
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Theoretical and experimental study of peroxy and alkoxy radicals in the NO3-initiated oxidation of isoprene†
L.
Vereecken
 *a,
P. T. M.
Carlsson
a,
A.
Novelli
a,
F.
Bernard‡
b,
S. S.
Brown
cd,
C.
Cho
a,
J. N.
Crowley
e,
H.
Fuchs
a,
W.
Mellouki
b,
D.
Reimer
a,
J.
Shenolikar
e,
R.
Tillmann
a,
L.
Zhou§
b,
A.
Kiendler-Scharr
a and
A.
Wahner
a
*a,
P. T. M.
Carlsson
a,
A.
Novelli
a,
F.
Bernard‡
b,
S. S.
Brown
cd,
C.
Cho
a,
J. N.
Crowley
e,
H.
Fuchs
a,
W.
Mellouki
b,
D.
Reimer
a,
J.
Shenolikar
e,
R.
Tillmann
a,
L.
Zhou§
b,
A.
Kiendler-Scharr
a and
A.
Wahner
a
aInstitute for Energy and Climate Research, Forschungszentrum Jülich GmbH, 52428 Jülich, Germany. E-mail: l.vereecken@fz-juelich.de
bInstitut de Combustion, Aérothermique, Réactivité et Environnement (ICARE), CNRS (UPR 3021)/OSUC, 1C Avenue de la Recherche Scientifique, 45071 Orléans CEDEX 2, France
cNOAA Chemical Sciences Laboratory, 325 Broadway, Boulder, CO 80305, USA
dDepartment of Chemistry, University of Colorado Boulder, Boulder, CO 80209, USA
eAtmospheric Chemistry Department, Max-Planck-Institut für Chemie, 55128 Mainz, Germany
First published on 2nd March 2021
Abstract
The initial stages of the nitrate radical (NO3) initiated oxidation of isoprene, in particular the fate of the peroxy (RO2) and alkoxy (RO) radicals, are examined by an extensive set of quantum chemical and theoretical kinetic calculations. It is shown that the oxidation mechanism is highly complex, and bears similarities to its OH-initiated oxidation mechanism as studied intensively over the last decade. The nascent nitrated RO2 radicals can interconvert by successive O2 addition/elimination reactions, and potentially have access to a wide range of unimolecular reactions with rate coefficients as high as 35 s−1; the contribution of this chemistry could not be ascertained experimentally. The chemistry of the alkoxy radicals derived from these peroxy radicals is affected by the nitrate moiety, and can lead to the formation of nitrated epoxy peroxy radicals in competition with isomerisation and decomposition channels that terminate the organic radical chain by NO2 elimination. The theoretical predictions are implemented in the FZJ-NO3-isoprene mechanism for NO3-initiated atmospheric oxidation of isoprene. The model predictions are compared against peroxy radical (RO2) and methyl vinyl ketone (MVK) measurements in a set of experiments on the isoprene + NO3 reaction system performed in the SAPHIR environmental chamber (IsopNO3 campaign). It is shown that the formation of NO2 from the peroxy radicals can prevent a large fraction of the peroxy radicals from being measured by the laser-induced fluorescence (ROxLIF) technique that relies on a quantitative conversion of peroxy radicals to hydroxyl radicals. Accounting for the relative conversion efficiency of RO2 species in the experiments, the agreement between observations and the theory-based FZJ-NO3-isoprene model predictions improves significantly. In addition, MVK formation in the NO3-initiated oxidation was found to be suppressed by the epoxidation of the unsaturated RO radical intermediates, allowing the model-predicted MVK concentrations to be in good agreement with the measurements. The FZJ-NO3-isoprene mechanism is compared against the MCM v3.3.1 and Wennberg et al. (2018) mechanisms.
1 Introduction
Of the volatile organic compounds (VOCs) emitted to the atmosphere, isoprene is among the most important, contributing half of the mass of non-methane VOCs released.1 Its tropospheric oxidation is predominantly initiated by hydroxyl (OH) radicals during the daytime, but at nighttime the lack of photolytic sources of OH shifts the main initiation channels to the nitrate radical, NO3, and ozone, O3.2–6 Although isoprene emissions are predominantly biogenic and light-driven, residual isoprene is left during the night, allowing a sizable fraction of the emitted isoprene to be oxidized by NO3 in the nocturnal boundary layer.7–9 Furthermore, under some conditions NO3 oxidation can still contribute for a sizable fraction to daytime atmospheric VOC oxidation.10,11The OH-initiated reaction of isoprene has been well-studied, where the last decade has seen many studies examining the complex oxidation pathways, the formation of oxygenated products, and the regeneration of OH radicals.4,12–22 The oxidation was shown to comprise a rapidly branching mechanism where correct treatment of the site-specificity of the initial OH addition and subsequent (reversible) O2 addition is critical:

| isoprene + OH → HO-isoprene radicals (4 isomers) | (R1) |
| HO-isoprene + O2 ↔ HO-isoprene-OO˙ peroxy radicals (8 isomers) | (R2) |
Although the reaction of the NO3 radical with isoprene can be expected to show the same complexity and to proceed through similar pathways, currently available mechanisms such as the Master Chemical Mechanism34–36 or the recent model by Wennberg et al.4 tend to include only a simplified version with a selected set of intermediates. Similar to the OH-initiated oxidation though, analyses based on the full branched reaction mechanism are necessary to elucidate the chemistry, and to decide which reactions can be omitted in specific reaction conditions. As indicated in a recent perspective on mechanism development,37 the formulation of such (near-)explicit models is an important tool in tackling modern challenges facing atmospheric sciences. Literature data38 suggests that nitrate substitution leads to slower radical chemistry compared to OH-substitution, and the relative importance of the individual channels can be expected to be different between OH- and NO3-initiated isoprene chemistry. A recent review by Wennberg et al.4 gives a good overview of the status quaestionis of isoprene oxidation by OH and NO3, based on the available literature on the NO3-initiated oxidation of isoprene up to now.39–48
In this work, we investigate several aspects of the first stages of the NO3-initiated oxidation of isoprene, and of measurements of nitrated RO2 radicals, as summarized in Fig. 1. First, we present a combined theoretical and experimental study on the initial steps in the isoprene + NO3 + O2 reaction mechanism, i.e. the formation and destruction of the initial set of 8 nitrated alkylperoxy radical isomers (called nitrate-RO2 hereafter). The subsequent chemistry of the isoprene-NO3 alkylperoxy and alkoxy radicals is likewise examined in detail. Their fate as governed by the competition between unimolecular and bimolecular reactions is important not only for the atmospheric chemistry of isoprene, but also plays a critical role in the experimental detection of the nitrate-RO2 radicals by a ROxLIF instrument. For the latter, we extend the recent work by Novelli et al.49 on nitrate-substituted alkoxy radicals (called nitrate-RO hereafter) to the nitrate-RO derived from isoprene. The chemistry of these unsaturated alkoxy radicals can proceed by reversible epoxidation and subsequent O2 capture, which affects both the detection of RO2 in the ROxLIF instrument, and the yield of MVK and MACR in the SAPHIR chamber and the atmosphere. The chemistry characterized is implemented in the FZJ-NO3-isoprene model.
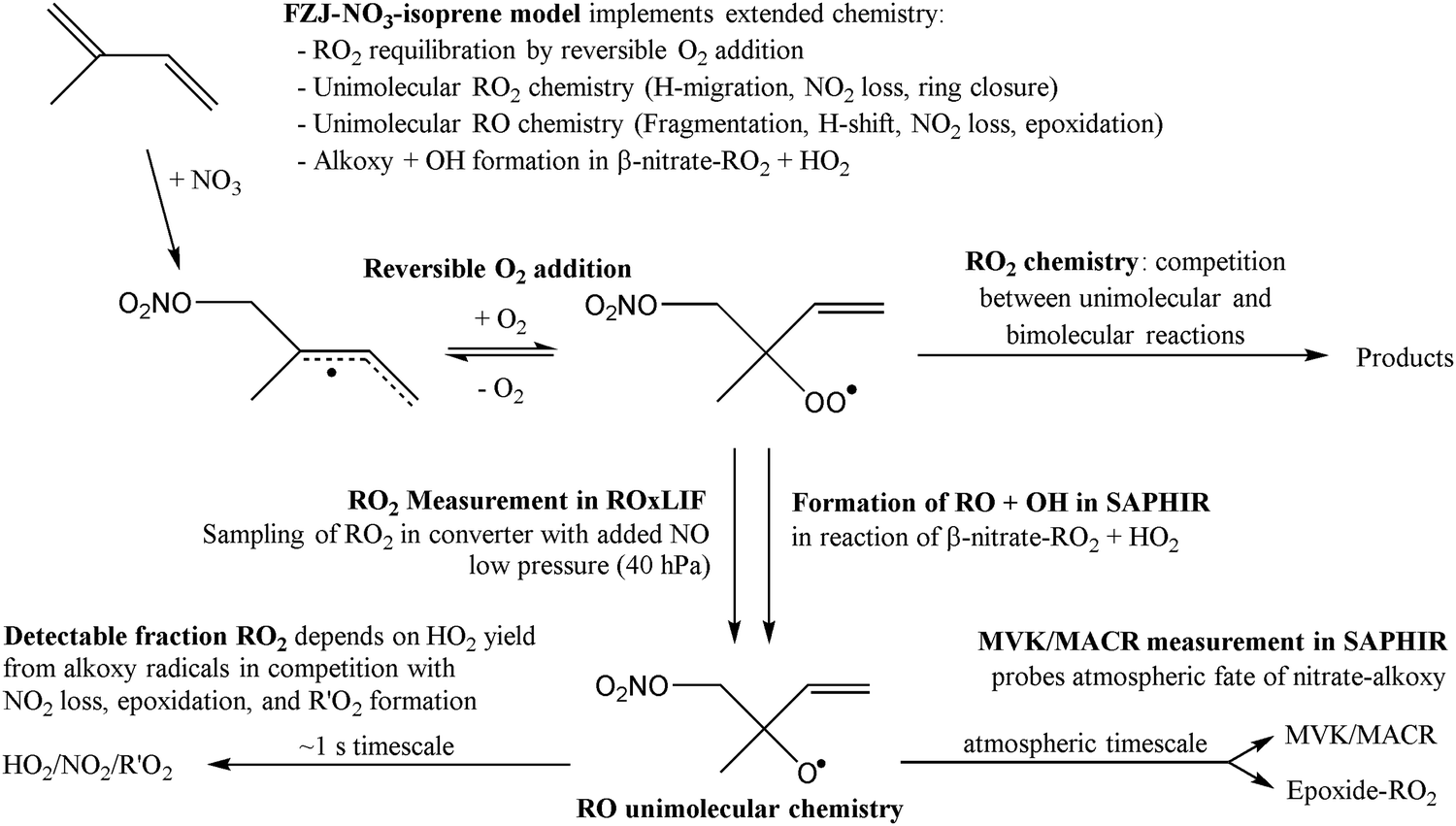 | ||
| Fig. 1 Summarizing scheme showing the investigated steps, which includes the NO3 and (reversible) O2 addition (site- and stereospecificity omitted), unimolecular chemistry of the nitrate peroxy and alkoxy radicals, and interpretation of the experimental RO2 and MVK/MACR measurements. All chemistry is implemented in the FZJ-NO3-isoprene model. | ||
2 Methodologies
2.1 Theoretical methodology
The nitrate-RO2 and nitrate-RO radicals derived from isoprene + NO3 were first optimized using the M06-2X/cc-pVDZ level of quantum chemical theory,50,51 characterizing all conformers of reactants and transition states for the reactions studied. The resulting geometries were re-optimized at the M06-2X/aug-cc-pVTZ level of theory,50 and combined with single point energy calculations on the lowest conformers at the CCSD(T)/aug-cc-pVTZ level of theory.52 ZPE corrections are done at the M06-2X/aug-cc-pVTZ level of theory, with vibrational wavenumbers scaled by 0.971.53,54 All quantum chemical calculations were performed using the Gaussian-16 software suites.55The rate coefficients of the reactions are obtained using multi-conformer transition state theory, MC-TST,56,57 incorporating the characteristics of all conformers in a rigid rotor harmonic oscillator approximation, as obtained at the CCSD(T)/aug-cc-pVTZ//M06-2X/aug-cc-pVTZ level of theory. Tunneling is included using an asymmetric Eckart barrier correction.58,59 At the chosen level of theory used here, the rate coefficients are expected to have an accuracy of a factor of 2 to 4. We also compare the rate coefficient predictions against available structure–activity relationships (SARs) for alkoxy decomposition38,49 and H-migration,60 and RO2 radical H-migration.33 The analysis of nitrate-RO2 thermal equilibrium population was likewise based on the ratios of the multi-conformer RO2 partition functions. The method for calculating bulk rate coefficients across rapidly equilibrating groups of reactants is already described for conformer and isomer equilibration by internal rotation61 or H-migration;27,33 it is also applicable12,13 to RO2 populations equilibrated by O2 addition/elimination as in the nitrate-RO2 radicals discussed in this work. Note that the rate of O2 addition forming the RO2 intermediates, and their reverse O2 elimination reactions, cannot be predicted a priori with the necessary accuracy for the task at hand, owing to the difficulties in characterizing the (near-)barrierless transition state on the flat potential energy surface for recombination of a doublet radical with a triplet biradical. We will thus base our analysis for the R to RO2 interconversion on the more reliable equilibrium constants, as discussed below.
The (de-)epoxidation reactions characterized in this work are fast, nearing the collision limit, and deviations from the Boltzmann distribution might occur. Any chemical activation effects depend on temperature, pressure, and the bath gas, and would be different for each source reaction of the epoxidizing alkoxy radical as formed from the parent RO2 radical reacting with NO, NO3, HO2 and each of the individual R′O2. Nguyen and Peeters21 examined the reaction rate of a similar epoxidation reaction based on energy-specific versus Boltzmann population based paradigms, but found only small differences. A second important criterion for thermalization is the rate of effective loss of conformers out of the epoxidation systems. In this work, these are unimolecular reactions and recombination with O2 removing the interconverting alkoxy and epoxy-alkyl radicals. The effective loss rates are typically slower than the (de-)epoxidation reactions, allowing for substantially more collisions than would be expected solely from the (de-)epoxidation rates, and hence provide better adherence to the thermal Boltzmann distribution across all conformers. To estimate the impact of chemical activation, we examined the fraction of prompt decomposition of 1-NO3-isoprene-2-O˙ radicals, the main alkoxy radical in our system, to MVK + NO2 (see ESI†).62 At 1 bar as in the SAPHIR chamber, we find that prompt decomposition is negligible, and all of the 1-NO3-isoprene-2-O˙ radicals form epoxy-peroxy radicals. At 25 mbar, we find some prompt decomposition only at internal energies above 17 kcal mol−1, and reaching a contribution of 10% only at 21 kcal mol−1, while the internal energy is estimated to be on average less than that. As such, we estimate that chemical activation effects are not overly critical, and the product distributions predicted in this work are sufficiently accurate even if the absolute rate coefficient may carry a somewhat larger uncertainty, and the predicted detectability of the nitrate-RO2 are likely somewhat overestimated. Quantifying the chemical activation effects, even in the low-pressure converter, are thus considered outside the scope of this work.
2.2 Experimental methodology
The experiments described here were part of a larger campaign on the NO3-initiated oxidation of isoprene (IsopNO3 campaign) conducted in the atmospheric simulation chamber SAPHIR. More details about the campaign can be found in Dewald et al.46 The SAPHIR chamber is made of a double-wall Teflon (FEP) film that is inert, and is kept under slightly higher than ambient pressure (by ∼30 Pa) to avoid external air penetrating the chamber. Due to small leakages and air consumption by instruments, a steady replenishment flow keeps the over-pressure constant, causing trace gases to be diluted at a rate of ∼6% h−1. More details regarding the chamber can be found elsewhere.63–65For all three experiments described within this study, the chamber was cleaned before each experiment by flushing with more than 6 times its volume with ultra-pure synthetic air provided by mixing nitrogen and oxygen (Linde, >99.99990%). Isoprene, O3 and NO2 were injected several times over the course of one experiment. Between 1.8 and 6 ppbv of isoprene were injected directly from the liquid (99% purity, Sigma Aldrich), while O3 was produced by a silent discharge ozonizer (O3onia) and added to reach concentrations between 40 and 110 ppbv. NO2 was added via calibrated flow controllers as a dilution of 519 ppm in N2 for concentrations between 3 and 25 ppbv. Two fans in the chamber ensure complete mixing of trace gases within 2 min. The NO3 oxidant is formed rapidly in the reaction of O3 with NO2:
| NO2 + O3 → NO3 + O2 | (R3) |
For the measurement of RO2 radicals using the ROxLIF system, these RO2 are first converted into alkoxy radicals in a converter (∼25 hPa) by reactions with NO. Subsequent chemistry of these RO radicals leads to formation of HO2 radicals, which are sampled in the fluorescence cell (∼4 hPa) downstream of the converter and are detected by OH fluorescence described above. The conversion efficiency through the ROxLIF system is calibrated using a reference RO2 (CH3OO). The final signal also includes the contribution from HO2 radicals present in the air sampled from the chamber, and the RO2 concentration is derived by subtracting this contribution from the ROxLIF measurement based on the HO2 LIF measurement described higher. A recent paper by Novelli et al.49 highlights how the LIF technique is likely to be blind to specific nitrated RO2 radicals due to the necessity of an OH or an HO2 radical to be formed for an RO2 radical to be detected. Specifically, it was found that the LIF technique cannot measure nitrated RO2 radicals formed from cis-2-butene and 2,3-dimethyl-2-butene, as the corresponding alkoxy radicals decompose to NO2 instead of forming HOx radicals. Similarly, the theoretical rate predictions for isoprene alkoxy radical reactions by the SAR described in that paper indicate that many of the nitrate-RO2 radicals from isoprene would not form HO2, and thus would not be detectable by the ROxLIF system. In this work, we estimate the conversion efficiencies of the dominant RO2 to detectable OH by explicit modelling of the converter and fluorescence cell chemistry (see ESI†).
