
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Experimental and theoretical study on the impact of a nitrate group on the chemistry of alkoxy radicals†‡
A.
Novelli
 *,
C.
Cho
,
H.
Fuchs
,
A.
Hofzumahaus
,
F.
Rohrer
,
R.
Tillmann
,
A.
Kiendler-Scharr
,
A.
Wahner
and
L.
Vereecken
*
*,
C.
Cho
,
H.
Fuchs
,
A.
Hofzumahaus
,
F.
Rohrer
,
R.
Tillmann
,
A.
Kiendler-Scharr
,
A.
Wahner
and
L.
Vereecken
*
Institute for Energy and Climate Research, Forschungszentrum Jülich GmbH, 52428 Jülich, Germany. E-mail: A.Novelli@fz-juelich.de; L.Vereecken@fz-juelich.de
First published on 2nd March 2021
Abstract
The chemistry of nitrated alkoxy radicals, and its impact on RO2 measurements using the laser induced fluorescence (LIF) technique, is examined by a combined theoretical and experimental study. Quantum chemical and theoretical kinetic calculations show that the decomposition of β-nitrate-alkoxy radicals is much slower than β-OH-substituted alkoxy radicals, and that the spontaneous fragmentation of the α-nitrate-alkyl radical product to a carbonyl product + NO2 prevents other β-substituents from efficiently reducing the energy barrier. The systematic series of calculations is summarized as an update to the structure–activity relationship (SAR) by Vereecken and Peeters (2009), and shows increasing decomposition rates with higher degrees of substitution, as in the series ethene to 2,3-dimethyl-butene, and dominant H-migration for sufficiently large alkoxy radicals such as those formed from 1-pentene or longer alkenes. The slow decomposition allows other reactions to become competitive, including epoxidation in unsaturated nitrate-alkoxy radicals; the decomposition SAR is likewise updated for β-epoxy substituents. A set of experiments investigating the NO3-initiated oxidation of ethene, propene, cis-2-butene, 2,3-dimethyl-butene, 1-pentene, and trans-2-hexene, were performed in the atmospheric simulation chamber SAPHIR with measurements of HO2 and RO2 radicals performed with a LIF instrument. Comparisons between modelled and measured HO2 radicals in all experiments, performed in excess of carbon monoxide to avoid OH radical chemistry, suggest that the reaction of HO2 with β-nitrate alkylperoxy radicals has a channel forming OH and an alkoxy radical in yields of 15–65%, compatible with earlier literature data on nitrated isoprene and α-pinene radicals. Model concentrations of RO2 radicals when including the results of the theoretical calculations described here, agreed within 10% with the measured RO2 radicals for all species investigated when the alkene oxidation is dominated by NO3 radicals. The formation of NO2 in the decomposition of β-nitrate alkoxy radicals prevents detection of the parent RO2 radical in a LIF instrument, as it relies on formation of HO2. The implications for measurements of RO2 in ambient and experimental conditions, such as for the NO3-dominated chemistry during nighttime, is discussed. The current results appear in disagreement with an earlier indirect experimental study by Yeh et al. on pentadecene.
1. Introduction
During day time, the most important atmospheric oxidants for organic species are the hydroxyl radical (OH), chlorine atoms (Cl), and ozone, O3.1 In contrast, during nighttime, OH and Cl become insignificant as oxidants due to the lack of photochemical production, and the main oxidation burden shifts to nitrate radicals (NO3) and ozone.2–5 NO3 is generated mostly from reactions of ozone with nitrogen oxides (NO, NO2). Due to its rapid photolysis NO3 does not accumulate during the day time although recent studies show that it can still quantitatively contribute to the oxidation of organic species during daytime.6 At nighttime, NO3 can be found in the lower troposphere at concentrations of a few pptv (∼108 molecule per cm3), and it is a major oxidant for most unsaturated volatile organic compounds (VOCs) including isoprene, monoterpenes, and other terpenoids emitted in large quantities to the atmosphere from biogenic and anthropogenic sources.7 Though the reaction rate of NO3 with VOCs is typically lower than for OH radicals,8–11 VOC loss processes by NO3 still contribute a sizable mass flux in the atmospheric oxidation of organic compounds.12Despite the important role of NO3 as an oxidant, NO3-initiated oxidation processes of unsaturated compounds have received much less attention than e.g. those by OH or O3. As such, the corresponding degradation mechanisms of VOCs are significantly less understood, even for key VOCs like isoprene.5 Though there are parallels between OH- and NO3-initiated oxidation mechanisms, the available literature indicates that hydroxy- versus nitrate substituents have a rather distinct impact on the reaction kinetics of the peroxy (RO2) and alkoxy (RO) radicals. For example, an OH-substituent leads to much faster decomposition and H-migration reactions than an NO3-group.13 Furthermore, α-ONO2 alkyl radicals formed from a β-nitrate alkoxy radical (R1) are known to eliminate NO2, decomposing to a carbonyl compound and stopping the organic radical oxidation chain.14
![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) C(ONO2)–C(O˙) C(ONO2)–C(O˙)![[double bond splayed right]](https://www.rsc.org/images/entities/char_e00a.gif) → C˙(ONO2) + O → C˙(ONO2) + O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C → CO + NO2 + OC C → CO + NO2 + OC | (R1) |
A recent perspective highlighted the need for reliable rate coefficient estimates for atmospheric models.15 To our knowledge, there are no direct experimental studies of the kinetics of nitrate-substituted alkoxy radicals. Some theoretical work exists,13,16,17 and the impact of an –ONO2 group has been included in a structure–activity relationship (SAR) for alkoxy radical decomposition by Vereecken and Peeters.13 This SAR is based on theoretical methodologies that are considered less accurate by nowadays standards yet matches the available experimental data well, but the SAR does not account fully for interactions between multiple substituents on the carbons of the decomposing C–C bond. Based on an indirect experimental product study for 1-pentadecene + NO3, Yeh et al.18 suggested that the rate of decomposition of NO3-substituted alkoxy radicals (called nitrate-RO hereafter) is underestimated by that SAR by several orders of magnitude. As decomposition of RO radicals competes against H-migration or reaction with O2,19–23 the rate of decomposition has an important impact on the atmospheric fate of nitrated intermediates, and hence the products formed and the rate of oxidative removal of VOCs from the atmosphere. Also, nitrates are often found in aerosols,24–27 whereas a fast decomposition reaction forming more volatile compounds and destroying the –ONO2 moiety by release of NO2 would have an impact on formation and growth of particulate matter.
Decomposition of nitrate-RO forming NO2 can also have repercussions for the measurement of nitrated peroxy radicals (called nitrate-RO2 hereafter) in the atmosphere with the laser induced fluorescence (LIF) technique.28–30 With this technique, RO2 radicals are quantitatively measured only if they form HO2 or OH radicals upon reaction with nitrogen monoxide (NO) in the so-called converter. Carbon monoxide, which is also added in the converter, converts OH into HO2 radicals, so that only HO2 radicals are present at the converter outlet. After transfer into the fluorescence cell (∼4 hPa), the HO2 is converted by reaction with NO to OH radicals which are then detected spectroscopically.31 The majority of RO2 radicals from OH-initiated VOC oxidation are detectable with the LIF instrument as they form HO2 in an NO-rich environment. The differences in the detection sensitivity for specific RO2 radicals are mainly determined by the number of reaction steps needed.31 For NO3-initiated oxidation, the nitrate-RO have a high likelihood of forming NO2 instead, and the LIF instrument would not be able to measure the parent nitrate-RO2 radicals. Indeed, to rationalize the discrepancies observed between modelled and measured RO2 radical for some nights with fast oxidation of alkenes by NO3 radicals during the ClearfLo campaign performed in August 2012 in the city of in London, Whalley et al.30 suggested that the LIF instrument is not able to detect the nitrate-RO2 radicals from ethene and propene. Therefore, the yield of HO2 radicals from nitrate-RO2 radicals in the LIF instrument can be used to assess the decomposition rate of specific nitrate-RO with LIF experiments, as NO2 formation from nitrate-RO often competes against the well-known alkoxy radical reaction with O2 forming HO2.
In this study, we perform an extensive study of the reactivity of nitrate-RO, using a combination of experimental, theoretical, and modeling techniques. The nitrate-RO studied range from C2 to C6 compounds, chosen to probe a wide range of decomposition and isomerisation rates. The theoretical work encompasses a series of quantum chemical and theoretical kinetic calculations on nitrate-RO, which are compared to earlier SAR predictions and used to further improve the SAR predictive capabilities. The accompanying experiments, performed in the atmospheric simulation chamber SAPHIR, together with modeling studies, probe the fate of the nitrate-RO formed from a series of alkenes + NO3 reactions. These provide a measure for the relative rate of unimolecular decomposition and isomerisation for nitrate-RO. The implications of the results on the nighttime atmospheric degradation of VOCs, and our ability to measure atmospheric RO2 radicals using LIF during night-time conditions, are discussed.
2. Methodologies
2.1 Theory
The geometries of nitrate-RO radicals derived from a series of aliphatic hydrocarbons were optimized using the M06-2X/cc-pVDZ level of quantum chemical theory,32,33 exhaustively characterizing all conformers of reactant and transition states for all pathways studied. The resulting geometries were then re-optimized at the M06-2X/aug-cc-pVTZ level of theory,32 combined with single point energy calculations on the lowest conformers at the CCSD(T)/aug-cc-pVTZ level of theory.34 ZPE corrections are done at this M06-2X level of theory, with vibrational wavenumbers scaled by 0.971.35,36 To verify the reliability of the calculations, additional methodologies were applied to selected compounds: B3LYP/6-31G(d,p), B3LYP/cc-pVDZ, B3LYP/aug-cc-pVTZ, BH&H/aug-cc-pVTZ, M06-2X-D3/aug-cc-pVTZ, MN15/aug-cc-pVTZ, ωB97X-D/aug-cc-pVTZ, MPWKCIS1K/6-31+G(d,p), PMP2/6-31G(d,p), and CBS-QB3, combined with CCSD(T)/aug-cc-pVDZ and/or CCSD(T)/aug-cc-pVTZ single point energy calculations.37–44 All calculations were performed using the Gaussian-16 software suites.45The rate coefficients of the reactions are obtained using multi-conformer transition state theory, MC-TST,46,47 incorporating the characteristics of all conformers obtained at the CCSD(T)/aug-cc-pVTZ//M06-2X/aug-cc-pVTZ level of theory. Tunneling is included using an asymmetric Eckart barrier correction.48,49
2.2 Atmospheric simulation chamber SAPHIR
The experiments were conducted in the atmospheric simulation chamber SAPHIR at Forschungszentrum Jülich, Germany which was extensively described in previous studies.50,51 Briefly, the SAPHIR chamber is made of a double-wall Teflon (FEP) film that is inert and has a high transmittance for solar radiation.52 Though located outdoors to allow realistic photolysis experiments, it is equipped with a shutter system that, when closed, can be used to mimic nighttime conditions by preventing light from entering the chamber. The synthetic air provided to the chamber is mixed from ultra-pure nitrogen and oxygen (Linde, >99.99990%). Two fans in the chamber ensure a complete mixing of trace gases within two minutes. The pressure in the chamber is slightly higher than ambient (∼30 Pa) to avoid external air penetrating the chamber. Due to small leakages and air consumption by instruments, trace gases are diluted at a rate of ∼6% h−1 due to the replenishment flow.Before each experiment the chamber was cleaned by exchanging the chamber air 6 to 8 times with pure synthetic air. Before injection of any species, measurements are performed in the empty chamber (up to 20 min) to scout for possible contaminations which can be identified with the value of the OH reactivity (background OH reactivity). Each alkene investigated was then injected in the clean empty and dark chamber. After ∼15 min, carbon monoxide (CO, AirLiquide, 10% in N2, purity N47) was injected in the chamber to reach a concentration of 200 ppm to scavenge the OH radical, followed by NO2 (Linde, 500 ppmv in N2, purity N41) and O3 injections, to concentrations of ∼40 ppbv and between 15 and 30 ppbv, respectively. CO and NO2 were injected by a mass flow controller while O3 was produced with a silent discharge ozonizer (O3onia). The reaction between NO2 and O3 was used to produce the NO3 radicals. Depending on the alkene investigated, one or two additional injections of the alkene followed by addition of NO2 to boost the NO3 production were performed. Ethene (AirLiquide, 10% in N2, purity N30), propene (AirLiquide, 99.999%, purity 5.0), cis-2-butene (AirLiquide, 1% in N2, purity N24), 2,3-dimethyl-2-butene (Sigma Aldrich, purity 99%), 1-pentene (Sigma Aldrich, purity 99%) and trans-2-hexene (Sigma Aldrich, purity 99%) were investigated within this study.
2.3 Instrumentation
The concentrations of OH, HO2 and RO2 radicals were measured with the laser induced fluorescence (LIF) instrument permanently in use at the SAPHIR chamber and described previously.53,54 Without going into the details of the LIF technique the components of interest for this study are highlighted. Within a LIF instrument, air is expanded through a nozzle into a low pressure chamber (∼4 hPa, fluorescence cell), where the OH radicals are detected by pulsed laser excited fluorescence at 308 nm, while both the HO2 and RO2 radicals need to be converted to OH radicals before detection.HO2 radicals are detected by adding NO in the fluorescence cell, converting HO2 to OH radicals for detection. Several studies have proven that RO2 radicals originating from OH oxidation from large alkanes (C4), alkenes (including isoprene), and aromatics, can cause an interference signal in the HO2 radicals measurement.54–57 A practical approach for avoiding the interference during the HO2 measurement is to lower the concentration of NO reacting with the sampled air inside the measurement cell. During this study, the NO concentration used was low (∼2.5 × 1013 cm−3) to minimize the possibility of an interference as described in Fuchs et al.54 Under the operative conditions of the LIF instrument, i.e. lower pressure, lower NO concentrations, and much shorter reaction time (∼0.2 ms) than in the converter (see below), none of the RO2 radicals in the reaction mixture are expected to yield sufficient HO2 to interfere with the LIF HO2 measurement.
The RO2 instrument (also called ROx-LIF, schematic in Fig. S22, ESI‡) measures the sum of atmospheric RO2, HO2 and OH radicals (ROx = RO2 + HO2 + OH), where under the current reaction conditions the contribution by OH is negligible. The RO2 radicals are first converted to HO2 in a so called converter (∼25 hPa), where a mixture of NO and CO is added to the sampled air. The reaction of RO2 radicals with NO leads to the formation of HO2 and OH radicals, while the reaction with CO converts OH back to HO2 radicals. The air is then transferred from the converter into the fluorescence cell (∼4 hPa), where the HO2 radicals are transformed by an excess of NO to OH for LIF detection. RO2 concentrations are determined from the difference of the ROx signal and separate measurements of HO2 and OH radicals. The conditions and residence time in the converter are chosen such that aliphatic peroxy radicals with small carbon numbers (C1 to C3) are almost quantitatively converted into HO2 radicals.31 The low pressure in the reactor slows down H-abstraction by O2, a direct path of formation of HO2 radicals, as the oxygen concentration is ∼40 times lower than in ambient conditions. The large majority of the alkoxy radicals formed will either decompose or isomerize13,58–60 and their detectability will be determined by their yield of HO2 radicals. A recent study on methyl vinyl ketone (MVK),61 one of the main products from the OH-initiated oxidation of isoprene, showed that only a partial detection of the RO2 radicals generated was achieved due to the “slow” formation of HO2 radicals.
The OH reactivity (kOH), the inverse lifetime of OH, was measured by a pump and probe technique coupled with a time-resolved detection of OH by LIF.62,63cis-2-Butene, 1-pentene and trans-2-hexene concentrations were measured by a proton-transfer-reaction time-of-flight mass spectrometer (PTR-ToF-MS, Ionicon).64,65 In addition, the PTR-ToF-MS provided absolute concentrations for acetone. Carbon monoxide and water vapour were measured by an instrument applying cavity ring-down spectroscopy (CRDS, Picarro). NO and nitrogen dioxide (NO2) were measured by chemiluminescence (CL, Eco Physics), and O3 by UV absorption (Ansyco). Table S3 (ESI‡) summarizes the instruments available during the experiments, giving time resolution, accuracy, and precision for each instrument,19,20 while Fig. S22 (ESI‡) shows a more detailed drawing of the ROxLIF instrument, and the pertaining chemistry.
2.4 Kinetic chemical model at ambient pressure
The measured radicals and trace gases were modelled with a zero-dimensional box model using chemical mechanistic information from the Master Chemical Mechanism downloaded via website: http://mcm.leeds.ac.uk/MCM, with nitrate-RO2, nitrate-RO, and ozonolysis chemistry updated as described below. Chamber-specific properties were also implemented in the model. First, a dilution rate was calculated from the measured replenishment flow into the chamber, and applied to all the trace gases present in the model to account for the dilution. For all the experiments shown in this study, except ethene and 2,3-dimethyl-2-butene, no background OH reactivity was observed. For the ethene experiment a background of 1 s−1 was measured in the empty chamber, which was parametrized with a co-reactant Y added to the model, which converts OH to HO2 in the same way as CO does.66–68 For the 2,3-dimethyl-2-butene experiment, a failure during the night-time flushing resulted in the presence of ∼2 ppb of O3, 0.7 ppb of NO2 and 2.8 ppm of CO culminating in a background OH reactivity of ∼14 s−1. For NO3 and N2O5, a wall loss of 1.6 × 10−3 s−1 and 3.3 × 10−4 s−1, respectively, was included in the model. These values were taken from a recent study by Dewald et al.69 which focused on a one-month long campaign (NO3Isop) investigating the oxidation of isoprene by NO3 radicals and was performed in the SAPHIR chamber during August 2018. A sensitivity test performed with both wall loss rates changed by a factor of two resulted in a change of less than 5% for the model calculations for HO2 and RO2 radicals. Unless mentioned explicitly, each model was constrained only for temperature, pressure and water vapor mixing ratio. O3, NO2, CO, and the alkene injections were implemented in the model by applying a source only active at the time of the injection (between 1 and 10 minutes), with a source strength adjusted to match the measured species.As no absolute calibration standards were available, the alkenes concentrations were derived as follows: (i) for ethene the concentration was derived from the measured OH reactivity (kOH); (ii) the propene concentration was determined from an optimal fit to the time-dependent depletion of O3. This method carries a larger uncertainty (±15%) as the ozone profile has only a moderate sensitivity to the propene concentration; (iii) for 2,3-dimethyl-2-butene the concentration was derived by an optimal fit to the O3 and acetone concentration profiles, where 2,3-dimethyl-2-butene is the only known and assumed acetone source in this system; (iv) for cis-2-butene, 1-pentene and trans-2-hexene, the concentrations were derived from the relative PTR-ToF-MS signal, calibrated against the measured kOH at the first injection.
Although the experiments were designed to minimize the reaction between O3 and the alkene, ozonolysis contributed on average between 7 and 60% to the chemical loss rate of the VOC investigated. The ozonolysis reaction schemes for all the alkenes investigated, except for 2,3-dimethyl-2-butene, were updated compared to the MCM v3.1.1, based on recent findings in the literature. The rate coefficients for the reactions of the stabilized Criegee intermediate CH2OO with NO2 and CO, and for the products formed, were also updated. In addition, updated oxidation schemes for OCHCH(OO˙)CH2CH3 (butanal-2-peroxy, named BUTALAO2 in the MCM v3.3.1) and OCHCH2OO˙ (ethanal-2-peroxy, named CHOCH2O2 in the MCM v3.3.1), formed in the ozonolysis of the alkenes, were included. All changes applied are discussed and referenced in the ESI‡ Section A.
