
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMethanol oxidation on Au(332): an isothermal pulsed molecular beam study†
Christoph D.
Feldt
 a,
Thorren
Gimm
ab,
Raphaell
Moreira
a,
Wiebke
Riedel
*a and
Thomas
Risse
*a
a,
Thorren
Gimm
ab,
Raphaell
Moreira
a,
Wiebke
Riedel
*a and
Thomas
Risse
*a
aInstitut für Chemie und Biochemie, Freie Universität Berlin, Arnimallee 22, 14195 Berlin, Germany. E-mail: wiebke.riedel@fu-berlin.de; risse@chemie.fu-berlin.de
bHelmholtz-Zentrum Berlin für Materialien und Energie, Hahn-Meitner-Platz 1, 14109 Berlin, Germany
First published on 24th September 2021
Abstract
Isothermal molecular beam experiments on the methanol oxidation over the stepped Au(332) surface were conducted under well-defined ultra-high vacuum conditions. In the measurements, a continuous flux of methanol at excess in the gas phase and pulses of atomic oxygen were provided to the surface kept at 230 K. The formation of the partial oxidation product methyl formate under the applied conditions was evidenced by time-resolved mass spectrometry, and accumulation of formate species, which resulted in a deactivation of the surface for methyl formate formation, was followed by in situ Infrared Reflection Absorption Spectroscopy measurements. The results suggest a different reactivity of oxygen accumulated during the oxygen pulses and atomic oxygen for the competing reaction pathways in the oxidation of methanol to the desired partial and the unwanted overoxidation products.
Introduction
Gold catalysis has received much attention since the first report by Haruta and co-workers.1–3 In recent years, a renewed interest emerged due to the unusual reactivity of nanoporous gold (np-Au) catalysts:4–7 While it was reported that only very small Au nanoparticles (<5–7 nm) on oxide supports are catalytically active,1,2,8,9 np-Au catalysts exhibit a high activity at low temperatures without an oxidic support and despite relatively large ligament sizes. Np-Au is formed by etching a less noble metal from an alloy with Au resulting in the formation of a porous structure. The resulting material contains mostly Au, but also residuals of the less noble metal, most often Ag, and consists of ligaments exhibiting both a number of low-coordinated sites as well as close-packed surfaces.10 In specific, these np-Au catalysts have shown a high selectivity at high conversion for the partial oxidation of methanol to methyl formate.6 Residual Ag is suggested to play an important role in the activation of molecular oxygen on these catalysts which is typically the rate limiting step for oxidation reactions on gold.6,11 While a variety of oxygen species, including also ordered and disordered oxidic phases, with different reactivities have been reported depending on the applied reaction conditions,12,13 the concentration of activated oxygen under typical reaction conditions is believed to be rather low due to a small number of active sites.11,14 In methanol oxidation, short contact times were reported to favor formation of formaldehyde over the coupling reaction to methyl formate,11 while an increase in oxygen or residual Ag content was connected to an enhancement in total oxidation products.6,11,12,14As the microscopic understanding of the processes on these catalysts remains incomplete, a number of studies on simplified model systems, such as Au single crystal surfaces, under well-defined ultra-high vacuum (UHV) conditions have been conducted.15–17 In agreement with results for np-Au, Temperature Programmed Reaction (TPR) measurements on Au(111) showed an enhanced selectivity to total oxidation for increased oxygen coverages as well as a lower methyl formate production.15 Moreover, TPR measurements indicated a lowered methanol reactivity in the presence of extended oxidic gold (AuOx) phases for Au(110).17 Besides CO2 and methyl formate, also formaldehyde and formic acid were reported as products, their ratio depending on the applied reaction conditions and surface coverages.15,16,18,19 Based on TPR measurements, a reaction mechanism for methyl formate formation by partial methanol oxidation in the presence of activated oxygen was proposed proceeding via a methoxy intermediate and a subsequent β-elimination to formaldehyde as rate-limiting step.15,18,20 Moreover, the formation of a dioxymethylene intermediate and formate species, which are formed at temperatures ≤200 K and stable up to at least 255 K, were reported as precursors for total oxidation products as well as for formic acid.15,18–21 However, the model studies are mainly focused on low-index surfaces and reactivity has been mostly studied by TPR measurements on surfaces pre-covered with activated oxygen species whereas catalytic activity of np-Au was studied under isothermal conditions using molecular oxygen as the oxidizing agent. Residual metals such as Ag or Cu present in np-Au catalysts were shown to play an important role in oxygen activation.6,11,12,14 In addition, low-coordinated sites present in large number on the ligaments of np-Au are considered to influence the catalytic properties as well, however, experimental evidence for this is scarce.22,23
In this study, an Au(332) surface exhibiting densely packed [110]-oriented steps separated by 6 atom wide (111) terraces is used as a model system. Previous structural investigations have shown that the steps may contain kinks depending on the crystal miscut.24 On this surface, pulsed molecular beam (MB) experiments under well-defined single collision conditions are conducted to investigate the kinetics of the methanol oxidation under isothermal conditions. As gold single crystal surfaces cannot activate molecular oxygen under ultra-high vacuum conditions, pulses of atomic oxygen supplied by a thermal cracker were used to allow for the oxidation reactions. The evolution of the gas phase products is monitored by time-resolved mass spectrometry, and in situ Infrared Reflection Absorption Spectroscopy (IRAS) is used for the detection of adsorbed surface species.
