
Analysis of structural organization and interaction mechanisms of detonation nanodiamond particles in hydrosols
Andrey A.
Knizhnik
ab,
Yulia G.
Polynskaya
 *b,
Alexander S.
Sinitsa
ab,
Nikita M.
Kuznetsov
a,
Sergey I.
Belousov
a,
Sergei N.
Chvalun
a and
Boris V.
Potapkin
ab
*b,
Alexander S.
Sinitsa
ab,
Nikita M.
Kuznetsov
a,
Sergey I.
Belousov
a,
Sergei N.
Chvalun
a and
Boris V.
Potapkin
ab
aNational Research Center “Kurchatov Institute”, Moscow, Russia
bKintech Lab Ltd, Moscow, Russia. E-mail: yupol@kintechlab.ru
First published on 18th December 2020
Abstract
Structural organization of hydrogen and oxygen functionalized nanodiamond (ND) particles in hydrosols was investigated using a cryo-TEM method. The formation of chain-like structures was observed for hydrogen functionalized NDs while oxygen functionalized NDs tend to form more compact structures. In order to understand possible interaction mechanisms between NDs in hydrosols and to explain these experimental results, first-principles calculations were performed. Charged H-terminated ND particles and particles with partially dissociated hydroxyl and carboxyl groups on the surface were investigated as models of a real ND particle in solution. For positively charged H-terminated particles, it was established that charge distribution is determined by the values of valence band maximum for the particle facets. For negatively charged oxygen functionalized particles, the charge is localized near functional groups. In both cases, interaction is determined by the interplay between Coulomb interaction and van der Waals attraction. For detailed analysis of the ND interaction, the continual model considering this interplay was developed. The results obtained with this model indicate that the formation of chain structures from linked ND particles is caused by charge separation inside the ND particle. For the H-terminated ND particles in water solution, strongly anisotropic distribution of electrostatic potential around the particles promotes formation of non-compact chain structures of particles via interaction between facets with opposite charges. This effect of charge separation is lower in the oxygen functionalized particles as the charge is localized at the uniformly distributed O-containing functional groups, thus, more compact structures can be formed. These general qualitative statements address the problem of interactions between the large number of ND particles and explain the presented cryo-TEM experimental results.
Introduction
Colloidal diamond nanoparticles (nanodiamonds, ND) are attracting significant interest due to their excellent mechanical and optical properties, high surface areas, tunable surface structures and non-toxicity.1 These properties open great perspectives in different technical2 and medical3,4 applications for different forms of ND materials, e.g., as a dispersion phase in suspension, as a filler in composites or polymers, etc.5 To date, there is a wide range of ND synthesis techniques (see ref. 6 for a review). Two main industrial technologies for ND synthesis are high-pressure high-temperature (HPHT) technology7 and detonation synthesis from explosives (detonation nanodiamonds, DND).8 In both cases, commercially available powders consisting of diamond nanoparticles with a size around several nanometers are produced. Colloidal suspensions were one of the first forms of ND material that became available.9 While for HPHT diamond particles in colloidal suspensions the smallest average particle size is around 10–20 nm,10 detonation method allows to obtain diamond nanoparticles with unusually narrow size in the range of 4–5 nm.2,11,12 Synthesized DND particles in powder or in colloidal solution exist only as agglomerates of interconnected single-crystal particles.13 So, for DND, there is a problem of deagglomeration of nanodiamond particles which is important for various applications11 and has been unsolved yet.To overcome this issue, several experimental methods forcing deagglomeration were introduced: stirred medium milling,14,15 salt-assisted ultrasonic deagglomeration,16 annealing in hydrogen17 and air18 atmosphere followed by powerful ultrasonic treatment. Hydrosols of deagglomerated DND particles obtained by these methods show similar but unusual properties: black color, high viscosity and long-time stability over a wide pH range. The last one was explained due to the high surface charge of the ND particles associated with the presence of functional groups on their surface. The emergence of surface charge on the ND particles may be caused by electrolytic dissociation in water13,19 or by diffusion of electrons from the ND particles into the solvent.20 These effects lead to formation of charged electrical double layers around the particles preventing their coagulation.21
The ability of ND particles to agglomerate in solvents may lead to several interesting phenomena like sol–gel transition in ND hydrosol demonstrated by Vul et al.22 For certain concentrations of ND particles, a special reversible state characterized by giant viscosity was found. The formation of this state was explained in the frame of the model that assumes non-isotropic surface charge distribution. A later analysis of the obtained hydrosols23 demonstrated the existence of chains of linked nanodiamond particles. CryoTEM analysis revealed that ND particles disaggregated by hydrogen treatment form chain structures in hydrosols, while ND particles disaggregated by oxygen treatment form more compact clusters.24 A recent study25 also observed the lace network of ND particles and suggested a theoretical model of agglomeration based on electrostatic interactions between faceted nanodiamond particles. Opletal et al.26 investigated the meso-structure of aggregated faceted polyhedral nanodiamonds and demonstrated that aggregation is determined by multi-polar facet-dependent surface electrostatic potential. From a theoretical point of view, the stability of ND hydrosols was in general explained in accordance to the Deryagin–Landau–Verwey–Overbeek (DLVO) theory.27 However, general theories like DLVO considers only isotropic charge distribution, so it cannot explain the formation of chain structures. It is generally accepted that agglomeration must be related to the charge distribution and interaction potential of a single DND particle, but the nature of this charge anisotropy remains unclear.
Attempts to understand this nature were undertaken using various theoretical methods, in particular, semi-empirical methods like the density functional based tight binding method with self-consistent charges (SCC DFTB),28–31 empirical molecular dynamic32 and density functional theory (DFT) methods.33 Barnard et al.31 showed that the (100) faces and (100)/(111) edges have a high positive surface electrostatic potential (SEP), and faces (111) – negative SEP, and found that the aggregation is due to strong Coulombic interactions depending upon the crystallographic index of the surface facets. Lai et al.30 using the same DFTB calculations demonstrated that the same model is applicable to H-terminated ND particles. Clearly, the nature of electrostatic potential distribution and its overall value is strongly affected by the state of a single particle surface. This state, apparently, is determined by surface termination by hydrogen or any other functional groups. Theoretical studies of functionalized ND particles34,35 showed that chemisorption of different functional groups is facet-selective, but there is still a question of how adsorbed functional groups affect surface charge distribution and, thus, interaction between ND particles. Another general drawback of the semi-empirical methods used in ND interaction studies28,30,33,35 is significant error in estimation of effective atomic charges.
On the other hand, some studies suggested that the nanodiamond aggregation is the result of van der Waals forces.36–38 Xu et al.33 investigated electrostatic and van der Waals interactions between two unfunctionalized nanodiamonds using the SCC-DFTB method and found that the van der Waals forces are much stronger than the electrostatic forces for these nanodiamonds. Unfortunately, quantitative analysis of Coulomb and van der Waals interactions between the functionalized nanodiamonds has not been performed so far in the literature. So, the mechanism of the aggregation of ND particles into organized chain networks is still a debated topic and should be analyzed using more reliable and accurate theoretical methods.