For all VOCs examined, the MCM v3.3.1 rate coefficients for the decomposition of the respective nitrate-RO were replaced with the theoretical predictions derived in this work (Table 2). For 1-pentene and trans-2-hexene, isomerization by H-migration is the dominant loss path for the nitrate-RO (Fig. S8 and S10, ESI‡). These paths, currently not present in the MCM v3.3.1, were included based on the theoretical data, and the subsequent chemistry of the products was derived based on SARs.13,59,60 Additional loss paths for the nitrate-RO listed in Table 2 were not implemented in the model as they contributed less than 2% of the total loss rate. Though contribution by OH-initiated oxidation is very small, isomerisation by H-migration and subsequent chemistry for radicals generated from the OH-initiated chemistry of 1-pentene and 2-hexene were also implemented based on SAR predictions (Fig. S7 and S9, ESI‡).
To summarize, the base model (M0) as shown within this study originates from the MCM v3.3.1 with the following enhancements:
– Nitrate-RO chemistry as characterized in this study (Table 2)
– Isomerization reactions for all nitrate-RO2 (Fig. S9 and S11, ESI‡) radicals formed from 1-pentene and trans-2-hexene.
– Chamber dilution.
– Updated ozonolysis chemistry for all alkenes investigated (except 2,3-dimethyl-2-butene) (ESI,‡ Section A).
– Updated oxidation schemes for ozonolysis products, i.e. CH2OO (ESI,‡ Section A), and OCHCH(OO˙)CH2CH3 and OCHCH2OO˙ radicals (Fig. S6 and S7, ESI‡).
– Isomerization reactions for all OH–RO2 (Fig. S8 and S10, ESI‡) radicals formed from 1-pentene and trans-2-hexene.
2.5 RO2 + HO2 radicals products yield in model M1
The chemical conditions of the experiments presented in this study allow for a correct interpretation of the comparison between modelled and measured RO2 radicals only if the HO2 radicals are correctly reproduced by the model used. Due to the virtually zero nitrogen monoxide concentration in the chamber, the HO2 concentrations are high, and HO2 radicals become the dominant loss process for the RO2 radicals. In addition, the excess of CO can cause a large imbalance in the modelled HO2 radical concentration if the OH radical production rate is not correctly predicted in the model as most of the OH radical generated will react with CO to produce HO2 radicals.Indeed, it was found that the base model M0 showed a systematic underestimation of the HO2 concentrations for all compounds, which suggests missing chemistry. Based on the product traces and the available reactant concentrations, we propose that the missing reaction is the formation of OH in the reaction of β-nitrate-alkylperoxy radicals with HO2. Such OH formation is already known to occur with high yields in the acylperoxy + HO2 reaction (R2),70–73 and is driven by the H-bonding between the carbonyl moiety and the hydrotetroxide H-atom in the RC(O)OOOOH adduct.
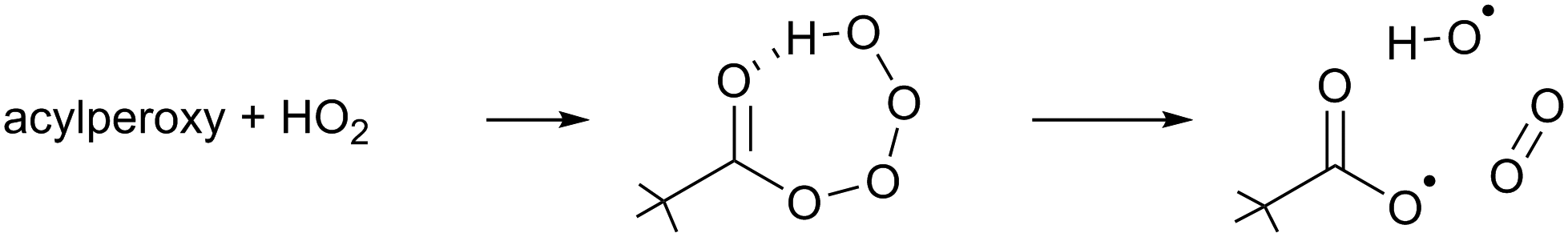 | (R2) |
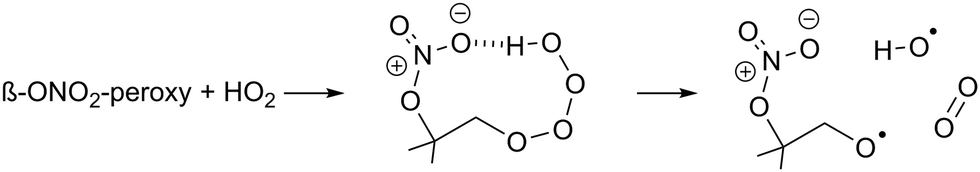 | (R3) |
β-Nitrate-hydrotetraoxides can have similar H-bonding (R3), and should thus be considered as potential OH sources. Though OH yields are known to be lower in e.g. β-OH or β-oxo RO2 radicals,70,73–75 the nitrate group is large and can reach the tetroxide H-atom more readily, with H-bonding aided by the partial charge separation in the nitrate moiety owing to the dative nitrogen–oxygen bond. The β-nitrate-RO2 + HO2 reaction forming nitrate-RO + OH has been proposed before in the context of isoprene oxidation, with OH yields from 22 to 58%,5,12,26,76 and α-pinene oxidation, with a 55 to 85% OH yield.17 The OH radicals formed thus are mostly converted to HO2 after reaction with CO, present as an OH scavenger in the experiments. The reaction was implemented in kinetic model M1 for all β-nitrate-RO2 radicals, retaining the RO2 + HO2 reaction rate as already available in MCM v3.3.1, but branching part of the products away from the traditional ROOH + O2 product towards RO + OH + O2. As no reliable data is available on this reaction, the yield of this channel (15–67%, Table S3, ESI‡) was taken as the only adjustable kinetic parameter in our model. This approach potentially underestimates the reaction rate somewhat, as similarly to the acylperoxy + HO2 reaction, the RO + OH product channel (occurring on the singlet electronic surface) does not compete against the ROOH channel (proceeding on the triplet surface) but is rather an additional reaction pathway that enhances the total reaction rate.77–79 As we are only sensitive to the total amount of HO2 formed, not the rate coefficient or HO2 yield separately, we cannot investigate this aspect further. It should be noted that the analysis of the detectability of the RO2 radicals (see below) yields very similar results if, instead of adding the above reaction, the HO2 concentration in the model is directly constrained to the measured HO2; the results on the nitrate-RO described below are thus independent of the exact nature of the HO2 source in the chamber proposed here.
2.6 Modeling of the RO2 to HO2 conversion in the LIF converter
Once the RO2 radicals are sampled into the converter, they rapidly react with NO, present in a ∼0.7 ppmv concentration, forming alkoxy radicals. The converter operates at a lower pressure of ∼25 hPa, such that most reactions, with the exclusion of reactions with O2, NO and unimolecular processes, become negligible. At this pressure, the concentration of O2 is a factor of ∼40 below ambient, reducing the pseudo-first order rate coefficient for H-abstraction from alkoxy radicals by O2 to k(298 K) ∼ 1.3 × 103 s−1. Decomposition and isomerization reactions therefore dominate the chemistry of most alkoxy radicals. Specifically, for the nitrate alkoxy investigated within this study, the detection of the nitrate RO2 radical is determined by the probability of HO2versus NO2 formation at chain termination, either from decomposition, isomerization, or RO + O2 reactions. To determine the relative conversion efficiency of each nitrate-RO2, the chemistry in the converter was modelled with its operational conditions during the experiments described in this study (25 hPa, 0.55 ppmv of NO, 0.17% CO, residence time ∼0.6 s). We define the relative conversion efficiency as the ratio between the OH formed from the RO2 of interest and the OH formed from the reference RO2 (CH3OO, used to calibrate the ROxLIF), at the point where its fluorescence is detected in the fluorescence cell (Table 1). We then distinguish between the RO2 concentration obtained from the sum of the modelled RO2 (labeled “total RO2” hereafter), against “detectable RO2” indicating the sum of all RO2 weighted by their relative conversion efficiency. For all RO2 not included in Table 1, the conversion efficiency is assumed to be equal to one; these are only present in minor concentrations.| Nitrate-RO2 | 298 K |
|---|---|
| Reference RO2 (CH3OO˙) | 1.0 |
| 2-ONO2-1-ethylperoxy | 0.90 |
| 2-ONO2-1-propylperoxy | 0.65 |
| 1-ONO2-2-propylperoxy | 0.50 |
| 3-ONO2-2-butylperoxy | 1.7 × 10−2 |
| 3-ONO2-2,3-diMe-2-butylperoxy | 1.2 × 10−7 |
| 1-ONO2-2-pentylperoxy | 1.0 |
| 2-ONO2-1-pentylperoxy | 1.0 |
| 2-ONO2-3-hexylperoxy | 1.0 |
| 3-ONO2-2-hexylperoxy | 1.0 |
| CH2(NO2)OO˙ | 3.4 × 10−7 |
3. Theoretical results
Table 2 lists the barrier heights and rate coefficients for the decomposition (R4) and isomerisation (R5) of the reactions of a set of β-ONO2 alkoxy radicals, and a set of β-epoxidized RO. The alkoxy radicals considered span a wide range of alkyl substituents, but also cover unsaturated and heterosubstituents. H-migration reactions are also included for those RO that allow 1,5- or 1,6-H-shifts; migrations over shorter spans are known to be too slow to compete.59 | (R4) |
 | (R5) |
| Reactant | Products | k(298 K) | A | n | E a | E b | SAR Eb | SAR* Eb |
|---|---|---|---|---|---|---|---|---|
| a Tabulated rate is per CH3 group; total rate for CH3 elimination is twice the given rate coefficient. b Preferential product decomposition is scission in the –ONO2 group, not the –OOH or –OOR group (see text). c Preferential product decomposition is scission in the –ONO group, not the –ONO2 group (see text). d The α-nitrate α-epoxy alkyl radical decomposes to NO2 with an epoxy-carbonyl coproduct (see table and text). e The barrier height after ZPE corrections is less then zero. | ||||||||
| Alkoxy radicals derived from ethene and propene | ||||||||
| ˙OCH2–CH2ONO2 | OCH2 + CH2O + ˙NO2 |
3.3 × 101 | 1.59 × 108 | 1.89 | 7805 | 15.9 | 15.1 | 15.3 |
| ˙OCH2–CH(CH3)ONO2 | OCH2 + CH(CH3)O + ˙NO2 |
5.6 × 102 | 2.40 × 109 | 1.41 | 6950 | 14.1 | 11.7 | 13.8 |
| ˙OCH(CH3)–CH2ONO2 | OCHCH3 + CH2O + ˙NO2 |
1.3 × 103 | 1.11 × 108 | 1.74 | 6379 | 13.2 | 12.8 | 13.0 |
| ˙CH3 + OCH–CH2ONO2 |
1.6 × 101 | 1.45 × 105 | 2.75 | 7381 | 15.7 | 15.6 | ||
| Alkoxy radicals derived from 2-butene | ||||||||
| (S,S)-˙OCH(CH3)–CH(CH3)ONO2 | OCHCH3 + CH(CH3)O + ˙NO2 |
5.9 × 104 | 5.29 × 109 | 1.19 | 5426 | 11.2 | 9.4 | 11.6 |
| ˙CH3 + OCH-CH(CH3)ONO2 |
2.3 × 101 | 3.86 × 107 | 1.95 | 7583 | 15.9 | 15.6 | ||
| (S,R)-˙OCH(CH3)–CH(CH3)ONO2 | OCHCH3 + CH(CH3)O + ˙NO2 |
3.6 × 104 | 1.64 × 109 | 1.45 | 5658 | 11.5 | 9.4 | 11.6 |
| ˙CH3 + OCH–CH(CH3)ONO2 |
1.7 × 101 | 1.08 × 106 | 2.58 | 7687 | 16.0 | 15.6 | ||
| Alkoxy radicals derived from isobutene and 2,3-dimethyl-2-butene | ||||||||
| ˙OCH2–C(CH3)2ONO2 | OCH2 + C(CH3)2O + ˙NO2 |
1.6 × 104 | 3.34 × 109 | 1.43 | 6070 | 12.5 | 8.3 | 12.3 |
| ˙OC(CH3)2–CH2ONO2 | OC(CH3)2 + CH2O + ˙NO2 |
2.1 × 104 | 2.57 × 107 | 2.04 | 5574 | 11.9 | 10.3 | 10.5 |
| ˙CH3 + OC(CH3)–CH2ONO2a |
9.9 × 102 | 3.93 × 105 | 2.81 | 6548 | 14.2 | 13.3 | ||
| ˙OC(CH3)2–C(CH3)2ONO2 | OC(CH3)2 + C(CH3)2O + ˙NO2 |
5.1 × 107 | 4.38 × 109 | 1.32 | 3578 | 7.6 | 4.3 | 7.7 |
| ˙CH3 + OC(CH3)-C(CH3)2ONO2a |
1.1 × 104 | 3.84 × 106 | 2.24 | 5541 | 12.0 | 13.3 | ||
| Alkoxy radicals derived from 1-pentene | ||||||||
| ˙OCH2–CH(CH2CH2CH3)ONO2 | OCH2 + CH(C3H7)O + ˙NO2 |
9.7 × 102 | 2.31 × 1010 | 1.14 | 7002 | 14.1 | 11.7 | 13.8 |
| HOCH2-CH(CH2C˙HCH3)ONO2 | 7.1 × 106 | 1.70 × 10−11 | 7.31 | 317 | 8.5 | N/A | ||
| HOCH2-CH(CH2CH2C˙H2)ONO2 | 5.5 × 104 | 1.04 × 10−19 | 9.82 | 390 | 10.7 | N/A | ||
| ˙OCH(CH2CH2CH3)–CH2ONO2 | OCHC3H7 + CH2O + ˙NO2 |
3.9 × 103 | 1.00 × 108 | 1.87 | 6208 | 12.9 | 12.8 | 13.0 |
| ˙C3H7 + OCH-CH2ONO2 |
6.7 × 103 | 4.95 × 106 | 2.30 | 5878 | 12.5 | 12.2 | ||
| HOCH(CH2CH2C˙H2)–CH2ONO2 | 4.5 × 106 | 8.94 × 10−12 | 7.30 | 238 | 8.2 | N/A | ||
| Alkoxy radical derived from 2-hexene | ||||||||
| (S,R)-˙OCH(CH3)–CH(C3H7)ONO2 | OCHCH3 + CH(CH2CH2CH3)O + ˙NO2 |
8.0 × 104 | 4.12 × 108 | 1.67 | 5376 | 11.0 | 9.4 | 11.6 |
| ˙CH3 + OCH–CH(CH2CH2CH3)ONO2 |
2.8 × 101 | 5.07 × 106 | 2.39 | 7673 | 15.7 | 15.6 | ||
| HOCH(CH3)–CH(CH2C˙HCH3)ONO2 | 2.3 × 107 | 1.94 × 10−7 | 5.99 | 517 | 7.0 | N/A | ||
| HOCH(CH3)–CH(CH2CH2C˙H2)ONO2 | 1.8 × 105 | 3.61 × 10−19 | 9.66 | 137 | 9.6 | N/A | ||
| (S,S)-˙OCH(CH3)–CH(C3H7)ONO2 | OCHCH3 + CH(CH2CH2CH3)O + ˙NO2 |
1.1 × 105 | 8.53 × 1010 | 0.95 | 5661 | 11.3 | 9.4 | 11.6 |
| ˙CH3 + OCH-CH(CH2CH2CH3)ONO2 |
2.2 × 101 | 1.26 × 106 | 2.57 | 7627 | 16.0 | 15.6 | ||
| HOCH(CH3)–CH(CH2C˙HCH3)ONO2 | 1.1 × 107 | 3.06 ×10−9 | 6.63 | 586 | 8.1 | N/A | ||
| HOCH(CH3)–CH(CH2CH2C˙H2)ONO2 | 1.2 × 105 | 2.21 × 10−22 | 10.7 | −146 | 10.4 | N/A | ||
| (S,R)-˙OCH(C3H7)–CH(CH3)ONO2 | OCHCH2CH2CH3 + CH(CH3)O + ˙NO2 |
3.7 × 104 | 1.93 × 108 | 1.71 | 5444 | 11.1 | 9.4 | 11.6 |
| ˙CH2CH2CH3 + OCH–CH(CH3)ONO2 |
4.5 × 103 | 5.83 × 107 | 1.99 | 6198 | 12.7 | 12.2 | ||
| HOCH(CH2CH2C˙H2)–CH(CH3)ONO2 | 3.6 × 106 | 8.75 × 10−16 | 8.59 | −248 | 8.2 | N/A | ||
| (S,S)-˙OCH(C3H7)–CH(CH3)ONO2 | OCHCH2CH2CH3 + CH(CH3)O + ˙NO2 |
5.3 × 104 | 2.01 × 1010 | 1.09 | 5687 | 11.5 | 9.4 | 11.6 |
| ˙CH2CH2CH3 + OCH–CH(CH3)ONO2 |
2.2 × 103 | 8.19 × 107 | 1.89 | 6339 | 13.1 | 12.2 | ||
| HOCH(CH2CH2C˙H2)–CH(CH3)ONO2 | 4.9 × 105 | 1.90 × 10−19 | 9.87 | 12 | 9.8 | N/A | ||
| Nitrate-RO radicals with various β-substituents | ||||||||
| ˙OCH2–CH(CH = CH2)ONO2 | OCH2 + CH(CHCH2)O + ˙NO2 |
7.1 × 104 | 1.35 × 108 | 1.54 | 4869 | 10.5 | 5.7 | 10.5 |
| ˙OCH2–CH(OH)ONO2 | OCH2 + CH(OH)O + ˙NO2 |
1.5 × 102 | 1.24 × 1010 | 1.37 | 7757 | 15.6 | 7.6 | 14.4 |
| ˙OCH2–C(OH)(CH3)ONO2 | OCH2 + CH(OH)(CH3)O + ˙NO2 |
2.1 × 104 | 7.15 × 109 | 1.34 | 6083 | 11.8 | 4.6 | 12.9 |
| ˙OCH2–CH(OCH3)ONO2 | OCH2 + CH(OCH3)O + ˙NO2 |
2.2 × 104 | 1.95 × 108 | 1.72 | 5631 | 11.7 | 7.9 | 11.7 |
| ˙OCH2–CH(NO)ONO2 | OCH2 + CH(NO)O + ˙NO2 |
2.8 × 104 | 1.46 × 103 | 3.30 | 4722 | 11.8 | 1.8 | 11.8 |
