propargyl Claisen rearrangement/carbonyl migration reactions. Allenyl aldehydes were isolated as alcohol after reduction with sodium borohydride in methanol. | ||
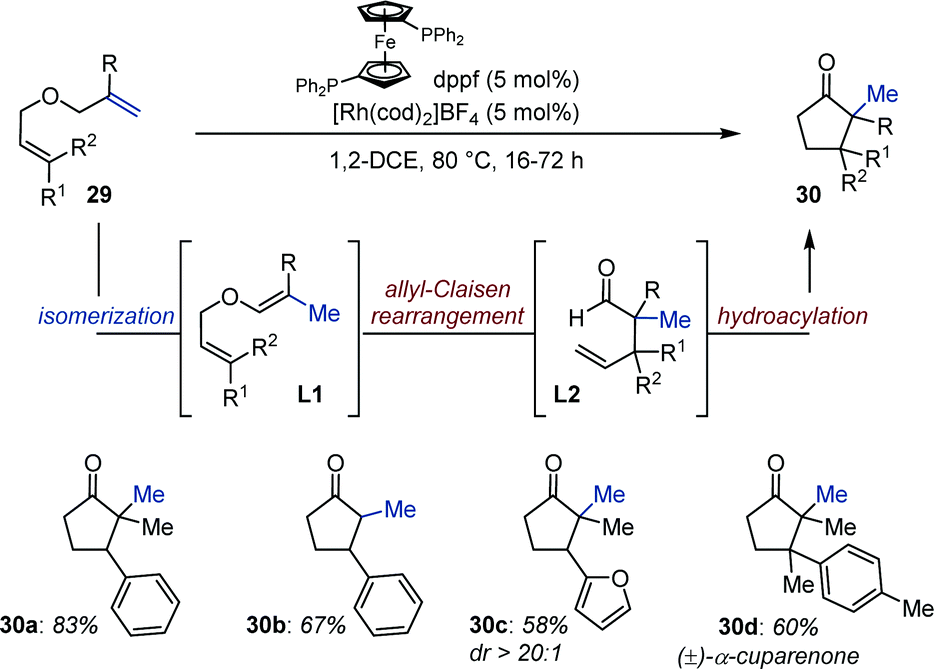
As a direct continuation of this study and inspired by seminal work from Eilbracht,39 Tanaka disclosed a Rh-catalyzed olefin isomerization/allyl Claisen rearrangement/intramolecular hydroacylation of diallyl ethers (29) (Fig. 12).40 The authors propose that chemoselective isomerization of the 1,1-disubstituted alkene in 29 produces alkenyl ethers L1, which subsequently undergo allyl Claisen rearrangement. Rhodium-catalyzed intramolecular hydroacylation of the pendant allyl ether moiety in L2 delivers substituted cyclopentanones (30). Of note, both L1 and L2 were observed by conducting the reaction at room temperature. Isotopically labelled substrates provided additional evidences in support of this mechanistic hypothesis. Overall, the catalytic system tolerates a variety of functional group and is compatible with heteroaryl-containing bis-allyl ethers. Cyclopentanones with two contiguous quaternary centers are accessible using this method as established with the synthesis of a naturally occurring terpenoid rac-α-cuparenone 30d.
 | ||
| Fig. 12 Rh-Catalyzed auto-tandem isomerization/Claisen rearrangement/hydroacylation of bis-allyl ethers. | ||
3.2 Domino processes
In 1977, Reuter and Salomon showed that non-symmetrical bis-allyl ethers (31) were competent substrates for the Ru-catalyzed isomerization/Claisen rearrangement (ICR) to produce γ,δ-unsaturated aldehydes and ketones (33) (Fig. 13A).41 Metal-catalyzed isomerization of the less substituted alkene occurs preferentially and poises the CC bond in a position favorable for a subsequent thermal Claisen rearrangement (M). The scope of this method was later extended by the Dixneuf group with the use of in situ generated [(NHC)Ru] complexes.42 Cadierno showed that similar reactions could be performed in refluxing water employing a Ru–arene precatalyst.43 Between 2003 and 2007, Nelson developed a series of elegant and practical protocols using iridium catalysis and optically active bis-allyl ethers (31) to afford syn-2,3-dialkyl-4-pentenal derivatives (33) with high diastereoselectivity and enantiospecificity (Fig. 13B).44 While the transition metal catalyzed isomerization occurs readily at room temperature, the [3,3] sigmatropic rearrangement requires temperatures that never exceed 40 °C. This contrasts markedly with the ruthenium-based catalytic systems for which temperatures ranging from 100 to 200 °C are necessary.41–43 Of important note, addition of triphenylphosphine (3 mol%) was hypothesized to attenuate the Lewis acid character of iridium in order to avoid epimerization of the aldehyde α-stereocenter. The origin of the high levels of diastereocontrol were attributed to the highly E-selective isomerization, which occurred exclusively at the less substituted allyl ether. The same group published a similar method for the construction of vicinal quaternary and tertiary stereocenters starting from 1,1-disubstituted allylic ethers, and later showed that anti-2,3-disusbtituted-pentenals were accessible by means of a simple Pd-catalyzed Claisen rearrangement protocol.44d,45 The efficiency of the iridium-catalyzed ICR protocol developed by Nelson was recognized by the Trost and Chen groups, who employed it in the total syntheses of communiol A and (+)-antroquinonol respectively.46 Of note, in addition to ruthenium and iridium, Stork, and Echavarren reported examples of domino isomerization/Claisen rearrangement using palladium catalysts.47
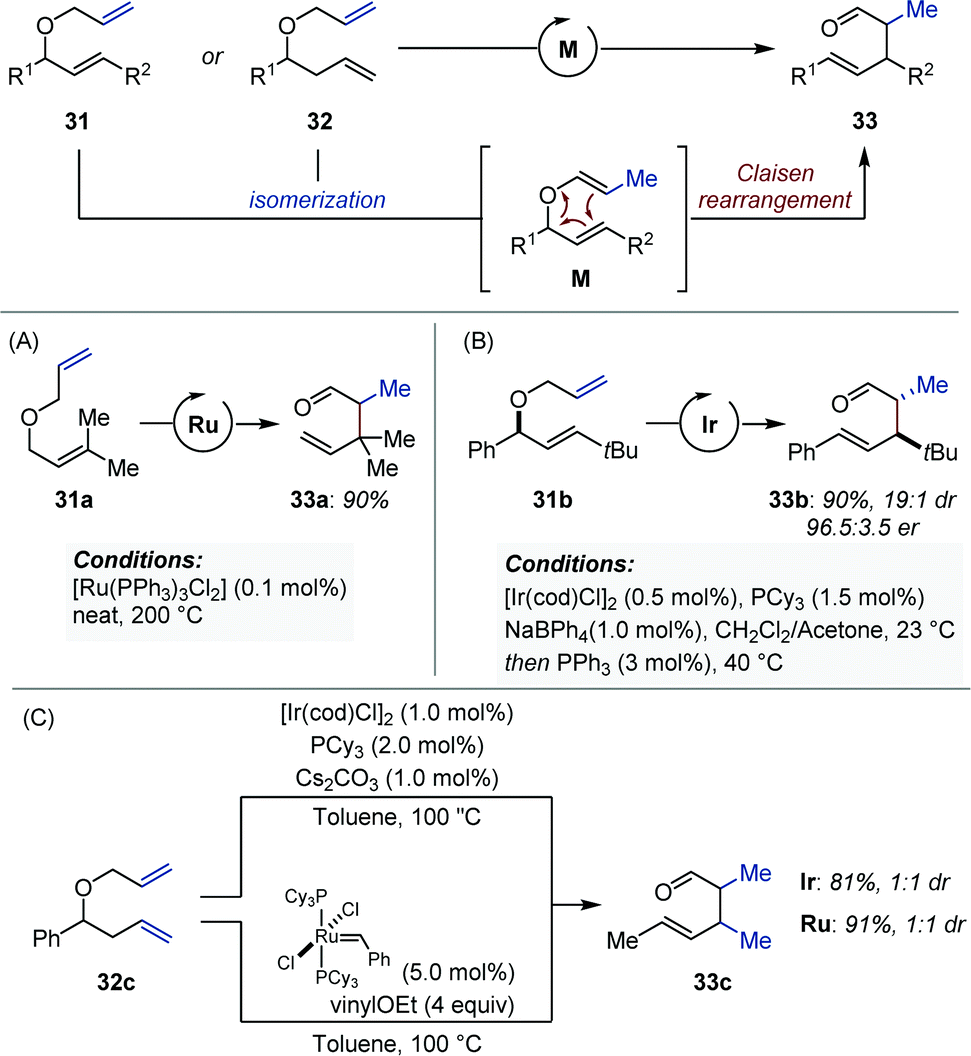 | ||
| Fig. 13 Metal-catalyzed isomerization/Claisen rearrangement of non-symmetrical bis-allyl ethers. | ||
Ishii and Schmidt independently achieved the domino metal-catalyzed isomerization/Claisen rearrangement of unsymmetrical allylic/homoallylic ethers (32) using iridium and ruthenium catalysts respectively (Fig. 13C).48,49 Although the products are identical to those obtained from bis-allyl ethers (vide supra), these reactions involve a double CC bond migration prior to the thermal [3.3] sigmatropic rearrangement. The absence of diastereoselectivity in either case can be attributed to the lack of stereocontrol upon olefin isomerization.
