
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSystematic optimization of a fragment-based force field against experimental pure-liquid properties considering large compound families: application to oxygen and nitrogen compounds†
Marina
P. Oliveira
 and
Philippe H.
Hünenberger
*
and
Philippe H.
Hünenberger
*
Laboratorium für Physikalische Chemie, ETH Zürich, ETH-Hönggerberg, HCI, CH-8093 Zürich, Switzerland. E-mail: phil@igc.phys.chem.ethz.ch; Tel: +41 44 632 5503
First published on 19th July 2021
Abstract
The CombiFF approach is a workflow for the automated refinement of force-field parameters against experimental condensed-phase data, considering entire classes of organic molecules constructed using a fragment library via combinatorial isomer enumeration. One peculiarity of this approach is that it relies on an electronegativity-equalization scheme to account for induction effects within molecules, with values of the atomic hardness and electronegativity as electrostatic parameters, rather than the partial charges themselves. In a previous article [M. P. Oliveira, M. Andrey, S. R. Rieder, L. Kern, D. F. Hahn, S. Riniker, B. A. C. Horta and P. H. Hünenberger, J. Chem. Theory. Comput. 2020, 16, 7525], CombiFF was introduced and applied to calibrate a GROMOS-compatible united-atom force field for the saturated acyclic (halo-)alkane family. Here, this scheme is employed for the construction of a corresponding force field for saturated acyclic compounds encompassing eight common chemical functional groups involving oxygen and/or nitrogen atoms, namely: ether, aldehyde, ketone, ester, alcohol, carboxylic acid, amine, and amide. Monofunctional as well as homo-polyfunctional compounds are considered. A total of 1712 experimental liquid densities ρliq and vaporization enthalpies ΔHvap concerning 1175 molecules are used for the calibration (339 molecules) and validation (836 molecules) of the 102 non-bonded interaction parameters of the force field. Using initial parameter values based on the GROMOS 2016H66 parameter set, convergence is reached after five iterations. Given access to one processor per simulated system, this operation only requires a few days of wall-clock computing time. After optimization, the root-mean-square deviations from experiment are 29.9 (22.4) kg m−3 for ρliq and 4.1 (5.5) kJ mol−1 for ΔHvap for the calibration (validation) set. Thus, a very good level of agreement with experiment is achieved in terms of these two properties, although the errors are inhomogeneously distributed across the different chemical functional groups.
1 Introduction
The ability of classical molecular dynamics (MD) simulation to represent accurately the properties of a given system depends crucially on the quality of the underlying potential-energy function or force field.1–7 One may tentatively distinguish three main strategies in the design of condensed-phase force fields:8(1) In fragment-based force fields9–13 (FBFF), the covalent and non-bonded parameters are specified within molecular fragments representative of the relevant chemical functional groups. Molecular topologies are built by assembling these fragments and invoking an assumption of transferability. The non-bonded parameters are calibrated primarily by fitting against experimental thermodynamic data for small organic compounds in the condensed phase.
(2) In hybrid force fields14,15 (HYFF), the covalent and van der Waals parameters are selected in a FBFF fashion, but the partial charges are derived based on quantum-mechanics (QM) calculations involving the target molecule, typically via electrostatic-potential fitting14–28 or electron-density partitioning.29–38 The assumption invoked in this case is that of a separability between van der Waals coefficients and partial charges. Given a selected charge-derivation scheme, the van der Waals parameters are again calibrated primarily against experimental condensed-phase data for small compounds.
(3) In QM-derived force fields39–46 (QDFF), the covalent and non-bonded parameters are determined simultaneously based on QM calculations involving the target molecule. The calculation schemes for the partial charges are the same as in the HYFF case. For the van der Waals coefficients, the derivation typically involves electron-density partitioning as a starting point.47–57 The assumption invoked here is that of a compatibility between the non-bonded interaction parameters appropriate for the isolated molecule and for the molecule in the condensed phase.
The HYFF and QDFF schemes are very popular nowadays, in particular because they: (i) benefit from fast QM calculation methods;58–65 (ii) promise an exhaustive coverage of the chemical space;66,67 (iii) take into account induction effects on the partial charges24,33,68 and, possibly, on the van der Waals coefficients;44,45,51–53,56,57 (iv) are comparatively easy to automate in terms of topology construction and parameter derivation. However, compared to the FBFF approach, they also present some major shortcomings: (i) the need to specify a reference structure (molecular conformation) and environment (e.g. vacuum or continuum solvent) in the QM calculations leads to an implicit dependence of the topological information on configurational information; (ii) the partial charges and van der Waals coefficients are not strictly speaking QM observables, i.e. they result from ad hoc derivation recipes rather than physics-based rules, and their values may also strongly depend on the choice of a QM level of theory and basis set;21–25,69–75 (iii) in practice, some parameters must still be optimized empirically,21,26,27,40,46,74,75e.g. van der Waals coefficients in HYFF and van der Waals repulsion coefficients38,44,45,56,57,76,77 in QDFF.
In contrast to the HYFF and QDFF approaches, the FBFF scheme fully acknowledges that the non-bonded parameters are truly empirical quantities. They compensate in a mean-field fashion for all sorts of deficiencies in the selected potential-energy function, and they are correlated with a number of associated choices,78 including those of the model resolution (e.g. united- vs. all-atom), van der Waals combination rules, cutoff distances, and treatment of the long-range interactions. As a result, the connection between these empirical non-bonded parameters and QM-inferred single-molecule properties may in fact be rather weak. However, for a FBFF approach to be useful in practice, it must achieve: (i) a sufficiently broad (even if not exhaustive) coverage of the chemical space; (ii) an appropriate representation of induction effects; (iii) a high degree of automation in the topology construction and parameter optimization. In a recent article,8 we introduced a scheme called CombiFF that presents these features.
The goal of CombiFF is the automated refinement of force-field parameters against experimental condensed-phase data, considering entire classes of organic molecules constructed using a fragment library via combinatorial isomer enumeration. The main steps of the scheme are: (i) definition of a molecule family; (ii) combinatorial enumeration of all isomers; (iii) query for experimental data; (iv) automatic construction of the molecular topologies by fragment assembly; (v) iterative refinement of the force-field parameters considering the entire family. This scheme borrows from earlier work on isomer enumeration79–82 and topology construction,83–95 as well as on automated single-compound force-field optimization approaches such as the POP scheme,96,97 the ForceBalance scheme,46,98–105 and other related schemes.106–111 One key feature of CombiFF is that once the time-consuming task of target-data selection/curation has been performed, the optimization of a force field is entirely automatic and, given access to a sufficient number of processors, only requires a few days of wall-clock computating time. For this reason, CombiFF also represents an ideal framework for assessing the impact of specific functional-form decisions on the accuracy of a force field at an optimal level of parametrization.
As a first application, CombiFF was used in ref. 8 to design a GROMOS-compatible united-atom force field for the saturated acyclic (halo-)alkane family. A total of 749 experimental liquid densities ρliq and vaporization enthalpies ΔHvap concerning 486 haloalkane molecules were considered for the calibration (228 molecules) and validation (258 molecules) of 32 non-bonded interaction parameters.
An important aspect of this force field is that the atomic partial charges are not specified explicitly within the fragments,112 but determined implicitly using an electronegativity-equalization (EE) scheme,8,113 which permits to account for electronic induction effects within molecules. The corresponding atomic parameters, i.e. hardness and electronegativity, are expected to factor out these induction effects and to be much less dependent than the partial charges themselves on the covalent environment of the atoms. Note that although the EE scheme serves to generate the atomic partial charges, its function is fundamentally distinct from the electrostatic-potential fitting14–28 or electron-density partitioning29–38 procedures used in HYFF and QDFF force fields. Whereas the latter procedures aim at deriving partial charges based on a QM calculation, the EE scheme is only used here as a physically-motivated parametric-fitting device for the electrostatic interactions in the liquid, in which the partial charges are solely determined by the condensed-phase properties (no QM input).
For the (halo-)alkane force field,8 the parameter calibration resulted in root-mean-square deviations from experiment of 49.8 (27.6) kg m−3 for ρliq and 2.7 (1.8) kJ mol−1 for ΔHvap considering the calibration (validation) set. The values are lower for the validation set, because it contains larger molecules (stronger influence of purely aliphatic interactions). The trends in the optimized parameters along the halogen series and across the compound family were found to be in line with chemical intuition based on considerations related to size, polarizability, softness, electronegativity, induction, and hyperconjugation. This is remarkable considering that the force-field calibration did not involve any QM calculation.
The goal of the present study is to extend the application of CombiFF to the calibration of a GROMOS-compatible united-atom force field for saturated acyclic compounds encompassing eight common chemical functional groups involving oxygen and/or nitrogen atoms, namely ether, aldehyde, ketone, ester, alcohol, carboxylic acid, amine, and amide. Considering compounds of up to ten carbon atoms including up to four occurrences of the same functional group, the corresponding family of molecules is referred to in this article as the O + N family.
The GROMOS-compatible 2016H66 force field112 is used as a starting point for the optimization and as a reference for performance comparison. Considering 62 small organic molecules, this parameter set was calibrated and validated against condensed-phase experimental data not only for ρliq and ΔHvap, but also for the hydration free energy ΔGwat and the solvation free energy ΔGche in cyclohexane. Note that in addition to 43 compounds of the O + N family, 2016H66 also included 19 molecules representative for thiols, sulfides, disulfides, aromatic compounds, and nucleic-acid bases, which are not considered here.
The present force-field reoptimization using CombiFF relies on a much higher observable-to-parameter ratio. Here, a total of 1712 experimental values for ρliq and ΔHvap concerning 1175 representative molecules of the O + N family are extracted from nine data sources,114–122 and used as target for the calibration and validation of 102 non-bonded interaction parameters.
2 Methodology
The CombiFF workflow for calibrating the parameters of a force field based on experimental data concerning a given compound family is illustrated in Fig. 1. For a detailed description, the reader is referred to our previous article,8 where the scheme was introduced (see in particular Appendix A therein). The present section only summarizes its main features, and provides information on its application to the O + N family. For the ease of reference, a few key numbers (symbols and values) relevant to this optimization are summarized in Table 1.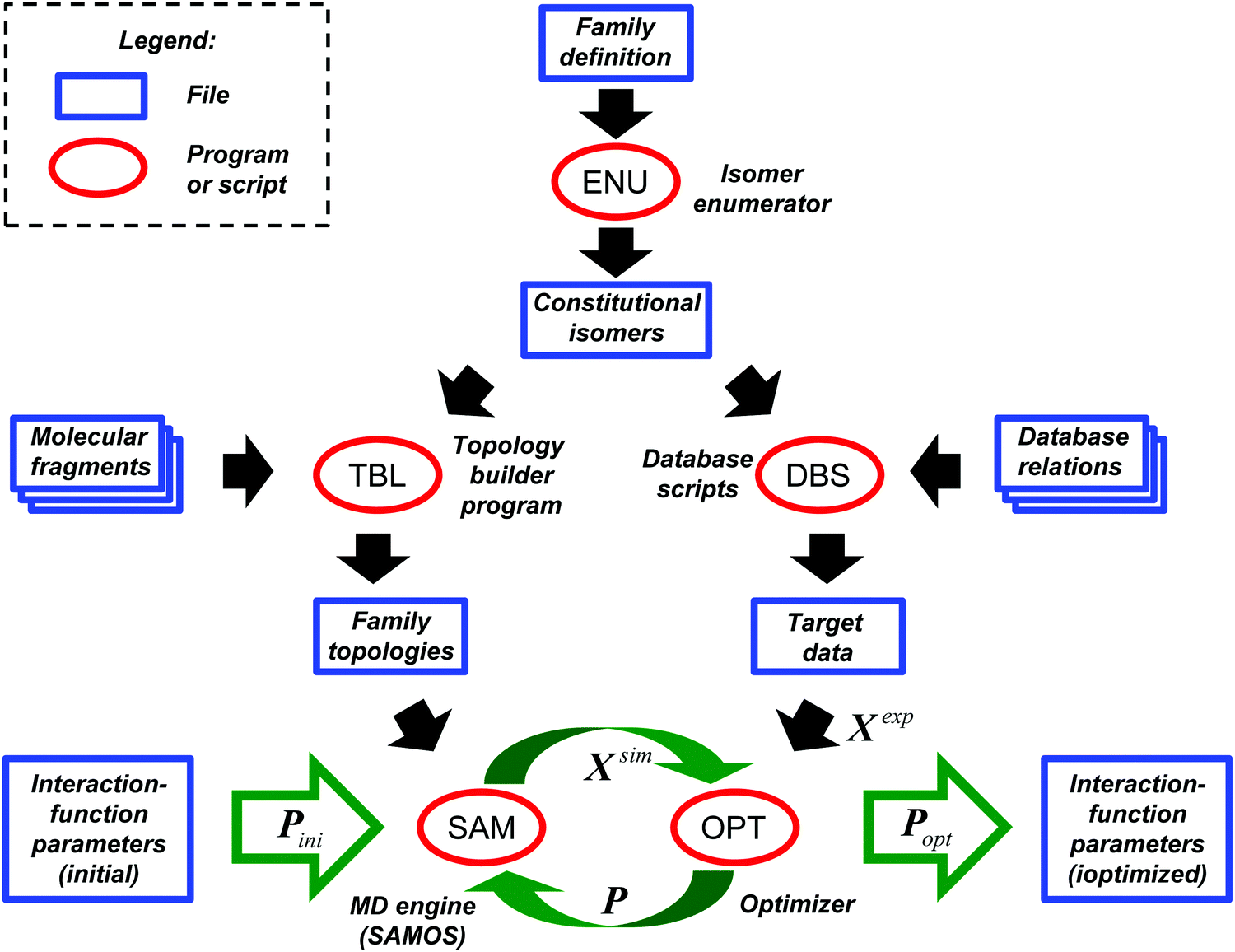 | ||
| Fig. 1 CombiFF workflow for calibrating the parameters of a force field based on experimental data concerning a given compound family. Based on the definition of the family, the program ENU enumerates all possible constitutional isomers. The program TBL is then used to construct the molecular topologies of the corresponding compounds, and the DBS scripts to extract available target experimental data pertaining to these molecules from an in-house database. By alternating simulations (MD engine SAM) to calculate the vector Xsim of simulated observables (as well as its derivative relative to all force-field parameters), and variations of the parameter vector P (optimization script OPT) designed to bring these simulated observables closer to their experimental target vector Xexp, the initial parameters Pini are progressively refined into optimal parameters Popt. | ||
| Number | Value | Meaning |
|---|---|---|
| N totiso | 57![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 905 905 |
Total number of constitutional isomers in the O + N family |
| N simiso | 1175 | Total number of isomers with available experimental data (= Ncaliso + Nvaliso) |
| N totexp | 1712 | Total number of experimental data points (= Ncalexp + Nvalexp) |
| N totsim | 1405 | Total number of distinct P,T-points in this data (= Ncalsim + Nvalsim) |
| N caliso | 339 | Compounds included in the calibration set |
| N calexp | 579 | Experimental data points for the calibration set (= Ncalρ + NcalΔH) |
| N cal ρ | 314 | Experimental ρliq data points for the calibration set |
| N calΔH | 265 | Experimental ΔHvap data points for the calibration set |
| N calsim | 408 | Distinct compounds and P,T-points (i.e. simulations) for the calibration set |
| N valiso | 836 | Compounds included in the validation set |
| N valexp | 1133 | Experimental data points for the validation set (= Nvalρ + NvalΔH) |
| N val ρ | 765 | Experimental ρliq data points for the validation set |
| N valΔH | 368 | Experimental ΔHvap data points for the validation set |
| N valsim | 997 | Distinct compounds and P,T-points (i.e. simulations) for the validation set |
| N EEatt, Natt | 47 | Number of EE-types (or, equivalently, atom types) |
| N LJatt | 12 | Number of LJ-types |
| N totprm | 233 | Total number of force-field parameters (= Ncovprm + Nattprm) |
| N covprm | 94 | Number of covalent parameters |
| N nbdprm | 139 | Number of non-bonded parameters |
| N calprm | 102 | Number of parameters that are optimized |
The O + N family of compounds is defined as the union of the 27 subfamilies listed in Table 2. It includes molecules of up to ten carbon atoms representative for eight chemical functional groups: ether, aldehyde, ketone, ester, alcohol, carboxylic acid, amine, and amide. Depending on the subfamily, the given functional group may occur up to four times in the compound. However, molecules combining different types of functional groups are not considered here. The Ntotiso = 57905 constitutional isomers of the O + N family were enumerated as canonical SMILES strings80,123 using an in-house program (ENU). The numbers Niso of isomers in each of the subfamilies are reported in Table 2.