| ˙OCH2–CH(NO2)ONO2 | OCH2 + CH(NO2)O + ˙NO2 |
3.2 × 10−2 | 2.14 × 109 | 1.48 | 9947 | 20.4 | 15.5 | 20.4 |
| ˙OCH2–C(=O)ONO2 | OCH2 + CO2 + ˙NO2 |
2.1 × 102 | 5.37 × 107 | 2.17 | 7395 | 15.6 | 6.6 | 15.5 |
| ˙OCH2–C(CH2)ONO2 |
OCH2 + CH2CO + ˙NO2 |
8.5 × 10−5 | 6.96 × 109 | 1.59 | 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 251 251 |
24.4 | 20.1 | 24.4 |
| 2-ONO2-2,3-epoxy-1-propyl | 4.5 × 105 | 1.83 × 1010 | 0.72 | 4389 | 9.5 | N/A | ||
| ˙OCH(OH)–CH2ONO2 | OCHOH + CH2O + ˙NO2 |
2.5 × 109 | 2.93 × 1010 | 0.78 | 2064 | 4.4 | 6.3 | 6.5 |
| ˙OCH2–CH(OOH)ONO2 | OCH2 + CH(OOH)O + ˙NO2b |
7.6 × 103 | 1.09 × 108 | 1.56 | 5496 | 11.7 | 5.9 | 11.7 |
| ˙OCH2–CH(OOCH3)ONO2 | OCH2 + CH(OOCH3)O + ˙NO2b |
1.1 × 102 | 9.73 × 104 | 3.04 | 7164 | 15.0 | 7.9 | 15.0 |
| ˙OCH2–CH(ONO)ONO2 | OCH2 + CH(=O)ONO2 + ˙NOc |
1.1 × 101 | 2.67 × 103 | 3.68 | 7889 | 16.4 | 9.1 | 16.4 |
| 2-ONO2-2,3-epoxy-1-propoxy | CH2O + 1-ONO2-1,2-epoxy-ethyld |
4.1 × 10−2 | 1.31 × 109 | 1.72 | 10118 |
20.1 | N/A | 20.1 |
| Cyclic β-ONO2-RO radicals | ||||||||
| 1-ONO2-cyclopropyl-CH2O˙ | ˙NO2 + cyclopropanone + CH2O |
3.9 × 10−1 | 4.17 × 1010 | 1.15 | 9529 | 18.8 | 17.5 | 18.9 |
| 1-ONO2-cyclobutyl-CH2O˙ | ˙NO2 + cyclobutanone + CH2O |
1.8 × 103 | 1.02 × 1012 | 0.49 | 6851 | 13.4 | 10.9 | 13.4 |
| 1-ONO2-cyclopentyl-CH2O˙ | ˙NO2 + cyclopentanone + CH2O |
1.1 × 104 | 2.30 × 1010 | 1.12 | 6236 | 12.6 | 8.1 | 12.6 |
| 1-ONO2-cyclohexyl-CH2O˙ | ˙NO2 + cyclohexanone + CH2O |
5.4 × 103 | 3.48 × 1010 | 1.15 | 6619 | 13.2 | 8.1 | 13.2 |
| syn-2-ONO2-cyclopropoxy | ˙NO2 + propanedial | N/Ae | 0e | 0.2 | 0e | |||
| anti-2-ONO2-cyclopropoxy | ˙NO2 + propanedial | N/Ae | 0e | 0.2 | 0e | |||
| syn-2-ONO2-cyclobutoxy | ˙NO2 + butanedial | 2.3 × 1011 | 8.36 × 1011 | 0.42 | 1102 | 2.4 | 1.5 | 2.4 |
| anti-2-ONO2-cyclobutoxy | ˙NO2 + butanedial | 9.7 × 1010 | 1.79 × 1010 | 0.96 | 1129 | 2.5 | 1.5 | 2.4 |
| syn-2-ONO2-cyclopentoxy | ˙NO2 + pentanedial | 1.4 × 107 | 2.26 × 1010 | 0.84 | 3620 | 7.4 | 6.4 | 8.3 |
| anti-2-ONO2-cyclopentoxy | ˙NO2 + pentanedial | 1.6 × 106 | 1.43 × 109 | 1.44 | 4470 | 9.2 | 6.4 | 8.3 |
| syn-2-ONO2-cyclohexoxy | ˙NO2 + hexanedial | 3.2 × 104 | 7.86 × 109 | 1.13 | 5620 | 11.5 | 8.8 | 11.1 |
| anti-2-ONO2-cyclohexoxy | ˙NO2 + hexanedial | 9.9 × 104 | 2.31 × 109 | 1.30 | 5209 | 10.7 | 8.8 | 11.1 |
| Epoxy-RO radicals | ||||||||
| 2,3-Epoxy-1-propoxy | CH2O + 1,2-epoxy-ethyl |
1.2 × 100 | 8.88 × 106 | 2.31 | 8628 | 17.7 | N/A | 17.6 |
| 2-Me-2,3-epoxy-1-propoxy | CH2O + 1,2-epoxy-2-propyl |
3.6 × 101 | 4.60 × 1010 | 1.22 | 8322 | 16.4 | N/A | 14.2 |
| anti-2,3-Epoxy-1-butoxy | CH2O + 1,2-epoxy-1-propyl |
1.5 × 100 | 2.63 × 109 | 1.41 | 8729 | 17.5 | N/A | 17.6 |
| syn-2,3-Epoxy-1-butoxy | CH2O + 1,2-epoxy-1-propyl |
1.5 × 100 | 7.04 × 108 | 1.58 | 8623 | 17.3 | N/A | 17.6 |
| 4-OH-2,3-epoxy-1-butyl | 2.5 × 105 | 6.83 × 10−21 | 10.2 | −198 | 10.1 | N/A | ||
| 3,4-Epoxy-2-butoxy | CH3CHO + 1,2-epoxy-ethyl |
5.9 × 101 | 1.69 × 109 | 1.56 | 7766 | 15.6 | N/A | 15.3 |
| CH3 + 2,3-epoxy-propanal | 2.1 × 102 | 1.01 × 107 | 2.05 | 6679 | 14.2 | 15.6 | ||
| 3,4-Epoxy-1-butoxy | CH2O + 2,3-epoxy-1-propyl |
1.6 × 102 | 3.29 × 108 | 1.67 | 7171 | 14.8 | 14.5 | |
| 4-OH-1,2-epoxy-1-butyl | 9.7 × 104 | 1.64 × 10−24 | 11.2 | −715 | 10.9 | N/A | ||
| 1,2-Epoxy-ethoxy | ˙CH2OCHO |
N/A | 0e | N/A | ||||
| ˙OCH2CHO |
N/A | 0.8 | N/A | |||||
| Alkyl radicals | ||||||||
| 1-ONO2-1,2-epoxy-ethyl | ˙NO2 + 1-oxo-1,2-epoxy-ethane | 6.4 × 1011 | 3.54 × 109 | 1.25 | 579 | 1.6 | N/A | |
| 1,2-Epoxy-ethyl | 2-oxo-ethyl | 1.7 × 103 | 2.83 × 10−26 | 12.8 | 2035 | 14.2 | N/A | |
| 1-ONO2-1-cyclopropyl | ˙NO2 + cyclopropanone | 4.3 × 1010 | 1.24 × 108 | 1.80 | 1308 | 3.4 | N/A | |
| 1-ONO2-1-cyclobutyl | ˙NO2 + cyclobutanone | ≤2 | N/A | |||||
3.1 Barrier heights and rate coefficients
The results for the decomposition reactions are compared with the predictions of the decomposition SAR by Vereecken and Peeters,13 where applicable, to assess the reactivity trends induced by the nitrate substituent. This SAR predicts the rate of decomposition based on the substituents on the α- and β-atoms involved in the C–C bond scission (R6), using a linear additive scheme (E1) affecting the barrier height Eb for reaction based on theory-derived parameters Fs for each type of substituent s including alkyl groups and –ONO2.13| Cα (O˙)–C → CαO + Cβ˙ | (R6) |
| Eb = 17.9 kcal mol−1 + ΣFs | (E1) |
Of the many substituents covered in the alkoxy decomposition SAR by Vereecken and Peeters,13 the nitrate substituent on the β-carbon is among the most likely to show non-additive effects with other substituents. In particular, the C˙–ONO2 product radical formed after the alkoxy radical bond scission is a transient species that itself decomposes spontaneously to a carbonyl + NO2,14 and this secondary decomposition process already starts to some extent during the initial alkoxy radical bond breaking. This is also reflected in the transition state geometries, where for example we see an elongation of the CO–NO2 bond, 1.42 Å, in the TS geometry for 2-ONO2-1-propoxy compared to 1.38 Å for the nitrate-RO reactant, as well as a contraction of the C–ONO2 bond from 1.43 Å in the reactant to 1.37 Å in the TS. This also implies that the TS is likely most favorable when the relative orientation of the moieties provides a planar C–O–NO2 geometry amenable to formation of a CO double bond on the sp2-hybridized central O-atom, and we indeed find that TS conformers with this feature have the lowest relative energies.
The results show that many of the decomposition reactions leading to a C˙–ONO2 product radical, and thus leading to secondary fragmentation to CO + NO2, are proceeding slower than anticipated based on the additive reactivity trends described in Vereecken and Peeters,13 suggesting that the SAR needs to be updated to include second-order parameters that are conditional to the presence of this specific substituent, thus accounting for the impact of the secondary dissociation. To our knowledge, the current set of data is the first systematic study on this aspect of alkoxy radical decomposition. Though this is not treated in detail in the current work, preliminary results show similar effects for hydroperoxy-substituted alkoxy radicals, where an –OOH group on the β-carbon undergoes a similar decomposition to CO + OH,80,81 and where a SAR based on linear additivity, i.e. where all substituent parameters are independent, seems to be incomplete. For example, the difference in the calculated barrier height for decomposition of ˙OCH2–CH2OOH, 8.3 kcal mol−1, compared to ˙OCH2–CH(CH3)OOH, 7.5 kcal mol−1, suggests that the methyl substituent is likewise hampered by the spontaneous decomposition of the –OOH group in lowering the barrier height for the parent alkoxy radical, partially negating its SAR-predicted impact of −3.4 kcal mol−1. A similar effect is anticipated for –OOR substitution, which likewise shows decomposition. For the C˙H2ONO radical, we also find a (near-)barrierless decomposition to HCHO + NO (see ESI‡), making nitrite-substituted alkoxy radicals also likely to exhibit cross-substituent interactions.
The related alkoxy H-migration SAR by Vereecken and Peeters59 did not cover nitrated substituents, and can thus not be compared directly to our theoretical predictions. However, for the compounds in Table 2, none of the H-migrations have the nitrate group implanted on the carbon bearing the migrating H-atom, and as such the spectator nitrate functionality should have only a minor impact on the rate coefficient and can be neglected. Within this approximation, we find that the rate coefficient predicted by the H-migration SAR matches the directly calculated values in Table 2 on average within a factor of 5, and a maximum deviation of a factor 14 at 298 K, in agreement with the order-of-magnitude uncertainty postulated for the H-migration SAR. The current set of values then confirms the predictions of the H-migration SAR within its uncertainty, and indicate that the nitrate functionality has only moderate impact on the H-migration rate coefficient when in a spectator position to the alkoxy radical O-atom. This also implies that the H-bond between the β-nitrate group and the newly formed hydroxyl functionality is still weak in the transition state. We currently have no data for migration of α-ONO2 H-atoms.
Table 2 also shows the result for the epoxidation reaction in a β-unsaturated alkoxy radical, ˙OCH2–C(CH2)ONO2, where the rate of epoxidation, k(298 K) = 4.5 × 105 s−1, exceeds the rate of decomposition, k(298 K) = 8.5 × 10−5 s−1, by several order of magnitude, and is comparable in magnitude to many of the decomposition rates for more favorably substituted nitrate-RO. In the OH-initiated oxidation of multi-unsaturated compounds, such as isoprene, the hydroxyl group greatly enhances the rate of alkoxy radical decomposition,13,19 and epoxidation is typically not considered. The inhibiting effect of the nitrate group on the decomposition, in contrast, could tip the balance in favor of epoxidation reactions in the alkoxy radicals derived from conjugated alkadienes, such as isoprene.
3.2 Validation of the SAR for non-nitrated decomposition
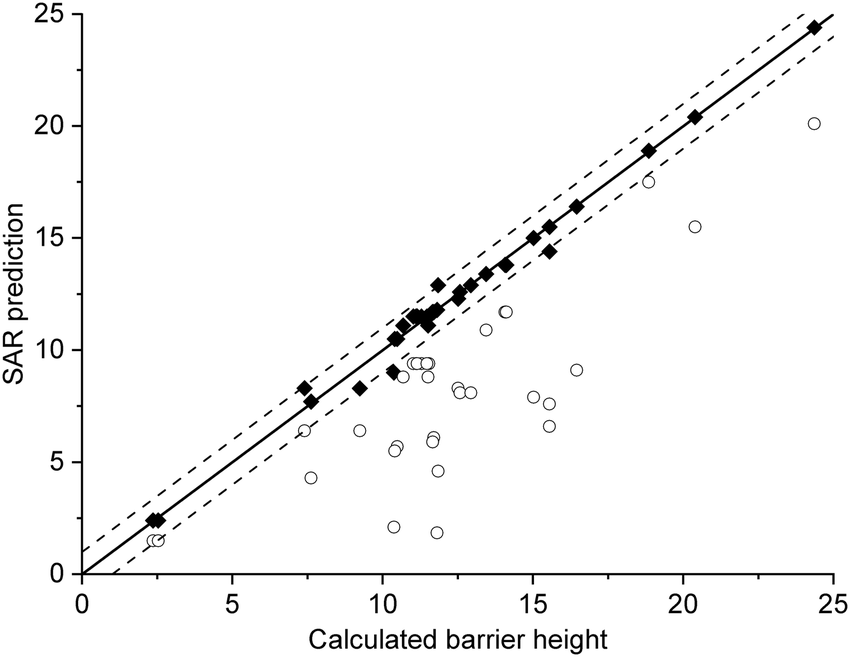
The current data set allows for validation of the barrier heights for decomposition for non-nitrated RO as predicted by the original SAR by Vereecken and Peeters,13i.e. we can verify that the SAR predicts barrier heights comparable to the direct theoretical predictions provided here, within the SAR uncertainty of ∼1 kcal mol−1. The new data is derived at a significantly more reliable level of theory, CCSD(T)//M06-2X versus B3LYP, than the first-generation SAR. Also, it allows validation of our assessment that deviations from the SAR for nitrate-RO is due to the –ONO2 moiety decomposition, as indeed other decomposition reaction with a spectator nitrate group, i.e. not leading to secondary fragmentation, should remain unaffected. The validation data set includes all decompositions from Table 2 where the –ONO2 moiety is not bonded to the product radical carbon, as well as the reactions where the leaving radical fragment is –C˙H2ONO2, i.e. the base case upon which the original SAR is built and where no interaction with other β-substituents exists. β-epoxy-alkoxy radicals are omitted as these are not covered by the Vereecken and Peeters SAR.For this subset of the data, Fig. 1 compares the barrier heights obtained by the SAR against the new set of data. As can be seen, the SAR performs very well, with excellent recovery over the entire barrier height range. The largest deviation, 2 kcal mol−1, was found for ˙OCH(OH)–CH2ONO2, where the SAR prediction does not account for the effect of internal H-bonding; note that the original SAR explicitly mentions such unaccounted-for H-bonding (e.g. in dihydroxy-substituted alkoxy radicals) as one of the most important sources of errors. All other deviations are less than 1.3 kcal mol−1, in agreement to the uncertainty of 1 kcal mol−1 asserted13 for the SAR. In this respect, one should also consider that the barrier height differences within each SAR class can differ by 1 kcal mol−1 and more, even between stereoisomers with otherwise identical molecular framework, and that striving for a SAR with sub-kcal mol−1 precision would require an impractically large number of higher-order correction terms to account for all possible substituent permutations.
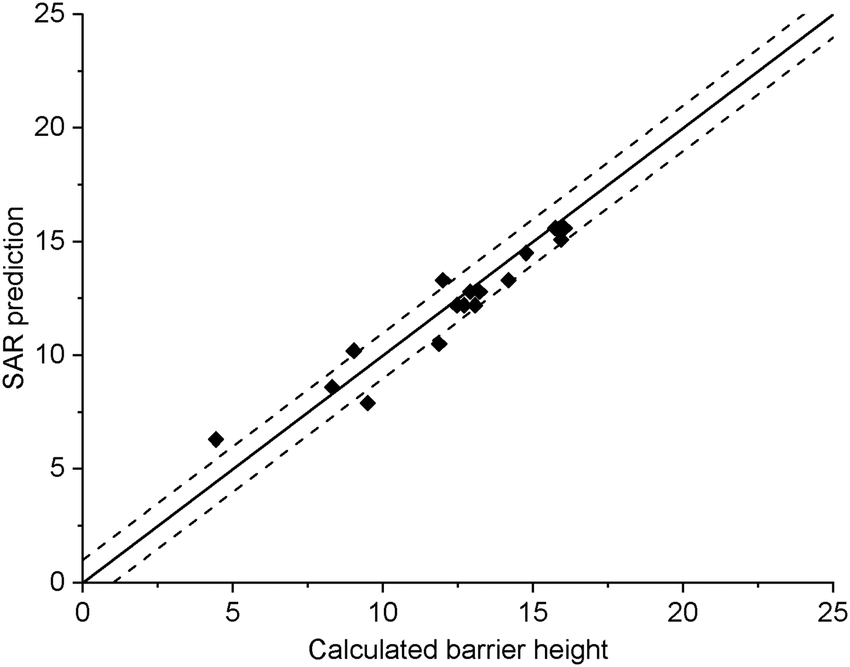 | ||
| Fig. 1 Comparison of the predictions by the SAR for alkoxy decomposition by Vereecken and Peeters,13 compared against the validation subset from Table 2 containing only the alkoxy decomposition reactions without a β-ONO2 substituent, or with a β-CH2ONO2 leaving moiety. The solid line indicates perfect agreement between calculated and SAR-predicted barrier heights (kcal mol−1), with the dashed lines showing a 1 kcal mol−1 deviation. This plot uses Fs(β-ONO2) = −2.8 kcal mol−1 for SAR predictions; the graph with the updated value Fs = −2.6 kcal mol−1 is visually indistinguishable. | ||
From this data set on nitrate-RO, we thus conclude that the SAR by Vereecken and Peeters13 performs very well for alkoxy radicals fragmentation without interactions between an –ONO2 group and other β-substituents. The only correction might be to increase the parameter for a β-ONO2 substituent in a –CH2ONO2 group from Fs(β-ONO2) = −2.8 to −2.6 kcal mol−1; however, such a correction is negligible compared to the scatter on the data.