3.3 Sequential processes
While tandem and domino reactions involving enol ethers occur preferentially intramolecularly, intermolecular transformations have been achieved by sequential multi-catalysis. Romano and Mazet identified conditions for the one-pot multimetallic conversion of allyl methylethers (34) in alkenes (35) with high degree of stereocontrol (Fig. 14).50 In the first step, a cationic iridium precatalyst is activated by molecular hydrogen and isomerizes 34 in vinyl methylethers (N) at room temperature. Two complementary [(NHC)Ni] catalysts were identified for the subsequent cross-coupling between the in situ-generated C–O electrophiles and a variety of Grignard reagents at elevated temperatures.51 The method is compatible with sensitive functional groups and a several olefinic substitution patterns. The newly generated alkenes were systematically obtained with high to very high levels of stereocontrol (E/Z: 4.5:1 to >25:1). Control experiments showed that the Ir-catalyzed process is stereoconvergent in nature and that the stereochemistry set during isomerization is preserved upon cross-coupling. Finally, the one-pot isomerization/cross-coupling sequence was conducted with a prochiral substrate and a chiral iridium precatalyst (Ir2), originally developed for the isomerization of allylic alcohols into aldehydes.52 The final olefinic products were isolated in moderate yield but with particularly high levels of stereo- and enantiocontrol (35e).50
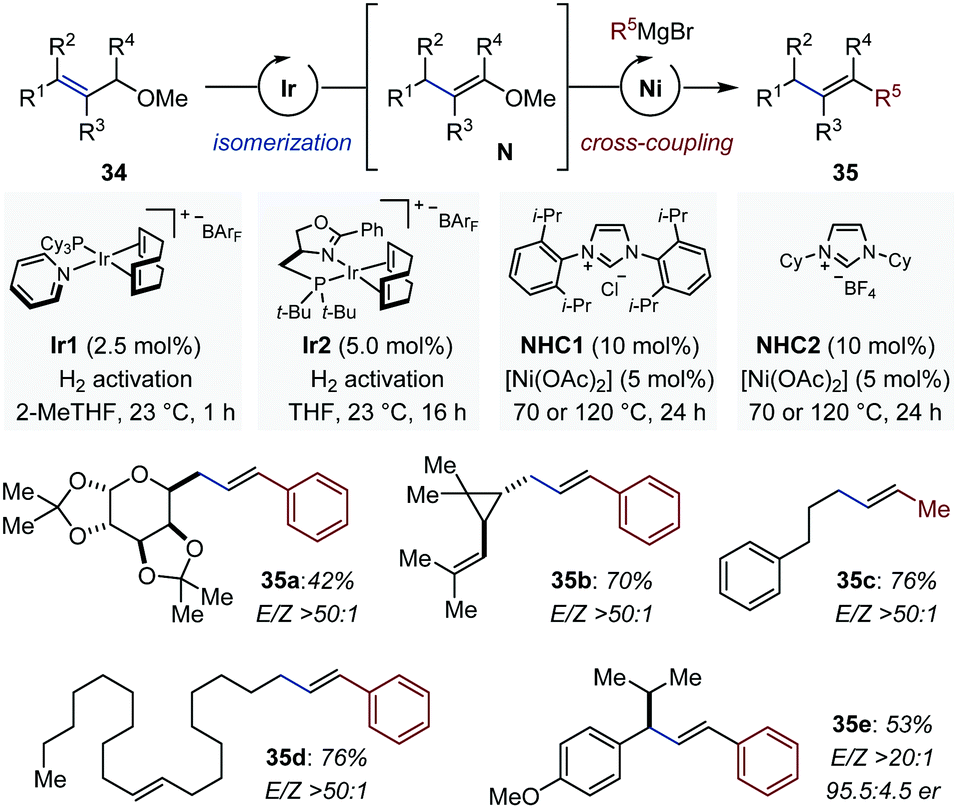 | ||
| Fig. 14 One-pot sequential Ir-catalyzed isomerization/Ni-catalyzed cross coupling to convert allyl methylethers in substituted alkenes. | ||
The inability of the iridium catalysts to displace CC bonds over more than one carbon atom prompted the same group to evaluate alternative systems for the development of a related long-range isomerization/cross-coupling sequence (Fig. 15).
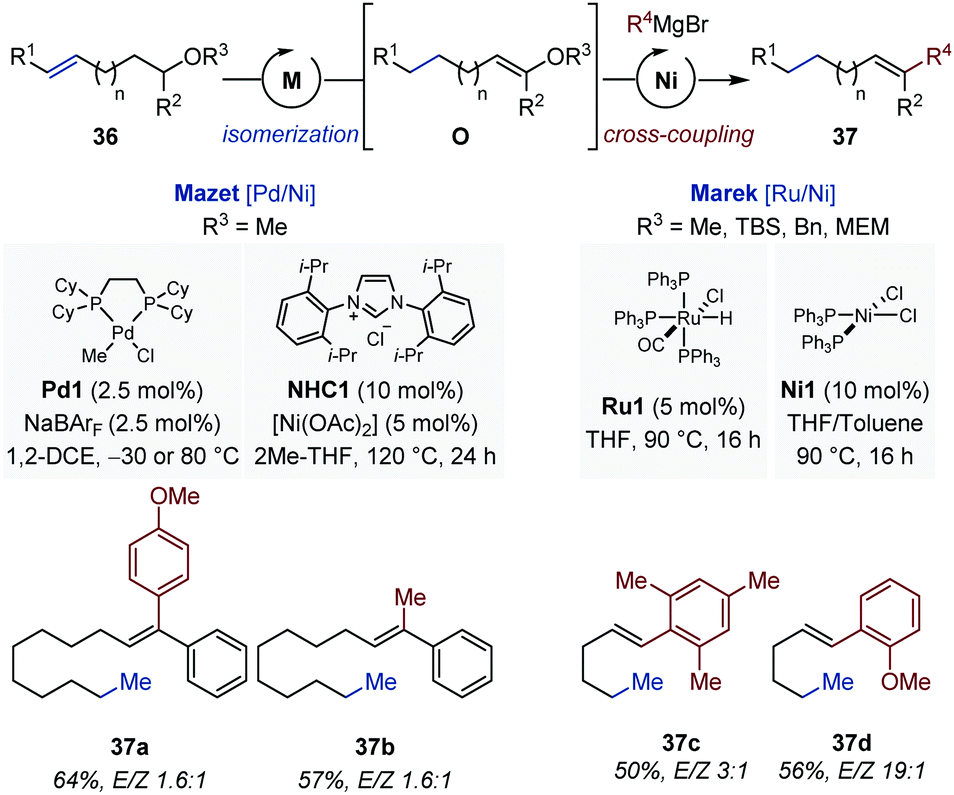 | ||
| Fig. 15 One-pot [Pd/Ni]- and [Ru/Ni]-catalyzed remote isomerization/cross coupling sequences for the synthesis of (poly)substituted alkenes. | ||
Thus, a [Pd/Ni] catalytic sequence was identified where the palladium catalysts enabled isomerization of the remote CC bond in several alkenyl methyl ethers (36).50,53 Solvent exchange between the isomerization step and the cross-coupling reaction was necessary. The final di- and trisubstituted alkenes were obtained in good yield but with low level of stereocontrol (originating from the isomerization step). Concomitantly, the Marek group reported a complementary [Ru/Ni] catalytic combination for the remote isomerization/cross-coupling of alkenyl benzyl-, silyl-, methoxyethoxy-methyl ethers leading to 1,2-disubstituted alkenes (37).54 While the isomerization was performed in refluxing THF, addition of toluene was found to be beneficial for the cross-coupling reaction (1:1 v/v). Only C(sp2) Grignard reagents were disclosed, and higher levels of stereocontrol were achieved in comparison with the [Pd/Ni] system (1:1 to 19:1).
Subsequently, the same group disclosed a related sequential catalytic isomerization/Suzuki cross-coupling strategy in which the vinyl boronic ester intermediates generated via isomerization act as a transmetalating agent rather than electrophilic coupling partner.55 Overall, 1,2-disubstituted styrenes were prepared with similar efficiency and stereocontrol than with the sequential alkenyl ether isomerization/Kumada cross-coupling approach.
4. Enamines as transient intermediates
4.1 Tandem processes
Enamines are typically prepared by Schiff-base condensation between a carbonyl compound and a secondary amine. Nonetheless, they can also be accessed through transition metal-catalyzed isomerization of allylic amines. In this context, the Hull laboratory developed an elegant enantioselective auto-tandem process consisting of Rh-catalyzed isomerization of allylamines (38), followed by enamine exchange and subsequent Rh-catalyzed oxidation to chiral β-branched amides (39) (Fig. 16).56 While the first step takes inspiration from the Rh-catalyzed enantioselective isomerization optimized by the Takasago Company for the production of menthol,57 the exchange process is based on the Rh-catalyzed conversion of allylic alcohols into amides under oxidative conditions identified in their own laboratory.58 In the present system, under basic reaction conditions the in situ generated enamines (P1) is converted to a hemiaminal (P2) in presence of an external amine nucleophile and water. Rh-catalyzed oxidation of P2 into amides 39 is proposed to occur next. A sacrificial hydrogen acceptor is used regenerate the initial Rh(I) complex. Overall, this approach enables the installation of a stereocenter and an amide functionality in a single step under mild conditions from readily available starting material. Excellent diastereo- and enantioselectivity were obtained using a broad set of allylic amines and amine nucleophiles such as primary amines, secondary amines or even anilines. In the following years, a similar protocol was documented using primary, secondary or tertiary alcohols as external nucleophiles giving access to β-branched esters in excellent yield and enantioselectivity starting from simple allylic amines.59 Finally, an assisted tandem Rh-catalyzed isomerization/reductive amination of allylic amines was also reported.60 The rhodium isomerization catalyst was converted into a hydrogen transfer catalyst by addition of formic acid at the final stage of the tandem process. Overall, this method delivers enantioenriched β-branched amines.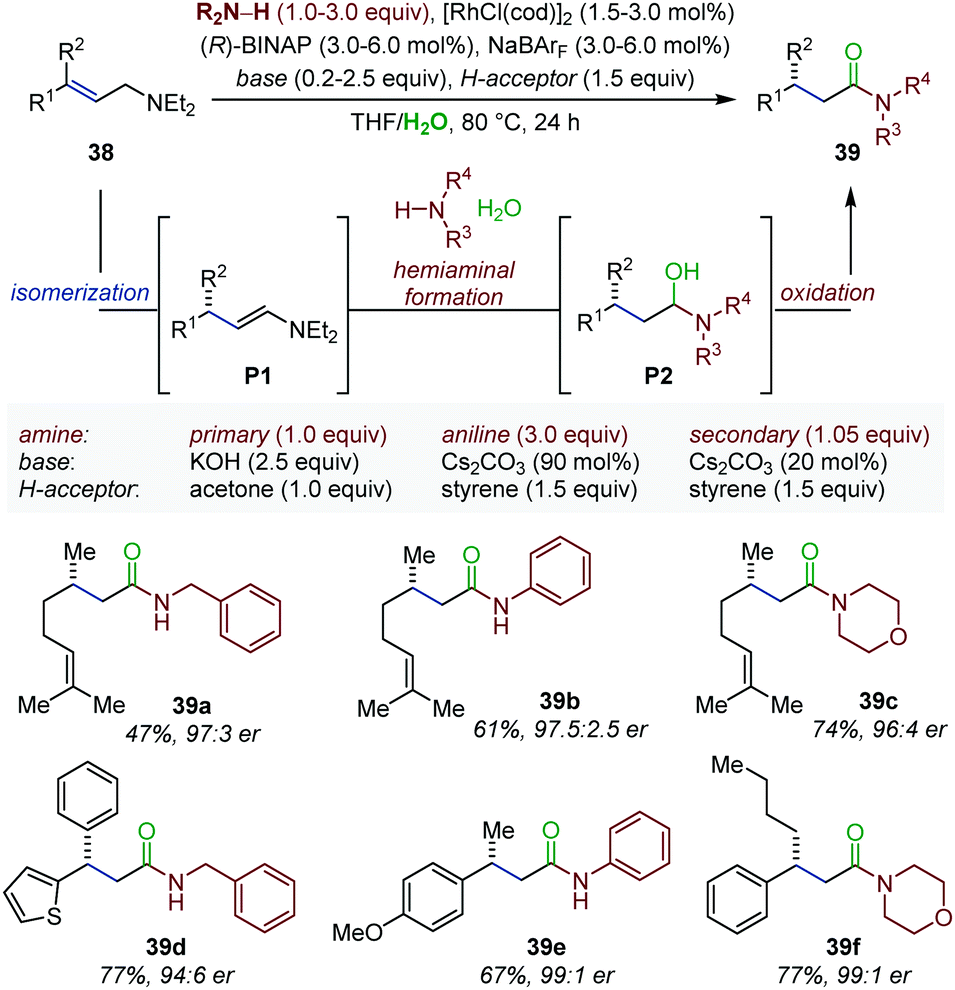 | ||
| Fig. 16 Enantioselective auto-tandem Rh-catalyzed isomerization/oxidation of allylic amines to β-chiral amides. | ||
4.2 Sequential processes
While in the auto-tandem approaches devised by the Hull laboratory, enamines are employed in intermolecular reactions, Arisawa and Nishida showed that sequential processes offer the possibility to exploit transient enamines for intramolecular heterocycle synthesis. In 2006, they established that a well-defined ruthenium hydride Ru3 is quantitatively generated upon treatment of Grubbs second-generation catalyst Ru2 with vinyloxytrimethylsilane (Fig. 6).27 In their seminal study, an isomerization/metathesis sequence using both ruthenium complexes for the preparation of a handful of protected indoles was developed.Later, Arisawa reported that commercially available ruthenium hydride Ru1 could be associated with Hoveyda–Grubbs metathesis catalyst Ru5 in a sequence consisting of a one-carbon isomerization/ring-closing metathesis/oxidation/1,3-dipolar addition using benzoquinone to deliver isoindolo[1,2-a]isoquinoline 41 (Fig. 17).61 This particular example highlights the versatility of their sequential isomerization/metathesis approach for heterocycle synthesis starting from simple N-allyl anilines.