| Function/acronym | Char. | n | m | N iso | N sim | Subfamily description |
|---|---|---|---|---|---|---|
| Ethers | ||||||
| ROR | O | 1–10 | 1 | 817 | 85 | C1–C10 mono-ethers |
| O | 1–10 | 2 | 2544 | 46 | C1–C10 di-ethers (including acetals and ketals) | |
| O | 1–10 | 3 | 4936 | 17 | C1–C10 tri-ethers (including acetals and ketals) | |
| O | 1–10 | 4 | 6614 | 6 | C1–C10 tetra-ethers (including acetals and ketals) | |
| Aldehydes | ||||||
| RCOH | A | 1 | 1 | 1 | 1 | Formaldehyde |
| A | 2–10 | 1 | 372 | 35 | C2–C10 mono-aldehydes | |
| A | 2–10 | 2 | 551 | 4 | C2–C10 di-aldehydes | |
| Ketones | ||||||
| RCOR | K | 1–10 | 1 | 335 | 85 | C1–C10 mono-ketones |
| K | 1–10 | 2 | 463 | 18 | C1–C10 di-ketones | |
| K | 1–10 | 3 | 379 | 2 | C1–C10 tri-ketones | |
| Esters | ||||||
| HCOOR | F | 1–10 | 1 | 372 | 16 | C1–C10 mono-esters (only formates) |
| HCOOR | F | 1–10 | 2 | 550 | 2 | C1–C10 di-esters (only formates) |
| RCOOR | E | 1–10 | 1 | 662 | 146 | C1–C10 mono-esters (without formates) |
| RCOOR | E | 1–10 | 2 | 1364 | 49 | C1–C10 di-esters (without formates) |
| Alcohols | ||||||
| ROH | L | 1–10 | 1 | 879 | 280 | C1–C10 mono-ols |
| L | 1–10 | 2 | 3670 | 101 | C1–C10 di-ols | |
| L | 1–10 | 3 | 11249 |
6 | C1–C10 tri-ols | |
| Carboxylic acids | ||||||
| RCOOH | C | 1 | 1 | 1 | 1 | Formic acid |
| C | 2–10 | 1 | 372 | 51 | C2–C10 mono-carboxylic acids | |
| C | 3–10 | 2 | 550 | 6 | C3–C10 di-carboxylic acids | |
| Amines | ||||||
| RNH2 | M | 1–10 | 1 | 879 | 52 | C1–C10 mono-primary-amines |
| RNHR | N | 1–10 | 1 | 817 | 42 | C1–C10 mono-secondary-amines |
| RNR2 | R | 1–10 | 1 | 420 | 43 | C1–C10 mono-tertiary-amines |
| RN2* | N | 1–10 | 2 | 17665 |
51 | C1–C10 di-amines |
| Amides | ||||||
| RCONH2 | D | 1–10 | 1 | 372 | 9 | C1–C10 mono-primary-amides |
| RCONHR | D | 1–10 | 1 | 662 | 7 | C1–C10 mono-secondary-amides |
| RCONR2 | D | 1–10 | 1 | 409 | 14 | C1–C10 mono-tertiary-amides |
| Total | — | — | — | 57905 |
1175 | Total over the 27 subfamilies |
To collect available experimental data, the SMILES strings must be mapped to various equivalent identifiers that can be found in the different experimental sources. For example, the SMILES string CC(O)C may appear under the alternative identifiers 2-propanol, propan-2-ol, isopropyl alcohol, isopropanol, or CAS 67-63-0. The PubChem database124 was used to obtain such alternative identifiers. About one quarter of the isomers (15592 molecules) were found in PubChem, and about one fifth of these (3425 molecules) were associated with a CAS registry number. The experimental database (DBS) maintained in our group was queried for ρliq and ΔHvap values pertaining to all these compounds, giving priority to a match by CAS (when available) over a match by name. The nine data sources accessed were ref. 114–122. Note that the data points from ref. 121 that are marked as “estimated” were discarded. The ΔHvap values from this source for the alcohols and carboxylic acids were excluded as well due to inconsistencies, unless a similar value was found in another source. This resulted in ρliq and/or ΔHvap values concerning Nsimiso = 1175 compounds, which were distributed into a calibration set of Ncaliso = 339 molecules and a validation set of Nvaliso = 836 molecules based on the number of carbon atoms (1–6 vs. 7–10). The structures of these compounds are shown in ESI,† Section S1 (Fig. S1 and S2). The acronyms employed for the individual molecules involve one letter and four digits. The letter is representative of the chemical function (see Table 2). The first digit stands for the number of carbon atoms, with the number ten mapped to the digit zero. Finally, the last three digits form a sequential index, further distinguishing compounds for which the first two symbols are identical.
The GROMOS-compatible molecular topologies of these compounds were generated automatically based on the SMILES strings using an in-house program (TBL), by linking the fragments shown in Fig. 2via bond overlap. A total of 54 fragments are required to construct all the molecules of the O + N family (as well as the saturated acyclic alkanes beyond methane). Note, however, that these fragments are only sufficient to generate molecules containing one or more occurrence of a single type of functional group.
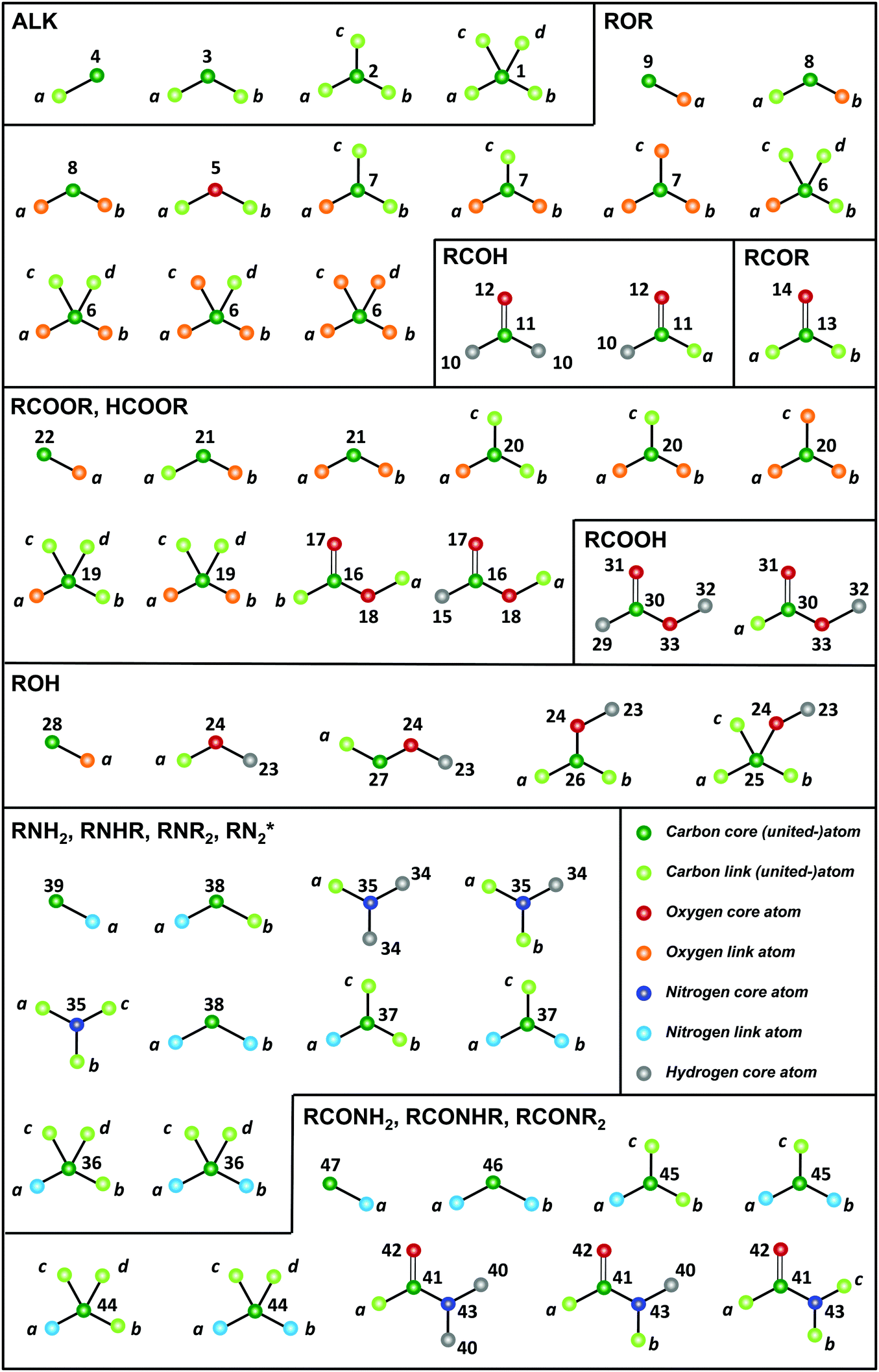 | ||
| Fig. 2 Molecular-topology fragments required for the representation of the O + N family. The 54 fragments represented are required for the construction of the saturated acyclic alkanes beyond methane (ALK) plus the molecules of the O + N family. Note that these fragments are only sufficient to generate molecules containing one ore more occurrence of a single type of functional group. The acronyms used for the different molecule groups are explicited in Table 2. Note that, for simplicity, formaldehyde HCOH and formic acid HCOOH are included in RCOH and RCOOH, respectively. The atoms of a fragment that can be linked (link atoms) are labeled with lower-case letters. The other atoms within the fragment (core atoms) are labeled with their atom types, referring to the numbering of Table 3. A linkage between two fragments is performed by overlapping a core-link bond of the two fragments. | ||
The experimental-data vector Xexp corresponding to the calibration set has the dimension Ncalexp = 579. It encompasses Ncalρ = 314 values for ρliq and NcalΔH = 265 values for ΔHvap, and requires Ncalsim= 408 independent simulations (i.e. distinct compounds and P,T-points) for its evaluation. The corresponding vector for the validation set has the dimension Nvalρ = 1133, encompasses Nvalρ = 765 values for ρliq and NvalΔH = 368 values for ΔHvap, and requires Nvalsim = 997 independent simulations for its evaluation. The reference experimental values retained for ρliq and/or ΔHvap, along with the associated P,T-points, are listed in ESI,† Section S2 (Table S1). This material can be downloaded freely from the internet under ref. 125, where the version 1.0 corresponds to the published article (further versions will include revisions and/or expansions of the data set).
Most of the state points considered are within 10 K of the standard temperature T− = 298.15 K (70% of the points) and within 0.2 bar of the atmospheric pressure P0 = 1 bar (78% of the points). The values for the remaining points range from 250 to 537 K and from 0.002 to 9.06 bar. The impact of using a (limited) fraction of target values at temperatures differing from T− as well as the ability of the force field to reproduce experimental data at temperatures differing (reasonably) from T− have already been investigated for the (halo-)alkane family8 (see Section S13 in the ESI,† of this previous article). There, it was shown that: (i) at the calibration level, there is no systematic correlation between the temperature and the error (calculation vs. experiment) for ρliq and ΔHvap; (ii) at the validation level (considering 50 experimental curves for T-dependent properties spanning a range of about 350 K), there is no correlation between the temperature and the error for ρliq, and only a weak positive correlation for ΔHvap.
The principles of the force-field representation employed here are compatible with those of the GROMOS force field126–131 in its 2016H66 variant,112 except for one important difference. The atomic partial charges are determined for each molecule based on an EE scheme.113 Similarly to our previous work8 (see Appendix A.4 therein), charge flows between atoms are only allowed within overall neutral charge groups, and intramolecular Coulombic effects (J-terms in the EE scheme) are only included for first and second covalent neighbors, as the corresponding interatomic distances can be considered (essentially) configuration-independent. The corresponding terms for more remote covalent neighbors are not necessarily negligible, but their inclusion would lead to conformation-dependent charges and require the application of an expensive on-the-fly EE scheme during the simulations. Since the EE scheme is only a parametric-fitting device for the charges, its effective parameters are expected compensate at least in part for possible deficiencies in the representation. The validity of this assumption is ultimately supported by the observation that an excellent fit to the experimental data can be obtained.
The charge groups relevant for the molecules of the O + N family are illustrated in Fig. 3. All the aliphatic (united-)atoms of the molecule (atom types CH0, CH1, CH2 and CH3 in Table 3) that are not explicitly included in one of these charge groups define separate one-particle charge groups with a charge of zero. The charge groups are used in GROMOS for the application of the non-bonded interaction cutoff, which performs a group-based truncation in terms of the centers of geometry of the two charge groups. The Gaussian-cloud interaction accounting for intramolecular Coulombic effects in the EE scheme (J-terms) between first and second covalent neighbors within a charge group relies on effective interatomic distances ![[r with combining macron]](https://www.rsc.org/images/entities/i_char_0072_0304.gif) calculated based on the reference bond lengths and angles of the covalent force-field parameters, along with effective radii that are set to the Lennard-Jones collision diameters σ of the involved (united-)atoms.
calculated based on the reference bond lengths and angles of the covalent force-field parameters, along with effective radii that are set to the Lennard-Jones collision diameters σ of the involved (united-)atoms.
