
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIdentification of the surface species in electrochemical promotion: ethylene oxidation over a Pt/YSZ catalyst†
Arafat
Toghan
ab,
Mark
Greiner
c,
Axel
Knop-Gericke
d and
Ronald
Imbihl
 *a
*a
aInstitut für Physikalische Chemie und Elektrochemie, Leibniz-Universität Hannover, Callinstrasse 3A, D-30167 Hannover, Germany. E-mail: imbihl@pci.uni-hannover.de
bChemistry Department, Faculty of Science, South Valley University, 83523 Qena, Egypt
cMax-Planck-Institut für Chemische Energiekonversion, Stiftstrasse 34 – 36, 445470 Mülheim an der Ruhr, Germany
dFritz-Haber-Institut der Max-Planck Gesellschaft, Abteilung Anorganische Chemie, Faradayweg 4-6, D-14195 Berlin, Germany
First published on 6th September 2021
Abstract
The electrochemical promotion of the C2H4 + O2 total oxidation reaction over a Pt catalyst, interfaced to yttrium stabilized zirconia (YSZ), has been studied at 0.25 mbar and T = 650 K using near ambient pressure X-ray photoelectron spectroscopy (NAP-XPS) as an in situ method. The electrochemical promoter effect is linked to the presence of a several layers thick graphitic overlayer that forms on the Pt surface in the presence of C2H4. Our NAP-XPS investigation reveals that electrochemical pumping of the Pt/YSZ catalyst, using a positive potential, leads to the spillover of oxygen surface species from the YSZ support onto the surface of the Pt electrode. Based on the XP spectra, the spillover species on Pt is identical to oxygen chemisorbed from the gas-phase.
Introduction
Porous noble metal electrodes interfaced to a solid electrolyte allow for a very efficient electrochemical promotion of heterogeneously catalyzed reactions (EPOC).1–5The original idea goes back to C. Wagner6 but the realization of the EPOC effect has been pioneered by Vayenas et al. who demonstrated this effect for about 100 reaction systems reaching an electrochemically induced rate enhancement up to a factor of 200.3,5 The physical basis for the EPOC effect is the electrochemically-induced spillover of the transported ionic species, after discharge from the solid electrolyte, onto the surface of the metal electrode. This spillover process has been demonstrated spectroscopically and with spatially resolved methods for the O2− conducting yttrium stabilized zirconia (YSZ) and for the Na+ conducting β′′-Al2O3 (β-alumina).7–12In general, the EPOC effect is non-Faradaic—that is, an electrochemically induced rate increase, Δr, can differ drastically from the rate increase predicted by the number of transported ions. In the case of ethylene oxidation over a Pt/YSZ catalyst a record value of 300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 was obtained for the so-called Λ–factor, defined by Λ = Δr/(I/2F), where I is the electric current and F is the Faraday constant.13 Accordingly, each transported O2− ion catalyzes the oxidation of about 100000 C2H4 molecules. The origin of the non-Faradaic behavior has been discussed controversially in the literature.2–5,14–18 In order to explain this striking non-Faradaicity the concept of a special oxygen spillover species, Oδ−(δ ≈ 2), has been introduced.14 Compared to regular chemisorbed oxygen this species should exhibit a reduced reactivity, and owing to its large dipole moment it should influence the reactivity of co-adsorbed species, thus acting similar to a promoter in classical heterogeneous catalysis. Alternatively, an ignition mechanism has been proposed in which the electrochemically generated oxygen spillover species triggers the reactive removal of a carbonaceous CHx layer that forms on the catalyst surface, inhibits oxygen adsorption and thereby poisons the catalyst.17,18
000 was obtained for the so-called Λ–factor, defined by Λ = Δr/(I/2F), where I is the electric current and F is the Faraday constant.13 Accordingly, each transported O2− ion catalyzes the oxidation of about 100000 C2H4 molecules. The origin of the non-Faradaic behavior has been discussed controversially in the literature.2–5,14–18 In order to explain this striking non-Faradaicity the concept of a special oxygen spillover species, Oδ−(δ ≈ 2), has been introduced.14 Compared to regular chemisorbed oxygen this species should exhibit a reduced reactivity, and owing to its large dipole moment it should influence the reactivity of co-adsorbed species, thus acting similar to a promoter in classical heterogeneous catalysis. Alternatively, an ignition mechanism has been proposed in which the electrochemically generated oxygen spillover species triggers the reactive removal of a carbonaceous CHx layer that forms on the catalyst surface, inhibits oxygen adsorption and thereby poisons the catalyst.17,18
A key obstacle in settling the questions regarding the nature of the oxygen spillover species and the origin of the non-Faradaic behavior has always been that no in situ measurements were available for characterizing the state of the Pt/YSZ catalyst under the conditions of a typical EPOC experiment—namely, at a pressure in the mbar range.19 In the first XPS (X-ray photoelectron spectroscopy) study of Pt/YSZ, conducted at 10−9 mbar and with a porous Pt layer, electrochemical pumping caused the appearance of an additional oxygen species that was characterized by an O 1s binding energy (BE) at 528.8 eV.9 As this BE was distinctly different from the O 1s BE of regular chemisorbed oxygen on Pt (at about 530 ± 0.5 eV), it was taken as evidence for a special oxygen spillover species.20–24 However, subsequent spatially resolving XPS measurements with a microstructured Pt/YSZ sample under an UHV environment revealed that the spillover species is identical with oxygen chemisorbed from the gas phase.7
This latter measurement clarified the question regarding the nature of the oxygen spillover species under high vacuum conditions. However, this measurement was not taken under reaction conditions and it did not settle the issue for the conditions of typical EPOC experiments which are conducted in the mbar range.1–5 Different thermodynamic conditions present at higher pressure/higher coverage might enable the formation of an oxygen species that is unstable at lower pressure. Studying the spillover processes at electrodes interfaced to YSZ at high p might also turn out to be crucial for understanding the performance of high temperature fuel cells.25,26
In order to elucidate the nature of the oxygen spillover species that forms during electrochemical promotion at higher pressures, in situ experiments using a differentially pumped X-ray photoelectron spectrometer were conducted at 0.25 mbar total pressure, thus bridging a large part of the pressure gap between UHV and EPOC experiments. Since these experiments allow us to identify the present surface species during electrochemical promotion, they should permit us to discriminate between the various proposed promotion mechanisms, i.e. the sacrificial promoter versus the ignition mechanism.