Measurements of isoprene (Fig. S11, ESI†), as well as the sum of methyl vinyl ketone (MVK) and methacrolein (MACR) were performed by VOCUS PTR-MS (Tofwerk AG and Aerodyne Research Inc.). The VOCUS isoprene concentration profiles were calibrated against the change in kOH and kNO3 (total OH and NO3 reactivity measurements as shown for these experiments in Dewald et al.46) at the time of injection, to correct for any dependence of the VOCUS signal on relative humidity. Our kinetic model (see below) finds MVK and MACR to be present in similar concentrations. Therefore, we convert the signal strength measured by the VOCUS to the summed concentration of MVK + MACR by applying the mean of their respective instrumental sensitivities. NO3 concentrations (Fig. S11, ESI†) were measured by a custom-built cavity ring down spectroscopy instrument as described in Dewald et al.46
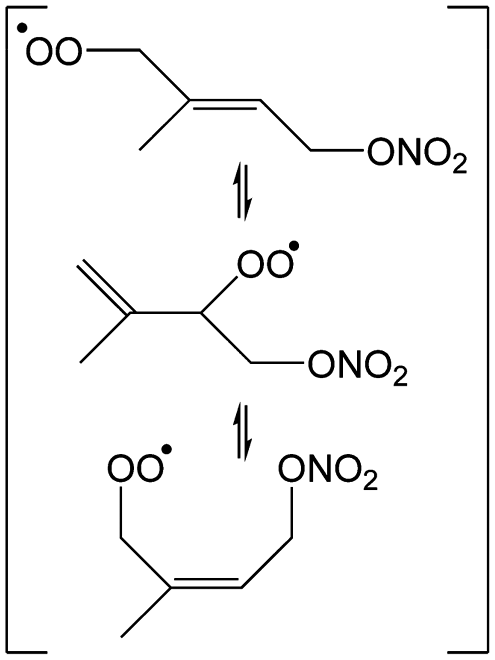
3 Theoretical results on the initial NO3 and O2 addition on isoprene
The site- and stereo-specific NO3 and O2 addition chemical scheme for isoprene is reminiscent of the LIM12,13 mechanism for isoprene + OH, i.e. both OH and NO3 addition leads to allyl-resonance stabilized alkyl radicals, which can reversibly add O2 on several radical sites forming distinct RO2 radicals. Such schemes for isoprene + OH are already implemented in the Master Chemical Mechanism, MCMv3.3.1,36 and in the recent isoprene mechanism described by Wennberg et al.4Fig. 2 depicts the scheme for the dominant NO3 radical addition channel on carbon C1 in isoprene, which constitutes4 87% of the reaction flux. The ESI† has the corresponding schemes for addition on carbon C4, with a contribution4 of 13%, and on the inner carbons C2 and C3, which have only a minor contribution and are neglected here. The addition reaction is exothermic by ∼32 kcal mol−1 for the formation of resonance-stabilized alkyl radicals. The redissociation of NO3-isoprene adducts to the reactants is negligible in our reaction conditions. Stereo-isomerisation of the cis- and trans-alkyl radicals at thermal energies is slow compared to O2 addition and has no significant influence. Theoretical work by Peeters et al.12,13 and Dibble69 showed however that the internal rotation is fast at the internal energies available through the addition of the radical oxidant. As discussed in more detail in the ESI,† we adopt an initial distribution of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for cis:trans 1-NO3-isoprenyl, and 7:3 for cis:trans 4-NO3-isoprenyl, based on these studies.
:1 for cis:trans 1-NO3-isoprenyl, and 7:3 for cis:trans 4-NO3-isoprenyl, based on these studies.
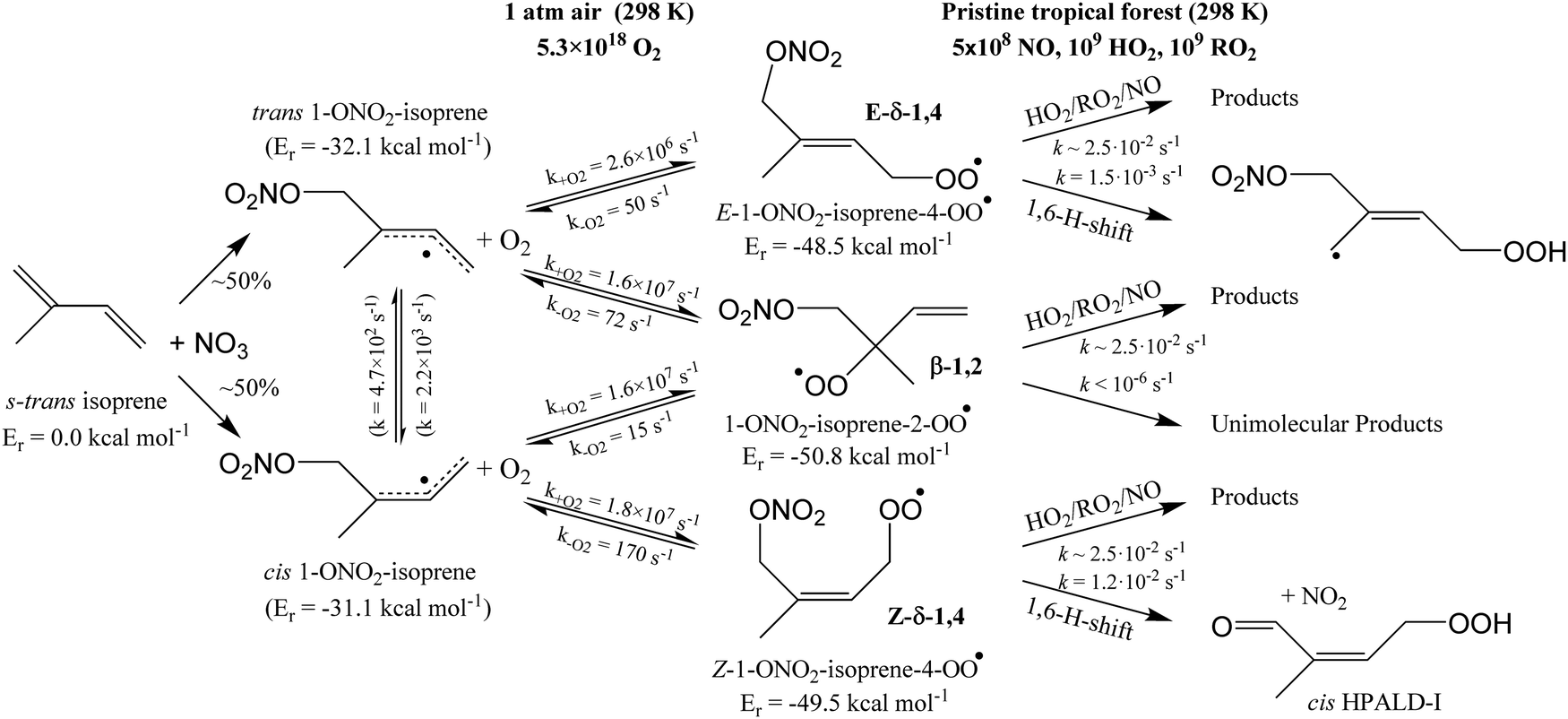 | ||
| Fig. 2 Reaction scheme for the site-specific addition of NO3 on C1 of isoprene, and the subsequent stereo-specific addition of O2. The ESI† includes schemes for NO3-addition on carbons C2, C3 and C4. Co-reactant concentrations are taken as in Peeters et al.12 | ||
Similar to OH-adducts of isoprene, the O2 addition is not overly exothermic, ≤20 kcal mol−1, and the O2 addition is reversible, allowing re-equilibration of the RO2 isomers by repetitive O2 elimination/addition events. This re-equilibration occurs in competition with unimolecular and bimolecular loss processes of the RO2 radicals, as discussed in a later section. An a priori characterization of the O2 addition and elimination reaction rates at sufficient accuracy for a kinetic model is computationally very expensive due to the (nearly) barrierless addition process,12 and the complexity in the quantum chemical description of the spin states of the many unpaired electrons. At this time, we therefore choose not to predict rate coefficients for these reactions directly. Instead, we adopt O2 addition rate coefficients as for the OH-isoprenyl + O2 reaction in the MCM v3.3.1 as based on the available literature data12,13,18,36 and evaluated by Novelli et al.14 The detailed analysis of Novelli et al.14 showed that a suitable ratio of formation to destruction rates is critical for a correct product distribution. The rate coefficients for O2 elimination from NO3-isoprene-O2 radicals are then obtained from the forward O2-addition reaction rates and theory-derived equilibrium constants, similar to the approach by Peeters et al.13 More details, the temperature-dependent rate coefficients, and the equilibrium constants are provided in the ESI.†
4 Theoretical results on nitrate-RO2
4.1 Elementary reactions
Table 1 summarizes the rate coefficients obtained for the unimolecular reactions of RO2 radicals formed in the isoprene + NO3 + O2 reaction. The theoretical results are compared against SAR predictions in the ESI.†| Reactant | Mechanism and products | E b | k(298 K) | A | n | E a |
|---|---|---|---|---|---|---|
| a Energies at the M06-2X/aug-cc-pVTZ level of theory. Given the barrier height, CCSD(T) and rate calculations were not done. b Less favorable as the adjacent syn-positioned –CH3 and –ONO2 groups induce more geometric hindrance. | ||||||
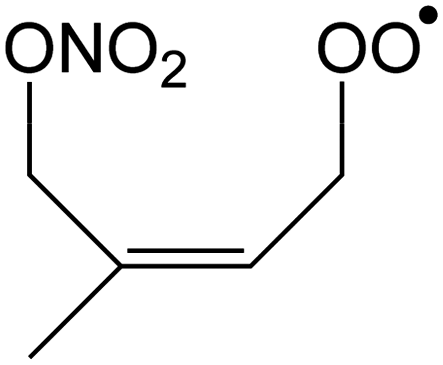
|
1,6-Migration of CH2ONO2 H-atom forming OCHC(CH3)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCH2OOH (HPALD I) + NO2 CHCH2OOH (HPALD I) + NO2 |
23.4 | 1.9 × 10−2 | 7.72 × 10−78 | 28.02 | −4158 |
| Ringclosure to 5-membered ring | 27.5 | 2.1 × 10−9 | 2.07 × 109 | 0.42 | 13062 |
|
| 1,6-Migration of CH3 H-atom forming O2NOCH2C(C˙H2)CHCH2OOH |
50.4a | |||||
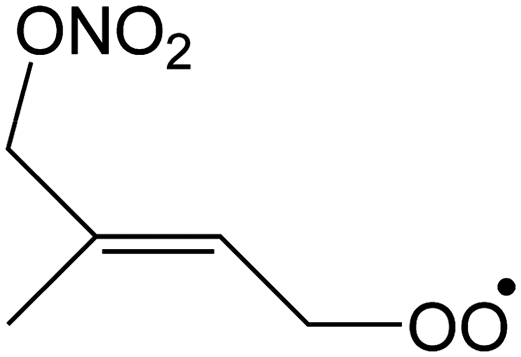
|
1,6-Migration of CH3 H-atom forming O2NOCH2C(C˙H2)CHCH2OOH |
23.3 | 1.5 × 10−3 | 2.58 × 10−84 | 30.21 | −4128 |
| Ringclosure to 5-membered ring | 28.6 | 7.2 × 10−11 | 3.43 × 104 | 1.90 | 13292 |
|
| 1,6-Migration of CH2ONO2 H-atom forming OCHC(CH3)CHCH2OOH (HPALD I) + NO2 |
46.9a | |||||
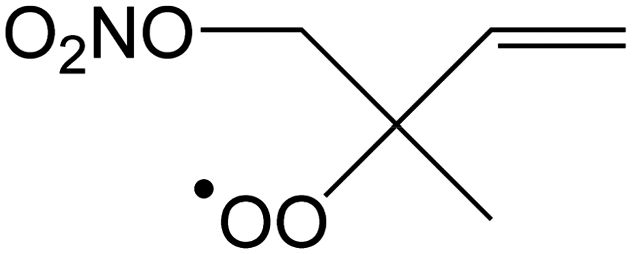
|
Ringclosure to 5-membered ring | 26.6 | 2.2 × 10−8 | 3.87 × 104 | 2.13 | 12028 |
| 1,4-Migration of CH2ONO2 H-atom forming NO2 + OCHC(CH3)(OOH)CHCH2 |
33.1 | 1.8 × 10−8 | 8.78 × 10−73 | 26.48 | 827 | |
| 1,4-Migration of CH3 H-atom forming O2NOCH2C(C˙H2)(OOH)CHCH2 |
35.9 | 1.6 × 10−10 | 4.82 × 10−75 | 27.35 | 2170 | |
| HO2 elimination CH2ONO2 H-atom forming O2NOCHC(CH3)CHCH2 |
26.3 | 3.5 × 10−7 | 4.09 × 10−38 | 16.41 | 6650 | |
| HO2 elimination CH3 H-atom forming O2NOCH2C(CH2)CHCH2 |
28.8 | 6.1 × 10−9 | 1.65 × 10−32 | 14.48 | 8418 | |
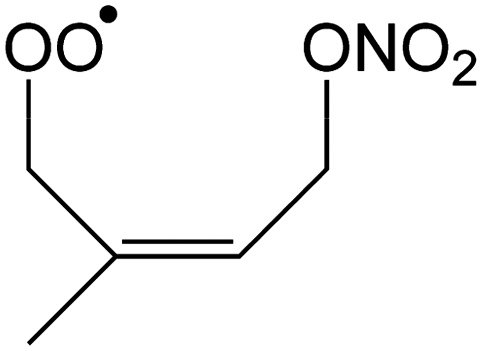
|
1,6-Migration of CH2ONO2 H-atom forming HOOCH2C(CH3)CHCHO (HPALD II) + NO2 |
22.3 | 5.4 × 10−2 | 2.52 × 10−75 | 27.06 | −4374 |
| 1,5-Migration of CH3 H-atom forming HOOCH2C(C˙H2)CHCH2ONO2 |
25.2 | 3.3 × 10−4 | 1.07 × 10−96 | 34.33 | −5178 | |
| Ringclosure to 5-membered ring | 27.4 | 3.1 × 10−9 | 9.51 × 108 | 0.50 | 12845 |
|
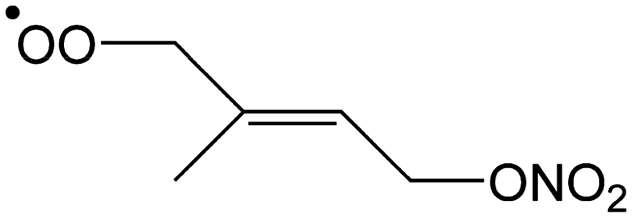
|
1,5-Migration of CH3 H-atom forming HOOCH2C(C˙H2)CHCH2ONO2 |
24.4 | 4.8 × 10−4 | 2.94 × 10−97 | 34.68 | −5088 |
| Ringclosure to 5-membered ring | 26.7 | 2.0 × 10−9 | 5.01 × 107 | 0.74 | 12514 |
|
| 1,6-Migration of CH2ONO2 H-atom forming HOOCH2C(CH3)CHCHO (HPALD II) + NO2 |
47.1a | |||||
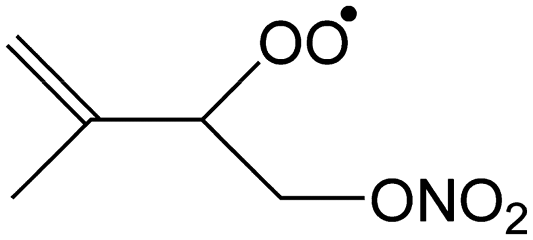
|
1,5-Migration of CH3 H-atom forming CH2C(C˙H2)CH(OOH)CH2ONO2 |
24.0 | 8.5 × 10−4 | 5.58 × 10−89 | 32.05 | −4050 |
| Ringclosure to 5-membered ring | 27.7 | 5.1 × 10−9 | 1.03 × 107 | 1.31 | 12722 |
|
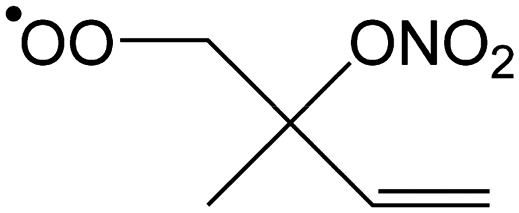
|
Ringclosure to 5-membered ring | 14.4 | 7.7 × 100 | 1.04 × 107 | 1.19 | 6218 |
| (R,S/S,R)-Product (syn) | 14.5 | 3.5 × 100 | 1.63 × 107 | 1.04 | 6348 | |
| (R,R/S,S)-Product (anti) | 14.4 | 4.2 × 100 | 6.94 × 106 | 1.12 | 6176 | |
| Ringclosure to 6-membered ring | 14.7 | 2.1 × 100 | 2.69 × 107 | 0.91 | 6420 | |
| 1,5-Migration of CH3 H-atom forming HOOCH2C(C˙H2)(ONO2)CHCH2 |
25.3 | 8.9 × 10−6 | 6.45 × 10−40 | 16.30 | 4235 | |
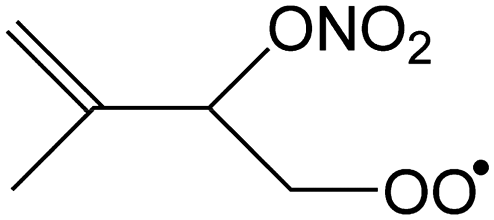
|
Ringclosure to 5-membered ring | 13.4 | 4.1 × 101 | 9.60 × 109 | 0.12 | 5951 |
| (R,S/S,R)-Product (anti) | 13.4 | 3.5 × 101 | 6.28 × 1011 | −0.57 | 6060 | |
| (R,R/S,S)-Product (syn)b | 14.7 | 5.5 × 100 | 4.44 × 1011 | −0.43 | 6748 | |
| Ringclosure to 6-membered ring | 15.7 | 1.5 × 100 | 2.47 × 1013 | −1.00 | 7384 | |
| 1,6-Migration of CH3 H-atom forming CH2C(C˙H2)CH(ONO2)CH2OOH |
22.8 | 1.7 × 10−2 | 8.97 × 10−67 | 24.55 | −2426 | |
1,6-H-Migration of the α-nitrooxy H-atom (–CH2ONO2) is the most favorable transition state, followed by migration of the methyl H-atoms. 1,4-H-Migrations are never competitive, nor are H-migrations between trans-substituents of the double bond. HO2 elimination with either the α-nitrooxy or the methyl H-atom was only examined for a single compound (1-ONO2-isoprene-2-OO˙) but as expected this reaction has sizable barriers exceeding 26 kcal mol−1, in agreement with earlier work,70–73 and can be neglected. Ring closure reactions should be divided into three categories: formation of 5-membered peroxide rings with an endo-cyclic alkyl radical site, formation of 5-membered rings with an exo-cyclic radical site, and formation of 6-membered rings (here always with an endo-cyclic radical site). Most nitrate-RO2 discussed here only have access to the first category, which typically has higher barriers ≥25 kcal mol−1, making them non-competitive. The barriers are a direct result of the ring strain in the TS, where the double bond is still intact and reaching the outer alkenic carbon with a 5-membered ring structure induces significant ring strain. If, on the other hand, the ring closure occurs on the inner carbon of the double bond, as e.g. in 2-ONO2-isoprene-1-OO˙ or 3-ONO2-isoprene-4-OO˙ formed in the minor isoprene + NO3 addition channels, the ring strain in the TS is significantly less and ring closure TS energies of ∼13–15 kcal mol−1 are found even for 5-membered rings. For 6-membered ring closure, here possible only in the latter two nitrate-RO2, the ring strain is significantly reduced despite the endo-cyclic double bond in the TS, and attack on the outer alkenic carbon also has low barriers, 14–16 kcal mol−1.