Experimental details
All experiments were conducted in an UHV setup consisting of two chambers previously described in detail.25 One chamber contains a sputter gun (IQE 11/35, SPECS), a low-energy electron diffraction (LEED) system (Omicron MCP LEED), an Auger spectrometer (PHI 11-010, PerkinElmer), and a quadrupole mass spectrometer (QMS, Prisma, Pfeiffer) with a Feulner cup for temperature programmed desorption (TPD) measurements. The second chamber is equipped with two effusive molecular beams26 and a thermal atomic oxygen source (Dr Eberl MBE-Komponenten GmbH), that can be modulated by automated valves and shutters, as well as with a stagnation flow monitor with a high precision ion gauge (360 Stabil-Ion, Granville-Phillips) to measure the pressure at and its distribution over the sample. A quadrupole mass spectrometer (MAX-500HT, Extrel) is used to monitor the evolution of gas phase species (CH2O+ fragment at m/z = 30; H3COCHO+ at m/z = 60; CO2+ at m/z = 44) over time in the pulsed isothermal molecular beam experiments. For in situ IRAS measurements in grazing reflection geometry, an IR spectrometer (IFS 66v, Bruker) is used (256 scans, nominal resolution of 4 cm−1, zero filling factor of 16). The Au(332) single crystal (10 mm diameter, 2 mm thick, Mateck) is mounted by Mo-clamps onto a boron nitride heater (HT-01, Momentive) which is attached to a home-made Mo-holder connected to a liquid nitrogen cooled Cu block allowing for sample cooling down to 100 K. The crystal temperature is measured by a type K thermocouple fixed in a 0.2 mm hole in the edge of the Au crystal. The thermocouple voltage is monitored by a commercial PID controller (3508, Eurotherm) allowing also for control of the sample temperature in TPD and isothermal experiments.The Au(332) surface was cleaned by repeated cycles of Ar+ ion sputtering (1000 V, 5–7 µA, 15 min) and subsequent annealing to 1000 K i. vac. for 10 min until no impurities could be detected by Auger spectroscopy and a sharp LEED image as expected for the (332) surface27 was observed (see Fig. S1, ESI†). Before use, methanol (Roth, ≥99.98%; dried over molecular sieve, 3 Å) and methyl formate (Sigma Aldrich, ≥99.8%) were cleaned by repeated freeze–pump–thaw cycles and applied to the sample surface through an effusive molecular beam. Oxygen (Air Liquide, 99.998%) was used as received. The pressure on the sample as function of the inlet-pressure of the beam was calibrated by a beam monitor at the sample position using Ar gas. Atomic oxygen was supplied by means of a thermal cracker (T = 1615 °C, 12.45 V, 12.60 A). The flux of atomic oxygen was calibrated by TPD measurements in the same UHV apparatus comparing the integrated signal intensities of O2 desorption from an Au(332) surface with respect to O2 desorption from Pt(111) after O2 exposure at 300 K resulting in p(2 × 2) structure with a coverage of 0.25 ML.28,29 A saturation coverage of 2.1 ML (1 ML corresponding to 1.4 × 1015 cm−2, thus, to one O atom per Au surface atom) was obtained on the Au(332) surface by exposure to atomic oxygen from the thermal cracker. The QMS signal intensity for methyl formate formation was quantified by calibration measurements with varying methyl formate fluxes dosed into the chamber using a well-defined effusive molecular beam source applying a non-reactive flag at the sample position to ensure comparable scattering conditions.