Experimental
Sample preparation and electrochemical set-up
For the preparation of the sample, we used a square-shaped (111) oriented YSZ single crystal (13 mol% yttrium, MaTeck GmbH, Germany) with a thickness of 1.5 mm and dimensions of 16 × 16 mm2. One side of the crystal was polished to a surface roughness <0.5 nm. The working electrode (WE) was prepared by applying Pt paste (A1118 Demetron) on the polished side of single crystal YSZ (111), and the counter (CE) by depositing Pt paste on the unpolished side of the YSZ substrate, followed by calcination at 1120 K in air for several hours (≈9 h) to remove the organic compounds. A porous network film with a thickness on the order of a few μm was obtained (Fig. 1c). | ||
| Fig. 1 (a) Schematic experimental set-up, WE = working electrode, CE = counter electrode. (b) Sketch of the porous Pt structure of the WE on top of YSZ (yttrium stabilized zirconia). (c) SEM micrograph of a Pt WE electrode on a YSZ (111) single crystal after annealing for 3 h in air at 1120 K. Top: top view, width of imaged area: 24 μm; bottom: cross section, thickness Pt layer: 8 μm. | ||
XPS and data analysis
The XPS experiments were performed using monochromatized synchrotron radiation at the ISSIS beamline in the synchrotron radiation facility BESSY II of the HZB in Berlin. A specially designed, differentially-pumped XPS system was utilized in order to minimize the path of the photoelectrons through the gas-phase.27 Details of the reaction cell and the differentially-pumped electrostatic lens system are described elsewhere.28 X-Rays were admitted to the experimental cell through a 50 nm-thick Si3N4 window. The photoelectrons, emitted under normal emission, entered a differentially pumped electrostatic lens system and were focused onto the entrance slit of a standard electron energy analyzer, where high vacuum conditions were maintained by an additional pumping stage. This set-up allowed a variation of the total pressure in the reaction cell between 10−7 and 0.5 mbar.As indicated in Fig. 1a, the working electrode is at ground potential and all binding energies are therefore referenced to the Fermi level of the working electrode. The excitation (photon energy) for Pt 4f, C 1s, and O 1s core level spectra were 220, 435, and 680 eV, respectively, resulting in the same high surface sensitivity, and an inelastic mean free path (IMFP) of the photoelectrons of about 0.86 nm in carbon and 0.4 nm in platinum. The Pt 4f, C 1s and O 1s envelopes were fitted using Casa XPS software after subtraction of Shirley background. The fitting of the spectra was done using Gaussian–Lorentzian functions, by fixing the peak position within ±0.1 eV.
Reaction experiments
Under reaction conditions, the cell was operated as a continuous flow reactor. Gases were introduced via mass flow controllers. Reaction products were monitored with a differentially-pumped quadruple mass spectrometer (QMS). The YSZ sample was heated from the backside through a 100 nm-thick SiC window with an infrared laser. The sample temperature was measured via a K-type Ni/NiCr thermocouple spotwelded to the WE on the front side of the sample. All experiments were conducted at T = 650 K.Prior to the experiments, the Pt film was cleaned by repeated cycles of mild Ar+ ion bombardment (t = 20 min, E = 1.5 keV, p(Ar) = 1 × 10−4 mbar). After repeated cycles of cleaning, the photoemission spectra indicated that the only remaining contaminants were silicon and oxygen (Fig. S1 in ESI†). During the experimental measurements, the sample was held at 650 K, in an atmosphere with a total pressure fixed at 0.25 mbar and varying ratios C2H4 and O2. The spectral regions of Pt 4f, O 1s and C 1s were recorded under reaction conditions. For background subtraction of the reaction rate, the experiments with the Pt/YSZ sample were repeated at room temperature. The background production of CO2 at 300 K is less than 1% of the CO2 production rate at 650 K.
