
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn augmented (multi-descriptor) grouping algorithm to optimize chemical ordering in nanoalloys†
Davide
Fioravanti
 a,
Giovanni
Barcaro
*a and
Alessandro
Fortunelli
*b
a,
Giovanni
Barcaro
*a and
Alessandro
Fortunelli
*b
aCNR-IPCF, CNR Research Area, Via Moruzzi 1, 56124 Pisa, Italy. E-mail: giovanni.barcaro@cnr.it
bCNR-ICCOM, CNR Research Area, Via Moruzzi 1, 56124 Pisa, Italy. E-mail: alessandro.fortunelli@cnr.it
First published on 21st September 2021
Abstract
We propose the Augmented Grouping Approach (AugGA) and its deployment in the Augmented Grouping GO (AugGGO) scheme, for an efficient exploration of the chemical ordering (or compositional structure) of multi-component (alloyed) nanoparticles. The approach is based on a ‘grouping’ strategy (previously proposed for high-symmetry structures) by which the number of compositional degrees of freedom of the system is decreased by defining sets of atoms (groups, or orbits, or shells) that are constrained to be populated by the same element. Three fundamental advances are here included with respect to previous proposals: (i) groups are defined on the basis of descriptors (no point-group symmetry is assumed), (ii) bulk groups can exploit general chemical ordering patterns taken from databases, and (iii) sub-grouping is realized via a multi-descriptor strategy (here using two basic descriptors: the atomic energy and a few types of geometry patterns). The AugGGO approach is applied to two prototypical examples of binary nanoalloys: Pd–Pt and Ag–Cu, with a size between ≈500 and ≈1300 atoms, in different configurations, and the convex hull of the mixing energy as a function of composition is derived. It is shown how the three advances here proposed decisively extend the power and scope of the grouping approach: (i) making it applicable to any generic structural framework, (ii) achieving a thorough sampling of the core regions of nanoparticles, and (iii) catching exotic/unexpected chemical ordering arrangements, at a computational cost which is 1–2 orders of magnitude smaller than that of traditional Monte Carlo single-exchange techniques.
1. Introduction
Optimizing the properties (and thus the performance) of a given system by moving in the space of chemical variables1 is a challenging task, due to the exponential increase in the number of possible structures and compositions.2 The task can be restricted to exploring only the composition (stoichiometry) degrees of freedom of isomers sharing a fixed structural framework. This decreases the complexity of the problem, which however remains of combinatorial, and therefore non-algorithmic, complexity: the number of homotops equals MN, where N is the total number of atoms and M is the number of components; this number corresponds to 2N for binary systems, 3N for ternary systems, etc. In the field of multi-component metallic nanoparticles or nanoalloys,3,4 on which we focus in the present work, these isomers are named ‘homotops’,5 and the arrangement of the different elements in the given structural framework is named ‘chemical ordering’ or ‘compositional structure’. At the computational level, a systematic search over the full set of homotops is practically unfeasible only for systems with a number of atoms larger than a few tens, such that MN remains computationally affordable. Developing methods to accelerate the exploration of the combinatorial phase space of larger systems still maintaining thoroughness over the physically and chemically significant isomers is mandatory to make simulations affordable and predictive.Although a systematic exploration of all possible homotops is feasible only for smaller systems, for larger systems one can use Global Optimization (GO) techniques, in which a stochastic search over a selection of structures and transformation moves is performed. The GO search can be made as extensive as far as it is compatible with available computational resources, and the goal of GO methods is to direct the sampling towards low-energy isomers.6 Many variants of GO have been devised, typically differing in the choice of transformation moves. Popular techniques are the basin-hopping7 and genetic algorithm8 methods, in which transformation moves are accepted or rejected on the basis of a Metropolis9 criterion based on the difference in the energy (or some transformed function7,10) between the starting and final configurations (see ref. 11 for a unified view of GO approaches). Possible approaches to GO in the chemical ordering space are traditionally based on individual-site moves, such as an exchange of two elements on two different sites of the structural framework,12,13 or an exchange of an element on a given site with an external reservoir.14 GO based on these individual-site chemical-ordering moves however exhibits a slow convergence of the search to the global minimum.
An alternative way to achieve an effective acceleration of the chemical ordering search is based on collective moves, involving simultaneous changes of many atoms, as realized in the class of ‘grouping’ approaches.15–17 In these approaches, atoms are grouped into equivalence sets (indifferently called ‘groups’ or ‘orbits’ or ‘shells’, but here we will mostly use the term ‘group’ to underline the generality of the approach), and the search is restricted to homotops in which the different groups are homogeneously populated, i.e., all the sites belonging to a given group, or orbit, or shell, are populated with the same element. If the number of orbits is Norbs and the total number of atoms is N, the configurational space of a binary system is reduced from 2N to 2Norbs. Except for the fact that collective variables are used, the principles and techniques of structural exploration remain the usual ones: sampling can be conducted in an exhaustive way (i.e., systematic sampling),15,16 or based on stochastic searches (the Grouping Global Optimization, GGO, approach,17 in the various variants and flavours of GO algorithms). GGO approaches have proven to be computationally efficient, enabling predictive sampling of the chemical ordering of systems composed of thousands of atoms when using empirical potentials, e.g., (Ag–Cu)4033,17 or hundreds of atoms when coupled with first-principles methods.18 Thus far, grouping techniques have been limited to high-symmetry structural frameworks, as the selection of orbits or shells was based on point-group symmetry, i.e., by grouping atoms equivalent by symmetry.15–18 Additionally, the stability of the algorithm with respect to broken-symmetry perturbations was also envisaged by considering sub-groups, i.e., by partitioning orbits into sub-orbits as obtained by reducing the point-group symmetry with respect to a symmetry axis or spherical symmetry.17 Despite ensuring a huge increase in computational efficiency, the applicability of GGO grouping methods thus far has suffered from being limited to high-symmetry configurations, which represent only a tiny fraction of the exponential number of amenable structures, and possible issues in predicting ordered patterns in the core of nanoparticles. To accelerate the search for low-symmetry structures and exhaustively sample bulk patterns a more general approach is needed.