One of the main goals of our study was comprehensive analysis of the nature of agglomeration of nanodiamonds in water solution. At the first step of this work, we performed a theoretical study of physicochemical properties and interaction mechanisms of two ND particles at the DFT/PBE level of theory. Using Bader analysis, the charge distribution and surface electrostatic potential (SEP) were calculated for both unfunctionalized and functionalized by hydrogen and oxygen containing functional groups ND particles. The contribution of van der Waals and electrostatic forces in the interaction energy of two ND particles was analyzed. For a more general analysis of the interaction of ND particles using the results of quantum-chemistry modeling we built up a continual model based on a numerical solution of the Poisson equation around ND particles. The results obtained with the developed model provided additional insight into the structural organization of ND particles in water solution allowing to explain the experimental results.
Computational details and experimental methods
Structural optimization and energy calculations based on the density functional theory approach were carried out using the PBE (Perdew–Burke–Ernzerhof) functional39 as implemented in the VASP code.40 The interaction of valence electron-core states was described by the Projector-Augmented Wave method (PAW).41 The kinetic energy of the plane wave basis set was 400 eV. The Brillouin-zone integration was performed using a Monkhorst–Pack k point sampling. The 23 × 23 × 23 Å (for single ND particle) and 23 × 23 × 43 Å (for two interacting ND particles) unit cells with periodic boundary conditions are considered with Gamma point. The first order Methfessel–Paxton smearing method with a width of 0.2 eV was used. All energies of functionalized nanodiamonds were calculated using D3-dispersion correction method with Becke–Jonson damping.42 Atom positions were optimized without any constrains until forces on the atoms were less than 0.03 eV Å−1. Structure optimization of a single pure ND particle resulted in surface reconstructions including graphitization of the (111) facets.Based on previous studies,43–45 atomistic model structures of ND particles were constructed using the shape of truncated octahedron. In our calculations, we used the C323 particle as a basic ND model (see Fig. 1) and a larger ND particle with 765 atoms (∼2 nm diameter) in order to understand the role of the ND size. The C323 particle with a size of 1.5 nm has facets with (001) and (111) diamond surfaces. This model allowed us to estimate both the surface electrostatic potential and interaction energies of two ND nanoparticles. To analyze H-functionalized ND particles, we considered the C323 structure after relaxation and passivated all dangling bonds by hydrogen atoms making the C323H136 ND particle. To investigate the NDs with the OH/COOH functionalized groups, we replaced one hydrogen atom on the (001) or (111) facets in the C323H136 particle by the OH- or COOH-group.
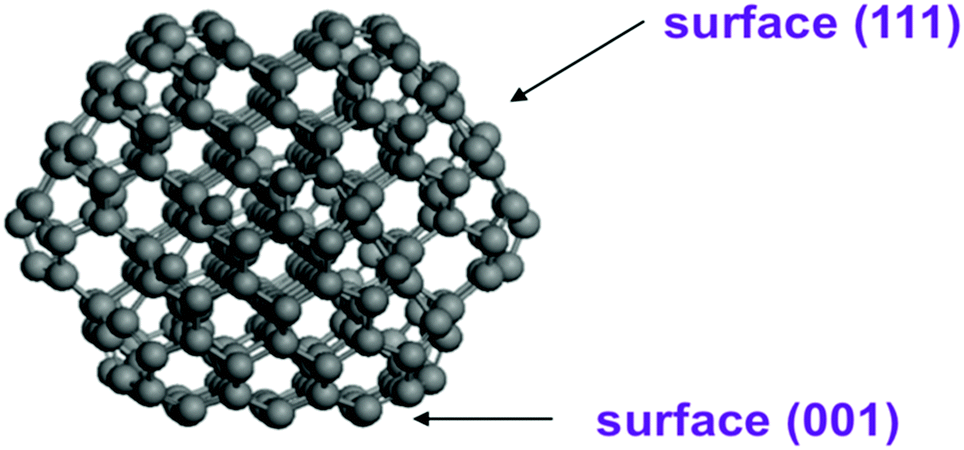 | ||
| Fig. 1 Optimized structure of truncated octahedron (C323) nanodiamonds. | ||
The Bader analysis46 of atomic charges was performed on the calculated charge density grid using the Bader Charge Analysis Code.47 Based on the calculated atomic charges, the surface electrostatic potentials (SEP) of ND particles were estimated.
The interaction energy (Eint) between the two ND particles was calculated as:
| Eint = END1–ND2 − END1 − END2 | (1) |
The continual model was developed to characterize the role of water environment in the aggregation of ND particles. In this model, a numerical solution of Poisson equation around the model ND particle was performed, assuming dipole layers on the particle surface and equipotential distribution inside the particle.
Cryo-TEM images of NDs hydrosols were obtained by Titan Krios 60–300 TEM/STEM (Thermo Fisher Scientific, USA) CryoEM, equipped with the detector Falcon II (Thermo Fisher Scientific, USA) and Cs image corrector (CEOS, Germany), at an accelerating voltage of 300 kV. The sample preparation procedure was performed as routine. The portion of 1 wt% hydrosol (3 ml) was applied to the glow discharged lacey carbon grid, blotted for 1.5 s and then plunge-freezed into liquid ethane cooled by liquid nitrogen in Vitrobot Mark IV (Thermo Fisher Scientific, USA).
Results and discussion
Experimental study
In order to characterize the structural organization of ND particles in water solutions, the cryo-TEM analysis of hydrosols with hydrogen and oxygen functionalized ND particles was performed. Such particles have a positive or negative ζ-potential in an aqueous medium, respectively.13Fig. 2 shows cryo-TEM images of detonation ND hydrosols with different surface functionalization at 1 wt% concentration. | ||
| Fig. 2 Cryo-TEM images of 1 wt% detonation ND hydrosols at various magnifications. (a and c) Are hydrogenated and (b and d) are oxidized NDs, respectively. | ||
It can be seen that in the case of hydrogenated particles (Fig. 2a and c), a percolation network of interacting particles forms, but for oxidized particles (Fig. 2b and d), a primary formation of compact structures is observed and percolation is not achieved at the studied concentration. A similar result was published by us earlier where we explained the differences in the rheological behavior of the particles with various types of surface functionalization.24 The formation of compact structures from the dispersion phase can be comprehended from the standpoint of the standard DLVO theory. However, in the case of NDs, the classical theory cannot explain the difference in the structural organization of particles with different ζ-potentials observed by the cryo-TEM method. This problem motivated the current theoretical study.
Apparently, the observed aggregation of ND particles in water solutions is determined by interaction forces between these particles. For functionalized ND particles, these forces are due to non-covalent interaction, such as long-range van der Waals and coulomb forces. Therefore, at first analysis of the charge and electrostatic potential distribution on pure and differently functionalized ND particles was performed using ab initio calculations and then interactions between these particles was investigated with a more general continual model. Finally, the obtained results were used for the interpretation of experimental data on ND aggregation in water solutions.