Results and discussion
For the electrochemical promotion experiments, a YSZ single crystal with porous Pt layers as electrodes is used as depicted in Fig. 1. In this two-electrode setup, a potentiostat controls the potential of the grounded working electrode relative to the counter electrode (CE). In the differentially pumped XPS, the Pt/YSZ catalyst is exposed to an O2/ethylene atmosphere of varying composition but with a fixed total pressure of 0.25 mbar.Upon application of a positive potential to the WE (anodic polarization), oxygen ions are transported through the YSZ solid electrolyte. After discharge at the three-phase-boundary (tpb) gas phase/Pt/YSZ the atomic oxygen, Otpb, can spill over onto the surface of the Pt electrode where this species can take part in a catalytic reaction:
| OYSZ2− − 2e−→ Otpb → Oad |
As will be shown below, the spillover oxygen species is identical to chemisorbed oxygen. For this reason, the spillover species in the above equation has been denoted as Oad.
With oxygen, ethylene undergoes total oxidation to CO2 on a Pt catalyst according to
| C2H4 + 3O2 → 2H2O + 2CO2 |
The products of the reaction are followed with a quadrupole mass spectrometer (QMS) while simultaneously the state of the WE surface is monitored with XPS.27,28
Fig. 2 displays the CO2 production rate, together with the carbon 1s signal, as the ratio p(C2H4)/p(O2) is varied at T = 650 K. At low p(C2H4) the surface is essentially carbon free and the CO2 production rate rises proportionally with p(C2H4) because the reactants adsorb and react uninhibitedly in this range. When a critical value of p(C2H4) is reached, a high carbon coverage builds up and simultaneously the reaction rate drops to a very low level. This transition is rather sharp and occurs completely at a critical value of p(C2H4).
 | ||
| Fig. 2 Carbon coverage and kinetics of ethylene oxidation without and with electrochemical promotion. Experimental conditions: T = 650 K, p(total) = 0.25 mbar. (a) CO2 production rate, r(CO2), and carbon coverage in numbers of graphitic layers vs. percentage of C2H4 in the gas-phase for OC conditions and with an applied electric potential VWC = 2 V. Since the carbon signal did not change during electrochemical promotion, both signals are represented by a single line. The noise level in the rate measurements was below 0.2% of the CO2 signal. The ionic current, IWC, is in the range 5–10 mA during electrochemical pumping. (b) Relative rate increase (r − r0)/r0 with r denoting the CO2 production rate with electrochemical promotion and r0 denoting the rate under open circuit conditions. Beyond 70% C2H4 in the feed, the background subtraction of CO2 production is not reliable enough for determining meaningful values of the relative rate increase. (c) C 1s photoelectron spectrum recorded under reaction conditions for a ratio p(C2H4)/p(O2) = 2:1 with an applied electric potential VWC = 2 V. For comparison, the spectra in pure ethylene and in pure O2 are displayed as well, both under OC conditions. The C 1s spectra were fitted with 3 components, in accordance with the procedure of Estrade-Szwarckopf,34 with the following parameters (position, FWHM, asymmetry): Graphitic (284.3 eV, 0.7 eV, DS[0.03,80]), defect (284.6 eV, 1.3 eV, symmetric), C–O (286.2 eV, 1.6 eV, symmetric). | ||
The main component of the C 1s spectra shown in Fig. 2c at 284.3 eV can be assigned to sp2-bonded C. For a thin graphitic layer, produced by ethylene decomposition on Pt(111), the same C 1s BE of 284.3 eV has been reported previously.29 A slightly higher value of 284.6 eV was found for graphitic carbon on polycrystalline Pt (Pt black).30 The additional components shown in Fig. 2c at 284.6 eV and 286.2 eV can be attributed to graphitic carbon or defects in the graphite and C–O species, respectively.30
The thickness of the graphitic overlayer was determined from the attenuation of the Pt 4f signal that occurs with carbon film formation. The thickness was calculated using standard equations for non-destructive XPS depth profiling,31 and an inelastic mean free path of 0.56 nm for electrons of kinetic energy 320 eV moving through graphite.32 With this method, and assuming uniform surface coverage and a graphite layer spacing of 0.35 nm,33 we estimate that a maximum of 8 layers of graphitic carbon are formed.
Upon application of an anodic (positive) potential, VWC, to the working electrode, the reaction rate rises by 15–20% as indicated in Fig. 2a. Since the electrochemical promotion effect occurs only right of the rate maximum, the EPOC effect is evidently linked to the presence of a substantial carbon coverage on the Pt surface. Remarkably, the electrochemical pumping does not lead to a detectable change in the C 1s signal. From the measured rate increase, and from the electric current of about 5 mA which flows upon electrochemical pumping at a ratio p(C2H4)/p(O2) = 1:1, one calculates a Λ-factor of ∼10, i.e. the reaction is moderately non-Faradaic.