Here we propose just such an approach, the Augmented Grouping Approach (AugGA), and in particular its deployment in an Augmented Grouping GO (AugGGO) scheme. The AugGA approach is still based on a ‘grouping’ philosophy but including three basic advances: (i) groups (or orbits, or shells) here rely on general descriptors – in other words sites in the structural framework are grouped according to common values of a general descriptor function (at variance with equivalence based on point-group symmetry as used in our previous work17), (ii) grouping is generalized to include the possibility of populating groups with compositional arrangements taken from chemical ordering databases (at variance with populating groups each with a single-element as realized so far), and (iii) sub-grouping is realized via a multi-descriptor strategy (the present proof-of-principle investigation is limited to basically two descriptors at a time, but the approach can be extended to as many descriptors as needed). These three advances decisively extend the power and scope of the grouping methods. By the first advance (i), point-group symmetry is not invoked and the method is applicable to any generic structural framework, which enables its exploitation in basically all existing GO codes to deal with any system. By the second advance (ii), collective moves are further generalized to include transformations in which selected orbits are populated with chemical ordering motifs taken from a wide (potentially continuously updated) database (incidentally, this technique can also be seen as a form of pre-defined sub-grouping): this advance is particularly beneficial to achieve a thorough sampling of the core regions of nanoparticles, avoiding missing ordered compositional structures that are difficult to catch for entropic reasons (a tiny number of realizations) such as L10, etc.19–21 By the third advance (iii), we formalize the previously defined combination of grouping and sub-grouping according to several (i.e., >1) descriptors or selection criteria: this significantly increases the thoroughness of the search especially in the case of exotic and unexpected surface chemical ordering arrangements, and is also very useful to catch and/or discover unusual chemical ordering motifs in the bulk of the particles.
In the following, we will demonstrate the power of the proposed Augmented Grouping Approach (AugGA), specifically in the form of the Augmented Grouping GO (AugGGO) scheme, by applying it to two prototypical examples of binary nanoalloys: Pd–Pt and Ag–Cu, deriving the convex hull16 of the mixing energy15,22 as a function of composition. More specifically, for both Pd–Pt and Ag–Cu alloys, three different configurations have been investigated: one at size 609 atoms, corresponding to a high-symmetry motif, and two at size 500 atoms, corresponding to two low-symmetry motifs.
2. Methods
As discussed in the introduction, the Augmented Grouping GO (AugGGO) approach here developed includes three basic advances: (i) groups are defined on the basis of general descriptor functions; (ii) grouping moves are generalized to include transformations to compositional arrangements taken from chemical ordering databases; and (iii) sub-grouping is realized via a multi-descriptor strategy. Let us discuss these advances in sequence.2.1 Descriptor-based groups (i)
The fundamental strategy of grouping consists in applying the same transformation (i.e., the same move) to atoms that share a common physics. This is by definition the case of atoms located on sites equivalent via operations of the point-group symmetry of the structural framework. In such a case, atoms within a given symmetry orbit will by definition also share common values of any, however-defined local descriptor function. In the case of low-symmetry structures, instead, none of the atoms is in principle exactly equivalent to any other atom (in the case of C1 point-group symmetry of the structural framework), but, yet, it is usual to find many atoms that share a similar (although not exactly identical) physics up to some fuzziness criterion. It is then reasonable and technically easy to select a local descriptor function and generalize the grouping approach to define groups (sets of sites) in the structural framework according to a similar value of this descriptor function.Let us give a definite example: the local descriptor function that we will use in the following is the atomic energy as defined via an empirical potential, the second-moment-approximation (SMA) potential.23 Note in passing that when using the atomic energy as a descriptor to define groups we calculate atomic energies by freezing the structural framework, whereas when we calculate the total energy of each chemical ordering representation within successive GO runs we allow for local geometry relaxation. In the top-left of Fig. 1, we show a Leary tetrahedron (Leary Td) made of 609 atoms, while the top-right of the same figure reports the corresponding population (density) of its atomic energies. We stress that the choice of the Leary Td is taken as a specific example, but the approach is completely general. The class of Leary tetrahedra, found for the first time as the putative global minimum (GM) of the 98 atom Lennard-Jones cluster,24 corresponds to a hybrid fcc-decahedral motif made of a crystalline core exhibiting a single (or double25) tetrahedron on the faces of which further growth in a stacking-fault fashion gives rise to five-fold axes running parallel to the edges of the core. The resulting shape benefits from a reduced surface energy thanks to the local non-crystalline environment at the surface edges and by an overall quasi-spherical shape. It is pictorially apparent (and it is further clarified by the arrows in Fig. 1) how the local energies of the atoms in this structure group into well-distinct peaks according to their different physics, i.e., their position in the framework. Note that to calculate the atomic energies to define the orbits we do not necessarily need to relax the geometry (that we can assume has been extracted from a structural GO search for some pure or alloy nanoparticle or from some structural database): we populate all sites with the same element, we calculate the SMA atomic energies, we plot their density as in Fig. 1, and we distinguish the peaks thus defining the groups, and then we populate the groups with different elements using as the only constraint the criterion that the elemental population is constant within each orbit. The idea is that the difference in the local coordination environment implies that the corresponding atomic energies are different and therefore correctly distinguishes the relevant orbits. It should also be stressed that in the present proof-of-principle implementation we selected the atomic energy as the most natural choice of a descriptor, but one could use equally well other descriptors (or order parameters), such as those derived from a common-neighbour analysis (CNA).26 Incidentally, we note that, in the CNA, one will have the option of distinguishing sites populated with different elements (the original formulation of the CNA does not distinguish elements but there is a clear advantage in distinguishing sites populated with different elements especially in the case of size-mismatched nanoalloys). Finally, peak recognition is here done manually, but visual recognition techniques27 can of course be employed, especially useful to automate the AugGGO scheme within a general GO code.