3.3 Improvement of the SAR for decomposition of β-nitrate-RO
The data in Table 2 show large deviations from the SAR for nitrate-RO decomposition with a β-ONO2 functionality and one or more additional substituents on the β-carbon, in line with the above rationalization. The inhibiting effect of the β-ONO2 group was not detected by Vereecken and Peeters:13 only a single compound was studied that is anticipated to show the impact to some extent, ˙OCH2-CH(CH3)ONO2 (2-nitrooxy-1-propoxy), and the lower-level B3LYP/6-31G(d,p) calculation for this compound underestimates the energy barrier by 1 to 2 kcal mol−1, masking the effect. The ESI‡ tabulates calculations at several levels of DFT, PMP2, CBS-QB3, and CCSD(T) for this compound, showing that while low-level energy predictions vary significantly, the results after higher-level CCSD(T) energy calculations all agree on the somewhat higher energy barrier found in this work.In all reactions in this class the energy barrier for decomposition is found to be higher than expected from the additive SAR by Vereecken and Peeters (Fig. 2), supporting the view that the dissociation of the nitrooxy group hampers the ability of other substituents to stabilize the forming radical site on the β-carbon. The impact of this barrierless secondary decomposition of the nitrate moiety on the energy barrier of the alkoxy decomposition reaction is implemented in the SAR by a second set of parameters for each additional β-substituent examined, i.e. β-alkyl, β-CC, β-OH, β-OR, β-OOH, β-OOR, β-NO, β-ONO, β-NO2, βO, and βCH2 (Table 3), to be used if the leaving radical moiety has an –ONO2 group; we incorporate the slightly updated Fs value for β-ONO2 (see above). We also include activity factors for cyclic compounds (Tables 4 and 5) where, similar to non-nitrated alkoxy radicals, we find that the impact of ring strain is most pronounced for the 3- and 4-membered rings, reduces strongly for 5-membered ring, and becomes comparable to a non-strained hydrocarbon chain for 6-membered rings. For the radical products 1-ONO2-cyclopropyl and 1-ONO2-cyclobutyl, we find that the nitrate group does not decompose spontaneously to NO2, owing to the additional ring strain in the cycloketone co-product. The barriers for decomposition to NO2 + cycloketone, however, remain very low, ≤3.5 kcal mol−1, leading to very fast decomposition at rates ≥4 × 1010 s−1, preventing recombination of the 1-nitrooxy-cycloalkyl product with O2 under atmospheric conditions (pseudo-first order rate coefficient ∼107 s−1).
 | ||
| Fig. 2 Performance of the SAR for alkoxy radical decomposition for β-nitrate-RO radicals with multiple β-substituents. Open symbols: original barrier height SAR predictions (kcal mol−1) by Vereecken and Peeters.13 Closed symbols: SAR with updated parameters accounting for substituent interaction (see Tables 3–5). The solid line indicates perfect agreement between calculated and SAR-predicted barrier heights, with the dashed lines showing a 1 kcal mol−1 deviation. | ||
| Substituent | F s | Substituent | F s without β-ONO2 | F s with β-ONO2 |
|---|---|---|---|---|
|
a If only 1 substituent is present on the α-carbon of the form –CHO, –CH2OR, –CH2OOH, or –CH2OOR (R = alkyl), use Fs = −0.7 eR=alkyl.
b Compounds of the form C(O˙)–NO spontaneously decompose to CO + ˙NO (this work).
c Product radicals of the form C˙OOH and C˙OOR spontaneously decompose to CO + OH/OR,80,81 while C˙ONO radicals decompose to CO + NO (this work). This decomposition could affect other Fs parameters (see text).
d Product radicals of the form C˙ONO2 spontaneously decompose to CO + NO2.14 If an –OOH or –OOR group is also present, the dominant pathway is decomposition of the –ONO2 group, leaving the OOH/OOR group intact (this work); if an –ONO group is present, this group will preferentially decompose, leaving the ONO2 group intact (this work).
e R = alkyl.
|
||||
| α-alkyl | −2.3a | β-alkyl | −3.4 | −1.5 |
| αO |
−12.7 | βO |
−8.5 | +0.2 |
| α-OH | −8.9 | β-OH | −7.5 | −0.9 |
| α-ORe | −9.2 | β-ORe | −9.1 | −3.6 |
| α-OOH | −8.9 | β-OOHc,d | −9.3 | −3.6 |
| α-OORe | −6.4 | β-OORc,d,e | −7.2 | −0.3 |
| α-NO | N/Ab | β-NO | −16.0 | −3.5 |
| α-NO2 | −2.2 | β-NO2 | +0.4 | +5.1 |
| α-ONO | −4.2 | β-ONOc,d | −6.0 | +1.1 |
| α-ONO2 | −3.8 | β-ONO2d | N/A | −2.6 |
| αC |
+21.5 | βC |
+5.0 | +9.1 |
| α-CC |
−4.9 | β-CC |
−9.6 | −4.8 |
Fig. 2 shows the performance of this adjusted SAR for the compounds studied in this work, compared to the original SAR. Note that similar to the original SAR we do not support bicyclic compounds as the change in ring strain is highly dependent on the molecular frame and can have very specific impact on the chemistry.17,83 Combinations of a β-ONO2 substituent with a β-OOH, β-OOR, or β-ONO group are not examined in great detail, as these functionalities similarly dissociate after the alkoxy decomposition. Our current set of calculations suggests that the β-OOH and β-OOR functionalities are somewhat more stable than the nitrate group, staying intact after nitrate-RO decomposition and instead allowing decomposition of the nitrate group to a carbonyl + NO2 functionality. For the β-nitrite group, we find the opposite, i.e. the –ONO group preferentially dissociates to carbonyl + NO, leaving the nitrate group intact. It is, however, hard to judge how much these predictions are affected by the energy distribution in the molecule, and the (non-statistical) energy flow when moving down the potential energy surface from transition state to products. In view of the lack of experimental data on these reactions, one should also consider the possibility that either of the β-OOH, β-OOR, β-ONO, and β-ONO2 substituents could dissociate, with a product yield that depends on temperature and pressure. In the atmosphere, no obvious formation pathways exist forming such gemini-substituted compounds, making these reactions less important.
3.4 Extension of the SARs to epoxy-RO
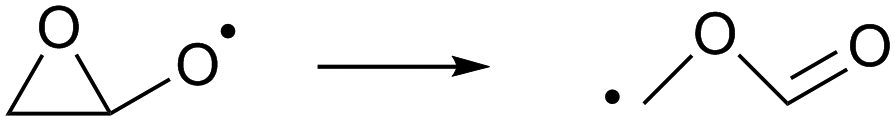
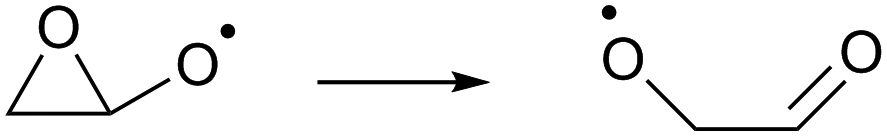
As the calculations listed in Table 2 show that epoxidation might be an important pathway for β-unsaturated nitrate-RO radicals, it is worthwhile extending the RO decomposition SAR to include the epoxy-functionality; while not important in this work, this will be needed in our companion paper on NO3-initiated oxidation of isoprene.82 To estimate the rate coefficients for decomposition (R7)–(R9) and isomerisation (R10) and (R11) of β-epoxydized alkoxy radicals, a set of calculations were performed on simple epoxy-RO radicals (Table 2); these results are interpreted in the framework of the SARs by Vereecken and Peeters.13,59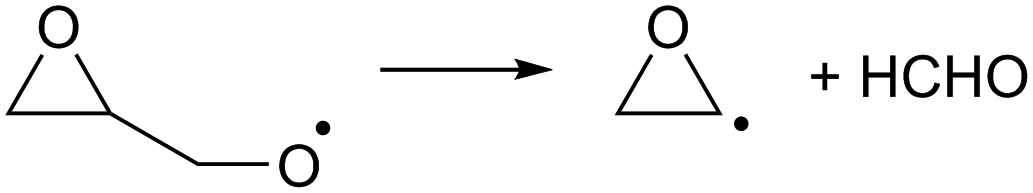 | (R7) |
 | (R8) |
 | (R9) |
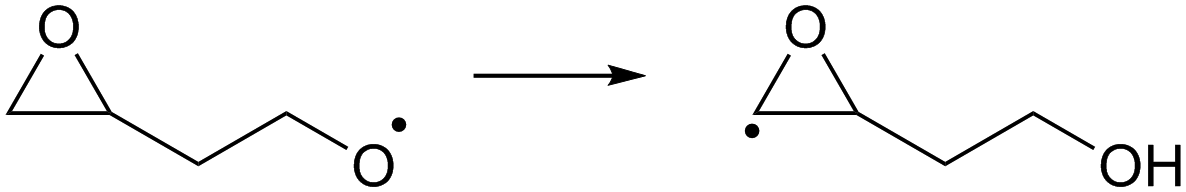 | (R10) |
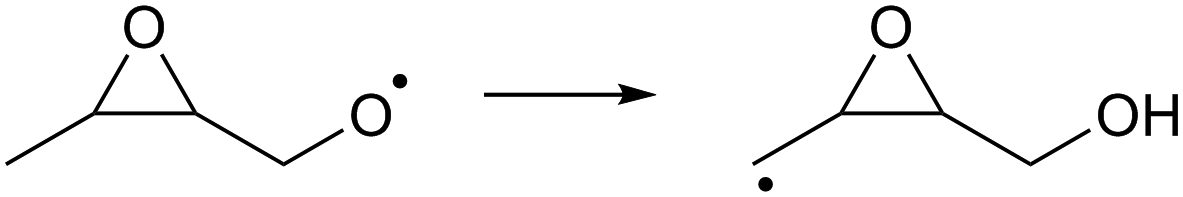 | (R11) |
Despite the presence of an oxygen atom substituent on the β-carbon atom, decomposition of β-epoxy-alkoxy radicals ((R7) and substituted analogues) is comparatively slow. The barrier heights for these reactions are high due to the increase in ring strain in the α-epoxy-alkyl radical product, and epoxy-RO decomposition forming these epoxyl radicals is unlikely to be competitive against other alkoxy reactions, including the reaction with O2 forming a carbonyl product + HO2. The substituent-specific parameter for the SAR by Vereecken and Peeters,13 which includes the impact of oxygen atom and the two carbons bearing the epoxy bridge, is determined as Fs(β-epoxy)= −0.3 kcal mol−1 (Table 4). An alkoxy radical implanted directly on an epoxy group (R8) has no barrier (after ZPE correction) for decomposition by epoxy-C–C bond scission, spontaneously forming an ester. Breaking of the epoxy-C–O bond, forming a β-carbonyl alkoxy radical (R9) is predicted to have a small barrier of 0.8 kcal mol−1. Migration of an α-epoxy-H atom (R10) is likewise not more favorable than migration of an aliphatic H-atom, as the ring strain negates any energetic advantage of having an oxygenated substituent on the migrating-H-bearing carbon. The rate coefficient at 298 K for 1,5-H-migration of a secondary α-epoxyl H-atom is slightly slower than that for a primary H-atom in –CH3. E.g. 1,5-H-migration of the secondary α-epoxyl H-atom in 3,4-epoxy-1-butoxy (R10) has a calculated rate coefficient k(298 K) = 9.7 × 104 s−1, whereas 1,5-migration of the primary H-atoms in 1-butoxy is predicted as k(298 K) = 3.2 × 105 s−1 by the Vereecken and Peeters SAR.59 Based on the current scarce data, we propose to estimate rate coefficients as being a factor three lower than predicted by the SAR59 for migration of an aliphatic H-atom for a similar migration span but with an H-atom rank of one order lower than on the epoxy ring, as in the example above. Further extensions of that SAR will be necessary if this class of H-migration is found to be important in the atmosphere.
| Substituent | F s | Substituent | F s without β-ONO2 | F s with β-ONO2 |
|---|---|---|---|---|
| a The 3-membered ring in α-substituted cyclopropoxy breaks without barrier. b The C–C bond in the 3-membered ring in α-epoxy-alkoxy radicals breaks without barrier. | ||||
| α-Cyc-prop | N/Aa | β-Cyc-prop | +2.4 | +3.6 |
| α-Cyc-but | −2.0 | β-Cyc-but | −4.2 | −1.9 |
| α-Cyc-pent | −2.0 | β-Cyc-pent | −7.0 | −2.7 |
| α-Cyc-hex | −2.0 | β-Cyc-hex | −7.0 | −2.1 |
α-Epoxy: 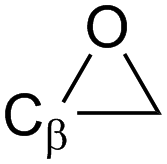 |
N/Ab | β-Epoxy: 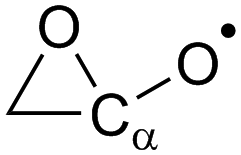 |
−0.3 | +4.8 |
| Ring size | F s without β-ONO2 | F s with β-ONO2 |
|---|---|---|
| a The 3-membered ring in β-ONO2-substituted cyclopropoxy breaks without barrier. b The C–C bond in the 3-membered ring in α-epoxy-alkoxy radicals breaks without barrier. | ||
| 3-Membered ring | −24.6 | N/Aa |
| 4-Membered ring | −17.1 | −14.8 |
| 5-Membered ring | −8.7 | −7.0 |
| 6-Membered ring | −6.3 | −4.2 |
Epoxy-ring: 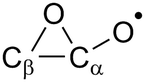 |
N/Ab | N/Ab |
The rate of H-migration across the syn-substituents of an epoxy-ring (R11) is comparable to a traditional aliphatic H-migration of the same span and order of the H-atom, e.g. a 1,5-H-migration in syn-2,3-epoxy-1-butoxy has a similar rate, k(298 K) = 1.6 × 105 s−1 (Table 2) as the analogous H-migration in 1-butoxy as predicted by the SAR (see above). As such, we propose at this time to directly apply the Vereecken and Peeters SAR59 for H-migrations between syn- or gemini-substituents. Note that the epoxy-ring geometrically prevents migration between anti-substituents, except for very long migrations spans which tend to have an unfavorable entropy factor and are unlikely to be competitive. Stereochemistry must thus be explicitly accounted for in mechanism development involving epoxy-RO.
In the unlikely event that an α-epoxy-alkyl radical is formed, we find that opening of the epoxy group forming a β-carbonyl alkyl radical (R12) has a large energy barrier (Table 2), and concomitantly a low rate of reaction that will not be able to compete against other loss processes such as addition of O2 on the radical site.
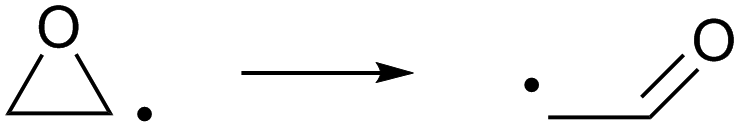 | (R12) |
This ring opening forms a vinoxy-stabilized radical but this resonance stabilization is not yet active in the transition state for ring opening due to the still unfavorable orbital overlap in the triangular TS geometry, and does not help in reducing the TS barrier height. For completeness, we also examined the presence of a nitrate-substituent on the epoxy group (R13). As for other β-ONO2-substituted alkoxy radicals we find that the nitrate-group affects the impact of the epoxy-substituents on the barrier height for decomposition (Table 3).
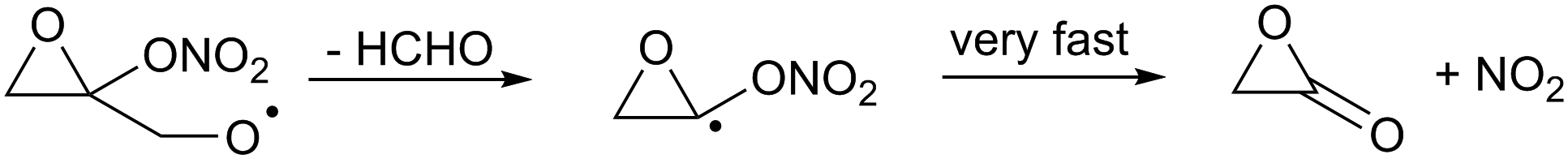 | (R13) |
Furthermore, we find that the α-ONO2-α-epoxy-alkyl radical formed in (R13) does not dissociate spontaneously, similar to what was found for cyclopropyl and cyclobutyl. Still, the barrier for decomposition, 1.6 kcal mol−1, and the resulting very fast decomposition rate, k(298 K) = 6 × 1011 s−1, precludes competing reactions and will lead to a strained lactone + NO2(R13).
4. Experimental results
4.1 Model and measurement comparison for the HO2 radicals
The measurement of HO2 radicals in the chamber using the LIF instrument (Fig. 3) allows for a direct comparison against the HO2 concentrations predicted by the kinetic model, as formed at the timescales of the SAPHIR chamber kinetics. Hence, an underestimation by the modelled chamber HO2 compared to the measurement for the experimental condition of the experiments shown here (large excess of CO) indicates a missing source of HO2 or OH, where the latter is mainly converted to HO2 by reaction with the excess CO added to the chamber reaction mixture. Information on the potential source reaction can be obtained from the dependence of the HO2 discrepancy on the evolution of the reaction mixture in the chamber, specifically the varying concentrations of the primary alkene and secondary products, the contributions of OH, NO3 and O3 in the oxidation of these VOCs, and the changing RO2 concentration and speciation.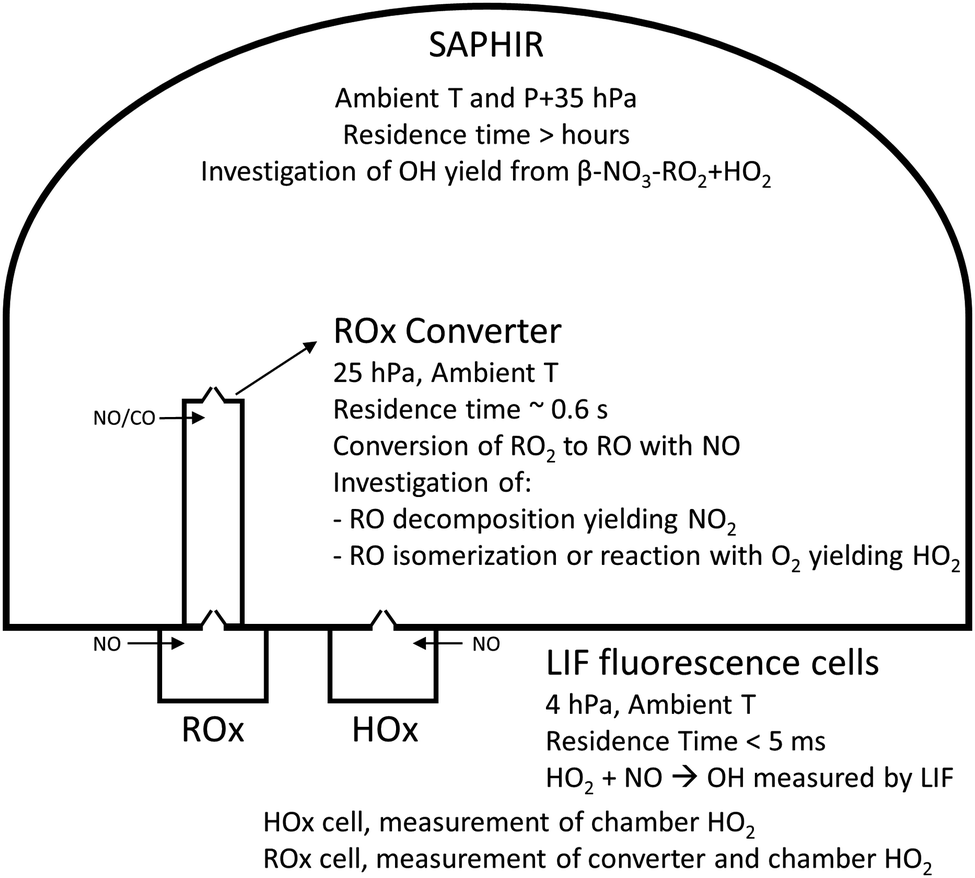 | ||
| Fig. 3 Schematic showing the experimental setup (not to scale), the various measurement devices, and the chemistry investigated. | ||
Fig. 4a depicts the comparison between measured and modelled HO2 radicals for the cis-2-butene experiment. Model M0 is largely underestimating the measured concentration by about up to a factor of three by the end of experiment. As highlighted in Section 2.4, all the experiments described in this study were performed in excess of CO except for the experiment performed with ethene where only 600 pptv of CO were present in the chamber and where the measured HO2 radicals could be reproduced by M0 within 5% (Fig. S16, ESI‡). This strongly suggested that, instead of a missing source of HO2 radicals, the large discrepancy observed could be caused by an underestimation of the OH radicals. As described in Section 2.5, due to their structure, the β-nitrate RO2 radicals observed in this study should be considered as a potential OH source via their reaction with HO2 radicals. This was implemented within M1 where the products of the reaction between nitrate-RO2 and HO2 radical were changed to include a fraction of OH and nitrate-RO radical, in addition to some of the traditional nitrate hydroperoxide product. The yield for the OH radical within M1 was adjusted so that an agreement within 10% between modelled and measured HO2 radicals could be achieved; this required yields ranging from 0.15 to 0.65 depending on the alkenes investigated (Table S2, ESI‡). Once this reaction is introduced within the model (M1), a good agreement can be found for all alkenes investigated (Fig. 4a and Fig. S17–S21, ESI‡). The good agreement typically extends across the duration of the experiment with a single optimized yield, suggesting that the time profiles of the reactants generating the missing HO2 must be similar to that of the modelled RO2 and HO2 concentrations. Although the yield for the OH radical formation carries a large uncertainty as it is currently fitted to match the observed HO2 radicals, an additional confirmation on the viability of this approach was offered by the measured acetone concentration within the 2,3-dimethyl-2-butene experiment (Fig. 4b). The measured acetone shows a sharp increase directly at the injection of 2,3-dimethyl-2-butene and then remains relatively constant as its main oxidant, the OH radical, is scavenged away by the excess of CO. Model M0, without nitrate-RO formation in the reaction of nitrate-RO2 + HO2 as a source of acetone, cannot reproduce the observed acetone yield. Also, independently of the absolute value, M0 shows a much slower increase in acetone concentrations, missing the observed sharp rise. In contrast, the formation of 3-ONO2-2,3-diMe-2-butoxy and OH radical included in M1 for the reaction between its parent nitrate-RO2 with HO2 radicals (OH yield = 0.67) drastically improves the agreement between measured and modelled acetone. The sharp increase in acetone formation in M1 is caused by the decomposition of 3-ONO2-2,3-diMe-2-butoxy radicals generating two molecules of acetone, one directly and one after NO2 elimination. While these observations could also be caused by some unknown acetone source in the experiments, acetone formation through this proposed channel matches well with the proposed mechanism for HO2 formation.