Results and discussion
Fig. 1 displays the results of a pulsed isothermal molecular beam experiment on the partial oxidation of methanol to methyl formate on the Au(332) surface. The reaction is conducted at 230 K by applying a constant flux of methanol (1.6 × 10−7 mbar, 4.2 × 1013 s−1 cm−2) and pulsing (200 s on, 300 s off) atomic oxygen (2.6 × 10−3 ML s−1, 0.4 × 1013 s−1 cm−2, approx. 0.5 ML per pulse) onto the surface. In these experiments, the methanol flux onto the surface is about one order of magnitude higher than that of atomic oxygen. The surface temperature of 230 K was chosen above the desorption temperature of methanol, formaldehyde and methyl formate from Au surfaces to allow for gas phase detection and prevent build-up of ice layers, but low compared to temperatures used in np-Au studies to ensure sufficiently long residence times of methanol or formaldehyde to allow for the coupling reaction to methyl formate under the applied single collision conditions. The methyl formate formation rate is followed by time-resolved QMS, while surface adsorbed species are investigated by in situ IRAS measurements. It can be seen in Fig. 1a that methyl formate is produced on Au(332) at 230 K under the applied isothermal, single collision conditions. Based on the quantitative calibrations for the oxygen atom flux and the methyl formate formation rate, the initial methyl formate formation rate amounts to approx. 4 × 1011 s−1 cm−2 corresponding to a selectivity of approx. 20% with respect to the supplied oxygen atoms. Fragmentation signals of methanol and methyl formate hampered the quantification of formaldehyde formation which is expected to compete effectively with the coupling reaction to methyl formate at 230 K under single collision conditions. Similarly, quantification of the total oxidation product CO2 was hindered by fragmentation signals of methyl formate and also by signals due to background reactions in the chamber. Across the pulse sequence, the methyl formate formation rate decreases roughly linearly indicating a surface deactivation for this reaction. In situ IRAS measurements (duration approx. 3 min) were conducted both during the oxygen pulse (start approx. 5–10 s after beginning of the oxygen pulse) and during the delay times (start after approx. 