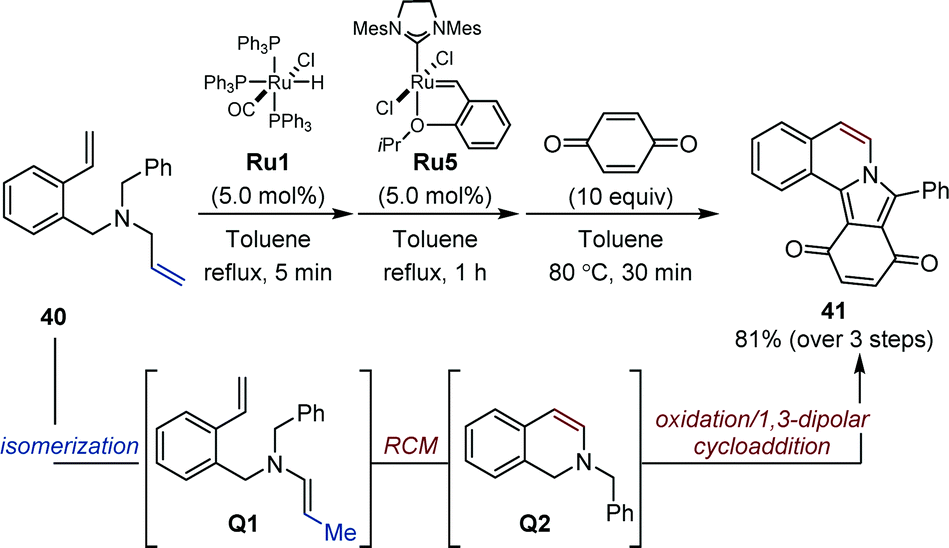 | ||
| Fig. 17 Sequential Ru-catalyzed isomerization/ring-closing metathesis/oxidation/1,3-dipolar cycloaddition. | ||
5. Carbonyls as transient intermediates
The conversion of allylic alcohols into carbonyl compounds is a cornerstone of transition metal-catalyzed olefin isomerization.52,62 Not only is the process thermodynamically favorable, but rapid increase in molecular complexity can be achieved from readily available starting materials. Several research groups have also combined this isomerization in one-pot processes by capitalizing on the ability of the carbonyl intermediate either to undergo α-functionalization or to direct C–H activation. Examples where the carbonyl intermediates are engaged in situ in reductive or oxidative transformations have also been reported.5.1 Tandem processes
Jun and co-workers reported a Rh-catalyzed auto-tandem isomerization/hydroacylation yielding non-symmetric aliphatic ketones (43) from simple allylic alcohols (42) and alkenes (Fig. 18).63 In the first step, aldehydes R1 are produced upon isomerization of 42. Schiff-base condensation with 2-amino-4-picoline in presence of benzoic acid gives aldimine R2in situ. Subsequent chelation-assisted hydroiminoacylation of the appropriate alkene followed by hydrolysis of the resulting ketamine affords the targeted ketone. Noticeably, this strategy (i) prevents aldol condensation, (ii) reduces catalyst deactivation upon decarbonylation, two processes often observed in hydroacylation employing aliphatic aldehydes. The reaction is critically influenced by the alkene substitution pattern. For instance, α,β-unsaturated ketones were observed as by-products in the isomerization/hydroacylation of trisubstituted allylic alcohols.64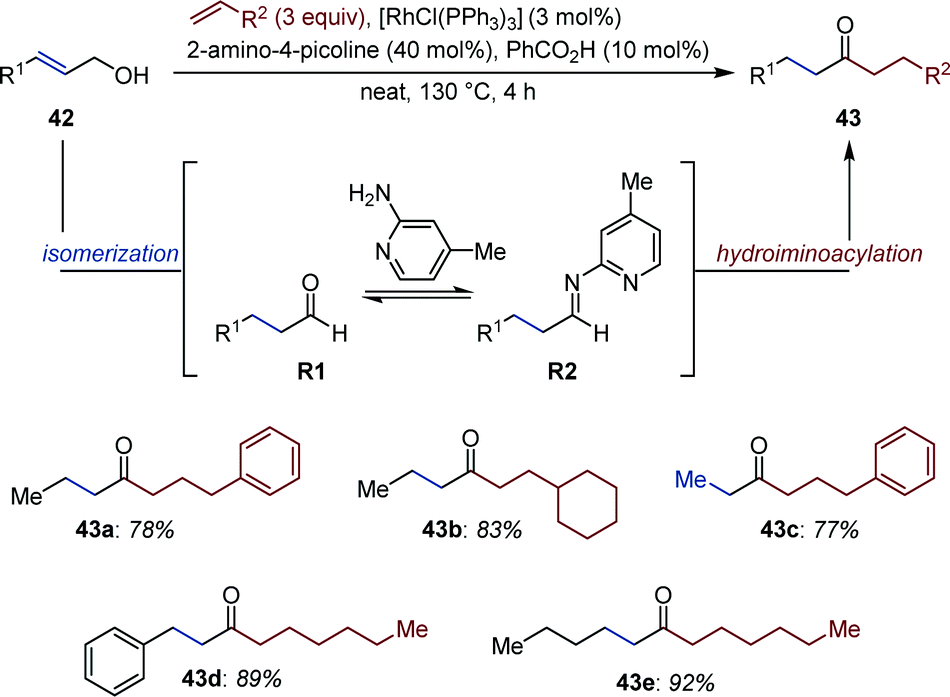 | ||
| Fig. 18 Rh-Catalyzed auto-tandem allylic alcohol isomerization/chelation-assisted hydroacylation. | ||
Bartoszewicz and Martín-Matute documented a Ru-catalyzed auto-tandem isomerization/C–H activation starting from secondary allylic alcohols.65 In a likely mechanistic scenario, the ketone intermediate (S1) generated upon isomerization directs the subsequent ortho-C–H activation (Fig. 19). Migratory insertion of the [Ru–H] (S2) across triethoxyvinylsilane, followed by reductive elimination affords 45. While sodium formate acts as both a base and a reducing agent, reproducibility issue were circumvented by addition of a catalytic amount of a bulky, electron-rich trialkylphosphine. The amount of double C–H activation product varied between ca. 1:1 to 1:0 depending upon the structure of the allylic alcohol (44). One example using a homoallylic alcohol was provided, indicating olefin migration can be sustained over several carbon atoms (45c).
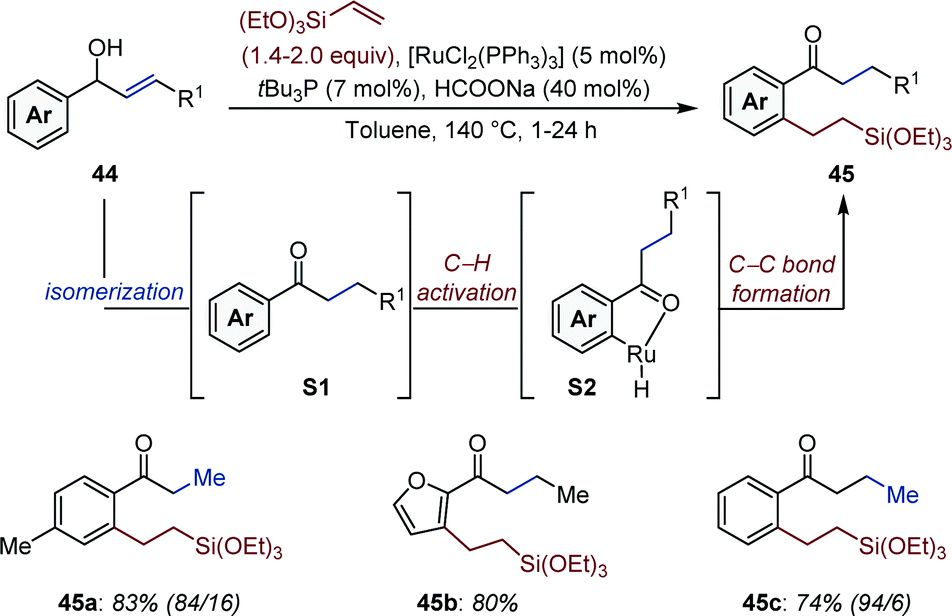 | ||
| Fig. 19 Ru-Catalyzed auto-tandem isomerization/C–H activation of secondary allylic alcohols. In parenthesis is given the ratio of the products of mono- and bis-functionalization. | ||
In the period 2007–2009, Cadierno, Gimeno and co-workers developed a series of Ru-catalyzed isomerization/transfer hydrogenation systems of primary and secondary allylic alcohols (Fig. 20).66 The tandem process consists first in the isomerization of the allylic alcohol (46), followed by transfer hydrogenation of the resulting carbonyl (T). The system operates under basic conditions using iso-propanol as hydrogen donor and [{(η6-C6Me6)RuCl(μ-Cl)}2] Ru6 as precatalyst and is applicable to a broad array of substrates, including derivatives with a trisubstituted olefin. The products are obtained in quasi-quantitative yield within relatively short reaction times. The overall transformation offers an attractive alternative to alkene hydrogenation because it circumvents the use of molecular hydrogen. It is also highly chemoselective as illustrated with the isomerization/transfer hydrogenation of geraniol leading exclusively to citronellol (47b). In the same contribution, the authors showed that the reaction could be carried out in water with similar efficiency when sodium formate was employed as hydrogen source. They later found that glycerol could act as both solvent and hydrogen donor.67 In a remarkable study, Bizet et al. disclosed an analogous tandem isomerization/transfer hydrogenation of γ-trifluoromethylated allylic alcohols using [(Ph3P)3RuCl2] as catalyst. The saturated alcohols were obtained in excellent yield and could be accessed from β-trifluromethylated enones with similar performances.68
