
Biamphiphilic ionic liquid based aqueous microemulsions as an efficient catalytic medium for cytochrome c†
Manvir
Kaur
,
Harmandeep
Kaur
,
Manpreet
Singh
,
Gagandeep
Singh
and
Tejwant Singh
Kang
 *
*
Department of Chemistry, UGC Sponsored Centre for Advanced Studies-II, Guru Nanak Dev University, Amritsar-143005, India. E-mail: tejwantsinghkang@gmail.com; tejwant.chem@gndu.ac.in; Tel: +91-183-2258802, ext. 3291
First published on 8th December 2020
Abstract
Considering the remarkable applicability of ionic liquids (ILs) in bio-catalysis involving enzymes, herein, we report new IL based aqueous microemulsions as a catalytic reactor for cytochrome c (Cyt-c). Microemulsions (μEs), comprising water as the polar component, imidazolium (cation) and dioctylsulfosuccinate (AOT) (anion) based biamphiphilic ionic liquid (BAIL) as the surfactant and a hydrophobic ionic liquid (HIL) as the non-polar component have been prepared and characterized. The use of BAIL has promoted the formation of μEs without any co-surfactant, owing to its higher surface activity. The effect of ester- or amide-functionalization of the alkyl chain of the imidazolium cation of BAILs on the phase behavior of μEs has been investigated. The prepared μEs have been characterized via conductivity, dynamic light scattering (DLS), UV-vis absorption and steady-state fluorescence (using external polarity probes) techniques. The prepared μEs have been employed as nano-reactors for exploring the catalytic activity of Cyt-c. The formed BAIL–water nano-interfaces in reverse μEs have exerted a positive effect on the catalytic activity of Cyt-c stored in a water pool of reverse μEs. A five-fold higher rate constant in μEs as compared to buffer establishes μEs as a better catalytic medium. Furthermore, the differing nature of nano-interfaces created by BAILs and water in reverse μEs, depending on the functionalization of the alkyl chain of the cationic part of BAIL, has exerted varying influence on the catalytic activity of Cyt-c. It is expected that the present work will result in providing a versatile platform for the creation of new IL and water based μEs for bio-catalytic applications.
1. Introduction
Ionic liquids (ILs)1 have attracted great interest from the scientific community due to their magnificent physicochemical properties and diverse applications.2–5 In particular, the negligible vapor pressure, good solvation ability and high thermal stability of ILs render them as a potential substitute for volatile organic solvents (VOSs).6–10 In the last decade, many ILs have emerged as a suitable medium for the dissolution and stabilization of proteins and enzymes.11–13 Enzymes when present in ILs have exhibited enhanced catalytic activity when compared to that observed in organic solvents.14–17 However, the lower solubility of enzymes in most of the ILs limits their biotechnological applications. To realize the maximum benefits of enzymes in conjunction with ILs, it is necessary to enhance the solubility of enzymes in ILs. In the past, many attempts have been made in this regard. Immobilization of enzymes using poly(ethylene oxide) (PEO), and ceramic Toyonite carrier etc.,16,18,19 in ILs has been achieved. Water has also been utilized as a co-solvent to enhance the solubilization of enzymes in ILs. However, the conformational changes in enzymes in IL–water systems, particularly in IL rich IL–water mixtures (water <5%), have resulted in low catalytic activity.20 On the other hand, enhanced enzymatic activity of enzymes has been observed when present in the colloids of water and hydrophilic ILs or surface active ILs (SAILs).21–23 Another self-assembled system of interest is microemulsion (μE), in which enzymes can be solubilized and stabilized in the polar domains.Therefore, considering the thermodynamic stability and applicability of reverse μEs24–28 towards protein stability,28,29 we have conceptualized the utilization of micro-domains of water in IL based reverse μEs for enzyme catalysis. Such μE would act as an efficient catalytic reactor for solubilization and enhancing the catalytic activity of enzymes. This would also overcome the issues related to limited solubility of enzymes in neat ILs,16 conformational changes in IL–water mixtures15,16 and the large amount of water used in colloidal systems.30,31 Earlier, hydrophobic ILs (HILs) were used as the non-polar component to create μEs and such μEs have been explored for enhanced catalytic activity of enzymes such as lipase,32,33 horseradish peroxide,34 and laccase35etc. In most of these reports, conventional surfactants in conjunction with co-surfactants have been used to stabilize IL based μEs.32–35 The use of co-surfactants many times may lead to reduced catalytic activity via interactions with enzymes at the polar-surfactant interface.33,36 Therefore, due to the better surface active properties of biamphiphilic ILs (BAILs) in comparison to conventional ionic surfactants,37 it would be advantageous to utilize BAILs as the surfactants for creating aqueous reverse μEs using HIL as the nonpolar medium.