Compared to the OH-substituted RO2 radicals formed in isoprene + OH, the unimolecular rate coefficients for nitrate-RO2 are generally slower. For example, the NO2-forming 1,6-H-migration in Z-1-ONO2-isoprene-4-OO˙ forming HPALD has a calculated rate coefficient k(298 K) = 1.9 × 10−2 s−1, whereas the analogous OH-regenerating H-shift in Z-1-OH-isoprene-4-OO˙ has a predicted rate of ∼0.5 s−1,12–14,74 a factor of 25 faster. The few RO2 for which faster reactions are accessible, such as ring closure in 2- and 3-ONO2-isoprene-RO2, are formed only in minor to negligible NO3 addition channels on isoprene. Unimolecular reactions of nitrate-RO2 will thus be less important than for OH-RO2 under atmospheric conditions, and become negligible even for only moderately elevated NOx, HO2, and RO2 concentrations. In the current experiments, the OH reaction still contributes for ∼10% of the isoprene loss, and given the expected small yields through unimolecular channels in the NO3-isoprene system, and the possibility of formation of isobaric products in other channels, products measurements such as for HPALD can likely not be used to verify a product yield prediction.
4.2 Equilibrium populations and bulk rate coefficients
As already discussed in detail in the literature on OH-initiated oxidation of isoprene and aromatic compounds,12,13,75 a key feature of the atmospheric chemistry of allylic radicals is the reversibility of the O2 addition. For isoprene in particular this means that the RO2 radicals can interconvert by redissociation and O2 addition, in competition with unimolecular and bimolecular RO2 loss processes. In this context, three RO2 populations are of interest. The first is the nascent RO2 distribution formed directly from the isoprene + NO3 + O2 reaction sequence. This distribution governs the product distribution at high rates of RO2 loss, e.g. in experiments with very high co-reactant concentrations. The second is the equilibrium RO2 population, established when the loss processes are negligible compared to re-equilibration by O2 elimination/addition. Finally, there is the instantaneous RO2 distribution, intermediate between the above two limiting distributions and determined by the competition between RO2 formation, reactive loss, and re-equilibration as governed by the specific reaction conditions; this distribution can be time-dependent if the environment (e.g. co-reactant concentrations) changes at a time scale comparable to the rate of population change.These distributions are derived and discussed in more detail the ESI.† Briefly: the nascent RO2 distribution can in principle be derived from measured product ratios at high co-reactant concentrations (e.g. NO), where the re-equilibration is overwhelmed, but no sufficiently detailed data are available. In this work, we estimate the nascent distribution by assuming a cis:trans ratio for NO3-isoprenyl radicals as calculated by Peeters et al.12,13 for the HO-isoprenyl radicals. Reaction rates of the subsequent O2 addition are adopted from the MCM v3.3.1 for OH-isoprenyl + O2,36 which are calibrated against the experimental data on RO2 decomposition from Teng et al.18 and evaluated in Novelli et al.14 Combined, this yields the nascent RO2 population (see ESI†). The equilibrium RO2 populations can be predicted directly from the theoretical data. Strictly speaking, this distribution cannot be reached experimentally even in dilute conditions due to (slight) perturbations of the population due to unimolecular and bimolecular reactions. For the OH-initiated oxidation, it was found that in atmospheric conditions the instantaneous isoprene-RO2 concentration is intermediate between the nascent and equilibrium populations, and that kinetic models need to include all chemical pathways.12–14 For nitrated RO2, unimolecular loss is slower and the instantaneous population is expected to be close to equilibrium distributions under atmospheric conditions, and under the conditions in our experiments.
To aid in the characterization of the nitrate-RO2, we provide the theoretically derived temperature-dependent equilibrium contributions to the population in Table 2, and shown in Fig. 3 (see ESI† for more populations). At temperatures below 450 K, the population is dominated by 1-ONO2-isoprene-2-OO˙ isomers, the most stable of the RO2 isomers formed after the dominant NO3 addition on isoprene. The 4-ONO2-isoprene RO2 are not dominated by a single RO2 isomer. The distribution of the RO2 has its impact on the phenomenological rate coefficient, i.e. the bulk rate coefficient for product formation from the total pool of RO2 isomers. For unimolecular reactions, these differ from the elementary reaction rates as the re-equilibration allows access to all reaction channels independent of the starting RO2 isomer, and because the effective reactant concentration for any particular channel is only a fraction of the total RO2 concentration. In Table 3, we provide the bulk rate coefficients for the nitrated RO2 in an equilibrium population; the experimental bulk rate coefficient can differ as the RO2 distribution and re-equilibration will be different.
| Reactant | E rel | f(298 K) | F | n | E a |
|---|---|---|---|---|---|
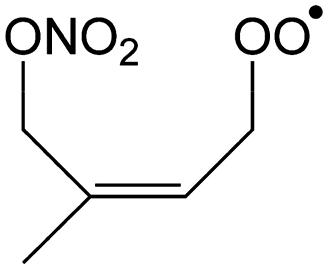
|
1.3 | 0.08 | 2.59 × 102 | −0.96 | 788 |
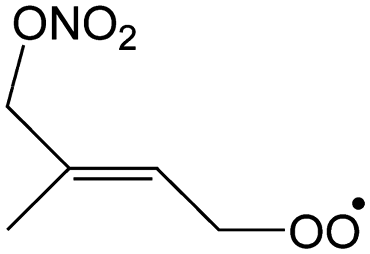
|
2.3 | 0.18 | 6.55 × 109 | −3.18 | 1851 |
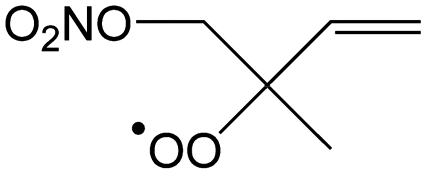
|
0.0 | 0.74 | 4.02 × 105 | −2.10 | 365 |
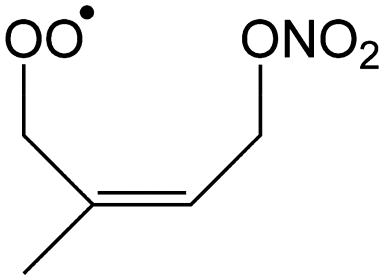
|
0.3 | 0.20 | 1.03 × 10−3 | 0.90 | −34 |
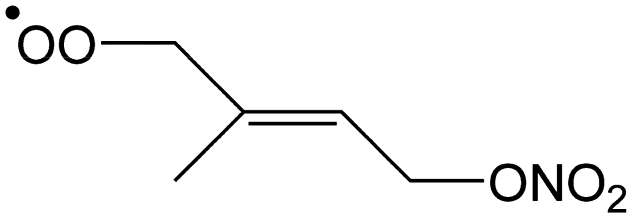
|
0.8 | 0.40 | 2.72 × 102 | −0.92 | 373 |
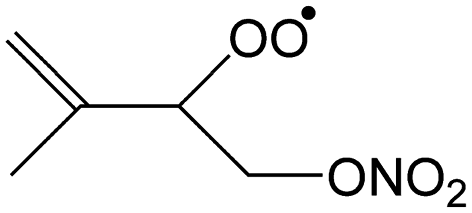
|
0.0 | 0.40 | 5.44 × 10 | −0.50 | −67 |
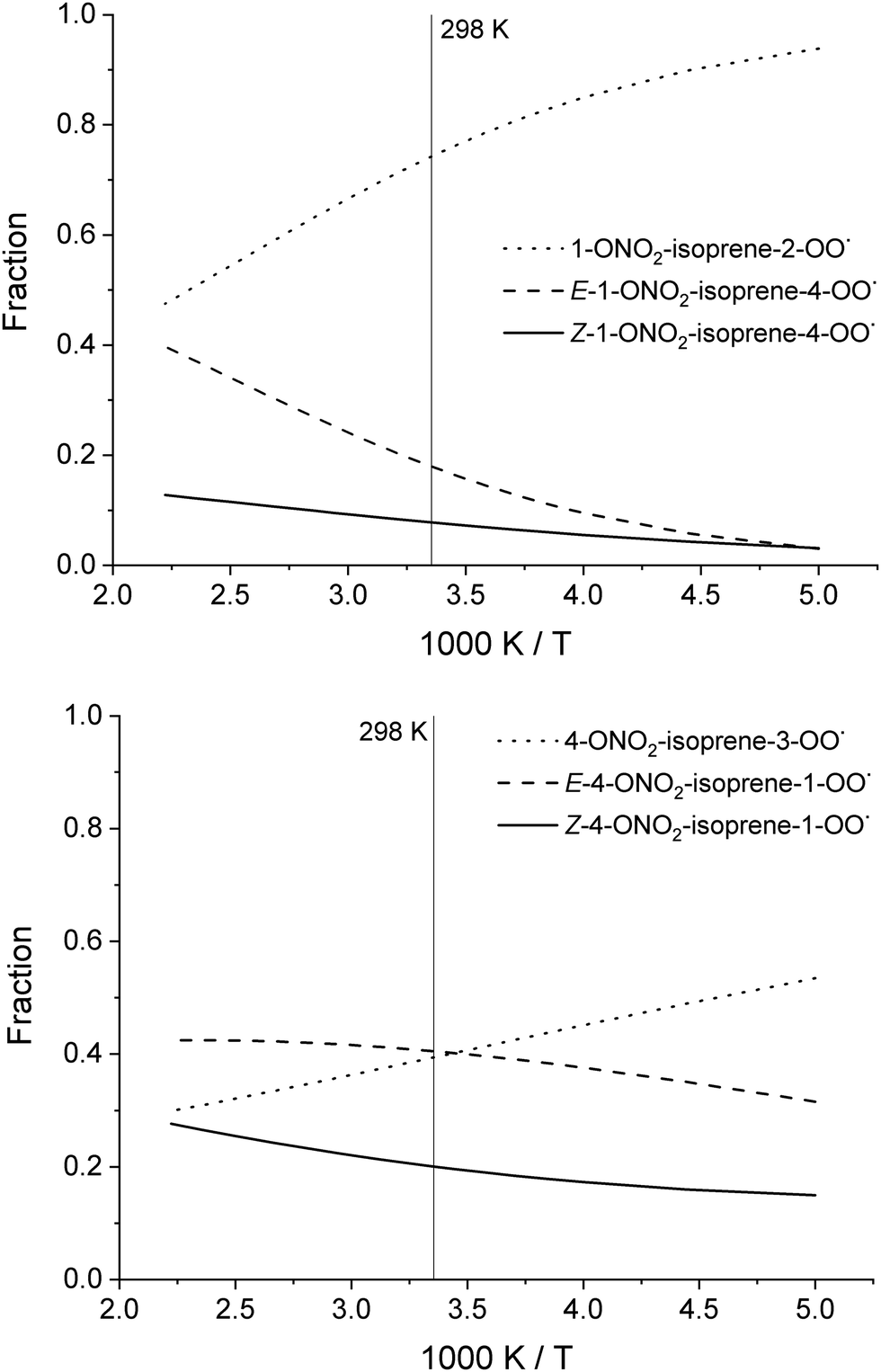 | ||
| Fig. 3 Temperature-dependent equilibrium population contribution for the individual RO2 isomers formed after NO3 addition to C1 (top) or C4 (bottom) of isoprene. | ||
| Reactant | Mechanism and products | E b | k(298 K) | A | n | E a |
|---|---|---|---|---|---|---|

|
Migration of CH2–ONO2 H-atoms forming NO2 + OCHC(CH3)CHCH2OOH (HPALD I) |
24.7 | 1.5 × 10−3 | 2.00 × 10−75 | 27.06 | −3369 |
| Migration of CH3 H-atoms forming O2NOCH2C(C˙H2)CHCH2OOH |
25.6 | 2.6 × 10−4 | 1.69 × 10−74 | 27.03 | −2275 | |
| Total | 1.7 × 10−3 | 1.25 × 10−79 | 28.62 | −3663 | ||
|
Migration of CH2ONO2 H-atoms forming NO2 + HOOCH2C(CH3)CHCHO (HPALD II) |
22.7 | 1.1 × 10−2 | 2.60 × 10−78 | 27.97 | −4410 |
| Migration of CH3 H-atoms forming HOOCH2C(C˙H2)CHCH2ONO2 |
25.2 | 2.0 × 10−4 | 8.02 × 10−95 | 33.76 | −4718 | |
| Migration of CH3 H-atoms forming CH2C(C˙H2)CH(OOH)CH2ONO2 |
23.9 | 3.4 × 10−4 | 3.04 × 10−88 | 31.55 | −4116 | |
| Total | 1.1 × 10−2 | 3.24 × 10−82 | 29.36 | −4732 |
5 Theoretical results on nitrate-RO
Table 4 lists the predicted rate coefficients for the most important reaction pathways of the alkoxy radicals formed from the primary nitrate-RO2 from isoprene + NO3. The theoretical results are compared against SAR predictions in the ESI.† Decomposition reactions eliminating the nitrate-bearing moiety, and H-migration of the γ-ONO2 hydrogen atoms are among the most important channels. These reactions lead to fragmentation of the nitrate moiety in the product γ-ONO2 alkyl radicals, forming NO2 and aborting the organic radical oxidation chain. In two cases, migration of a methyl H-atom is among the faster reactions, though these H-shifts are typically slower than migration of the γ-ONO2 hydrogen atoms. All the above nitrate-RO reactions are typically faster than the reaction of the nitrate-RO with O2, forming a carbonyl and HO2, thus often forgoing chain termination forming HO2 by formation of NO2 instead. It is important to note that reactions of the nitrate-RO are typically slower than the analogous reactions of hydroxylated alkoxy radicals formed in the OH-initiated oxidation of isoprene, enabling alternative reactions to become competitive. In particular, the adjacent double bond allows for epoxidation reactions (see Table 4), leading to the formation of nitrated epoxy alkyl radicals, which are isoenergetic with the parent nitrate-RO radicals within a few kcal mol−1. Epoxidation reactions in unsaturated compounds has been studied before, mainly in the atmospheric oxidation of aromatic compounds,75,76 and are found to be fast and reversible (see below). The competitive epoxidation has important ramifications for the predicted product distribution. For example, the main fate of 1-NO3-isoprene-2-O˙ is now predicted to be formation of the epoxide, hampering formation of methyl vinyl ketone (MVK), a well-known product of atmospheric isoprene oxidation. This epoxidation channel would also preclude MVK formation as proposed by Wennberg et al.4 in the reaction of 1-NO3-isoprene-2-OO˙ with HO2. As the epoxidation is reversible, the final product formation from the nitrate-RO can only be assessed through an explicit chemical mechanism. Contrary to the ring closure reaction in nitrate-RO2, ring closure in the isoprene-derived nitrate-RO radicals across longer spans than epoxidation is not competitive.| Reactant | Mechanism and products | E b | k(298 K) | A | n | E a |
|---|---|---|---|---|---|---|
|