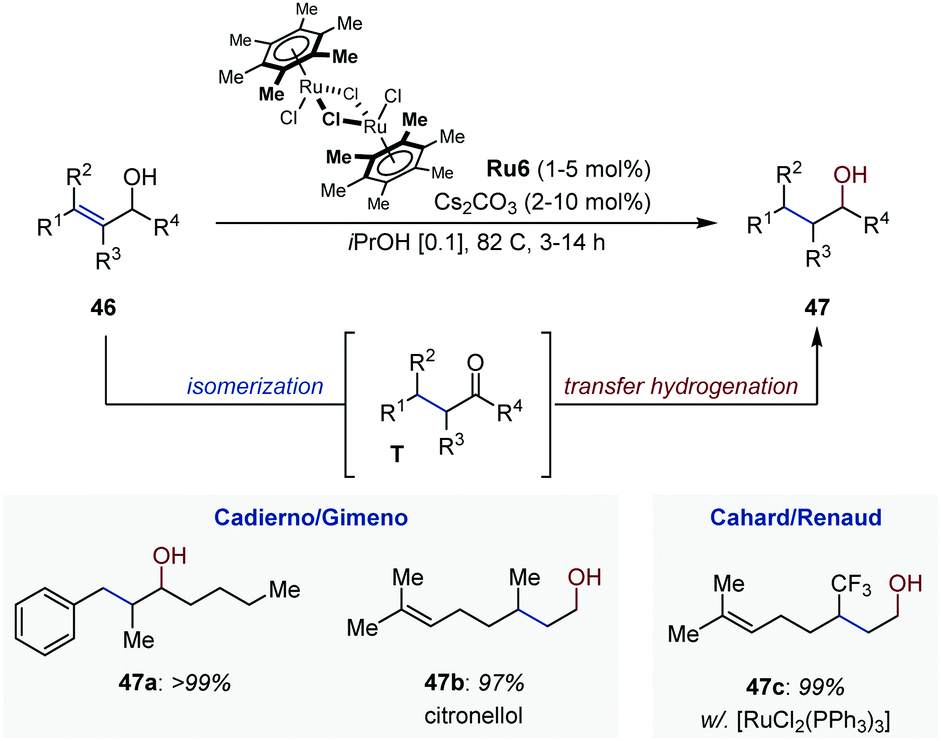 | ||
| Fig. 20 Ru-Catalyzed auto-tandem isomerization/transfer hydrogenation of allylic alcohols. | ||
In 2012, Sowa disclosed the first version of the Ru-catalyzed enantioselective isomerization/transfer hydrogenation of various allylic alcohols (Fig. 21).69 The highest enantioselectivities were obtained for geraniol and nerol for which (S)-Tol-Binap and (S,S)-iPrDuphos were found to be the most effective chiral ligands. In analogy with related hydrogenation reactions, the system was found to be stereospecific with (S)-Tol-Binap and stereoconvergent with (S,S)-iPrDuphos (Fig. 21).70 On the basis of deuterium labelling experiments, the authors proposed that the isomerization proceeds via a 1,3-H shift mechanism and is the enantio-determining step of the tandem process. The same group and the Adolfsson lab concurrently reported a related tandem isomerization/asymmetric transfer hydrogenation of secondary allylic alcohols using either diamine or α-amino acid hydroxyamide chiral ligands.71
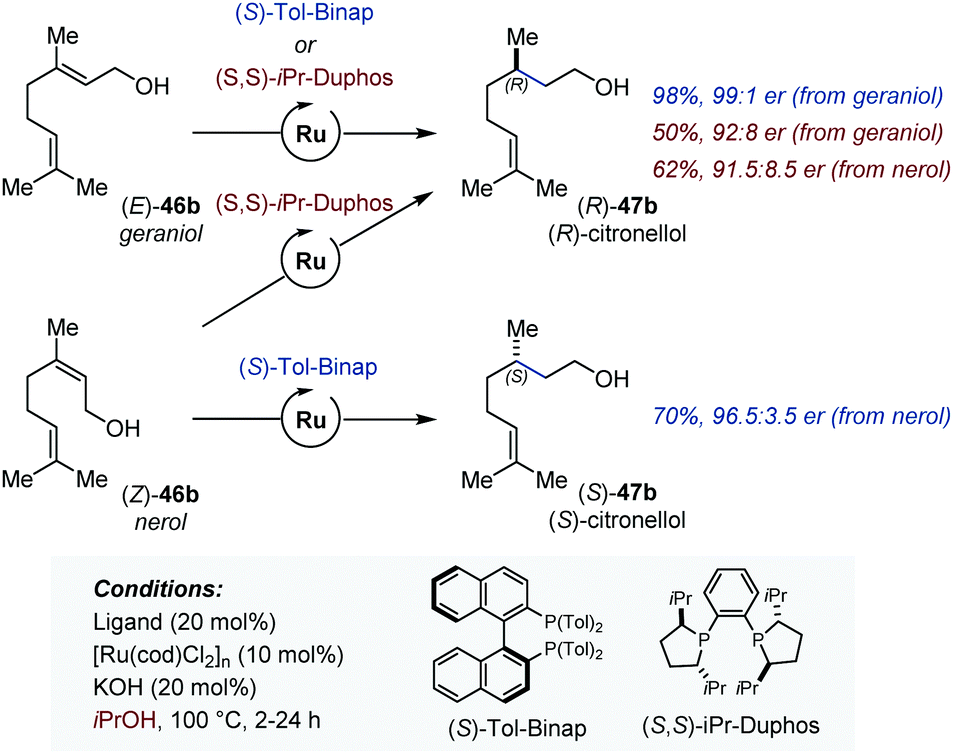 | ||
| Fig. 21 Ru-Catalyzed enantioselective auto-tandem isomerization/transfer hydrogenation of allylic alcohols. | ||
Bruneau established that saturated amines could be accessed from allylic alcohols via Ru-catalyzed auto-tandem isomerization/reductive amination of primary allylic alcohols using arene-ruthenium complex Ru7 at elevated temperatures (Fig. 22).72 Under the optimized reaction conditions, secondary amines were reacted in excellent yields, while primary anilines suffered from parasitic dialkylation. The reaction proceeds in presence of formic acid, which serves as reducing agent. Under the optimized reaction conditions, a broad range of primary and secondary amines was isolated in moderate to good yield. Of particular note, aniline 49e could be generated in 71% yield from a mixture of five different monoterpenoids. Beyond the synthetic interest of working with complex mixtures, this result indicates that hydrogen (auto-)transfer occurs from different alcohols and aldehydes and converges toward the formation of a single product. A related process was developed shortly after for allylic and homoallylic alcohols using a light-activated Fe catalyst and polymethylhydrosiloxane as reducing agent.73
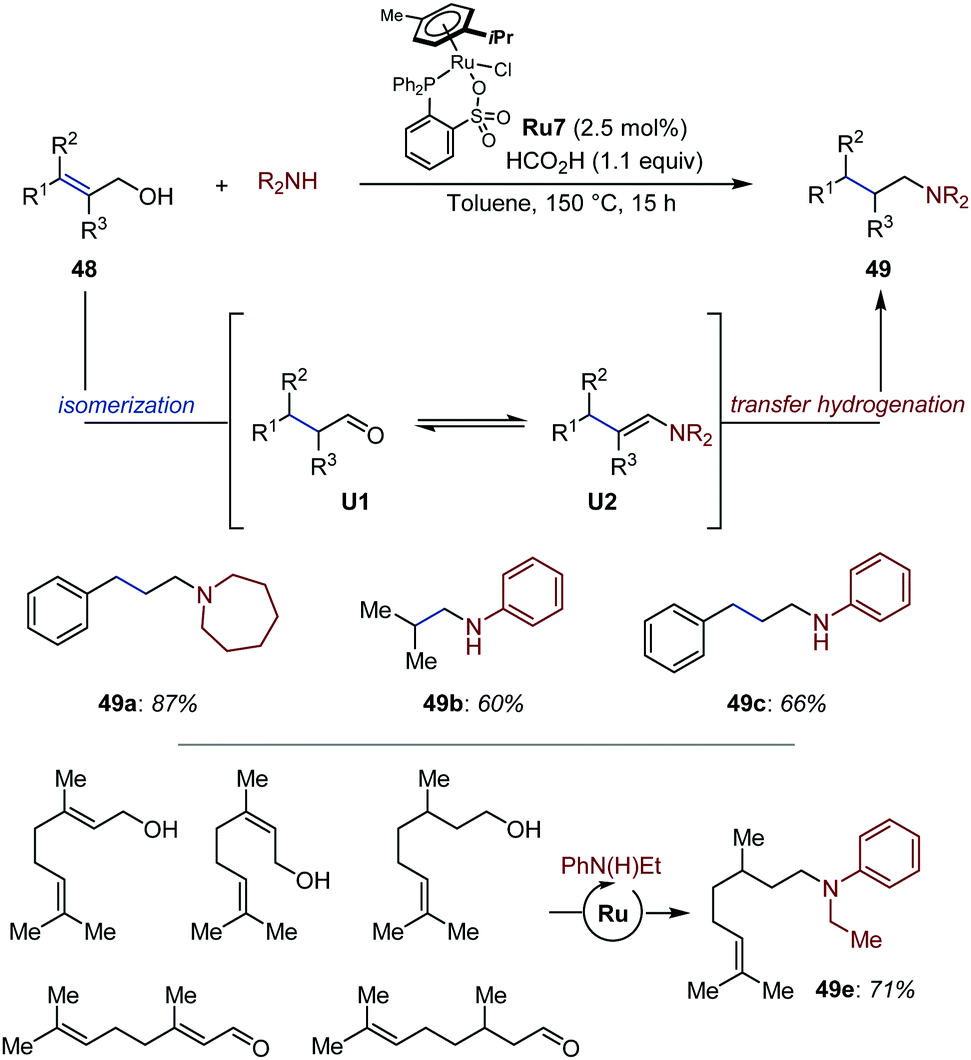 | ||
| Fig. 22 Ru-Catalyzed auto-tandem reductive amination of allylic alcohols. | ||
As part of their program directed to the synthesis of valuable amides from simple starting materials, Wu and Hull disclosed a Rh-catalyzed tandem oxidative amidation of allylic alcohols using a wide array of primary and secondary amines with acetone or styrene as hydrogen acceptor.58 The system served as a basis for the enantioselective tandem oxidative amidation of allylic amines presented in Section 4.1. Given the mechanistic similarities, it will not be discussed in further details in this section.
Based on a related strategy, Li and co-workers developed a Ru-catalyzed isomerization/acceptorless dehydrogenation of allylic alcohols for the synthesis of quinazolinones (51) using o-aminobenzamide nucleophiles (Fig. 23).74 The system is general and requires only low loading of the commercially available [(p-cymene)RuCl2]2 complex under microwave irradiations (130 °C). A plausible mechanism has been proposed based on a series of control experiments. After initial Ru-catalyzed isomerization, condensation of o-aminobenzamide to the resulting aldehydes (V1) generates 2,3-dihydroquinazolinones (V2). Observation of gas evolution and NMR studies provided evidences for a Ru-catalyzed acceptorless dehydrogenation producing quinazolinones (51).