The O 1s spectra shown in Fig. 3, recorded under varying reaction conditions, reveal the presence of several oxygen species on the Pt surface. Species A at 529. 4 eV is assigned to chemisorbed oxygen, Oad.7,20–24 A higher-lying state, species B, at 530.5 eV occurs in a region where the OHad species is found;35 a contribution from an additional state of chemisorbed oxygen cannot be excluded because the values reported in the literature for oxygen chemisorbed on Pt spread over a range of about 1 eV.20–24 A third state, species C, at 531.7 eV BE, is due to SiOx (x close to 2), contamination that accumulates on the sample, and is present in all experiments conducted in the high pressure XPS chamber.36 Finally, the fourth state, D, at 532.7 eV BE, is located in a region where C–O species are found.37 Species A, at 529.4 eV, can be unambiguously assigned to chemisorbed oxygen, Oad, because the same component develops when oxygen from the gas phase is adsorbed as demonstrated by spectrum (iii) in (b). In this experiment after having the species A removed by clean-off reactions with the residual gas (C2H4, H2, CO) the sample is exposed under open circuit conditions to gaseous oxygen at p(O2) = 0.25 mbar leading to spectrum (iii). Under all reaction conditions applied here, no changes in the Pt 4f region were detected, which would have been indicative of Pt oxide formation.
 | ||
| Fig. 3 Photoelectron spectra of the O 1s region characterizing the different oxygen species on the Pt surface under open circuit (OC) reaction conditions and during electrochemical pumping. The ratio p(C2H4)/p(O2) is indicated. Experimental conditions: T = 650 K, p(total) = 0.25 mbar. (a) O 1s spectra showing the effect of electrochemical pumping under reaction conditions close to the rate maximum (Fig. 2a). The O 1s spectra were fitted with 4 components: A: chemisorbed oxygen; B: OH species + additional surface oxygen species; C: SiOx+ C=O species, D: C–O species. The following parameters were used for fitting (position, FWHM), all with zero asymmetry and 50% Gauss: A (529.4 eV, 1.4 eV); B (530.5 eV, 1.5 eV); C (531.7 eV, 1.6 eV); D (532.7 eV, 1.7 eV). (b) Spectrum of the sample in pure O2 with p(O2) = 0.25 mbar (iii) and under reaction conditions with a large excess of ethylene (iv), both recorded under OC conditions. (c) Electrochemically generated spillover oxygen in vacuum at p ≈ 10−8 mbar (vi). For comparison, a spectrum of the sample prior to electrochemical pumping is shown (v). | ||
The O 1s spectrum (v) shown in Fig. 3c exhibits the different oxygen species which remain on the surface after pumping off the gases coming from the high rate branch of the reaction (Fig. 2a). Species A representing regular chemisorbed oxygen has been almost completely removed by clean-off reactions with the residual gas. During electrochemical pumping in a vacuum with VWC = 2 V, it is this species A which strongly grows as shown by spectrum (vi). This demonstrates that the spillover species is identical with species A and therefore with regular chemisorbed oxygen adsorbed from the gas phase. The growth of the oxygen coverage in both experiments, electrochemical pumping in (vi) and adsorption from the gas-phase in (iii), can be described by the same component in the O 1s region, species A. Furthermore, the relative peak areas are nearly the same when the sample is in a vacuum with an applied voltage as when the sample is in pure O2. All this is consistent with electrochemically produced spillover oxygen being identical to chemisorbed oxygen species adsorbed from the gas phase. Remarkably, nearly the same BE's of O 1s have been found in a UHV type study of Pt/YSZ where the values of 529.5 eV and 530.4 eV have been reported for the electrochemically induced oxygen spillover species.38 An overview of the different O 1s binding energies reported in the literature for chemisorbed oxygen on Pt obtained in adsorption experiments and in electrochemical spillover experiments is given in Table 1. The same value of 530.4 eV has been found under UHV for the spillover/chemisorbed oxygen species on a microstructured Pt film on a YSZ substrate.7 Evidently, the O 1s BE of chemisorbed/spillover oxygen varies over a range of about 1 eV but this is what the data in the literature also show.20–24,38–41 Structural differences and presumably also the amount of oxygen dissolved in the bulk can shift the O 1s binding energy of chemisorbed oxygen on Pt by this amount.
| System | O 1s BE [eV] | Assignment | References |
|---|---|---|---|
| Pt/YSZ | 528.8 | Oδ−(δ ≈ 2) | 9 |
| Pt/YSZ | 530.4 | Oad | 7 |
| Pt/YSZ | 529.5, 530.4 | Oδ−(δ ≈ 2), Oad | 38 |
| Pt/YSZ | 529.4 | Oad | This work |
| Ptpoly/O2 | 530.0 | Oad | 21 |
| Pt(111)/O2 | 530.2 | Oad | 20 |
| Pt(111)/O2 | 529.8 | Oad | 22 |
| Pt(111)/Oatomic | 530.8 | Oad | 39 |
| Pt(533)/O2 | 529.7 | Oad | 40 |
| Pt(332)/O2 | 529.5 | Oad | 23 |
| Pt(531)/O2 | 529.5 | Oad | 41 |
Under reaction conditions, the amount of electrochemically produced spillover oxygen depends strongly on the composition of the gas phase (i.e. on whether oxidizing or reducing conditions prevail). As demonstrated in Fig. 4a, with a large excess of oxygen, there is hardly any detectable growth in oxygen coverage due to electrochemical pumping. This is not too surprising because under these conditions the surface is already fully covered with oxygen. The largest increase in oxygen coverage caused by electrochemical pumping is found just at the position of the rate maximum in Fig. 4a. In the region to the right of the rate maximum, which corresponds to a carbon-covered surface, the amount of chemisorbed oxygen is very small under open circuit conditions. With electrochemical pumping, a small amount of chemisorbed oxygen can be generated. As shown in Fig. 4b, the effect of electrochemical pumping on all the other oxygen species, besides chemisorbed oxygen, is small.
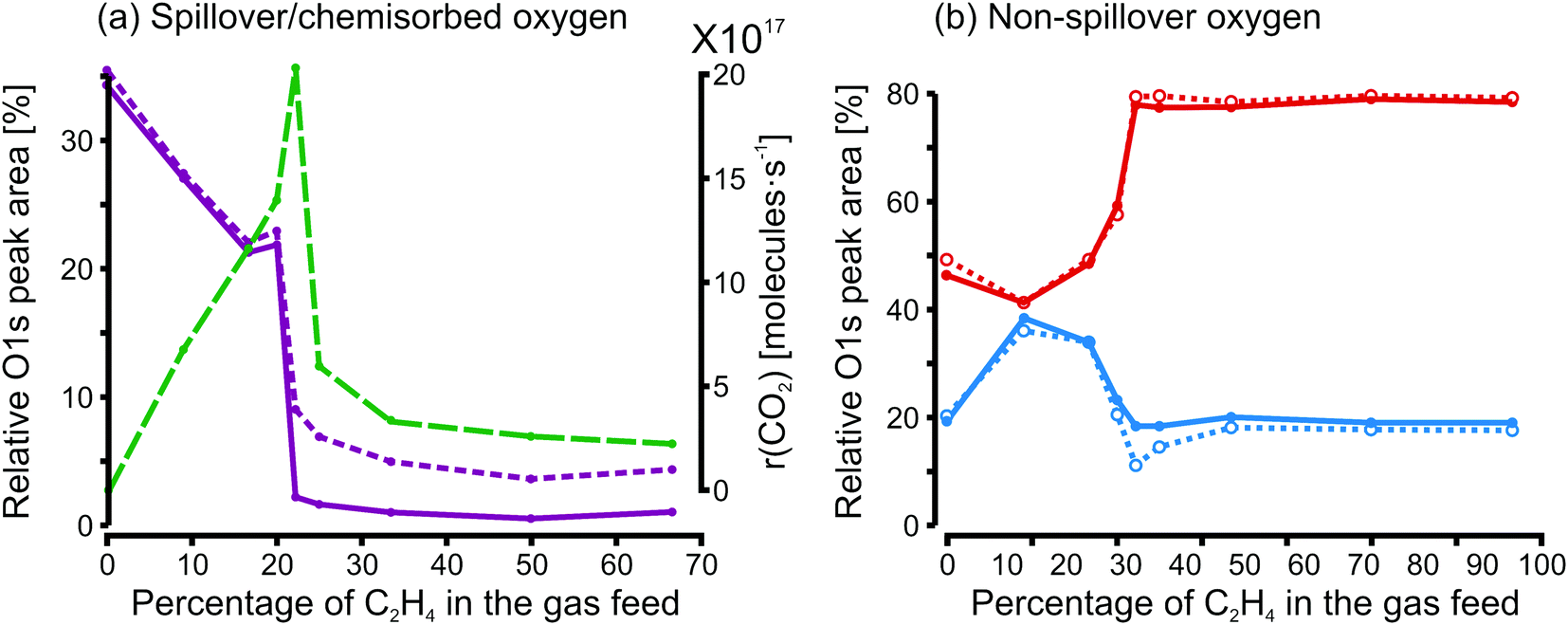 | ||
| Fig. 4 Relative contributions of the different oxygen species during varying reaction conditions under OC conditions (full lines) and during electrochemical activation (dashed lines). (a) Variation of the chemisorbed oxygen/spillover oxygen species (species A) under open circuit conditions and with electrochemical pumping. The reaction rate under OC conditions (green line) has been included in the plot. (b) Variation of the non-spillover oxygen species under open circuit conditions and with electrochemical pumping. Separately shown is the B component (OHad + additional surface oxygen species, blue lines) while the remaining oxygen species, C and D, have been combined (red lines). | ||
From the experimental results, the following conclusions can be drawn.
(i)The electrochemically induced spillover species is identical with regular chemisorbed oxygen, not only under UHV but also under high pressure conditions (0.25 mbar).
(ii)Under ethylene-rich conditions the catalytic activity of the Pt surface is poisoned due to the formation of a several layers thick graphitic overlayer, which inhibits O2 adsorption.
(iii)The occurrence of an electrochemical promotion effect in ethylene oxidation is linked to the presence of a carbon overlayer on the Pt surface.
The results here are in full agreement with earlier studies of the same reaction system conducted in the 10−5 and 10−6 mbar range under an UHV environment.17,18 The measurements also confirm an explanation already made soon after the discovery of the EPOC effect, namely that electrochemically induced spillover is the physical basis of the promotion effect. For explaining the non-Faradaic nature of the EPOC effect, basically, three models have been proposed: (i) the sacrificial promoter concept;3,14 (ii) the ignition model;12,17,18 and (iii) a chain reaction model16,42 which lacks experimental support but, since it results in autocatalytic kinetics, is similar to (ii). The data here do not support the sacrificial promoter model because this model is based on the existence of a special spillover species with properties distinctly different from regular chemisorbed oxygen. Whether such a species can be found at higher pressure remains an open question.
In the ignition model, spillover oxygen eats a hole into the carbon layer thus circumventing the inhibition of oxygen adsorption from the gas phase.8,12,17,18 Since oxygen from the gas phase can now adsorb and react, the hole in the carbonaceous overlayer will be enlarged, thus giving rise to a process with autocatalytic behavior. The experimental observation that the EPOC effect is linked to the presence of a carbon overlayer on Pt finds a simple explanation in the ignition mechanism. From the ignition mechanism, one would expect to observe a decrease of the carbon signal upon electrochemical pumping, as carbon should be reacted away by spillover oxygen and oxygen from the gas phase. However, in our experiments, the carbon signal practically does not change at all during electrochemical pumping.
This discrepancy can be resolved if one considers the actual physical situation at the Pt/YSZ interface, as sketched in Fig. 1b. With a porous Pt electrode of about 8 μm thickness, the three-phase boundary (TPB) gas/metal/solid electrolyte is not directly detectible by the XPS measurements, but only the outer surface plane. However, it is the tpb and the surrounding Pt area where electrochemical pumping can influence catalytic activity because there the spillover oxygen concentration is the highest. The spillover oxygen can reactively remove the carbon, thus activating the Pt surface.8 However, if the reaction fronts created at the tpb do not propagate throughout the porous Pt layer reaching the outer surface plane, but rather remain restricted to the vicinity of the TPB, then the carbon signal measured by XPS will remain constant. As indicated by the moderate rate increase in the reaction rate upon electrochemical pumping, such an incomplete ignition is evidently the case here. The fact that some spillover of oxygen is also seen on the low rate branch of the kinetics where the surface is carbon covered (Fig. 4) can be attributed to the heterogeneity of the porous Pt layer.18
The relatively long diffusion path that spillover oxygen has to take in order to get from the TPB, where it originates, to the outer surface plane where it is detected, also explains why a substantial amount of electrochemically induced spillover oxygen is only found around the rate maximum as shown in Fig. 4a. A high oxygen coverage and a high carbon coverage will both suppress the diffusion of oxygen. The best chance to reach the outer surface plane will be if the total adsorbate coverage is at a minimum, a condition fulfilled at the rate maximum. Using the parameters determined by Mutoro et al. for oxygen diffusion on Pt/YSZ one calculates that oxygen atoms at T = 650 K can cover a distance of about 30 μm in one second.43 This means that oxygen originating at the TPB can easily reach the outer surface plane of a porous Pt electrode provided that enough vacant sites on the Pt surface are available so that oxygen is able to diffuse.
By applying the Nernst equation to the oxygen electrode, it is seen that the pumping potentials of 1–2 V correspond to an enormous (virtual) p(O2), which in turn should suffice to generate Pt oxide at T = 650 K.44 In fact in recent experiments the formation of Pt oxide was detected at the interface Pt/YSZ.45 From the observation made here, that no sign of Pt oxide can be detected with XPS, one has to conclude that the chemical potential of oxygen falls off rapidly with increasing distance from the TPB due to oxygen losses by chemical reaction and thermal desorption. As a consequence, Pt oxide only forms at the interface Pt/YSZ and near the TPB.
If one takes a look at the published EPOC studies of ethylene oxidation on Pt/YSZ, one realizes that even for seemingly identical catalysts and reaction conditions the so-called Λ-factors describing the non-Faradaicity of the promotion effect vary enormously: the values 80–330,46 188,47 865,48 1.5 × 10432 and 3 × 10549 have been reported. This demonstrates, first of all, that there is a preparation problem in the sense that it is obviously difficult to obtain highly active Pt/YSZ catalysts in a reproducible way. The finding of nearly ten monolayers thick carbon deposits on Pt can at least partially explain these difficulties because carbon layers of such a thickness will render ignition by reaction fronts rather difficult. Secondly, there is also a problem in the control of the experimental conditions. It has been suspected that in some of the experiments that were conducted in the mbar pressure range, the reaction is no longer isothermal, due to the heat generated by the reaction.4,15,17 A thermal ignition connected with a temperature increase of several tens of Kelvin would of course explain the enormous magnitude of some of the Λ-factors reported in the literature. This remains to be verified in future, by well-defined experiments. Currently, no such data are available.
In a recent combined experimental and quantum chemical study the electrochemical promotion of ethylene oxidation over a RuO2 surface was investigated.50 However, since dissociation occurs via bonding of the C2H4 molecule to the O-species on the surface, the mechanism is different than the one discussed here.
Conclusions
In conclusion, we applied near ambient pressure XPS to study in situ the surface species formed during the electrochemical promotion of ethylene oxidation over a Pt/YSZ catalyst under conditions close to that of typical EPOC experiments. The EPOC effect was shown to be linked to the presence of a graphitic overlayer on the Pt surface. We found that electrochemically induced oxygen spillover occurs and that the spillover species is identical with oxygen chemisorbed from the gas phase. No indication of the existence of a special oxygen spillover species was detected.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Helmholtz-Zentrum Berlin for beam time at the electron storage ring BESSY II and the authors thank the BESSY staff for help and support. Financial support from the Deutsche Forschungsgemeinschaft (DFG) is gratefully acknowledged.Notes and references
- C. G. Vayenas, S. Bebelis and S. Ladas, Nature, 1990, 343, 625 CrossRef CAS.
- C. G. Vayenas; M. M. Jaksic; S. I. Bebelis; S. G. Nephytides, ed. J. O. M. Bockris, et al., Modern Aspects of Electrochemistry, Plenum Press, New York, 1996, vol. 29, p. 57 Search PubMed.
- C. G. Vayenas; S. Bebelis; C. Pliangos; S. Brosda; D. Tsiplakides, Electrochemical Activation of Catalysis: Promotion, Electrochemical Promotion, and Metal-Support Interactions, Kluwer Academic/Plenum Publishers, New York, 2001 Search PubMed.
- R. Imbihl, Prog. Surf. Sci., 2010, 85, 240 CrossRef.
- P. Vernoux, et al. , Chem. Rev., 2013, 113, 8192 CrossRef CAS PubMed.
- C. Wagner, Adv. Catal., 1970, 21, 32 Search PubMed.
- B. Luerßen, S. Günther, H. Marbach, M. Kiskinova, J. Janek and R. Imbihl, Chem. Phys. Lett., 2000, 316, 331 CrossRef.
- B. Luerßen, E. Mutoro, H. Fischer, S. Günther, R. Imbihl and J. Janek, Angew. Chem., Int. Ed., 2006, 45, 1473 CrossRef PubMed.
- S. Ladas, S. Kennou, S. Bebelis and C. G. Vayenas, J. Phys. Chem., 1993, 97, 8845 CrossRef CAS.
- I. V. Yentekakis, A. Palermo, N. C. Filkin, M. S. Tikhov and R. M. Lambert, J. Phys. Chem. B, 1997, 101, 3759 CrossRef CAS.
- F. J. Williams, A. Palermo, S. Tracey, M. S. Tikhov and R. M. Lambert, J. Phys. Chem. B, 2002, 106, 5668 CrossRef CAS.
- A. Toghan, R. Arrigo, A. Knop-Gericke and R. Imbihl, J. Catal., 2012, 296, 99 CrossRef CAS.
- S. Bebelis and C. G. Vayenas, J. Catal., 1989, 118, 125 CrossRef CAS.
- C. G. Vayenas, S. Brosda and C. Pliangos, J. Catal., 2003, 216, 487 CrossRef CAS.
- C. G. Vayenas and P. Vernoux, ChemPhysChem, 2011, 12, 1761 CrossRef CAS PubMed; R. Imbihl and A. Toghan, ChemPhysChem, 2011, 12, 1764 CrossRef.
- V. A. Sobyanin, V. I. Sobolev, V. D. Belyaev and O. A. Mar,ina, Catal. Lett., 1993, 18, 153 CrossRef CAS.
- A. Toghan, L. M. Rösken and R. Imbihl, ChemPhysChem, 2010, 11, 1452 CrossRef CAS PubMed.
- A. Toghan, L. M. Rösken and R. Imbihl, Phys. Chem. Chem. Phys., 2010, 12, 9811 RSC.
- In ref. 12 ambient pressure XPS at 0.25 mbar has been conducted with the system YSZ/Pt,Ag/C2H4 + O2 but due to the presence of several metallic constituents the spillover results with electrochemical pumping were difficult to interpret.
- M. Peuckert and H. P. Bonzel, Surf. Sci., 1984, 145, 239 CrossRef CAS.
- P. Legare, L. Hilaire and G. Maire, Surf. Sci., 1984, 141, 604 CrossRef CAS.
- C. Puglia, A. Nilsson, B. Hernnas, O. Karis, P. Bennich and N. Martensson, Surf. Sci., 1995, 342, 119 CrossRef CAS.
- J. G. Wang, et al. , Phys. Rev. Lett., 2005, 95, 256102 CrossRef CAS PubMed.