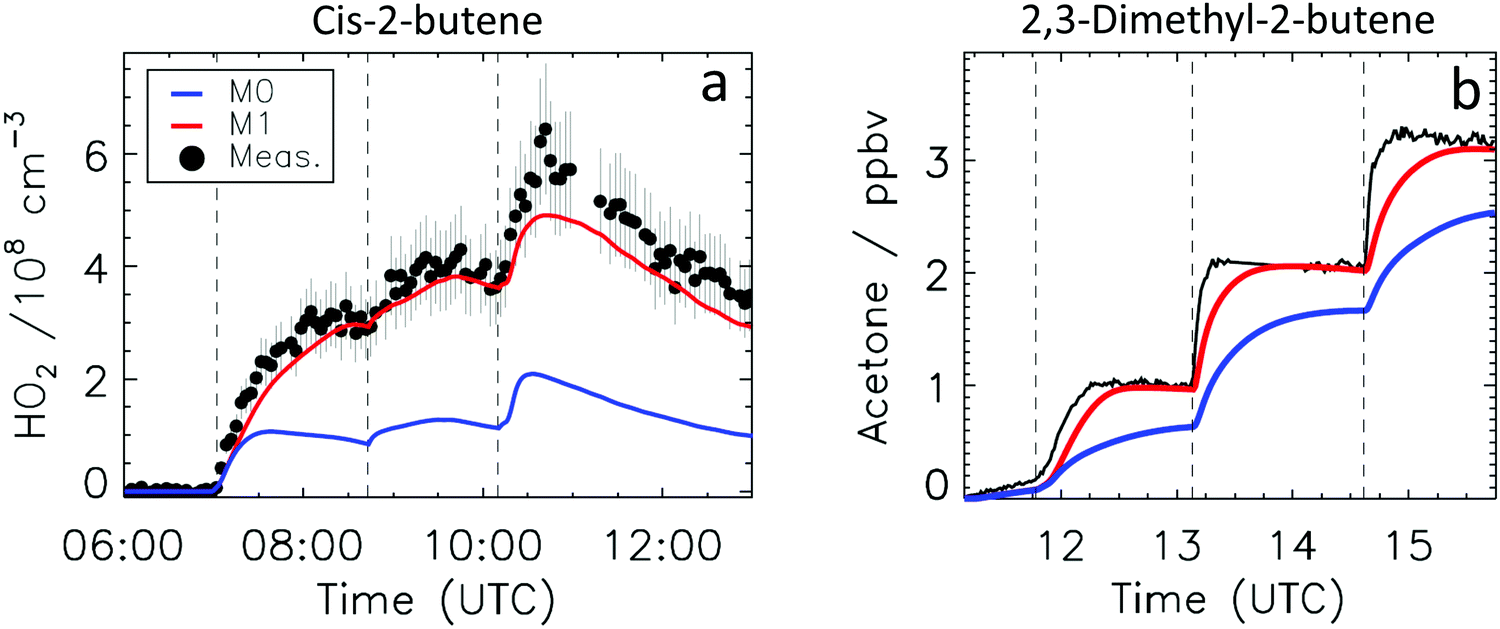 | ||
| Fig. 4 Comparison of the experimental data against models M0 (without nitrate-RO + OH-forming channel in the β-ONO2-RO2 + HO2 reaction), and M1 (including an optimized nitrate-RO + OH yield for the β-ONO2-RO2 + HO2 reaction) (a) time-dependent HO2 concentrations in the cis-2-butene experiment, with 45% OH yield in M1; the improved agreement for model M1 is also found for all other alkenes; (b) time-dependent acetone concentrations in the 2,3-dimethyl-2-butene experiment, with 67% nitrate-RO + OH yield in M1. Vertical dashed lines indicate the times when reactants were added. | ||
To investigate the detectability of the nitrate RO2 radicals by LIF technique and to validate the theoretical results within this study, a good reproduction of the measured HO2 radicals by the model used is central owing to their role in the consumption of the RO2 radicals. For this reason, only M1 will be further considered.
4.2 Investigation of nitrate-RO radical chemistry
The goal of the experiments described in the following sections was twofold. First, they support the theoretical calculations presented in this study by comparing the detectable RO2 radicals by LIF against the result of a model calculation. The choice of alkenes investigated spanned from ethene to trans-2-hexene to cover nitrated alkoxy radicals which do not decompose or where the decomposition is slow (ethene and propene), over alkoxy radicals where the decomposition is dominating (cis-2-butene and 2,3-dimethyl-2-butene), to alkoxy radicals where the isomerization is the most important chemical path (1-pentene and trans-2-hexene). Second, the experiments show that the different behavior of the nitrate alkoxy radical should translate in a complete, partial or null detectability of the parent nitrate RO2 radical by ROxLIF technique, as a function of the substitution, thus affecting RO2 LIF measurements in laboratory and field measurements.The kinetics of the nitrate-RO radical chemistry, and the detectability of nitrate-RO2 using the ROxLIF instrument is determined by measuring the HO2 concentration at the end of the ROxLIF converter originating from the nitrate-RO2 sampled from the SAPHIR chamber (Fig. 3). The reaction mixture in the chamber is changing very slowly compared to the reaction time in the converter (minutes to hours versus < 1 s), and the chamber thus acts primarily as a source of RO2 being sampled into the converter. The model prediction of the total RO2 concentration should be accurate, as it depends mostly on literature alkene + NO3 reaction rates, measured concentrations of alkene, O3, NO2, NO3, and HO2, and on the kinetics of RO2 + RO2/HO2/NO3 as described by SARs available in the literature. The RO2 radicals, once sampled into the converter, are converted to alkoxy radicals by reaction with added NO. These RO radicals will then undergo unimolecular reactions, i.e. decomposition or isomerisation, as described in this work, in competition with their reaction with O2. As RO decomposition yields NO2 as the product fragment, while reaction with O2 produces HO2, the concentration of HO2 measured probes the relative rate of alkoxy decomposition against reaction of the alkoxy radical with O2, where the rate of the latter is generally accepted to be similar for all alkoxy radicals19,20 and thus acts as the reference rate. For larger nitrate-RO formed from 1-pentene and 2-hexene, isomerisation and decomposition are both faster than reaction with O2, but isomerisation forms HO2 whereas decomposition does not. For these compounds the yield of HO2 thus shows the competition of isomerisation against decomposition. Due to the reduced pressure in the converter, 25 hPa, and the limited reaction time in the converter, ∼0.6 s, bimolecular reactions other than the reaction of RO2 and HO2 with NO, the reaction of alkyl and RO radicals with O2, and the reaction of OH radicals with CO (see elsewhere), are negligible and cannot interfere. The agreement between the theoretical results and the experimental data across the range of compounds studied is then a measure of the reliability of the relative rate predictions. Furthermore, irrespective of the absolute rate coefficients, the experimental detectability of nitrate-RO2 by a ROxLIF instrument has direct repercussions for ambient RO2 measurements.
4.3 Ethene
Fig. 5 shows the comparison between measured ozone, NO2 and kOH against model results for the ethene experiment, where only a single injection of alkene was performed. Good agreement can be seen for all observables with no differences among M0 and M1. Although the NO3 radical concentration was not measured during the experiments shown in this study, the good agreement observed between modelled and measured NO2 and O3 indicates that the production rate of NO3 was well represented within the model. The NO3 loss rate is driven by reaction with the alkene, and by reaction with NO2 forming N2O5, with the latter being lost on the chamber walls (see Section 2.4). Fig. 6 shows the comparison between the measured and the modelled RO2 and HO2 radicals, where a very good agreement (within 5%) can be found for both species. Although only 33% of ethene reacted with NO3, the model predicts that 50% of the total RO2 consists of 2-nitrate-1-ethylperoxy radicals, with the remainder arising from the reaction of OH radicals with ethene. Within the LIF converter, the 2-ONO2-1-ethylperoxy radical is converted by reaction with NO to 2-ONO2-1-ethoxy, which is theoretically predicted to decompose at a rate of 3.3 × 10−1 s−1 at 298 K (Table 1), much slower than its reaction with O2, ∼1.2 × 103 s−1. Decomposition of the 2-ONO2-1-ethoxy radical, which forms NO2 undetectable by the LIF instrument, then plays a marginal role (less than 3% of the total alkoxy loss), leaving the reaction with O2 forming detectable HO2 radicals as the dominant nitrate-RO loss. The good agreement between predicted detectable RO2 and measured RO2 concentration, confirms the dominance of HO2 formation, in agreement with the slow decomposition rate as calculated by theory (Table 2).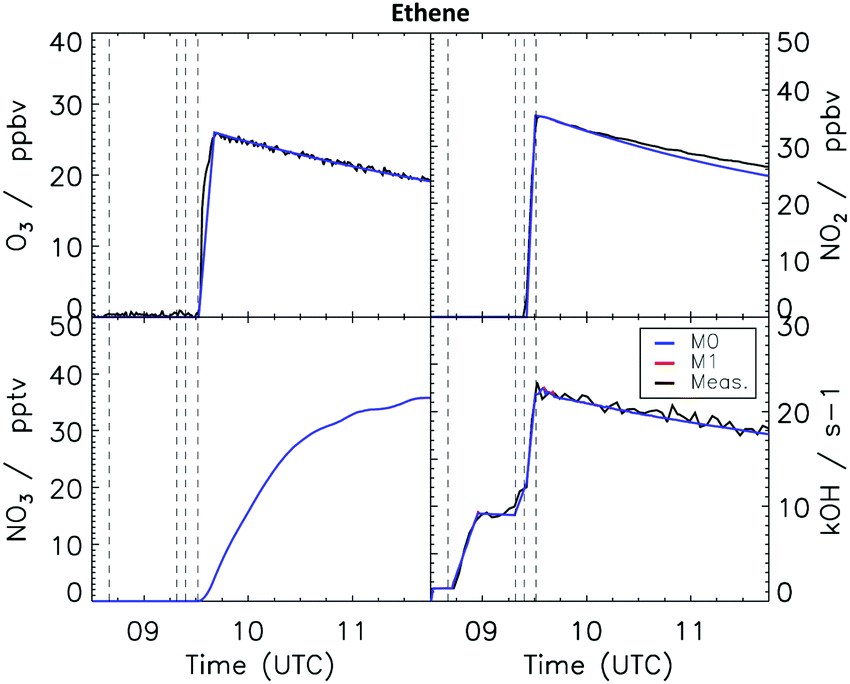 | ||
| Fig. 5 Comparison of modelled and measured trace gases for the ethene experiment. No measurements were available for the NO3 radicals. Vertical dashed lines indicate the times when different species where injected in the SAPHIR chamber. No difference is observed when using the updated MCM v3.3.1 (M0) or the modified model M1 including formation of alkoxy radical from the reaction between nitrate-RO2 and HO2 radicals. | ||
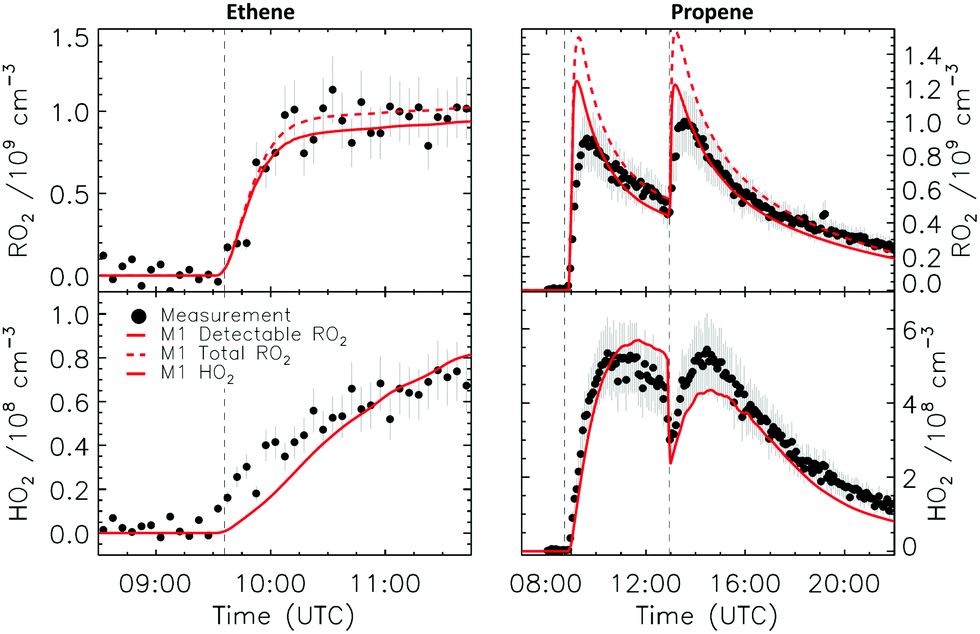 | ||
| Fig. 6 Comparison of modelled and measured RO2 and HO2 radicals for the ethene and propene experiments. Vertical dashed lines indicate the times when reactants were added. | ||
4.4 Propene
Fig. 6 shows measured and modelled RO2 and HO2 radicals for the propene experiments. Similarly to the ethene experiment, only a small difference between the total RO2 and the detectable RO2 radicals is predicted by the model (∼10%). Propene has two distinguishable NO3 addition sites, and thus leads to two distinct nitrate-RO2 and hence two nitrate-RO radicals in the converter. Of these nitrate-RO, 1-ONO2-2-propoxy is formed with a yield of 0.35 as predicted by the MCM v3.3.1, and has a calculated rate of decomposition at 298 K of 1.3 × 103 s−1 (Table 2), directly comparable to the pseudo-first order rate for H-abstraction by O2, 1.2 × 103 s−1, in the converter. Compared to ethene, a larger discrepancy between total RO2 and detectable RO2 is therefore to be expected as decomposition forms NO2 instead of HO2 radicals. The other nitrate-RO radical, 2-ONO2-1-propoxy, decomposes slower (Table 2) and thus yields more HO2 (Table 1). Despite the larger contribution of NO2versus HO2 formation in the nitrate-RO radical chemistry in the converter compared to ethene, the difference between total and detectable RO2 in the experiment still remains rather limited, as the reaction between NO3 radical and propene contributes only 35% of the propene chemical loss, with the bulk of the RO2 radicals comprising detectable RO2 arising from the ozone-initiated oxidation of propene. A somewhat larger uncertainty than for the other experiments analyzed pertains to the propene experiment due to the lack of direct measurement of the propene concentration combined with a failure of the kOH instrument. The propene concentration was instead estimated by matching the observed and modelled decay of O3, where ozone loss with NO2 is well captured by the model simulation, and reaction with propene is the only other important loss for O3 (Fig. S12, ESI‡). Using this method, the propene concentration could be estimated at 110 ppbv for both injections, respectively, carrying a ±15% uncertainty. Model sensitivity calculations performed using the lower and upper limiting concentrations showed a negligible impact of this uncertainty on the model-estimated concentration of HO2 and RO2 radicals.4.5 cis-2-Butene
Fig. 7 shows the comparison between measured and modelled RO2 and HO2 radicals for the 2-butene experiment. Modelled detectable RO2 radical concentration are in very good agreement (within 10%) with the experimentally observed RO2 radicals, while the predicted total RO2 radical concentrations are a factor of two larger than the measurement. This result is in very good agreement with the theoretical calculations. The calculated decomposition rate for 3-ONO2-2-butoxy at 298 K is 4.6 × 104 s−1 (Table 2), roughly one order of magnitude faster than the pseudo-first-order reaction with oxygen in the converter, and the LIF instrument is not able to detect most of the nitrate-RO2 (Table 1). In the conditions of the experiment within this study, 2-ONO2-3-butylperoxy radicals constitutes ∼50% of the total RO2 radicals formed and therefore the detected RO2 should be roughly 50% of the total RO2 radical.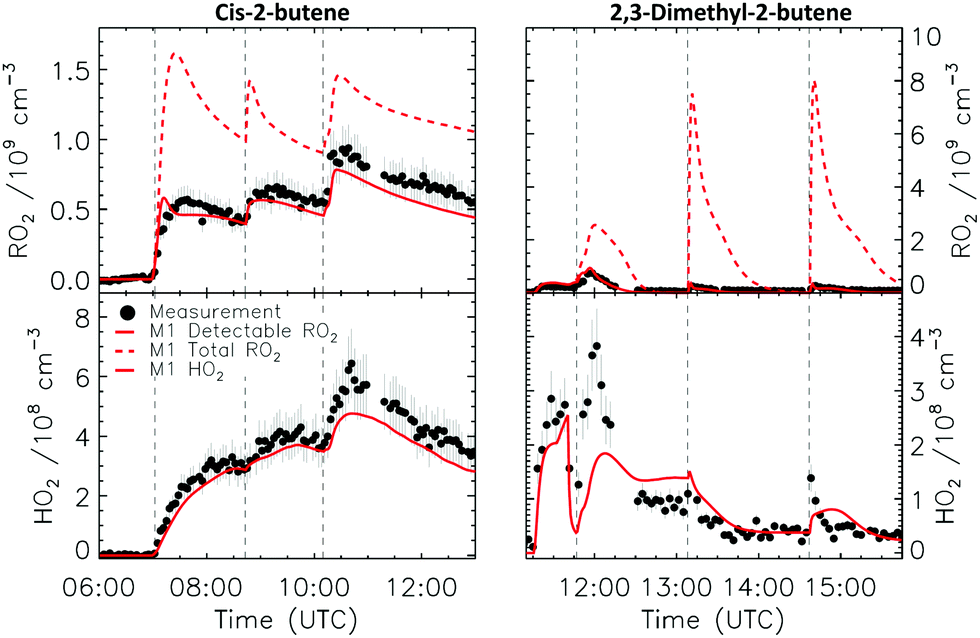 | ||
| Fig. 7 Comparison of modelled and measured RO2 and HO2 radicals for the cis-2-butene and 2,3-dimethyl-2-butene experiments. Vertical dashed lines indicate the times when reactants were added. | ||
For this experiment, cis-2-butene time series from the PTR-ToF-MS are available and shown in Fig. S13 (ESI‡). The good agreement observed between the model results and measurements gives additional confidence that the concentration of NO3 radicals is well represented within the model as both production species (NO2 and O3) and loss reactions (cis-2-butene) are well captured.