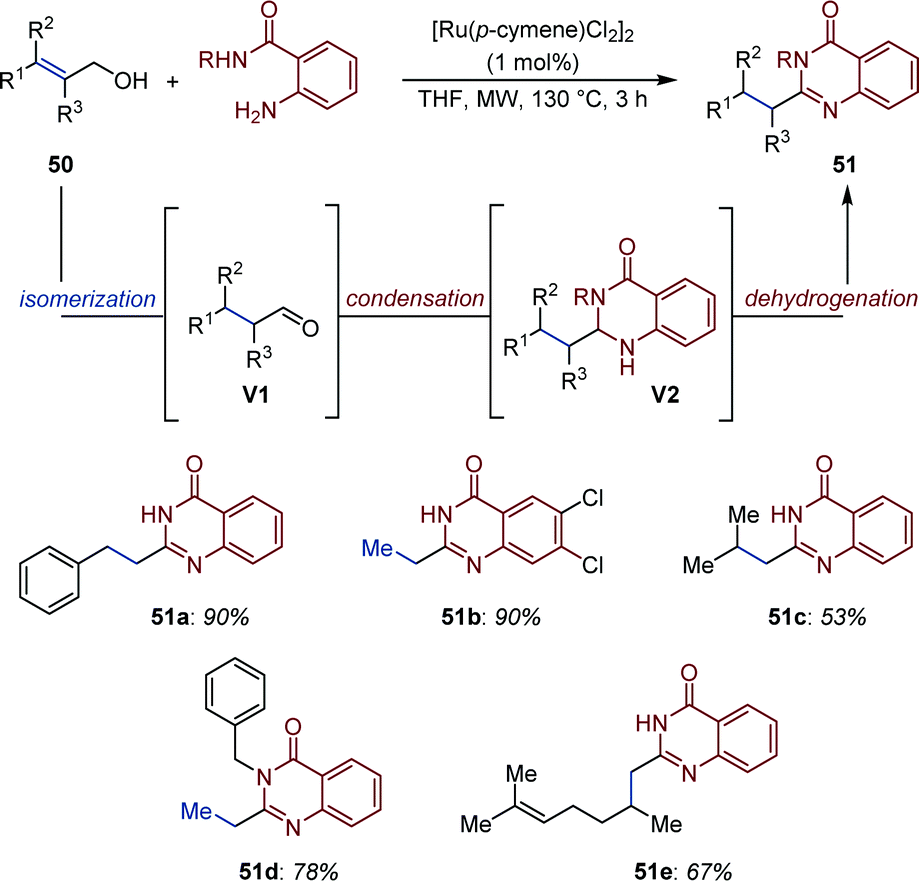 | ||
| Fig. 23 Synthesis of quinazolinones via Ru-catalyzed auto-tandem redox isomerization/acceptorless dehydrogenation. | ||
6. Transition metal enolates as transient intermediates
While the formation of main group enolates typically requires enolization of a carbonyl compound under basic conditions, transition metal enolates are usually prepared by exchange with preformed alkali enolates. Alternatively, transition metal enolates can be generated by oxidative addition into α-halo carbonyls, by decomposition of α-azo carbonyls or by conjugate reduction of α,β-unsaturated carbonyls.75 A different approach consists in generating transient transition metal enolates by olefin isomerization before engaging them in a subsequent transformation. Examples of this strategy are discussed below.6.1 Tandem processes
Building on the observation that alkenyl alcohols undergo isomerization/aldol dimerization under Ru-catalysis,76 Ryu and co-workers followed a related C–C bond formation strategy for the construction of 2-alkyl substituted 1,3-diketones (53) starting from non-conjugated enones (52) and aldehydes (Fig. 24).77 Initially, the ruthenium hydride Ru1 reconjugates the remote CC bond with the carbonyl group (W1). Subsequent hydro-ruthenation generates Ru-enolate W2, which undergoes aldol condensation with the aldehyde to produce β-keto alkoxy-ruthenium intermediate W3, which upon β-H elimination delivers the product.16 This sequence is supported by preliminary mechanistic experiments performed with isotopically labelled aldehydes. The method was applied to a handful of aliphatic and aromatic aldehydes using hex-5-en-2-one as model substrate. The expected 1,3-diketones were isolated in good to excellent yield.
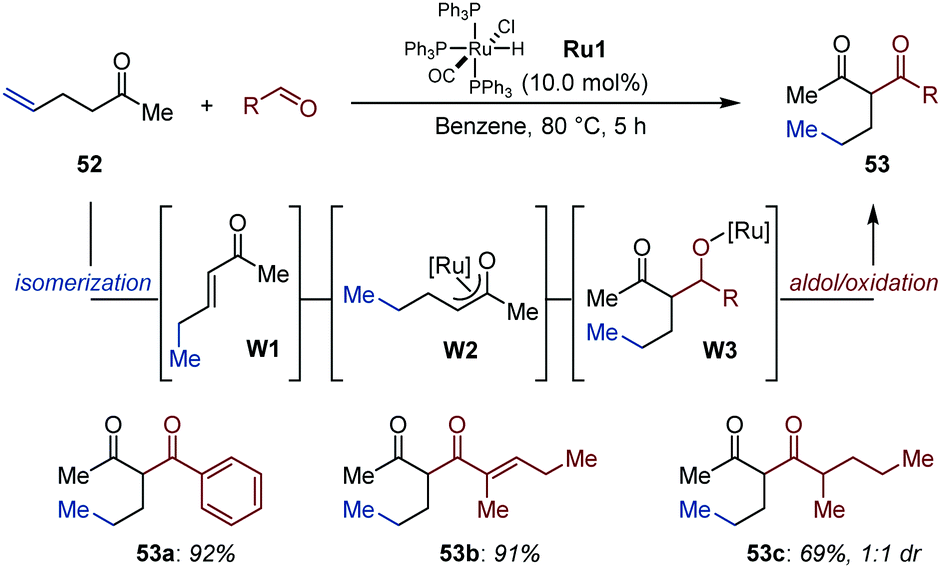 | ||
| Fig. 24 Auto-tandem Ru-catalyzed isomerization/aldol addition/oxidation of hex-5-en-2-one with aldehydes. | ||
A joint effort from the Lautens and Taylor groups led to the development of an elegant Rh-catalyzed auto-tandem isomerization/allylation of unsymmetrical diallyl carbonates for the synthesis of aldehydes possessing a α-quaternary center (Fig. 25).78 The protocol displays high chemoselectivity, as ketones, Michael acceptors and heteroaromatics are all well-tolerated. The reactions is also compatible with substrates possessing an enolizable position (55c). Competition and crossover experiments together with reaction progress kinetic analysis (RPKA) shed light on the operating mechanism of this intriguing catalytic process. The concomitant formation of a nucleophilic Rh-enolate (X1) and an electrophilic Rh-allyl species (X2) is proposed to precede bimetallic C–C bond formation to produce 55. While formation of X2 is believed to proceed via an oxidative addition/decarboxylation sequence, X1 is presumably generated via isomerization of the most hindered CC bond. Moreover, the reaction was found to be second order in rhodium, which is consistent with the C–C bond formation step operating through a rate-limiting bimetallic pathway. Of note, controlled experiments ruled out the possibility of a Claisen rearrangement being involved in product formation.
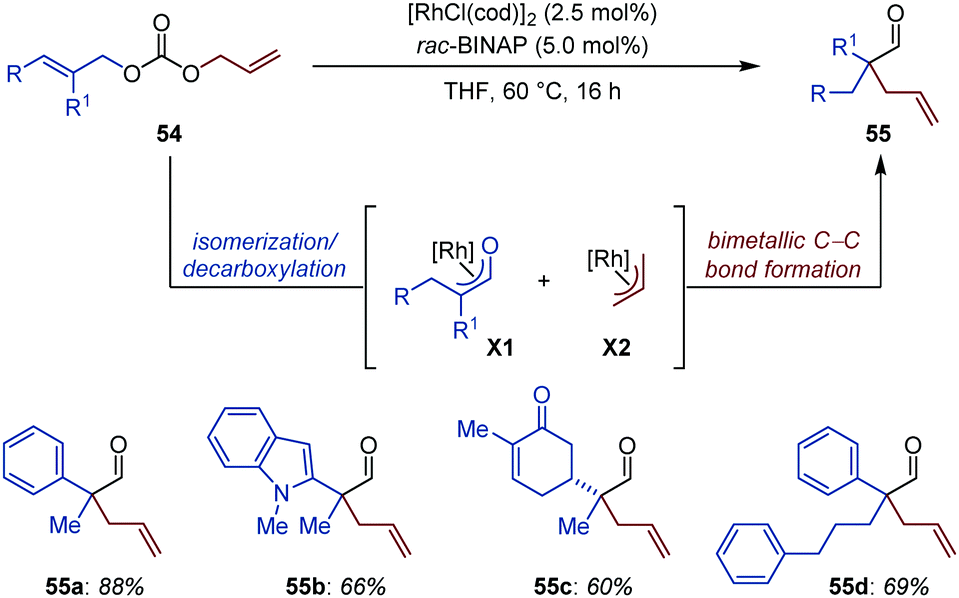 | ||
| Fig. 25 Rh-Catalyzed auto-tandem isomerization/allylation of diallyl carbonates. | ||
6.2 Domino processes
Pioneering studies from Bosnich and Motherwell established the potential of enolates in situ generated by metal-catalyzed isomerization of allylic alcohols to engage in subsequent C–C bond-forming domino aldol or Mannich processes.79 Quite remarkably, with this approach formation of the metal enolate does not require the presence of a carbonyl group or of a carbonyl group flanked with a reactive α-functionality. The reader is invited to consult the exhaustive review written by Martín-Matute to appreciate the quantity and quality of the work accomplished in this area.80In recent year, the field has witnessed the elaboration of related domino reactions in which sources of electrophilic halides have been employed as quenching agent of the transient enolate (Fig. 26). The selective synthesis of monosubstituted α-iodo- (57a), α-bromo- (57b), α-chloro- (57c) and α-fluoroketones (57d) has been accomplished under mild and base-free reaction conditions using cyclopentadienyl–iridium complex Ir1 and aqueous solvent systems.81 Acyloins (57e) have been prepared following the same approach using an oxoammonium tetrafluoroborate salt derived from TEMPO.82 Finally, an ingenious variant based on an isomerization/umpolung functionalization strategy has been published by the Martín-Matute group.83 The combination of the appropriate iodine(III) reagent in methanol provided access to the targeted α-alkoxy ketones.