10–20 s after end of the oxygen pulse, when the methyl formate formation rate was roughly decreased to background level). These in situ IRAS measurements shown in Fig. 1b demonstrate the appearance of a strong signal centered at 1332 cm−1 and weaker signals around 2832 cm−1 and 2907 cm−1. The intensity of the peaks grows across the pulse sequence, while their positions remain largely constant and no clear additional peaks occur. While the observed IRAS signals are shifted with respect to signals attributed by Xu et al. to formate species on Au(111),15 the positions are overall comparable to signals reported by Senanayake et al. at 1332 cm−1, 2824 cm−1 and 2896 cm−1 assigned to formate prepared by adsorption of formic acid on O pre-covered Au(111) and attributed to the νs(OCO) stretching, νaCH and a combination of the νs(OCO) and νaCH modes of bidentate formate species, respectively.30 The signals attributed to formate species remain stable on the surface after the pulsed isothermal MB experiment conducted at 230 K, in agreement with previous literature reports for Au surfaces.15,18,30 Thus, despite the about 10 fold excess of methanol flux, an overoxidation of methanol, as compared to the desired product methyl formate (or formaldehyde), occurs under the applied conditions.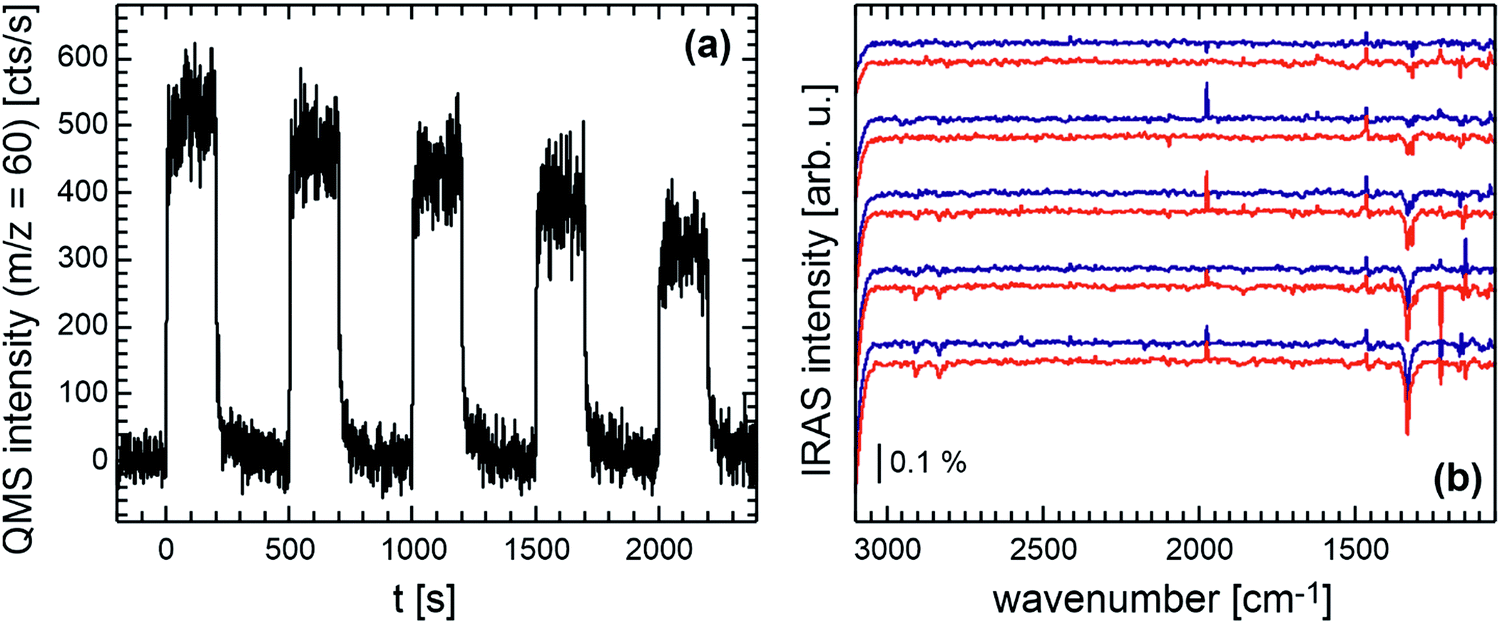 | ||
| Fig. 1 Pulsed isothermal molecular beam experiment on the methanol oxidation on Au(332) at 230 K: (a) methyl formate formation rate (m/z = 60) and (b) in situ IRAS measurements during the oxygen pulse (blue) and in the delay times (red) across the pulse sequence (from top to bottom). | ||
Considering the transient behavior of the methyl formate formation rate in more detail (Fig. 1a), it can be seen that the rate upon oxygen exposure rapidly increases and subsequently approaches a (quasi) steady state value for each oxygen pulse. It should be noted that no clear rate decrease during the pulse duration is detected which may be expected, if the surface deactivates during the pulse e.g. due to the formation of formate. After switching off the oxygen supply, the methyl formate formation rate decreases initially very fast and levels back to baseline within about 100 s. For the first pulse, the corresponding amount of methyl formate desorbing during the delay time is about 5% of the amount produced during the pulse. This suggests that a fraction of the deposited oxygen atoms is still present at the end of the oxygen pulse. The methyl formate formation rate for the subsequent oxygen pulse is, however, significantly decreased indicating the progressing deactivation for methyl formate formation to occur rather in the delay time between the oxygen pulses. Concomitantly, the IRAS signal intensity of the formate related peaks (Fig. 1b) is found to increase mostly during the delay times between the oxygen pulses. The increase in formate concentration during the delay times clearly shows that some of the oxygen present at the end of the pulse results in the formation of the unwanted overoxidation product.