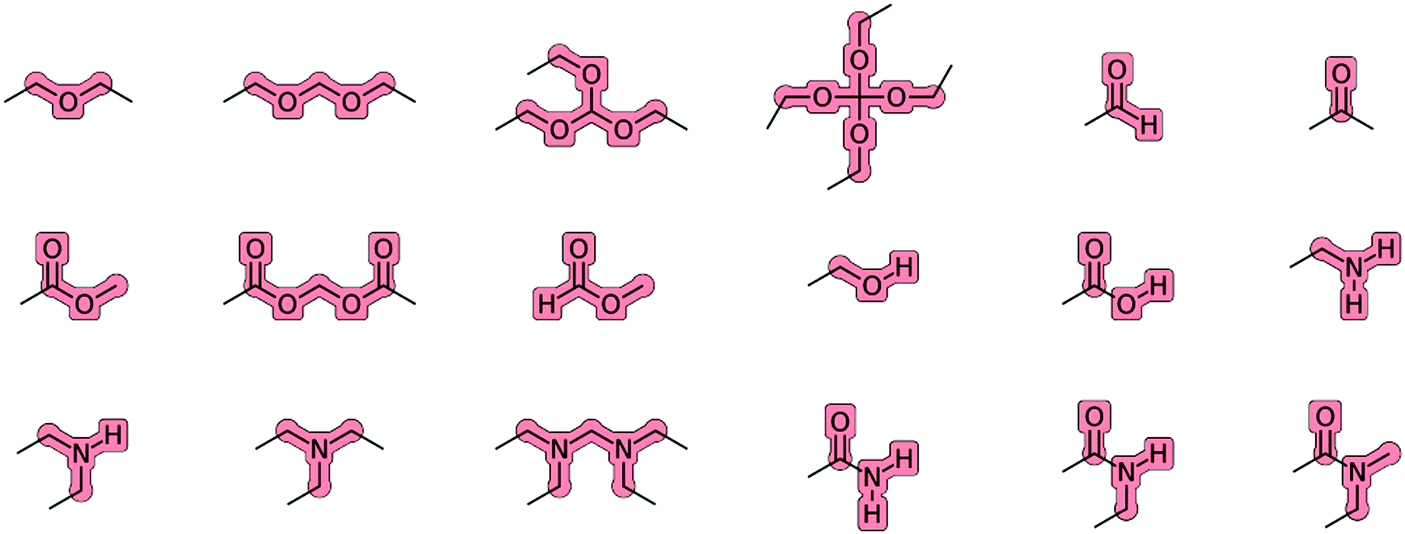 | ||
| Fig. 3 Charge groups relevant for the compounds of the O + N family. Charge flows in the EE scheme are only permitted between atoms belonging to the same overall neutral charge group. All the aliphatic (united-)atoms of the molecule (atom types CH0, CH1, CH2 and CH3 in Table 3) that are not explicitly included in one of these charge groups define separate one-particle charge groups with a charge of zero. | ||
| Idx | Atom type (EE-type) | LJ-type | η [e−1 V] | χ [V] | Usage |
|---|---|---|---|---|---|
| Aliphatic carbon (united-)atoms | |||||
| 1 | CH0 | CH0 | — | — | CH0 carbon atom (methanetetryl group) |
| 2 | CH1 | CH1 | — | — | CH1 carbon united-atom (methanetriyl group) |
| 3 | CH2 | CH2 | — | — | CH2 carbon united-atom (methylene group) |
| 4 | CH3 | CH3 | — | — | CH3 carbon united-atom (methyl group) |
| Ether | |||||
| 5 | O_eth | OR | 35.447 | 37.782 | Ether oxygen atom |
| 6 | CH0_O_eth | CH0 | 2.814 | 21.953 | Alkoxylated CH0 atom |
| 7 | CH1_O_eth | CH1 | 6.568 | 19.993 | Alkoxylated CH1 united-atom |
| 8 | CH2_O_eth | CH2 | 13.981 | 16.921 | Alkoxylated CH2 united-atom |
| 9 | CH3_O_eth | CH3 | 22.115 | 14.233 | Alkoxylated CH3 united-atom |
| Aldehyde | |||||
| 10 | H_CO_ald | HC | 17.156 | 21.368 | Aldehyde hydrogen atom |
| 11 | C_ald | C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O O |
15.981 | 16.008 | Aldehyde carbonyl carbon atom |
| 12 | O_ald | OC |
8.821 | 23.815 | Aldehyde carbonyl oxygen atom |
| Ketone | |||||
| 13 | C_ket | CO |
36.256 | 15.724 | Ketone carbonyl carbon atom |
| 14 | O_ket | OC |
17.326 | 38.611 | Ketone carbonyl oxygen atom |
| Ester | |||||
| 15 | H_CO_est | HC | 16.648 | 24.832 | Formate ester hydrogen atom |
| 16 | C_est | CO |
21.158 | 11.713 | Ester carbonyl carbon atom |
| 17 | O_est | OC |
12.815 | 28.520 | Ester carbonyl oxygen atom |
| 18 | O_R_est | OR | 25.554 | 29.741 | Ester acylated oxygen atom |
| 19 | CH0_O_est | CH0 | 9.033 | 19.163 | Ester oxygen-linked CH0 atom |
| 20 | CH1_O_est | CH1 | 12.401 | 18.349 | Ester oxygen-linked CH1 united-atom |
| 21 | CH2_O_est | CH2 | 36.940 | 13.503 | Ester oxygen-linked CH2 united-atom |
| 22 | CH3_O_est | CH3 | 19.577 | 16.726 | Ester oxygen-linked CH3 united-atom |
| Alcohol | |||||
| 23 | H_ol | HB | 35.794 | 17.095 | Hydroxyl hydrogen atom |
| 24 | O_ol | OH | 31.057 | 46.647 | Hydoxyl oxygen atom |
| 25 | CH0_O_ol | CH0 | 36.145 | 19.963 | Hydroxylated CH0 atom |
| 26 | CH1_O_ol | CH1 | 32.298 | 18.392 | Hydroxylated CH1 united-atom |
| 27 | CH2_O_ol | CH2 | 30.423 | 17.200 | Hydroxylated CH2 united-atom |
| 28 | CH3_O_ol | CH3 | 29.995 | 17.135 | Hydroxylated CH3 united-atom |
| Carboxylic acid | |||||
| 29 | H_CO_acd | HC | 28.828 | 12.561 | Formic acid hydrogen atom |
| 30 | C_acd | CO |
29.109 | 14.269 | Carboxylic acid carbonyl carbon atom |
| 31 | O_acd | OC |
36.286 | 45.338 | Carboxylic acid carbonyl oxygen atom |
| 32 | H_O_acd | HB | 31.782 | 14.733 | Carboxylic acid hydroxyl hydrogen atom |
| 33 | O_H_acd | OH | 41.291 | 36.891 | Carboxylic acid hydroxyl oxygen atom |
| Amine | |||||
| 34 | H_N_amn | HB | 44.559 | 7.467 | Amine hydrogen atom |
| 35 | N_amn | N_amn | 39.441 | 47.379 | Amine nitrogen atom |
| 36 | CH0_N_amn | CH0 | 33.307 | 13.667 | Aminated CH0 atom |
| 37 | CH1_N_amn | CH1 | 31.777 | 15.046 | Aminated CH1 united-atom |
| 38 | CH2_N_amn | CH2 | 36.009 | 12.981 | Aminated CH2 united-atom |
| 39 | CH3_N_amn | CH3 | 31.313 | 13.650 | Aminated CH3 united-atom |
| Amide | |||||
| 40 | H_N_amd | HB | 29.386 | 14.179 | Amide nitrogen-linked hydrogen atom |
| 41 | C_amd | CO |
30.320 | 14.492 | Amide carbonyl carbon atom |
| 42 | O_amd | OC |
28.316 | 42.678 | Amide carbonyl oxygen atom |
| 43 | N_amd | N_amd | 30.230 | 37.064 | Amide acylated nitrogen atom |
| 44 | CH0_N_amd | CH0 | (35.585) | (15.614) | Amide nitrogen-linked CH0 atom (estimated) |
| 45 | CH1_N_amd | CH1 | 35.544 | 19.373 | Amide nitrogen-linked CH1 united-atom |
| 46 | CH2_N_amd | CH2 | 36.828 | 13.164 | Amide nitrogen-linked CH2 united-atom |
| 47 | CH3_N_amd | CH3 | 34.383 | 14.305 | Amide nitrogen-linked CH3 united-atom |
The covalent interaction parameters relevant for the O + N family were taken or ported by analogy from the 2016H66 parameter set,112 and kept unaltered. The corresponding information is summarized in ESI,† Section S3 (Table S2 and Fig. S3). Only the non-bonded interaction parameters were subjected to refinement, and solely a subset thereof.
The atomic partial charges are determined indirectly via the EE scheme based on two types of atomic parameters, the hardness η and the electronegativity χ. Owing to the use of a geometric-mean combination rule132,133 for the Lennard-Jones (LJ) interactions,134 the corresponding pairwise coefficients are also constructed based on two types of atomic parameters, the collision diameter σ and the well depth ε. Following the GROMOS design principle, the values σ and ε are only used in the combination rule for non-hydrogen-bonding LJ-type pairs (corresponding to the LJ parameters C6 and C12,I in GROMOS). For hydrogen-bonding LJ-type pairs, GROMOS relies on a modified set of LJ parameters with slightly enhanced repulsion. In this case, alternative values ![[small sigma, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e10d.gif) and
and ![[small epsilon, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0de.gif) are used instead (corresponding to the LJ parameters C6 and C12,II in GROMOS). For simplicity, the value of the dispersion coefficient C6 is kept identical in the two sets. As result, only needs to be specified, while can be deduced as = εσ6/6. Finally, for third covalent neighbors, yet another pair of values σ* and ε* is used in the combination rule. Each atom type of the force field is thus associated with a unique selection for six (non-hydrogen-bonding type) or seven (potentially hydrogen-bonding type) parameters. However, the same σ and ε parameters are often used for different atom types of the same element. As a result, the present force field for the O + N family relies on a number Natt = 47 of atom types, which are equivalent to EE-types (NEEatt = Natt), but involves a smaller number NLJatt = 12 of LJ-types. The final (optimized) values of the EE parameters for the 47 atom types (or EE-types) are reported in Table 3, along with a LJ-type. The latter refers to the entries of Table 4, where the final values of the LJ parameters are reported for the 12 LJ-types. The correspondence between elements, LJ-types, atom-types (EE-types), and chemical functional groups involving the latter atom types is also illustrated schematically in Fig. 4.
are used instead (corresponding to the LJ parameters C6 and C12,II in GROMOS). For simplicity, the value of the dispersion coefficient C6 is kept identical in the two sets. As result, only needs to be specified, while can be deduced as = εσ6/6. Finally, for third covalent neighbors, yet another pair of values σ* and ε* is used in the combination rule. Each atom type of the force field is thus associated with a unique selection for six (non-hydrogen-bonding type) or seven (potentially hydrogen-bonding type) parameters. However, the same σ and ε parameters are often used for different atom types of the same element. As a result, the present force field for the O + N family relies on a number Natt = 47 of atom types, which are equivalent to EE-types (NEEatt = Natt), but involves a smaller number NLJatt = 12 of LJ-types. The final (optimized) values of the EE parameters for the 47 atom types (or EE-types) are reported in Table 3, along with a LJ-type. The latter refers to the entries of Table 4, where the final values of the LJ parameters are reported for the 12 LJ-types. The correspondence between elements, LJ-types, atom-types (EE-types), and chemical functional groups involving the latter atom types is also illustrated schematically in Fig. 4.
and associated with the LJ-types of the two involved atoms are used instead. The value of C6 is common, so that only is specified in the table, while can be deduced as = εσ6/6 (0.359, 0.614, 0.619, 0.619, and 0.504 kJ mol−1 for OR, OC, OH, N_amn, and N_amd, respectively). The hydrogen-bonding LJ-type pairs are those involving a potential hydrogen-bond donor (OH, N_amn, or N_amd) and a potential hydrogen-bond acceptor (OR, OC, OH, N_amn, or N_amd). Finally, for third covalent neighbors, the alternative values σ* and ε* are to be used instead. Note that the ten LJ parameters σ and ε corresponding to the aliphatic (united-)atom types CH0 to CH3 and to the polar hydrogen atom HB were not subject to optimization, but taken directly from the 2016H66 parameter set.112 The same applies to all the σ* and ε* parameters. The value of OR (between parentheses) is kept identical to σ in the absence of parametrization target (no hydrogen bonding is possible in molecules containing only ether groups). The initial values of the LJ parameters (used to start the optimization) can be found in ESI,,† Table S4
| LJ type | σ |
|
σ* | ε | ε* | Usage |
|---|---|---|---|---|---|---|
| [nm] | [kJ mol−1] | |||||
| Carbon | ||||||
| CH0 | 0.664 | — | 0.336 | 0.007 | 0.406 | CH0 carbon atom (methanetetryl group) |
| CH1 | 0.502 | — | 0.330 | 0.095 | 0.567 | CH1 carbon united-atom (methanetriyl group) |
| CH2 | 0.407 | — | 0.316 | 0.411 | 1.176 | CH2 carbon united-atom (methylene group) |
| CH3 | 0.375 | — | 0.309 | 0.867 | 1.946 | CH3 carbon united-atom (methyl group) |
| CO |
0.345 | — | 0.336 | 0.326 | 0.406 | Carbonyl carbon atom |
| Oxygen | ||||||
| OR | 0.301 | (0.301) | 0.287 | 0.359 | 1.011 | Ether oxygen atom |
| OC |
0.296 | 0.313 | 0.262 | 0.857 | 1.725 | Carbonyl oxygen atom |
| OH | 0.287 | 0.298 | 0.287 | 0.776 | 1.011 | Hydoxyl oxygen atom |
| Nitrogen | ||||||
| N_amn | 0.312 | 0.308 | 0.298 | 0.572 | 0.877 | Amine nitrogen atom |
| N_amd | 0.320 | 0.312 | 0.298 | 0.429 | 0.877 | Amide nitrogen atom |
| Hydrogen | ||||||
| HC | 0.223 | — | 0.223 | 0.119 | 0.119 | Carbonyl-linked hydrogen atom |
| HB | 0.000 | — | 0.000 | 0.000 | 0.000 | Oxygen- or nitrogen-linked hydrogen atom |
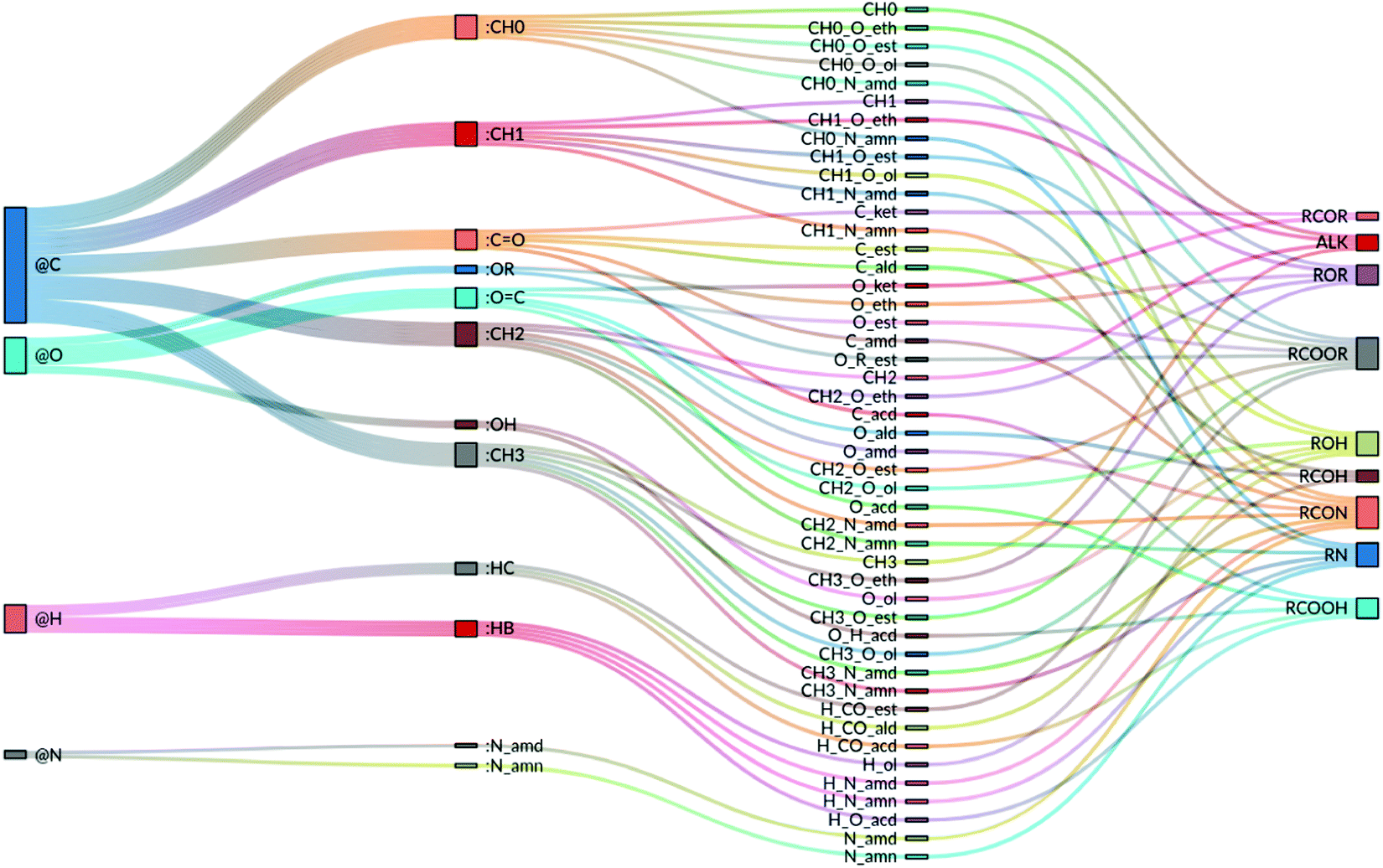 | ||
| Fig. 4 Correspondence between the 4 elements, the 12 LJ-types, the 47 atom-types (EE-types) and 9 chemical functional groups involving the latter atom types in the proposed GROMOS-compatible force field for the O + N family. The first column refers to the elements, the second to the LJ-types (see Table 4), the third to the atom- or EE-types (see Table 3), and the fourth to the chemical functional groups (see Table 2; note that ALK was added for alkanes and that RCOOR, RCON and RN group all the esters, amides and amines, respectively, of Table 2). | ||
The four aliphatic atom types (CH0, CH1, CH2 and CH3) have no EE parameters, as their charge is always zero. The LJ parameters of these atom types,135–137 along with those of the polar hydrogen atom (type HB, zero in GROMOS), as well as all the third-neighbor LJ interaction parameters, were also excluded from the optimization, i.e. kept as in the 2016H66 set.112 Note also that, in the absence of parametrization target, the η and χ values of EE-type CH0_N_amd are estimated (no experimental data found for a compound involving this type), and that the value of the LJ-type OR is kept identical to σ (no hydrogen bonding is possible in molecules containing only ether groups). The initial parameter values selected to start the optimization are reported in ESI,† Section S4 (Tables S3 and S4). For the LJ parameters, they were ported from the 2016H66 force field.112 For the EE parameters, they were fitted to best reproduce the atomic partial charges of this force field.