4.6 2,3-Dimethyl-2-butene
Fig. 7 shows the comparison between measured and modelled HO2 and RO2 radicals for the 2,3-dimethyl-2-butene experiment. A difference between the total and detectable RO2 radicals of almost one order of magnitude is predicted, with the latter in much better agreement with the measured RO2 radical concentrations (within 20%). The 3-ONO2-2,3-diMe-2-butoxy radicals formed in the converter during the 2,3-dimethyl-2-butene experiment is expected to decompose extremely rapidly, k(298 K) = 5.1 × 107 s−1 (Table 2) without formation of HO2 radicals, and is therefore undetectable by the LIF technique. Furthermore, due to the experimental conditions (Fig. S14, ESI‡) and the high rate coefficient predicted for the reaction between 2,3-dimethyl-2-butene and NO3 radicals (5.7 × 10−11 cm3 s−1 in the MCM v3.3.1), the nitrate-RO2 formed from this reaction are the dominant RO2 radical, and a large difference between total RO2 and observed RO2 is expected and observed.4.7 1-Pentene and trans-2-hexene
Fig. 8 compares the model results and measurement of RO2 and HO2 radicals for 1-pentene and trans-2-hexene. Owing to their longer chain, isomerization reactions are potentially competitive for the RO2 and RO radicals formed after reaction of the alkenes with both NO3 and OH radicals, and needed to be added to the model (see Section 2.4). For the 1-pentene experiment, a better agreement can be observed between measured and modelled RO2 radicals for the second injection when the impact of ozonolysis is limited (see Section 4.8). The dominant path for all nitrate-RO radicals formed after the reaction of the nitrate-RO2 radicals with NO is isomerization (1,5-H-migration), with high calculated rate coefficients in excess of 3 × 106 s−1 (Table 2). The RO2 radical formed after this H-migration, in the conditions of the LIF reactor and therefore in excess of NO, will again form alkoxy radicals (R14) which will likewise undergo rapid H-migration of the α-OH hydrogen atoms, with the resulting α-OH alkyl radical reacting very quickly with O2 to form HO2 radicals with a carbonyl coproduct. Migration of the α-ONO2 H-atom is much slower and is not competitive.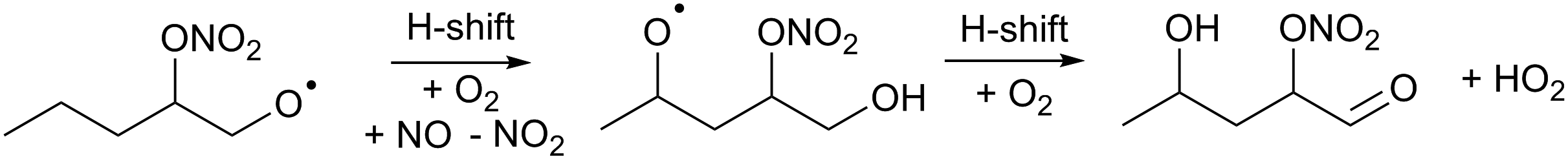 | (R14) |
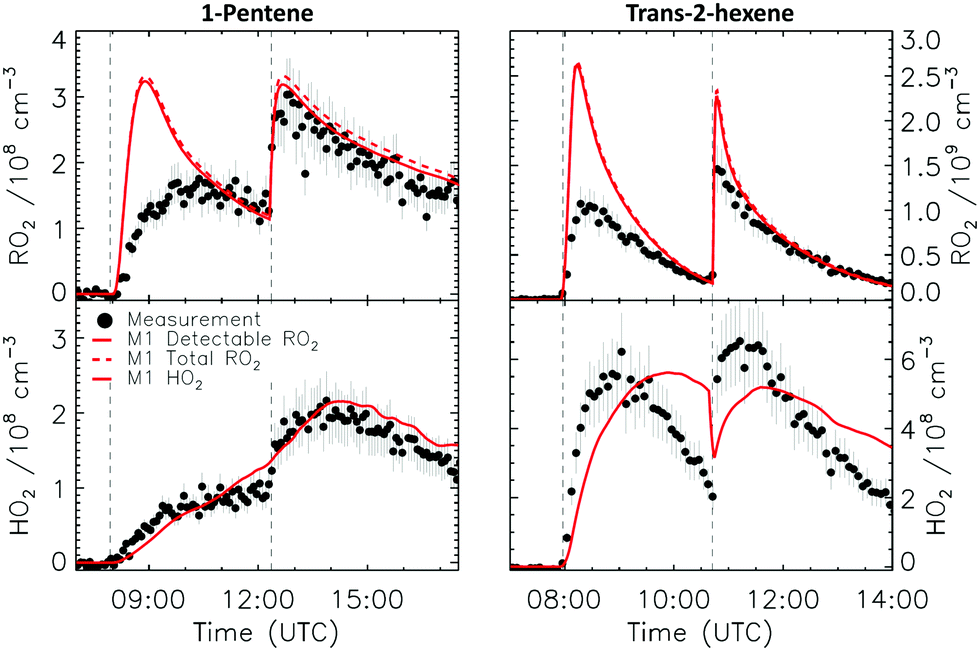 | ||
| Fig. 8 Comparison of modelled and measured RO2 and HO2 radicals for the 1-pentene and trans-2-hexene experiments. Vertical dashed lines indicate the times when additional reactants were added. | ||
This chemistry is shown in full in Fig. S9 and S11 (ESI‡) for 1-pentene and trans-2-hexene, respectively. Hence, the RO2 radicals in these systems should all be detectable. The small fraction of RO2 radicals which should not be detected originates from the CH2OO + NO2 reaction (see ESI‡ and Table 1), and lies well within the measurement uncertainty (5%).
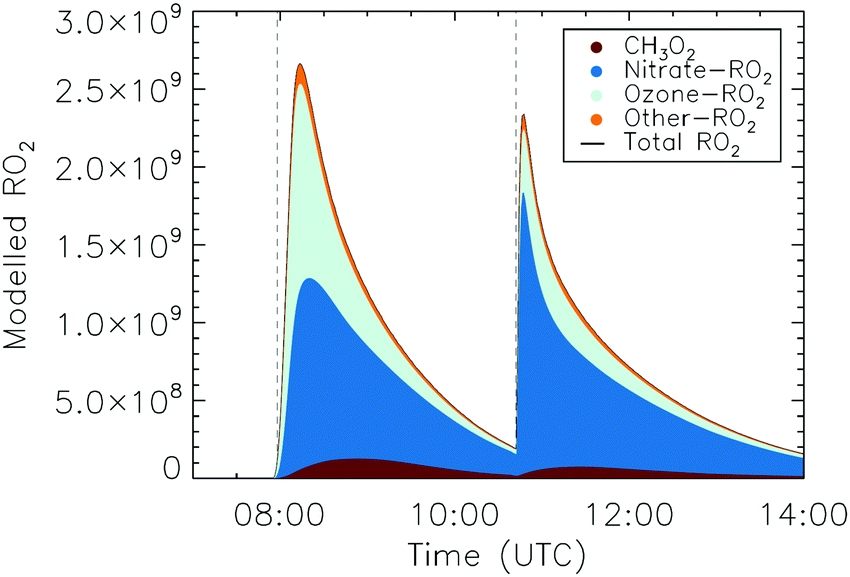
For the trans-2-hexene experiment (Fig. 8) the rate coefficient for the reaction with NO3 radicals needed to be increased by a factor 2 from the value currently used in the MCM v3.3.1 to obtain a good agreement between the measured and the modelled trans-2-hexene concentration decay (Fig. S16, ESI‡). The faster rate is responsible for the small peak in the nitrate-RO2 radicals as predicted for the second injection of trans-2-hexene (Fig. 8 and 9), but which is not observed in the measurement. Similarly to 1-pentene, a negligible difference between modelled detectable RO2 radicals and the measurement is observed, aside from this initial peak, confirming the theoretical calculations.
4.8 Remaining uncertainties
Due to the presence of O3 in the reaction system as a source of NO3 radicals in its reaction with NO2, the chemistry in the chamber is complex as it is initiated by NO3 but also by O3. The ozonolysis is important predominantly at the start of the experiment, or following an additional injection of ozone, while its impact is reduced at later times especially after the second alkene injection. Fig. 9 shows the expected speciation of the modelled RO2 radicals for the trans-2-hexene experiment, which highlights a peak for the first trans-2-hexene injection caused predominantly by RO2 radicals generated in the ozonolysis model, in particular RO2 radicals such as OCHCH2OO˙ and OCHCH(OO˙)CH2CH3 formed as co-products of OH in the so-called vinyl-hydroperoxide (VHP) channel. Most of the measurements (Fig. 6–8) show this extraneous peak in the RO2 traces at the start of the experiments. This indicates that the model is currently not able to properly describe ozonolysis chemistry, either due to an incorrect assignment of the products formed in the ozonolysis reaction (see ESI‡), or due to an incorrect implementation of the subsequent chemistry of the peroxy and alkoxy radicals formed from these products despite our efforts to improve the mechanism for ethanal-2-peroxy and butanal-2-peroxy radicals. As this work does not aim to improve the ozonolysis chemistry, we mostly ignore this initial overestimation of the RO2 concentration, and focus instead on the agreement between model and measurement in the later stages of the experiment, where the chemistry is mostly driven by NO3. The poor performance of the ozonolysis model chemistry still has some influence even at these later reaction stages, and is one of the sources of the remaining discrepancies between model and measurement.
 | ||
| Fig. 9 Time-dependent RO2 concentrations in the trans-2-hexene experiment, lumped by their source from nitrate- or ozone-initiated chemistry. Vertical dashed lines indicate the times when reactants were added. | ||
There is also a discrepancy observed at the second injection of trans-2-hexene, where the model shows a sharp increase in RO2 concentration that is more dampened in the observations, and is not due solely to ozone-derived RO2. This suggests that the chemistry in the chamber seems to be delayed compared to what the model predicts at times of drastic reactant concentrations. The reason for this discrepancy is unclear as perfect mixing is achieved in the chamber within two minutes, and the time resolution of the RO2 radical LIF, ∼180 seconds, is sufficiently fine such that the peak should be observable. The shape of the concentration time profiles suggests the temporary formation of reservoir species at times of high reactant concentrations, but despite deeper examination of species such as N2O5 or PANs in the model the issue could not be resolved at this time.
Finally, it should be noted that our model is based on purely a priori theoretical predictions for the dominant alkoxy radicals, and otherwise relies only on literature data, SARs, and the MCM v3.1.1 implementation for most of the remaining chemistry, without fitting the kinetic parameters in the model to the observations (except the unknown OH yields for β-nitrate-RO2 + HO2, and the NO3 + trans-2-hexene rate coefficient). An optimization of some of the rate coefficients would lead to even better agreement between observations and model; however, the values thus updated would likely remain well within the uncertainty of the predictions and observations, and would then not lead to improved chemical understanding.
5. Comparison to literature experimental data
To our knowledge, no direct measurements of nitrate-RO or epoxy-RO decomposition or isomerisation reactions are available in the literature. Yeh et al.18 describe an experiment on NO3-initiated oxidation of 1-pentadecene, where the products are collected on filters and analyzed. Based on the observed products, they conclude that isomerisation and decomposition reactions of nitrate-RO radicals are comparable in rate, while the expectation from SARs is that H-migration is significantly faster. They propose that the decomposition SAR by Vereecken and Peeters13 is incorrect, overestimating the barrier heights for β-ONO2-substituted alkoxy radicals by ∼3.7 kcal mol−1; this reduction in barrier height would allow decomposition to be competitive against H-migration as predicted by the SAR by Vereecken and Peeters.59The current theoretical results remain in clear disagreement with the result of Yeh et al.18 The higher-level calculations presented in this work support the barrier heights for decomposition predicted by the SAR by Vereecken and Peeters,13 and even find that they should be slightly higher than the predictions by this SAR for 1-ONO2-2-pentadecoxy. While we did not examine pentadecene-derived alkoxy radicals directly due to the computational cost, the H-migration rates in 1-pentene and 2-hexene-derived nitrate-RO remain in agreement with the H-migration SAR by Vereecken and Peeters,59 dominating over the decomposition by several orders of magnitude. Though our experiments do not directly measure absolute rate coefficients, the experimental data presented in this work likewise finds rate coefficient comparable to the theoretical predictions. Specifically, the ratio of NO2versus HO2 formation from the alkoxy radicals in the converter probes the relative rate of alkoxy decomposition, isomerisation, and reaction of the alkoxy radical with O2, where the latter is generally considered to be similar for all alkoxy radicals,19,20 and thus acts as the reference rate.
To illustrate the impact of the faster decomposition rate proposed by Yeh et al., we performed model simulations replacing our theory-predicted decomposition rates with values based on their findings. In this sensitivity study, we set decomposition for nitrate-RO from 1-pentene and 2-hexene to a rate 2.5 times faster than the theory-predicted isomerisation rate, mimicking the 0.397 ratio of isomerisation to decomposition by Yeh et al. For propene, we calculated decomposition rate coefficients from the SAR by Vereecken and Peeters13 with an additional 3.7 kcal mol−1 reduction in barrier height as proposed by Yeh et al.18 As this strong increase in rate also changes the competition of decomposition against the RO + O2 reaction and hence the HO2 balance in the chamber, model runs were performed with the HO2 constrained to the measurements, to avoid secondary effects. The results are depicted in Fig. 10. Using the faster decomposition proposed by Yeh et al., the RO2 radicals in the propene experiment would then yield very little HO2 in the converter (Table S5, ESI‡) as decomposition would overwhelm reaction with O2, in disagreement with our observations. In a similar vein, if the rate of isomerisation and decomposition would be of similar magnitude as proposed by Yeh et al., more than half of the nitrate-RO2 radicals in the 1-pentene experiment would not be observable in the converter, whereas the observation requires that virtually all RO2 radicals are converted to HO2. The more competitive Yeh et al. decomposition rate for nitrate-RO from hexene would likewise mask most of the RO2 from detection as HO2 in the converter, whereas the observations support HO2 formation by isomerisation to remain the dominant channel as in 1-pentene, despite the additional methyl group in hexene accelerating alkoxy decomposition.
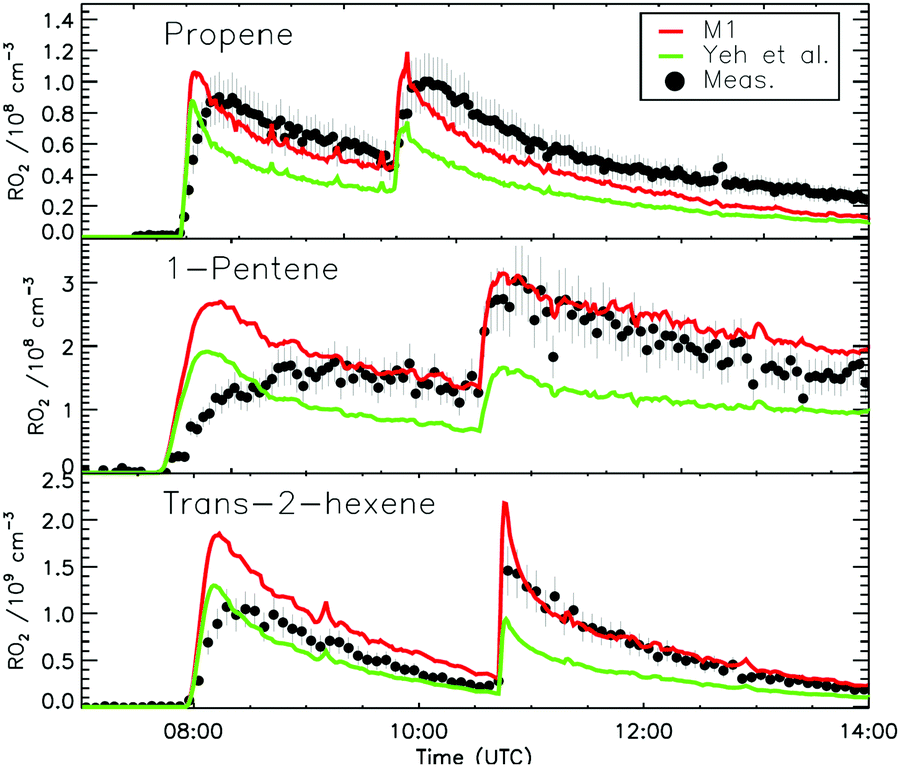 | ||
| Fig. 10 Comparison of modelled detectable RO2 and measured RO2 radicals for the propene, 1-pentene and trans-2-hexene experiments in model M1 or when considering the fast decomposition rate for the nitrate-RO radicals as suggested by Yeh et al.18 The HO2 radical concentration is constrained to the measurement within all model runs shown. | ||
It is evident that the fast decomposition rate for the nitrate-RO as suggested by Yeh et al. results in a modelled detectable RO2 radical concentration which is too small and underestimates the measured RO2 radicals for all the alkenes during those times when the chemistry is dominated by the NO3 radicals, such as after the second VOC injection. The theoretically calculated rates in this study as used in M1 result in a much better agreement with the measurement. As theoretical work is also in agreement with other experimental data on alkoxy radical decomposition and isomerisation, it appears that reducing the barrier height in the nitrate-RO decomposition SAR may not be the most appropriate solution in bringing agreement between the SARs and the Yeh et al. experiments. The complexity of the Yeh et al. experiments, involving product capture on filters and increased temperatures during their analysis, makes it very difficult to assess why their observations are so different from our combined theoretical and experimental study. We can’t exclude that the very long aliphatic chains in the Yeh et al. experiment, C15, exhibit reaction pathways that do not exist for shorter chains, ≤C6.
6. Implications for ambient LIF measurements of RO2 during night time
The NO3 radical is often the dominant oxidant during nighttime due to its faster rate coefficient with alkenes as compared to ozone. This is true for both pristine6,84,85 and more polluted environments.12,86 Therefore, the inability of the LIF technique to detect certain nitrate-RO2 radicals has repercussion in the way models and measurements need to be compared to correctly assess the nighttime chemistry of RO2 radicals.Concentrations of RO2 radicals measured with LIF instruments are reported for two megacities, i.e. London, UK,30 and Beijing, China,28,87,88 and for two rural locations in China.29,89 It is not easy to incorporate the findings from the current study to the provided interpretation of those campaign observations. Some of those studies28,29 utilize lumped models (e.g. RACM) where the chemistry initiated by NO3 radicals is not very detailed, making it difficult to assess the performances of the LIF instrument. The studies in Beijing87,88 were characterized by very high NO (up to 100 ppbv) concentrations also in the night, which decreased the lifetime of both NO3 and RO2 radicals. The study in London30 is a good example where during several nights with high NO3 reactivity the used model (MCM v3.2) predicted very high concentrations of RO2 radicals, almost an order of magnitude higher than what was measured by the LIF instrument. The authors argued that the sensitivity of the LIF towards nitrate-RO2 radicals from ethene and propene might be lower than unity as they might not produce an HO2 radical for each alkoxy radical formed. Indeed, this suggestion is broadly in agreement with the results in this study, although we find that the large difference observed would not arise from ethene or propene but rather be due to other alkenes whose nitrate-RO decompose fast. Examples include 2,3-dimethyl-2-butene and cis-2-butene, where the latter was measured during the campaign (∼20 ppt). Larger concentrations of isoprene and α-pinene (∼200 ppt for both) were also observed and applying the SAR provided in this study suggests that the nitrate-RO2 radicals originating from isoprene would not be fully detectable in the ROxLIF instrument.