Herein, we have conceptualized and developed IL based aqueous μEs for investigating the catalytic activity of cytochrome c (Cyt-c) in reverse micelles of HIL stabilized with BAILs. For this, BAILs based on dioctylsulfosuccinate (AOT) as the anion and imidazolium as the cation have been synthesized and characterized. AOT has been selected as the component of BAIL owing to its widespread use as a surfactant in the preparation of μEs.38 However, to realize the maximum benefits of HILs as a continuous medium, it is necessary to dissolve AOT in an adequate amount, the solubility of which is very limited in ILs.39 The high solubility of AOT has been achieved by encompassing AOT as a component of BAIL in conjunction with different imidazolium cations. Hence, AOT based BAILs, 1-hexyl-3-methylimidazolium dioctylsulfossuccinate, [C6mim][AOT], its ester-, [C6Emim][AOT] and amide-, [C6Amim][AOT], -functionalized counterparts have been synthesized and characterized. 1-Ethyl-3-methylimidazolium bis-(trifluorosulfonyl)imide, [C2mim][Tf2N], has been utilized as the non-polar component owing to its low viscosity and higher ionic conductivity.40 Moreover, [Tf2N] based HILs are among the category of most stable ILs and have been explored for obtaining higher catalytic activity of enzymes like α-chymotrypsin, lipase, and Cyt-c etc.16 However, in reported studies, the enzymes remained dispersed in the HIL. To increase the solubility and feasibility of bio-catalysis, we have employed water as the polar medium. Thus, the prepared μEs have been further utilized as the catalytic reactor for encapsulating Cyt-c that shows ∼5 times enhanced catalytic activity as compared to that observed in buffer.
2. Materials
n-Hexanol (>99%), hexylamine (>99%), bromoactyl bromide (>98%), 1-hexyl-3-methylimidazolium chloride (>98%), sodium dioctylsulfosuccinate (>97%), methyl orange and coumarin-153 were purchased from Sigma Aldrich and used without further purification. [C2mim][Tf2N] (99%) was purchased from IOLITEC. Different BAILs were synthesized employing the procedure as reported by Rao et al.41 The synthesized BAILs were characterized using 1H NMR spectroscopy and the characterization data are provided in Annexure S1 of the ESI.† Cyt-c from horse heart and guaiacol (98%) has been purchased from SRL Pvt. Ltd. Hydrogen peroxide (H2O2) (30% w/v) was purchased from Fischer Scientific. Di-potassium hydrogen phosphate (99%) and potassium di-hydrogen phosphate (99%) were purchased from SD Fine-Chem Ltd and used for the preparation of buffer solution (10 mM) in degassed Millipore grade water.3. Methods
3.1. Preparation and characterization of microemulsions
The molecular structures of different BAILs and [C2mim][Tf2N] (HIL) employed in this study are provided in Scheme 1. Phase diagrams of ternary systems comprising water/BAIL/HIL were constructed using cloud phase titration. For each titration, the amount of respective BAIL and HIL was fixed and the transition from transparent to turbid phase was monitored on the addition of water. The formed IL based aqueous μEs were characterized by using conductivity measurements to probe the presence of different μE regions.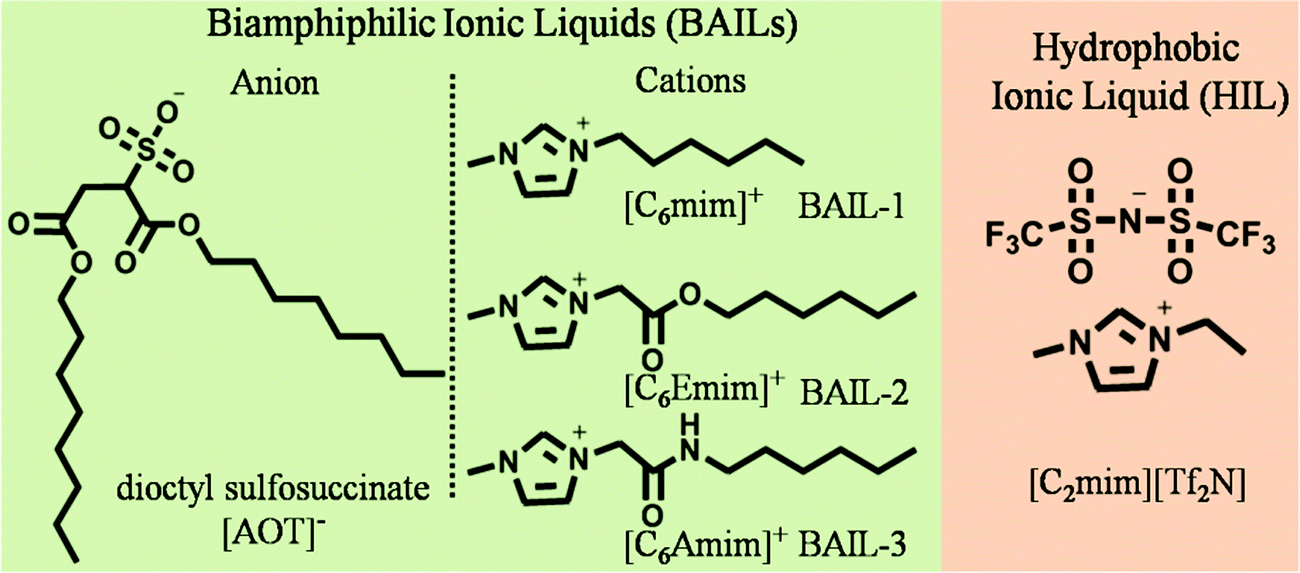 | ||
| Scheme 1 The molecular structure of different BAILs and HILs used in μEs. | ||
A digital conductivity meter (systronics 308) was used to measure the specific conductance (κ) using a cell of unit cell constant. Before the measurements, the conductivity meter was calibrated using aqueous KCl of different concentrations at 25 °C. The conductivity measurements were made in triplicate and found to be accurate within an uncertainty of ±0.7%. For conductivity measurements, depending upon the μE region, the amount of BAIL and HIL was fixed at 70% and 30%, respectively, and the solutions were titrated against water. The hydrodynamic diameter of the formed micro-droplets was explored using dynamic light scattering (DLS) measurements using a light scattering apparatus (Zetasizer, Nanoseries, Nano-ZS, Malvern Instruments) equipped with a built-in temperature controller having an accuracy of ±0.1 K at a scattering angle of 173°. An average of 5 measurements was considered as