- R. Arrigo, M. Hävecker, M. E. Schuster, C. Ranjan, E. Stotz, A. Knop-Gericke and R. Schlögl, Angew. Chem., Int. Ed., 2013, 52, 11660 CrossRef CAS PubMed.
- S. Adler, Chem. Rev., 2004, 104, 4791–4843 CrossRef CAS PubMed.
- C. Zhang, et al. , J. Am. Chem. Soc., 2013, 135, 11572 CrossRef CAS PubMed.
- D. F. Ogletree, H. Bluhm, G. Lebedev, C. S. Fadley, Z. Hussain and M. Salmeron, Rev. Sci. Instrum., 2002, 73, 3872 CrossRef CAS.
- A. Knop-Gericke, E. Kleimenov, M. Hävecker, R. Blume, D. Teschner, S. Zafeiratos, R. Schlögl, V. I. Bukhtiyarov, V. V. Kaichev, I. P. Prosvirin, A. I. Nizovskii, H. Bluhm, A. Barinov, P. Dudin and M. Kiskinova, Adv. Catal., 2009, 52, 213 CAS.
- N. Freyer, G. Pirug and H. P. Bonzel, Surf. Sci., 1983, 126, 487 CrossRef CAS.
- N. M. Rodriguez, P. E. Anderson, A. Wootsch, U. Wild, R. Schlögl and Z. Paàl, J. Catal., 2001, 197, 365 CrossRef CAS.
- M. C. López-Santos, F. Yubero, J. P. Espinos and A. R. Gonzalez-Elipe, Anal. Bioanal. Chem., 2010, 396, 2757 CrossRef PubMed.
- S. Tanuma, C. J. Powell and D. R. Penn, Surf. Interface Anal., 1994, 21, 165 CrossRef.
- G. E. Bacon, Acta Crystallogr., 1951, 4, 558–561 CrossRef.
- H. Estrade-Szwarckopf, Carbon, 2004, 42, 1713 CrossRef.
- T. Schiros, Thesis, Stockholm, 2008 CrossRef; G. B. Fisher and J. L. Gland, Surf. Sci., 1980, 94, 446 CrossRef.
- S. Günther, A. Scheibe, H. Bluhm, M. Haevecker, E. Kleimenov, A. Knop-Gericke, R. Schlögl and R. Imbihl, J. Phys. Chem. C, 2008, 112, 15382 CrossRef.
- G. Beamson; D. BriggsHigh Resolution XPS of Organic Polymers - The Scienta ESCA300 Database, Wiley Interscience, 1992, Appendices 3.1 and 3.2. http://(http://www.xpsfitting.com/2013/08/oxygen-1s-for-organic-compounds.html) Search PubMed.
- D. J. Davis, G. Kyriakou, R. B. Grant, M. S. Tikhov and R. M. Lambert, J. Phys. Chem. C, 2007, 111, 1491 CrossRef CAS.
- C. R. Parkinson, M. Walker and C. F. McConville, Surf. Sci., 2003, 545, 19 CrossRef CAS.
- S. Günther, et al. , J. Phys. Chem. C, 2008, 112, 15382 CrossRef.
- G. Held, L. B. Jones, E. A. Seddon and D. A. King, J. Phys. Chem. B, 2005, 109, 6159 CrossRef CAS PubMed.
- V. D. Belyaev, T. I. Politova and V. A. Sobyanin, Solid State Ionics, 2000, 136/137, 721 CrossRef.
- E. Mutoro, C. Hellwig, B. Luerssen, S. Guenther, W. Bessler and J. Janek, Phys. Chem. Chem. Phys., 2011, 13, 12798 RSC.
- T. Jacob, J. Electroanal. Chem., 2007, 607, 158 CrossRef CAS.
- H. Pöpke, E. Mutoro, C. Raiß, B. Luerßen, M. Amati, M. K. Abyaneh, L. Gregoratti and J. Janek, Electrochim. Acta, 2011, 56, 10668 CrossRef.
- S. Peng-ont, S. Souentie, S. Assabumrungrat, P. Praserthdam, S. Brosda and C. G. Vayenas, Catal. Lett., 2013, 143, 445 CrossRef CAS.
- C. G. Vayenas, A. Ioannides and S. Bebelis, J. Catal., 1991, 129, 67 CrossRef CAS.
- C. Koutsodontisa, A. Katsaounisa, J. C. Figueroab, C. Cavalcab, J. Carmo, J. Pereirab and C. G. Vayenas, Top. Catal., 2006, 38, 157 CrossRef.
- E. I. Papaioannou, S. Souentie, F. M. Sapountzi, A. Hammad, D. Labou, S. Brosda and C. G. Vayenas, J. Appl. Electrochem., 2010, 40, 1859 CrossRef CAS.
- Y. M. Hajar, L. Treps, C. Michel, E. A. Baranova and S. N. Steinmann, Catal. Sci. Technol., 2019, 9, 5915 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: XPS survey spectrum of Pt working electrode after cleaning, kinetics of ethylene oxidation under conditions of oxygen excess. See DOI: 10.1039/d1cp02757c |
| This journal is © the Owner Societies 2021 |