Following from the above choices, the present force field for the O + N family involves Ntotprm = 233 parameters, namely Ncovprm = 94 covalent parameters along with Nnbdprm = 139 non-bonded parameters (2 × 43 relevant EE-types + 4 × 7 non-hydrogen-bonding LJ-types + 5 × 5 potentially hydrogen-bonding LJ-types), among which Ncalprm = 102 are subject to optimization (omitted are 2 EE parameters for CH0_N_amd, 2 × 12 third-neighbor LJ parameters, 2 × 5 LJ parameters for CH0 to CH3 and HB, and 1 LJ parameter for the potentially hydrogen-bonding OR). The optimization of these parameters against Ncalexp = 579 experimental data points in the calibration set involves an observable-to-parameter ratio of 5.7 (up to 16.8 when also considering the Nvalexp = 1133 data points in the validation set). This ratio is further analyzed for each of the EE- and LJ-types separately in ESI,† Section S5 (Tables S5 and S6). A favorable observable-to-parameter ratio is observed in most cases, although three EE-types (CH3_O_ol, H_CO_acd, and CH1_N_amd) only occur in a single representative molecule, and one (CH0_N_amd) is not represented at all.
The search for optimal parameters was performed as in our previous work8 (see Appendix A.7 therein), by minimizing an objective function Q(P;Xexp) of the parameter vector P accounting for the deviation of the simulated-data vector Xsim(P) relative to the experimental-data vector Xexp. This function is defined as
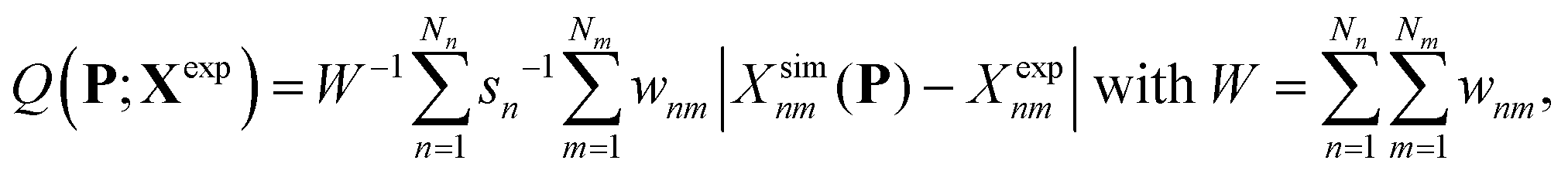 | (1) |
The optimization is performed by assuming that Xsim(P) is approximately linear in parameter changes within a small trust region around a reference point P0 in parameter space, i.e. using the local first-order approximation ![[Q with combining tilde]](https://www.rsc.org/images/entities/i_char_0051_0303.gif) (P;P0,Xexp) to Q(P;Xexp) defined by
(P;P0,Xexp) to Q(P;Xexp) defined by
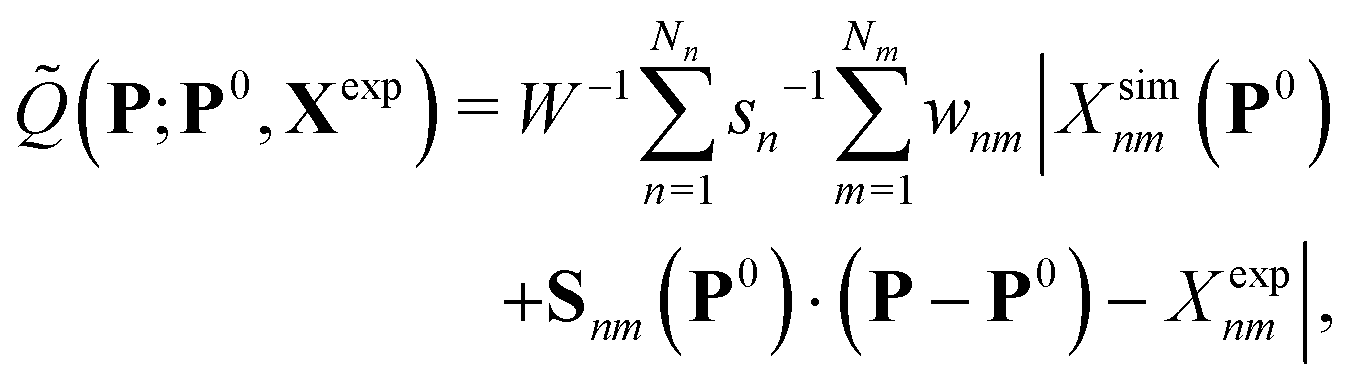 | (2) |
![[S with combining low line]](https://www.rsc.org/images/entities/b_char_0053_0332.gif) (P0) is the sensitivity matrix defined by the variations of the different molecule/observable combinations with respect to variations of the Nk parameters around the point P0, i.e.
(P0) is the sensitivity matrix defined by the variations of the different molecule/observable combinations with respect to variations of the Nk parameters around the point P0, i.e.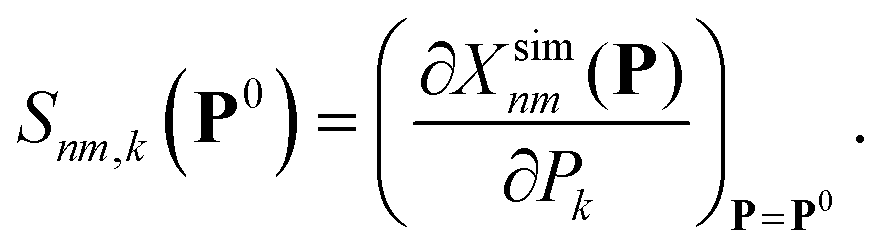 | (3) |
within a specified trust region around P0. This region is defined here in terms of maximal allowed relative changes in each of the parameters over an iteration, set to 5% for all parameters optimized. Since the function is convex, the point P* is unique. Once determined, it is selected as a new point P0 to carry out simulations in view of a next iteration. Note that convexity only applies to the local approximation of eqn (2), but not to the objective function Q of eqn (1) itself, which will generally present many local minima in parameter space.
In practice, the optimization algorithm is carried out by an optimizer script (OPT), and involves the following steps: (1) select an initial guess for P0i at iteration i = 0; (2) perform Ncalsim simulations to get the simulated-data vector Xsim along with the sensitivity matrix S at P0i; (3) calculate the real value Qreali = Q(P0i;Xexp) of the objective function at this point in parameter space; (4) minimize (P;P0i,Xexp) in eqn (2) with respect to P starting from P0i and remaining within the trust radius, leading to Pi*; (5) calculate the predicted value Qpredi+1 = (Pi*;P0i,Xexp) of the objective function; (6) set P0i+1 to Pi*, increment i, and iterate to step (2) until convergence.
Step (2) is the expensive part of the calculation. In contrast, step (4) is inexpensive, and carried out in practice using a simplex minimization. Considering steps (3) and (5), from i = 1 onward, the real value Qreali at iteration i can be compared to the predicted value Qpredi from the previous iteration, giving a hint about the accuracy of the linearization in eqn (2) within the imposed trust radius. If the algorithm terminates at iteration imax, the final parameter set is 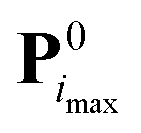 with the value
with the value 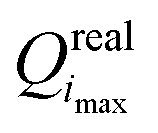 of the objective function. Although
of the objective function. Although 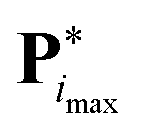 and
and 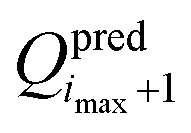 are available, it is preferable to stop at a force field with an explicitly calculated objective function. In the present application to the O + N family, a total of imax = 5 iterations were performed to reach convergence. Given the setup adopted in the simulations and access to 408 processors (3 GHz Intel Xeon), i.e. one for each of the Ncalsim simulations to be carried out at each iteration, the full optimization required about three days of wall-clock computing time. The non-bonded parameters of the final force field are reported in Tables 3 and 4, along with ESI,† Table S2 for the covalent terms.
are available, it is preferable to stop at a force field with an explicitly calculated objective function. In the present application to the O + N family, a total of imax = 5 iterations were performed to reach convergence. Given the setup adopted in the simulations and access to 408 processors (3 GHz Intel Xeon), i.e. one for each of the Ncalsim simulations to be carried out at each iteration, the full optimization required about three days of wall-clock computing time. The non-bonded parameters of the final force field are reported in Tables 3 and 4, along with ESI,† Table S2 for the covalent terms.
The simulations were performed using an in-house GROMOS-compatible simulation engine in C++ called SAMOS (SAM). The pure-liquid MD simulations were carried out under periodic boundary conditions based on cubic computational boxes containing 512 molecules, and in the isothermal–isobaric ensemble at the reference pressures P and temperatures T listed in ESI,† Table S1. The temperature was maintained close to its reference value using a Nosé–Hoover thermostat139 with a coupling time of 0.1 ps. The pressure was maintained close to its reference value using an Andersen barostat140 with a coupling time of 0.5 ps. The isothermal compressibility was set to 4.575 × 10−4 kJ−1 mol nm3 for compatibility with the standard GROMOS setup.112 The ideal-gas simulations relied on stochastic dynamics141–145 (SD). They were also carried out under periodic boundary conditions based on cubic computational boxes containing 512 molecules but with all intermolecular interactions turned off, and in the canonical ensemble at the same temperatures as the corresponding pure-liquid simulations. The friction coefficient was set to 2 ps−1.
Newton's equations of motion (MD) or Langevin's equations of motion (SD) were integrated using the leap-frog scheme141,146 with a timestep of 2 fs. Constraints on all bond lengths were enforced by application of the SHAKE procedure147 with a relative geometric tolerance of 10−5. The non-bonded interactions were calculated using a twin-range scheme148 based on charge-group distances with short- and long-range cutoff radii set to 0.8 and 1.4 nm, respectively, and an update frequency of 5 timesteps for the short-range pairlist and intermediate-range interactions. In the pure-liquid simulations, the mean effect of the omitted electrostatic interactions beyond the long-range cutoff distance was reintroduced using a reaction-field correction.149,150 The corresponding permittivities are listed in ESI,† Table S1.
Additional details about the simulation protocols can be found in ref. 8. The protocol applied here is essentially the same, except for the use of a doubled friction coefficient in SD and a SHAKE tolerance reduced by a factor of ten. These modifications were found necessary for a better temperature control in the case of molecules with explicit hydrogen atoms. The GROMOS-compatible molecular topologies and equilibrated liquid configurations for the Ntotsim = 1175 molecules considered here can be downloaded freely from the internet under ref. 125 (version 1.0).