 | ||
| Fig. 1 (Top) Illustration of the operation of generating groups for the 609-atom Leary tetrahedron. The atoms are coloured according to their different atomic (site) energy and the resulting groups are reported in the DOS (Density Of States) plot of atomic energies on the right-side. Below, a further partition of the groups according to a second (structural) descriptor is shown. (Bottom) Optimal orientation of the Leary structure according to the use of the C4, C5 or C6 symmetry axis – the z-axis is perpendicular to the plane of the figure. | ||
We also note in passing that the AugGA technique, similarly to the original grouping scheme, can exploit any given approach to the computation of the energy and its derivatives (forces, needed to optimize the structures), such as an empirical potential as in the present work,23 but also more general machine-learning potentials,28 or effective Hamiltonian methods,13,29 or even first-principles methods such as Density-Functional Theory (DFT), as also done in our previous work.18 Indeed, the same descriptor here used, i.e., the atomic energy calculated via an empirical potential, can be used also in conjunction with DFT30 or any other method to calculate the energy and forces. The underlying idea is that the descriptor is able to distinguish as precisely as possible the physics of the system and therefore the physically significant orbits; clearly, the more precisely this physics is described the more predictive the search will be, but in our experience simple empirical-potential (SMA) atomic energies are usually sufficient for the purpose. More precisely, according to SMA, the total energy of the i-th atom of the system is partitioned into an attractive many-body contribution, Ea(i), and a repulsive contribution, Er(i), whose analytical expressions are the following:
 | (1) |
 | (2) |
2.2 Database-derived moves (ii) and multi-descriptor grouping (iii)
We describe advances (ii) and (iii) together since, in the present proof-of-principle investigation, our databases rely on symmetry recognition and we limit ourselves to two descriptors at a time: the atomic energy and an element of local symmetry, so that both databases (ii) and multi-descriptor (iii) involve local symmetry recognition, the topic of the following section. | ||
| Fig. 2 Scheme of the grouping approach employing one or two descriptors. | ||
A low-symmetry structure must be properly centred and re-oriented before grouping atoms. The protocol we use for this purpose is as follows. The central atom is selected on the basis of maximum spherical symmetry: each atom is scrutinized as the candidate centre of the cluster, the corresponding spherical groups (orbits, built by choosing the distance from the centre as a grouping criterion) are counted, and the atom corresponding to the minimum number of orbits is selected as the cluster centre. In general, the central atom is typically close to the centre of mass of the cluster and lies on a symmetry axis. After selecting the central atom, a proper orientation of the Cartesian axes is selected, an operation of pivotal importance to exploit grouping schemes based on symmetry breaking. Due to the presence of several possible different symmetry axes in the main structural motif, such as C3 (or C6), C4 and C5 axes (see for example Fig. 1, bottom panel, referring to the 609-atom Leary tetrahedron), orienting the z-axis along one of these axes achieves the optimal strategy for an efficient grouping. An angle sweeping in polar coordinates is used to estimate the presence of the symmetry axis for each sampled orientation and select the preferred one.
Note in passing that the AugGGO exploration keeps track of all the patterns sampled by the algorithm (each one with its energy) and the so-accumulated database provides, for any size and composition, a spectrum of lowest-energy and higher-energy patterns which could be used in a statistical way to build up the partition function of the system and derive thermodynamic functions. Moreover, by performing AugGGO on different structural motifs with the same number of atoms one can investigate the competition among structural families, or one can also compare a range of different sizes and investigate the cross-over among structural families as a function of the number of atoms, as done in previous work.15–17
To conclude this section, we underline that the present multi-descriptor approach, here deployed in the proof-of-principle case of only two descriptors at a time, can naturally be extended to exploit as many descriptors as useful. This is particularly promising if combined with structural searches within a general GO scheme and specifically within the Parallel-Walker (PW) approach.12 Incidentally, to implement the PW approach, one needs to define a general function to recognize the similarity in chemical ordering between two configurations. We propose that a simple and effective recognition function corresponds to the scalar product between normalized ‘strings’, ranging between a maximum of 1 for the same string and a minimum of 0 when no sub-groups of the framework are populated with the same element.
3. Results and discussion
In the following, we will present two sets of results.(1) The first set focuses on high-symmetry motifs: our goal in this set is to confirm the accuracy of the method by comparing with previous results on similar systems obtained using the original GGO approach, provide an estimate of its computational efficiency, and show that, even in the case of high-symmetry motifs, the database-including (ii) and multi-descriptor (iii) advances here proposed offer significant advantages over the original GGO.
(2) The second set focuses on low-symmetry motifs, to demonstrate that the descriptor-based-orbit advance (i) here proposed can be applied to generic structural frameworks without any symmetry or other restrictions.
For definitiveness, we will then focus on three structures: one at size 609 atoms, corresponding to the high-symmetry motif shown in Fig. 1, and two at size 500 atoms, corresponding to two low-symmetry motif configurations found as low-energy structures in unbiased structural searches conducted for the two nanoalloys.