Charge distribution and surface potential for nanodiamond
The neutral, positive and negative charged C323 clusters (see Fig. 3a–c) were considered to investigate the distribution of the surface electrostatic potential (SEP) around ND particles. The (111) surface in the neutral unfunctionalized C323 ND exhibits a negative value of SEP (about −0.5 V), whereas the (001) surface possess the positive potential (about 0.2 V). The SEP distribution on different facets for neutral C323 ND is in qualitative agreement with previous theoretical works,31,33 but the calculated values of surface electrostatic potential are significantly smaller than in previously published results. This quantitative discrepancy can be explained by the difference of the calculation methods: the DFT method in this study is more accurate with respect to the prediction of atomic charges than the density functional-based tight binding (DFTB) method with the self-consistent charge which was used in the previous works. For negatively and positively charged C323 particles, there is no significant difference between the SEP values for (001) and (111) facets with respect to neutral ND. The overall physical picture, namely, SEP distribution over (001) and (111) facets, its sign and value range, is the same for C323 and C765 ND particles (see Fig. 3d). So, one can assume that our further statements are valid for ND particles with larger sizes.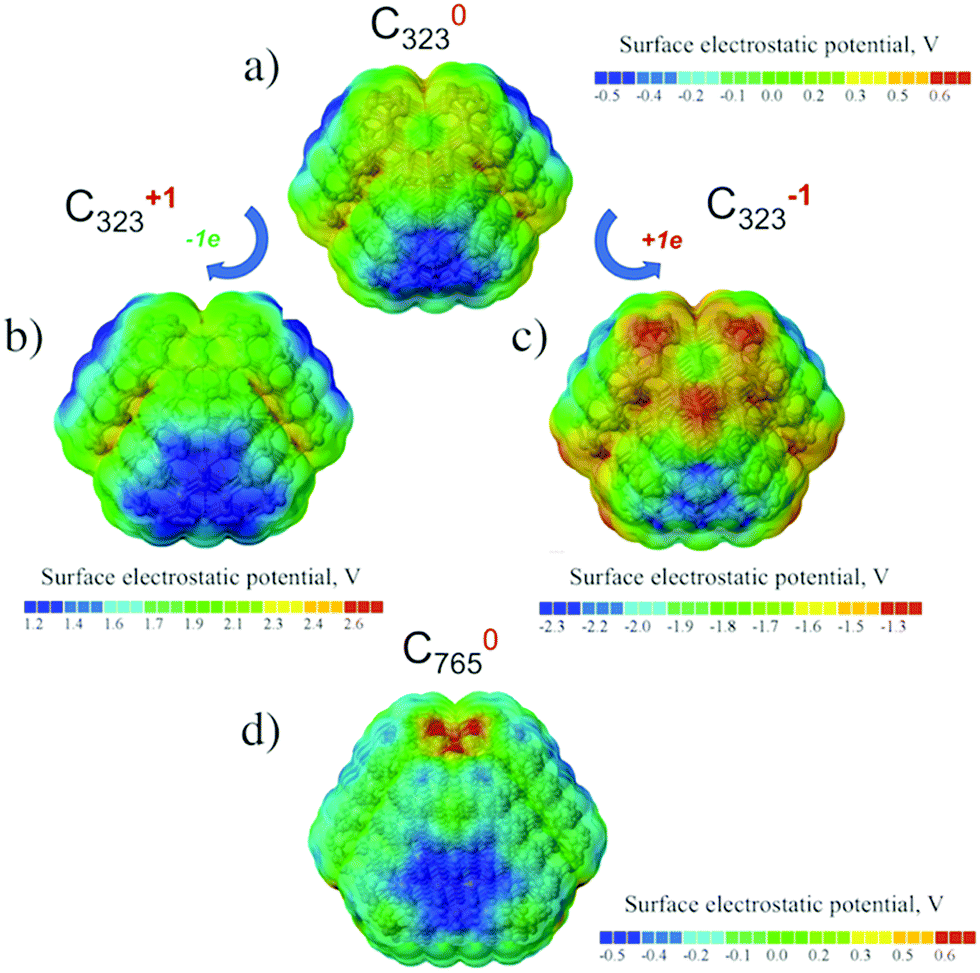 | ||
| Fig. 3 Surface electrostatic potential distribution for the optimized structures of the neutral (a), positively (b) and negatively (c) charged C323, (d) neutral C765 nanodiamonds. Corresponding absolute values of SEP in volts are given in colored scales below the structures. | ||
Real DND particles disaggregated by hydrogen or oxygen treatment should have functional groups on the surface. Therefore, in order to understand the experimental results for colloidal DND particles, it is more relevant to analyze the potential distribution for functionalized NDs.
For hydrogen-treated ND particles, all dangling bonds on the particle surface were passivated by hydrogen atoms. The calculated SEP distribution for the H-terminated C323 particle is shown in Fig. 4a. One can see that the difference of SEP values on the (111) and (001) facets is much smaller than for the unfunctionalized particle and cannot be determined reliably from this figure. Thus, hydrogen termination results in isotropic distribution of charge and electrostatic potential on the ND particle surfaces.