3 Results and discussion
The evolution of the objective function Q against the iteration number i is illustrated in Fig. 5. The graph shows both the real value Qreali at iteration i calculated according to eqn (1) and its predicted value Qpredi calculated based on the simulations at iteration i − 1 according to eqn (2). After two iterations, the predicted and real values agree very well, indicating that the linear approximation is accurate within the chosen trust region for the parameter variations. During the first three iterations, the objective function drops sharply. It is essentially converged after four iterations, and the fifth one brings no further improvement. The final force field corresponds to iteration i = 5 and is associated with a value of 1.69 for Q.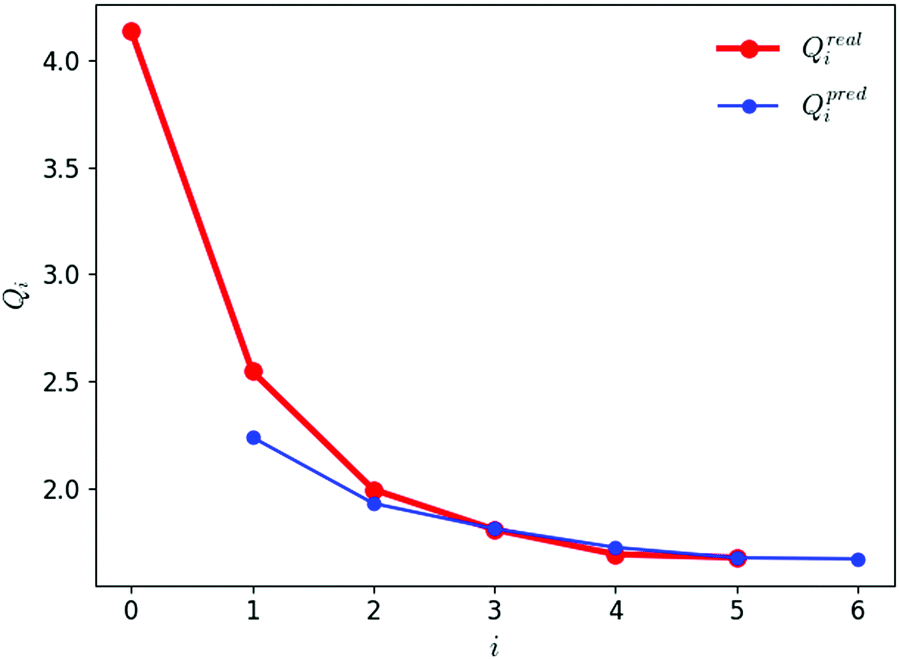 | ||
| Fig. 5 Evolution of the predicted and real values of the objective function against the iteration number during the force-field parameter optimization. The real value Qreali at iteration i is calculated according to eqn (1). The predicted value Qpredi is calculated according to eqn 2 based on the simulations at iteration i − 1. The first simulations at i = 0 using the initial parameter set leads to a first real value Qreal0 and a first predicted value Qpredi. The last simulations at i = 5 using the final (optimized) parameter set leads to the final real value Qreal5 (and a predicted value Qpred6, which is discarded). | ||
The evolution of the Ncalprm = 102 non-bonded interaction parameters subject to calibration against the iteration number i is shown in Fig. 6 and 7 for the LJ and EE parameters, respectively. The largest parameter changes typically occur within the first three iterations, where the decrease of Q is most pronounced. However, the magnitudes of these changes still remain limited. This is not unexpected considering that the initial values selected to start the optimization (ESI,† Section S4) are ported from the 2016H66 parameter set,112 and thus already expected to perform reasonably well.
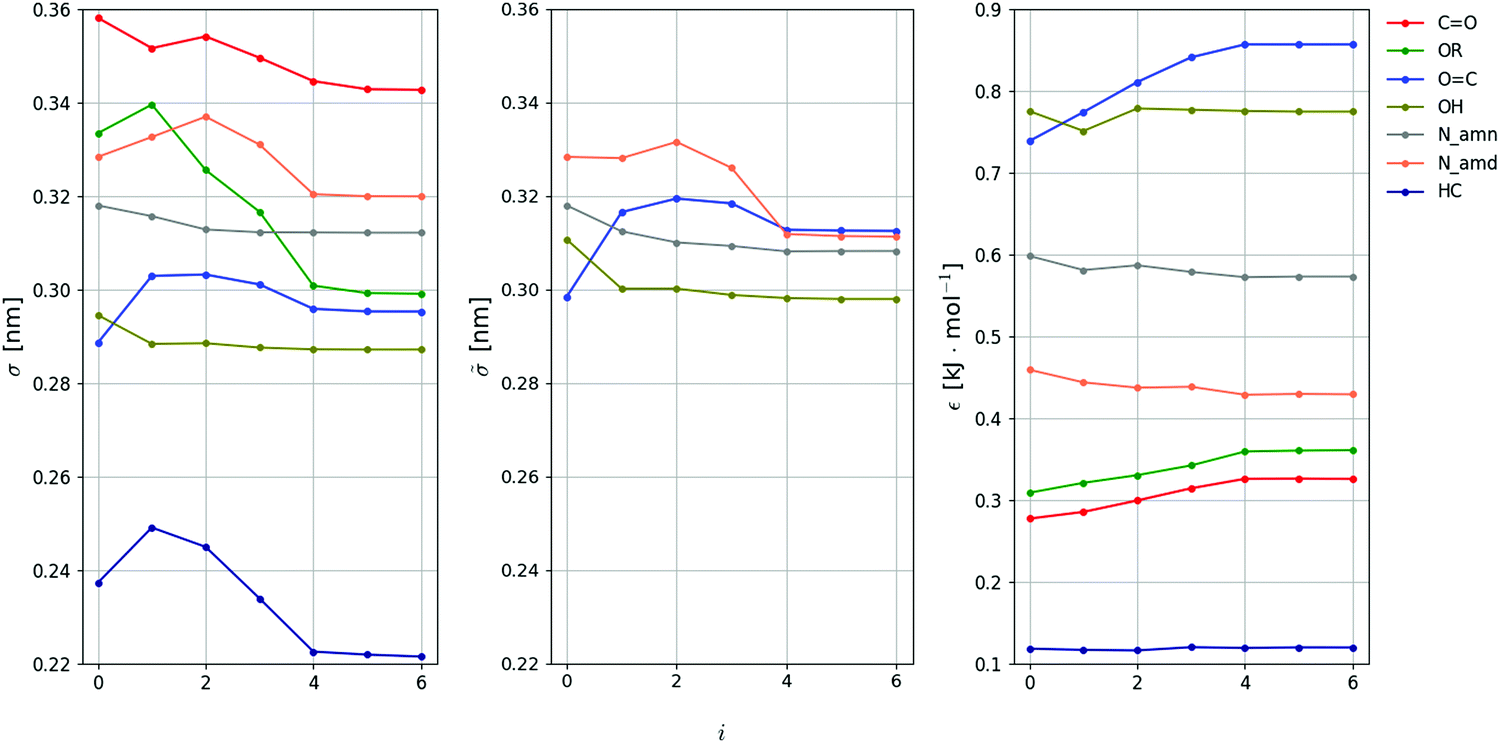 | ||
| Fig. 6 Evolution of the LJ interaction parameters against the iteration number during the force-field parameter optimization. The value of each parameter is reported at iteration i. The 18 parameters considered are the collision diameter σ and well depth ε for 7 non-hydrogen-bonding LJ-types, along with the collision diameter for 4 (out of the 5) potentially hydrogen-bonding LJ-types, see Table 4. Since the value of C6 is common to the two sets, only is displayed, while can be deduced as = εσ6/6. The value of the LJ-type OR (not shown) is kept identical to σ in the absence of parametrization target (no hydrogen bonding is possible in molecules containing only ether groups). The final force-field parameters are those corresponding to iteration i = 5 (the values at i = 6 correspond to proposed changes for a next iteration, and are discarded). | ||
 | ||
| Fig. 7 Evolution of the EE interaction parameters against the iteration number during the force-field parameter optimization. The value of each parameter is reported at iteration i. The 84 parameters considered are the hardness η and electronegativity χ for the 42 EE-types subject to optimization, see Table 3. The final force-field parameters are those corresponding to iteration i = 5 (the values at i = 6 correspond to proposed changes for a next iteration, and are discarded). | ||
The level of agreement between the optimized force field and experiment in terms of ρliq and ΔHvap is illustrated in Fig. 8 for both the calibration (top panels) and validation (bottom panels) sets. The corresponding numerical values can be found in ESI,† Section S6 (Table S.7). The statistics per compound types is provided in Table 5 and illustrated graphically in Fig. 9 for both the calibration (top panels) and validation (bottom panels) sets, distinguishing between molecules presenting one, two, three, or four occurrences of the specific functional group. In this statistics, two classes of compounds are also considered separately, namely the non-hydrogen-bonding ones (NHB), including ethers, aldehydes, ketones, esters, tertiary amines, and tertiary amides, and the hydrogen-bonding ones (HBD), including alcohols, carboxylic acids, the other amines, and the other amides.
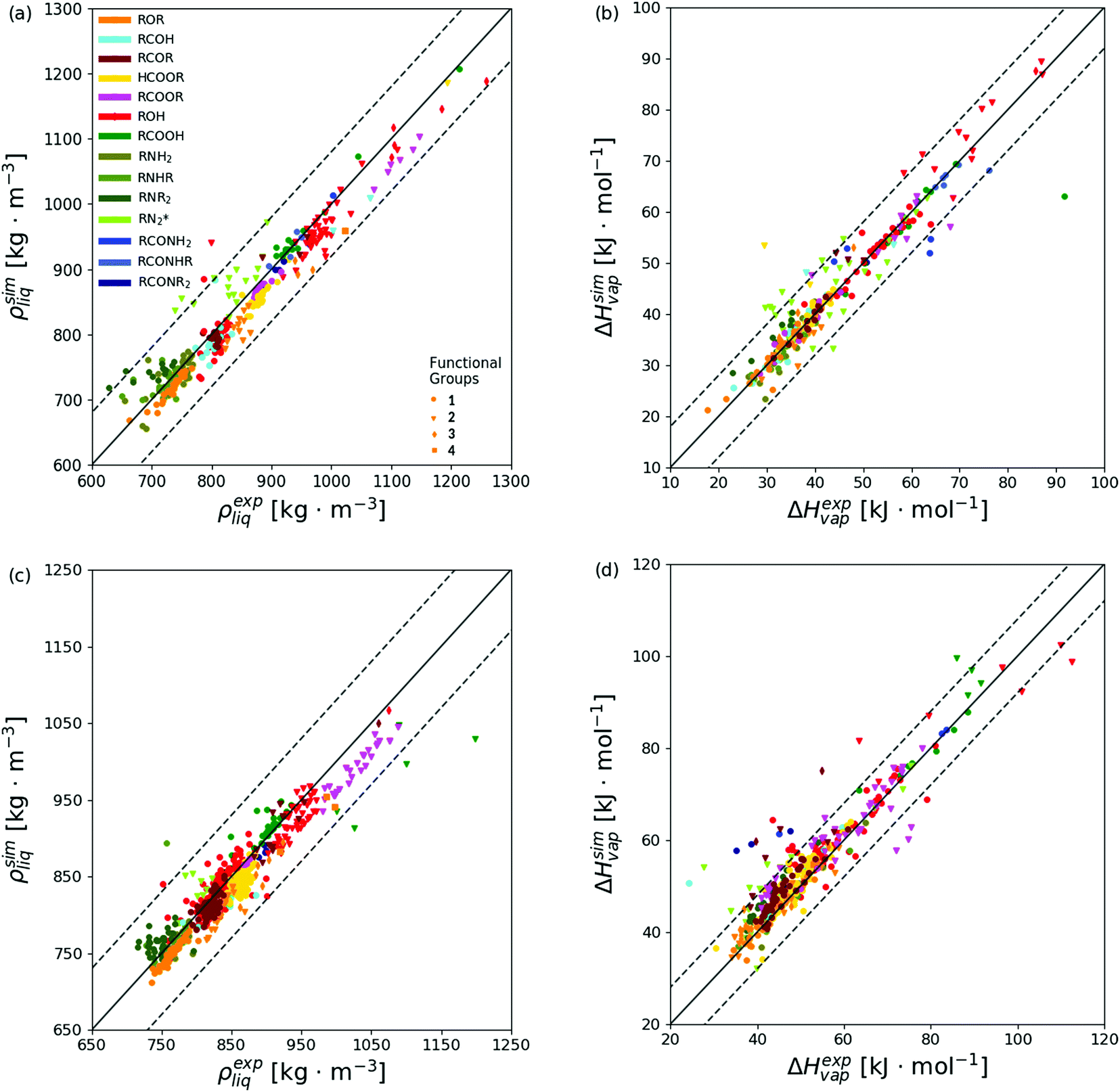 | ||
| Fig. 8 Simulated versus experimental properties based on the optimized force field. The results are reported for the calibration set (a and b) and the validation set (c and d), considering the pure-liquid density ρliq and vaporization enthalpy ΔHvap. The colors are selected according to the 14 groups of molecules defined in Table 2. The symbols are selected according to the number of occurrences of the functional group in the molecule. The diagonal solid lines indicate perfect agreement, and the ranges between the two dashed lines indicate agreement within ±80.0 kg m−3 for ρliq or ±8.0 kJ mol−1 for ΔHvap (scaling factors sn used in eqn (1) multiplied by 4 and 8, respectively). The corresponding numerical values can be found in ESI,† Section S6, the statistics per groups of molecules in Table 5 and Fig. 9, and the information about the main outliers (points outside the ranges defined by the dashed lines in any of the four panels) in ESI,† Section S7. | ||
| Code | m | Calibration | Validation | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N cal ρ | ρ liq [kg m−3] | N calΔH | ΔHvap [kJ mol−1] | N val ρ | ρ liq [kg m−3] | N valΔH | ΔHvap [kJ mol−1] | ||||||
| RMSD | AVED | RMSD | AVED | RMSD | AVED | RMSD | AVED | ||||||
| ROR | 1 | 28 | 15.7 | −13.8 | 23 | 1.8 | 0.5 | 56 | 15.4 | −14.6 | 19 | 1.5 | 0.1 |
| 2 | 11 | 37.8 | −34.8 | 12 | 2.7 | −1.8 | 28 | 30.4 | −28.2 | 17 | 2.1 | 0.1 | |
| 3 | 3 | 51.5 | −48.5 | 4 | 3.5 | 3.0 | 6 | 39.0 | −36.9 | 11 | 3.7 | 2.8 | |
| 4 | 1 | 64.2 | −64.2 | 2 | 0.6 | 0.6 | 3 | 42.1 | −40.7 | 3 | 1.5 | 1.2 | |
| 1–4 | 43 | 28.4 | −22.8 | 41 | 2.3 | 0.1 | 93 | 24.0 | −21.0 | 50 | 2.3 | 0.8 | |
| RCOH | 1 | 16 | 27.3 | −9.4 | 13 | 1.7 | 0.6 | 21 | 21.9 | −16.1 | 8 | 9.6 | 5.2 |
| 2 | 2 | 50.1 | −49.8 | 2 | 7.3 | 3.8 | 0 | — | — | 0 | — | — | |
| 1–2 | 18 | 30.7 | −13.9 | 15 | 3.1 | 1.0 | 21 | 21.9 | −16.1 | 8 | 9.6 | 5.2 | |
| RCOR | 1 | 11 | 15.2 | −8.4 | 11 | 0.9 | −0.1 | 73 | 12.8 | −7.4 | 49 | 3.3 | 2.8 |
| 2 | 3 | 24.8 | −0.5 | 3 | 5.1 | 4.1 | 11 | 18.0 | −5.2 | 6 | 10.5 | 9.2 | |
| 3 | 0 | − | − | 0 | − | − | 1 | 9.6 | −9.6 | 2 | 20.2 | 20.2 | |
| 1–3 | 14 | 17.7 | −6.7 | 14 | 2.5 | 0.8 | 85 | 13.5 | −7.1 | 57 | 5.9 | 4.1 | |
| HCOOR | 1 | 12 | 12.2 | −11.3 | 10 | 1.6 | 0.5 | 4 | 2.6 | −2.4 | 3 | 0.8 | 0.3 |
| 2 | 1 | 7.5 | −7.5 | 2 | 17.6 | 15.5 | 0 | — | — | 0 | — | — | |
| 1–2 | 13 | 11.9 | −11.0 | 12 | 7.3 | 3.0 | 4 | 2.6 | −2.4 | 3 | 0.8 | 0.3 | |
| RCOOR | 1 | 20 | 24.1 | −22.0 | 19 | 1.1 | 0.5 | 115 | 21.1 | −18.8 | 70 | 3.2 | 1.9 |
| 2 | 6 | 45.4 | −45.3 | 9 | 4.9 | −0.6 | 27 | 37.5 | −36.5 | 38 | 5.9 | 2.0 | |
| 1–2 | 26 | 30.4 | −27.3 | 28 | 2.9 | 0.2 | 142 | 25.1 | −22.2 | 108 | 4.3 | 1.9 | |
| ROH | 1 | 33 | 28.4 | −14.0 | 32 | 2.1 | −0.2 | 247 | 16.1 | −3.1 | 55 | 5.1 | 2.3 |
| 2 | 45 | 34.5 | −17.9 | 12 | 5.2 | 2.9 | 52 | 21.6 | −15.6 | 10 | 8.7 | 0.9 | |
| 3 | 5 | 38.4 | −27.0 | 1 | 1.9 | 1.9 | 1 | 8.2 | −8.2 | 0 | — | — | |
| 1–3 | 83 | 32.5 | −16.9 | 45 | 3.2 | 0.7 | 300 | 17.2 | −5.3 | 65 | 5.8 | 2.0 | |
| RCOOH | 1 | 17 | 12.0 | 6.3 | 13 | 8.0 | −2.4 | 35 | 17.1 | 11.7 | 7 | 3.1 | 1.1 |
| 2 | 0 | — | — | 0 | — | — | 5 | 107.6 | −98.4 | 4 | 8.1 | 6.8 | |
| 1–2 | 17 | 12.0 | 6.3 | 13 | 8.0 | −2.4 | 40 | 41.3 | −2.1 | 11 | 5.5 | 3.2 | |
| RNH2 | 1 | 34 | 20.2 | −2.0 | 22 | 2.1 | −0.3 | 17 | 14.1 | −5.9 | 9 | 2.2 | 0.3 |
| RNHR | 1 | 25 | 22.0 | 5.4 | 19 | 1.4 | −0.5 | 16 | 36.2 | 7.8 | 10 | 1.8 | 0.0 |
| RNR2 | 1 | 13 | 46.1 | 39.4 | 12 | 3.6 | 3.2 | 31 | 25.6 | 18.4 | 17 | 4.9 | 4.7 |
| RN2* | 2 | 16 | 57.8 | 48.1 | 31 | 6.1 | 2.1 | 9 | 36.2 | 28.0 | 19 | 8.9 | 5.1 |
| 1–2 | 88 | 34.9 | 15.3 | 84 | 4.1 | 1.0 | 73 | 27.8 | 11.6 | 55 | 6.0 | 3.3 | |
| RCONH2 | 1 | 4 | 15.6 | −8.0 | 4 | 8.7 | −2.0 | 1 | 2.2 | −2.2 | 3 | 9.5 | 5.9 |
| RCONHR | 1 | 4 | 10.7 | 3.4 | 7 | 3.1 | −1.4 | 0 | — | — | 0 | — | — |
| RCONR2 | 1 | 4 | 8.7 | −6.1 | 2 | 0.5 | −0.1 | 6 | 11.6 | −9.0 | 8 | 12.5 | 9.6 |
| 1 | 12 | 12.0 | −3.6 | 13 | 5.4 | −1.4 | 7 | 10.8 | −8.1 | 11 | 11.8 | 8.6 | |
| NHB | — | 131 | 29.0 | −12.9 | 124 | 3.5 | 0.9 | 382 | 22.3 | −14.5 | 251 | 5.2 | 2.7 |
| HBD | — | 183 | 30.6 | −2.6 | 141 | 4.6 | 0.2 | 383 | 22.4 | −3.6 | 117 | 6.1 | 2.4 |
| All | — | 314 | 29.9 | −6.9 | 265 | 4.1 | 0.5 | 765 | 22.4 | −9.1 | 368 | 5.5 | 2.6 |
 | ||
| Fig. 9 Statistics concerning the discrepancies between simulated and experimental properties based on the optimized force field. The results are reported for the calibration set (a and b) and the validation set (c and d). The values of the root-mean square deviation (RMSD, transparent bar) and average deviation (AVED, solid bar) in ρliq (a and c) and ΔHvap (b and d) for selected groups of molecules (Table 2) are compared. The successive bars in each group correspond to compounds containing 1, 2, 3 or 4 occurrences of the functional group. The last three bars refer to non-hydrogen-bonding (NHB), hydrogen-bonding (HBD), and the entire set (All) of molecules. The corresponding numerical values can be found in Table 5. | ||
Considering all compounds of the calibration set, the optimized force field has an overall root-mean-square deviation (RMSD) relative to experiment of 29.9 kg m−3 for ρliq and 4.1 kJ mol−1 for ΔHvap. Thus, given the choice of the scaling factors sn in eqn (1), namely 20 kg m−3 for ρliq and 1 kJ mol−1 for ΔHvap, the final value of 1.69 for is dominated by the ΔHvap errors. A similar and somewhat stronger bias is also observed in the validation set, with final RMSD values of 22.4 kg m−3 for ρliq and 5.5 kJ mol−1 for ΔHvap. This observation contrasts with the results of the previous application of CombiFF to the saturated haloalkanes,8 where the contributions of the two types of observables were of comparable magnitudes, with RMSD values of 49.8 (27.6) kg m−3 for ρliq and 2.7 (1.8) kJ mol−1 for ΔHvap over the calibration (validation) set. For the O + N family, one also observes a tendential underestimation of the density and overestimation of the vaporization enthalpy, with average deviations (AVED) relative to experiment of −6.9 (−9.1) kg m−3 for ρliq and +0.5 (+2.6) kJ mol−1 for ΔHvap over the calibration (validation) set. The corresponding values for the saturated haloalkanes8 were comparatively smaller in magnitude, namely +1.2 (+5.3) kg m−3 for ρliq and 0.0 (0.0) kJ mol−1 for ΔHvap.
In both calibration and validation sets, the errors are not distributed homogeneously across the different chemical groups. The ρliq values are nearly systematically underestimated, but the monocarboxylic acids, the amines (except primary), and the secondary amides present positive deviations instead. The underestimation is particularly pronounced for the dicarboxylic acids in the validation set (AVED value of −98.4 kg m−3). Similarly, the ΔHvap values are nearly systematically overestimated, but a few compound groups in the calibration set present slightly negative deviations, all below 1.0 kJ mol−1 in magnitude except for the diethers, the monocarboxylic acids, and the primary and secondary amides (down to −2.4 kJ mol−1). The overestimation is particularly pronounced for the formic diesters and the triketones (AVED values of +15.5 and +20.2 kJ mol−1, respectively). For both ρliq and ΔHvap, the deviations relative to experiment within a given group of molecules are also seen to nearly systematically increase upon increasing the number of occurrences of the functional group in the molecule.
The most prominent outliers of both calibration and validation sets, namely the 58 molecules (62 simulations) with deviations larger than 80.0 kg m−3 for ρliq and/or larger than 8.0 kJ mol−1 for ΔHvap, are discussed in ESI,† Section S7 (Table S8 and Fig. S4). Among these 58 molecules, 40 encompass two functional groups (two notable exceptions being methanol and trimethylamine). Methanol was also used in the calibration of 2016H66. Similarly to the results shown here, ρliq and ΔHvap are overestimated, but 2016H66 leads to significantly lower errors in terms of ρliq.
Finally, a comparison between the present force field and the original 2016H66 parameter set112 is presented in ESI,† Section S8 (Tables S9–S11). After optimization of the non-bonded interaction parameters, the overall agreement with experiment is enhanced, although not very pronouncedly. Moreover, it is also non-systematic across the different groups of molecules. In particular, the accuracy enhancement is most significant for polyfunctional molecules compared to monofunctional ones, although these molecules still present comparatively large deviations after the parameter refinement (see above).
4 Conclusions
In this article, the CombiFF scheme8 (Fig. 1) was applied to the calibration of a GROMOS-compatible united-atom force field for the O + N family of compounds (Table 2). This force field relies on 47 atom types (equivalent to EE-types) distributed over 12 LJ-types. The aliphatic (united-)atom EE- and LJ-types are common with the previously optimized (halo-)alkane force field.8 The calibration of the 102 non-bonded interaction parameters was performed here against 579 experimental liquid densities ρliq and vaporization enthalpies ΔHvap concerning 339 small molecules, which represents an observable-to-parameter ratio of 5.7. A collection of 1133 additional ρliq and ΔHvap values concerning 836 other molecules was used for subsequent validation, leading to an effective observable-to-parameter ratio of 16.8. The calibration of the force field required five iterations, corresponding to about three days of wall-clock computing time using 408 processors.The optimized (final) force-field parameters (reported in Tables 3 and 4, along with ESI,† Table S2) lead to RMSD (AVED) values of 29.9 (−6.9) kg m−3 for ρliq and 4.1 (0.5) kJ mol−1 for ΔHvap over the calibration set. The corresponding values over the validation set are 22.4 (−9.1) kg m−3 and 5.5 (2.6) kJ mol−1, respectively. Considering the two sets together (1175 molecules, 1712 data points), the relevant RMSD values are 24.8 kg m−3 for ρliq and 4.9 kJ mol−1 for ΔHvap. This shows that a very good level of agreement with experiment can be achieved for the O + N family using a simple united-atom force field, and that the overall observable-to-parameter ratio of 16.8 is sufficient to define an appropriate set of parameters. However, appropriateness does not imply uniqueness. As observed previously,8 the solution reached upon optimization is essentially unique for the LJ interaction parameters, but a larger extent of degeneracy is observed for the EE parameters. This degeneracy leads to a significant variability in these parameters, whereas the atomic partial charges themselves are not significanltly affected.151
A more detailed analysis of the errors reveals three main trends: (i) the residual errors are biased towards larger discrepancies in terms of ΔHvap relative to ρliq (compared to our previous work on haloalkanes,8 where the errors were balanced between the two observables); (ii) the ρliq values are nearly systematically underestimated and the ΔHvap values are nearly systematically overestimated relative to experiment; (iii) the deviations nearly systematically increase with the number of occurrences of the functional group in a molecule. These observations may result in part from the following three factors.
First, the ΔHvap comparison is likely to be affected by intrinsically larger experimental and computational errors compared to the ρliq comparison. Experimentally, the ΔHvap values result from more complicated measurements (temperature-dependence of the vapor pressure or calorimetry at the boiling point for ΔHvapvs. simple volumetric measurement for ρliq), and are potentially also affected by more significant ambiguities related to the measurement pressure (isochoric measurement at the vapor pressure of the liquid vs. isobaric measurement in the presence of an inert gas) and the possible need for and/or application of real-gas corrections. Computationally, the ΔHvap values probe a less local and more collective property (energetics for ΔHvapvs. packing for ρliq), and are potentially also affected by more significant calculation ambiguities (influence of the treatment of long-range interactions, neglect of the difference in polarization between liquid-phase and gas-phase molecules when using a non-polarizable force field).
Second, there appears to be conflicting requirements imposed by the two types of observables. If a given parameter variation enhances the tightness of molecular packing, it will generally also increase the strength of attractive intermolecular interactions. As a result, one generally expects the changes in ρliq and ΔHvap induced by a given parameter variation to be positively correlated. In the optimized force field proposed here for the O+N family, a compromise is reached and the residual negative errors for ρliq and positive errors for ΔHvap cannot be relieved by further parameter variations. This situation may arise from inaccurate experimental data (incompatibility between ρliq and ΔHvap observables) and/or from a lack of flexibility of the force-field functional form (e.g. limitations of the LJ combination rules or of the EE charge scheme, absence of explicit electronic polarizability).
Third, the lower force-field accuracy for polyfunctional compounds may result from a combination of multiple factors: (i) insufficient number of polyfunctional compounds in the calibration set (e.g. dicarboxylic acids and triketones are only included in the validation set); (ii) inaccurate representation of the conformational properties of the molecules (torsional-dihedral and third-neighbor parameters ported rather crudely from the 2016H66 force field without reoptimization, see ESI,† Fig. S3); (iii) inaccurate representation of the charge transfers within the molecules (charge transfers in the EE scheme are only allowed here within charge groups).
Compared to the 2016H66 parameter set,112 the calibration/validation of which involved 43 monofunctional molecules of the O + N family, the present reoptimization leads to a general improvement in terms both ρliq and ΔHvap, especially for polyfunctional molecules. However, this improvement is neither extremely pronounced nor entirely systematic for the monofunctional compounds. This suggests that the 2016H66 parametrization was already close to optimal for these 43 compounds.
A key component of the selected force-field representation is that the atomic partial charges are generated using an EE scheme, which permits to take into account induction effects. Thus, the atomic partial charges can change according to the chemical environment within the considered molecules. For example, primary, secondary and tertiary alcohols have different charge sets in the present force field. In contrast, in the 2016H66 set,112 the charges were fixed for a given functional group, irrespective of its chemical environment. This feature is expected to enhance significantly the model flexibility, especially when multiple functional groups are present within the same molecule. However, the requirement to define small neutral charge groups and the corresponding definitions adopted in this work still represent a limitation on the representation of inductive effects. For example, in compounds involving a carbonyl group, the Cα carbon (united-)atom (i.e. directly attached to the carbonyl group) has a charge of zero (which is not the case for a Hα). This is consistent with the charge-group choices made in the 2016H66 set,112 but clearly at odds with chemical intuition.
Generally speaking, a number of the force-field representation choices made here will have to be addressed again in future work, and the parameter optimization repeated accordingly. This will include in particular a reconsideration of: (i) the parameters that were not reoptimized here, i.e. the bond-stretching and bond-angle bending parameters, the torsional-dihedral and third-neighbor interaction parameters, and the non-bonded interaction parameters of the aliphatic types; (ii) the choice of the EE- and LJ-type sets, e.g. by introducing distinct LJ-types for alcohol vs. acid as well as ether vs. ester oxygen atoms; (iii) the choice of model resolution, i.e. united- vs. all-atom; (iv) the choice of a combination rule, e.g. geometric-mean vs. others, and its possible (partial) by-passing; (v) the restriction of EE charge flows to small neutral charge groups (see above). The latter restriction could for example be alleviated by using a more general EE scheme8 (see Conclusion section therein) involving damped charge transfers throughout the entire molecule, applied with a smooth atom-based truncation of the non-bonded interactions in the simulations.152 Efforts are currently in progress along these different lines.
A possible further development of CombiFF would be the design of a polarizable force field of the fluctuating-charge type,153–163 where an on-the-fly EE scheme would incorporate the effect of the configuration-dependent (i.e. local and instantaneous) electric potential on the atomic partial charges of each molecule during the MD simulation (now also including the J-terms for intramolecular Coulombic effects beyond first- and second-neighbors). A particularly appealing feature of this development is that it would not require any additional force-field parameters, but a mere CombiFF recalibration of the existing ones under application of the fluctuating-charge scheme.