We have chosen two different alloys, PdPt and AgCu (where the latter nano-alloy pair is immiscible in the bulk20), that have two distinct behaviours at the nanoscale: PdPt exhibits a tendency to the formation of ordered mixing patterns,18,32 whereas AgCu retains its tendency to segregation in the form of core–shell or Janus motifs.33–36 These nanoalloys have attracted much attention in recent years thanks to their interesting catalytic properties.37–44
The energy and forces are computed via the SMA (Second Moment Approximation) potential using parametrization schemes reported in the literature.16,23
Before going into detail, let us recall two basic concepts in the nanoalloy field: the mixing energy15,17,22 and the convex hull.16 The mixing energy is defined as:
| Δ[NA, NB] = Ealloy[NA, NB] − NAEA[N]/N − NBEB[N]/N | (3) |
Once the mixing energy is calculated for a set of compositions, its convex hull46,47 can be estimated to derive nanoalloy phase diagrams.16 It can be noted in passing, in analogy with the theory of bulk alloy phase diagrams, that, when the envelope of lowest mixing energies as a function of composition has a concave (not convex) profile, the possibility of bistability and phase segregation exists.
3.1 High-symmetry motifs
To investigate high-symmetry motifs, we have chosen to investigate chemical ordering in the same 609-atom Leary tetrahedron shown in Fig. 1. A partial fragment of this motif has been singled out in GO structural searches of a PdPt nanoalloy with 500 atoms (with various compositions) and manually completed at size 609 atoms to realize a high-symmetry structure. As already discussed, this hybrid motif has been already discussed in the literature for this alloy16,25 and can be described as made of a crystalline (tetrahedral) core on the faces of which further growth in a stacking fault fashion gives rise to a local 5-fold axis along the directions of the six edges of its core. This combination of a crystalline core and surface 5-fold symmetry is effective in lowering the surface energy in both pure and alloyed systems, especially in the case of alloys made of elements (such as Pd and Pt) with a reduced size-mismatch (the experimental nearest-neighbour distances in the bulk are 2.75 and 2.77 Angstrom, respectively). For the AgCu alloy, which is characterized by a pronounced size mismatch (the experimental nearest-neighbour distances in the bulk are 2.88 and 2.49 Angstrom, respectively), this motif is not stable in the size range around 500 atoms. As we directly observed in an unbiased structural search of this alloy, highly non-crystalline motifs based on icosahedral units are stable instead.35,45 Nevertheless, we decided to test the ordering in the Leary motif also for the AgCu alloy to demonstrate that the efficiency of the AugGGO approach is not limited to low-lying structural motifs. We will consider much more stable motifs for the AgCu alloy in the results on the low-symmetry motif structure in Section 3.2.PdPt alloy In Fig. 3 (top panel) we have reported the mixing energy curves for the 609-atom Leary Td by considering 5 different grouping schemes: (a) one spherical (exploiting the full symmetry of the cluster, black line), (b) three layered on the basis of the values of the atom z-coordinates by choosing the three different orientations shown in the lower panel of Fig. 1 (purple line for C4, green line for C5 and cyan line for C6), and (c) one cylindrical on the basis of the distance of each atom from the z-axis (orange line). We have spanned a wide range of compositions from about 10% to about 90% in Pd content with a fine grid (the incremental Pd content between two consecutive points is 5 atoms). By looking at the curves, we can see that the lowest-energy one corresponds to spherical symmetry grouping; the other schemes tend to mimic (or reproduce) the spherical scheme with more success when choosing the orientation along the C4 axis (for both layered and cylindrical schemes, which practically coincide) and give the worst result when orienting along the C5 axis, due to the fact that C5 axes are found only in peripheral regions of this structure. In the lower panel of Fig. 3 we have reported the lowest-energy patterns for compositions corresponding to a Pd content of 20%, 40%, 60% and 80%, by decomposing each structure in concentric layers. We can see that, in the whole compositional range, multi-shell arrangements dominate with the tendency of Pd to populate, at first, the cluster surface and then the internal layers. On the contrary, Pt has the tendency to occupy the most internal sites with a strong tendency to segregate in the sub-surface layer at high Pd content. This tendency to show a multi-shell arrangement, which is the typical mixing pattern of elements with a strong mixing tendency, is akin to the patchy multi-shell pattern which has been predicted for PdPt nanoalloys at smaller sizes and whose stability has been proven by first-principles calculations18 (actually, the tendency to show patchy multi-shell patterns is actually strengthened by first-principles approaches with respect to the less accurate SMA empirical potential); patchy patterns can indeed be easily recognized in the internal shells at both 40% and 80% Pd content. These results are in overall agreement with previous investigations on this nanoalloy and confirm the ability of the AugGGO approach in devising the preferred pattern of chemical ordering.