The observation of a higher effective formation rate of formate species under rather oxygen-deficient conditions, i.e. between oxygen pulses, is unexpected at first glance, as the formation of overoxidation products, such as formate in the absence of an oxygen supply, i.e. conditions of low oxygen availability, is counterintuitive. It can be understood assuming that formate is not a spectator species, i.e. the formate cannot only be formed but also removed, in particular during the oxygen pulses, which allows their concentration to stay almost constant during the pulse. The increase of the formate concentration in the delay times thus points to a significant reduction of the decomposition channel under rather oxygen-deficient conditions. To verify that formate can be further oxidized already at 230 K during conditions of the oxygen pulses, a formate covered Au(332) surface, as obtained after a pulsed isothermal methanol oxidation experiment discussed above, was exposed to atomic oxygen (same flux without additional methanol exposure) and IRAS measurements were conducted to follow the evolution of the formate signals. The IRAS measurements, displayed in Fig. 2, clearly show that the IRAS intensity of the formate signals (e.g. the band around 1332 cm−1) decreases and even vanishes for a prolonged exposure to atomic oxygen. As some of the oxygen is expected to be consumed in the oxidative decomposition of formate, an oxygen TPD (Fig. S2, ESI†) was conducted after the experiment displayed in Fig. 2. Desorption of residual oxygen shows that not all oxygen is consumed by the oxidative decomposition of formate species. However, the amount of desorbing oxygen is lower than expected for a clean Au(332) surface (Fig. S2, ESI†). Moreover, for a formate surface coverage, for which the IRAS signal intensity is saturated, the reduction in oxygen desorption is consistent with an overall high surface coverage of the formate species. A further quantification by monitoring the presumable product CO2 of the formate decomposition is prevented by fragmentation signals of methyl formate as well as non-negligible background reactions in the chamber.
 | ||
| Fig. 2 In situ IRAS measurements of a formate pre-covered Au(332) surface obtained after isothermal methanol oxidation experiment (i) directly after formate formation and after exposure with atomic oxygen (0.4 × 1013 s−1 cm−2) at 230 K for (ii) 230 s, (iii) 410 s and (iv) 610 s. | ||
The analysis of an isothermal experiment with 15 oxygen pulses shows that the formation of formate species as quantified by the IRAS signal intensity of the formate species at 1332 cm−1 is clearly correlated with the decrease in the methyl formate formation rate (Fig. 3a, see also Fig. S3, ESI†). After the 10th pulse of the pulse sequence, the methyl formate formation rate levels below 10% of the initial rate and the IRAS intensity saturates. This suggests that the accumulating formate species block surface sites required for the coupling reaction and thereby deactivate the surface for methyl formate formation.
 | ||
| Fig. 3 Results of a pulsed isothermal experiment on the methanol partial oxidation on Au(332) at 230 K using conditions identical to Fig. 1. (a) Methyl formate formation rate (m/z = 60) (see also Fig. S3, ESI†) as function of the integrated IRAS intensity of the formate signal centered at 1332 cm−1 obtained from measurements during the first 10 oxygen pulses and (b) intensity of the QMS signals m/z = 60 (black circles), m/z = 18 (blue squares) and m/z = 30 (red triangles) as a function of the number of O pulses. | ||
While the methyl formate formation is strongly reduced across the pulse sequence, the surface remains active for other reaction channels as indicated by the evolution of the signal intensity of m/z = 30 (formaldehyde) and m/z = 18 (water) during the pulse sequence (Fig. 3b). Even though a quantification of these products is not possible, the data show a clear increase of the signal intensity of m/z = 30 (red triangles) during the first 6 pulses which exhibits only a slight decrease towards the end of the experiment. This indicates a shift in selectivity from the coupling reaction yielding methyl formate towards desorption of formaldehyde, as the surface becomes covered with formate species. The H2O formation (m/z = 18, black squares) remains, after an initial drop, constant until the m/z = 30 signal reaches its maximum. Afterwards, the m/z = 18 signal decreases to a constant, but slightly lower intensity for the last pulses in this series.
How to reconcile this behavior? To this end, it is important to recall that partial oxidation of methanol to formaldehyde and methyl formate results in the same amount of water per oxygen atom, while the formation of higher oxidation products such as formate or CO2 produces less water.
| 2CH3OH + 2Oact → H3COCHO + 2H2O | (R1) |
| 2CH3OH + 2Oact → 2CH2O + 2H2O | (R2) |
| CH3OH + 3Oact → CO2 + 2H2O | (R3) |
| 2CH3OH + 5Oact → 2HCOO + 3H2O | (R4) |
Thus, the almost constant water formation after the initial drop may result from a compensation of the decreasing methyl formate formation with the increasing formaldehyde production. However, towards the end of the experiment, the water formation drops to a reduced level which is accompanied by a slight reduction of formaldehyde formation. This is consistent with a selectivity shift to overoxidation where the fraction of oxygen that is incorporated into C-containing products, e.g. formate, increases, resulting in a decrease of the amount of water that can be produced per oxygen atom. The observation is thus consistent with a formation and subsequent oxidative decomposition of formate during the oxygen pulses (see Fig. 2) which competes with methanol oxidation to methyl formate and formaldehyde under the applied conditions.