7. Discussion and conclusions
In this study, we described a combined theoretical, experimental, and modeling study of the fate of nitrated alkoxy radicals under atmospheric conditions. The high-level theoretical results find that the nitrate group slows down alkoxy radical decomposition when positioned on the β-carbon of the breaking C–C bond, compared to earlier data by Vereecken and Peeters.13 The main mechanism behind this hindrance is the partial decomposition of the –ONO2 moiety in the transition state for alkoxy decomposition, with partial formation of a carbonyl functionality. This feature hampers the ability of other β-substituents to stabilize the forming alkyl radical moiety. The alkoxy decomposition towards a β-ONO2 group is always followed by a direct secondary decomposition of the nitrate group, forming a carbonyl group and the NO2 product. The structure–activity relationship by Vereecken and Peeters13 was amended with a second set of parameters for use when a β-ONO2 substituent is present, incorporating the interaction effects. The SAR was also extended to cover α- and β-epoxy-substituents, following the observation that epoxidation could be a competitive channel for unsaturated nitrate-RO due to their slower decomposition. For all other alkoxy decomposition reactions, the SAR was found to be in excellent agreement with the current set of high-level calculations, typically with rate coefficients within the SAR uncertainty of a factor ∼10. The SAR for H-migration in alkoxy radicals by Vereecken and Peeters59 remains in similarly good agreement with the current theoretical set of data.The experimental RO2 radicals data collected in chamber experiments could be well reproduced by a model incorporating the rate of decomposition and isomerization for the nitrate alkoxy radicals as calculated in this study. It should be stressed that the model utilized in this study was based only on currently available literature data and our theoretical results. Specifically, no fitting optimization was performed on the key kinetic parameters in the model, and only the OH yield in the β-nitrate-RO2 + HO2 reaction and the rate coefficient for the trans-2-hexene + NO3 reaction needed to be quantified based on the observations. An alternative approach constraining HO2 in the model to the experimental measurements yielded very similar results. The model agreement to the observations was within the instrumental uncertainties for ethene, propene, cis-2-butene, and 2,3-dimethyl-2-butene. The agreement between the model and observations in the 2-pentene and trans-2-hexene experiments was somewhat less good particularly at the start of the experiment where the RO2 concentration is dominated by products formed from ozonolysis rather than NO3 chemistry. The chemical model here is more complex due to the introduction of the chemistry following the isomerization reactions for the formed RO2 radicals, and is likely less reliable as we have no direct theoretical or experimental results but rather rely on SAR predictions. Although isomerisation by H-migration is not the dominant loss for the RO2 radicals produced from the oxidation by NO3 and OH radicals, the current predictions suggest it dominates the loss of the OCHCH(OO˙)CH2CH3 and OCHCH2(OO˙) radicals formed in the vinyl-hydroperoxide channels of the ozonolysis reactions, and should be a general feature after OH formation in many ozonolysis reactions. For both the 1-pentene and trans-2-hexene experiments, the concentration of RO2 radicals at the beginning of the first VOC degradation period sees a contribution of up to ∼40% from this latter radical and its descendants, and constitutes the dominant driver for the disagreement between experiment and model. Once the ozone concentration decreases and NO3 increases, the nitrate-RO2 radicals become a larger fraction of the total RO2 radicals and good agreement between measured and modelled RO2 radicals can be observed. This strongly suggests that the chemistry of the ozone-derived RO2, and their behavior in the LIF converter, is not fully understood yet and is not correctly reflected by the current model. As this study focuses on the chemistry of nitrate alkoxy, no additional effort was expended on elucidating the ozone-derived RO2 chemistry at this time. Overall, then, we find that the model describes the nitrate-RO2 and nitrate-RO chemistry well in the chamber and converter across a wide range of compounds, with good prediction of the impact of the nitrate-RO chemistry on the LIF detection. The experimental data supports the theoretical results, with a slow decomposition rate for the smallest nitrate-RO, accelerating rapidly as more substituents are added on the breaking bond. For nitrate-RO large enough to undergo H-migration reactions, this isomerisation become strongly dominant over the decomposition, shifting the fate of the nitrate-RO in the converter from NO2 formation to HO2 formation. This reactivity trend is summarized in Fig. 11. The good correspondence did not require any tuning of the rate coefficients, though small adjustments within the uncertainties of the theoretical data or literature sources would lead to even better results.
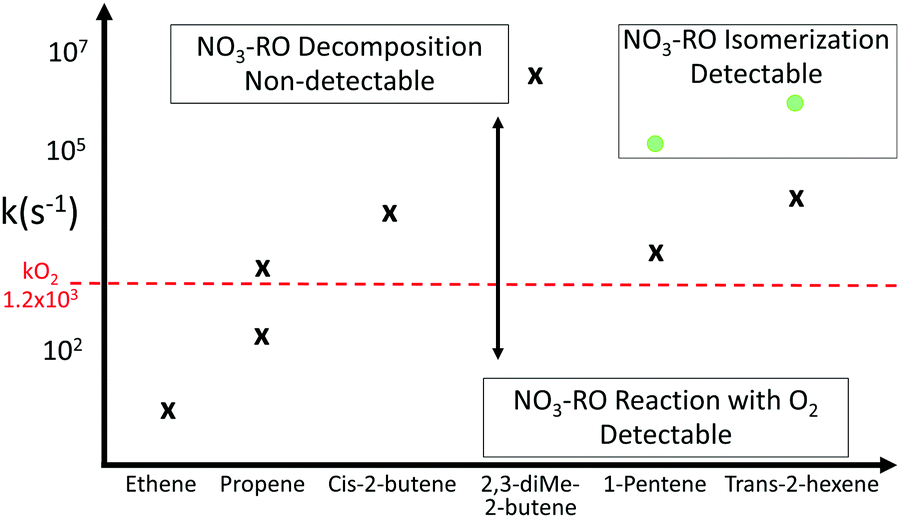 | ||
| Fig. 11 Visualization of the reactivity trend across a range of β-nitrate-RO radicals, showing the rate of decomposition and isomerisation against the reaction with O2 at the pressure in the converter. Fast decomposition reactions yield NO2, (partially) masking the nitrate-RO2 from detection in the ROxLIF instrument; if reaction with O2 or isomerisation is dominant, HO2 is formed and the nitrate-RO2 can be detected by ROxLIF. | ||
We also incorporated the reaction of β-nitrate-RO2 radicals with HO2 partially forming OH and β-nitrate-RO radicals, as earlier described in the literature for isoprene- and α-pinene derived RO2. Comparison between model and observations suggests this channel is critical for a correct description of the HOx chemistry for all compounds due to the excess of CO at which the experiments were performed. As no reliable data are available, we optimized the OH yields in the β-nitrate-RO2 + HO2 reaction, finding values ranging from 0.15 to 0.65 depending on the VOC investigated. The yields are comparable to the value of 22 to 85% suggested by literature data5,12,17,26,76 for isoprene- and α-pinene-derived RO2 (Table S3, ESI‡).
The current set of experimental and theoretical data is in disagreement with the experimental data by Yeh et al.,18 who concluded that the decomposition and isomerisation of aliphatic nitrate-RO have comparable rates, and that the alkoxy radicals SARs by Vereecken and Peeters13,59 thus strongly underestimate the rate of decomposition relative to isomerisation. Our current set of high-level theoretical results still finds that decomposition is slower, and that isomerisation dominates over decomposition, in agreement with the original SARs. Likewise, the experimental data is in agreement with the current predicted rate coefficients for decomposition and isomerisation, and provides a direct probe of the relative rates of alkoxy decomposition, H-migration, and reaction with O2. The good agreement between theory and experiment strongly supports our current results. In contrast, implementing the suggestion by Yeh et al. to reduce the barriers for decomposition such that the latter reaction is competitive against isomerisation leads to systematic underestimation of the LIF signal relative to our direct experimental observations. It is unclear why the Yeh et al. results deviate from the collective experimental and theoretical data set on alkoxy radical chemistry. We refrain from speculating at this time as the Yeh et al. experiments are complex, requiring several steps to analyze the results, and without additional data we cannot determine where the observations start to deviate. We recommend the use of the updated SAR parameters in this work for the prediction of nitrate-RO decomposition rates.
The results found in this work have several important atmospheric implications. Firstly, we find that the nitrate-RO decompose slower than similarly substituted alkoxy radicals formed from the OH-initiated oxidation of alkenes. This reduces the impact of chain fragmentation, and hence retains longer carbon chains with lower vapor pressure. Combined with the oxygenated nitrate group, this suggests that nighttime chemistry initiated by the NO3 radical could be more amenable to the formation and growth of highly oxygenated molecules (HOMs) and aerosols. A second implication is that field measurements of RO2 radicals formed in NO3-initiated chemistry performed with LIF instruments might underestimate the RO2 radical concentration, and a careful investigation of the VOC present will be needed to correctly compare measurements and modelled results. Due to the sparse number of campaigns including RO2 radical measurements and their stronger focus on the daytime chemistry, it is hard to assess the impact of this study on previous campaigns. Future field campaigns with measurements of RO2 radicals by LIF should be performed in environments with high loads of unsaturated VOC, such as isoprene, and high concentration of NO3 radicals, to validate the current findings in a natural environment. Finally, the current results indicate that the NO3-initiated atmospheric oxidation of several important biogenic VOCs such as isoprene and monoterpenes could be different from what is currently described in the literature. To explore the implications, we will examine the chemistry of alkoxy and alkylperoxy radicals from the NO3-initiated oxidation of isoprene in an upcoming publication.82
Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 Research and Innovation Program (SARLEP grant agreement no. 681529).References
- B. J. Finlayson-Pitts and J. N. Pitts, Chemistry of the Upper and Lower Atmosphere: Theory, Experiments, and Applications, Academic Press, San Diego, 1999 Search PubMed.
- S. S. Brown and J. Stutz, Nighttime radical observations and chemistry, Chem. Soc. Rev., 2012, 41, 6405–6447 RSC.
- J. Lelieveld, S. Gromov, A. Pozzer and D. Taraborrelli, Global tropospheric hydroxyl distribution, budget and reactivity, Atmos. Chem. Phys., 2016, 16, 12477–12493 CrossRef CAS.
- R. P. Wayne, I. Barnes, P. Biggs, J. P. Burrows, C. E. Canosa-Mas, J. Hjorth, G. Le Bras, G. K. Moortgat, D. Perner, G. Poulet, G. Restelli and H. Sidebottom, The nitrate radical: Physics, chemistry, and the atmosphere, Atmos. Environ., 1991, 25, 1–203 CrossRef.
- P. O. Wennberg, K. H. Bates, J. D. Crounse, L. G. Dodson, R. C. McVay, L. A. Mertens, T. B. Nguyen, E. Praske, R. H. Schwantes, M. D. Smarte, J. M. St Clair, A. P. Teng, X. Zhang and J. H. Seinfeld, Gas-Phase Reactions of Isoprene and Its Major Oxidation Products, Chem. Rev., 2018, 118, 3337–3390 CrossRef CAS.
- J. Liebmann, E. Karu, N. Sobanski, J. Schuladen, M. Ehn, S. Schallhart, L. Quelever, H. Hellen, H. Hakola, T. Hoffmann, J. Williams, H. Fischer, J. Lelieveld and J. N. Crowley, Direct measurement of NO3 radical reactivity in a boreal forest, Atmospheric, Chem. Phys., 2018, 18, 3799–3815 CAS.
- A. B. Guenther, X. Jiang, C. L. Heald, T. Sakulyanontvittaya, T. Duhl, L. K. Emmons and X. Wang, The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 2012, 5, 1471–1492 CrossRef.
- R. Atkinson, Kinetics and Mechanisms of the Gas Phase Reactions of the NO3 Radical with Organic Compounds, J. Phys. Chem. Ref. Data, 1991, 20, 459–507 CrossRef CAS.
- IUPAC Subcommittee on Atmospheric Chemical Kinetic Data Evaluation, Evaluated Kinetic Data, IUPAC, 2017.
- A. Mellouki, G. Le Bras and H. Sidebottom, Kinetics and Mechanisms of the Oxidation of Oxygenated Organic Compounds in the Gas Phase, Chem. Rev., 2003, 103, 5077–5096 CrossRef CAS.
- NASA Panel for Data Evaluation, 2017.
- R. H. Schwantes, A. P. Teng, T. B. Nguyen, M. M. Coggon, J. D. Crounse, J. M. St Clair, X. Zhang, K. A. Schilling, J. H. Seinfeld and P. O. Wennberg, Isoprene NO3 Oxidation Products from the RO2 + HO2 Pathway, J. Phys. Chem. A, 2015, 119, 10158–10171 CrossRef CAS.
- L. Vereecken and J. Peeters, Decomposition of substituted alkoxy radicals—part I: a generalized structure–activity relationship for reaction barrier heights, Phys. Chem. Chem. Phys., 2009, 11, 9062–9074 RSC.
- L. Vereecken, Computational study of the stability of α-nitroxy-substituted alkyl radicals, Chem. Phys. Lett., 2008, 466, 127–130 CrossRef CAS.
- L. Vereecken, B. Aumont, I. Barnes, J. W. Bozzelli, M. J. Goldman, W. H. Green, S. Madronich, M. R. McGillen, A. Mellouki, J. J. Orlando, B. Picquet-Varrault, A. R. Rickard, W. R. Stockwell, T. J. Wallington and W. P. L. Carter, Perspective on Mechanism Development and Structure-Activity Relationships for Gas-Phase Atmospheric Chemistry, Int. J. Chem. Kinet., 2018, 50, 435–469 CrossRef CAS.
- M. P. Perez-Casany, I. Nebot-Gil and J. Sanchez-Marin, Ab initio study on the mechanism of tropospheric reactions of the nitrate radical with alkenes: Propene, J. Phys. Chem. A, 2000, 104, 6277–6286 CrossRef CAS.
- T. Kurtén, K. H. Møller, T. B. Nguyen, R. H. Schwantes, P. K. Misztal, L. Su, P. O. Wennberg, J. L. Fry and H. G. Kjaergaard, Alkoxy Radical Bond Scissions Explain the Anomalously Low Secondary Organic Aerosol and Organonitrate Yields From alpha-Pinene + NO3, J. Phys. Chem. Lett., 2017, 8, 2826–2834 CrossRef.
- G. K. Yeh, M. S. Claflin and P. J. Ziemann, Products and Mechanism of the Reaction of 1-Pentadecene with NO3 Radicals and the Effect of a -ONO2 Group on Alkoxy Radical Decomposition, J. Phys. Chem. A, 2015, 119, 10684–10696 CrossRef CAS.
- R. Atkinson, Rate constants for the atmospheric reactions of alkoxy radicals: An updated estimation method, Atmos. Environ., 2007, 41, 8468–8485 CrossRef CAS.
- R. Atkinson, Gas-phase tropospheric chemistry of organic compounds: a review, Atmos. Environ., 2007, 41(Supplement), 200–240 CrossRef.
- T. S. Dibble and J. Chai, in Advances in Atmospheric Chemistry: Volume 2: Organic Oxidation and Multiphase Chemistry, ed. J. R. Barker, A. L. Steiner and T. J. Wallington, World Scientific Publishing Co. Pte. Ltd, Singapore, 1st edn, 2019, vol. 1, pp. 377–527 Search PubMed.
- J. J. Orlando, G. S. Tyndall and T. J. Wallington, The Atmospheric Chemistry of Alkoxy Radicals, Chem. Rev., 2003, 103, 4657–4690 CrossRef CAS.
- L. Vereecken and J. S. Francisco, Theoretical studies of atmospheric reaction mechanisms in the troposphere, Chem. Soc. Rev., 2012, 41, 6259–6293 RSC.
- T. Nah, J. Sanchez, C. M. Boyd and N. L. Ng, Photochemical Aging of alpha-pinene and beta-pinene Secondary Organic Aerosol formed from Nitrate Radical Oxidation, Environ. Sci. Technol., 2016, 50, 222–231 CrossRef CAS.
- A. E. Perring, S. E. Pusede and R. C. Cohen, An Observational Perspective on the Atmospheric Impacts of Alkyl and Multifunctional Nitrates on Ozone and Secondary Organic Aerosol, Chem. Rev., 2013, 113, 5848–5870 CrossRef CAS.
- A. W. Rollins, A. Kiendler-Scharr, J. L. Fry, T. Brauers, S. S. Brown, H.-P. Dorn, W. P. Dube, H. Fuchs, A. Mensah, T. F. Mentel, F. Rohrer, R. Tillmann, R. Wegener, P. J. Wooldridge and R. C. Cohen, Isoprene oxidation by nitrate radical: alkyl nitrate and secondary organic aerosol yields, Atmospheric, Chem. Phys., 2009, 9, 6685–6703 CAS.
- N. L. Ng, S. S. Brown, A. T. Archibald, E. Atlas, R. C. Cohen, J. N. Crowley, D. A. Day, N. M. Donahue, J. L. Fry, H. Fuchs, R. J. Griffin, M. I. Guzman, H. Herrmann, A. Hodzic, Y. Iinuma, J. L. Jimenez, A. Kiendler-Scharr, B. H. Lee, D. J. Luecken, J. Mao, R. McLaren, A. Mutzel, H. D. Osthoff, B. Ouyang, B. Picquet-Varrault, U. Platt, H. O. T. Pye, Y. Rudich, R. H. Schwantes, M. Shiraiwa, J. Stutz, J. A. Thornton, A. Tilgner, B. J. Williams and R. A. Zaveri, Nitrate radicals and biogenic volatile organic compounds: oxidation, mechanisms, and organic aerosol, Atmospheric, Chem. Phys., 2017, 17, 2103–2162 CAS.
- Z. Tan, F. Rohrer, K. Lu, X. Ma, B. Bohn, S. Broch, H. Dong, H. Fuchs, G. I. Gkatzelis, A. Hofzumahaus, F. Holland, X. Li, Y. Liu, Y. Liu, A. Novelli, M. Shao, H. Wang, Y. Wu, L. Zeng, M. Hu, A. Kiendler-Scharr, A. Wahner and Y. Zhang, Wintertime photochemistry in Beijing: observations of ROx radical concentrations in the North China Plain during the BEST-ONE campaign, Atmos. Chem. Phys., 2018, 18, 12391–12411 CrossRef CAS.
- Z. Tan, H. Fuchs, K. Lu, A. Hofzumahaus, B. Bohn, S. Broch, H. Dong, S. Gomm, R. Haeseler, L. He, F. Holland, X. Li, Y. Liu, S. Lu, F. Rohrer, M. Shao, B. Wang, M. Wang, Y. Wu, L. Zeng, Y. Zhang, A. Wahner and Y. Zhang, Radical chemistry at a rural site (Wangdu) in the North China Plain: observation and model calculations of OH, HO2 and RO2 radicals, Atmos. Chem. Phys., 2017, 17, 663–690 CrossRef CAS.
- L. K. Whalley, D. Stone, R. Dunmore, J. Hamilton, J. R. Hopkins, J. D. Lee, A. C. Lewis, P. Williams, J. Kleffmann, S. Laufs, R. Woodward-Massey and D. E. Heard, Understanding in situ ozone production in the summertime through radical observations and modelling studies during the Clean air for London project (ClearfLo), Atmos. Chem. Phys., 2018, 18, 2547–2571 CrossRef CAS.
- H. Fuchs, F. Holland and A. Hofzumahaus, Measurement of tropospheric RO2 and HO2 radicals by a laser-induced fluorescence instrument, Rev. Sci. Instrum., 2008, 79, 084104 CrossRef.
- T. H. Dunning, Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- G. D. Purvis and R. J. Bartlett, A full coupled-cluster singles and doubles model: The inclusion of disconnected triples, J. Chem. Phys., 1982, 76, 1910 CrossRef CAS.
- I. M. Alecu, J. Zheng, Y. Zhao and D. G. Truhlar, Computational Thermochemistry: Scale Factor Databases and Scale Factors for Vibrational Frequencies Obtained from Electronic Model Chemistries, J. Chem. Theory Comput., 2010, 6, 2872–2887 CrossRef CAS.
- J. L. Bao, J. Zheng, I. M. Alecu, B. J. Lynch, Y. Zhao and D. G. Truhlar, Database of Frequency Scale Factors for Electronic Model Chemistries (Version 4), http://comp.chem.umn.edu/freqscale/index.html.
- A. D. Becke, Density-functional thermochemistry. II. The effect of the Perdew–Wang generalized-gradient correlation correction, J. Chem. Phys., 1992, 97, 9173–9177 CrossRef CAS.
- J.-D. Chai and M. Head-Gordon, Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- L. Goerigk, A. Hansen, C. Bauer, S. Ehrlich, A. Najibi and S. Grimme, A look at the density functional theory zoo with the advanced GMTKN55 database for general main group thermochemistry, kinetics and noncovalent interactions, Phys. Chem. Chem. Phys., 2017, 19, 32184–32215 RSC.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the Damping Function in Dispersion Corrected Density Functional Theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- J. A. Montgomery, M. J. Frisch, J. W. Ochterski and G. A. Petersson, A complete basis set model chemistry. VII. Use of the minimum population localization method, J. Chem. Phys, 2000, 112, 6532–6542 CrossRef CAS.
- H. S. Yu, X. He, S. L. Li and D. G. Truhlar, MN15: A Kohn-Sham global-hybrid exchange-correlation density functional with broad accuracy for multi-reference and single-reference systems and noncovalent interactions, Chem. Sci., 2016, 7, 5032–5051 RSC.
- Y. Zhao, N. Gonzalez-Garcia and D. G. Truhlar, Benchmark database of barrier heights for heavy atom transfer, nucleophilic substitution, association, and unimolecular reactions and its use to test theoretical methods, J. Phys. Chem. A, 2005, 109, 2012–2018 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Eara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Mntgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision B.01, Gaussian Inc., Wallington CT, 2016 Search PubMed.
- D. G. Truhlar, B. C. Garrett and S. J. Klippenstein, Current Status of Transition-State Theory, J. Phys. Chem., 1996, 100, 12771–12800 CrossRef CAS.
- L. Vereecken and J. Peeters, The 1,5-H-shift in 1-butoxy: A case study in the rigorous implementation of transition state theory for a multirotamer system, J. Chem. Phys., 2003, 119, 5159–5170 CrossRef CAS.
- C. Eckart, The penetration of a potential barrier by electrons, Phys. Rev., 1930, 35, 1303–1309 CrossRef CAS.
- H. S. Johnston and J. Heicklen, Tunneling corrections for unsymmetrical Eckart potential energy barriers, J. Phys. Chem., 1962, 66, 532–533 CrossRef.
- F. Rohrer, B. Bohn, T. Brauers, D. Bruning, F. J. Johnen, A. Wahner and J. Kleffmann, Characterisation of the photolytic HONO-source in the atmosphere simulation chamber SAPHIR, Atmos. Chem. Phys., 2005, 5, 2189–2201 CrossRef CAS.
- E. Schlosser, B. Bohn, T. Brauers, H.-P. Dorn, H. Fuchs, R. Haeseler, A. Hofzumahaus, F. Holland, F. Rohrer, L. O. Rupp, M. Siese, R. Tillmann and A. Wahner, Intercomparison of two hydroxyl radical measurement techniques at the atmosphere simulation chamber SAPHIR, J. Atmos. Chem., 2007, 56, 187–205 CrossRef CAS.
- B. Bohn and H. Zilken, Model-aided radiometric determination of photolysis frequencies in a sunlit atmosphere simulation chamber, Atmos. Chem. Phys., 2005, 5, 191–206 CrossRef CAS.
- F. Holland, A. Hofzumahaus, R. Schafer, A. Kraus and H. W. Patz, Measurements of OH and HO2 radical concentrations and photolysis frequencies during BERLIOZ, J. Geophys. Res.: Atmos., 2003, 108, 8246 CrossRef.
- H. Fuchs, B. Bohn, A. Hofzumahaus, F. Holland, K. D. Lu, S. Nehr, F. Rohrer and A. Wahner, Detection of HO2 by laser-induced fluorescence: calibration and interferences from RO2 radicals, Atmos. Meas. Tech., 2011, 4, 1209–1225 CrossRef CAS.
- R. S. Hornbrook, J. H. Crawford, G. D. Edwards, O. Goyea, R. L. Mauldin, J. S. Olson and C. A. Cantrell, Measurements of tropospheric HO2 and RO2 by oxygen dilution modulation and chemical ionization mass spectrometry, Atmos. Meas. Tech., 2011, 4, 735–756 CrossRef CAS.
- L. K. Whalley, M. A. Blitz, M. Desservettaz, P. W. Seakins and D. E. Heard, Reporting the sensitivity of laser-induced fluorescence instruments used for HO2 detection to an interference from RO2 radicals and introducing a novel approach that enables HO2 and certain RO2 types to be selectively measured, Atmos. Meas. Tech., 2013, 6, 3425–3440 CrossRef CAS.
- M. M. Lew, S. Dusanter and P. S. Stevens, Measurement of interferences associated with the detection of the hydroperoxy radical in the atmosphere using laser-induced fluorescence, Atmos. Meas. Tech., 2018, 11, 95–109 CrossRef.
- B. Nozière and L. Vereecken, Direct Observation of Aliphatic Peroxy Radical Autoxidation and Water Effects: an Experimental and Theoretical Study, Angew. Chem., Int. Ed., 2019, 58, 13976–13982 CrossRef.
- L. Vereecken and J. Peeters, A structure–activity relationship for the rate coefficient of H-migration in substituted alkoxy radicals, Phys. Chem. Chem. Phys., 2010, 12, 12608–12620 RSC.
- L. Vereecken and B. Nozière, H migration in peroxy radicals under atmospheric conditions, Atmos. Meas. Tech., 2020, 20, 7429–7458 CAS.
- H. Fuchs, S. Albrecht, I. Acir, B. Bohn, M. Breitenlechner, H.-P. Dorn, G. I. Gkatzelis, A. Hofzumahaus, F. Holland, M. Kaminski, F. N. Keutsch, A. Novelli, D. Reimer, F. Rohrer, R. Tillmann, L. Vereecken, R. Wegener, A. Zaytsev, A. Kiendler-Scharr and A. Wahner, Investigation of the oxidation of methyl vinyl ketone (MVK) by OH radicals in the atmospheric simulation chamber SAPHIR, Atmos. Chem. Phys., 2018, 18, 8001–8016 CrossRef CAS.
- S. Lou, F. Holland, F. Rohrer, K. Lu, B. Bohn, T. Brauers, C. C. Chang, H. Fuchs, R. Haeseler, K. Kita, Y. Kondo, X. Li, M. Shao, L. Zeng, A. Wahner, Y. Zhang, W. Wang and A. Hofzumahaus, Atmospheric OH reactivities in the Pearl River Delta - China in summer 2006: measurement and model results, Atmos. Chem. Phys., 2010, 10, 11243–11260 CrossRef CAS.
- H. Fuchs, A. Novelli, M. Rolletter, A. Hofzumahaus, E. Y. Pfannerstill, S. Kessel, A. Edtbauer, J. Williams, V. Michoud, S. Dusanter, N. Locoge, N. Zannoni, V. Gros, F. Truong, R. Sarda-Esteve, D. R. Cryer, C. A. Brumby, L. K. Whalley, D. Stone, P. W. Seakins, D. E. Heard, C. Schoemaecker, M. Blocquet, S. Coudert, S. Batut, C. Fittschen, A. B. Thames, W. H. Brune, C. Ernest, H. Harder, J. B. A. Muller, T. Elste, D. Kubistin, S. Andres, B. Bohn, T. Hohaus, F. Holland, X. Li, F. Rohrer, A. Kiendler-Scharr, R. Tillmann, R. Wegener, Z. Yu, Q. Zou and A. Wahner, Comparison of OH reactivity measurements in the atmospheric simulation chamber SAPHIR, Atmos. Meas. Tech., 2017, 10, 4023–4053 CrossRef CAS.
- W. Lindinger, A. Hansel and A. Jordan, On-line monitoring of volatile organic compounds at pptv levels by means of proton-transfer-reaction mass spectrometry (PTR-MS) medical applications, food control and environmental research, Int. J. Mass Spectrom. Ion Processes, 1998, 173, 191–241 CrossRef CAS.
- A. Jordan, S. Haidacher, G. Hanel, E. Hartungen, L. Maerk, H. Seehauser, R. Schottkowsky, P. Sulzer and T. D. Maerk, A high resolution and high sensitivity proton-transfer-reaction time-of-flight mass spectrometer (PTR-TOF-MS), Int. J. Mass Spectrom., 2009, 286, 122–128 CrossRef CAS.
- M. Kaminski, H. Fuchs, I.-H. Acir, B. Bohn, T. Brauers, H.-P. Dorn, R. Häseler, A. Hofzumahaus, X. Li, A. Lutz, S. Nehr, F. Rohrer, R. Tillmann, L. Vereecken, R. Wegener and A. Wahner, Investigation of the β-pinene photooxidation by OH in the atmosphere simulation chamber SAPHIR, Atmos. Chem. Phys., 2017, 17, 6631–6650 CrossRef CAS.
- A. Novelli, M. Kaminski, M. Rolletter, I.-H. Acir, B. Bohn, H.-P. Dorn, X. Li, A. Lutz, S. Nehr, F. Rohrer, R. Tillmann, R. Wegener, F. Holland, A. Hofzumahaus, A. Kiendler-Scharr, A. Wahner and H. Fuchs, Evaluation of OH and HO2 concentrations and their budgets during photooxidation of 2-methyl-3-butene-2-ol (MBO) in the atmospheric simulation chamber SAPHIR, Atmos. Chem. Phys., 2018, 18, 11409–11422 CrossRef CAS.
- H. Fuchs, I.-H. Acir, B. Bohn, T. Brauers, H.-P. Dorn, R. Haeseler, A. Hofzumahaus, F. Holland, M. Kaminski, X. Li, K. Lu, A. Lutz, S. Nehr, F. Rohrer, R. Tillmann, R. Wegener and A. Wahner, OH regeneration from methacrolein oxidation investigated in the atmosphere simulation chamber SAPHIR, Atmos. Chem. Phys., 2014, 14, 7895–7908 CrossRef.
- P. Dewald, J. M. Liebmann, N. Friedrich, J. Shenolikar, J. Schuladen, F. Rohrer, D. Reimer, R. Tillmann, A. Novelli, C. Cho, K. Xu, R. Holzinger, F. Bernard, L. Zhou, W. Mellouki, S. S. Brown, H. Fuchs, J. Lelieveld and J. N. Crowley, Evolution of NO3 reactivity during the oxidation of isoprene, Atmos. Chem. Phys., 2020, 20, 10459–10475 CrossRef CAS.
- C. B. M. Groβ, T. J. Dillon and J. N. Crowley, Pressure dependent OH yields in the reactions of CH3CO and HOCH2CO with O2, Phys. Chem. Chem. Phys., 2014, 16, 10990–10998 RSC.
- F. A. F. Winiberg, T. J. Dillon, S. C. Orr, C. B. M. Groß, I. Bejan, C. A. Brumby, M. J. Evans, S. C. Smith, D. E. Heard and P. W. Seakins, Direct measurements of OH and other product yields from the HO2 + CH3C(O)O2 reaction, Atmospheric, Chem. Phys., 2016, 16, 4023–4042 CAS.
- A. O. Hui, M. Fradet, M. Okumura and S. P. Sander, Temperature Dependence Study of the Kinetics and Product Yields of the HO2 + CH3C(O)O2 Reaction by Direct Detection of OH and HO2 Radicals Using 2f-IR Wavelength Modulation Spectroscopy, J. Phys. Chem. A, 2019, 123, 3655–3671 CrossRef CAS.
- M. E. Jenkin, R. Valorso, B. Aumont and A. R. Rickard, Estimation of rate coefficients and branching ratios for reactions of organic peroxy radicals for use in automated mechanism construction, Atmos. Chem. Phys., 2019, 19, 7691–7717 CrossRef CAS.
- I. Bridier, B. Veyret, R. Lesclaux and M. Jenkin, Flash-Photolysis Study of the UV Spectrum and Kinetics of Reactions of the Acetonylperoxy Radical, J. Chem. Soc., Faraday Trans., 1993, 89, 2993–2997 RSC.
- M. E. Jenkin, G. D. Hayman, T. J. Wallington, M. D. Hurley, J. C. Ball, O. J. Nielsen and T. Ellermann, Kinetic and Mechanistic Study of the Self-Reaction of CH3OCH2O2 Radicals, J. Phys. Chem., 1993, 97, 11712–11723 CrossRef CAS.
- A. J. Kwan, A. W. H. Chan, N. L. Ng, H. G. Kjaergaard, J. H. Seinfeld and P. O. Wennberg, Peroxy radical chemistry and OH radical production during the NO3-initiated oxidation of isoprene, Atmos. Chem. Phys., 2012, 12, 7499–7515 CrossRef CAS.
- IUPAC Subcommittee on Atmospheric Chemical Kinetic Data Evaluation, 2017.
- A. S. Hasson, G. S. Tyndall, J. J. Orlando, S. Singh, S. Q. Hernandez, S. Campbell and Y. Ibarra, Branching Ratios for the Reaction of Selected Carbonyl-Containing Peroxy Radicals with Hydroperoxy Radicals, J. Phys. Chem. A, 2012, 116, 6264–6281 CrossRef CAS.
- J.-P. Le Crâne, M.-T. Rayez, J.-C. Rayez and E. Villenave, A reinvestigation of the kinetics and the mechanism of the CH3C(O)O2+ HO2 reaction using both experimental and theoretical approaches, Phys. Chem. Chem. Phys., 2006, 8, 2163–2171 RSC.
- O. M. Anglada, R. Crehuet and J. S. Francisco, The Stability of alpha-Hydroperoxyalkyl Radicals, Chem. – Eur. J., 2016, 22, 18092–18100 CrossRef.
- L. Vereecken, T. L. Nguyen, I. Hermans and J. Peeters, Computational study of the stability of α-hydroperoxyl- or α-alkylperoxyl substituted alkyl radicals, Chem. Phys. Lett., 2004, 393, 432–436 CrossRef CAS.
- L. Vereecken, P. Carlsson, F. Bernard, S. S. Brown, C. Cho, N. Friedrich, H. Fuchs, J. M. Liebmann, W. Mellouki, A. Novelli, D. Reimer, R. Tillmann, L. Zhou, A. Kiendler-Scharr and A. Wahner, Theoretical and experimental study of peroxy and alkoxy radicals in the NO3-initiated oxidation of isoprene, Phys. Chem. Chem. Phys., 2020 10.1039/d0cp06267g.
- M. S. Claflin and P. J. Ziemann, Identification and Quantitation of Aerosol Products of the Reaction of β-Pinene with NO 3 Radicals and Implications for Gas- and Particle-Phase Reaction Mechanisms, J. Phys. Chem. A, 2018, 122, 3640–3652 CrossRef CAS.
- D. Mogensen, S. Smolander, A. Sogachev, L. Zhou, V. Sinha, A. Guenther, J. Williams, T. Nieminen, M. K. Kajos, J. Rinne, M. Kulmala and M. Boy, Modelling atmospheric OH-reactivity in a boreal forest ecosystem, Atmos. Chem. Phys., 2011, 11, 9709–9719 CrossRef CAS.
- J. M. Liebmann, J. B. A. Muller, D. Kubistin, A. Claude, R. Holla, C. Plass-Duelmer, J. Lelieveld and J. N. Crowley, Direct measurements of NO3 reactivity in and above the boundary layer of a mountaintop site: identification of reactive trace gases and comparison with OH reactivity, Atmos. Chem. Phys., 2018, 18, 12045–12059 CrossRef CAS.
- P. M. Edwards, K. C. Aikin, W. P. Dube, J. L. Fry, J. B. Gilman, J. A. de Gouw, M. G. Graus, T. F. Hanisco, J. Holloway, G. Huber, J. Kaiser, F. N. Keutsch, B. M. Lerner, J. A. Neuman, D. D. Parrish, J. Peischl, I. B. Pollack, A. R. Ravishankara, J. M. Roberts, T. B. Ryerson, M. Trainer, P. R. Veres, G. M. Wolfe, C. Warneke and S. S. Brown, Transition from high- to low-NOx control of night-time oxidation in the southeastern US, Nat. Geosci., 2017, 10, 490–495 CrossRef CAS.
- L. K. Whalley, E. J. Slater, R. Woodward-Massey, C. Ye, J. D. Lee, F. Squires, J. R. Hopkins, R. E. Dunmore, M. Shaw, J. F. Hamilton, A. C. Lewis, A. Mehra, S. D. Worrall, A. Bacak, T. J. Bannan, H. Coe, B. Ouyang, R. L. Jones, L. R. Crilley, L. J. Kramer, W. J. Bloss, T. Vu, S. Kotthaus, S. Grimmond, Y. Sun, W. Xu, S. Yue, L. Ren, W. J. F. Acton, C. N. Hewitt, X. Wang, P. Fu and D. E. Heard, Evaluating the sensitivity of radical chemistry and ozone formation to ambient VOCs and NOx in Beijing, Atmos. Chem. Phys. Discuss., 2020, 21, 2125–2147 CrossRef.
- E. J. Slater, L. K. Whalley, R. Woodward-Massey, C. Ye, J. D. Lee, F. Squires, J. R. Hopkins, R. E. Dunmore, M. Shaw, J. F. Hamilton, A. C. Lewis, L. R. Crilley, L. Kramer, W. Bloss, T. Vu, Y. Sun, W. Xu, S. Yue, L. Ren, W. J. F. Acton, C. N. Hewitt, X. Wang, P. Fu and D. E. Heard, Elevated levels of OH observed in haze events during wintertime in central Beijing, Atmos. Chem. Phys. Discuss., 2020, 20, 14847–14871 CrossRef CAS.
- Z. Tan, K. Lu, A. Hofzumahaus, H. Fuchs, B. Bohn, F. Holland, Y. Liu, F. Rohrer, M. Shao, K. Sun, Y. Wu, L. Zeng, Y. Zhang, Q. Zou, A. Kiendler-Scharr, A. Wahner and Y. Zhang, Experimental budgets of OH, HO2, and RO2 radicals and implications for ozone formation in the Pearl River Delta in China 2014, Atmos. Chem. Phys., 2019, 19, 7129–7150 CrossRef CAS.
Footnotes |
| † The data of the experiments in the SAPHIR chamber used in this work are available on the EUROCHAMP data home page (https://data.eurochamp.org/). |
| ‡ Electronic supplementary information (ESI) available: The supplement related to this article is available online, and contains updated and new NO3, O3, and OH-initiated reaction schemes, calculations on radical decomposition and isomerisation, plots and tables on experimental conditions and instrumentation, tabulated estimated OH yields in the RO2 + HO2 reaction, comparison of measured and modelled HO2 concentrations, and the raw quantum chemical results for all structures calculated in this work. See DOI: 10.1039/d0cp05555g |
| This journal is © the Owner Societies 2021 |