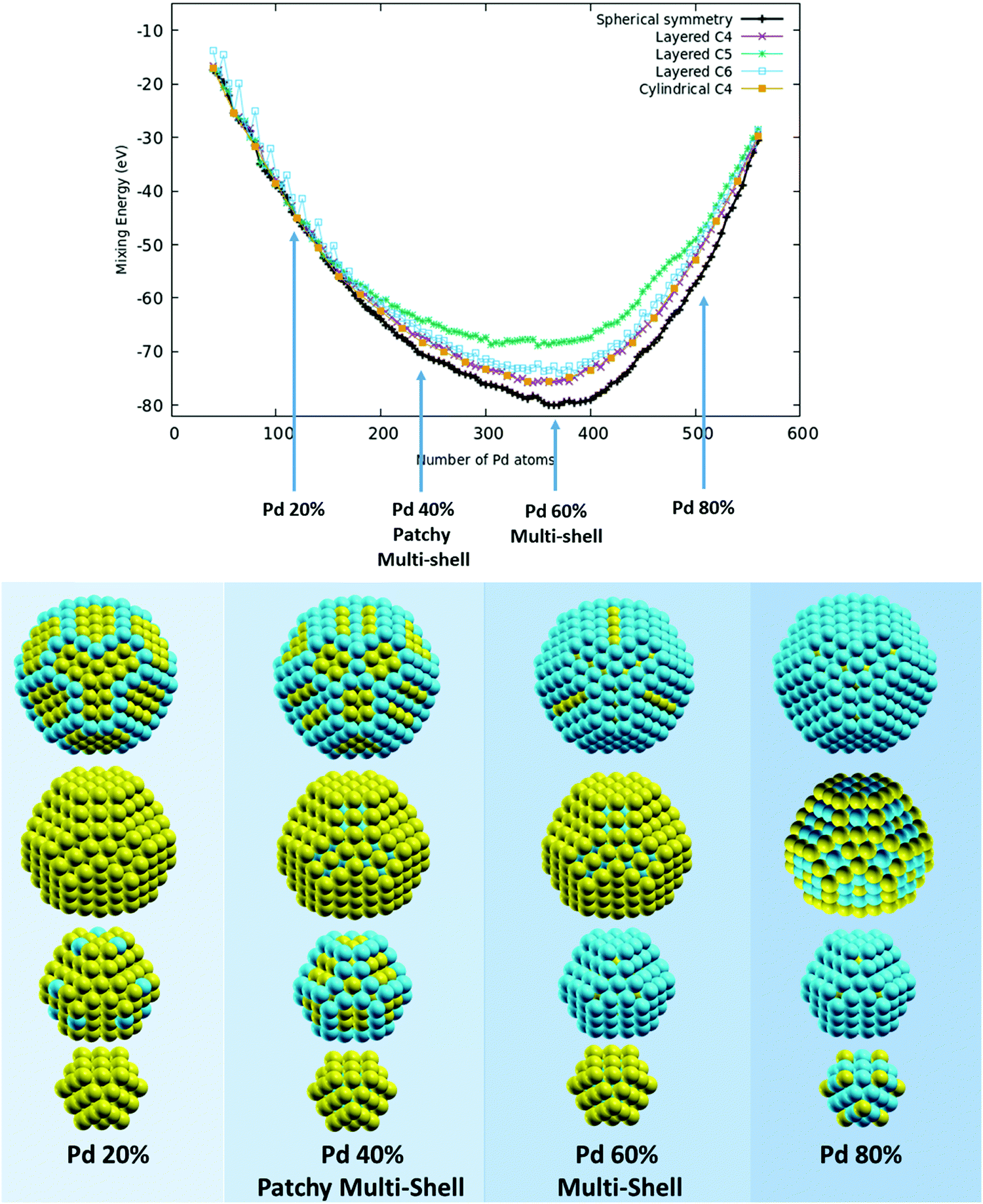 | ||
| Fig. 3 (top) Excess energy curves for the PdPt alloy in the 609-atom Leary tetrahedron by invoking spherical symmetry (black) or broken symmetry according to a layering scheme by choosing the best C4 orientation (purple), C5 orientation (green) or C6 orientation (light blue); the curve corresponding to a cylindrical grouping scheme along a C4 orientation is also reported (orange). (bottom) Chemical ordering patterns corresponding to an increasing concentration of Pd; for each composition, each structure is decomposed in layers, see the main text for further details; Pd atoms in cyan, Pt atoms in yellow. | ||
 | ||
| Fig. 4 (top) Excess energy curves for the AgCu alloy in the 609-atom Leary tetrahedron by invoking spherical symmetry (black) or broken symmetry according to a layering scheme by choosing the best C4 orientation (purple), C5 orientation (green) or C6 orientation (light blue); the curve corresponding to a cylindrical grouping scheme along a C4 orientation is also reported (orange). (bottom) Chemical ordering patterns corresponding to an increasing concentration of Ag; for each composition, each structure is visualized by using big spheres (first row) and smaller balls for Ag to highlight the internal ordering of Cu. Ag atoms in white, Cu atoms in red. | ||
From this section, we can conclude that the new implementation of the grouping algorithm has guaranteed (i) a synergic enhancement of the efficiency of the automatic grouping scheme generation, which in turn has led to (ii) a deeper comprehension of the physics of the investigated systems, highlighting for the PdPt alloy a competition between pure and patchy multi-shell ordering, whereas for AgCu the stabilization in the mixing region of core/shell regular (low Ag content) and distorted (chiral) structures (intermediate Ag content).
3.2 Low-symmetry motifs
 | ||
| Fig. 5 Excess energy curves for two selected low-symmetry structural motifs for both the PdPt alloy (for a 40% and 60% Pd composition, top panel) and the AgCu alloy (for a 40% and 80% Ag composition, bottom panel). The view style for the two alloys is the same used in Fig. 2 and 3, respectively. | ||
3.3 Assessment of the computational efficiency of the AugGGO approach
An important aspect of the present proposal regards the efficiency of the AugGGO algorithm. To assess it, we have compared the evolution of the total energy in GO runs using the AugGGO approach or a BH algorithm based on single exchange moves, as implemented in the code by Rossi et al.,12 on selected structures, diverse in size, motif and type of alloy (note that, to make the comparison exhaustive, we included sizes and structures not previously discussed). The results of this comparison are shown in Fig. 6; in each plot the evolution of the total energy as a function of the number of GO steps is reported for the two approaches (AugGGO vs. BH exchange) in the case of four examples: two for PdPt and two for AgCu, at different sizes. In all four cases, the curves prove that a remarkable reduction in the number of BH steps needed to reach the low-energy regions of the PES (Potential Energy Surface) is achieved when using the new AugGGO approach. This reduction in the number of BH steps amounts to 1–2 orders of magnitude. The much increased efficiency of AugGGO is independent of the alloy nature, the size of the cluster and of the structural motif. The plots in Fig. 6 also implicitly contain a quantitative validation of our predictions. First, it can be observed that the AugGGO and the BH-exchange curves reach asymptotic values differing in energy by only 1–2 hundredths of an eV. Moreover, we add that the putative AugGGO and the BH-exchange global minima also correspond to the same chemical-ordering patterns (not shown). This technically validates the AugGGO approach, as it demonstrates that our approach is able to reproduce the global minimum (both energy and chemical order pattern) in the phase space of the chemical ordering produced by the BH exchange approach, an approach unbiased and free of grouping constraints. In terms of the physics, we recall once more that the goal of AugGGO is not to perform a full structural GO, but one limited to the variables related to chemical ordering, irrespective of whether the investigated structural framework pertains to the absolute global minimum (indeed we know for sure that in some cases it does not). | ||
| Fig. 6 Comparison of the performance of the AugGGO approach vs. the BH-exchange approach: plots of the energy of the systems as a function of the MC steps for selected structures of the PdPt alloy (top row) and of the AgCu alloy (bottom row) for different sizes and structural models. Three different BH runs and three different grouping runs are reported in each panel. | ||
To conclude this section, we highlight the following points. (i) The AugGGO approach seems to be able to single out the preferred (global minimum) chemical ordering pattern of both high-symmetry and low-symmetry structures. Incidentally, we see in the investigated examples that the physics of the high- and low-symmetry structures is similar, implying that the high-symmetry configurations are good models to mimic real nanoclusters, although peculiar finite-size effects do occur when releasing the high-symmetry constraint. (ii) The computational efficiency of AugGGO is much improved with respect to other GO tools, such as BH-exchange, achieving the prediction of the global minimum chemical ordering in a few tens of MC steps, thus 1–2 orders of magnitude smaller than those needed in the BH-exchange algorithm. This makes the exploration of much larger structures and the simulation of complicated systems composed of several metallic elements computationally affordable and feasible.