1,5-Shift of O2NOCH2 H-atom forming NO2 + OCHC(CH3)CHOH |
7.9 | 5.1 × 107 | 8.93 × 10−30 | 13.31 | −2629 |
| Epoxidation forming 1-NO3-3,4-epoxy-isoprene-2-yl | 5.6 | 2.3 × 108 | 1.62 × 1012 | −0.01 | 2622 | |
|
1,5-Shift of CH3 H-atom forming O2NOCH2C(C˙H2)CH2OH |
9.7 | 2.2 × 106 | 2.08 × 10−37 | 15.82 | −2675 |
| Epoxidation forming 1-NO3-3,4-epoxy-isoprene-2-yl | 5.9 | 1.2 × 108 | 1.24 × 1010 | 0.77 | 2682 | |
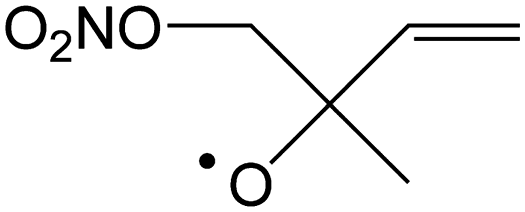
|
Decomposition forming NO2 + CH2O + MVK | 9.5 | 2.7 × 105 | 2.69 × 106 | 2.24 | 4491 |
| Decomposition forming ˙CH3 + O2NOCH2C(O)CHCH2 |
13.6 | 8.0 × 102 | 1.00 × 106 | 2.47 | 6325 | |
| Epoxidation forming syn-1-NO3-2,3-epoxy-isoprene-4-yl | 5.0 | 4.7 × 108 | 8.96 × 109 | 0.78 | 2207 | |
| Epoxidation forming anti-1-NO3-2,3-epoxy-isoprene-4-yl | 4.7 | 9.8 × 108 | 4.24 × 109 | 0.92 | 1996 | |
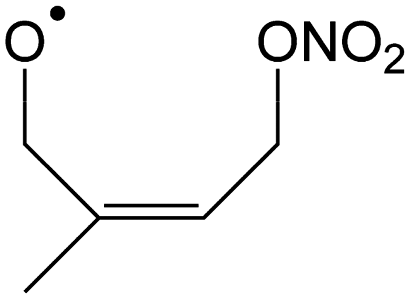
|
1,5-Shift of CH2ONO2 H-atom forming HOCH2C(CH3)CHCH2O + NO2 |
7.9 | 2.7 × 107 | 1.35 × 10−27 | 12.67 | −1997 |
| Epoxidation forming 4-NO3-1,2-epoxy-isoprene-3-yl | 6.3 | 3.9 × 107 | 1.15 × 109 | 1.12 | 2906 | |
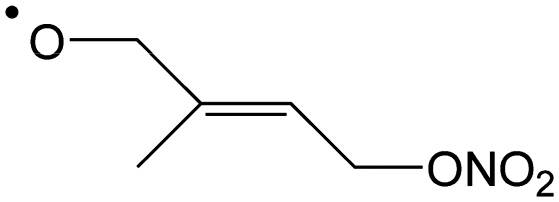
|
Epoxidation forming 4-NO3-1,2-epoxy-isoprene-3-yl | 7.3 | 8.6 × 106 | 8.91 × 108 | 1.09 | 3233 |
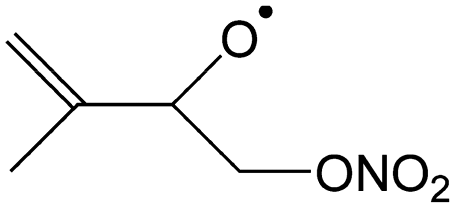
|
Decomposition forming MACR + CH2O + NO2 | 9.0 | 3.3 × 105 | 8.06 × 105 | 2.19 | 3981 |
| Epoxidation syn-4-NO3-2,3-epoxy-isoprene-1-yl forming | 6.2 | 1.4 × 108 | 1.19 × 1012 | 0.06 | 2795 | |
| Epoxidation anti-4-NO3-2,3-epoxy-isoprene-1-yl forming | 6.3 | 5.2 × 107 | 2.31 × 1010 | 0.54 | 2736 | |
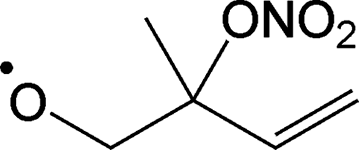
|
Decomposition forming CH2O + MVK + NO2 | 10.4 | 1.6 × 105 | 1.98 × 108 | 1.64 | 4901 |
1,5-Ring closure forming 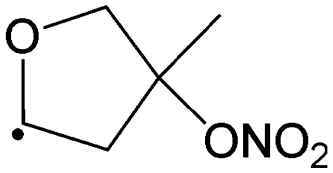 |
14.9 | 8.8 × 100 | 6.92 × 108 | 0.87 | 6905 | |
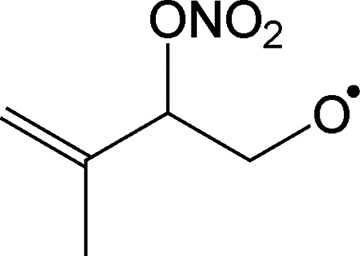
|
Decomposition forming MACR + NO2 + CH2O | 10.4 | 8.6 × 104 | 1.16 × 109 | 1.24 | 4931 |
| 1,5-shift of CH3 H-atom forming CH2C(C˙H2)CH(ONO2)CH2OH |
10.2 | 3.4 × 105 | 5.53 × 10−33 | 14.16 | −1882 | |
1,5-Ring closure forming 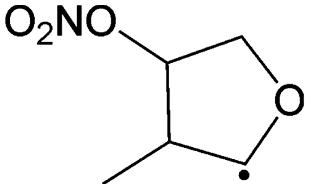 |
14.9 | 1.6 × 101 | 7.31 × 1011 | −0.08 | 7187 | |
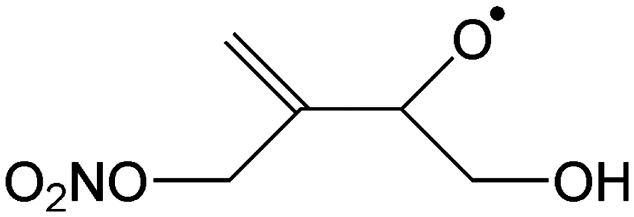
|
Epoxidation forming anti-5-NO3-isoprene-2,3-epoxide-4-OH-1-yl | 7.4 | 3.4 × 107 | 5.74 × 1013 | −0.49 | 3440 |
| Epoxidation forming syn-5-NO3-isoprene-2,3-epoxide-4-OH-1-yl | 8.5 | 4.9 × 106 | 2.09 × 1016 | −1.40 | 4227 | |
| Decomposition forming nitrooxy-methyl-acrolein + C˙H2OH | 4.0 | 2.8 × 109 | 1.39 × 106 | 2.11 | 1314 |
6 Theoretical results for epoxy radicals
Chemical schemes for epoxidation and subsequent chemistry are shown in Fig. 4. Due to the highly branching nature of the chemistry, not all pathways were theoretically characterized, but rely on SAR predictions instead. The theoretical predictions available are discussed below.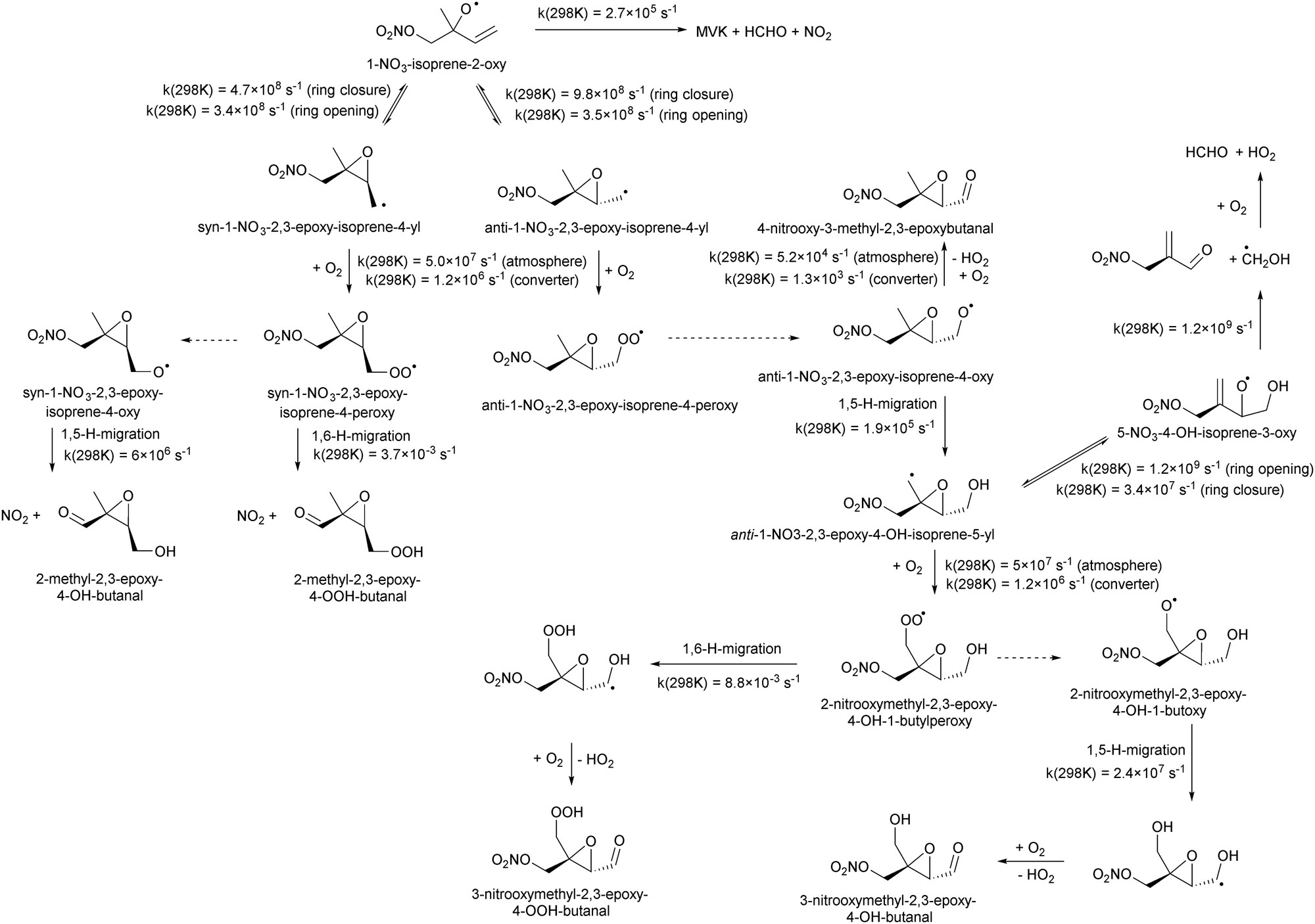 | ||
| Fig. 4 Oxidation scheme showing the dominant pathways for 1-NO3-isoprene-2-O˙ radicals formed in the NO3-initiated oxidation of isoprene. The dotted arrows indicates alkoxy radical formation in the reaction of RO2 with NO, NO3, HO2, or R′O2; these reactions are not depicted. In the high-NO low-pressure conditions of the converter, anti-stereoisomers will be detectable by partially forming HO2, while syn-isomers will eliminate NO2 and can not be detected by ROxLIF. | ||
6.1 Epoxy-alkyl radicals: ring breaking versus O2 addition
The epoxy-alkyl radicals formed from the nitrate-RO have two reaction pathways available: ring opening back to the nitrate-RO, or addition of an O2 molecule, forming an epoxy-nitrate-RO2. The epoxy-alkyl radicals have an interesting impact on the stereo-specificity of the reaction, where the Z- and E-1-NO3-isoprene-4-O˙ radicals form the same alkyl-epoxy radical (Table 5 and ESI†). As the internal rotation of the epoxy-moiety is comparable in rate or faster than the epoxy ring opening,21 this enables Z/E-stereo-isomerisation by reversible epoxidation; similar stereo-isomerisation has been reported earlier by Nguyen and Peeters21 for other unsaturated alkoxy radicals. A different type of stereo-chemistry is found for the 1-NO3-isoprene-2-O˙ radical, which leads to two stereo-specific alkyl-epoxy radicals with distinct chemistry (Table 5 and Fig. 4). Similar stereo-specific considerations apply to the intermediates formed after NO3 addition on C4 of isoprene (see ESI†). As the epoxidation is essentially energy-neutral and the barriers for ring opening/closing are low, the ring opening is as fast as the epoxidation reaction, k(298 K) ∼ 108 s−1, enabling fast interconversion between the nitrate-RO and the epoxy-alkyl radical (Tables 4 and 5). The ultimate fate of the nitrate-RO is then determined by the competition between O2 addition on the epoxy-alkyl radical, versus the dissociation or H-migration in the nitrate-RO. Recombination reactions of larger alkyl radicals with O2 are comparatively fast, k(298 K) ∼ 5.6 × 10−13 to 1.7 × 10−11 cm3 molecule−1 s−1.77–80 Contrary to the nitrate-RO2 discussed above, the product epoxy-RO2 are not expected to redissociate as the parent epoxy-alkyl radicals are not resonance stabilized. Adopting a value for O2 addition of k(298 K) = 7.5 × 10−12 cm3 molecule−1 s−1 leads, under atmospheric conditions (0.2 atm O2), to a pseudo-first order rate coefficient of k(298 K) ≈ 5 × 107 s−1. For many of the nitrate-RO radicals formed from isoprene, the main fate is then formation of an epoxy-nitrate-RO2 radical.| Reactant | Products | E b | k(298 K) | A | n | E a |
|---|---|---|---|---|---|---|
| 1-NO3-3,4-epoxy-isoprene-2-yl | Z-1-NO3-isoprene-4-O˙ | 6.0 | 7.3 × 107 | 7.51 × 108 | 1.10 | 2556 |
| E-1-NO3-isoprene-4-O˙ | 6.3 | 1.5 × 108 | 3.86 × 1010 | 0.72 | 2877 | |
| syn-1-NO3-2,3-epoxy-isoprene-4-yl | 1-NO3-isoprene-2-O˙ | 5.6 | 3.4 × 108 | 7.42 × 109 | 0.95 | 2535 |
| anti-1-NO3-2,3-epoxy-isoprene-4-yl | 1-NO3-isoprene-2-O˙ | 5.6 | 3.5 × 108 | 1.57 × 1010 | 0.80 | 2489 |
| 4-NO3-1,2-epoxy-isoprene-3-yl | Z-4-NO3-isoprene-1-O˙ | 6.9 | 4.9 × 107 | 3.46 × 1010 | 0.77 | 3270 |
| E-4-NO3-isoprene-1-O˙ | 7.6 | 2.8 × 107 | 9.63 × 1011 | 0.37 | 3745 | |
| syn-4-NO3-2,3-epoxy-isoprene-1-yl | 4-NO3-isoprene-3-O˙ | 5.4 | 8.1 × 108 | 8.15 × 1010 | 0.65 | 2475 |
| anti-1-NO3-2,3-epoxy-isoprene-1-yl | 4-NO3-isoprene-3-O˙ | 5.0 | 8.5 × 108 | 5.53 × 109 | 0.93 | 2142 |
| anti-5-NO3-isoprene-2,3-epoxide-4-OH-1-yl | 5-NO3-isoprene-4-OH-3-O˙ | 4.7 | 1.2 × 109 | 1.66 × 108 | 1.33 | 1652 |
| syn-5-NO3-isoprene-2,3-epoxide-4-OH-1-yl | 5-NO3-isoprene-4-OH-3-O˙ | 5.4 | 5.4 × 108 | 4.04 × 1011 | 0.24 | 2377 |
The rate of opening of the epoxy-ring is of the same order of magnitude as the O2 addition in most cases, such that bulk rate coefficients across the alkoxy/epoxy system are not valid. Still, for atmospheric modeling purposes, some model simplification remains possible. Between 250–350 K and 0.2 to 1 atm air, 1-NO3-isoprene-2-O˙ radicals convert to 68 ± 2% anti-1-NO3-2,3-epoxy-isoprene-4-OO˙, with the remainder forming syn-1-NO3-2,3-epoxy-isoprene-4-OO˙, while direct alkoxy radical reaction with O2 or alkoxy dissociation to NO2 constitute less than 0.1%. For 4-NO3-isoprene-3-O˙ radicals we find 26 ± 3% anti-4-NO3-2,3-epoxy-isoprene-1-OO˙, with the remainder forming syn-4-NO3-2,3-epoxy-isoprene-1-OO˙, while direct reactions with O2 and dissociation to NO2 constitute less than 0.4%. For the 1-NO3-2,3-epoxy-4-OH-isoprene-5-yl and 5-NO3-4-OH-isoprene-3-O˙ radicals (Fig. 4) the near-exclusive fate is formation of nitrated MACR + HCHO + HO2, owing to the fast decomposition in the alkoxy radical. For the other nitrate-RO2, the unimolecular reactions are more competitive against O2 addition in the epoxy-alkyl radical, and the final product distribution shows stronger temperature- and pressure dependencies that are not readily reduced to a single value.
It is interesting to note that unsaturated, hydroxylated RO radicals are less likely to form epoxy-RO2 radicals than unsaturated nitrate-RO radicals. Compared to a nitrate group, an –OH substituent tends to lead to faster isomerisation and decomposition reactions,38,49,60 which often dominate over the O2 addition in the epoxy-alkyl intermediate. Examples include the fast elimination of C˙H2OH radicals from the β-OH-alkoxy radical in Fig. 4, or the fast H-migration in the δ-OH-alkoxy radical studied by Nguyen and Peeters.21 This suggests that formation of epoxides could be more prevalent in nighttime NO3-initiated oxidation of terpenoids, compared to the OH-initiated oxidation dominant during daytime, which may have repercussions for aerosol formation and growth by reactive uptake. In judging the impact of epoxidation, one should also consider that subsequent chemistry of the epoxy radicals formed could form β-epoxy-alkyl radicals that de-epoxidize, and thus reduce the yield of epoxidized products. Fig. 4 shows an example for 1-NO3-isoprene-2-O˙ radicals: about two thirds of this nitrate-RO radical forms anti-substituted epoxy-RO2, but subsequent H-migration in the product alkoxy radical allows for rapid de-epoxidation and fragmentation forming nitrated MACR + HCHO + HO2 as the main product under oxidative conditions.