the data. Micro-polarity of the investigated μEs was explored using UV-Visible and Fluorescence measurements. UV-Visible measurements were made using a UV-Vis spectrophotometer (UV-1800 SHIMADZU) in the wavelength range of 200–800 nm employing methyl orange (MO) as an absorption probe at a concentration of 2 × 10−6 M using a quartz cuvette of path length 1 cm. Steady-State fluorescence measurements were performed with coumarin-153 (c-153) as an external fluorescent probe (1 × 10−6 M) using a PerkinElmer LS-55 spectrophotometer using a quartz cuvette of path length 1 cm at an excitation wavelength of 482 nm. For spectroscopic measurements, the amount of BAIL and HIL was fixed at 70 and 30% w/w, respectively, and the solutions were titrated against water similar to that performed for conductivity measurements. The concentration of the respective components has been decided according to the μE region (line ‘a’ and ‘b’ in Fig. 1A and B, respectively) obtained in the phase diagram.
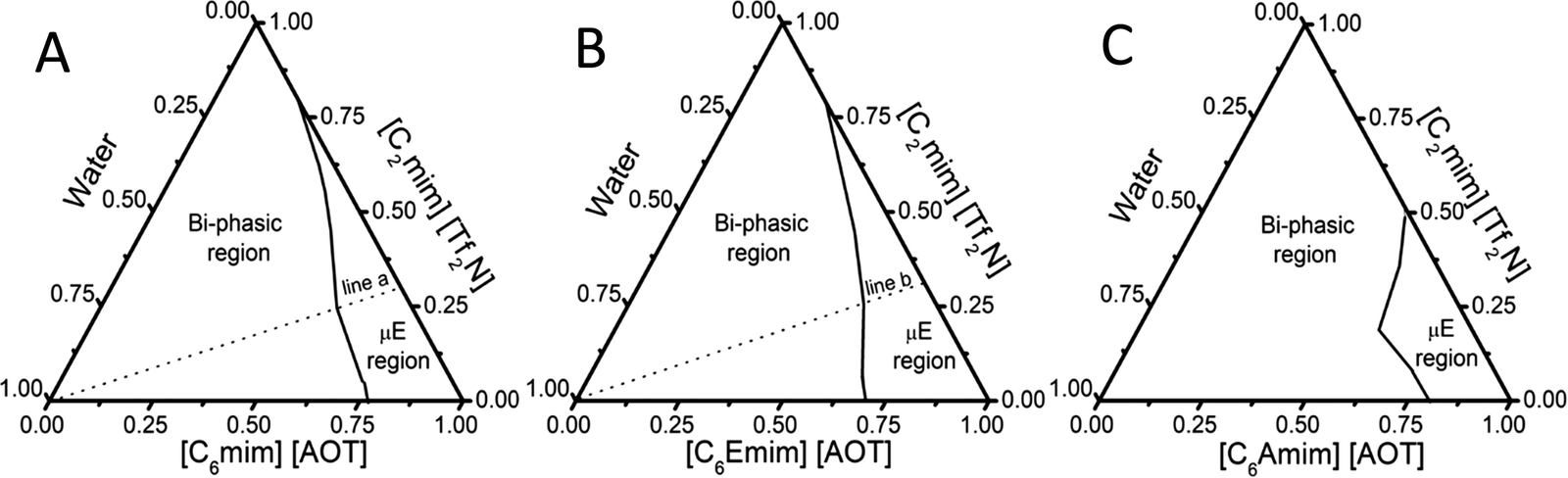 | ||
| Fig. 1 (A–C) The obtained phase diagrams of (A) μE-1; (B) μE-2 and (C) μE-3 at 298.15 K. | ||
3.2. Catalytic activity of cytochrome c in microemulsions
Stock solution of Cyt-c (80 μM) in 10 mM phosphate buffer was used to prepare μEs instead of neat water. The final concentration of Cyt-c loaded in the reverse aqueous domains of μEs was found to be 5 μM. Conformational changes of the peptide chain surrounding the heme cleft upon encapsulation in reverse micelles of the investigated μEs were explored using UV-Vis measurements in the wavelength range of 200–800 nm employing a quartz cuvette of path length 1 cm using a UV-1800 SHIMADZU spectrophotometer. For the investigated μEs, the weight ratio of BAIL and HIL was fixed at 70% w/w and 30% w/w, respectively, according to the single-phase region of μEs. Following that, the stock solution of Cyt-c in buffer was added to the mixture of BAIL and HIL. Alterations in the tertiary structure of Cyt-c in reverse micelles were monitored using circular dichroism (CD) measurements performed using a Jasco J-810 spectrometer with a quartz cuvette of path length 1 mm. The functional stability of Cyt-c in an aqueous pool of μEs was determined using guaiacol as the substrate and H2O2 as the oxygen donor. For catalytic activity, 1.6 g of the investigated μEs having 65% w/w and 29% w/w of BAIL and HIL, respectively, was taken in a vial along with 6% w/w of aqueous reverse pool containing Cyt-c (5 μM). In this ternary mixture containing Cyt-c, 50 mg of 20 mM guaiacol solution was added followed by the addition of 50 mg of 50 mM H2O2 solution. The control experiment was performed in buffer under similar conditions.4. Results and discussion
4.1. Phase behavior, regions, size and micropolarity of microemulsions
The construction of a ternary phase diagram is the foremost step to study the formation of microemulsions (μEs). As shown in Scheme 1, the different BAILs employed for the construction of μEs have been abbreviated as BAIL-1 (non-functional), BAIL-2 (ester-functionalized) and BAIL-3 (amide-functionalized). [C2mim][Tf2N] has been used as the hydrophobic IL and is abbreviated as HIL. Ternary phase diagrams containing water/BAILs/HIL are shown in Fig. 1. The investigated ternary systems formed by using BAIL-1, BAIL-2 and BAIL-3 are abbreviated as μE-1, μE-2 and μE-3, respectively. As shown in Fig. 1, the ternary phase diagrams of μEs consist of two regions: a μE-region and a bi-phasic region. The μE-region corresponds to the single-phase transparent region that indicates the formation of μE, while the bi-phasic region points out the phase separation in the respective ternary systems. A relatively lesser μE region as compared to that of other IL41–43 or IL/surface active ionic liquid (SAIL)28,44 based μEs has been observed in the present work. This is assigned to the less solubility of AOT based BAILs in water as well as in HILs as compared to that of previously employed SAILs.28,44The area under the observed μE region for different systems follows the order: μE-2 > μE-1 > μE-3. Varying sets of interactions such as electrostatic, hydrogen-bonding (H-bonding) and hydrophobic interactions play their role in the formation of μEs. Relatively more single-phase region, observed in the case of μE-2 as compared to that observed for μE-1 and μE-3, is assigned to the presence of ester moiety in BAIL-2. The presence of a H-bonding prone ester-moiety in the alkyl chain of the cation of BAIL-2 near the imidazolium head group synergistically with ester-moieties present in AOT as well as ion–dipole interactions of water with SO3− in the anionic counterparts of the BAIL-2 increases the extent of encapsulation of water.45 On the other hand, μE-3 is associated with a very small μE region despite having H-bond donor and acceptor –NH and –CO– groups, respectively, in the cation. It is natural to assume that the anion and cation of BAIL-3 not only undergo electrostatic interactions but also exhibit inter-molecular H-bonding interactions. Such inter-molecular H-bonding interactions hinder the water molecules to interact with BAIL-3 efficiently. This is supported by the gel formation in the case of μE-3 even before the phase separation. Therefore, due to the gelation in the case of μE-3 along with a very low μE region, μE-3 is only investigated for its phase behavior.
Different regions of μEs, i.e., water-in-HIL (W/HIL), bicontinuous (BC), and HIL-in-water (HIL/W) have been probed (Fig. S1, ESI†) via specific conductance (κ) measurements (Fig. 2). Initial increase in κ values with the addition of water indicates the redistribution of ionic moieties due to the growth of polar domains in W/HIL μE. This increase is more in the case of μE-1 as compared to μE-2. The only structural difference between BAIL-1 and BAIL-2 is the presence of a H-bond donor ester moiety in the cation of BAIL-2. Therefore, a relatively large increase in κ in the case of μE-1 as compared to μE-2 is attributed to the presence of H-bonding interactions between the water and ester-moiety of the anionic as well as the cationic part of BAIL-2, which reduce the mobility of ions in μE-2 as supported by DFT calculations (Fig. S2, ESI†).
 | ||
| Fig. 2 (A) The dependence of specific conductivity (κ) of μEs on water w/w%; (B and C) the calculation of percolation threshold (Wp) from the derivative plot of κ versus water w/w% for (B) μE-1 and (C) μE-2 (insets of figure B and C show the data fitting performed to calculate the scaling parameter s on the points below Wp). | ||
Further addition of water leads to the formation of the BC region, which is generally observed in continuation with the W/HIL phase.46 The presence of BC phase has been established with the help of percolation theory.46,47 The percolation threshold (Wp), which is the transition point where one region transforms to another region with the addition of polar component, corresponds to the inflection point in the graph of d(κ)/d(water wt%) vs. water wt% (Fig. 2B and C). κ, below and after Wp follows the scaling rules:47
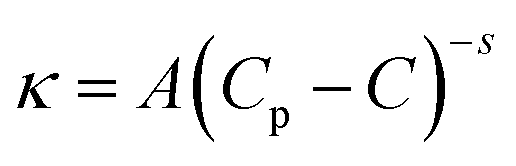
where κ is the specific conductivity, Cp is the percolation threshold (Cp = Wp), C is the amount of polar component, s and t are the scaling parameters, and A and B are the constants. The obtained values of the scaling parameter, s, are −0.699 and −0.802 for μE-1 and μE-2, respectively (Fig. 2B and C). The obtained s values for the present μEs are close to the scaling parameters obtained for the reported IL based μEs.28,46,47 The s value <1 suggests the presence of static percolation that is associated with the existence of BC phase in μEs.47κ continues to increase with a relatively smaller slope in the BC region as compared to W/HIL (Fig. 2A). Such a small increase in κ in the BC region is assigned to the formation of continuous channels of different components that restrict the mobility of ions. Further addition of water results in the formation of a bi-phasic system. Generally, the HIL/W region is observed in continuation to the BC region but here in the concerned μEs the HIL/W region is not found in the μE-region due to much less single phase region. However, κ continues to rise even in the bi-phasic region that could be assigned to the formation of the HIL/W phase due to the redistribution of ionic moieties in the region under investigation.
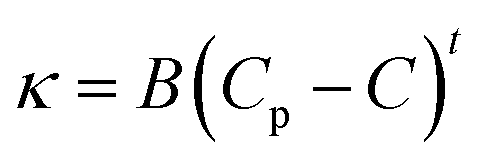
Hydrodynamic diameter (Dh) of the micro-droplets in ternary systems (Fig. 3A) along line a and b (Fig. 1A and B) has been monitored as a function of R value ([water]/[BAIL] = R). There is a linear increase in the size of micro-droplets with increasing R value (Fig. 3A and Fig. S3, ESI†), which is in line with the ‘swelling law of reverse micelles’.45,48 Generally, micro-droplets swell when water forms a polar pool in reverse μEs via strong interactions with the surfactant interface. Therefore, an increase in the Dh of micro-droplets indicates the formation of non-interacting water micro-droplets in W/HIL μE. This also suggests the different nature of water present in reverse μEs than the bulk water as suggested in earlier reports.45 As observed from Fig. 3A, μE-2 possesses a larger size when compared to μE-1. This could be assigned to the presence of H-bonding prone ester functionality in the cationic head group of BAIL-2. This could result in establishing H-bonding interactions between BAIL-2 and water accompanied by water penetration in the stern layer of reverse micelles, which leads to the relative swelling of the reverse micelle. The assertion has been supported by DFT calculations (Fig. S2, ESI†), which established that water present towards the water–BAIL interface undergoes H-bonding interactions with the ester group appended to the imidazolium cation as well as the AOT anion. On the other hand, water penetrated in the stern layer undergoes dipole–dipole interactions (Fig. S2, ESI†). It has also been established that AOT does not interact strongly with imidazolium cations.45 Hence, it is natural to assume that the cationic head group could stay in closer vicinity of the polar pool of reverse micelles as compared to AOT.45 This along with water penetration results in increase in the size of μE-2.
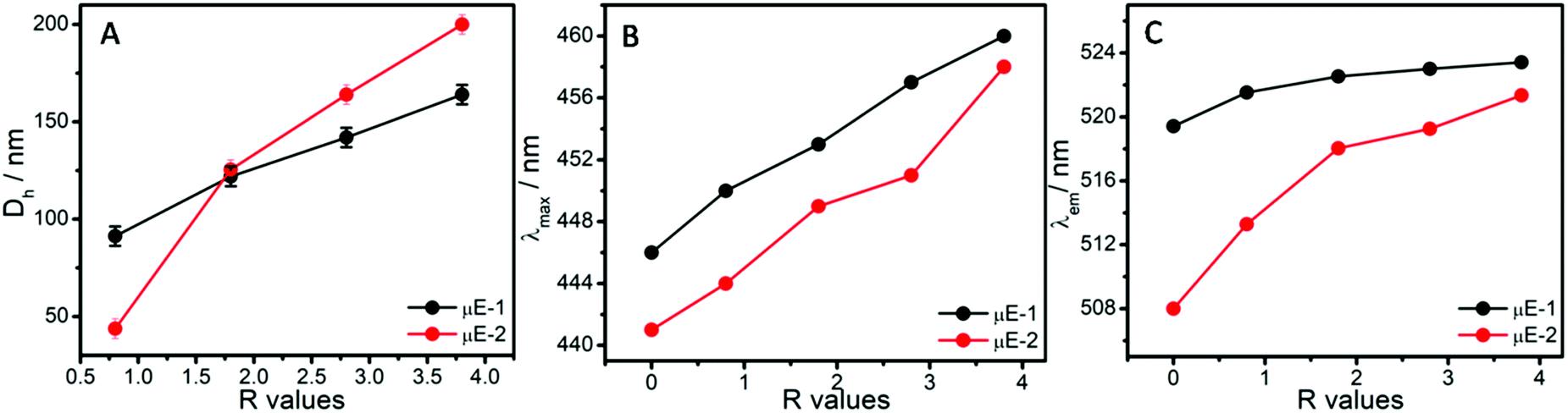 | ||
| Fig. 3 The variation in (A) hydrodynamic size (Dh) of the micro-droplets, (B) λmax of MO and (C) λem of c-153, in μEs as a function of R values. | ||
The micro-polarity of the ternary systems has been investigated using methyl orange (MO) and Coumarin-153 (c-153) as the polarity probes. The variation in absorption maxima (λmax) of MO in μEs has been monitored as a function of R value as shown in Fig. 3B and Fig. S4, ESI.† The λmax of MO in water and HIL is 464 and 424 nm, respectively.44 Because of the polarity sensitive nature, MO gives a blue-shifted λmax at 424 nm in the case of pure HIL as compared to water. This is associated with the lowered energy of the LUMO of MO in water.49,50 Despite the hydrophobic nature of HIL, MO gets solubilized in it due to complexation with HIL as suggested in the literature.49 Being an anionic dye, MO would preferably interact with the cationic part of HIL having a dipole moment ∼1.65 D (Table S1, ESI†), which is relatively lower than that with water (1.85 D).51 Thus, it would stabilize the excited state of MO to a relatively lower extent as compared to water. This results in the blue shift of λmax of MO in the case of HIL as compared to water. λmax of MO is found to be 445 nm and 442 nm in the mixtures of BAIL and HIL in the case of μE-1 and μE-2, respectively. The dipole moment of the investigated BAIL ions is found to be larger than that of HIL (Table S1, ESI†). Thus, it results in the red shift of λmax of MO in the mixtures of BAIL and HIL and suggests the presence of MO in the relatively polar region. Considering the anionic nature of MO, it is natural to assume that MO resides near the polar head group of BAILs and interacts with imidazolium cations. Furthermore, with increase in the R value, red shift in the λmax of MO is observed in both μEs. This indicates the migration of MO towards a more polar environment that establishes the interaction of water with BAILs.49 The obtained dipole moment of the investigated BAIL ions is even higher than that of water, which is natural because electronic polarizability increases with the increase in molecular size. However, the number density of ionic moieties decreases with the increase in molecular size and is comparatively lower than that of molecular solvents.52 Therefore, here it is difficult to explain the results solely on the basis of dipole moment. At the highest R value, λmax of MO in μE-1 and μE-2 comes out to be ∼460 and 458 nm, respectively, which is less than that observed in bulk water (∼464 nm). This reveals the presence of MO at the BAIL/water interface and signifies the different behavior of water in the constrained environment of reverse μE as compared to bulk water due to H-bonding and ion–dipole interactions with BAILs. Furthermore, a relatively lower λmax of MO in the case of μE-2, as compared to μE-1, divulges the existence of the cationic head group of BAIL-2 in close vicinity of water as compared to that in BAIL-1 as discussed earlier.
This is further supported by the variation in the emission maxima (λem) of c-153 in μEs with the change in R values (Fig. 3C and Fig. S5, ESI†). The emission spectra of c-153 depend upon the H-bonding ability, polarity and viscosity of the microenvironment. Therefore, due to the polarity sensitive nature, the c-153 probe is generally employed to study the micro-polarity of heterogeneous systems like μEs.44 In the case of water and HIL, the λem values of c-153 are 552 and 525 nm, respectively. This blue shift of the λem of c-153 is associated with the lower dielectric constant of HIL (∼12.3) when compared to that of water (∼80).53 In BAIL and HIL mixtures, the λem values of c-153 are 518 and 508 in the case of μE-1 and μE-2, respectively (Fig. 3C). This indicates the presence of c-153 in a relatively more hydrophobic region, which could be the hydrophobic alkyl chain region of BAILs (Scheme 2). Furthermore, a red shift in λem of c-153 is observed on addition of water (Fig. 3C). This suggests the migration of probe molecules towards a more polar microenvironment that signifies the interaction of water with BAIL and reveals the growth of a polar pool in μEs. The μE-2 possesses lower λem values as compared to that observed in μE-1. A similar observation has been made using UV-Vis absorbance measurements, which supports the stronger interactions of BAIL-2 with water at the interface of the reverse micelle. The different λem values of c-153 in μEs (∼524 and 520 nm) as compared to those in bulk water (∼552) suggest the altered H-bonding network of water present at the water–BAIL interface μEs.
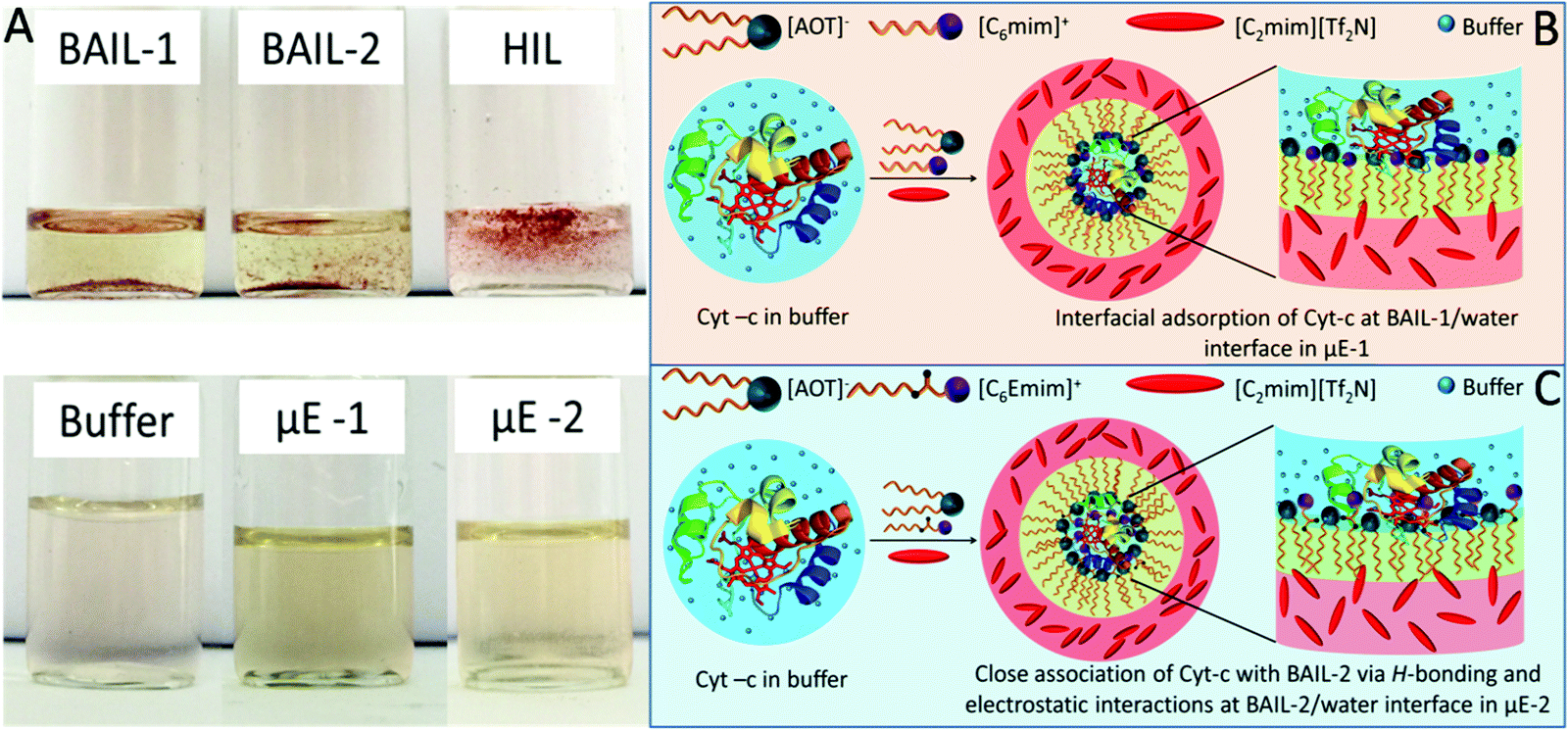 | ||
| Scheme 2 (A) Photographs showing the suspension of Cyt-c in BAILs and HIL, solubilization of Cyt in buffer and μEs; and schematic representation of adsorption of Cyt-c at the interface of (B) μE-1 and (C) μE-2. | ||
4.2. Catalytic stability of cytochrome c in microemulsions
The pool of water in reverse micelles of μEs has been employed as the catalytic medium for Cyt-c. Firstly, the solubility of Cyt-c in different components of μEs was monitored by naked eye examination. Cyt-c remains suspended in BAILs and HIL as shown in Scheme 2A. Therefore, the stock solution of Cyt-c in buffer (as a polar medium in place of water) is used for loading the enzyme in μEs. To avoid any phase separation during the enzymatic reaction, appropriate concentration of different components of μE has been chosen depending upon the single-phase region of μE. The oxidation of guaiacol to form tetraguaiacol in the presence of Cyt-c as the catalyst is confirmed by the appearance of a brown color with time (Fig. 4A).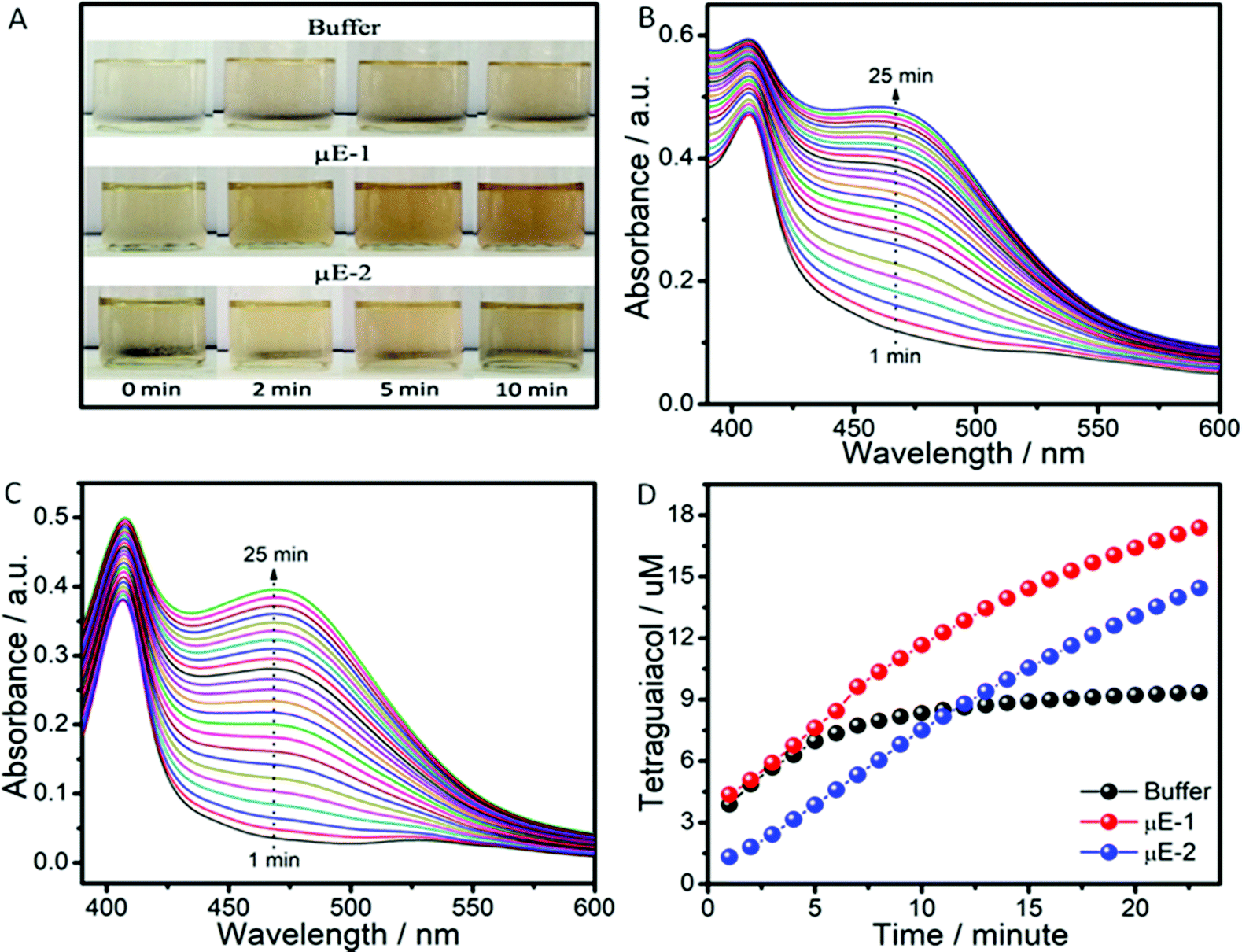 | ||
Fig. 4 (A) The photographs showing the catalytic activity of Cyt-c against guaiacol in buffer, μE-1 and μE-2; absorption spectra of tetraguaiacol in a mixture of (B) μE-1 and (C) μE-2 with ethanol in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio; and (D) catalytic activity of Cyt-c in a mixture of buffer, μE-1 and μE-2 with ethanol in a 1:1 ratio. :1 ratio; and (D) catalytic activity of Cyt-c in a mixture of buffer, μE-1 and μE-2 with ethanol in a 1:1 ratio. | ||
The appearance of a relatively more intense color in the case of μE-1 indicates the higher catalytic efficiency of Cyt-c in μE-1 as compared to that observed in the buffer as well as in μE-2 (Fig. 4A). However, due to its hydrophobic nature, tetraguaiacol prefers to dissolve in HIL or at the interface of HIL/BAIL, which results in the non-uniform concentration of teraguaicaol in μEs, and reliable absorption spectra are not obtained (Fig. S6, ESI†). The presence of an isobestic point in the absorbance spectra of μEs between Cyt-c and teraguaicaol suggests the complex formation. This could also lead