4. Conclusions
Optimizing the functionalities of materials systems by changing their composition is an important part of any materials rational design strategy. To achieve this goal, efficient tools to explore the compositional space are strongly needed at both the experimental and computational levels, which is an extremely difficult task since this space is combinatorial, and thus exponentially increasing with the system size. Here we restrict ourselves to sampling at the computational level the stoichiometry degrees of freedom of isomers sharing a fixed structural framework (homotops) but with a different ‘chemical ordering’ or ‘compositional structure’: this sampling is still of combinatorial, and therefore non-algorithmic, complexity, as the number of homotops equals MN, where N is the total number of atoms and M is the number of components. Within this constraint, we develop an original Augmented Grouping Approach (AugGA), specifically in the form of the Augmented Grouping GO (AugGGO) scheme.The AugGA approach is based on a previously proposed ‘grouping’ philosophy, in which the number of compositional degrees of freedom of the system is decreased by defining sets of atoms (groups, or orbits, or shells) that are constrained to be populated by the same element. However, the power and scope of the grouping methods is here decisively extended by including three basic advances: (i) groups are defined on the basis of descriptors rather than point-group symmetry, (ii) chemical ordering patterns from databases are exploited, and (iii) sub-grouping is realized via a multi-descriptor strategy (here: the atomic energy and a few types of geometry pattern recognition). The first advance (i) makes the approach applicable to any generic structural framework, thus overcoming the major limitation of previous grouping methods. The use of descriptors in the definition of groups also makes the approach much more flexible and opens a wealth of possibilities in finely tuning the degrees of freedom of the proposed technique. The second advance (ii) improves the chance of achieving a thorough sampling of the core regions of nanoparticles. Finally, both the second (ii) and third (iii) advances improve the chance of catching exotic/unexpected chemical ordering arrangements, easily overlooked because they may be strongly penalized by entropic factors due to a tiny number of possible realizations, although energetically favoured.
By focusing on paradigmatic examples of two binary alloys, PdPt and AgCu, at sizes spanning from ≈500 to ≈1300 atoms, and exhibiting diverse structural motifs, here we show how AugGGO (i.e., AugGA using a Monte Carlo stochastic search in the compositional phase space) enables one to accelerate the exploration of the combinatorial phase space of large systems still maintaining thoroughness over the physically and chemically significant isomers – a mandatory step to make simulations affordable and predictive. Specifically, we demonstrate that AugGGO can predict the same global minimum compositional structure of all the investigated nanoalloys within a few tens of Monte Carlo steps and thus at a computational cost which is 1–2 orders of magnitude smaller than that of traditional Monte Carlo single-exchange techniques, i.e., the traditional approach based on individual-site moves.
Finally, we underline how the AugGA/AugGGO approach lends itself to interesting perspectives and possible further developments.
In particular, in the present work we have used the atomic energy calculated via an empirical potential as a descriptor function (or order parameter), but as discussed in Section 2 the approach is completely general and could employ any local function of atomic coordinates. This freedom will also facilitate the next development of AugGGO, that is, combining chemical ordering sampling with structural sampling into one general GO scheme12 (work is in progress along these lines). In terms of other descriptor functions (or order parameters), one could use for example those derived from a common-neighbour analysis (CNA).26
In this respect, it is interesting to note that CNA is usually applied to mono-elemental systems, but it could be easily generalized to binary or multicomponent systems.
A further, straightforward and promising perspective is the application of AugGGO to ternary, quaternary, etc. nanoalloys, high-entropy alloys,52 super-conductors,53 quasi-crystals,54etc.
The automated recognition of the best descriptor(s) to use in the definition of groups is finally a third promising advance for a user-friendly distribution of our code and to investigate physically very complex cases (this is also in progress in our lab).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Computational support from the Cineca Supercomputing Center within the ISCRA programme is gratefully acknowledged. A. F. gratefully acknowledges the contribution of the International Research Network IRN on Nanoalloys (CNRS).Notes and references
- P. Kirkpatrick and C. Ellis, Chemical space, Nature, 2004, 432, 823 Search PubMed.
- D. J. Wales and J. P. K. Doye, Energy landscapes applications clusters biomolecules and glasses, Cambridge University Press, 2003 Search PubMed.
- R. Ferrando, Structure and Properties of Nanoalloys, Elsevier, 2016 Search PubMed.