6.2 H-Migration reactions in epoxidized nitrate-RO2 radicals
Table 6 lists a set of calculations on epoxy-nitrate-RO2 radicals derived from isoprene. H-Migration of an aliphatic or an α-ONO2-H-atom across the epoxide functionality is an order of magnitude faster than the predictions by the SAR by Vereecken and Nozière for corresponding H-migrations in non-cyclic RO2 radicals. This contrasts with the results for epoxidized alkoxy radicals by Novelli et al.,49 who found little influence of the ring structure for H-migration across an epoxide ring. Similar to the findings of Novelli et al., we find that migration of an α-epoxy H-atom is not very favorable, despite the presence of an oxygen atom, and is only as fast as migration of an aliphatic H-atom. All the calculated H-migrations are slow, k(298 K) ≤ 10−3 s−1, and do not contribute significantly in our experimental conditions.| Reactant | Mechanism and products | Stereoa | E b | k(298 K) | A | n | E a |
|---|---|---|---|---|---|---|---|
| a Stereo-designator and stereo-specific rate coefficient for molecules with 2 chiral centers. Also indicated is the geometric average over the stereoisomers, as used in the kinetic model. | |||||||
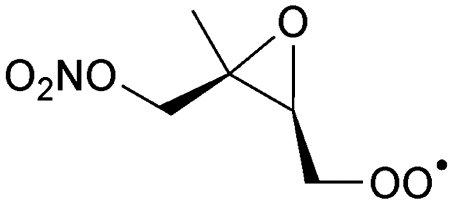
|
1,6-H-shift of O2NOCH2 H-atom | 24.0 | 3.7 × 10−3 | 1.77 × 10−83 | 30.09 | −4035 | |
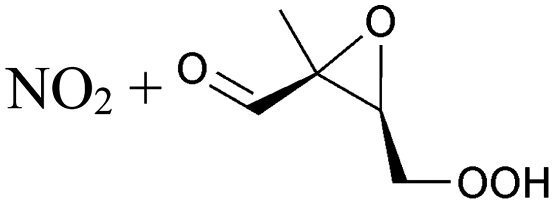
|
|||||||
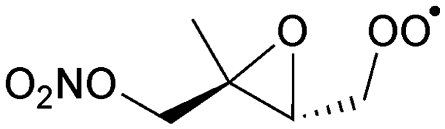
|
1,6-H-shift of CH3 H-atom | 24.8 | 1.2 × 10−4 | 1.71 × 10−96 | 34.52 | −4434 | |
|
|||||||
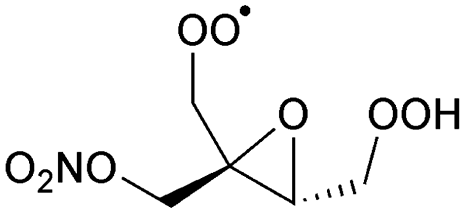
|
1,5-H-shift of α-epoxy H-atom | (R,S)/(S,R) | 24.5 | 3.6 × 10−4 | 1.35 × 10−93 | 33.70 | −4165 |
|
(R,R)/(S,S) | 25.2 | 9.7 × 10−5 | 7.30 × 10−98 | 35.03 | −4442 | |
| Geom. ave. | 1.9 × 10−4 | 9.92 × 10−96 | 34.36 | −4304 | |||
6.3 Isomerisation and decomposition of epoxidized nitrate-RO radicals
The rate coefficients for decomposition and isomerisation of a set of β-epoxidized alkoxy radicals are listed in Table 7. Decomposition forming an α-epoxy-alkyl radical is slow, due to high barriers owing to the increase in ring strain in these products. This is in agreement with our earlier work that found that a β-epoxy-moiety is less effective in lowering the barrier height for decomposition than alkyl or oxygenated substituents.49 Likewise, the barrier heights for the unfavorable elimination of a bare methyl radical product are comparatively high, making this reaction slow. Elimination of a ˙CH2ONO2 radical, where possible, is the most favorable decomposition; this radical readily decomposes further to HCHO + NO2. The decomposition barriers for all these processes are reproduced within 1 kcal mol−1 by the SARs by Vereecken and Peeters38 and Novelli et al.38,49 The fastest reactions for the set of alkoxy radicals listed in Table 7 are syn-1,5-H-migrations across the epoxy moiety, either of a methyl H-atom, or more favorably of a –CH2ONO2 hydrogen leading to NO2 elimination. As already anticipated by Novelli et al.,49 the rate coefficients for H-migration across the epoxy moiety are within a factor of 2 of the rate of isomerisation in aliphatic alkoxy radicals (see SAR by Vereecken and Peeters57,60), where the α-ONO2 substituent has a similar impact on the rate as an alkyl group.| Reactant | Mechanism and products | Stereoa | E b | k(298 K) | A | n | E a |
|---|---|---|---|---|---|---|---|
| a Stereo-designator and stereo-specific rate coefficient for molecules with 2 chiral centers. Also indicated is the geometric average over the stereoisomers, as used in the kinetic model. | |||||||
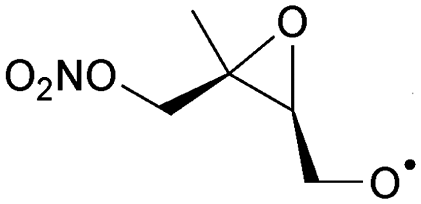
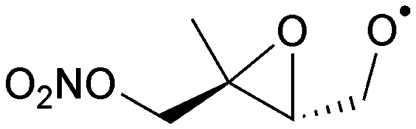
|
1,5-H-Shift of O2NOCH2 H-atom | 8.8 | 6.0 × 106 | 4.14 × 10−16 | 8.68 | −477 | |
|
|||||||
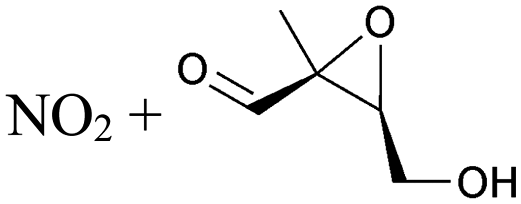
| Decomposition | 17.9 | 1.5 × 100 | 3.65 × 1010 | 1.02 | 8861 | ||
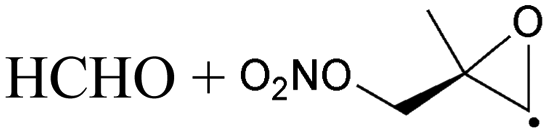
|
|||||||
|
1,5-H-Shift of CH3 H-atom | 10.6 | 1.9 × 105 | 3.23 × 10−23 | 10.99 | −400 | |
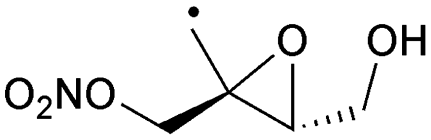
|
|||||||
| Decomposition | 18.3 | 4.3 × 10−1 | 9.28 × 108 | 1.61 | 9134 | ||
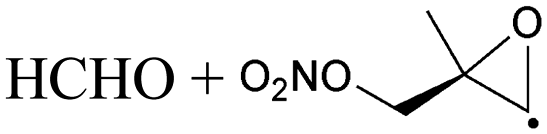
|
|||||||
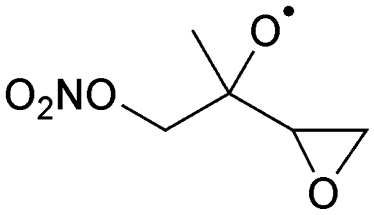
|
Decomposition | (R,S)/(S,R) | 9.9 | 4.0 × 104 | 2.44 × 105 | 2.28 | 4416 |
| NO2 + HCHO + 1,2-epoxy-3-butanone | (R,R)/(S,S) | 11.4 | 1.1 × 104 | 2.33 × 107 | 1.83 | 5376 | |
| Geom. ave. | 2.1 × 104 | 2.38 × 106 | 2.06 | 4896 | |||
| Decomposition | (R,S)/(S,R) | 14.5 | 1.7 × 102 | 5.12 × 106 | 2.22 | 6852 | |
| CH3 + 1-ONO2-3,4-epoxy-2-butanone | (R,R)/(S,S) | 13.9 | 2.4 × 102 | 5.22 × 102 | 3.52 | 6209 | |
| Geom. ave. | 2.0 × 102 | 5.17 × 104 | 2.87 | 6530 | |||
| Decomposition | (R,S)/(S,R) | 13.0 | 3.1 × 103 | 1.33 × 109 | 1.48 | 6372 | |
| 1,2-Epoxy-ethyl + 1-ONO2-2-propanone | (R,R)/(S,S) | 12.5 | 5.5 × 103 | 3.84 × 108 | 1.56 | 5978 | |
| Geom. ave. | 4.1 × 103 | 7.14 × 108 | 1.52 | 6175 | |||
7 Chemical mechanism implementation
The reactions and their theory-derived rate coefficients described in the earlier sections embedded in extended oxidation mechanisms for the relevant RO2 and RO radicals, are implemented in a zero-dimensional box model, called FZJ-NO3-isoprene henceforth, which can be used for modeling the trace gas concentration time series in the experiments.As discussed higher, the rates of the re-equilibration reactions of the primary nitrate-RO2 radicals by O2-elimination/re-addiction are optimized to reproduce the theoretically derived RO2 equilibrium distribution in the absence of other loss processes. Since the system of variables is under-determined, we choose to approximate the oxygen addition rates to have the same rates as for the alkyl radicals in the OH + isoprene system.12–14 The ESI† lists k(T) values for all isoprene-derived RO2 radicals, while the resulting rates at 298 K are illustrated in Fig. 2 for the main addition channel. Note that this approach only yields correct rate ratios between O2 addition and elimination, where the absolute magnitude of the values is inherited from the isoprene + OH + O2 system. The site- and stereo-specificity of the initial NO3 addition is obtained from literature data (see above).
Unimolecular reaction rates for nitrate-RO2, nitrate-RO, and epoxy-RO radicals from isoprene were taken from theoretical calculations in this work. Where necessary to derive extended oxidation schemes (see Fig. 4 and ESI†), these were supplemented by application of the pertinent SARs.33,38,49,60 The RO2 bimolecular rate coefficients and product distributions were derived using the structure–activity relationship (SAR) by Jenkin et al.,81 which does not explicitly incorporate the effect of the nitrate substituent. However, for the reaction rates for nitrate-RO2 + NO3, we used the data by Dewald et al.46 In addition, an OH-product channel is added to the reaction of β-ONO2-RO2 + HO2, as introduced in earlier work.4,41–43,49,82
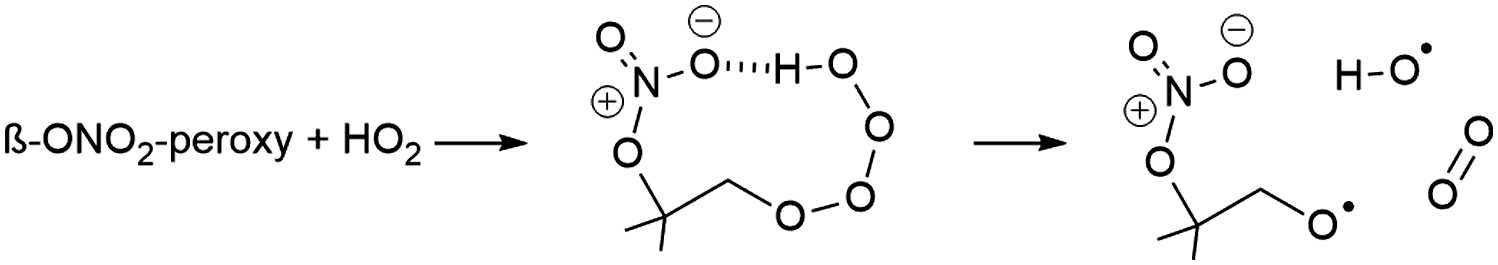 | (R4) |
As we focus mainly on the formation and consumption of the initial RO2, the subsequent chemistry of the products following RO2 uni- or bimolecular reactions is included following the CalTech mechanism or, if none is provided there, we adopt the chemistry as described by the Master Chemical Mechanism v3.3.134–36 (available at http://mcm.leeds.ac.uk). The isoprene ozonolysis mechanism is updated based on the literature data and is described in detail in the ESI.† The chemistry following the NO3-addition on the central carbons of isoprene is not fully expanded in our current model. The yields for these channels are expected to be low, and we currently do not have product information that would allow determining these yields. These channels are rendered inoperable in the current model with a zero yield. The resulting FZJ-NO3-isoprene model is provided in the ESI.†
For modeling the current set of experiments, the model also incorporates chamber-specific processes. A dilution coefficient was added for all trace gases in the model. This is necessary due to the replenishment flow required to keep the over-pressure in the chamber (see Experimental methodology). Isoprene, O3 and NO2 injections in the experiment were incorporated into the model as a short-period source reaction optimized to yield concentrations in agreement with their respective measurements, leaving these species concentrations otherwise unconstrained; Fig. S11 (ESI†) shows a comparison of the measured against modelled isoprene concentrations. The concentration of NO3 was constrained to the measurement in order to reduce the impact of uncertainty in NO3 and/or N2O5 loss on the chamber wall, which can be significant46 and variable in time. Temperature, pressure and water content were also constrained to the measurements. As discussed in detail in the ESI,† there is an unresolved discrepancy between the modelled HO2 concentration in the simulations, and the HO2 measurement from the LIF system, with the latter higher by up to a factor of 10. Two limiting cases are considered: (i) the discrepancy is assumed to be solely due to missing sources of HO2 radicals in the models, and therefore the modelled HO2 concentration is constrained to the measured HO2 radicals, and (ii) the discrepancy is assumed to be fully due to RO2 radicals interfering in the LIF HO2 measurement,67 and the HO2 and RO2 measurement are corrected for accordingly. These two limiting cases allows bracketing the impact of the uncertainty on the HO2 radical concentrations in the experiments described. It is important to underline that the chemical conditions within the experiments described in this study, specifically the lack of NO, result in much larger RO2 radical concentrations as compared to the HO2 radicals (a factor of 10 more). Therefore, even a small interference (less than 10%) from the RO2 radicals would drastically affect the measured HO2 radicals (Fig. S9, ESI†).
The main differences between the MCM v3.3.1, CalTech, and FZJ-NO3-isoprene models can be summarized as follows. The CalTech mechanism by Wennberg et al. ignores the stereospecificity of the δ-NO3-isoprene-OO isomers, lumping the Z- and E-isomers into a single species despite their distinct chemistry both at the RO2 and the subsequent RO stage. After NO3 addition on isoprene, the CalTech model favors O2 addition on the δ-position over the β-position, whereas the FZJ-NO3-isoprene model predicts a much higher contribution of the β-ONO2-RO2 radicals based on the theoretical Boltzmann distribution (Fig. 3). The CalTech mechanism also does not include the reversible epoxidation reactions for β-unsaturated nitrate-RO. Finally, the CalTech mechanism applies an OH-forming channel in the nitrate-RO2 + HO2 reaction irrespective of the distance between the –ONO2 and –OO˙ substituents, whereas FZJ-NO3-isoprene considers this path viable only for β-nitrate-RO2. The MCM v3.3.1 model incorporates only a single nitrate-RO2 isomer with limited subsequent chemistry. As such, it does not yet include nuances in chemistry such as the RO2 re-equilibration (which is implemented for isoprene + OH), RO2 unimolecular reactions, epoxidation in nitrate-RO, or the OH-channel in nitrate-RO2 + HO2.
8 Prediction of the ROxLIF detectable RO2 fraction
The RO2 radicals are sampled from the SAPHIR chamber in the ROxLIF converter at a lower pressure (25 hPa), and NO is added; due to the low pressure, bimolecular reactions other than with NO and O2 are negligible. During the residence time in the ROxLIF converter, ∼0.6 s, the RO2 radicals react with NO forming RO radicals, which in turn are expected to convert to HO2, as described below. Finally, the ROxLIF system measures the HO2 concentration by sampling in a fluorescence cell, where HO2 gets converted to OH in reaction with NO, and detected by LIF. The ROxLIF system thus measures the sum of HO2 formed from RO2 in the converter, and the HO2 radicals already present in the sampled SAPHIR chamber air; subtracting the SAPHIR HO2 and OH concentrations as measured directly by two additional and separate fluorescence cells (OH and HOx cells) then yields the HO2 signal attributable to conversion of RO2. Calibration of this signal relative to a known CH3O2 standard allows determination of absolute total RO2 concentrations, thus accounting for residence time, wall losses, and other characteristics of the converter.The observation of RO2 radicals in the ROxLIF instrument thus hinges on the formation of OH or HO2 radicals from the alkoxy radicals derived from these RO2 radicals under the high-NO conditions of the converter. Aliphatic and hydroxylated alkoxy radicals react with O2(R5) and (R6), possibly after one or more decomposition steps, to form HO2 and are thus typically detectable. However, as already studied in detail by Novelli et al.,49 nitrate-substituted RO2 radicals can undergo chain termination by decomposition of the nitrate group (R7), forming NO2, and are thus undetectable in the ROxLIF measurement.