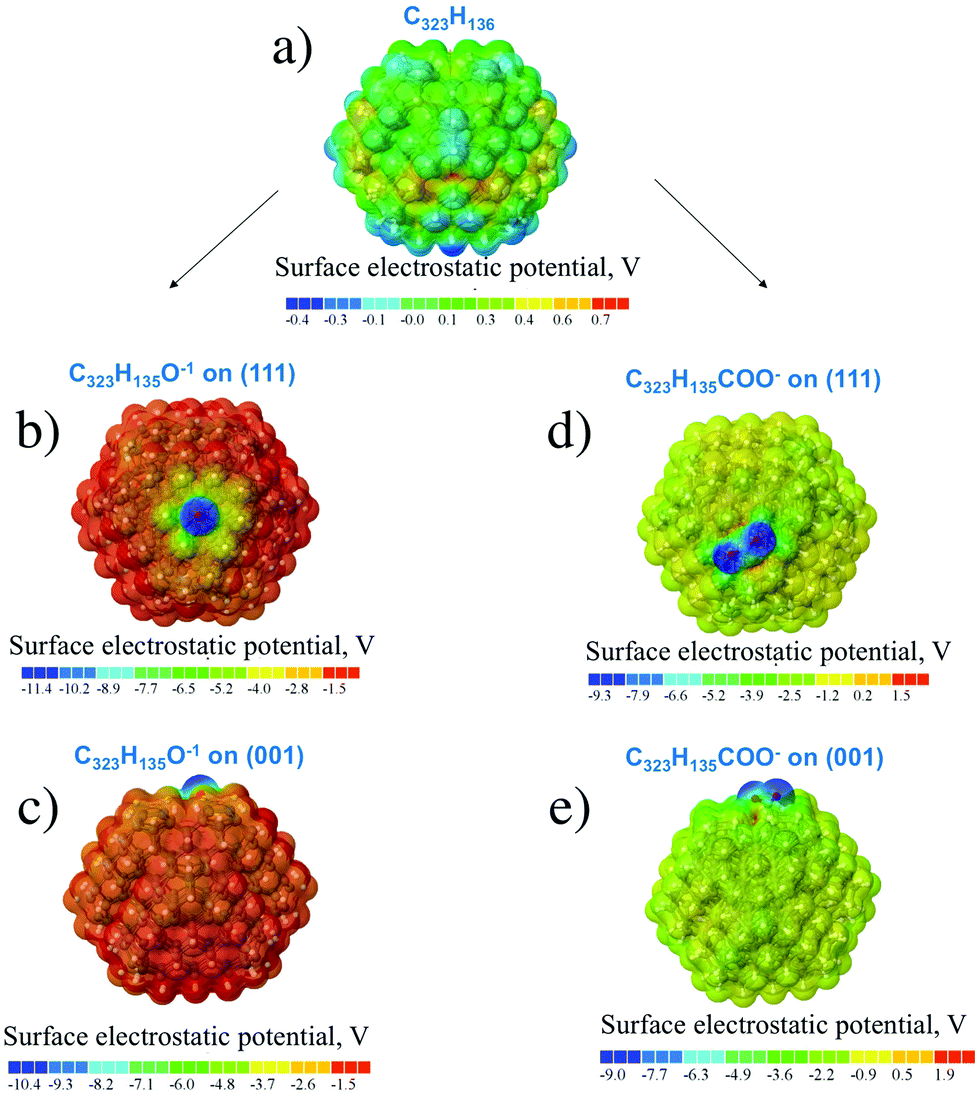 | ||
| Fig. 4 Calculated distribution of surface electrostatic potentials in the H-terminated nanodiamond particle C323H136 (a) and functionalized nanodiamond particles C323H135O− (b and c) and C323H135COO− (d and e) structures. Corresponding absolute values of SEP in volts are given in colored scales below the structures. | ||
For oxygen treated ND particles, there are different oxygen-containing functional groups on the surface, mainly hydroxyl and carboxyl groups.36 The electrolytic dissociation of these groups in hydrosol leads to negative charging of these particles in water solutions. Therefore, the distribution of surface electrostatic potential for OH- and COOH-functionalized NDs with partially dissociated functional groups O−/COO−, C323H135O− and C323H135COO− in the negative charged state was calculated. From Fig. 4b–e it can be seen that the negative SEP is localized on the oxygen atoms for all considered cases. First-principles calculations showed that the formation energy of the COO−-group on the (001) surface is close to that on the (111) surface as the calculated formation energy difference is only 0.1 eV. Previously, Lai et al.35 using the SCC-DFTB method investigated the COOH-group adsorption on the different ND surfaces and found that the (001) and (110) facets provide a more stable adsorption of carboxyl-group than the (111) facet with formation energy differences of about several eV. This can be related to the semi-empirical DFTB method used in that work, while a more accurate DFT method is used here. Moreover, in the work35 only non-charged COOH-groups on the diamond were considered. Thus, it can be assumed that (COO)− charged carboxyl groups should have homogeneous distribution on the ND surface.
Analysis of surface electrostatic potential of charged ND particles functionalized with OH- and COOH-groups showed that negative charge is localized near the considered oxygen-containing group after their dissociation in water solution. Moreover, the OH- and COOH-groups should have a homogeneous distribution on the ND surface due to similar formation energies on the (111) and (001) surfaces.
We want to note that the presented quantum-chemistry calculations of surface electrostatic potentials were made in vacuum, as calculations in the real environment are even more computational-costly and lie beyond current computational powers for considered particles with <1000 atoms, so direct comparison of the obtained values with the experimental data is incorrect. For rough comparison, the water environment can be taken into account if we divide the SEP value from QC calculations by the dielectric constant of water. The experimental values of ζ-potential of ND particles with a size around 4–5 nm varies from 0.02 to 0.05 V.17,48 The average ζ-potential based on the QC-calculated values is ∼2 eV (see Fig. 4b–e). This gives an extrapolated value (taking into account the dielectric constant of water ε ∼ 81) of about 0.01–0.02 eV, which is in fine agreement with the experimental data.
Interaction of nanodiamond particles
In this section, the interaction of ND particles with different charges and functionalizations using DFT calculations was studied. It was found that the interaction energy Eint of two unfunctionalized C323 NDs is about −3.59 eV (see eqn (1)). Comparative analysis of van der Waals and covalent contributions to Eint showed that the covalent contribution is about −2.86 eV in this case. Thus, covalent bonding is responsible for the unfunctionalized NDs interaction. However, as mentioned above, in water solutions, ND particles should be functionalized and the nature of interaction of such NDs is still unclear. So, for explaining the presented experimental data, we focused on the investigation of non-covalent interaction between functionalized NDs.At first, the interaction energies for a pair of identical neutral C323H136 NDs with different orientations were calculated. The interaction between the (001)/(001), (001)/(111), and (111)/(111) facets was considered (see Fig. 5). The relaxed shortest distances (d) between hydrogen atoms of two ND particles with different facet matching were similar in all optimized structures (≈2.5 Å) as well as interaction energies Eint ∼ 0.6 eV (see Table 1). The difference between interaction energies does not exceed 0.15 eV. One can also see from Table 1 that the van der Waals interaction EvdW gives the largest (about 90%) contribution to the total interaction energy Eint. These results are in agreement with the DFT-based study by Xu et al.,33 where it was also concluded that the interaction between two NDs is driven by the van der Waals forces with their contribution of ~90% to the interaction energy. Such significant contribution of the van der Waals interaction and relatively low difference between interaction energies allow us to conclude that the interaction between neutral H-terminated NDs is isotropic.