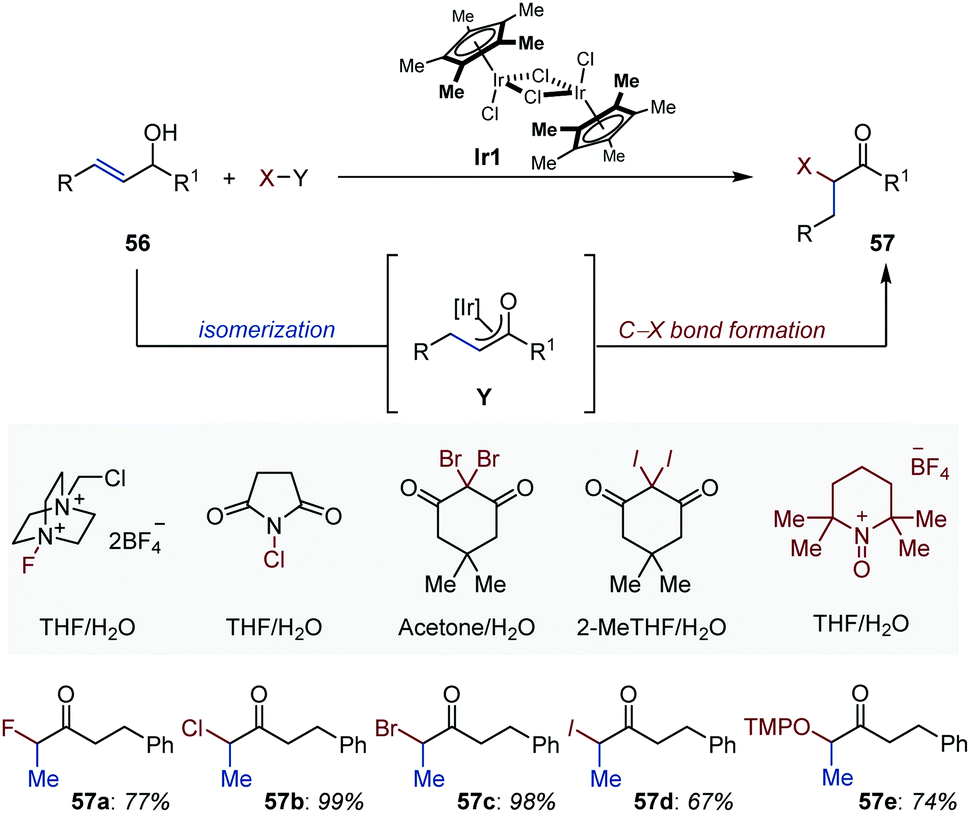 | ||
| Fig. 26 Ir-Catalyzed domino isomerization/C–X and C–O bond formation processes. | ||
6.3 Sequential processes
The development of iterative enantioselective cross-aldol reactions between two distinct aldehydes is particularly challenging because it necessitates to differentiate their role as donor and acceptor in order to avoid synthesis of 1,3-polyols of random composition.84 In a remarkable study, Lin et al. described the Cu-catalyzed aldol reaction between aldehydes (acceptor) and boron enolates (donor, (Z1)) in situ generated by Ir-catalyzed isomerization of the allylic oxyboronates (58) (Fig. 27).85 The proof of concept was validated for the single aldol reaction. Under the optimized conditions, the iridium catalyst is activated with molecular hydrogen at 0 °C in acetone and the aldol reaction is accomplished with 5 mol% of a [(R)-(DTBM-SEGPHOS)CuMes] catalyst in the presence of 1 equivalent of isopropanol in THF at −60 °C. Aliphatic, aryl, and heteroaryl aldehydes all gave the cross-aldol products in moderate to excellent yields with consistently high diastereo- and enantioselectivity (after reduction (59a–d)). By adjustment of the nature and stoichiometry of the boronate, the alcohol and the nature of the amine additive, the aldol process can be repeated by using the aldol products as acceptor in the subsequent aldol addition. One-pot diastereo- and enantioselective double, triple- and quadruple aldol reactions were successfully achieved following this strategy (59e–h). Moreover, the stereodivergency of the process was demonstrated by simply using the chiral bisphosphine ligand of opposite handedness (59e–f).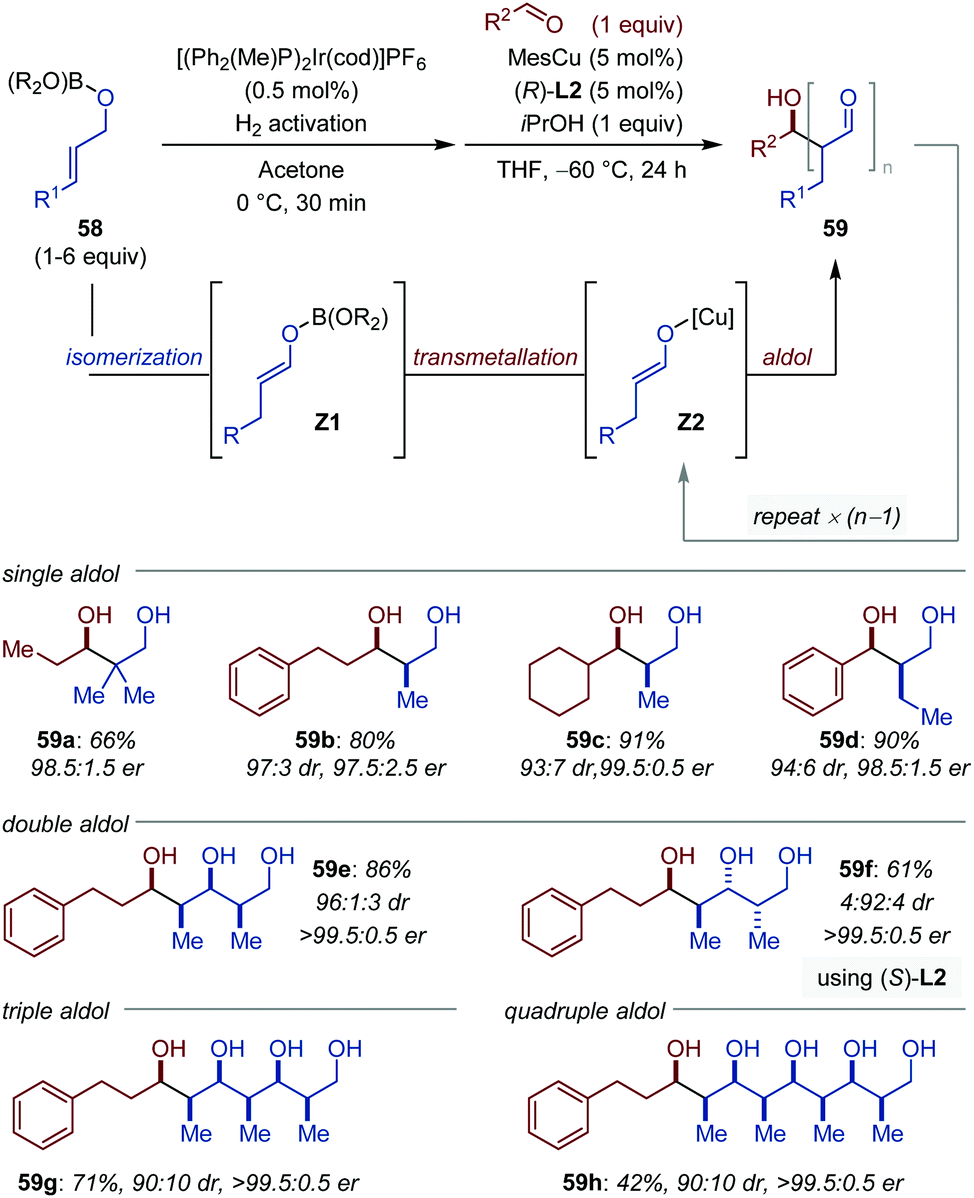 | ||
| Fig. 27 Sequential [Ir/Cu]-catalyzed isomerization/iterative aldol reaction. L2 = DTBM-SEGPHOS (see Fig. 9). Products were isolated after reduction. | ||
7. Conclusions
One-pot processes based on transition metal-catalyzed isomerization of olefins enable chemists to convert commodity chemicals into more complex molecular architectures with maximum efficiency, be it in terms of atom-, step-, and redox-economy. Over the last decades, a large number of novel transformations have been developed using either tandem, domino or sequential approaches. Adjusting and controlling the reactivity of a distal position by catalytic migration of an alkene offers new perspectives for retrosynthetic analysis. For instance, remote and apparently inert sp3 hybridized carbon atoms can now be functionalized by judicious design of isomerization/functionalization processes. In the same vein, ethers and tertiary amines can be converted into far more reactive entities upon transition metal-catalyzed olefin isomerization (i.e. enol ethers and enamines) and subsequently engaged in one-pot transformations that would not be conceivable with traditional approaches. Electrophilic carbonyls or nucleophilic enolates can also be generated by alkene isomerization and used in situ for the functionalization of previously inaccessible carbon atoms. In addition to providing access to novel reactivity patterns, recent reports have highlighted the possibility to exert exquisite relative and absolute stereocontrol in complex systems based on remote functionalization strategies or iterative sequences. In spite of these remarkable achievements, there is certainly much ingenious one-pot catalytic reactions that can be developed and novel reactivity that can be unveiled.86 For instance, the latest reports on zirconium-catalyzed olefin isomerization augurs well for the development of tandem approaches exploiting early transition metal catalysis.87 Moreover, the intermediacy of alkyl-boron or silicon compounds, generated by well-established isomerizing hydrofunctionalization of alkenes, could open new synthetic opportunities, if coupled with a sequential catalytic functionalization of the C–B and C–Si bond.88 If one looks beyond the field of organic synthesis, extension of tandem or sequential catalysis to chain-walking polymerization processes represent an exciting venue for polymer chemistry.89 We hope this Review will gather interest and inspire the design of novel one-pot processes based on transition metal-catalyzed olefin isomerization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the University of Geneva and the Swiss National Science Foundation for financial support (grants 200020_175489 and 200021_188490).Notes and references
- (a) E. Larionov, H. Li and C. Mazet, Chem. Commun., 2014, 50, 9816–9826 RSC; (b) A. Vasseur, J. Bruffaerts and I. Marek, Nat. Chem., 2016, 8, 209–219 CrossRef CAS; (c) H. Sommer, F. Juliá-Hernández, R. Martin and I. Marek, ACS Cent. Sci., 2018, 4, 153–165 CrossRef CAS; (d) T. Kochi, S. Kanno and F. Kakiuchi, Tetrahedron Lett., 2019, 60, 150938 CrossRef CAS.
- (a) B. M. Trost, Science, 1991, 254, 1471–1477 CrossRef CAS; (b) B. M. Trost, Angew. Chem., Int. Ed. Engl., 1995, 34, 259–281 CrossRef CAS; (c) P. A. Wender, V. A. Verma, T. J. Paxton and T. H. Pillow, Acc. Chem. Res., 2008, 41, 40–49 CrossRef CAS; (d) N. Z. Burns, P. S. Baran and R. W. Hoffmann, Angew. Chem., Int. Ed., 2009, 48, 2854–2867 CrossRef CAS; (e) J. J. Molloy, T. Morack and R. Gilmour, Angew. Chem., Int. Ed., 2019, 58, 13654–13664 CrossRef CAS.
- Y. Hayashi, Chem. Sci., 2016, 7, 866–880 RSC.
- (a) S. F. Mayer, W. Kroutil and K. Faber, Chem. Soc. Rev., 2001, 30, 332–339 RSC; (b) I. Oroz-Guinea and E. García-Junceda, Curr. Opin. Chem. Biol., 2013, 17, 236–249 CrossRef.
- (a) O. J. Pamies and E. Bäckvall, Chem. Rev., 2003, 103, 3247–3262 CrossRef CAS; (b) Z. Shao and H. Zhang, Chem. Soc. Rev., 2009, 38, 2745–2755 RSC; (c) M. Rueping, R. M. Koenigs and I. Atodiresei, Chem. – Eur. J., 2010, 16, 9350–9365 CrossRef CAS; (d) L. M. Ambrosini and T. H. Lambert, ChemCatChem, 2010, 2, 1373–1380 CrossRef CAS; (e) A. Galván, F. J. Fañanás and F. Rodríguez, Eur. J. Inorg. Chem., 2016, 1306–1313 CrossRef.
- D. E. Fogg and E. N. Dos Santos, Coord. Chem. Rev., 2004, 248, 2365–2379 CrossRef CAS.
- (a) J. M. Lee, Y. Na, H. Han and S. Chang, Chem. Soc. Rev., 2004, 33, 302–312 RSC; (b) J. C. Wasilke, S. J. Obrey, R. T. Baker and G. C. Bazan, Chem. Rev., 2005, 105, 1001–1020 CrossRef CAS; (c) C. Robert and C. M. Thomas, Chem. Soc. Rev., 2013, 42, 9392–9402 RSC; (d) A. Behr, A. J. Vorholt, K. A. Ostrowski and T. Seidensticker, Green Chem., 2014, 16, 982–1006 RSC; (e) H. Pellissier, Adv. Synth. Catal., 2020, 362, 2289–2325 CrossRef CAS.