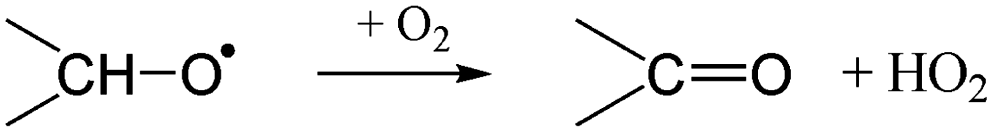 | (R5) |
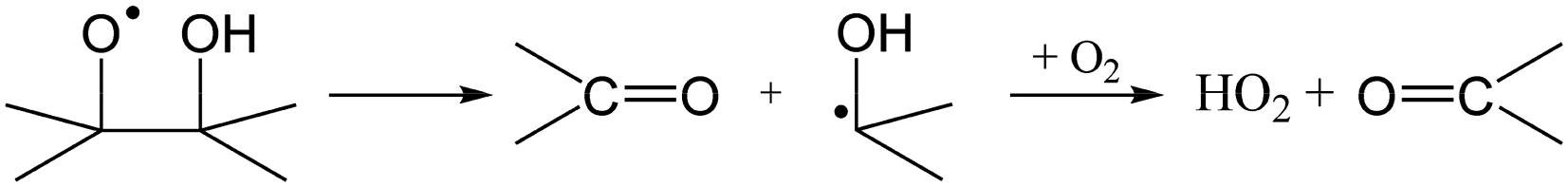 | (R6) |
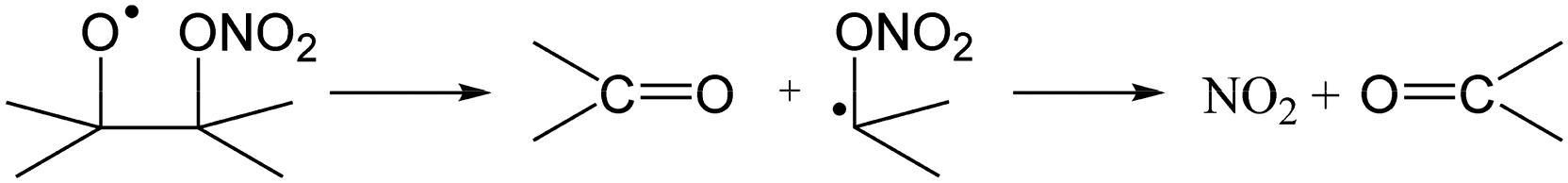 | (R7) |
To aid the interpretation of the RO2 measurements in the NO3-initiated oxidation experiments on isoprene, we modelled the fate of the RO2 radicals under the reaction conditions in the converter (25 hPa, 0.55 ppmv of NO, 0.17% CO, residence time ∼0.6 s),85 using the relevant subsets of the FZJ-NO3-isoprene model for the target RO2 radicals. From these model calculations, we find that the isoprene-derived nitrate-RO2 radicals, and the RO2 radicals formed in their subsequent reactions in the SAPHIR chamber, are not converted fully to OH or HO2 radicals but rather generate NO2 under the conditions in the converter, and will thus remain partially invisible in the LIF measurement of RO2. For example, at 298 K the main nitrate-RO2 species 1-NO3-isoprene-2-OO˙ and 1-NO3-isoprene-4-OO˙ are estimated to produce only 25% and 11–15% HOx, respectively, in the fluorescence cell relative to the calibrating CH3O2 radical, with the remainder forming undetectable NO2 or remaining as one of the intermediate RO2. For the nitrate-RO2 formed in the minor addition channel, 4-NO3-isoprene-3-OO˙ and 4-NO3-isoprene-1-OO˙, less than 3% are predicted to be detectable. The temperature-dependent detectability for all relevant nitrate-RO2 and nitrate-epoxy-RO2 is listed in Table S2 in the ESI.† The ROxLIF instrument in use at the SAPHIR chamber is thus not expected to return the sum of all RO2 concentrations (called “total RO2” hereafter) as predicted in the model, but measurements instead correspond to the summation of all RO2 concentrations weighted by their speciated HOx yields to obtain the so-called “detectable RO2” sum. For non-nitrated RO2, we adopt unity HO2 formation efficiency relative to the calibrating CH3O2 radical.85 As the relative nitrate-RO2 radical detectability depends on the conditions in the converter and fluorescence cell, e.g. NO concentration, pressure, etc., the detectability can't be generalized to other ROxLIF instruments, and each system needs to be experimentally characterized. The a priori predictions of the detectability in the current work do not benefit from any such experimental calibration, and are thus affected by the combined uncertainties on temperature, pressure, converter and fluorescence cell residence time, wall losses, rate coefficients, chemical activation, and possible unknown reactions in a complex reaction mechanism. Several of these aspects are discussed in more detail in the ESI.† Overall, we estimate an uncertainty of about a factor of 2 to 3 on the estimated relative conversion efficiency governing the detectability of RO2, mostly due to the uncertainties on the various rate coefficients in the kinetic model used to derive these conversion efficiencies. It must be stressed that the reduced O2 concentrations in the converter affects the chemistry rather significantly. Under atmospheric conditions, formation of epoxy-RO2 is expected to be more prevalent, whereas in the converter, direct alkoxy decomposition and isomerisation reactions are more competitive.
9 Comparison to experimental observations
9.1 Peroxy radical concentration
Modelling the experiments using the FZJ-NO3-isoprene mechanism indicates that oxidation of isoprene by NO3 was the dominant degradation pathway, with 10–15% of isoprene oxidized each by ozone and hydroxyl radicals. Isoprene ozonolysis forms mainly stable products and has only a small contribution to the RO2 pool; these ozone-RO2 are not discussed further in this work. RO2 radicals from the oxidation of isoprene by OH radicals contribute about 20% to the total RO2 according to our model, and all these RO2 are detectable in the ROxLIF instrument.38,49,60 The bulk of the RO2 radicals in the reaction mixture of the experiments, however, is nitrate-RO2 radicals which, as discussed above, are only partially detectable (see Fig. S14 in the ESI†). As shown in Fig. 5 and additional figures in the ESI,† we predict for all three experiment days large discrepancies between the total RO2 and the RO2 detected by the ROxLIF instrument. The modelled detectable RO2, however, differs from the measured RO2 concentration only by about a factor of 2, if the FZJ-NO3-isoprene chemical mechanism is applied. The fate of the nitrate-RO2 is strongly affected by the O2-driven RO2 re-equilibration as implemented in the FZJ-NO3-isoprene model. This re-equilibration is rapid compared to the rate of RO2 loss processes, such that the instantaneous RO2 population is always close to the equilibrium fractions, with 1-NO3-isoprene-2-peroxy radicals being the dominant RO2 species (Fig. 3), with 70 to 75% contribution in the 1-NO3-isoprene peroxy radical pool, depending on the temperature. This tertiary peroxy radical acts as a reservoir species, owing to its comparatively slow bimolecular loss processes (k ∼ 1.6 × 10−2 s−1 for HO2/RO2/NO3 combined, see below),81 and combined with the slow unimolecular reactions (k ∼ 1.8 × 10−3 s−1, Table 3) this leads to lifetimes of about 2 min for 1-NO3–RO2 radicals. In contrast, bimolecular and unimolecular reactions of 4-nitrooxy RO2 radicals have overall higher (pseudo-first order) rate coefficients (respectively 2.1 × 10−2 and 1.1 × 102 s−1, Table 3), have lifetimes of around 1 min, and contribute significantly less to the RO2 concentration. Table 8 list the contributions of the respective loss processes for each nitrate-RO2. Overall, the majority of the nitrate-RO2 are lost by reaction with HO2 and NO3. RO2 + RO2 reactions are never dominant, even for the experiment with the highest modelled peak RO2 concentrations in excess of 8 × 109 molecule cm−3. Some nitrate-RO2 have a fast unimolecular loss channel, which can become the dominant loss path for these specific isomers but has only a small overall contribution.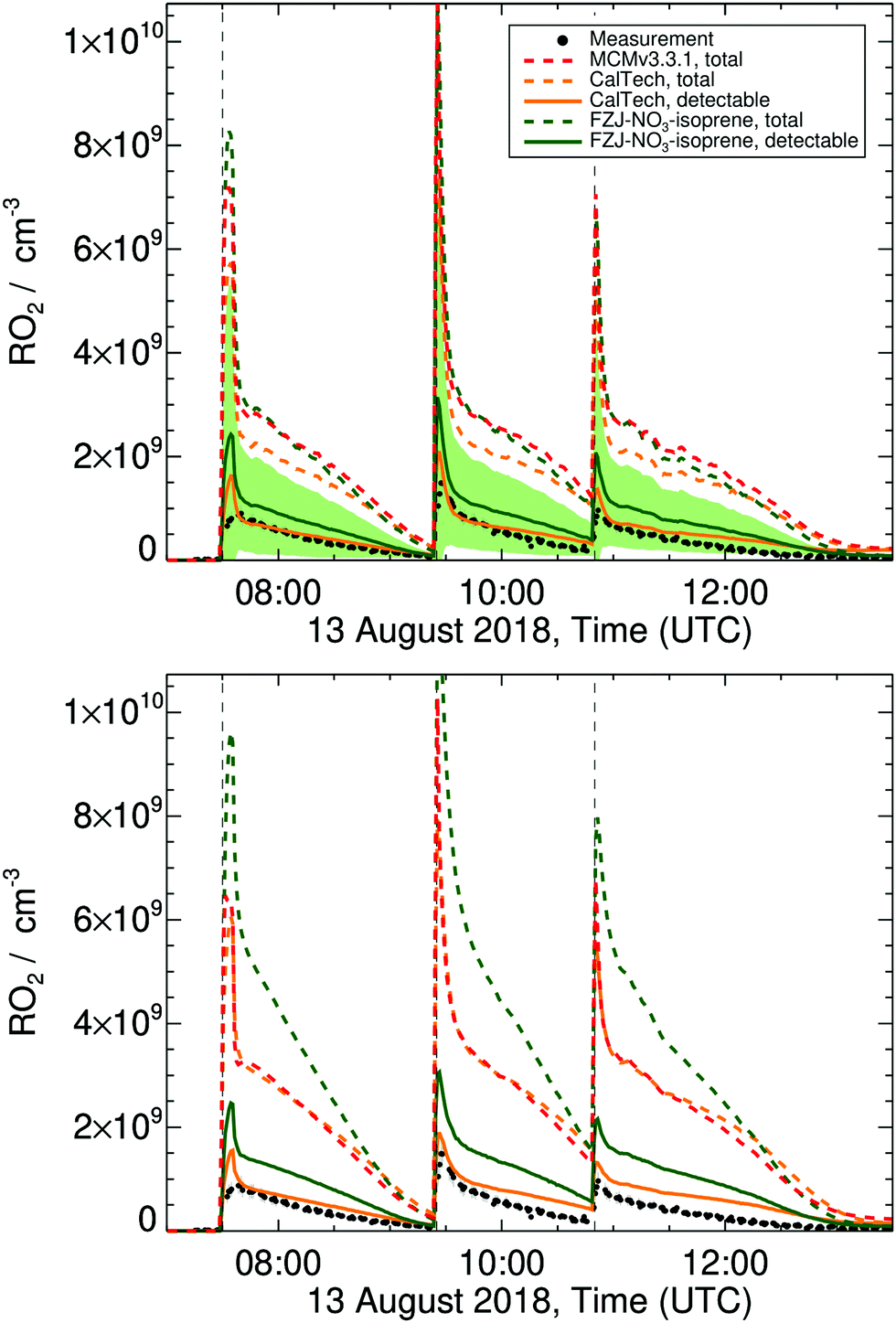 | ||
| Fig. 5 Measured and modelled RO2 concentration time profiles for the experiment on 13 Aug 2018, as described in Dewald et al.,46 with reaction conditions shown in ESI† Section K. Dashed lines represent the total RO2 predicted by the models; upper panel: model results with HO2 constrained to measurements, bottom panel: unconstrained HO2. For the more detailed CalTech and FZJ-NO3-isoprene models, additional traces (solid lines) show the estimated detectable RO2, which can be compared to the measurements. The shaded area shows a factor of 2 uncertainty on the estimated conversion efficiency factors of the detectable RO2 predicted by the FZJ-NO3-isoprene model. | ||
| Reactant | Unimolecular | HO2 | NO3 | RO2 |
|---|---|---|---|---|
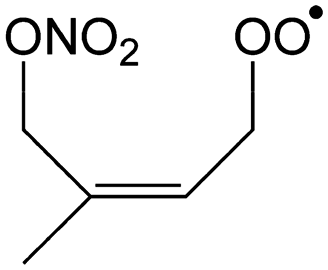
|
46–79%/49–82% | 9–21%/2–4% | 5–12%/5–12% | 6–21%/10–35% |
|
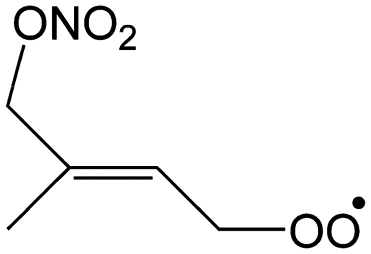
— | 36–44%/8–13% | 20–25%/22–28% | 31–44%/59–70% |
|
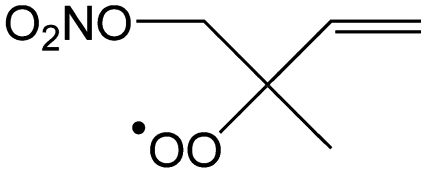
— | 63–64%/26–32% | 35–38%/67–72% | 0–1%/1–2% |
| All 1-NO3-isoprene-RO2 average lifetime: ∼113/164 s | 4–6%/4–7% | 54–57%/20–27% | 30–34%/53–59% | 6–10%/12–17% |
|
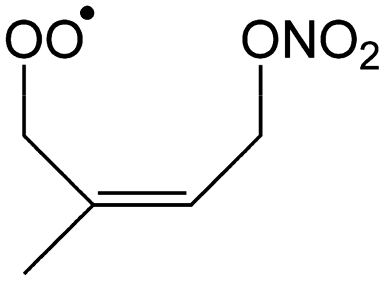
72–92%/76–93% | 3–11%/1–2% | 2–6%/2–7% | 2–10%/3–16% |
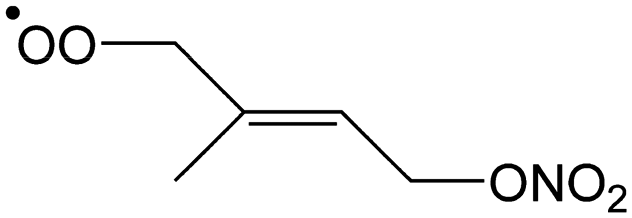
|
— | 41–44%/9–14% | 23–28%/27–32% | 28–36%/51–65% |
|
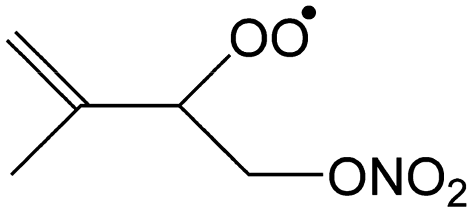
— | 53–55%/17–22% | 31–35%/49–53% | 11–15%/24–35% |
| All 4-NO3-isoprene-RO2 average lifetime: ∼65/73 s | 14–18%/15–19% | 38–42%/10–15% | 22–26%/30–36% | 16–22%/30–43% |
| All isoprene-RO2 average lifetime: ∼107/153 s | 5–8%/5–8% | 52–55%/19–25% | 29–33%/51–56% | 7–12%/14–20% |
If the MCM v3.3.1 mechanism instead of FZJ-NO3-isoprene is used in the model, the nitrate-RO2 population is represented by a single RO2 isomer (E-1-NO3-isoprene-4-OO˙) and at room temperature only 15% of this RO2 isomer would be detectable in the ROxLIF. However, due to the reduction in the MCM to only one RO2 isomer, measured and modelled RO2 concentrations can hardly be compared.
If the CalTech mechanism is applied, the predicted detectable RO2 also reproduces the experimental measurements well (Fig. 5), indicating that the more refined description of the nitrate-RO2 radicals similar to the FZJ-NO3-isoprene model is beneficial. The lower ratio of 1-NO3-isoprene-2-OO˙ to 1-NO3-isoprene-4-OO˙ radicals in the CalTech mechanism compared to the FZJ-NO3-isoprene mechanism leads to a shorter chemical lifetime of the nitrate-RO2 and hence lower RO2 concentrations. Given the uncertainty of the estimated detectability, however, we cannot confirm that these lower predicted detectable RO2 concentrations are in better agreement with the experiment. The RO2 concentrations predicted with the CalTech mechanism are more constant over time, while the FZJ-NO3-isoprene mechanism follows a more gradual decrease in RO2 concentration (Fig. 5). This suggests that the modeling of the RO2 population may be further improved by explicitly incorporating the distinct stereo-specific chemistry of Z- and E-nitrate-RO2 radicals, and basing the nitrate-RO2 populations on the theory-derived equilibria, as done in FZJ-NO3-isoprene. Without accurate calibration of the detectability of the nitrate-RO2, it is hard to further characterize the relative performance of the CalTech and FZJ-NO3-isoprene mechanisms on the RO2 concentration. This is especially true for later times in the experiment where secondary chemistry becomes increasingly important, and the uncertainty in the modelled value increases as the chemical mechanism for these species has not yet been updated to the latest chemical insights.
9.2 Methyl vinyl ketone and methacrolein yields
Prominent stable products from isoprene oxidation are methyl vinyl ketone (MVK) and methacrolein (MACR), produced from and destroyed by various reaction channels such as ozonolyis, NO3 and OH reactions.4 Therefore, differences between measured and modelled MVK and MACR concentrations can be caused by incorrect production or loss from either one of the pathways. For the conditions of the experiments in this work, 80% of isoprene is lost in the reaction with NO3. In the FZJ-NO3-isoprene model, however, less than 1% of the modelled MVK and MACR concentrations originate from the NO3 + isoprene oxidation. The remaining part is formed through the isoprene + OH chemistry (80%) and through ozonolysis (19%) although these two channels represent only ∼10–15% each of the isoprene loss. The FZJ-NO3-isoprene mechanism reproduces the measured MVK + MACR concentrations within 25% (Fig. 6 and additional figures in the ESI†). This difference is close to the uncertainty of the MVK and MACR yields in the mechanism and the measurement uncertainty (±14%). The latter is partially due to the difference in sensitivities for MVK and MACR in the VOCUS PTR-MS, such that the comparison depends on the knowledge of the MACR to MVK ratio.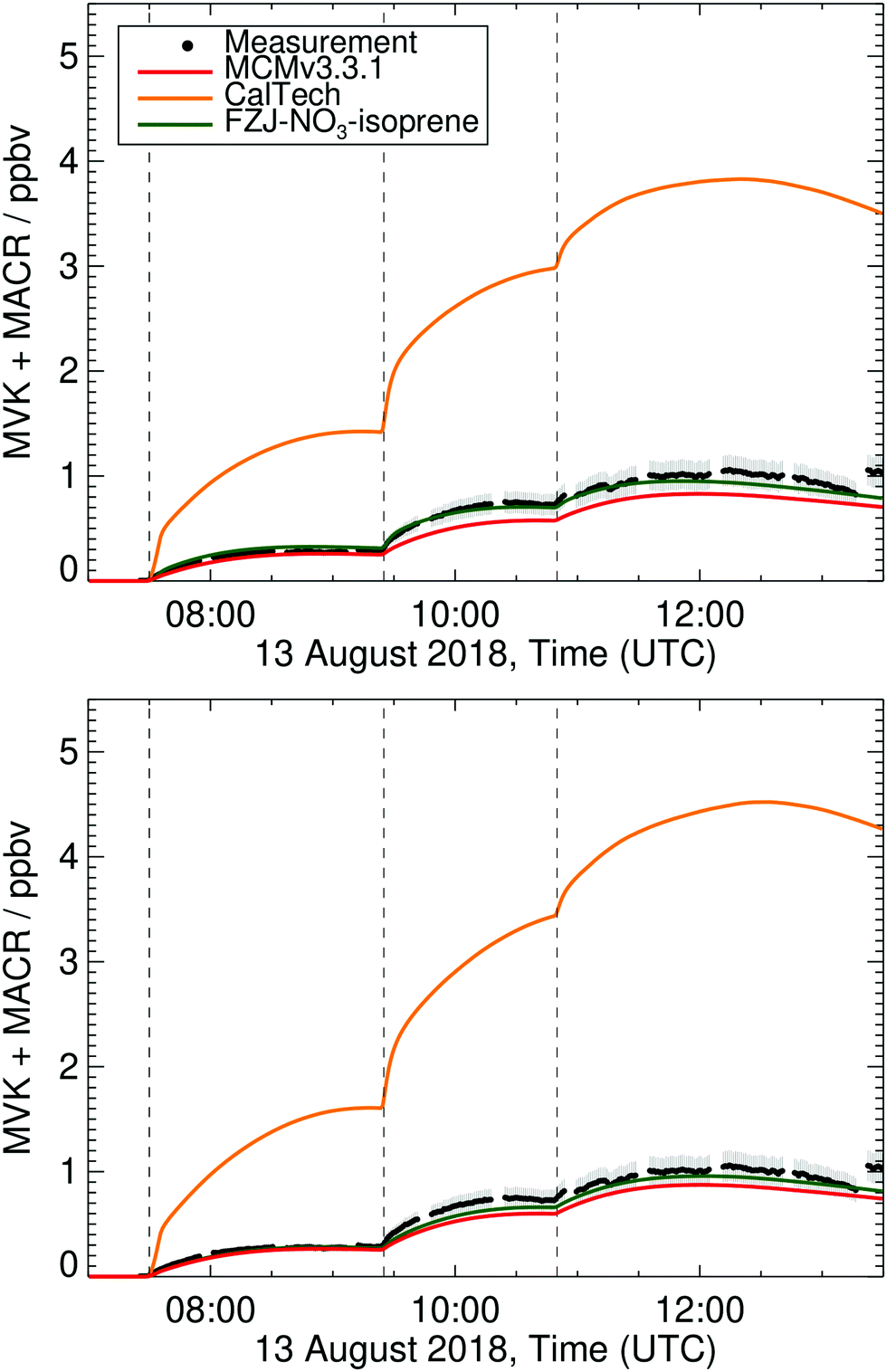 | ||
| Fig. 6 Measured and modelled methyl vinyl ketone + methacrolein (MVK + MACR) concentration time profiles for the experiment on 13 Aug 2018 as described in Dewald et al.,46 with reaction conditions shown in ESI† Section K. Upper panel: Model results with HO2 constrained to measurements; bottom panel: unconstrained HO2. The measurement assumes equal amounts of MVK and MACR are present to account for the differing sensitivities. For the conditions present in these experiments, this assumption is validated by the modelling. | ||
Applying the MCM v3.3.1 mechanism, MVK + MACR concentrations are reproduced within 15%, even though no formation of these compounds from NO3 chemistry is included. This is compensated by a higher MVK yield from isoprene ozonolysis, compared to the FZJ-NO3-isoprene scheme (see ESI†). In contrast to the FZJ-NO3-isoprene and MCM v3.3.1 mechanisms, the CalTech mechanism overestimates MVK + MACR concentrations by between 250 to 400%, mainly due to the complete conversion of 1-NO3-isoprene-2-O˙ radicals, formed in the 1-NO3-isoprene-2-OO˙ + HO2/RO2/NO3 reactions, to MVK. In the FZJ-NO3-isoprene model these nitrate-RO radicals are also formed, but their decomposition to MVK is overwhelmed by the epoxidation reactions described above. Without the competing epoxidation channel, the MVK + MACR concentration predicted by FZJ-NO3-isoprene would even exceed that predicted by CalTech model, owing to the larger fraction of 1-NO3-isoprene-2-OO˙ radicals in the nitrate-RO2 population. A prominent source of 1-NO3-isoprene-2-O˙ radicals is the reaction of nitrate-RO2 + HO2 forming nitrate-RO + OH (reaction (R4)). The need for OH formation in this channel as put to evidence by other experimental studies,41–43 combined with the low measured MVK + MACR concentrations in our experiments indirectly validates the proposed epoxidation with O2 addition chemistry in the FZJ-NO3-isoprene model. Due to the competing, faster bimolecular reactions and its lower yield, the decomposition of the 4-NO3-isoprene-3-OO˙ to MACR only plays a minor role.
10 Conclusions
The NO3-initiated chemistry of isoprene appears to be as complex as the OH-initiated oxidation of isoprene. The available theoretical and experimental data indicates that, similar to the LIM-mechanism for OH-initiated oxidation,12,13 the initial steps of the isoprene + NO3 mechanism must take the site-and stereo-specificity of the NO3 and O2 addition into account, as well as the re-equilibration of the RO2 isomers by O2 elimination/re-addition. The nitrate-RO2 radicals show several reaction channels that may be competitive in pristine environments. For the main NO3-addition channels on the outer carbons of isoprene, the dominant unimolecular reactions of the RO2 formed, with k(298 K) up to 5 × 10−2 s−1, are the migration of the CH2ONO2 H-atoms, forming HPALD + NO2, thus linking the mechanism to the OH-initiated oxidation of isoprene where HPALD is thought to be a key intermediate in OH regeneration.14,19,86 The rate predictions for RO2 H-migration are found to be in good agreement with a recent SAR by Vereecken and Nozière33 for H-migration in RO2 radicals. The rate coefficients for H-migration in nitrate-RO2 are generally slower than in hydroxylated RO2, such that the impact of unimolecular RO2 chemistry in the atmosphere will be significantly less for isoprene + NO3 than for isoprene + OH; at this time we have no experimental evidence for significant HPALD formation in the NO3-initiated oxidation. Only for the RO2 formed in the minor addition channels, i.e. with NO3 adding on the central carbons, are fast ring closure reactions with k(298 K) exceeding 1 s−1 possible that might prove competitive against bimolecular reactions, and might lead to highly oxygenated molecules, albeit in small yields. We observed no products we could assign uniquely to these channels.The chemistry of the nitrated alkoxy radicals shows several fast decomposition and isomerisation reactions leading to the fragmentation of the nitrate group, forming a carbonyl compound and NO2. Decomposition and isomerisation reactions of nitrate-RO radicals are typically slower than in the analogous OH-substituted alkoxy radicals,38,49,60 allowing epoxidation reactions in unsaturated nitrate-RO to become competitive. The role of these epoxidized compounds is not fully elucidated yet, but has important impact on the predicted product formation. For example, the theoretical predictions indicate a very small yield of methyl-vinyl-ketone (MVK) from 1-NO3-isoprene-2-O˙ radicals, contrary to expectations by Wennberg et al.4 The predicted MVK + MACR yields are in good agreement with the experimental observations, provided epoxidation is taken into account. The formation of epoxides could indicate a higher propensity for aerosol formation at nighttime due to reactive uptake on particles. Still, a strong contribution of epoxidation also does not necessarily imply a high yield of epoxidized products, as the epoxide may undergo ring opening in subsequent reaction steps. For example, the dominant epoxidation channel in the important 1-NO3-isoprene-2-O˙ radical still leads mainly to non-epoxidized nitrated MACR, due to ring reopening after a hydrogen shift (Fig. 4).
As predicted in a recent study by Novelli et al.,49 the observation of nitrated RO2 radicals formed from isoprene using a LIF instrument by their conversion to alkoxy radicals and ultimately HO2 with an excess NO is not straightforward, owing to the potential NO2 elimination. When considering this chemistry in the comparison between model and experiment, we find that less than a third of the nitrate-RO2 are detectable, and agreement within a factor ∼2 is obtained, subject to considerable uncertainties on the exact relative conversion efficiency.
Though the current work has characterized a large number of reactions important in the initial stages of the NO3-initiated oxidation of isoprene, further work is needed to elucidate the later stages of the chemistry. The theoretically characterized pathways may also be of importance in the NO3-driven atmospheric chemistry of other unsaturated organic compounds. Finally, though the results in this work are only weakly dependent on the HO2 concentration, further investigation of the cause of the difference between modelled and measured HO2 would be useful.
Author contributions
All authors contributed to the conceptualization, execution and discussion of the campaign on NO3-initiated isoprene oxidation. LV performed the quantum chemical and theoretical kinetic calculations, and derived the oxidation schemes. PC, LV and AN implemented the mechanisms and performed the modeling studies. HF and SSB planned the experiments and organized them. RT and DR provided isoprene and MVK + MACR data. FB, JC, SSB, WM, JS, and LZ provided NO3 data. AN and CC provided the RO2 and HO2 data. LV, AN, PC, and HF wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research has received support by Horizon 2020 Projects EUROCHAMP-2020 (grant no. 730997) and SARLEP (grant no. 681529).References
- A. B. Guenther, X. Jiang, C. L. Heald, T. Sakulyanontvittaya, T. Duhl, L. K. Emmons and X. Wang, The model of emissions of gases and aerosols from nature version 2.1 (MEGAN2.1): An extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 2012, 5, 1471–1492 CrossRef.
- R. P. Wayne, I. Barnes, P. Biggs, J. P. Burrows, C. E. Canosa-Mas, J. Hjorth, G. Le Bras, G. K. Moortgat, D. Perner, G. Poulet, G. Restelli and H. Sidebottom, The nitrate radical: Physics, chemistry, and the atmosphere, Atmos. Environ., 1991, 25, 1–203 CrossRef.
- R. Atkinson, Gas-phase tropospheric chemistry of organic compounds: A review, Atmos. Environ., 2007, 41(Supplement), 200–240 CrossRef.
- P. O. Wennberg, K. H. Bates, J. D. Crounse, L. G. Dodson, R. C. McVay, L. A. Mertens, T. B. Nguyen, E. Praske, R. H. Schwantes, M. D. Smarte, J. M. St Clair, A. P. Teng, X. Zhang and J. H. Seinfeld, Gas-phase reactions of isoprene and its major oxidation products, Chem. Rev., 2018, 118, 3337–3390 CrossRef CAS.
- A. Geyer, B. Alicke, S. Konrad, T. Schmitz, J. Stutz and U. Platt, Chemistry and oxidation capacity of the nitrate radical in the continental boundary layer near Berlin, J. Geophys. Res.: Atmos., 2001, 106, 8013–8025 CrossRef CAS.
- B. J. Finlayson-Pitts and J. N. Pitts, Chemistry of the Upper and Lower Atmosphere: Theory, Experiments, and Applications, Academic Press, San Diego, 1999 Search PubMed.
- J. Liebmann, E. Karu, N. Sobanski, J. Schuladen, M. Ehn, S. Schallhart, L. Quelever, H. Hellen, H. Hakola, T. Hoffmann, J. Williams, H. Fischer, J. Lelieveld and J. N. Crowley, Direct measurement of NO3 radical reactivity in a boreal forest, Atmos. Chem. Phys., 2018, 18, 3799–3815 CrossRef CAS.
- C. Warneke, J. A. de Gouw, P. D. Goldan, W. C. Kuster, E. J. Williams, B. M. Lerner, R. Jakoubek, S. S. Brown, H. Stark, M. Aldener, A. R. Ravishankara, J. M. Roberts, M. Marchewka, S. Bertman, D. T. Sueper, S. A. McKeen, J. F. Meagher and F. C. Fehsenfeld, Comparison of daytime and nighttime oxidation of biogenic and anthropogenic VOCs along the New England coast in summer during New England Air Quality Study 2002, J. Geophys. Res.: Atmos., 2004, 109, D10309 CrossRef.
- S. S. Brown, W. P. Dube, J. Peischl, T. B. Ryerson, E. Atlas, C. Warneke, J. A. de Gouw, S. te, L. Hekkert, C. A. Brock, F. Flocke, M. Trainer, D. D. Parrish, F. C. Feshenfeld and A. R. Ravishankara, Budgets for nocturnal VOC oxidation by nitrate radicals aloft during the 2006 Texas Air Quality Study, J. Geophys. Res.: Atmos., 2011, 116, D24305 Search PubMed.
- J. M. Liebmann, J. B. A. Muller, D. Kubistin, A. Claude, R. Holla, C. Plass-Duelmer, J. Lelieveld and J. N. Crowley, Direct measurements of NO3 reactivity in and above the boundary layer of a mountaintop site: Identification of reactive trace gases and comparison with OH reactivity, Atmos. Chem. Phys., 2018, 18, 12045–12059 CrossRef CAS.
- J. Liebmann, N. Sobanski, J. Schuladen, E. Karu, H. Hellén, H. Hakola, Q. Zha, M. Ehn, M. Riva, L. Heikkinen, J. Williams, H. Fischer, J. Lelieveld and J. N. Crowley, Alkyl nitrates in the boreal forest: formation via the NO3-, OH- and O3-induced oxidation of biogenic volatile organic compounds and ambient lifetimes, Atmos. Chem. Phys., 2019, 19, 10391–10403 CrossRef CAS.
- J. Peeters, T. L. Nguyen and L. Vereecken, HOx radical regeneration in the oxidation of isoprene, Phys. Chem. Chem. Phys., 2009, 11, 5935–5939 RSC.
- J. Peeters, J.-F. J. Müller, T. Stavrakou and V. S. Nguyen, Hydroxyl radical recycling in isoprene oxidation driven by hydrogen bonding and hydrogen tunneling: The upgraded LIM1 mechanism, J. Phys. Chem. A, 2014, 118, 8625–8643 CrossRef CAS.
- A. Novelli, L. Vereecken, B. Bohn, H.-P. Dorn, G. I. Gkatzelis, A. Hofzumahaus, F. Holland, D. Reimer, F. Rohrer, S. Rosanka, D. Taraborrelli, R. Tillmann, R. Wegener, Z. Yu, A. Kiendler-Scharr, A. Wahner and H. Fuchs, Importance of isomerization reactions for the OH radical regeneration from the photo-oxidation of isoprene investigated in the atmospheric simulation chamber SAPHIR, Atmos. Chem. Phys., 2020, 20, 3333–3355 CrossRef CAS.
- J. Lelieveld, T. M. Butler, J. N. Crowley, T. J. Dillon, H. Fischer, L. Ganzeveld, H. Harder, M. G. Lawrence, M. Martinez, D. Taraborrelli and J. Williams, Atmospheric oxidation capacity sustained by a tropical forest, Nature, 2008, 452, 737–740 CrossRef CAS.
- K. H. Møller, K. H. Bates and H. G. Kjaergaard, The importance of peroxy radical hydrogen-shift reactions in atmospheric isoprene oxidation, J. Phys. Chem. A, 2019, 123, 920–932 CrossRef.
- T. Berndt, Formation of carbonyls and hydroperoxyenals (HPALDs) from the OH radical reaction of isoprene for low-NOx conditions: Influence of temperature and water vapour content, J. Atmos. Chem., 2012, 69, 253–272 CrossRef CAS.
- A. P. Teng, J. D. Crounse and P. O. Wennberg, Isoprene peroxy radical dynamics, J. Am. Chem. Soc., 2017, 139, 5367–5377 CrossRef CAS.
- J.-F. Müller, T. Stavrakou and J. Peeters, Chemistry and deposition in the model of atmospheric composition at global and regional scales using inversion techniques for trace gas emissions (MAGRITTE v1.1) – Part 1: Chemical mechanism, Geosci. Model Dev., 2019, 12, 2307–2356 CrossRef.
- H. Fuchs, A. Hofzumahaus, F. Rohrer, B. Bohn, T. Brauers, H.-P. Dorn, R. Häseler, F. Holland, M. Kaminski, X. Li, K. Lu, S. Nehr, R. Tillmann, R. Wegener and A. Wahner, Experimental evidence for efficient hydroxyl radical regeneration in isoprene oxidation, Nat. Geosci., 2013, 6, 1023–1026 CrossRef CAS.
- V. S. Nguyen and J. Peeters, Fast (E)–(Z) isomerization mechanisms of substituted allyloxy radicals in isoprene oxidation, J. Phys. Chem. A, 2015, 119, 7270–7276 CrossRef CAS.
- J. Peeters and T. L. Nguyen, Unusually fast 1,6-H shifts of enolic hydrogens in peroxy radicals: Formation of the first-generation C2 and C3 carbonyls in the oxidation of isoprene, J. Phys. Chem. A, 2012, 116, 6134–6141 CrossRef CAS.
- J. D. Crounse, F. Paulot, H. G. Kjaergaard and P. O. Wennberg, Peroxy radical isomerization in the oxidation of isoprene, Phys. Chem. Chem. Phys., 2011, 13, 13607–13613 RSC.
- J. D. Crounse, L. B. Nielsen, S. Jørgensen, H. G. Kjaergaard and P. O. Wennberg, Autoxidation of organic compounds in the atmosphere, J. Phys. Chem. Lett., 2013, 4, 3513–3520 CrossRef CAS.