Work is also in progress to expand the CombiFF calibration/validation to other chemical families, to polyfunctional molecules mixing different types of functional groups, and to the consideration of further thermodynamic, transport, and dielectric properties of the liquid (as well as properties concerning the gas and solid phases). In particular, for calibration, it might be of interest to consider vapor pressures Pvap in addition to (or instead of) vaporization enthalpies ΔHvap as a target for probing the intermolecular energetics. The quantity Pvap is more readily available experimentally and easier to measure (thus likely affected by smaller uncertainties). The price to pay is that its calculation is more difficult than that of ΔHvap, as it corresponds to a free-energy (rather than energy) calculation. In terms of validation, it will be essential to assess the accuracy of the CombiFF force fields not only in terms of the optimization targets ρliq and ΔHvap, but also in terms of other properties. Such an assessment considering both the previously reported (halo-)alkane force field8 and the present O + N force field has already been performed in terms of nine additional properties (surface-tension coefficient γ, isothermal compressibility κT, isobaric thermal expansion coefficient αP, isobaric heat capacity cP, static relative dielectric permittivity ε, self-diffusion coefficient D, shear viscosity η, hydration free energy ΔGwat and free energy of solvation ΔGche in cyclohexane). The calculated values of these additional properties show reasonable to good agreement with experiment, except for cP, D and η, where larger discrepancies are observed, in large part related to the classical treatment of the vibrations and the use of united atoms. These results will be reported in a forthcoming article.
Conflicts of interest
There are no conflicts to declare.Note added after first publication
This article replaces the version published on 19th July 2021, which contained various errors.Acknowledgements
Financial support by the Swiss National Science Foundation (Grant No. 200021-175944) is gratefully acknowledged. The authors are also grateful to Bill Acree, Sadra Kashef Ol Gheta, Salomé Rieder, Alžbeta Kubincová, and Bruno Horta for insightful discussions and useful suggestions.References
- T. A. Halgren, Curr. Opin. Struct. Biol., 1995, 5, 205 CrossRef CAS PubMed.
- P. H. Hünenberger and W. F. van Gunsteren, Empirical classical interaction functions for molecular simulations, in Computer simulation of biomolecular systems, theoretical and experimental applications, ed. W. F. van Gunsteren, P. K. Weiner, A. J. Wilkinson, Kluwer/Escom Science Publishers, Dordrecht, The Netherlands, 1997, vol. 3, pp. 3–82 Search PubMed.
- P. H. Hünenberger and W. F. van Gunsteren, Empirical classical force fields for molecular systems, in Lecture notes in Chemistry, ed. A. F. Sax, Springer Verlag, Berlin, Germany, 1999, pp. 177–214 Search PubMed.
- D. A. MacKerell Jr, J. Comput. Chem., 2004, 25, 1584 CrossRef PubMed.
- L. Monticelli and D. P. Tieleman, Methods Mol. Biol., 2013, 924, 197 CrossRef CAS PubMed.
- P. S. Nerenberg and T. Head-Gordon, Curr. Opin. Struct. Biol., 2018, 49, 129 CrossRef CAS PubMed.
- S. Riniker, J. Chem. Inf. Model., 2018, 58, 565 CrossRef CAS PubMed.
- M. P. Oliveira, M. Andrey, S. R. Rieder, L. Kern, D. F. Hahn, S. Riniker, B. A. C. Horta and P. H. Hünenberger, J. Chem. Theory Comput., 2020, 16, 7525 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Am. Chem. Soc., 1981, 103, 335 CrossRef CAS.
- P. K. Weiner and P. A. Kollman, J. Comput. Chem., 1981, 2, 287 CrossRef CAS.
- B. R. Brooks, R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan and M. Karplus, J. Comput. Chem., 1983, 4, 187 CrossRef CAS.
- J. Hermans, H. J. C. Berendsen, W. F. van Gunsteren and J. P. M. Postma, Biopolymers, 1984, 23, 1513 CrossRef CAS.
- W. F. van Gunsteren and H. J. C. Berendsen, Groningen molecular simulation (GROMOS) library manual, BIOMOS, Groningen, The Netherlands, 1987 Search PubMed.
- C. I. Bayly, P. Cieplak, W. D. Cornell and P. A. Kollman, J. Phys. Chem., 1993, 97, 10269 CrossRef CAS.
- Y. Duan, C. Wu, S. Chowdhury, M. C. Lee, G. Xiong, W. Yang Zhang, R. Cieplak, P. Luo, R. Lee, T. Caldwell, J. Wang and J. Kollman, J. Comput. Chem., 2003, 24, 1999 CrossRef CAS PubMed.
- C. M. Breneman and K. B. Wilberg, J. Comput. Chem., 1990, 11, 361 CrossRef CAS.
- D. E. Williams, Rev. Comput. Chem., 1991, 2, 219 CAS.
- R. H. Henchman and J. W. Essex, J. Comput. Chem., 1999, 20, 483 CrossRef CAS.
- A. Jakalian, D. B. Jack and C. I. Bayly, J. Comput. Chem., 2002, 23, 1623 CrossRef CAS PubMed.
- C. Chipot, J. Comput. Chem., 2003, 24, 409 CrossRef CAS PubMed.
- M. Udier-Blagović, P. Morales de Tirado, S. A. Pearlman and W. L. Jorgensen, J. Comput. Chem., 2004, 25, 1322 CrossRef PubMed.
- A. Stachowicz, A. Styrcz and J. Korchowiec, J. Mol. Model., 2011, 17, 2217 CrossRef CAS PubMed.
- A. Stachowicz and J. Korchowiec, Struct. Chem., 2012, 23, 1449 CrossRef CAS.
- A. Ahmed and S. I. Sandler, J. Chem. Theory Comput., 2013, 9, 2774 CrossRef CAS PubMed.
- J. P. M. Jämbeck, F. Mocci, A. P. Lyubartsev and A. Laaksonen, J. Comput. Chem., 2013, 34, 187 CrossRef PubMed.
- D. S. Cerutti, J. E. Rice, W. C. Swope and D. A. Case, J. Phys. Chem. B, 2013, 117, 2328 CrossRef CAS PubMed.
- D. S. Cerutti, W. C. Swope, J. E. Rice and D. A. Case, J. Chem. Theory Comput., 2014, 10, 4515 CrossRef CAS PubMed.
- C. M. Ionescu, D. Sehnal, F. L. Falginella, P. Pant, L. Pravda, T. Bouchal, S. V. Vařeková, S. Geidl and J. Koča, J. Cheminform., 2015, 7, 50 CrossRef PubMed.
- F. L. Hirshfeld, Theor. Chim. Acta, 1977, 44, 129 CrossRef CAS.
- P. Bultinck, C. Van Alsenoy, P. W. Ayers and R. Carbó-Dorca, J. Chem. Phys., 2007, 126, 144111 CrossRef PubMed.
- O. Yakovenko, A. A. Oliferenko, V. G. Bdzhola, V. A. Palyulin and N. S. Zefirov, J. Comput. Chem., 2008, 29, 1332 CrossRef CAS PubMed.
- T. A. Manz and D. S. Sholl, J. Chem. Theory Comput., 2010, 6, 2455 CrossRef CAS PubMed.
- N. M. Garrido, M. Jorge, A. J. Queimada, J. R. B. Gomes, I. G. Economou and E. A. Macedo, Phys. Chem. Chem. Phys., 2011, 13, 17384 RSC.
- T. A. Manz and D. S. Sholl, J. Chem. Theory Comput., 2012, 8, 2844 CrossRef CAS PubMed.
- L. P. Lee, D. J. Cole, C.-K. Skylaris, W. L. Jorgensen and M. C. Payne, J. Chem. Theory Comput., 2013, 9, 2981 CrossRef CAS PubMed.
- L. P. Lee, N. Gabaldon Limas, D. J. Cole, M. C. Pyne, C.-K. Skylaris and T. A. Manz, J. Chem. Theory Comput., 2014, 10, 5377 CrossRef CAS PubMed.
- T. Verstraelen, S. Vandenbrande, F. Heidar-Zadeh, L. Vanduyfhuys, V. Van Speybroeck, M. Waroquier and P. W. Ayers, J. Chem. Theory Comput., 2016, 12, 3894 CrossRef CAS PubMed.
- A. Peérez de la Luz, J. A. Aguilar-Pineda, J. G. Méndez-Bermúdez and J. Alejandre, J. Chem. Theory Comput., 2018, 14, 5949 CrossRef PubMed.
- D. Reith and K. N. Kirschner, Comput. Phys. Commun., 2011, 182, 2184 CrossRef CAS.
- S. Grimme, J. Chem. Theory Comput., 2014, 10, 4497 CrossRef CAS PubMed.
- G. Prampolini, M. Campetella, N. De Mitri, P. R. Livotto and I. Cacelli, J. Chem. Theory Comput., 2016, 12, 5525 CrossRef CAS PubMed.
- J.-P. Piquemal and K. D. Jordan, J. Chem. Phys., 2017, 147, 161401 CrossRef PubMed.
- P. Xu, E. B. Guidez, C. Bertoni and M. S. Gordon, J. Chem. Phys., 2018, 148, 090901 CrossRef.
- J. T. Horton, A. E. A. Allen, L. S. Dodda and D. J. Cole, J. Chem. Inf. Model., 2019, 59, 1366 CrossRef CAS PubMed.
- A. E. A. Allen, M. J. Robertson, M. C. Payne and D. J. Cole, ACS Omega, 2019, 4, 14537 CrossRef CAS PubMed.
- S. M. Kantonen, H. S. Muddana, M. Schauperl, N. M. Henriksen and L. Wang, J. Chem. Theory Comput., 2020, 16, 1115 CrossRef CAS PubMed.
- X. Chu and A. Dalgarno, J. Chem. Phys., 2004, 121, 4083 CrossRef CAS PubMed.
- A. Olasz, K. Vanommeslaeghe, A. Krishtal, T. Veszprémi, C. Van Alsenoy and P. Geerlings, J. Chem. Phys., 2007, 127, 224105 CrossRef PubMed.
- A. J. Stone and A. J. Misquitta, Int. Rev. Phys. Chem., 2007, 26, 193 Search PubMed.
- A. J. Stone, Science, 2008, 321, 787 CrossRef CAS PubMed.
- A. Tkatchenko and M. Scheffler, Phys. Rev. Lett., 2009, 102, 073005 CrossRef PubMed.
- A. Tkatchenko, R. A. DiStasio Jr, R. Car and M. Scheffler, Phys. Rev. Lett., 2012, 108, 236402 CrossRef PubMed.
- V. V. Gobre and A. Tkatchenko, Nat. Commun., 2013, 4, 2341 CrossRef PubMed.
- T. Bučko, S. Lebègue, J. Hafner and J. G. Ángyán, J. Chem. Theory Comput., 2013, 9, 4293 CrossRef PubMed.
- T. Bučko, S. Lebègue, J. G. Ángyán and J. Hafner, J. Chem. Phys., 2014, 141, 034114 CrossRef PubMed.
- D. J. Cole, J. Z. Vilseck, J. Tirado-Rives, M. C. Payne and W. L. Jorgensen, J. Chem. Theory Comput., 2016, 12, 2312 CrossRef CAS PubMed.
- M. Mohebifar, E. R. Johnson and C. N. Rowley, J. Chem. Theory Comput., 2017, 13, 6146 CrossRef CAS PubMed.
- C.-K. Skylaris, P. D. Haynes, A. A. Mostofi and M. C. Payne, J. Chem. Phys., 2005, 122, 084119 CrossRef PubMed.
- I. S. Ufimtsev, N. Luehr and T. J. Martinez, J. Phys. Chem. Lett., 2011, 2, 1789 CrossRef CAS.
- D. R. Bowler and T. Miyazaki, Rep. Prog. Phys., 2012, 75, 036503 CrossRef CAS PubMed.
- J. Dziedzic, S. J. Fox, T. Fox, C. S. Tautermann and C.-K. Skylaris, Int. J. Quantum Chem., 2013, 113, 771 CrossRef CAS.
- K. Wilkinson and C.-K. Skylaris, J. Comput. Chem., 2013, 34, 2446 CrossRef CAS PubMed.
- K. A. Wilkinson, N. D. M. Hine and C.-K. Skylaris, J. Chem. Theory Comput., 2014, 10, 4782 CrossRef CAS PubMed.
- G. Lever, D. J. Cole, R. Lonsdale, K. E. Ranaghan, D. J. Wales, A. J. Mulholland, C.-K. Skylaris and M. C. Payne, J. Phys. Chem. Lett., 2014, 5, 3614 CrossRef CAS PubMed.
- C. A. Renison, K. D. Fernandes and K. J. Naidoo, J. Comput. Chem., 2015, 36, 1410 CrossRef CAS PubMed.
- L. Ruddigkeit, R. van Deursen, L. C. Blum and J.-L. Reymond, J. Chem. Inf. Model., 2012, 52, 2864 CrossRef CAS PubMed.
- J.-L. Reymond, Acc. Chem. Res., 2015, 48, 722 CrossRef CAS PubMed.
- W. L. Jorgensen, K. P. Jensen and A. N. Alexandrova, J. Chem. Theory Comput., 2007, 3, 1987 CrossRef CAS PubMed.
- A. J. Stone, J. Chem. Theory Comput., 2005, 1, 1128 CrossRef CAS PubMed.
- D. L. Mobley, E. Dumont, J. D. Chodera and K. A. Dill, J. Phys. Chem. B, 2007, 111, 2242 CrossRef CAS PubMed.
- P. Bultinck, P. W. Ayers, S. Fias, K. Tiels and C. Van Alsenoy, Chem. Phys. Lett., 2007, 444, 205 CrossRef CAS.
- O. Beckstein and B. I. Iorga, J. Comput.-Aided Mol. Des., 2012, 26, 635 CrossRef CAS PubMed.
- O. Beckstein, A. Fourrier and B. I. Iorga, J. Comput.-Aided Mol. Des., 2014, 28, 265 CrossRef CAS PubMed.
- J. Z. Vilseck, J. Tirado-Rives and W. L. Jorgensen, J. Chem. Theory Comput., 2014, 10, 2802 CrossRef CAS PubMed.
- L. S. Dodda, J. Z. Vilseck, K. J. Cutrona and W. L. Jorgensen, J. Chem. Theory Comput., 2015, 11, 4273 CrossRef CAS PubMed.
- E. Boulanger, L. Huang, C. Rupakheti, A. D. MacKerell Jr. and B. Roux, J. Chem. Theory Comput., 2018, 14, 3121 CrossRef CAS PubMed.
- K. M. Visscher and D. P. Geerke, J. Chem. Theory Comput., 2019, 15, 1875 CrossRef CAS PubMed.
- Y. M. H. Gonçalves, C. Senac, P. F. J. Fuchs, P. H. Hünenberger and B. A. C. Horta, J. Chem. Theory Comput., 2019, 15, 1806 CrossRef PubMed.
- J. R. Ullmann, J. ACM, 1976, 23, 31 CrossRef.
- D. Weininger, A. Weininger and J. L. Weininger, J. Chem. Inf. Comput. Sci., 1989, 29, 97 CrossRef CAS.
- R. Grund, Konstruktion molekularer Graphen mit gegebenen Hybridisierungen und überlappungsfreien Fragmenten, Lehrstuhl II für Mathematik der Universität Bayreuth, 1994.
- N. Schneider, R. A. Sayle and G. A. Landrum, J. Chem. Inf. Model., 2015, 55, 2111 CrossRef CAS PubMed.
- J. Wang, W. Wang, P. A. Kollman and D. A. Case, J. Mol. Graphics Modell., 2006, 25, 247 CrossRef CAS PubMed.