- F. Calvo, Nanoalloys – From Fundamentals to Emergent Applications, Elsevier, 2nd edn, 2020 Search PubMed.
- J. Jellinek and E. B. Krissinel, NinAlm alloy clusters: Analysis of structural forms and their energy ordering, Chem. Phys. Lett., 1996, 258, 283–292 Search PubMed.
- M. Sahimi and H. Hamzehpour, Efficient computational strategies for solving global optimization problems, Comput. Sci. Eng., 2010, 12, 74–82 Search PubMed.
- Z. Li and H. A. Scheraga, Monte Carlo-minimization approach to the multiple-minima problem in protein folding, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 6611–6615 Search PubMed.
- R. L. Johnston, Evolving better nanoparticles: Genetic algorithms for optimising cluster geometries, Dalton Trans., 2003, 4193–4207 Search PubMed.
- N. Metropolis, A. W. Rosenbluth, M. N. Rosenbluth, A. H. Teller and E. Teller, Equation of State Calculations by Fast Computing Machines, J. Chem. Phys., 2004, 21, 1087 Search PubMed.
- J. P. K. Doye and D. J. Wales, Thermodynamics of global optimization, Phys. Rev. Lett., 1998, 80, 1357–1360 Search PubMed.
- G. Barcaro, L. Sementa, F. R. Negreiros, I. O. Thomas, S. Vajda and A. Fortunelli, in Oxide Materials at the Two-Dimensional Limit, ed. F. P. Netzer and A. Fortunelli, Springer, Berlin Heidelberg, 2016, vol. 234, pp. 39–90 Search PubMed.
- G. Rossi and R. Ferrando, Searching for low-energy structures of nanoparticles: a comparison of different methods and algorithms, J. Phys.: Condens. Matter, 2009, 21, 084208 Search PubMed.
- S. M. Kozlov, G. Kovács, R. Ferrando and K. M. Neyman, How to determine accurate chemical ordering in several nanometer large bimetallic crystallites from electronic structure calculations, Chem. Sci., 2015, 6, 3868–3880 Search PubMed.
- S. V. Karewar, N. Gupta, A. Caro and S. G. Srinivasan, A concentration dependent embedded atom method potential for the Mg-Li system, Comput. Mater. Sci., 2014, 85, 172–178 Search PubMed.
- A. Fortunelli and A. M. Velasco, THEOCHEM, 1999, 487, 251–266 Search PubMed.
- L. O. Paz-Borbón, T. V. Mortimer-Jones, R. L. Johnston, A. Posada-Amarillas, G. Barcaro and A. Fortunelli, Structures and energetics of 98 atom Pd-Pt nanoalloys: Potential stability of the Leary tetrahedron for bimetallic nanoparticles, Phys. Chem. Chem. Phys., 2007, 9, 5202–5208 Search PubMed.
- G. Barcaro, L. Sementa and A. Fortunelli, A grouping approach to homotop global optimization in alloy nanoparticles, Phys. Chem. Chem. Phys., 2014, 16, 24256–24265 Search PubMed.
- G. Barcaro, A. Fortunelli, M. Polak and L. Rubinovich, Patchy multishell segregation in Pd–Pt alloy nanoparticles, Nano Lett., 2011, 11, 1766–1769 Search PubMed.
- S. Bajaj, M. G. Haverty, R. Arróyave, W. A. Goddard and S. Shankar, Phase stability in nanoscale material systems: Extension from bulk phase diagrams, Nanoscale, 2015, 7, 9868–9877 Search PubMed.
- ASM Handbook, Volume 03 – Alloy Phase Diagrams, ed. H. Okamoto, M. E. Schlesinger and E. M. Mueller, ASM International, 2016 Search PubMed.
- P. J. Spencer, A brief history of CALPHAD, CALPHAD: Comput. Coupling Phase Diagrams Thermochem., 2008, 32, 1–8 Search PubMed.
- R. Ferrando, A. Fortunelli and G. Rossi, Quantum effects on the structure of pure and binary metallic nanoclusters, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 085449 Search PubMed.
- F. Cleri and V. Rosato, Tight-binding potentials for transition metals and alloys, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 22–33 Search PubMed.
- R. H. Leary and J. P. K. Doye, Tetrahedral global minimum for the 98-atom Lennard-Jones cluster, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1999, 60, R6320 Search PubMed.
- L. O. Paz-Borbón, R. L. Johnston, G. Barcaro and A. Fortunelli, A mixed structural motif in 34-atom Pd–Pt clusters, J. Phys. Chem. C, 2007, 111, 2936–2941 Search PubMed.
- J. D. Honeycutt and H. C. Andersen, Molecular dynamics study of melting and freezing of small Lennard-Jones clusters, J. Phys. Chem., 1987, 91, 4950–4963 Search PubMed.
- C. Steger, M. Ulrich and C. Wiedemann, Machine vision algorithms and applications, Wiley-VCH, 2018 Search PubMed.
- J. Behler and M. Parrinello, Generalized neural-network representation of high-dimensional potential-energy surfaces, Phys. Rev. Lett., 2007, 98, 146401 Search PubMed.
- L. Rubinovich, M. I. Haftel, N. Bernstein and M. Polak, Compositional structures and thermodynamic properties of Pd–Cu, Rh–Pd, and Rh–Pd–Cu nanoclusters computed by a combined free-energy concentration expansion method and tight-binding approach, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 035405 Search PubMed.
- M. Jäger, R. Schäfer and R. L. Johnston, First principles global optimization of metal clusters and nanoalloys, Adv. Phys.: X, 2018, 3, 1077–1108 Search PubMed.
- L. D. Marks and L. D. Marks, Surface structure and energetics of multiply twinned particles, Philos. Mag. A, 1984, 49, 81–93 Search PubMed.