- G. da Silva, Oxidation of carboxylic acids regenerates hydroxyl radicals in the unpolluted and nighttime troposphere, J. Phys. Chem. A, 2010, 114, 6861–6869 CrossRef CAS.
- S. Wang, M. Riva, C. Yan, M. Ehn and L. Wang, Primary formation of highly oxidized multifunctional products in the OH-initiated oxidation of isoprene: A combined theoretical and experimental study, Environ. Sci. Technol., 2018, 52, 12255–12264 CrossRef CAS.
- B. Nozière and L. Vereecken, Direct observation of aliphatic peroxy radical autoxidation and water effects: An experimental and theoretical study, Angew. Chem., Int. Ed., 2019, 58, 13976–13982 CrossRef.
- E. Praske, R. V. Otkjær, J. D. Crounse, J. C. Hethcox, B. M. Stoltz, H. G. Kjaergaard and P. O. Wennberg, Intramolecular hydrogen shift chemistry of hydroperoxy-substituted peroxy radicals, J. Phys. Chem. A, 2019, 123, 590–600 CrossRef CAS.
- L. Xu, K. H. Møller, J. D. Crounse, R. Otkær, H. G. Kjaergaard and P. O. Wennberg, Unimolecular reactions of peroxy radicals formed in the oxidation of α-pinene and β-pinene by hydroxyl radicals, J. Phys. Chem. A, 2019, 123, 1661–1674 CrossRef CAS.
- R. V. Otkjær, H. H. Jakobsen, C. M. Tram and H. G. Kjaergaard, Calculated hydrogen shift rate constants in substituted alkyl peroxy radicals, J. Phys. Chem. A, 2018, 112, 8665–8673 CrossRef.
- A. Miyoshi, Systematic computational study on the unimolecular reactions of alkylperoxy (RO2), Hydroperoxyalkyl (QOOH), and hydroperoxyalkylperoxy (O2QOOH) radicals, J. Phys. Chem. A, 2011, 115, 3301–3325 CrossRef CAS.
- S. Sharma, S. Raman and W. H. Green, Intramolecular hydrogen migration in alkylperoxy and hydroperoxyalkylperoxy radicals: Accurate treatment of hindered rotors, J. Phys. Chem. A, 2010, 114, 5689–5701 CrossRef CAS.
- L. Vereecken and B. Nozière, H migration in peroxy radicals under atmospheric conditions, Atmos. Chem. Phys., 2020, 20, 7429–7458 CrossRef CAS.
- M. E. Jenkin, S. M. Saunders and M. J. Pilling, The tropospheric degradation of volatile organic compounds: A protocol for mechanism development, Atmos. Environ., 1997, 31, 81–104 CrossRef CAS.
- S. M. Saunders, M. E. Jenkin, R. G. Derwent and M. J. Pilling, Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): Tropospheric degradation of non-aromatic volatile organic compounds, Atmos. Chem. Phys., 2003, 3, 161–180 CrossRef CAS.
- M. E. Jenkin, J. C. Young and A. R. Rickard, The MCM v3.3.1 degradation scheme for isoprene, Atmos. Chem. Phys., 2015, 15, 11433–11459 CrossRef CAS.
- L. Vereecken, B. Aumont, I. Barnes, J. W. Bozzelli, M. J. Goldman, W. H. Green, S. Madronich, M. R. McGillen, A. Mellouki, J. J. Orlando, B. Picquet-Varrault, A. R. Rickard, W. R. Stockwell, T. J. Wallington and W. P. L. Carter, Perspective on mechanism development and structure–activity relationships for gas-phase atmospheric chemistry, Int. J. Chem. Kinet., 2018, 50, 435–469 CrossRef CAS.
- L. Vereecken and J. Peeters, Decomposition of substituted alkoxy radicals—part I: A generalized structure–activity relationship for reaction barrier heights, Phys. Chem. Chem. Phys., 2009, 11, 9062–9074 RSC.
- T. Berndt and O. Böge, Gas-phase reaction of NO3 radicals with isoprene: A kinetic and mechanistic study, Int. J. Chem. Kinet., 1997, 29, 755–765 CrossRef CAS.
- I. Suh, W. Lei and R. Zhang, Experimental and theoretical studies of isoprene reaction with NO3, J. Phys. Chem. A, 2001, 105, 6471–6478 CrossRef CAS.
- A. W. Rollins, A. Kiendler-Scharr, J. L. Fry, T. Brauers, S. S. Brown, H.-P. Dorn, W. P. Dube, H. Fuchs, A. Mensah, T. F. Mentel, F. Rohrer, R. Tillmann, R. Wegener, P. J. Wooldridge and R. C. Cohen, Isoprene oxidation by nitrate radical: Alkyl nitrate and secondary organic aerosol yields, Atmos. Chem. Phys., 2009, 9, 6685–6703 CrossRef CAS.
- R. H. Schwantes, A. P. Teng, T. B. Nguyen, M. M. Coggon, J. D. Crounse, J. M. St Clair, X. Zhang, K. A. Schilling, J. H. Seinfeld and P. O. Wennberg, Isoprene NO3 oxidation products from the RO2 + HO2 pathway, J. Phys. Chem. A, 2015, 119, 10158–10171 CrossRef CAS.
- A. J. Kwan, A. W. H. Chan, N. L. Ng, H. G. Kjaergaard, J. H. Seinfeld and P. O. Wennberg, Peroxy radical chemistry and OH radical production during the NO3-initiated oxidation of isoprene, Atmos. Chem. Phys., 2012, 12, 7499–7515 CrossRef CAS.
- E. S. C. Kwok, S. M. Aschmann, J. Arey and R. Atkinson, Product formation from the reaction of the NO3 radical with isoprene and rate constants for the reactions of methacrolein and methyl vinyl ketone with the NO3 radical, Int. J. Chem. Kinet., 1996, 28, 925–934 CrossRef CAS.
- D. Casanova, F. P. Rotzinger and M. Graetzel, Computational study of promising organic dyes for high-performance sensitized solar cells, J. Chem. Theory Comput., 2010, 6, 1219–1227 CrossRef CAS.
- P. Dewald, J. M. Liebmann, N. Friedrich, J. Shenolikar, J. Schuladen, F. Rohrer, D. Reimer, R. Tillmann, A. Novelli, C. Cho, K. Xu, R. Holzinger, F. Bernard, L. Zhou, W. Mellouki, S. S. Brown, H. Fuchs, J. Lelieveld and J. N. Crowley, Evolution of NO3 reactivity during the oxidation of isoprene, Atmos. Chem. Phys., 2020, 20, 10459–10475 CrossRef CAS.
- M. A. H. Khan, B.-L. Schlich, M. E. Jenkin, M. C. Cooke, R. G. Derwent, J. L. Neu, C. J. Percival and D. E. Shallcross, Changes to simulated global atmospheric composition resulting from recent revisions to isoprene oxidation chemistry, Atmos. Environ., 2021, 244, 117914 CrossRef CAS.
- J. Zhao and R. Zhang, A theoretical investigation of nitrooxyalkyl peroxy radicals from NO3-initiated oxidation of isoprene, Atmos. Environ., 2008, 42, 5849–5858 CrossRef CAS.
- A. Novelli, C. Cho, H. Fuchs, A. Hofzumahaus, F. Rohrer, R. Tillmann, A. Kiendler-Scharr, A. Wahner and L. Vereecken, Experimental and theoretical study on the impact of a nitrate group on the chemistry of alkoxy radicals, Phys. Chem. Chem. Phys., 2021 10.1039/d0cp05555g.
- T. H. Dunning, Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- G. D. Purvis and R. J. Bartlett, A full coupled-cluster singles and doubles model: The inclusion of disconnected triples, J. Chem. Phys., 1982, 76, 1910 CrossRef CAS.
- I. M. Alecu, J. Zheng, Y. Zhao and D. G. Truhlar, Computational thermochemistry: Scale factor databases and scale factors for vibrational frequencies obtained from electronic model chemistries, J. Chem. Theory Comput., 2010, 6, 2872–2887 CrossRef CAS.
- J. L. Bao, J. Zheng, I. M. Alecu, B. J. Lynch, Y. Zhao and D. G. Truhlar, Database of Frequency Scale Factors for Electronic Model Chemistries (Version 4), http://comp.chem.umn.edu/freqscale/index.html.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Eara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Mntgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision B.01, Gaussian Inc., Wallington, CT, 2016 Search PubMed.
- D. G. Truhlar, B. C. Garrett and S. J. Klippenstein, Current status of transition-state theory, J. Phys. Chem., 1996, 100, 12771–12800 CrossRef CAS.
- L. Vereecken and J. Peeters, The 1,5-H-shift in 1-butoxy: A case study in the rigorous implementation of transition state theory for a multirotamer system, J. Chem. Phys., 2003, 119, 5159–5170 CrossRef CAS.
- C. Eckart, The penetration of a potential barrier by electrons, Phys. Rev., 1930, 35, 1303–1309 CrossRef CAS.
- H. S. Johnston and J. Heicklen, Tunneling corrections for unsymmetrical Eckart potential energy barriers, J. Phys. Chem., 1962, 66, 532–533 CrossRef.
- L. Vereecken and J. Peeters, A structure–activity relationship for the rate coefficient of H-migration in substituted alkoxy radicals, Phys. Chem. Chem. Phys., 2010, 12, 12608–12620 RSC.
- L. Vereecken, A. Novelli and D. Taraborrelli, Unimolecular decay strongly limits concentration of Criegee intermediates in the atmosphere, Phys. Chem. Chem. Phys., 2017, 19, 31599–31612 RSC.
- L. Vereecken, G. Huyberechts and J. Peeters, Stochastic simulation of chemically activated unimolecular reactions, J. Chem. Phys., 1997, 106, 6564–6573 CrossRef CAS.
- F. Rohrer, B. Bohn, T. Brauers, D. Bruning, F. J. Johnen, A. Wahner and J. Kleffmann, Characterisation of the photolytic HONO-source in the atmosphere simulation chamber SAPHIR, Atmos. Chem. Phys., 2005, 5, 2189–2201 CrossRef CAS.
- D. Poppe, T. Brauers, H.-P. Dorn, M. Karl, T. Mentel, E. Schlosser, R. Tillmann, R. Wegener and A. Wahner, OH-Initiated degradation of several hydrocarbons in the atmosphere simulation chamber SAPHIR, J. Atmos. Chem., 2007, 57, 203–214 CrossRef CAS.
- E. Schlosser, B. Bohn, T. Brauers, H.-P. Dorn, H. Fuchs, R. Haeseler, A. Hofzumahaus, F. Holland, F. Rohrer, L. O. Rupp, M. Siese, R. Tillmann and A. Wahner, Intercomparison of two hydroxyl radical measurement techniques at the atmosphere simulation chamber SAPHIR, J. Atmos. Chem., 2007, 56, 187–205 CrossRef CAS.
- F. Holland, A. Hofzumahaus, R. Schafer, A. Kraus and H. W. Patz, Measurements of OH and HO2 radical concentrations and photolysis frequencies during BERLIOZ, J. Geophys. Res.: Atmos., 2003, 108, 8246 CrossRef.
- H. Fuchs, B. Bohn, A. Hofzumahaus, F. Holland, K. D. Lu, S. Nehr, F. Rohrer and A. Wahner, Detection of HO2 by laser-induced fluorescence: Calibration and interferences from RO2 radicals, Atmos. Meas. Tech., 2011, 4, 1209–1225 CrossRef CAS.
- L. K. Whalley, M. A. Blitz, M. Desservettaz, P. W. Seakins and D. E. Heard, Reporting the sensitivity of laser-induced fluorescence instruments used for HO2 detection to an interference from RO2 radicals and introducing a novel approach that enables HO2 and certain RO2 types to be selectively measured, Atmos. Meas. Tech., 2013, 6, 3425–3440 CrossRef CAS.
- T. S. Dibble, Isomerization of OH-isoprene adducts and hydroxyalkoxy isoprene radicals, J. Phys. Chem. A, 2002, 106, 6643–6650 CrossRef CAS.
- H. Fuchs, S. Albrecht, I. Acir, B. Bohn, M. Breitenlechner, H.-P. Dorn, G. I. Gkatzelis, A. Hofzumahaus, F. Holland, M. Kaminski, F. N. Keutsch, A. Novelli, D. Reimer, F. Rohrer, R. Tillmann, L. Vereecken, R. Wegener, A. Zaytsev, A. Kiendler-Scharr and A. Wahner, Investigation of the oxidation of methyl vinyl ketone (MVK) by OH radicals in the atmospheric simulation chamber SAPHIR, Atmos. Chem. Phys., 2018, 18, 8001–8016 CrossRef CAS.
- X. T. Le, T. V.-T. Mai, K. C. Lin and L. K. Huynh, Low temperature oxidation kinetics of biodiesel molecules: Rate rules for concerted HO2 elimination from alkyl ester peroxy radicals, J. Phys. Chem. A, 2018, 122(42), 8259–8273 CrossRef CAS.
- J. A. Miller and S. J. Klippenstein, The reaction between ethyl and molecular oxygen II: Further analysis, Int. J. Chem. Kinet., 2001, 33, 654–668 CrossRef CAS.
- S. M. Villano, L. K. Huynh, H.-H. Carstensen and A. M. Dean, High-pressure rate rules for alkyl + O2 reactions. 1. The dissociation, concerted elimination, and isomerization channels of the alkyl peroxy radical, J. Phys. Chem. A, 2011, 115, 13425–13442 CrossRef CAS.
- T. L. Nguyen, L. Vereecken and J. Peeters, HOx Regeneration in the oxidation of isoprene III: Theoretical study of the key isomerisation of the Z-δ-hydroxy-peroxy isoprene radicals, ChemPhysChem, 2010, 11, 3996–4001 CrossRef CAS.
- L. Vereecken, in Advances in Atmospheric Chemistry, Organic Oxidation and Multiphase Chemistry, ed. J. R. Barker, A. L. Steiner and T. J. Wallington, World Scientific Publishing Co. Pte. Ltd, Singapore, 1st edn, 2019, vol. 2, pp. 377–527 Search PubMed.
- L. Wang, R. Wu and C. Xu, Atmospheric oxidation mechanism of benzene. Fates of alkoxy radical intermediates and revised mechanism, J. Phys. Chem. A, 2013, 117, 14163–14168 CrossRef CAS.
- C. A. Whelan, M. A. Blitz, R. Shannon, L. Onel, J. P. Lockhart, P. W. Seakins and D. Stone, Temperature and pressure dependent kinetics of QOOH decomposition and reaction with O2: Experimental and theoretical investigations of QOOH radicals derived from Cl + (CH3)3COOH, J. Phys. Chem. A, 2019, 123, 10254–10262 CrossRef CAS.
- T. M. Lenhardt, C. E. Mcdade and K. D. Bayes, Rates of reaction of butyl radicals with molecular-oxygen, J. Chem. Phys., 1980, 72, 304–310 CrossRef CAS.
- IUPAC Subcommittee on Atmospheric Chemical Kinetic Data Evaluation, Evaluated Kinetic Data, 2017, available at: http://iupac.pole-ether.fr/index.html Search PubMed.
- J. Zádor, H. Huang, O. Welz, J. Zetterberg, D. L. Osborn and C. A. Taatjes, Directly measuring reaction kinetics of ˙QOOH – A crucial but elusive intermediate in hydrocarbon autoignition, Phys. Chem. Chem. Phys., 2013, 15, 10753–10760 RSC.
- M. E. Jenkin, R. Valorso, B. Aumont and A. R. Rickard, Estimation of rate coefficients and branching ratios for reactions of organic peroxy radicals for use in automated mechanism construction, Atmos. Chem. Phys., 2019, 19, 7691–7717 CrossRef CAS.
- T. Kurtén, K. H. Møller, T. B. Nguyen, R. H. Schwantes, P. K. Misztal, L. Su, P. O. Wennberg, J. L. Fry and H. G. Kjaergaard, Alkoxy radical bond scissions explain the anomalously low secondary organic aerosol and organonitrate yields from α-pinene + NO3, J. Phys. Chem. Lett., 2017, 8, 2826–2834 CrossRef.
- A. S. Hasson, G. S. Tyndall, J. J. Orlando, S. Singh, S. Q. Hernandez, S. Campbell and Y. Ibarra, Branching ratios for the reaction of selected carbonyl-containing peroxy radicals with hydroperoxy radicals, J. Phys. Chem. A, 2012, 116, 6264–6281 CrossRef CAS.
- J.-P. Le Crâne, M.-T. Rayez, J.-C. Rayez and E. Villenave, A reinvestigation of the kinetics and the mechanism of the CH3C(O)O2 + HO2 reaction using both experimental and theoretical approaches, Phys. Chem. Chem. Phys., 2006, 8, 2163–2171 RSC.
- H. Fuchs, F. Holland and A. Hofzumahaus, Measurement of tropospheric RO2 and HO2 radicals by a laser-induced fluorescence instrument, Rev. Sci. Instrum., 2008, 79, 084104 CrossRef.
- T. Berndt, N. Hyttinen, H. Herrmann and A. Hansel, First oxidation products from the reaction of hydroxyl radicals with isoprene for pristine environmental conditions, Chem. Commun., 2019, 2, 21 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional oxidation schemes, rate coefficients, experimental data, modeling results, RO2 speciation and conversion efficiencies, discussion of converter- and fluorescence cell specific chemistry, the full FZJ-NO3-isoprene model, and calculated geometries, frequencies and energies for all intermediates and transition states. See DOI: 10.1039/d0cp06267g |
| ‡ Present address: Laboratoire de Physique et Chimie de l'Environnement et de l'Espace (LPC2E), Centre National de la Recherche Scientifique (CNRS), Université d'Orléans, Observatoire des Sciences de l'Univers en Région Centre – Val de Loire (OSUC), Orléans, France. |
| § Present address: College of Architecture and Environment, Sichuan University, Chengdu, China. |
| This journal is © the Owner Societies 2021 |