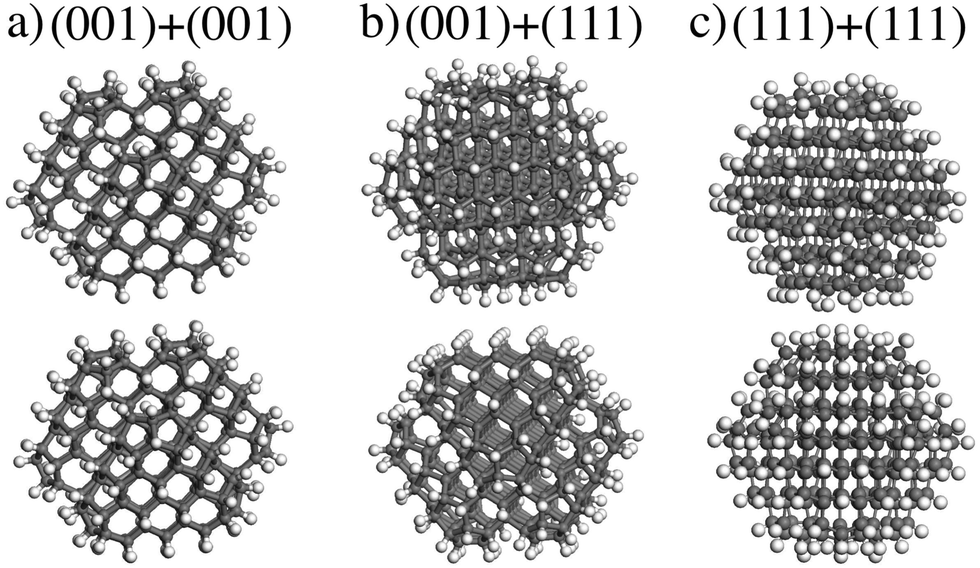 | ||
| Fig. 5 The optimized structures of two H-terminated C323H136 NDs interaction: the interaction between (a) (001)/(001), (b) (001)/(111), and (c) (111)/(111) ND particle facet. | ||
| Functionalization | Charge | Facet matching | d, Å | E int, eV | E vdW, eV |
|---|---|---|---|---|---|
| Hydrogen | 0 | (001)/(001) | 2.5 | −0.63 | −0.54 |
| (001)/(111) | 2.6 | −0.50 | −0.42 | ||
| (111)/(111) | 2.6 | −0.65 | −0.66 | ||
| OH-group | −2 | O−@(001)/(111) | 3.5 | −0.71 | −0.53 |
| (001)/O−@(111) | 3.5 | −0.48 | −0.59 | ||
| COOH-group | −2 | COO−@(001)/(111) | 3.5 | −0.38 | −0.49 |
| (001)/COO−@(111) | 3.5 | −0.51 | −0.41 | ||
Then, interactions of two charged NDs functionalized with hydroxyl and carboxyl groups were investigated. Two cases were considered: (i) interaction of the (001) surface with O−/COO−-group and the H-terminated (111) surface; (ii) interaction of the (111) surface with O−/COO−-group and H-terminated (001) surface (see Fig. 6). The interaction energies for these configurations are given in Table 1. For the O− group, the interaction energy between ND particles may increase with respect to H-terminated particles, which can be attributed to polarization interaction of the charged group with the neighboring dielectric particle. However, for the COO− group interaction, energies are the same or smaller than that for the H-terminated particle, which can be explained by a larger distance between particles (3.5 Å vs. 2.5 Å) because the carboxyl functional group acts as protrusions on the ND surface. Thus, calculations show that for two charged interacting ND particles with O−/COO− groups there is still an interplay between additional Coulomb interaction and decreased van der Waals attraction. Because OH- and COOH-groups should have homogeneous distribution on the ND surface due to similar formation energies, weak interaction anisotropy for such particles in experimental samples can be expected. Thus, these atomistic first-principles calculations do not give the full picture of formation of experimentally observed aggregates.
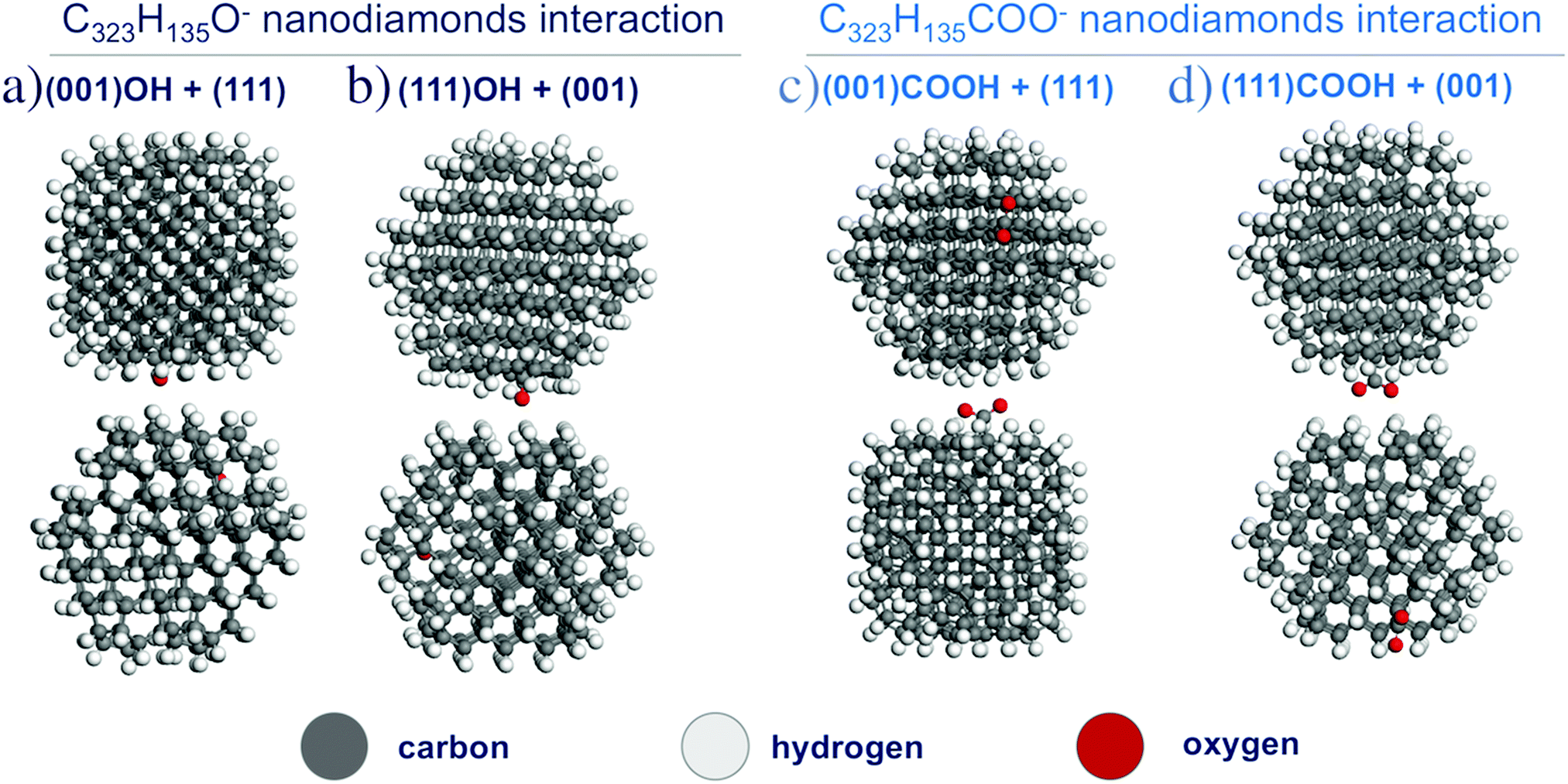 | ||
| Fig. 6 The optimized structures of two C323H135O− (a and b) and C323H135COO− (c and d) nanodiamond interactions. | ||
Interpretation of experimental data on ND aggregation in a water solution – continual model
The presented results of first-principles calculations of charge and electrostatic potential distribution for a single ND particle with electrolytically dissociated oxygen-containing groups demonstrated that negative charge is localized on oxygen atoms in these groups. Also, these calculations showed that energetics of (COO)− charged carboxyl groups is virtually the same on the (001) and (111) diamond surface. Thus, one can expect that (COO)− charged carboxyl groups are distributed uniformly and act as surface charge localizers.A more puzzling question is positive charging of hydrogen-treated ND particles in hydrosols. It was suggested in literature49 that surface transfer doping can take place for hydrogenated diamond surfaces in water solutions, resulting in the formation of positive ζ-potential. This effect is similar to surface conductivity of hydrogenated bulk diamond material under exposure to air or water.20 The surface transfer doping is determined by the position of valence band maximum (VBM) or electron affinity of a diamond surface. The larger the value of VBM depth is, the higher energy is needed to remove the electron from the particle.
In order to characterize charge transfer properties of different facets of H-terminated NDs, the VBM position with respect to the vacuum level of the infinite H-terminated (001) and (111) diamond surfaces was calculated using the DFT/PBE level of theory. The calculated VBM depth for the infinite H-terminated (001) surface is about 4.1 eV, which is higher by 0.6 eV than the VBM depth for H-terminated (111) diamond surface (3.5 eV). It is known from the experiment that the H-terminated (111) diamond surface has negative electron affinity,50 which is in agreement with the calculated value of VBM depth.
The difference of VBM positions with respect to vacuum level can be attributed to different values of C–H group dipoles on the (001) and (111) surfaces. The difference of the VBM depth of diamond surfaces is not important for neutral ND particles, since there are no free charge carriers in such particles to perform charge redistribution. However, for charged ND particles the difference of VBM depth of different ND facets should induce charge redistribution between different facets. In particular, the H-terminated (001) facet with deeper VBM should accept electrons from the (111) facet with higher VBM position, so the (001) facet should be negatively charged, while the (111) facet should be positively charged in order to equilibrate work functions of these surfaces.
The additional voltage drop induced by these surface charges should be equal to 1/2ΔVVBM difference of the VBM depth for these facets, which is about 0.5 V in accordance with the presented first-principles calculations. Thus, the additional electric field value induced by charge separation in a ND particle should be
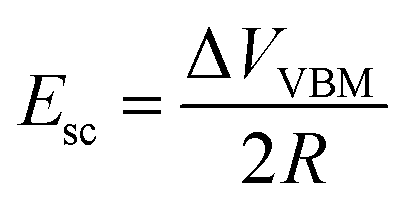 | (2) |
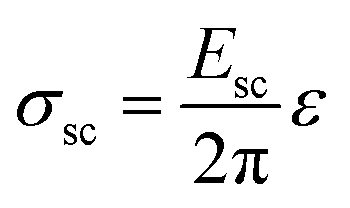 | (3) |
To illustrate this statement, the numerical solution of Poisson equation around the model ND particle with truncated octahedral shape, assuming dipole layers on the particle surface and equipotential distribution inside the particle, was performed. The assumption of equipotential ND particle is reasonable since typical charge density for charged ND is about 1021 cm−3 giving the corresponding Debye length of about 1 Å, which is significantly smaller than the ND radius. The difference of voltage drops in the dipole layers for the (001) and (111) facets was set to 0.5 eV, according to our QC calculations. The slice of the calculated distribution is shown in Fig. 7a for the plane passing through the centers of the (001) and (111) facets. Asymmetric perturbation of potential around the ND particle, localized at a scale of the facet size, is observed.
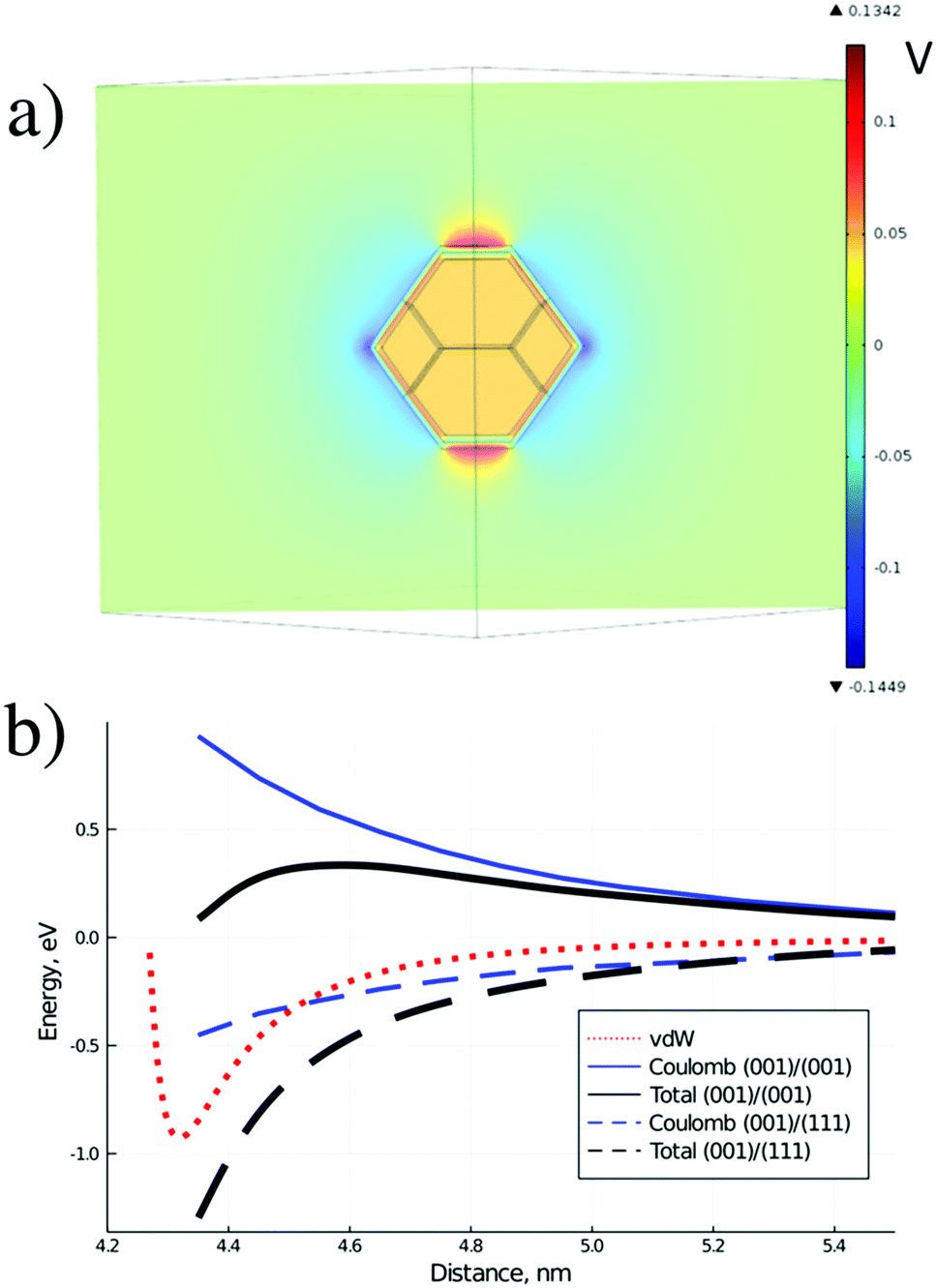 | ||
| Fig. 7 (a) Calculated distribution of electrostatic potential (in Volts) around an H-terminated ND particle in the plane passing through the centers of the (001) and (111) facets and through the particle center, and (b) estimated interaction potential between two H-terminated ND in water solution for different matching of ND facets. D is the distance between ND particle centers. | ||
Thus, in the case of H-terminated ND particles in water solution strongly anisotropic distribution of electrostatic potential around the particle is observed, opposite to the standard case of aggregation in aqueous dispersions, considered by the DLVO theory.51 Such distribution should promote the formation of non-compact chain structures of particles via interaction of facets with opposite charges.
One can estimate the additional interaction energy (attractive of repulsive) of ND particles with redistributed surface charges given by the above equation as
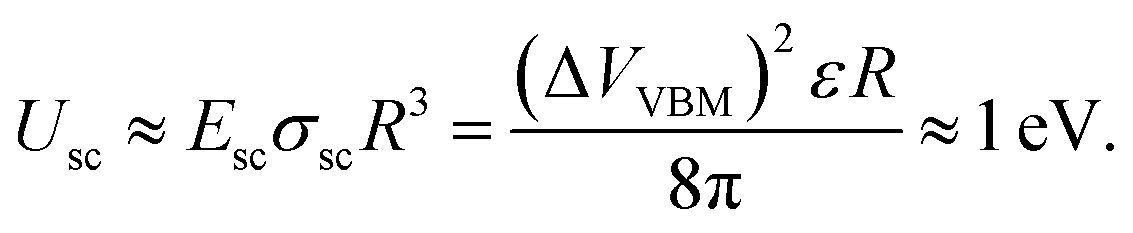 | (4) |
Numerically calculated Coulomb interaction potentials between two ND particles with different facet matching in water solution are shown in Fig. 7b together with the estimated total interaction potentials. The van der Waals interaction was calculated by integration of the atomic C–C Lennard-Jones potential, which has both attractive and repulsive parts, over volumes of two ND particles. Coulomb interaction was calculated using numerical solution of Poisson equation under the assumption of continuum dielectric media. The total interaction potential between NDs includes Coulomb and van der Waals contribution, estimated from first-principles calculations of specific interaction energy (about 10 meV Å−2) of H-terminated ND facets, reduced by diamond-water van der Waals interaction and rescaled to a 4 nm diameter ND particle. It is seen from Fig. 7b that for a ND pair with similar facets the Coulomb interaction results in a barrier for ND aggregation (similar to standard DLVO theory), while for a ND pair with different facets there is no barrier for the aggregation process. The calculated contribution of Coulomb potential for the ND pair with oppositely charged facets is about −0.5 eV at the equilibrium distance, which is in reasonable agreement with the above estimate. Thus, ND particles should prefer to form structures with matching of different facets (with opposite charges) at the contact due to charge separation inside the hydrogenated ND particles in water solutions.
This fact of charge separation in H-terminated ND particles can explain the formation of non-compact chain structures from ND particles (probably linked with different facets) in experiments. The effect of charge separation should be absent in the oxygen treated particles as charges are localized at the uniformly distributed oxygen-containing functional groups, meaning that more compact structures (with less selective linking) can be formed in this case.
Conclusions
Experimental data showed a difference in the structural organization of ND particles with different ζ-potentials in water solution: positively charged (hydrogen treated) ND particles form chain structures while negatively charged (oxygen-treated) ND particles form compact structures. In order to explain this difference and to investigate the physico-chemical properties and interaction mechanisms of ND particles in water solution, first-principles calculations were performed and a continual model was developed.Real DND particles disaggregated by hydrogen or oxygen treatment should have functional groups on the surface. Therefore, in order to explain the experimental results for colloidal DND particles, we performed DFT calculations to analyze the interaction between functionalized ND particles. Our DFT calculations showed that interplay between Coulomb interaction and van der Waals attraction is observed for charged functionalized ND particles. It was shown that for neutral H-terminated ND particles the surface potential is isotropic and cannot explain experimental data. For ND particles with partially dissociated oxygen-containing functional groups like O−/COO−, the charge is localized near these groups and its distribution is anisotropic. This allowed us to conclude that the nature of charge anisotropy in ND particles may be related to the presence of oxygen-containing functionalized groups on the ND surface. However, according to the performed DFT calculations, these groups should be distributed uniformly on the ND surface due to small (∼0.1 eV) formation energy difference for the (111) and (001) facets. For positively charged H-terminated particles, calculations showed that charge should be distributed in accordance to the values of valence band maximum on the particle facets. These conclusions derived from the performed DFT calculations cannot fully explain the observed difference in aggregation of the hydrogen-treated and oxygen-treated ND particles into chained or compact ND structures, respectively.
For further analysis of the ND interaction, a continual model taking into account the role of environment and actual size of the considered ND particles (∼4 nm) was developed. In this model, we used a numerical solution of the Poisson equation around the ND particle in a dielectric medium. Parameters for dipole layers on the particle surface were taken from first-principles calculations.
The results obtained with the continual model allowed us to suggest that the formation of chain structures from H-terminated ND particles is caused by charge separation. This model confirmed that H-terminated ND particles link to each other by different facets with opposite charges illustrating orientation dependency in ND interactions. However, this effect of charge separation should be absent in the oxygen treated particles, in which charges are localized at the O-containing functional groups, thus, more compact structures can be formed. These general qualitative statements address the problem of interactions between the large number of the ND particles and explain the presented cryoTEM experimental results. This continual model also allows us to conclude that the observed effects should noticeably depend on the nature of the solvent, in particular, its dielectric properties.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors sincerely acknowledge the valuable and profound comments by Prof. Alexander Vul’ during the manuscript preparation. This work has been carried out using computing resources of the federal collective usage center Complex for Simulation and Data Processing for Mega-science Facilities at the NRC “Kurchatov Institute”. The authors also acknowledge support from the resource center of probe and electron microscopy “Nanozond” of the NRC “Kurchatov Institute” for the cryo-TEM structural studies. This project was partially funded by the Russian Foundation for Basic Research (project 18-29-19117).Notes and references
- A. S. Barnard, Analyst, 2009, 134, 1751 RSC.
- A. Vul’ and O. Shenderova, Detonation Nanodiamonds, Pan Stanford Publising, New York, 2014 Search PubMed.
- D. Ho, Nanodiamons Application in Bioligy and Nanoscale Medicine, Springer US, New York, 2010 Search PubMed.
- R. Narayan, Diamond-based materials for biomedical applications, Woodhead Publishing, 2013 Search PubMed.
- A. V. Shvidchenko, E. D. Eidelman, A. Y. Vul’, N. M. Kuznetsov, D. Y. Stolyarova, S. I. Belousov and S. N. Chvalun, Adv. Colloid Interface Sci., 2019, 268, 64 CrossRef CAS PubMed.
- S. Kumar, M. Nehra, D. Kedia, N. Dilbaghi, K. Tankeshwar and K. Kim, Carbon, 2019, 143, 678 CrossRef CAS.
- J. Boudou, P. Curmi, F. Jelezko, J. Wrachtrup, P. Aubert, M. Sennour, G. Balasubramanian, R. Reuter, A. Thorel and E. Gaffet, Nanotechnology, 2009, 20, 235602 CrossRef PubMed.
- M. Dubois, K. Guerin, E. Petit, N. Batisse, A. Hamwi, N. Komatsu, J. Giraudet, P. Pirotte and F. Masin, J. Phys. Chem. C, 2009, 113, 10371 CrossRef CAS.
- B. M. Ozawa, M. Inaguma, M. Takahashi, F. Kataoka, A. Krüger and E. Osawa, Adv. Mater., 2007, 19, 1201 CrossRef.
- N. Nunn, M. Torelli, G. Mcguire and O. Shenderova, Curr. Opin. Solid State Mater. Sci., 2017, 21, 1 CrossRef CAS.
- O. Williams, Nanodiamond, Royal Society of Chemistry, 2014 Search PubMed.
- V. N. Mochalin, O. Shenderova, D. Ho and Y. Gogotsi, Nat. Nanotechnol., 2011, 7, 11 CrossRef PubMed.
- T. Dideikin, et al. , Carbon, 2017, 122, 737 CrossRef.
- E. Osawa, Pure Appl. Chem., 2008, 80, 1365 CAS.
- O. A. Shenderova and D. M. Gruen, Ultrananocrystalline Diamond Synthesis, Properties, and apllication, William Andrew Publishing, New York, 2006 Search PubMed.
- K. Turcheniuk, C. Trecazzi, C. Deeleepojananan and V. V. Mochalin, ACS Appl. Mater. Interfaces, 2016, 8, 25461 CrossRef CAS PubMed.
- O. A. Williams, J. Hees, Ch Dieker, W. Jäger, L. Kirste and E. Nebel, ACS Nano, 2010, 4, 4824 CrossRef CAS PubMed.
- A. E. Aleksenskiy, E. D. Eydelman and A. Y. Vul, Nanosci. Nanotechnol. Lett., 2011, 3, 68 CrossRef CAS.
- A. V. Shvidchenko, A. N. Zhukov, A. T. Dideikin, M. V. Baidakova, M. S. Shestakov, V. V. Shnitov and A. Y. Vul, Colloid J., 2016, 78, 235 CrossRef CAS.
- V. Chakrapani, J. C. Angus, A. B. Anderson, S. D. Wolter, B. R. Stoner and G. U. Sumanasekera, Science, 2007, 318, 1424 CrossRef CAS PubMed.
- A. V. Delgado, F. González-Caballero, R. J. Hunter, L. K. Koopal and J. Lyklema, J. Colloid Interface Sci., 2007, 309, 194 CrossRef CAS PubMed.
- A. Y. Vul, E. D. Eidelman, A. E. Aleksenskiy, A. V. Shvidchenko, A. T. Dideikin, V. S. Yuferev, V. T. Lebedev, Y. V. Kul'velis and M. V. Avdeev, Carbon, 2017, 114, 242 CrossRef CAS.
- N. M. Kuznetsov, S. I. Belousov, D. Y. Stolyarova, A. V. Bakirov, S. N. Chvalun, A. V. Shvidchenko, E. D. Eidelman and A. Y. Vul’, Diamond Relat. Mater., 2018, 83, 141 CrossRef CAS.
- N. M. Kuznetsov, S. I. Belousov, A. V. Bakirov, S. N. Chvalun, R. A. Kamyshinsky, A. A. Mikhutkin, A. L. Vasiliev, P. M. Tolstoy, A. S. Mazur, E. D. Eidelman, E. B. Yudina and A. Y. Vul, Carbon, 2020, 161, 486 CrossRef CAS.
- S. L. Y. Chang, et al. , Nanoscale, 2020, 12, 5363 RSC.
- G. Opletal, S. Chang and A. Barnard, Nanoscale, 2020, 12, 19870 RSC.
- A. C. Pierre, Carbon materials and technology, Springer US, New York, 2002 Search PubMed.
- A. S. Barnard and M. Sternberg, J. Mater. Chem., 2007, 17, 4811 RSC.
- A. S. Barnard and E. Osawa, Nanoscale, 2014, 6, 1188 RSC.
- L. Lai and A. S. Barnard, J. Phys. Chem. Lett., 2012, 3, 896 CrossRef CAS PubMed.
- A. S. Barnard, J. Mater. Chem., 2008, 18, 4038 RSC.
- Z. Ge and Y. Wang, J. Phys. Chem. B, 2017, 121, 3394 CrossRef CAS PubMed.
- Q. Xu and X. Zhao, J. Mater. Chem., 2012, 22, 16416 RSC.
- L. Lai and A. S. Barnard, Nanoscale, 2011, 3, 2566 RSC.
- L. Lai and A. S. Barnard, Nanoscale, 2014, 6, 14185 RSC.
- A. Krueger and D. Lang, Adv. Funct. Mater., 2012, 22, 890 CrossRef CAS.
- H. Huang, L. Dai, D. H. Wang, L. S. Tan and E. Osawa, J. Mater. Chem., 2008, 18, 1347 RSC.
- H. D. Wang, Q. Yang and C. H. Niu, Diamond Relat. Mater., 2012, 25, 73 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- G. Kresse and J. Furthmiiller, Comput. Mater. Sci., 1996, 6, 15 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 11 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- A. S. Barnard, Cryst. Growth Des., 2009, 9, 4860 CrossRef CAS.
- X. Duan, W. Tian, H. Zhang, H. Sun, Z. Ao, Z. Shao and S. Wang, ACS Catal., 2019, 9, 7494 CrossRef CAS.
- C. Wang, B. Zheng, W. T. Zheng, C. Q. Qu, L. Qiao, S. S. Yu and Q. Jiang, Diamond Relat. Mater., 2009, 18, 1310 CrossRef CAS.
- R. F. W. Bader, Acc. Chem. Res., 1985, 18, 9 CrossRef CAS.
- G. Henkelman, A. Arnaldsson and H. Jónsson, Comput. Mater. Sci., 2006, 36, 354 CrossRef.
- J. Hees, A. Kriele and O. A. Williams, Chem. Phys. Lett., 2011, 509, 12 CrossRef CAS.
- T. Petit, H. A. Girard, A. Trouvé, I. Batonneau-Gener, P. Bergonzo and J. Arnault, Nanoscale, 2013, 5, 8958 RSC.
- J. B. Cui, J. Ristein and L. Ley, Phys. Rev. Lett., 1998, 81, 429 CrossRef CAS.
- B. Derjaguin and L. Landau, Prog. Surf. Sci., 1993, 43, 30 CrossRef.
| This journal is © the Owner Societies 2021 |