- K. Vanommeslaeghe and A. D. MacKerell Jr, J. Chem. Inf. Model., 2012, 52, 3144 CrossRef CAS PubMed.
- K. Vanommeslaeghe, E. P. Raman and A. D. MacKerell Jr, J. Chem. Inf. Model., 2012, 52, 3155 CrossRef CAS PubMed.
- A. K. Malde, L. Zuo, M. Breeze, M. Stroet, D. Poger, P. C. Nair, C. Oostenbrink and A. E. Mark, J. Chem. Theory Comput., 2011, 7, 4026 CrossRef CAS PubMed.
- K. B. Koziara, M. Stroet, A. K. Malde and A. E. Mark, J. Comput.-Aided Mol. Des., 2014, 28, 221 CrossRef CAS PubMed.
- M. Stroet, B. Caron, K. M. Visscher, D. P. Geerke, A. K. Malde and A. E. Mark, J. Chem. Theory Comput., 2018, 14, 5834 CrossRef CAS PubMed.
- E. Krieger, G. Koraimann and G. Vriend, Proteins: Struct., Funct., Genet., 2002, 47, 393 CrossRef CAS PubMed.
- A. W. Schüttelkopf and D. M. F. van Aalten, Acta Crystallogr., 2004, D60, 1355 CrossRef PubMed.
- A. A. S. T. Ribeiro, B. A. C. Horta and R. B. de Alencastro, J. Braz. Chem. Soc., 2008, 19, 1433 CrossRef CAS.
- V. Zoete, M. A. Cuendet, A. Grosdidier and O. Michielin, J. Comput. Chem., 2011, 32, 2359 CrossRef CAS PubMed.
- C. Margreitter, D. Petrov and B. Zagrovic, Nucleic Acids Res., 2013, 41, W422 CrossRef PubMed.
- S. Jo, X. Cheng, S. M. Islam, L. Huang, H. Rui, A. Zhu, H. S. Lee, Y. Qi, W. Han, K. Vanommeslaeghe, A. D. MacKerell Jr, B. Roux and W. Im, Adv. Protein Chem. Struct. Biol., 2014, 96, 235 CAS.
- Y. Pevzner, E. Frugier, V. Schalk, A. Caflisch and H. L. Woodcock, J. Chem. Inf. Model., 2014, 54, 2612 CrossRef CAS PubMed.
- M. Di Pierro and R. Elber, J. Chem. Theory Comput., 2013, 9, 3311 CrossRef CAS PubMed.
- M. Di Pierro, M. L. Mugnai and R. Elber, J. Phys. Chem. B, 2015, 119, 836 CrossRef CAS PubMed.
- L.-P. Wang, J. Chen and T. van Voorhis, J. Chem. Theory Comput., 2013, 9, 452 CrossRef CAS PubMed.
- L.-P. Wang, T. Head-Gordon, J. W. Ponder, P. Ren, J. D. Chodera, P. K. Eastman, T. J. Martinez and V. S. Pande, J. Phys. Chem. B, 2013, 117, 9956 CrossRef CAS PubMed.
- L.-P. Wang, T. J. Martinez and V. S. Pande, J. Phys. Chem. Lett., 2014, 5, 1885 CrossRef CAS PubMed.
- R. Qi, L.-P. Wang, Q. Wang, V. S. Pande and P. Ren, J. Chem. Phys., 2015, 143, 014504 CrossRef PubMed.
- M. L. Laury, L.-P. Wang, V. S. Pande, T. Head-Gordon and J. W. Ponder, J. Phys. Chem. B, 2015, 119, 9423 CrossRef CAS PubMed.
- K. A. McKiernan, L.-P. Wang and V. S. Pande, J. Chem. Theory Comput., 2016, 12, 5960 CrossRef CAS PubMed.
- A. D. Wade, L.-P. Wang and D. J. Huggins, J. Chem. Inf. Model., 2018, 58, 1766 CrossRef CAS PubMed.
- Y. Qiu, P. S. Nerenberg, T. Head-Gordon and L.-P. Wang, J. Phys. Chem. B, 2019, 123, 7061 CrossRef CAS PubMed.
- J. Yin, A. T. Fenley, N. M. Henriksen and M. K. Gilson, J. Phys. Chem. B, 2015, 119, 10145 CrossRef CAS PubMed.
- J. Yin, N. M. Henriksen, H. S. Muddana and M. K. Gilson, J. Chem. Theory Comput., 2018, 14, 3621 CrossRef CAS PubMed.
- L. N. Naden and M. R. Shirts, J. Chem. Theory Comput., 2015, 12, 1806 CrossRef PubMed.
- M. Stroet, K. B. Koziara, A. K. Malde and A. E. Mark, J. Chem. Theory Comput., 2017, 13, 6201 CrossRef CAS PubMed.
- R. A. Messerly, S. M. Razavi and M. R. Shirts, J. Chem. Theory Comput., 2018, 14, 3144 CrossRef CAS PubMed.
- R. A. Messerly, M. S. Barhaghi, J. J. Potoff and M. R. Shirts, J. Chem. Eng. Data, 2019, 64, 3701 CrossRef CAS.
- B. A. C. Horta, P. T. Merz, P. Fuchs, J. Dolenc, S. Riniker and P. H. Hünenberger, J. Chem. Theory Comput., 2016, 12, 3825 CrossRef CAS PubMed.
- T. Verstraelen, V. Van Speybroeck and M. Waroquier, J. Chem. Phys., 2009, 131, 044127 CrossRef PubMed.
- W. Acree Jr and J. S. Chickos, J. Phys. Chem. Ref. Data, 2016, 45, 033101 CrossRef.
- M. Frenkel, X. Hong, R. C. Wilhoit and K. R. Hall, in Thermodynamic properties of organic compounds and their mixtures. Densities of alcohols, ed. K. R. Hall and K. N. Marsh, Landolt-Börnstein Series, Springer-Verlag, Berlin/Heidelberg, Deutschland, 2000, vol. IV/8G Search PubMed.
- M. Frenkel, X. Hong, R. C. Wilhoit and K. R. Hall, in Thermodynamic properties of organic compounds and their mixtures. Densities of esters and ethers, ed. K. R. Hall and K. N. Marsh, Landolt-Börnstein Series, Springer-Verlag, Berlin/Heidelberg, Deutschland, 2001, vol. IV/8H Search PubMed.
- M. Frenkel, X. Hong, Q. Dong, X. Yan and R. D. Chirico, in Thermodynamic properties of organic compounds and their mixtures. Densities of phenols, aldehydes, ketones, carboxylic acids, amines, nitriles, and nitrohydrocarbons, ed. K. R. Hall and K. N. Marsh, Landolt-Börnstein Series, Springer-VerlagSpringer-Verlag, Berlin/Heidelberg, Deutschland, 2002, vol. IV/8I Search PubMed.
- M. Frenkel, R. D. Chirico, V. Diky, Q. Dong, K. N. Marsh, J. H. Dymond, W. A. Wakeham, S. E. Stein, E. Königsberger and A. R. H. Goodwin, Pure Appl. Chem., 2006, 78, 541 CAS.
- C. Wohlfahrt, Static dielectric constants of pure liquids and binary liquid mixtures, Springer, Berlin, Germany, 2008, vol. IV/17 Search PubMed.
- J. R. Rumble, CRC Handbook of Chemistry and Physics, CRC Press/Taylor and Francis, Boca Raton, USA, 98th edn, 2018 Search PubMed.
- C. L. Yaws, Thermophysical properties of chemicals and hydrocarbons, Gulf Professional Publishing (Elsevier), Oxford, UK, 2nd edn, 2014 Search PubMed.
- Springer Nature, Springer Materials database. Available at: https://materials.springer.com, 2018.
- D. Weininger, J. Chem. Inf. Comput. Sci., 1988, 28, 31 CrossRef CAS.
- S. Kim, J. Chen, T. Cheng, A. Gindulyte, J. He, S. He, Q. Li, B. A. Shoemaker, P. A. Thiessen, B. Yu, L. Zaslavsky, J. Zhang and E. E. Bolton, Nucleic Acids Res., 2019, 47, D1102 CrossRef PubMed.
- P. H. Hünenberger, CombiFF Data Collection in the ETHZ Research Collection (tar-file CombiFF_saturated_O_and_N_compounds, version 1.0 corresponds to the published article), 2021 DOI:10.3929/ethz-b-000445271.
- W. F. van Gunsteren, S. R. Billeter, A. A. Eising, P. H. Hünenberger, P. Krüger, A. E. Mark, W. R. P. Scott and I. G. Tironi, Biomolecular simulation: The GROMOS96 manual and user guide, Verlag der Fachvereine, Zürich, Switzerland, 1996 Search PubMed.
- W. F. van Gunsteren, X. Daura and A. E. Mark, GROMOS force field, in Encyclopedia of computational chemistry, ed. P. Schleyer, John Wiley & Sons, Chichester, UK, 1998, vol. 2, pp. 1211–1216 Search PubMed.
- W. R. P. Scott, P. H. Hünenberger, I. G. Tironi, A. E. Mark, S. R. Billeter, J. Fennen, A. E. Torda, T. Huber, P. Krüger and W. F. van Gunsteren, J. Phys. Chem. A, 1999, 103, 3596 CrossRef CAS.
- C. Oostenbrink, A. Villa, A. E. Mark and W. F. van Gunsteren, J. Comput. Chem., 2004, 25, 1656 CrossRef CAS PubMed.
- M. Christen, P. H. Hünenberger, D. Bakowies, R. Baron, R. Bürgi, D. P. Geerke, T. N. Heinz, M. A. Kastenholz, V. Kräutler, C. Oostenbrink, C. Peter, D. Trzesniak and W. F. van Gunsteren, J. Comput. Chem., 2005, 26, 1719 CrossRef CAS PubMed.
- W. F. van Gunsteren, The GROMOS software for biomolecular simulation. Available at: http://www.gromos.net, 05/05/2011.
- A. T. Hagler, E. Huler and S. Lifson, J. Am. Chem. Soc., 1974, 96, 5319 CrossRef CAS PubMed.
- S. Lifson, A. T. Hagler and P. Dauber, J. Am. Chem. Soc., 1979, 101, 5111 CrossRef CAS.
- J. E. Lennard-Jones, Physica, 1937, 4, 941 CrossRef CAS.
- X. Daura, A. E. Mark and W. F. van Gunsteren, J. Comput. Chem., 1998, 19, 535 CrossRef CAS.
- L. D. Schuler and W. F. van Gunsteren, Mol. Simul., 2000, 25, 301 CrossRef CAS.
- L. D. Schuler, X. Daura and W. F. van Gunsteren, J. Comput. Chem., 2001, 22, 1205 CrossRef CAS.
- E. Bourasseau, M. Haboudou, A. Boutin, A. H. Fuchs and P. Ungerer, J. Chem. Phys., 2003, 118, 3020 CrossRef CAS.
- W. G. Hoover, Phys. Rev. A: At., Mol., Opt. Phys., 1985, 31, 1695 CrossRef PubMed.
- H. C. Andersen, J. Chem. Phys., 1980, 72, 2384 CrossRef CAS.
- W. F. van Gunsteren, H. J. C. Berendsen and J. A. C. Rullmann, Mol. Phys., 1981, 44, 69 CrossRef CAS.
- E. Guàrdia and J. A. Padró, J. Chem. Phys., 1985, 83, 1917 CrossRef.
- W. F. van Gunsteren and H. J. C. Berendsen, Mol. Simul., 1988, 1, 173 CrossRef.
- S. Yun-yu, W. Lu and W. F. van Gunsteren, Mol. Simul., 1988, 1, 369 CrossRef.
- W. F. van Gunsteren, Molecular dynamics and stochastic dynamics simulation: a primer, in Computer simulation of biomolecular systems, theoretical and experimental applications, ed. W. F. van Gunsteren, P. K. Weiner, A. J. Wilkinson, ESCOM Science Publishers, B.V., Leiden, The Netherlands, 1993, vol. 2, pp. 3–36 Search PubMed.
- R. W. Hockney, Methods Comput. Phys., 1970, 9, 135 Search PubMed.
- J.-P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, J. Comput. Phys., 1977, 23, 327 CrossRef CAS.
- H. J. C. Berendsen, W. F. van Gunsteren, H. R. J. Zwinderman and R. G. Geurtsen, Ann. N. Y. Acad. Sci., 1986, 482, 269 CrossRef CAS PubMed.
- J. A. Barker and R. O. Watts, Mol. Phys., 1973, 26, 789 CrossRef CAS.
- I. G. Tironi, R. Sperb, P. E. Smith and W. F. van Gunsteren, J. Chem. Phys., 1995, 102, 5451 CrossRef CAS.
- T. Verstraelen, P. Bultinck, V. Van Speybroeck, P. W. Ayers, D. Van Neck and M. Waroquier, J. Chem. Theory Comput., 2011, 7, 1750 CrossRef CAS PubMed.
- A. Kubincová, P. H. Hünenberger and M. Krishnan, J. Chem. Phys., 2020, 152, 104713 CrossRef PubMed.
- A. K. Rappé and W. A. Goddard, J. Phys. Chem., 1991, 95, 3358 CrossRef.
- B. A. Wells, C. De Bruin-Dickason and A. L. Chaffee, J. Phys. Chem. C, 2015, 119, 456 CrossRef CAS.
- S. W. Rick, S. J. Stuart and B. J. Berne, J. Chem. Phys., 1994, 101, 6141 CrossRef CAS.
- S. W. Rick and B. J. Berne, J. Am. Chem. Soc., 1996, 118, 672 CrossRef CAS.
- S. W. Rick, J. Chem. Phys., 2001, 114, 2276 CrossRef CAS.
- J. L. Banks, G. A. Kaminski, R. H. Zhou, D. T. Mainz, B. J. Berne and R. A. Friesner, J. Chem. Phys., 1999, 110, 741 CrossRef CAS.
- H. A. Stern, G. A. Kaminski, J. L. Banks, R. Zhou, B. J. Berne and R. A. Friesner, J. Phys. Chem. B, 1999, 103, 4730 CrossRef CAS.
- S. Patel and C. L. Brooks III, J. Comput. Chem., 2004, 25, 1 CrossRef CAS PubMed.
- S. Patel, A. D. MacKerell Jr and C. L. Brooks III, J. Comput. Chem., 2004, 25, 1504 CrossRef CAS PubMed.
- S. Patel and C. L. Brooks III, Mol. Simul., 2006, 32, 231 CrossRef CAS.
- Z.-Z. Yang, J.-J. Wang and D.-X. Zhao, J. Comput. Chem., 2014, 35, 1690 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Includes detailed information concerning: (i) the compounds in the calibration and validation sets of the O + N family; (ii) the experimental data; (iii) the covalent parameters; (iv) the initial values of the non-bonded parameters; (v) the observable-to-parameter ratio for the different atom types; (vi) the comparison with experiment; (vii) the list of outliers; (viii) the comparison with results using the 2016H66 parameter set. Additional material (molecule identifiers, coordinate and topology files, data tables for experimental and simulated values) can be downloaded freely from the internet under ref. 125, where version 1.0 corresponds to the published article (further versions will include revisions and/or expansions of the data set). See DOI: 10.1039/d1cp02001c |
| This journal is © the Owner Societies 2021 |