- (a) N. Shindoh, Y. Takemoto and K. Takasu, Chem. – Eur. J., 2009, 15, 12168–12179 CrossRef CAS; (b) J. E. Camp, Eur. J. Org. Chem., 2017, 425–433 CrossRef CAS.
- (a) T. L. Lohr and T. J. Marks, Nat. Chem., 2015, 7, 477–482 CrossRef CAS; (b) M. M. Lorion, K. Maindan, A. R. Kapdiand and L. Ackermann, Chem. Soc. Rev., 2017, 46, 7399–7420 RSC.
- (a) L. F. Tietze, Chem. Rev., 1996, 96, 115–136 CrossRef CAS; (b) C. Grondal, M. Jeanty and D. Enders, Nat. Chem., 2010, 2, 167–178 CrossRef CAS; (c) S. A. Snyder, Applications of Domino Transformations in Organic Synthesis, Science of Synthesis, Georg Thieme Verlag, Stuttgart, 2016, vol. 1–2 Search PubMed; (d) H. Pellissier, Adv. Synth. Catal., 2016, 358, 2194–2259 CrossRef CAS; (e) H. Pellissier, Adv. Synth. Catal., 2019, 361, 1733–1755 CrossRef CAS.
- For reviews on tandem and sequential processes that combine transition metal-catalyzed alkene isomerization and organic catalysis, see: (a) P. Kathe and I. Fleischer, ChemCatChem, 2019, 11, 3343–3354 CrossRef CAS; (b) P. S. Wang, D. F. Chen and L. Z. Gong, Top. Curr. Chem., 2020, 378 Search PubMed.
- J. Pollini, W. M. Pankau and L. J. Gooßen, Chem. – Eur. J., 2019, 25, 7416–7425 CrossRef CAS.
- (a) M. Vilches-Herrera, L. Domke and A. Börner, ACS Catal., 2014, 4, 1706–1724 CrossRef CAS; (b) V. Goldbach, P. Roesle and S. Mecking, ACS Catal., 2015, 5, 5951–5972 CrossRef CAS.
- For reviews, see: (a) V. Goldbach, P. Roesle and S. Mecking, ACS Catal., 2015, 5, 5951–5972 CrossRef CAS; (b) D. Janssen-Müller, B. Sahoo, S. Z. Sun and R. Martin, Isr. J. Chem., 2020, 60, 195–206 CrossRef . For selected examples, see: ; (c) J. V. Obligacion and P. J. Chirik, J. Am. Chem. Soc., 2013, 135, 19107–19110 CrossRef CAS; (d) P. Roesle, L. Caporaso, M. Schnitte, V. Goldbach, L. Cavallo and S. Mecking, J. Am. Chem. Soc., 2014, 136, 16871–16881 CrossRef CAS; (e) F. Julía-Hernández, T. Moragas, J. Cornella and R. Martin, Nature, 2017, 545, 84–88 CrossRef; (f) F. Chen, K. Chen, Y. Zhang, Y. He, Y.-M. Wang and S. Zhu, J. Am. Chem. Soc., 2017, 139, 13929–13935 CrossRef CAS; (g) X. Chen, Z. Cheng, J. Guo and Z. Lu, Nat. Commun., 2018, 9, 3939 CrossRef; (h) A. J. Borah and Z. Shi, J. Am. Chem. Soc., 2018, 140, 6062–6066 CrossRef CAS; (i) R. Sang, P. Kucmierczyk, K. Dong, R. Franke, H. Neumann, R. Jackstell and M. Beller, J. Am. Chem. Soc., 2018, 140, 5217–5223 CrossRef CAS; (j) Y. Liu, K. Dong, M. Beller and S. Mecking, ACS Catal., 2018, 8, 9232–9237 CrossRef CAS; (k) T. Kochi, K. Ichinose, M. Shigekane, T. Hamasaki and F. Kakiuchi, Angew. Chem., Int. Ed., 2019, 58, 5261–5265 CrossRef CAS; (l) P. M. Kathe and I. Fleischer, Org. Lett., 2019, 21, 2213–2217 CrossRef CAS; (m) J. Yang, J. Liu, H. Neumann, R. Franke, R. Jackstell and M. Beller, Science, 2019, 366, 1514–1517 CrossRef CAS; (n) J. Li, S. Qu and W. Zhao, Angew. Chem., Int. Ed., 2020, 59, 2360–2364 CrossRef CAS.
- Contributions for which a tandem catalytic process has been invoked but the mechanistic scenario not fully ascertained have been excluded from our selection. See: (a) W.-C. Lee, C.-H. Wang, Y.-H. Lin, W.-C. Shih and T.-G. Ong, Org. Lett., 2013, 15, 5358–5361 CrossRef CAS; (b) T. Yamakawa and N. Yoshikai, Chem. – Asian J., 2014, 9, 1242–1246 CrossRef CAS; (c) Y. Ebe, M. Onoda, T. Nishimura and H. Yorimitsu, Angew. Chem., Int. Ed., 2017, 56, 5607–5611 CrossRef CAS; (d) H. S. Li, G. Guo, R. Z. Zhang and F. Li, Org. Lett., 2018, 20, 5040–5043 CrossRef CAS.
- S. Omura, T. Fukuyama, J. Horiguchi, Y. Murakami and I. Ryu, J. Am. Chem. Soc., 2008, 130, 14094–14095 CrossRef CAS.
- H. Wakamatsu, M. Nishida, N. Adachi and M. Mori, J. Org. Chem., 2000, 65, 3966–9370 CrossRef CAS.
- F. Shibahara, J. F. Bower and M. J. Krische, J. Am. Chem. Soc., 2008, 130, 6338–6339 CrossRef CAS.
- (a) D. M. Ohlmann, L. J. Gooßen and M. Dierker, Chem. – Eur. J., 2011, 17, 9508–9519 CrossRef CAS; (b) L. J. Gooßen, D. M. Ohlmann and M. Dierker, US Pat., 8877958 B2, 2014 Search PubMed.
- T. Hayashi and K. Yamasaki, Chem. Rev., 2003, 103, 2829–2844 CrossRef CAS.
- X. Zhang, F. Jordan and M. Szostak, Org. Chem. Front., 2018, 5, 2515–2521 RSC.
- R. E. Murray, E. L. Walter and K. M. Doll, ACS Catal., 2014, 4, 3517–3520 CrossRef CAS.
- J. Zhao, L. Liu, S. Xiang, Q. Liu and H. Chen, Org. Biomol. Chem., 2015, 13, 5613–5616 RSC.
- M. Hassam, A. Taher, G. E. Arnott, I. R. Green and W. A. L. van Otterlo, Chem. Rev., 2015, 115, 5462–5569 CrossRef CAS.
- B. W. Michel, L. D. Steffens and M. S. Sigman, J. Am. Chem. Soc., 2011, 133, 8317–8325 CrossRef CAS.
- M. Liniger, Y. Liu and B. M. Stoltz, J. Am. Chem. Soc., 2017, 139, 13944–13949 CrossRef CAS.
- M. Arisawa, Y. Terada, K. Takahashi, M. Nakagawa and A. Nishida, J. Org. Chem., 2006, 71, 4255–4261 CrossRef CAS.
- T. Gaide, J. Bianga, K. Schlipköter, A. Behr and A. J. Vorholt, ACS Catal., 2017, 7, 4163–4171 CrossRef CAS.
- (a) D. M. Ohlmann, N. Tschauder, J.-P. Stockis, K. Gooßen, M. Dierker and L. J. Gooßen, J. Am. Chem. Soc., 2012, 134, 13716–13729 CrossRef CAS; (b) S. De, N. Sivendran, B. Maity, N. Pirkl, D. Koley and L. J. Gooßen, ACS Catal., 2020, 10, 4517–4533 CrossRef CAS.
- (a) E. Billig, A. G. Abatjoglou and D. R. Bryant, US Pat., 4668651, 1987 and 4769498, 1988 Search PubMed; (b) G. D. Cuny and S. L. Buchwald, J. Am. Chem. Soc., 1993, 115, 2066–2068 CrossRef CAS; (c) A. van Rooy, K. Goubitz, J. Fraanje, P. C. J. Kamer, P. W. N. M. van Leeuwen, N. Veldman and A. L. Spek, Organometallics, 1996, 15, 835–847 CrossRef CAS.
- F. M. S. Rodrigues, P. K. Kucmierczyk, M. Pineiro, R. Jackstell, R. Franke, M. M. Pereira and M. Beller, ChemSusChem, 2018, 11, 2310–2314 CrossRef CAS.
- Y. Yuki, K. Takahashi, Y. Tanaka and K. Nozaki, J. Am. Chem. Soc., 2013, 135, 17393–17400 CrossRef CAS.
- H. Sommer, T. Weissbrod and I. Marek, ACS Catal., 2019, 9, 2400–2406 CrossRef CAS.
- I. Massad and I. Marek, ACS Catal., 2020, 10, 5793–5804 CrossRef CAS.
- C. Romano, D. Fiorito and C. Mazet, J. Am. Chem. Soc., 2019, 141, 16983–16990 CrossRef CAS.
- V. Milata, S. Rádl and S. Voltrová, in 32.5.3 Product Subclass 3: Enol Ethers. Science of Synthesis, ed. J. Mulzer, Thieme, Stuttgart, 2008, vol. 32, p. 589 Search PubMed.
- J. V. Crivello and S. K. Rajaraman, Macromol. Symp., 1998, 132, 37–43 CrossRef CAS.
- K. Tanaka, E. Okazaki and Y. Shibata, J. Am. Chem. Soc., 2009, 131, 10822–10823 CrossRef CAS.
- P. Eilbracht, A. Gersmeir, D. Lennartz and T. Huber, Synthesis, 1995, 330–334 CrossRef CAS.
- R. Okamoto and K. Tanaka, Org. Lett., 2013, 15, 2112–2115 CrossRef CAS.
- J. M. Reuter and R. G. Salomon, J. Org. Chem., 1977, 42, 3360–3364 CrossRef CAS.
- (a) J. Le Nôtre, L. Brissieux, D. Sémeril, C. Bruneau and P. H. Dixneuf, Chem. Commun., 2002, 1772–1773 RSC; (b) J. Le Nôtre, R. Touzani, O. Lavastre, C. Bruneau and P. H. Dixneuf, Adv. Synth. Catal., 2005, 347, 783–791 CrossRef.
- B. Lastra-Barreira, J. Francos, P. Crochet and V. Cadierno, Organometallics, 2018, 37, 3465–3474 CrossRef CAS.
- (a) S. G. Nelson, C. J. Bungard and K. Wang, J. Am. Chem. Soc., 2003, 125, 13000–13001 CrossRef CAS; (b) B. D. Stevens and S. G. Nelson, J. Org. Chem., 2005, 70, 4375–4379 CrossRef CAS; (c) S. G. Nelson and K. Wang, J. Am. Chem. Soc., 2006, 128, 4232–4233 CrossRef CAS; (d) K. Wang, C. J. Bungard and S. G. Nelson, Org. Lett., 2007, 9, 2325–2328 CrossRef CAS.
- N. J. Kerrigan, C. J. Bungard and S. G. Nelson, Tetrahedron, 2008, 64, 6863–6869 CrossRef CAS.