- Q. Yuan, Z. Zhou, J. Zhuang and X. Wang, Pd-Pt random alloy nanocubes with tunable compositions and their enhanced electrocatalytic activities, Chem. Commun., 2010, 46, 1491–1493 Search PubMed.
- C. Langlois, Z. L. Li, J. Yuan, D. Alloyeau, J. Nelayah, D. Bochicchio, R. Ferrando and C. Ricolleau, Transition from core–shell to Janus chemical configuration for bimetallic nanoparticles, Nanoscale, 2012, 4, 3381 Search PubMed.
- D. Bochicchio and R. Ferrando, Morphological instability of core-shell metallic nanoparticles, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 165435 Search PubMed.
- I. Parsina and F. Baletto, Tailoring the structural motif of AgCo nanoalloys: Core/shell versus janus-like, J. Phys. Chem. C, 2010, 114, 1504–1511 Search PubMed.
- F. Baletto, Structural properties of sub-nanometer metallic clusters, J. Phys.: Condens. Matter, 2019, 31, 113001 Search PubMed.
- Z. Peng and H. Yang, Synthesis and oxygen reduction electrocatalytic property of Pt-on-Pd bimetallic heteronanostructures, J. Am. Chem. Soc., 2009, 131, 7542–7543 Search PubMed.
- A. X. Yin, X. Q. Min, W. Zhu, H. S. Wu, Y. W. Zhang and C. H. Yan, Multiply twinned Pt-Pd nanoicosahedrons as highly active electrocatalysts for methanol oxidation, Chem. Commun., 2012, 48, 543–545 Search PubMed.
- H. Zhang, M. Jin and Y. Xia, Enhancing the catalytic and electrocatalytic properties of Pt-based catalysts by forming bimetallic nanocrystals with Pd, Chem. Soc. Rev., 2012, 41, 8035–8049 Search PubMed.
- P. Zhang, Y. Hu, B. Li, Q. Zhang, C. Zhou, H. Yu, X. Zhang, L. Chen, B. Eichhorn and S. Zhou, Kinetically stabilized Pd@Pt core-shell octahedral nanoparticles with thin Pt layers for enhanced catalytic hydrogenation performance, ACS Catal., 2015, 5, 1335–1343 Search PubMed.
- A. Laghrissi and M. Es-Souni, Porous PtPd alloy nanotubes: Towards high performance electrocatalysts with low Pt-loading, Catal. Sci. Technol., 2019, 9, 4355–4364 Search PubMed.
- J. M. Conesa, M. V. Morales, C. López-Olmos, I. Rodríguez-Ramos and A. Guerrero-Ruiz, Comparative study of Cu, Ag and Ag-Cu catalysts over graphite in the ethanol dehydrogenation reaction: Catalytic activity, deactivation and regeneration, Appl. Catal., A, 2019, 576, 54–64 Search PubMed.
- K. Shin, D. H. Kim, S. C. Yeo and H. M. Lee, Catalysis Today, Elsevier, 2012, vol. 185, pp. 94–98 Search PubMed.
- X. Liu, D. Wang and Y. Li, Synthesis and catalytic properties of bimetallic nanomaterials with various architectures, Nano Today, 2012, 7, 448–466 Search PubMed.
- R. Ferrando, Symmetry breaking and morphological instabilities in core-shell metallic nanoparticles, J. Phys.: Condens. Matter, 2015, 27, 013003 Search PubMed.
- D. Hildebrandt and D. Glasser, Predicting phase and chemical equilibrium using the convex hull of the Gibbs free energy, Chem. Eng. J., 1994, 54, 187–197 Search PubMed.
- N. Perevoshchikova, B. Appolaire, J. Teixeira, E. Aeby-Gautier and S. Denis, A convex hull algorithm for a grid minimization of Gibbs energy as initial step in equilibrium calculations in two-phase multicomponent alloys, Comput. Mater. Sci., 2012, 61, 54–66 Search PubMed.
- D. Bochicchio and R. Ferrando, Size-dependent transition to high-symmetry chiral structures in AgCu, AgCo, AgNi, and AuNi nanoalloys, Nano Lett., 2010, 10, 4211–4216 Search PubMed.
- C. Langlois, Z. L. Li, J. Yuan, D. Alloyeau, J. Nelayah, D. Bochicchio, R. Ferrando and C. Ricolleauv, Transition from core-shell to Janus chemical configuration for bimetallic nanoparticles, Nanoscale, 2012, 4, 3381–3388 Search PubMed.
- A. Fortunelli and G. Barcaro, Density-Functional Theory of Free and Supported Metal Nanoclusters and Nanoalloys, in Metal Clusters and Nanoalloys, ed. M. M. Mariscal, O. A. Oviedo and E. P. Marcos Leiva, Springer, New York, NY, 2013, pp. 29–79 Search PubMed.
- G. Rossi, A. Rapallo, C. Mottet, A. Fortunelli, F. Baletto and R. Ferrando, Magic polyicosahedral core-shell clusters, Phys. Rev. Lett., 2004, 93, 105503 Search PubMed.
- Y. F. Ye, Q. Wang, J. Lu, C. T. Liu and Y. Yang, High-entropy alloy: challenges and prospects, Mater. Today, 2016, 19, 349–362 Search PubMed.
- P. Koželj, S. Vrtnik, A. Jelen, S. Jazbec, Z. Jagličić, S. Maiti, M. Feuerbacher, W. Steurer and J. Dolinšek, Discovery of a Superconducting High-Entropy Alloy, Phys. Rev. Lett., 2014, 113, 107001 Search PubMed.
- J.-M. Dubois, Properties- and applications of quasicrystals and complex metallic alloys, Chem. Soc. Rev., 2012, 41, 6760–6777 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp03583e |
| This journal is © the Owner Societies 2021 |