While the formal reaction eqn (R1)–(R4) can explain the observed changes of the different reaction products throughout the pulse series, they do not allow to gain insight into the mechanistic reason for this behavior. With respect to this, it is important to consider the activated oxygen species, globally denoted Oact in the equations, in more detail.
Xu et al. reported TPR experiments of methanol oxidation on O pre-covered Au(111) and observed a decreasing selectivity towards methyl formate formation with increasing oxygen pre-coverage.15 This is in line with reports on np-Au showing that in the presence of oxidic phases as e.g. prepared by an ozone treatment of the sample exposure to methanol at 150 °C leads to total oxidation,12,13,32 while partial oxidation to methyl formate or formaldehyde on ozone-treated np-Au is detected rather in the absence of the oxidic phases and in the presence of different “selective” oxygen species.12 However, a detailed microscopic understanding of the correlation between the different oxygen species (atom, oxygen islands, preferred decoration of step edges, or oxides as indicated by the high saturation coverage etc.) and their activity for the different reaction pathways is not yet available.12,13,17,31–33 The experiments presented above allow to shed some light on this question. To this end Fig. 4 provides a simplified reaction network based on the mechanism proposed in literature also indicating contributions of the different oxygen species to reaction channels.15,18,20
 | ||
| Fig. 4 Simplified reaction network for the partial oxidation of methanol on Au and competing reaction pathways i.e. desorption formaldehyde and overoxidation of formaldehyde to formate and subsequently CO2. Subscript (a) denotes adsorbed species; subscript (g) gas phase products. Methyl formate is highlighted because the importance of oxygen atoms for the kinetics of the different reaction steps is unknown. | ||
We basically observe constant methyl formate formation rates during the oxygen pulse (Fig. 1a) despite the fact that oxygen accumulates on the surface in this period. This strongly suggests that accumulated oxygen does not alter the activity of the surface for this reaction channel during the pulse, e.g. by blocking sites for methyl formate formation. In contrast to that, increasing amounts of adsorbed formate species correlate with a decreasing formation rate of methyl formate which points to the fact that adsorbed formate species can block sites active for the coupling reaction to methyl formate. As previously shown, oxygen atoms tend to form AuOx islands/phases whose reactivity and selectivity depend e.g. on the size of the islands,17,31,34 and is expected to be different than that of atomic oxygen supplied by the thermal cracker. From the results presented here it is possible to deduce that formate cannot only be formed but is also removed during the oxygen pulses presumably by the formation of CO2 which desorbs at the temperature chosen here. Hence, the final oxidation of formate to CO2 is at least significantly suppressed in the absence of oxygen atoms, while formate formation is readily possible by AuOx islands/phases. This is in line with TPR-like experiments in which CO2 and H2O desorption due to formate decomposition from Au(111) was reported around 280 K and on Au(110) around 340 K, i.e. at higher temperatures, despite the presence of residual oxygen.15,35 In case the reaction network sketched in Fig. 4 resembles the reaction mechanism, one can conclude that the formation of formaldehyde has to be feasible on AuOx islands/phases as well, because of the formation of formate in the delay times between the oxygen pulses.36
Previously, it has been shown that at least small AuOx islands/phases (low oxygen coverage) allow to form methyl formate with high selectivity on Au(111). As we accumulate oxygen on the surface and thus increase the number of possible reaction sites on such AuOx islands/phases, one would expect a change in the methyl formate formation rate in case this reaction channel would contribute considerably to the rate of methyl formate formation. This is not observed in the experiment. This suggests that atomic oxygen species are kinetically more important in the isothermal experiments performed here, however, their precise role for the different reaction steps is not known.