- (a) B. M. Trost and T. Zhang, Org. Lett., 2006, 8, 6007–6010 CrossRef CAS; (b) R. S. Sulake and C. Chen, Org. Lett., 2015, 17, 1138–1141 CrossRef CAS.
- (a) G. Stork and K. S. Atwal, Tetrahedron Lett., 1982, 23, 2073–2076 CrossRef CAS; (b) C. Nevado and A. M. Echavarren, Tetrahedron, 2004, 60, 9735–9744 CrossRef CAS.
- T. Higashino, S. Sakaguchi and Y. Ishii, Org. Lett., 2000, 2, 4193–4195 CrossRef CAS.
- B. Schmidt, Synlett, 2004, 1541–1544 CrossRef CAS.
- C. Romano and C. Mazet, J. Am. Chem. Soc., 2018, 140, 4743–4750 CrossRef CAS.
- (a) B. M. Rosen, K. W. Quasdorf, D. A. Wilson, N. Zhang, A.-M. Resmerita, N. K. Garg and V. Percec, Chem. Rev., 2011, 111, 1346–1416 CrossRef CAS; (b) J. Cornella, C. Zarate and R. Martin, Chem. Soc. Rev., 2014, 43, 8081–8097 RSC; (c) M. Tobisu and N. Chatani, Acc. Chem. Res., 2015, 48, 1717–1726 CrossRef CAS.
- H. Li and C. Mazet, Acc. Chem. Res., 2016, 49, 1232–1241 CrossRef CAS.
- For the original development of the Pd catalyst, see: (a) E. Larionov, L. Lin, L. Guénée and C. Mazet, J. Am. Chem. Soc., 2014, 136, 16882–16894 CrossRef CAS; (b) L. Lin, C. Romano and C. Mazet, J. Am. Chem. Soc., 2016, 138, 10344–10350 CrossRef CAS.
- G. Ho, L. Judkele, J. Bruffaerts and I. Marek, Angew. Chem., Int. Ed., 2018, 57, 8012–8016 CrossRef CAS.
- G.-M. Ho, L. Segura and I. Marek, Chem. Sci., 2020, 11, 5944–5949 RSC.
- Z. Wu, S. D. Laffoon, T. T. Nguyen, J. D. McAlpin and K. L. Hull, Angew. Chem., Int. Ed., 2017, 56, 1371–1375 CrossRef CAS.
- (a) K. Tani, T. Yamagata, S. Akutagawa, H. Kumobayashi, T. Taketomi, H. Takaya, A. Miyashita, R. Noyori and S. Otsuka, J. Am. Chem. Soc., 1984, 106, 5208–5217 CrossRef CAS; (b) S. Inoue, H. Takaya, K. Tani, S. Otsuka, T. Sato and R. Noyori, J. Am. Chem. Soc., 1990, 112, 4897–4905 CrossRef CAS; (c) M. Emura and H. Matsuda, Chem. Biodiversity, 2014, 11, 1688–1699 CrossRef CAS.
- Z. Wu and K. L. Hull, Chem. Sci., 2016, 7, 969–975 RSC.
- S. D. Laffoon, Z. Wu and K. L. Hull, Chem. Commun., 2018, 54, 7814–7817 RSC.
- Z. Wu, S. D. Laffoon and K. L. Hull, Nat. Commun., 2018, 9, 1185 CrossRef.
- Y. Fujii, T. Takehara, T. Suzuki, H. Fujioka, S. Shuto and M. Arisawa, Adv. Synth. Catal., 2015, 357, 4055–4062 CrossRef CAS.
- (a) R. C. van der Drift, E. Bouwman and E. J. Drent, Organomet. Chem., 2002, 650, 1–24 CrossRef CAS; (b) R. Uma, C. Crévisy and R. Grée, Chem. Rev., 2003, 103, 27–51 CrossRef CAS; (c) L. Mantilli and C. Mazet, Chem. Lett., 2011, 40, 341–344 CrossRef CAS; (d) P. Lorenzo-Luis, A. Romerosa and M. Serrano-Ruiz, ACS Catal., 2012, 2, 1079–1086 CrossRef CAS; (e) D. Cahard, S. Gaillard and J.-L. Renaud, Tetrahedron Lett., 2015, 56, 6159–6169 CrossRef CAS.
- D. Y. Lee, C. W. Moon and C. H. Jun, J. Org. Chem., 2002, 67, 3945–3948 CrossRef CAS.
- (a) M. C. Willis, Chem. Rev., 2010, 110, 725–748 CrossRef CAS; (b) M. Fernández and M. C. Willis, Rhodium(I)-catalyzed hydroacylation, in Rhodium catalysis in organic synthesis: Methods and reactions, ed. K. Tanaka, Wiley-VCH, 2019 Search PubMed.
- A. Bartoszewicz and B. Martín-Matute, Org. Lett., 2009, 11, 1749–1752 CrossRef CAS.
- (a) V. Cadierno, J. Francos, J. Gimeno and N. Nebra, Chem. Commun., 2007, 2536–2538 RSC; (b) V. Cadierno, P. Crochet, J. Francos, S. E. García-Garrido, J. Gimeno and N. Nebra, Green Chem., 2009, 11, 1992–2000 RSC.
- A. E. Díaz-Álvarez, P. Crochet and V. Cadierno, Catal. Commun., 2011, 13, 91–96 CrossRef.
- V. Bizet, X. Pannecoucke, J. L. Renaud and D. Cahard, Adv. Synth. Catal., 2013, 355, 1394–1402 CrossRef CAS.
- (a) R. Wu, M. G. Beauchamps, J. M. Laquidara and J. R. Sowa, Angew. Chem., Int. Ed., 2012, 51, 2106–2110 CrossRef CAS . See also: ; (b) V. Bizet, X. Pannecoucke, J.-L. Renaud and D. Cahard, Angew. Chem., Int. Ed., 2012, 51, 6467–6470 CrossRef CAS.
- Y. Sun, C. LeBlond, J. Wang, D. G. Blackmond, J. Laquidara and J. R. Sowa Jr., J. Am. Chem. Soc., 1995, 117, 12647–12648 CrossRef CAS.
- (a) T. Slagbrand, H. Lundberg and H. Adolfsson, Chem. – Eur. J., 2014, 20, 16102–16106 CrossRef CAS; (b) C. O. Shoola, T. DelMastro, R. Wu and J. R. Sowa, Eur. J. Org. Chem., 2015, 1670–1673 CrossRef CAS.
- Z. Sahli, B. Sundararaju, M. Achard and C. Bruneau, Org. Lett., 2011, 13, 3964–3967 CrossRef CAS.
- H. Li, M. Achard, C. Bruneau, J. B. Sortais and C. Darcel, RSC Adv., 2014, 4, 25892–25897 RSC.
- W. Zhang, C. Meng, Y. Liu, Y. Tang and F. Li, Adv. Synth. Catal., 2018, 360, 3751–3759 CrossRef CAS.
- (a) The Chemistry of Metal Enolates, in PATAI's Chemistry of Functional Groups, ed. J. Zabicky, Wiley, 2018, vol. 2 Search PubMed; (b) D. Vargová, I. Némethová, K. Plevová and R. Šebesta, ACS Catal., 2019, 9, 3104–3143 CrossRef.
- T. Doi, T. Fukuyama, S. Minamino, G. Husson and I. Ryu, Chem. Commun., 2006, 1875–1877 RSC.
- T. Fukuyama, T. Doi, S. Minamino, S. Omura and I. Ryu, Angew. Chem., Int. Ed., 2007, 46, 5559–5561 CrossRef CAS.
- J. Masson-Makdissi, Y. J. Jang, L. Prieto, M. S. Taylor and M. Lautens, ACS Catal., 2019, 9, 11808–11812 CrossRef CAS.
- (a) S. H. Bergens and B. Bosnich, J. Am. Chem. Soc., 1991, 113, 958–967 CrossRef CAS; (b) L. J. Gazzard, W. B. Motherwell and D. A. Sandham, J. Chem. Soc., Perkin Trans. 1, 1999, 979–993 RSC; (c) C. Schneider and M. Boomhoff, Aldol Reactions in Domino Processes, in Domino Reactions, ed. L. F. Tietze, Wiley-VCH, Weinheim, Germany, 2014, ch. 8, pp. 267–294 Search PubMed.
- N. Ahlsten, A. Bartoszewicz and B. Martín-Matute, Dalton Trans., 2012, 41, 1660–1670 RSC.
- (a) N. Ahlsten, A. B. Gõmez and B. Martín-Matute, Angew. Chem., Int. Ed., 2013, 52, 6273–6276 CrossRef CAS; (b) A. B. Gõmez, E. Erbing, M. Batuecas, A. Vázquez-Romero and B. Martín-Matute, Chem. – Eur. J., 2014, 20, 10703–10709 CrossRef; (c) A. Vázquez-Romero, A. B. Gómez and B. Martín-Matute, ACS Catal., 2015, 5, 708–714 CrossRef; (d) S. Martinez-Erro, A. Bermejo Gómez, A. Vázquez-Romero, E. Erbing and B. Martín-Matute, Chem. Commun., 2017, 53, 9842–9845 RSC; (e) A. Sanz-Marco, Š. Možina, S. Martinez-Erro, J. Iskra and B. Martín-Matute, Adv. Synth. Catal., 2018, 360, 3884–3888 CrossRef CAS.
- A. Sanz-Marco, S. Martinez-Erro and B. Martín-Matute, Chem. – Eur. J., 2018, 24, 11564–11567 CrossRef CAS.
- A. Sanz-Marco, S. Martinez-Erro, M. Pauze, E. Gómez-Bengoa and B. Martín-Matute, Nat. Commun., 2019, 10, 5244 CrossRef.
- (a) B. Schetter and R. Mahrwald, Angew. Chem., Int. Ed., 2006, 45, 7506–7525 CrossRef CAS; (b) P. B. Brady and H. Yamamoto, in Modern Methods in Stereoselective Aldol Reactions, ed. R. Mahrwald, Wiley-VCH, Weinheim, Germany, 2013, pp. 269–291 Search PubMed; (c) J. Feng, Z. A. Kasun and M. J. Krische, J. Am. Chem. Soc., 2016, 138, 5467–5478 CrossRef CAS.
- L. Lin, K. Yamamoto, H. Mitsunuma, Y. Kanzaki, S. Matsunaga and M. Kanai, J. Am. Chem. Soc., 2015, 137, 15418–15421 CrossRef CAS.
- (a) X. Liu, B. Liu and Q. Liu, Angew. Chem., Int. Ed., 2020, 59, 6750–6755 CrossRef CAS; (b) C. Yang, Y. Gao, S. Bai, C. Jiang and X. Qi, J. Am. Chem. Soc., 2020, 142, 11506–11513 CrossRef CAS.
- C. Matt, C. Kern and J. Streuff, ACS Catal., 2020, 10, 6409–6413 CrossRef CAS.
- S. Hanna, T. Wills, T. W. Butcher and J. F. Hartwig, ACS Catal., 2020, 10, 8736–8741 CrossRef CAS.
- (a) H. E. Basbug Alhan, G. R. Jones and E. Harth, Angew. Chem., Int. Ed., 2020, 59, 4743–4749 CrossRef CAS; (b) G. R. Jones, H. E. Basbug Alhan, L. J. Karas, J. I. Wu and E. Harth, Angew. Chem., Int. Ed., 2020, 59 DOI:10.1002/anie.202012400.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |