
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFirst principles characterisation of bio–nano interface†
Ian
Rouse
 a,
David
Power
a,
Erik G.
Brandt
b,
Matthew
Schneemilch
c,
Konstantinos
Kotsis
a,
Nick
Quirke
c,
Alexander P.
Lyubartsev
b and
Vladimir
Lobaskin
*a
a,
David
Power
a,
Erik G.
Brandt
b,
Matthew
Schneemilch
c,
Konstantinos
Kotsis
a,
Nick
Quirke
c,
Alexander P.
Lyubartsev
b and
Vladimir
Lobaskin
*a
aSchool of Physics, University College Dublin, Belfield, Dublin 4, Ireland. E-mail: vladimir.lobaskin@ucd.ie
bDepartment of Materials and Environmental Chemistry, Stockholm University, S-10691 Stockholm, Sweden
cDepartment of Chemistry, Imperial College, 301G Molecular Sciences Research Hub, White City Campus, 80 Wood Lane, London W12 OBZ, UK
First published on 10th June 2021
Abstract
Nanomaterials possess a wide range of potential applications due to their novel properties and exceptionally high activity as a result of their large surface to volume ratios compared to bulk matter. The active surface may present both advantage and risk when the nanomaterials interact with living organisms. As the overall biological impact of nanomaterials is triggered and mediated by interactions at the bio–nano interface, an ability to predict those from the atomistic descriptors, especially before the material is produced, can present enormous advantage for the development of nanotechnology. Fast screening of nanomaterials and their variations for specific biological effects can be enabled using computational materials modelling. The challenge lies in the range of scales that needs to be crossed from the material-specific atomistic representation to the relevant length scales covering typical biomolecules (proteins and lipids). In this work, we present a systematic multiscale approach that allows one to evaluate crucial interactions at the bionano interface from the first principles without any prior information about the material and thus establish links between the details of the nanomaterials structure to protein–nanoparticle interactions. As an example, an advanced computational characterization of titanium dioxide nanoparticles (6 different surfaces of rutile and anatase polymorphs) has been performed. We computed characteristics of the titanium dioxide interface with water using density functional theory for electronic density, used these parameters to derive an atomistic force field, and calculated adsorption energies for essential biomolecules on the surface of titania nanoparticles via direct atomistic simulations and coarse-grained molecular dynamics. Hydration energies, as well as adsorption energies for a set of 40 blood proteins are reported.
Introduction
Throughout the 21st century, Feynman's vision of the control of materials down to the atomistic level has begun to take shape in the form of nanotechnology: the study and use of materials of characteristic sizes on the order of 1–100 nanometres. At this scale, matter behaves significantly differently to bulk materials as a consequence of increased surface area, modified coordination of surface atoms, and different electronic band structures, amongst other properties. Consequently, these nanoparticles (NPs) may exhibit properties varying dramatically from the bulk materials, from absorbing specific wavelengths of light to increased catalytic activity. These effects may be tuned by manipulating the average size and shape of the NP, and as a result, nanoscale particles and fibres have found use in areas as diverse as food, medicine, cosmetics, and construction materials.In nanotechnology applications involving biological tissues there is an enormous diversity and complexity of responses and impacts, which are believed to be induced and steered by interactions at the NP surface. These biomolecules consist of a relatively small number of components, with only 20 amino acids present in tens of thousands of proteins, and only 6 elements – carbon, hydrogen, nitrogen, oxygen, phosphorus and sulphur – comprising most of the biomolecules. Given this highly structured and repetitive nature, it can be hoped that the bio–nano interactions are controlled by only a few material parameters. While there are many possible combinations of these atoms that may be biologically relevant, some of these combinations are far more frequent and may play the dominant role in the entire chain of molecular events following the contact of an NP with biological fluids and tissues. In particular, it can be expected that the interaction of NPs with water, amino acids, segments of lipids, sugars, and nucleic acids is especially important. The interactions with these representative fragments may constitute the material's fingerprint that is predictive of consecutive biological responses.1 Indeed, the NP–protein corona, the layer of adsorbed molecules formed upon immersion of the NP into a biological fluid, was found to be predictive of the NP biological activity such as association with live cells.2 It is reasonable to assume that the contents of this corona is highly dependent on the interaction of the NP with individual amino acid residues.
In contrast to the relatively stable and well-defined realm of biomolecules, the world of nanomaterials is heterogenous and subject to continuous innovation. Thousands of novel NP variations come to market annually. The materials are adjusted and modified for specific purposes through multiple methods. The optimisation of nanomaterials for the best performance and the lowest risk may require experiments with live cells or live animals, which often represents the most expensive and time-consuming part of the material development and approval for applications. Replacement of such experiments by in silico methods could not only help to save resources but would also enable a much deeper approach to material development that is based on the knowledge of the relationships between the material characteristics and their activities. In this way, one can create materials that are safe by design. An establishment of quantitative models relating the NP structure to specific activities is therefore a sacred goal of materials research.
The possibility to model and predict the biological activity of NPs from first principles seems to be beyond the capacity of even the most modern computers due to the typical time and length scale involved and complexity of the biological response. An alternative is the construction of a predictive system by combining physics-based modelling (up to the limits imposed by the system size) with a data-driven approach to relate the physical molecular events to the biological events of interest. Such physical molecular events (e.g. biomolecular adsorption on the NPs, biomembrane contact, penetration, or disruption) can be considered as initiating events of biological pathways, and thus be predictive of the outcomes.3,4 Although direct molecular simulation can reach the relevant system sizes that include a NP and proteins or lipid membranes, in practice obtaining results on meaningful timescales using atomistic models would take an infeasible amount of computational time, with even state-of-the-art techniques enabling only a few microseconds of the system's evolution. 5 To be able to discern between variations of NPs by their interaction with various biomolecules we therefore require a multiscale modelling scheme that can tackle the bio–nano interface and relate the respective interactions to the details of the NP structure derived from the first principles.
Recent literature presents multiple examples of models predicting biological activity of nanomaterials to their physicochemical properties. It is known that metal ion release rates from metal oxide NPs and conduction band gaps correlate with cytotoxicity.6–8 The overlap of conduction band energy levels with the cellular redox potential in such systems determines the ability of NPs to induce oxygen radicals, oxidative stress, and inflammation.7,8 Beside the hazard itself, the transport and exposure characteristics can also be related to the NP properties. Specifically, it has been shown that statistics of protein adsorption on NPs correlates with the NP cell association.9,10 Some of the properties, such as hydrophobicity, protein adsorption affinity, dissolution rates and the ability to generate reactive oxygen species, were identified by the nanosafety expert community as determinants of biological activity,2 and their provision is seen as a crucial step towards the development of predictive schemes. Where such properties are not available experimentally, materials modelling can be useful to provide the necessary data. From the point of view of rational materials design, such an approach would be most valuable if the modelling did not rely on any experimental characteristics and evaluated the materials’ characteristics from first principles.
In this work, we introduce a systematic method for characterizing the interactions between inorganic NPs and biomolecules from first principles using coupled quantum chemistry, atomistic molecular dynamics, and mesoscopic methods. We obtain results for both the rutile and anatase forms of TiO2 and demonstrate the calculation of the free energy of adsorption of a range of proteins to titania NPs of varying radius and surface potential. The methodology proposed here can straightforwardly be adapted to other nanomaterials and the force field obtained for atomistic simulations of titanium dioxide applied to its interactions with molecules other than the biomaterials considered here.
Methods
We propose here a systematic approach to generating a quantitative description of a specific bio–nano interface using materials modelling. Our multiscale method includes:• evaluation of detailed properties of the NP interface with water and parameterization of the atomistic force field for the material using electronic structure methods.
• calculation of interactions of the biomolecule building blocks (amino acids, lipid segments, DNA bases) with the surface of the material and interaction between the building blocks at the atomistic level at the specified conditions.
• parameterization of the coarse-grained (CG) force field for biomaterial building blocks and construction of the sample of arbitrary size and shape.
• CG modelling of interaction of entire biomolecules with the NP surface and calculation of preferred orientation and the mean adsorption energy.
We start with evaluation of intrinsic properties of the target nanomaterial, namely titania, using common electronic structure methods. We first evaluate the relevant material (TiO2) characteristics (band structure, ionisation potential, electronegativity, etc.) relating to the electronic structure using ab initio models. We further use ab initio molecular dynamics (MD) to quantify the hydration state of the material surface and derive parameters of an atomistic force field. We then perform atomistic MD simulations with the obtained force field to evaluate the immersion enthalpy for the material in water, as well as adsorption energy profiles (the potentials of mean force (PMF)) for essential biomolecules such as lipids and proteins, lipid bilayers, and molecular fragments. To compute further characteristics of the bionano interface we use these PMFs within the recently proposed CG United Atom approach (UA)11,12 to obtain binding free energies of proteins on NPs constructed from these materials over a range of radii and surface charges.
Semi-empirical, DFT and DFTB
We used density functional theory and semi-empirical methods to calculate the electronic structure descriptors of TiO2 materials. Semi-empirical calculations have been performed with the MOPAC computational code.13 All material properties (band gap, ionization potential, Mulliken electronegativity, absolute hardness, dispersion, polarizability and dipole moment) were obtained with the PM6 method using the D3 correction on the DFT optimized structures.14 Geometry optimizations of the rutile and anatase TiO2 bulk structures were performed with SCC-DFTB15 using the DFTB+ software16 with the parametrization tiorg-0-117 and with the SIESTA code18 with the PBE functional19 and a DZP (double-z polarized) basis. Troullier–Martins pseudopotentials20 were applied on the core electrons.Force field parameterization
The quality of the force field used in classical atomistic simulations is of primary importance for the simulation to produce reliable results. While there exist validated force fields describing bulk materials or aqueous solutions of organic and biomolecules, they are less developed for description of surface properties of (nano)materials. For modelling of metal or metal oxide surface in aqueous media an additional problem is adequate representation of the material surface which is modified (in comparison to the bulk material structure) by reactions with water building a surface-specific layer containing hydroxyl groups, bound molecular water or other surface modifications.Previously, a force field for classical MD simulations of TiO2 was developed by Matsui and Akaogi.21 In that force field Ti and O atoms interact by Buckingham and electrostatic potentials without bonded interactions. The force field of ref.21 was designed to model bulk materials and possible phase transformations, but its applicability to describe TiO2 hydrated surface and compatibility with biomolecular force fields remain unclear. Recently, some of us22 have developed TiO2 force field of a standard molecular-mechanical form with electrostatic, Lennard-Jones and bonded interactions, which was further modified by scaling of charges according to the electronic continuum correction theory.23 The force field of ref.22 was parametrized to fit available experimental data on crystal structures and water adsorption enthalpies. This use of only non-extensive experimental data left uncertainties in the definition of some parameters as well as about details of the hydrated surface structure. We therefore adopted a new strategy to derive force field for classical MD simulations of nanoparticles in aqueous biomolecular environment based on high quality ab initio modelling.
To parameterize the force field for titanium dioxide as employed in the atomistic simulations presented here, we used a multiscale approach. In this method, the detailed structure of the hydroxylated layer of a metal oxide surface, as well as parameters of the force field describing interactions of the fully hydrated surface with surrounding biomolecular solution are obtained from high-quality ab initio MD simulations. Ab initio MD simulations provide a representative set of snapshots correctly representing thermal fluctuations of the studied systems, which is used for the further analysis. The method of deriving force field parameters is based on partitioning the quantum mechanical electron density into atomic basins. We apply the population analysis method by Manz24 to partition the electron density among the atoms and extract individual net atomic charges and atomic volumes for individual atoms, as well as information on the bond orders for individual pairs. Briefly, the method consists of the following.
First, the net atomic charges (NAC), defined by the DDEC6 partitioning method,25 are computed:
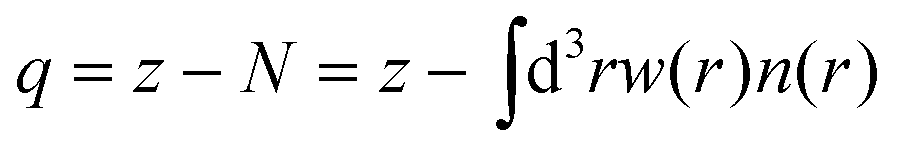 | (1) |
Second, the cubed atomic moment (CAM) are determined:
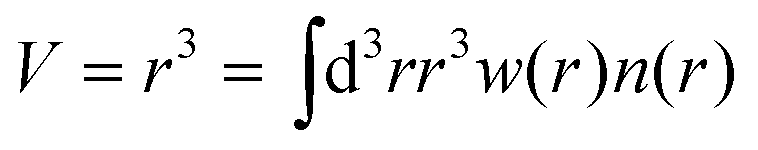 | (2) |
Third, the concept of bond order (BO), defined by26
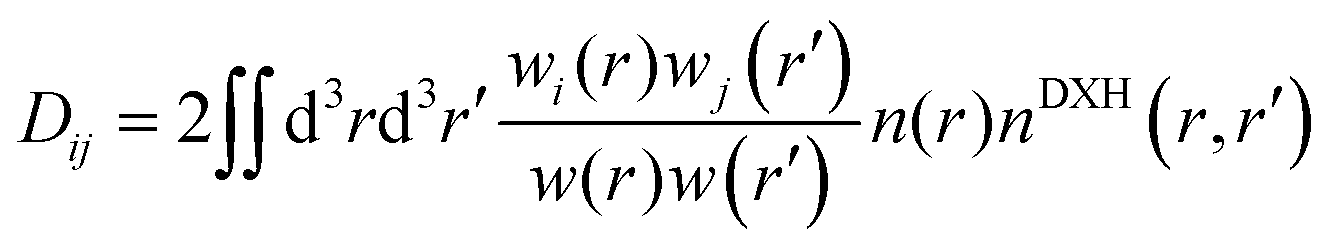 | (3) |
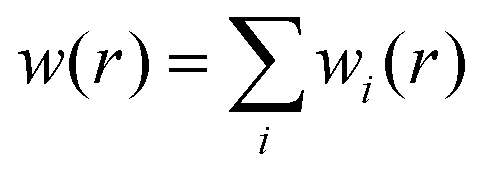 is the total spherical weight, nDXH(r, r′) is a normalized probability distribution (over r′ such as to exclude exactly one electron) that quantifies the so-called dressed exchange hole. The electron density close to the nucleus is depleted due to the exchange interaction. This concept can be modified for bond order by contraction/expansion of the density to align bond orders with the conventional view. With this definition the bond order between two atoms decreases smoothly to zero as the distance between them approaches infinity.
is the total spherical weight, nDXH(r, r′) is a normalized probability distribution (over r′ such as to exclude exactly one electron) that quantifies the so-called dressed exchange hole. The electron density close to the nucleus is depleted due to the exchange interaction. This concept can be modified for bond order by contraction/expansion of the density to align bond orders with the conventional view. With this definition the bond order between two atoms decreases smoothly to zero as the distance between them approaches infinity.
As a training ensemble, we have used trajectories generated in ab initio molecular dynamics simulations described previously.27 Briefly, six fully hydrated TiO2 planar surfaces of low surface energy (rutile 110, 100 and 001, and anatase 101, 100 and 001), each arranged in 2D periodic slab immersed in water, were simulated with DFT computed forces during 50 ps, and 20 snapshots for each surface taken each 1 ps from the last 20 ps of the simulations were taken for the analysis. This set of surfaces form a good representation of the surface of a rutile or anatase nanoparticle without requiring the significant computational time required to simulate an entire nanoparticle. Each member of the ensemble was subject to atom-in-molecule analysis with the Density-Derived and Chemical (DDEC6) method25 to determine net atomic charges, volumes, and bond order parameters using ChargeMol v3.5 software.28
We have used the calculated bond order to determine whether any two atoms are bound by setting a threshold value of 0.25. The local connectivity of atoms was used to determine the force field types. Disregarding atom coordinations which were observed in less than 1% cases, we identified two force field types for Ti-atoms, five types of oxygen atoms, and one type of hydrogen, as described in the Results section below. For each type of atom, we have computed average net atom charges, eqn (1), and cubic atomic volumes, eqn (2). Computed net atomic charges were used directly to determine partial atom charges of the force field, with a minor modification providing total zero charge for stochiochemical sample of TiO2. Net atomic volumes defined by eqn (2) and averaged over identified force field types were used to determine parameters B of the Lennard-Jones potential using the theory developed by Tkachenko and Scheffler,29 and B(V) dependences numerically computed and tabulated for each atom nucleus by Gould.30,31 Finally, the repulsive Lennard-Jones parameters A were determined by simple scaling relationships from the atomic volumes:
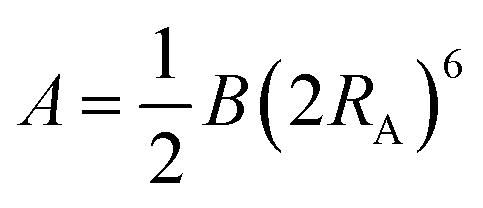 | (4) |
Lipid–NP interaction and heat of immersion
The interaction of NPs with lipids can be expected to differ depending on whether single lipids or larger structures are considered, and to parameterise these we consider both adhesion energies for a bilayer of lipids and the binding energy of single lipids, calculated through atomistic simulations as detailed previously.32 We additionally calculate heats of immersion as follows. The change in enthalpy upon immersion was estimated by conducting simulations of three systems: the surface in contact with water, the surface in a vacuum, and a simulation of bulk water containing an identical number of water molecules as the first system. The immersion enthalpy was calculated from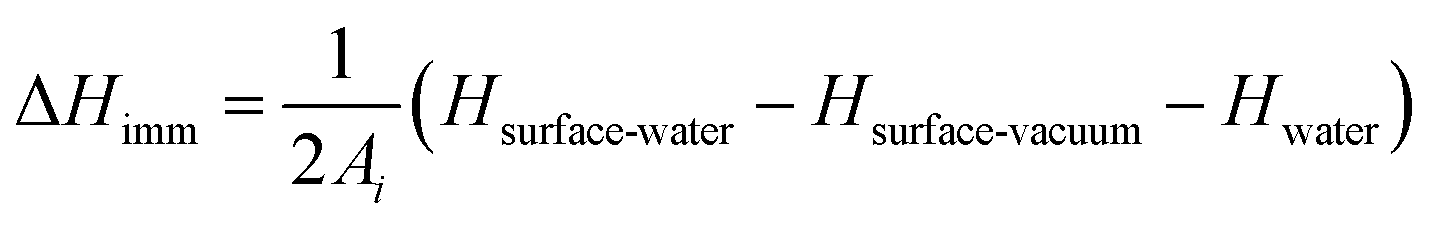 | (5) |
Adsorption of biomolecular fragments
To characterize interactions between NP surface and small biomolecular fragments, we have selected a set of 30 small molecules which represent all typical molecular fragments present in biomolecular fluids, and computed PMF and binding free energies of these fragments to NP. There are 20 naturally occurring amino acids of which the proteins of living organisms consist. Each full amino acid contains a peptide backbone fragment which is common to all amino acids. In order to avoid redundancy, we excluded the backbone fragment and considered the side chain analogues for all amino acids excluding glycine (for which the side chain analogue is just an H atom) and proline which has a different structure. This set of side chain analogues consists of 18 molecules. Histidine exists in two isomeric forms (denoted as HIE and HID) and we include both. These side chain analogues also have the same structure as hydrophobic lipid tails and certain common lipid head groups (phosphatidylserine, PS and sphingomyelin, SM), further extending the range of larger biomolecules covered by this set. Furthermore, components of headgroups of lipids and sugars are included. The full list of chosen biomolecules consists of:• Amino acid side chain analogues where backbone fragment is substituted by a hydrogen atom. We use notations of conventional amino acids for the side chain analogues.
• Glycine and proline amino acids with the backbone fragment (GLY, PRO).
• Modified charged forms of amino acids with pKa values between 4 and 10 (HIS+, GLU-protonated, CYS-, denoted HIP, GAN, CYM)
• Segments of the most abundant lipids: choline (CHL) and phosphate (PHO) group of phosphatidylcholine (PC) lipids; amino group (ETA) of ethanolamine lipids (PE), ester group (EST)
• D-Glucose representing sugars (DGL).
The list of molecules introduced here covers all the main types of chemical entities: hydrophobic, polar, aromatic, and charged, and represent all typical molecular fragments present in biofluids. Thus, the set of PMFs and binding free energies of these molecules makes a “fingerprint” of NP with respect to bio–nano interactions.
Calculations of PMFs and binding free energy for each small molecule from the set near a fully hydrated 2D-periodic TiO2 slab were performed in 300 ns adaptive well-tempered metadynamics simulations using simulation setup previously reported12 and summarized in the ESI.†
Biomolecular adsorption
Given the complexity of protein–NP systems, it is of interest to produce a scheme to produce adsorption free energies for large proteins based on these simple fragments. The amino acid PMFs can, in principle, be used to calculate the binding energy to nanoparticles for larger biomolecules (e.g. proteins) assembled from these fragments under the approximation of pairwise additivity. However, the PMFs include only interactions between the biomolecule and the nanomaterial up to a distance of about 1 nm, whereas the bulk of the nanomaterial interacts with these molecular fragments via the long-range van der Waals interaction which may remain significant at distances beyond the cut-off used for the calculation of the PMFs. To include these effects while avoiding the need to explicitly sum over the interaction between each atom in the NP and in the target biomolecule, we employ the continuum approximation and treat each AA bead as a sphere interacting with the spherical NP through the Hamaker approach. In this model, the bulk interaction between two spheres of radii R1, R2 with the surface–surface distance d is given by,33,34 | (6) |
 | (7) |
Once the PMFs and Hamaker constants for the set of amino acids and other fragments of interests have been calculated, we employ the UA methodology12 to calculate the binding energies of a set of reference proteins (see ESI†) onto spherical titania NPs. In this model, the protein is represented as a set of beads, with each bead representing one amino acid. The interaction potential between the NP and a bead consists of three components. The first is the PMF describing the short-range potential obtained through atomistic simulations as described in the previous section and corrected to take into account the radius of the NP by applying a distance-dependent scaling factor.12 Using this correction, a set of PMFs calculated for a planar slab of the material may be applied to all spherical NPs of this material, substantially reducing the computational time required to evaluate the binding energy for a set of NPs of the same material. To account for the bulk of the NP beyond the cut-off range of the PMF, the van der Waals interaction is added as the second component. This term is corrected to exclude the volume of the NP sufficiently close to the AA that it would be included in the PMF.
The final component is the electrostatic interaction in the Debye–Hückel approximation, which accounts for the interaction between the surface charge of the NP and charged residues and is specified in terms of the surface (zeta) potential.12 The total potential for a given bead type is calculated by summing over these three contributions, and then summed over all beads to produce the total interaction potential at a given orientation of the biomolecule relative to the surface of the NP, denoted U(z, ϕ, θ). Here, z gives the distance between the centre of the NP and the closest point of the protein. The binding energy for a protein on a spherical NP of radius R as a function of the orientation of the protein ϕ, θ is given by,11
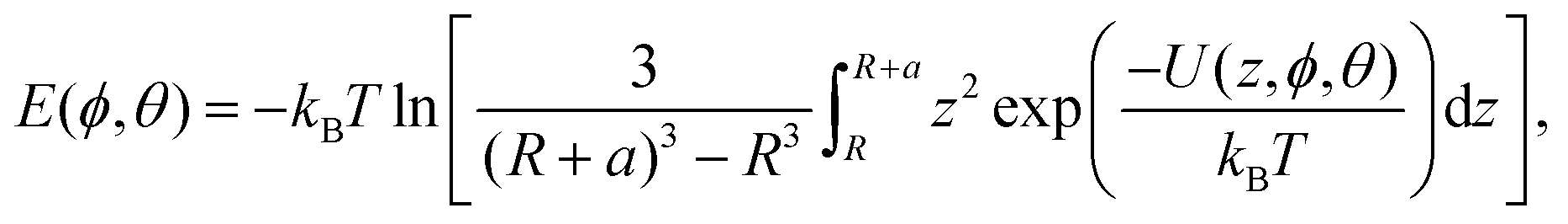 | (8) |
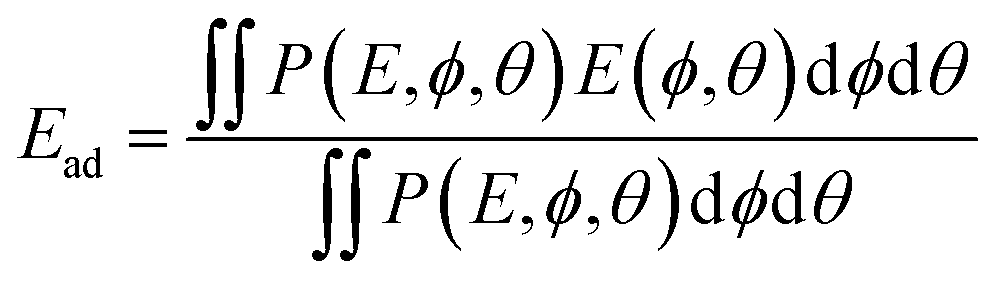 | (9) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θexp(−E(ϕ, θ)/kBT).
θexp(−E(ϕ, θ)/kBT).
Electrostatic potentials were calculated assuming a Debye length of 0.766 nm as calculated for an electrolyte concentration of 150 mmol in water at 300 K. Binding energies were calculated for radii in the interval 5–200 nm and for five values of the electrostatic surface potential between −50 and +50 mV.
Results & discussion
Intrinsic NP properties
Electronic properties of a specific nanomaterial can be obtained through computational techniques such as quantum mechanical semi-empirical calculations based on the Hartree–Fock formalism13 and density functional theory methods, with results for TiO2 shown in Table 1 and Table S1 of the ESI.† This allows for both the optimization of the structure of a given nanomaterial on the density functional tight binding (DFTB) level16 and the density functional theory (DFT) level and the calculation of physicochemical properties of this material by semi-empirical quantum mechanical calculations.13 Temperature and the size of the NP are important factors that determine the stability of the TiO2 forms. Here, we calculate electronic band gaps, ionization potentials, electronegativity, hardness, dispersion corrections, polarizability and the dipole moment for bulk anatase and rutile representing the core of a TiO2 NP at the semi-empirical level of theory in comparison to DFT and DFTB calculations for band gaps and ionization potentials. Such descriptors have been applied in statistical models to describe toxicity of metal oxide nanomaterials in relation to the core properties of a NP.8,36,37 Moreover, we employ DFT calculations (Table S1 in ESI†) using the SIESTA code18 where the unit cells of anatase and rutile TiO2 are used to describe the extended bulk structures by Monkhorst–Pack meshes for the point sampling of the Brillouin zone integration.38| TiO2 solid systems | Band gap, eV | Ionization potential– Valence band maximum energy, eV | Mulliken Electronegativity | Parr & Pople absolute hardness | Dispersion energy per atom, kJ mol-1 | Polarizability, Å3 | Dipole moment, Debye |
|---|---|---|---|---|---|---|---|
| Semi-empirical | |||||||
| Anatase | 9.41 | 6.67 | 1.96 | 4.70 | −4.92 | 133.01 | 0.34 |
| Rutile | 9.66 | 5.51 | 0.68 | 4.83 | −5.41 | 122.32 | 6.16 |
| DFTB | |||||||
| Anatase | 3.42 | 3.36 | — | — | — | — | — |
| Rutile | 2.68 | 3.64 | — | — | — | — | — |
| DFT | |||||||
| Anatase | 2.49 | 8.75 | — | — | — | — | — |
| Rutile | 2.25 | 9.46 | — | — | — | — | — |
Extrinsic NP properties
The first step in this parameterization is a development of a force field for atomistic simulations between biomolecular fragments and the NP. This issue was handled as discussed in the Methods section (part Force Field Parameterization) and a summary of the identified force field types and force field parameters are given in Tables 2 and 3.
| Atom type | Coordination | Description | Partial charge, e | σ, Å | ε, kJ mol−1 |
|---|---|---|---|---|---|
| H | H–O1 | Hydrogen | 0.417 | 0 | 0 |
| TiA | Ti–O6 | Bulk Ti | 2.248 | 1.99 | 13.79 |
| TiB | Ti–O5 | Surface Ti/defect | 2.159 | 1.9 | 13.79 |
| OA | O–Ti3 | Bulk TiO2 oxygen | −1.124 | 3.51 | 0.409 |
| OB | O–Ti2 | Bridge oxygen | −1.035 | 3.42 | 0.401 |
| OF | O–H1Ti1 | Hydroxyl oxygen | −0.913 | 3.29 | 0.389 |
| OG | O–H1Ti2 | Protonated oxygen bridge | −1.035 | 3.42 | 0.401 |
| OH | O–H2Ti1 | Adsorbed water | −0.923 | 3.151 | 0.634 |
| Bond | Equilibrium distance, Å | Force constant, kJ mol−1 Å−1 |
|---|---|---|
| Ti*–OA | 1.9 | 8000 |
| Ti*–OB | 1.9 | 8000 |
| Ti*–OF | 1.9 | 8000 |
| Ti*–OG | 1.9 | 8000 |
| Ti*–OH | 1.8 | 400. |
| OF–H | 1.0 | 3267 |
| OH–H | 1.0 | 3267 |
| Angle | Equilibrium angle, deg | Force constant, kJ mol1 deg−1 |
|---|---|---|
| OF–Ti*–OH | 90. | 500. |
| OH–Ti*–OH | 90. | 500. |
| Ti*–OF–H | 114.85 | 543. |
| Ti*–OG–H | 112.6 | 564. |
| Ti*–OH–H | 114.85 | 543. |
| H–OH–H | 104.2 | 628. |
Using the bond order concept, we have determined local connectivity of the atoms in AIMD trajectories of ref. 27, and identified the following atom types at the hydrated surface of TiO2: Ti–O6 and Ti–O5, which are Ti atoms coordinated by 6 respectively 5 oxygens; O–Ti3 which is oxygen coordinated by 3 Ti atoms, normally present in the bulk of material; O–Ti2 which is bridge oxygen at the TiO2 surface; O–H1Ti1 which is oxygen of a hydroxyl group attached to the surface; O–H1Ti2 which is protonated bridge oxygen, and O–H2Ti1, which is oxygen of the molecularly adsorbed water. Analysis of AIMD simulation results showed however very low fraction of protonated oxygen bridges. While formation of protonated oxygen bridges was observed in simulations of hydrated TiO2 surfaces using Reax force field,40 in ab initio simulations27 such protonated bridges were unstable, resulting in one of Ti–O bond cleavage and formation of additional OH-group. We did not include protonated oxygen bridges in classical atomistic simulations because of their low (about 1%) abundance in ab initio simulations, but we include force field parameters for them in Tables 2 and 3 having in mind that content of protonated bridges is pH-dependent and may increase at low pH.
As observed in ab initio simulations,27 undercoordinated surface Ti-atoms have either adsorbed water or hydroxyl groups bound to them. Each hydroxyl group contributes a charge of about −0.4e (taking in mind than if an OH group is attached to undercoordinated Ti-O5 atom, its force field type is changed to Ti–O6). We selected the fraction of hydroxyl groups at the surface as 30%, from the condition that the experimentally measured surface charge of TiO2 NPs at neutral pH is about −0.62e nm−2.41 In the atomistic simulations, the OH-groups were attached to Ti surface atoms in a random manner. Molecularly bound water was attached to all other sites. While such setup excludes eventual surface diffusion of OH-groups, ab initio simulations27 showed that after settling equilibrium structure further diffusion in the first hydration layer is practically absent on a ten-picoseconds time scale. Eventual diffusion of adsorbed water or protons on a longer time scale would only change the localization of the OH groups at the surface, which still can be well represented by a random localization of these groups at the surface. Thus, this diffusion will not have a major influence on binding of biomolecules to the surface.
After setting up hydroxyl groups and adsorbed water, the total charge of the TiO2 slab was brought to the nearest integer value by a minimal scaling of the atom charges, and the corresponding amount of Na+ counterions was added to the system. Furthermore, ions of NaCl salt were added to the solution in concentration 0.15 M.
Biomolecular adsorption on NPs
Using this force field, we computed PMFs for the range of biomolecular fragments to rutile (110) and anatase (101) planar surfaces. These surfaces are the lowest energy surfaces in the respective forms of TiO2, and by this reason one can expect that they represent most of the surface of the NPs. A similar model has been used previously to study adsorption of proteins on a surface of rutile NP.42 We extend the previous calculations by covering a wider range of biomolecular fragments and using a more advanced force field accounting for the difference between bulk and surface titania, allowing multiple chemical environments for oxygen atoms, and moreover taking into account structural differences between the anatase and rutile forms. The PMFs are depicted in the ESI,† and the computed binding free energies extracted from them are shown in Fig. 1, from which differences in the adsorption profile for the two forms of TiO2 can clearly be seen. The most strongly binding molecules for anatase are glutamic acid (GLU), cysteine anion (CYM) and aspartic acid (ASP), all these are negatively charged molecules with either carboxyl group or thiolate. None of these molecules show significant binding to rutile. The difference in binding could originate in the specific structure of the anatase surface where negatively charged groups of the molecules can coordinate favourably with hydrated positively charged titanium atoms. On the rutile surface, access to titanium atoms is screened by the bridging oxygen atoms, thus preventing the binding of anionic molecules. The different character of binding of small molecules have consequences for the binding of larger biomolecules and formation of the protein corona. The Hamaker constants associated with these fragments were calculated using parameters found from the literature and are listed in Table 4.43,44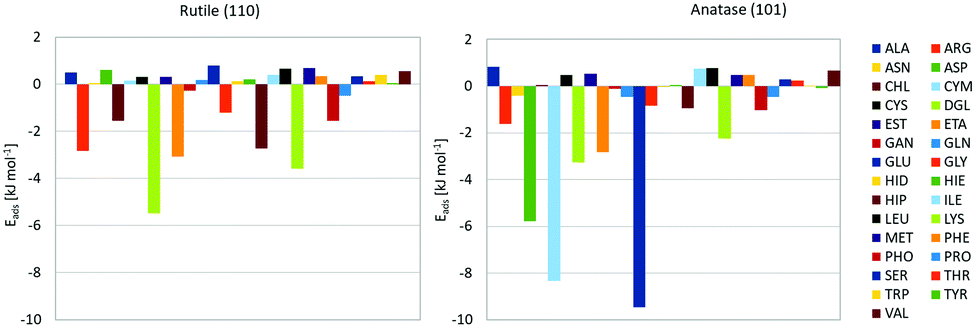 | ||
| Fig. 1 Adsorption free energies of biomolecule fragments to rutile (left) and anatase (right) TiO2 surfaces. | ||
| AA | ALA | ARG | ASN | ASP | CYS | GLN | GLU | GLY | HIS | ILE |
|---|---|---|---|---|---|---|---|---|---|---|
| A (anatase-AA) | 19.53 | 23.47 | 25.28 | 25.88 | 24.88 | 23.87 | 22.86 | 24.88 | 25.89 | 16.86 |
| A (rutile-AA) | 17.19 | 21.90 | 22.30 | 22.83 | 21.94 | 21.05 | 20.15 | 21.94 | 22.83 | 14.87 |
| AA | LEU | LYS | MET | PHE | PRO | SER | THR | TRP | TYR | VAL |
|---|---|---|---|---|---|---|---|---|---|---|
| A (anatase-AA) | 16.65 | 20.13 | 22.25 | 24.68 | 18.82 | 24.27 | 20.33 | 29.42 | 22.04 | 17.07 |
| A (rutile-AA) | 14.66 | 17.74 | 19.61 | 21.77 | 16.57 | 21.41 | 17.92 | 25.97 | 19.43 | 15.03 |
To further investigate the difference between the two materials, we calculated the binding energies of a set of 40 proteins on anatase and rutile NPs of a range of radii and zeta potentials using the coarse grained model discussed previously. The proteins were chosen primarily based on ones for which PDB structures exist for the human form of the protein with complete coverage of the sequence and include insulin (4EWZ), transferrin (1D3K) and an IgG domain (4HVW). To these, we have further added HSA (4K2C), BSA (4F5S), lung surfactant SP-D (3DBZ) and a lysozyme (1REX) as these are of biological interest. The resulting binding free energies, of which some are presented in Fig. 2 and the complete set provided in the ESI,† depend strongly on the radius of the NP and on a lesser extent on the value of zeta potential as depicted in Fig. 2(c and d).
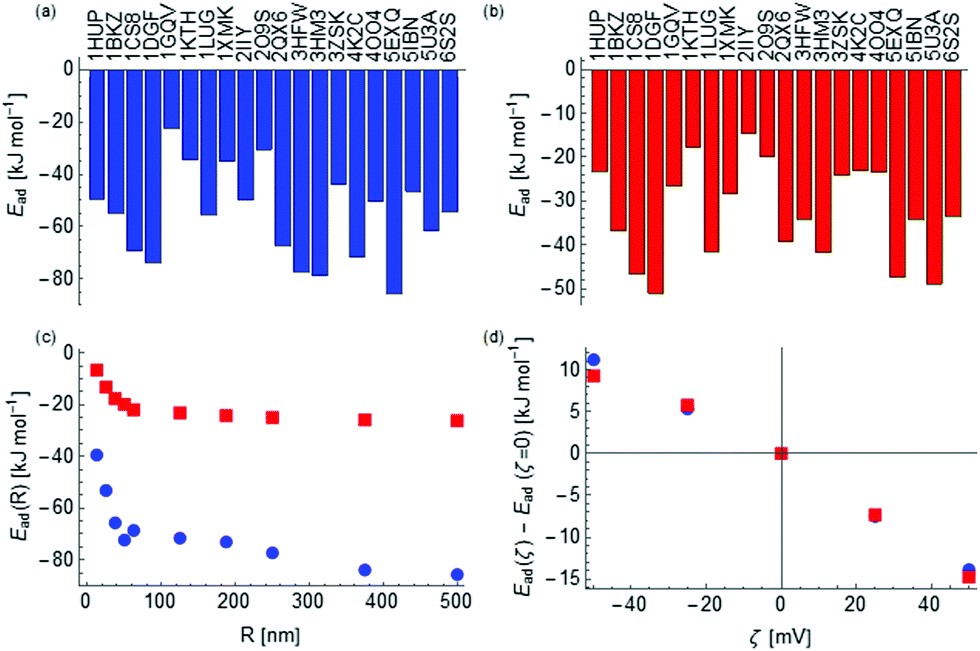 | ||
| Fig. 2 Adsorption free energies of a set of proteins (labelled by their Protein Databank identifier) onto spherical anatase (a) and rutile (b) NPs of radius 50 nm and zero zeta potential. The variation as a function of the radius for human serum albumin (PDB-ID: 4K2C) is shown in (c) and variation as a function of zeta potential for a particle of radius 50 nm in (d) for both anatase (blue) and rutile (red). | ||
Immersion and adhesion enthalpies, lipid wrapping
A key measure of the degree of interaction between NPs and membranes is the extent to which the NP adheres to or is wrapped by the membrane. According to the Helfrich membrane model45 applied by Deserno and Gelbart46 to the wrapping of spherical particles by membranes, the outcome is determined by whether the adhesion strength w (the free energy of adsorption per unit area) is sufficient to overcome the bending energy penalty associated with the required membrane deformation. Particles smaller than certain critical size will not adhere to the membrane. Larger particles will adhere and undergo wrapping; the extent to which wrapping occurs is determined by the membrane tension. With low tension and/or strong adhesion, particles will be completely engulfed, following which the particle may detach from the membrane leaving a membrane pore and potentially to cytotoxicity. The adhesion strength drives the particle wrapping process, so that any assessment of the NP interaction with cells must include an estimate of its adhesion strength w for a given membrane, which may be approximated by the adhesion strength with respect to the membrane lipids. Adhesion strength data can then be used to estimate the probability of passive NP uptake by human cells as well as pulmonary surfactant disruption due to the interaction with NPs in the alveolar spaces of lungs for evaluation of NP toxicity. A major obstacle to progress is the lack of quantitative experimental data for NP/lipid bilayer adsorption. We have previously proposed methods of calculating w from molecular simulation and applied these to a range of materials including gold,32 silica,47 and titania.48 Some data for DMPC lipids on titania surfaces are shown in Table 5, calculated through atomistic simulations using the force field developed here.| Polymorph | Rutile | Rutile | Rutile | Rutile | Anatase | Anatase | Anatase | Anatase |
|---|---|---|---|---|---|---|---|---|
| Miller index | 110 | 100 | 101 | 001 | 101 | 100 | 001 | 110 |
| Heat of immersion (mN m−1) | 1024 | 1009 | 847 | 1102 | 774 | 1176 | 392 | 1006 |
| Single lipid adsorption energy (kJ mol−1) | −1.8 | −3.4 | −13.8 | −0.4 | −1.7 | −0.2 | −0.1 | −1.9 |
| Adhesion strength (mN m−1) | −1.8 | −3.6 | −4.0 | −0.3 | −1.0 | −1.1 | −0.6 | −4.0 |
Discussion
In silico material characterization provides valuable information on the NP properties that may not be readily available from experiments. Yet, due to principal technical limitations such as the large required system size or time scale, the computations at the nanoscale are necessarily approximate and often cannot guarantee quantitative accuracy in reproducing experimentally observed properties in absolute terms. The possible mismatches with experimental data may be caused by unrealistic assumptions about the material such as its crystalline order, structure of the NP surface, material purity, or the coating. Unfortunately, such assumptions are unavoidable as experimental datasheets are often lacking the relevant information.In our calculations of intrinsic properties of titanium oxides, we used common quantum mechanical methods on anatase and rutile polymorphs of bulk TiO2. The methods show different accuracy with respect to different material properties. It is known that the band gap of rutile is lower than anatase TiO2 by ca. 0.2 eV49 and the ionization potential is larger for anatase than rutile. Our DFT and DFTB band gap results show the trend consistent with experiments, while semi-empirical calculations predict the opposite trend. For the ionization potential, the semi-empirical calculations give the trend consistent with experiments, while both DFT and DFTB show larger values for rutile. More accurate approaches have been used in literature to reproduce not only the relative trends between rutile and anatase TiO2 but accurate values of band gaps and band alignment in accordance with measurements.49
In the evaluation of extrinsic properties of titania NPs, such as interaction with water, lipids and proteins, we used the new force field developed in this work. All titania surfaces exhibited a large degree of hydrophilicity as reflected in the uniformly exothermic heats of immersion, which were an order of magnitude greater than the silica surfaces studied previously. A far greater range in heat of immersion was observed on the anatase surfaces than for rutile. From the data on the lowest energy surfaces, (110) for rutile and (101) for anatase, rutile appears to be more hydrophilic. Thus, as discussed in Section 2.2, the anatase surface is more strongly binding to the amino acid molecules than the rutile surface is, and this in turn leads to a difference in the calculated binding energies for the proteins considered as can be seen in Fig. 2, providing further evidence that a wide range of biomolecules should be selected for the calculation of predictive descriptors. The stronger binding of proteins to anatase correlates with the weaker binding of water as compared to rutile, as seen from the heats of immersion. The observed trends for the protein binding energies here are in agreement with experiments showing that anatase titania binds blood serum proteins more strongly than the rutile polymorph.50
For the majority of the surfaces, the energy of adsorption of single lipids was less than the thermal energy indicating that lipids do not spontaneously adsorb on these surfaces. The only surface to display strong adsorption was the rutile 101 surface with a minimum of −13.8 kJ mol−1. The average adhesion strength across the cleavage planes for rutile and anatase is relatively weak at −2.0 ± 0.4 mN m−1. However, rutile has two surface (100 and 101) which strongly bind lipids (−4 mN m−1), while anatase has only one (110). Since particle surfaces are expected to comprise a range of low energy cleavage planes, this observation suggests a slightly greater tendency for bilayers to wrap rutile NPs compared to anatase NPs, but both forms wrap less than amorphous silica.
The computed binding affinities of amino acids/proteins and lipids to TiO2 would allow further to address questions of cellular uptake of nanoparticles covered by biomolecular corona, and further evolution of corona after cellular uptake and encapsulation of nanoparticles in phagosomes. This can be done either by direct modelling of such processes within UA models, or by providing adsorption energies data as extrinsic descriptors in the nanoinformatics approaches.
As discussed previously,12 the UA model contains a number of approximations, which may cause systematic errors in the protein adsorption energies. The most significant of these is the assumption that all contributions to the NP–protein potential can be treated additively and that the orientation of AA side chains can be neglected. Moreover, charge regulation in both the protein and the NPs are neglected, and the protein is assumed to be fixed to its native structure and cannot relax due to binding to the surface of the NP. Finally, we note that the Hamaker constants required are typically not available ab initio and must be obtained from the literature, with different sources providing different values for these constants. Thus, the calculated adsorption energies do not necessarily predict the correct absolute value. We expect, however, that these factors are not significant due to the characteristic sizes of the NP–protein complex, and we expect it to produce the correct ranking of proteins by affinity to a particular NP. This, in turn, should enable the correct ranking of the corona abundances. Moreover, the simplifications in this model present substantial time savings in comparison to more computationally intense calculations of protein adsorption using atomistic simulations.5,51 These atomistic simulations are limited to providing binding energies for a single NP, whereas the method outline here enables the rapid calculation of binding energies for a whole class of NPs of the same material but varying sizes, shapes, and zeta potentials.
The set of descriptors given here is by no means definitive but are selected as a set of reasonably simple properties that can be evaluated using standard computational techniques and can be employed to further characterize the surface. As an example, we note that the binding energies calculated here may be of use in estimating the composition of the protein corona which forms around a NP by using these as input for simple models of the steady-state corona.52,53 They may additionally be of use for machine-learning models of more complex phenomena such as uptake by cells, as they provide a quantitative means to encode the chemical identity of the NP. In this work, we have presented results for titanium dioxide as this is a particularly important nanomaterial and is being extensively investigated by toxicological studies,54 however, we stress that the methodology used here is completely general and can be applied to a wide range of nanomaterials other than titanium dioxide, e.g. metals, metal oxides, silica and carbon-based nanomaterials. Likewise, the range of biomolecules that can be evaluated can readily be extended by identifying further basic structures and generating PMFs and Hamaker constants for these.
Conclusions
In this work, we have performed an in-depth parameterization of titanium dioxide nanoparticles on scales ranging from the atomic to mesoscopic. In particular, we have developed a novel forcefield for atomistic simulations in which multiple coordinate sites are considered, and we have demonstrated that the crystal phase of the titanium dioxide (anatase or rutile) leads to different affinities towards biomolecules. Our general methodology can be straightforwardly adapted to other nanomaterials, enabling their in-depth characterization and prediction of their interactions with a range of biomolecules.Author contributions
IR, DP, MS, EGB, KK, APL: software, investigation, analysis, visualisation; IR, KK, APL, NQ, VL: conceptualisation, writing; NQ, APL, VL: funding acquisition, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work has been funded by EU Horizon 2020 under grant agreements No. 686098 (SmartNanoTox project), No. 731032 (NanoCommons project), and 814572 (NanoSolveIT project), and by Science Foundation Ireland through grant 16/IA/4506. The computations of binding free energies were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC) at the Parallel Computer Center (PDC).Notes and references
- X.-R. Xia, N. A. Monteiro-Riviere and J. E. Riviere, Nat. Nanotechnol., 2010, 5, 671 CrossRef CAS PubMed.
- OECD-NANoREG-ProSafe joint meeting in Paris, Nov 29–Dec 1, 2016, http://www.h2020-prosafe.eu/?p=954.
- E. Valsami-Jones and I. Lynch, Science, 2015, 350, 388 CrossRef CAS PubMed.
- S. A. Love, M. A. Maurer-Jones, J. W. Thompson, Y.-S. Lin and C. L. Haynes, Annu. Rev. Anal. Chem., 2012, 5, 181 CrossRef CAS PubMed.
- H. Lee, Small, 2020, 17, 1906598 CrossRef PubMed.
- T. Puzyn, B. Rasulev, A. Gajewicz, X. Hu, T. P. Dasari, A. Michalkova, H. M. Hwang, A. Toropov, D. Leszczynsk and J. Leszczynski, Nat. Nanotechnol., 2011, 6, 175 CrossRef CAS PubMed.
- H. Zhang, Z. Ji, T. Xia, H. Meng, C. Low-Kam, R. Liu, S. Pokhrel, S. Lin, X. Wang, Y. P. Liao, M. Wang, L. Li, R. Rallo, R. Damoiseaux, D. Telesca, L. Mädler, Y. Cohen, J. I. Zink and A. E. Nel, ACS Nano, 2013, 6, 4349 CrossRef PubMed.
- A. Gajewicz, N. Schaeublin, B. Rasulev, S. Hussain, D. Leszczynska, T. Puzyn and J. Leszczynski, Nanotoxicology, 2015, 9, 313 CrossRef CAS PubMed.
- C. D. Walkey, J. B. Olsen, F. Song, R. Liu, H. Guo, D. W. H. Olsen, Y. Cohen, A. Emili and W. C. W. Chan, ACS Nano, 2014, 8, 2439 CrossRef CAS PubMed.
- P. Kamath, A. Fernandez, F. Giralt and C. R. Rallo, Top. Med. Chem., 2015, 15, 1930 CrossRef CAS PubMed.
- H. Lopez and V. Lobaskin, J. Chem. Phys., 2015, 143, 243138 CrossRef PubMed.
- D. Power, I. Rouse, S. Poggio, E. Brandt, H. Lopez, A. Lyubartsev and V. Lobaskin, Modell. Simul. Mater. Sci. Eng., 2019, 27, 084003 CrossRef CAS.
- J. J. P. Stewart, Computational Chemistry, Colorado Springs, CO, USA, 2016, http://HTTP://OpenMOPAC.net Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- M. Elstner, D. Porezag, G. Jungnickel, J. Elsner, M. Haugk, T. Frauenheim, S. Suhai and G. Seifert, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 7260 CrossRef CAS.
- B. Aradi, B. Hourahine and T. Frauenheim, J. Phys. Chem. A, 2007, 111, 5678 CrossRef CAS PubMed.
- G. Dolgonos, B. Aradi, N. H. Moreira and T. Frauenheim, J. Chem. Theory Comput., 2010, 6, 266 CrossRef CAS PubMed.
- J. M. Soler, E. Artacho, J. D. Gale, A. García, J. Junquera, P. Ordejón and D. Sánchez-Portal, J. Phys.: Condens. Matter, 2002, 14, 2745 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- N. Troullier and J. L. Martins, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 43, 1993 CrossRef CAS PubMed.
- M. Matsui and M. Akaogi, Mol. Simul., 1991, 6, 239 CrossRef.
- E. G. Brandt and A. P. Lyubartsev, J. Phys. Chem. C, 2015, 119, 18110 CrossRef CAS.
- D. Biriukov, O. Kroutil and M. Predota, Phys. Chem. Chem. Phys., 2018, 20, 23954 RSC.
- T. A. Manz and N. G. Limas, RSC Adv., 2016, 6, 47771 RSC.
- F. O. Kannemann and A. D. Becke, J. Chem. Phys., 2012, 136, 034109 CrossRef PubMed.
- T. A. Manz, RSC Adv., 2017, 7, 45552 RSC.
- L. Agosta, E. G. Brandt and A. P. Lyubartsev, J. Chem. Phys., 2017, 147, 024704 CrossRef PubMed.
- ChargeMol v3.5 software [https://sourceforge.net/projects/ddec/].
- A. Tkatchenko and M. Scheffler, Phys. Rev. Lett., 2009, 102, 073005 CrossRef PubMed.
- T. Gould, J. Chem. Phys., 2016, 145, 084308 CrossRef PubMed.
- T. Gould and T. Bučko, J. Chem. Theory Comput., 2016, 12, 3603 CrossRef CAS PubMed.
- M. Schneemilch and N. Quirke, Chem. Phys. Lett., 2016, 664, 199 CrossRef CAS.
- H. C. Hamaker, Physica, 1937, 4, 1058 CrossRef CAS.
- J. N. Israelachvili, Intermolecular and Surface Forces, 3rd edn, AP, 2011 Search PubMed.
- S. Alsharif, D. Power, I. Rouse and V. Lobaskin, Nanomaterials, 2020, 10(10), 1967 CrossRef CAS PubMed.
- K. Tämm, L. Sikk, J. Burk, R. Rallo, S. Pokhrel, L. Mädler, J. J. Scott-Fordsmand, P. Burk and T. Tämm, Nanoscale, 2016, 8, 16243 RSC.
- S. Kar, A. Gajewicz, T. Puzyn, K. Roy and J. Leszczynski, Ecotoxicol. Environ. Saf., 2014, 9, 162 CrossRef PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188 CrossRef.
- T. Cedervall, I. Lynch, S. Lindman, T. Berggård, E. Thulin, H. Nilsson and S. Linse, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2050 CrossRef CAS PubMed.
- Z. Futera and N. J. English, J. Phys. Chem. C, 2017, 121, 6701 CrossRef CAS.
- A. Vakurov, R. Drummond-Brydson, O. Ugwumsinachi and A. Nelson, J. Colloid Interface Sci., 2016, 473, 75 CrossRef CAS PubMed.
- S. Liu, X. Y. Meng, J. M. Perez-Aguilar and R. Zhou, Sci. Rep., 2016, 6, 37761 CrossRef CAS PubMed.
- M. N. Polyanskiy, Refractive Index Database, https://refractiveindex.info, accessed: 01, 2020.
- H. Zhao, P. H. Brown and P. Schuck, Biophys. J., 2011, 100, 2309 CrossRef CAS PubMed.
- W. Helfrich, Z. Naturforsch., C: Biochem., Biophys., Biol., Virol., 1973, 28, 693 CrossRef CAS PubMed.
- M. Deserno and W. M. Gelbart, J. Phys. Chem. B, 2002, 21, 5543 CrossRef.
- M. Schneemilch and N. Quirke, J. Chem. Phys., 2018, 148, 194704 CrossRef CAS PubMed ; Erratum, 2019, 150, 229901.
- M. Schneemilch and N. Quirke, J. Chem. Phys., 2019, 151, 134707 CrossRef CAS PubMed.
- D. O. Scanlon, C. W. Dunnill, J. Buckeridge, S. A. Shevlin, A. J. Logsdail, S. M. Woodley, R. R. A. Catlow, M. J. Powell, R. G. Palgrve, I. P. Parkin and G. W. Watson, Nat. Mater., 2013, 12, 798 CrossRef CAS PubMed.
- M. S. Zaqout, T. Sumizawa, H. Igisu, T. Higashi and T. Myojo, J. Occup. Health, 2011, 53, 75 CrossRef CAS PubMed.
- C. Ge, J. Du, L. Zhao, L. Wang, Y. Liu, D. Li, Y. Yang, R. Zhou, Y. Zhao, Z. Chai and C. Chen, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 16968 CrossRef CAS PubMed.
- D. Dell’Orco, M. Lundqvist, C. Oslakovic, T. Cedervall and S. Linse, PLoS One, 2010, 5, e10949 CrossRef.
- F. D. Sahneh, C. Scoglio and R. Riviere, PLoS One, 2013, 8, e64690 CrossRef CAS PubMed.
- P. H. Danielsen, K. B. Knudsen, J. Štrancar, P. Umek, T. Koklič, M. Garvas, E. Vanhala, S. Savukoski, Y. Ding, A. M. Madsen, N. R. Jacobsen, I. K. Weydahl, T. Berthing, S. S. Poulsen, O. Schmid, H. Wolff and U. Vogel, Toxicol. Appl. Pharmacol., 2020, 386, 114830 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp01116b |
| This journal is © the Owner Societies 2021 |