Conclusions
Pulsed isothermal molecular beam experiments were conducted to investigate the partial oxidation of methanol to methyl formate on the stepped Au(332) surface at 230 K. While methyl formate formation was observed under the applied single collision conditions, the formation rate was found to decrease across the pulse sequence due to the build-up of formate species as evidenced by in situ IRAS measurements. The concentration of the formate species was found to effectively increase in the delay times between the oxygen pulses, thus, under rather oxygen-deficient conditions during which methyl formate formation is rather limited. The lack of formate build-up during the pulses is due to a faster oxidative decomposition of formate species occurring even at the low surface temperature of 230 K during the exposure with atomic oxygen i.e. under more strongly oxidizing conditions. In contrast to formate species, accumulating oxygen expected to form AuOx-phases was not found to deactivate the methyl formate formation during the pulses. Hence, its presence does not change the selectivity of the reaction strongly towards total oxidation products, which is in contrast to TPR studies suggesting a strong dependence of the selectivity on the oxygen coverage. These results further suggest a lower reactivity of accumulated oxygen for the methyl formate formation and the oxidative decomposition of formate, while residual oxygen forms formate species effectively. Even though the ability to form methyl formate is rapidly lost under these conditions, the results indicate that the surface remains active for the formation of formaldehyde throughout the sequence, suggesting that the Au surface capability to do partial oxidation of methanol is at least transiently present during the oxygen pulses. However, the local concentration of methoxy species required for a coupling to methyl formate is reduced such that the formaldehyde desorption becomes the dominant channel.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We acknowledge the financial support from the German Research Foundation (DFG) within the framework of research unit 2231 “NAGOCAT” Project No. RI 1025/3-1(2). C. D. F. thanks the International Max-Planck Research School “Functional Interfaces in Physics and Chemistry” for support and the IMPRS for Elementary Processes in Physical Chemistry. R. M. thanks to the DAAD for support (Project No.: 290149/2014-2 DAAD/CNPq).References
- M. Haruta, T. Kobayashi, H. Sano and N. Yamada, Chem. Lett., 1987, 405–408 CrossRef.
- R. Meyer, C. Lemire, S. K. Shaikhutdinov and H. Freund, Gold Bull., 2004, 37, 72–124 CrossRef.
- A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896–7936 CrossRef.
- V. Zielasek, B. Jürgens, C. Schulz, J. Biener, M. M. Biener, A. V. Hamza and M. Bäumer, Angew. Chem., Int. Ed., 2006, 45, 8241–8244 CrossRef CAS PubMed.
- C. X. Xu, J. X. Su, X. H. Xu, P. P. Liu, H. J. Zhao, F. Tian and Y. Ding, J. Am. Chem. Soc., 2007, 129, 42–43 CrossRef CAS PubMed.
- A. Wittstock, V. Zielasek, J. Biener, C. M. Friend and M. Bäumer, Science, 2010, 327, 319–322 CrossRef CAS PubMed.
- B. S. Takale, X. J. Feng, Y. Lu, M. Bao, T. A. Jin, T. Minato and Y. Yamamoto, J. Am. Chem. Soc., 2016, 138, 10356–10364 CrossRef CAS PubMed.
- G. C. Bond, Catal. Today, 2002, 72, 5–9 CrossRef CAS.
- M. Sankar, Q. He, R. V. Engel, M. A. Sainna, A. J. Logsdail, A. Roldan, D. J. Willock, N. Agarwal, C. J. Kiely and G. J. Hutchings, Chem. Rev., 2020, 120, 3890–3938 CrossRef CAS PubMed.
- T. Fujita, P. F. Guan, K. McKenna, X. Y. Lang, A. Hirata, L. Zhang, T. Tokunaga, S. Arai, Y. Yamamoto, N. Tanaka, Y. Ishikawa, N. Asao, Y. Yamamoto, J. Erlebacher and M. W. Chen, Nat. Mater., 2012, 11, 775–780 CrossRef CAS.
- L. C. Wang, M. L. Personick, S. Karakalos, R. Fushimi, C. M. Friend and R. J. Madix, J. Catal., 2016, 344, 778–783 CrossRef CAS.
- B. Zugic, L. C. Wang, C. Heine, D. N. Zakharov, B. A. J. Lechner, E. A. Stach, J. Biener, M. Salmeron, R. J. Madix and C. M. Friend, Nat. Mater., 2017, 16, 558–564 CrossRef CAS.
- B. Zugic, M. A. van Spronsen, C. Heine, M. M. Montemore, Y. Y. Li, D. N. Zakharov, S. Karakalos, B. A. J. Lechner, E. Crumlin, M. M. Biener, A. I. Frenkel, J. Biener, E. A. Stach, M. B. Salmeron, E. Kaxiras, R. J. Madix and C. M. Friend, J. Catal., 2019, 380, 366–374 CrossRef CAS.
- L. C. Wang, C. M. Friend, R. Fushimi and R. J. Madix, Faraday Discuss., 2016, 188, 57–67 RSC.
- B. J. Xu, X. Y. Liu, J. Haubrich, R. J. Madix and C. M. Friend, Angew. Chem., Int. Ed., 2009, 48, 4206–4209 CrossRef CAS PubMed.
- B. J. Xu, C. G. F. Siler, R. J. Madix and C. M. Friend, Chem. – Eur. J., 2014, 20, 4646–4652 CrossRef CAS.
- F. Hiebel, S. Karakalos, Y. F. Xu, C. M. Friend and R. J. Madix, Top. Catal., 2018, 61, 299–307 CrossRef CAS.
- D. A. Outka and R. J. Madix, J. Am. Chem. Soc., 1987, 109, 1708–1714 CrossRef CAS.
- J. Gong, D. W. Flaherty, R. A. Ojifinni, J. M. White and C. B. Mullins, J. Phys. Chem. C, 2008, 112, 5501–5509 CrossRef CAS.
- B. J. Xu, J. Haubrich, T. A. Baker, E. Kaxiras and C. M. Friend, J. Phys. Chem. C, 2011, 115, 3703–3708 CrossRef CAS.
- S. P. Liu, P. Jin, D. H. Zhang, C. Hao and X. M. Yang, Appl. Surf. Sci., 2013, 265, 443–451 CrossRef CAS.
- W. L. Yim, T. Nowitzki, M. Necke, H. Schnars, P. Nickut, J. Biener, M. M. Biener, V. Zielasek, K. Al-Shamery, T. Klüner and M. Bäumer, J. Phys. Chem. C, 2007, 111, 445–451 CrossRef CAS.
- G. Tomaschun, W. Dononelli, Y. Li, M. Bäumer, T. Klüner and L. V. Moskaleva, J. Catal., 2018, 364, 216–227 CrossRef CAS.
- M. J. Prieto, E. A. Carbonio, S. Fatayer, R. Landers and A. de Siervo, Phys. Chem. Chem. Phys., 2014, 16, 13329–13339 RSC.
- R. Moreira, PhD thesis, Freie Universtität Berlin, 2018.
- J. Libuda, I. Meusel, J. Hartmann and H. J. Freund, Rev. Sci. Instrum., 2000, 71, 4395–4408 CrossRef CAS.
- M. J. Prieto, E. A. Carbonio, R. Landers and A. de Siervo, Surf. Sci., 2013, 617, 87–93 CrossRef CAS.
- K. Mortensen, C. Klink, F. Jensen, F. Besenbacher and I. Stensgaard, Surf. Sci., 1989, 220, L701–L708 CrossRef CAS.
- P. R. Norton, J. A. Davies and T. E. Jackman, Surf. Sci., 1982, 122, L593–L600 CrossRef CAS.
- S. D. Senanayake, D. Stacchiola, P. Liu, C. B. Mullins, J. Hrbek and J. A. Rodriguez, J. Phys. Chem. C, 2009, 113, 19536–19544 CrossRef CAS.
- B. K. Min and C. M. Friend, Chem. Rev., 2007, 107, 2709–2724 CrossRef CAS PubMed.
- M. L. Personick, B. Zugic, M. M. Biener, J. Biener, R. J. Madix and C. M. Friend, ACS Catal., 2015, 5, 4237–4241 CrossRef CAS.
- Y. Li, W. Dononelli, R. Moreira, T. Risse, M. Bäumer, T. Klüner and L. V. Moskaleva, J. Phys. Chem. C, 2018, 122, 5349–5357 CrossRef CAS.
- B. K. Min, A. R. Alemozafar, D. Pinnaduwage, X. Deng and C. M. Friend, J. Phys. Chem. B, 2006, 110, 19833–19838 CrossRef CAS PubMed.
- D. A. Outka and R. J. Madix, Surf. Sci., 1987, 179, 361–376 CrossRef CAS.
- B. C. Krüger, G. B. Park, S. Meyer, R. J. V. Wagner, A. M. Wodtke and T. Schäfer, Phys. Chem. Chem. Phys., 2017, 19, 19896–19903 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Temperature Programmed Desorption measurements of oxygen from bare and formate pre-covered Au(332). Methyl formate formation rate as measured in a pulsed isothermal MB experiment with a prolonged pulse sequence. See DOI: 10.1039/d1cp03436g |
| This journal is © the Owner Societies 2021 |