to the underestimation of the formed teraguaicaol (Fig. S6, ESI†). Therefore, to acquire appropriate absorption spectra of tetraguiacol (Fig. 4B, C and Fig. S7, ESI†), ethanol is added in μEs and in buffer containing Cyt-c. The concentration of tetraguaiacol (Fig. 4D) formed during the catalytic reaction is obtained by using the absorbance values at 470 nm at different intervals of time during the reaction using molar extinction coefficient ∼2.66 × 10−4 M−1.54
At the initiation of the reaction, the concentration of tetraguaiacol obtained in the case of buffer and μE-1 is found to be similar. However, in the case of μE-2, the obtained concentration of tetraguaiacol is quite a bit less when compared to that observed in buffer and μE-1. This is attributed to the alteration in the tertiary structure of Cyt-c in μE-2 due to close association with BAIL-2 via H-bonding and electrostatic interactions as shown in Scheme 2. With time, the product formation increases in both the investigated μEs while it saturates in the case of buffer. Rate constant values for the catalytic reactions in buffer, μE-1 and μE-2 are found to be 0.2 s−1, 1.157 M−1 s−1 and 1.122 M−1 s−1, respectively (Fig. S8, ESI†). Therefore, μEs exhibit a ∼5 fold greater rate of formation as compared to that of buffer. This could be assigned to the difference in the behavior of Cyt-c in μEs as compared to that in buffer. Furthermore, the difference in the rate of formation in μE-1 and μE-2 is associated with the varying degree of denaturation, that is, the coordination distance of the methionine residue to heme iron, in the investigated μEs as discussed later. Moreover, Cyt-c is found to be stable for at least ten days in μEs and exhibits a good peroxidase activity (Fig. S9 and S10, ESI†). Although the activity of Cyt-c decreases with time in buffer and μEs over an extended period, the Cyt-c in μEs possesses a higher rate constant than that in buffer even after ten days (Fig. S10D, ESI†).
Conformational changes around the heme cleft of the enzyme in μEs have been explored by employing UV-Vis and CD measurements (Fig. 5). Native Cyt-c shows an intense soret band at 409 nm due to the π–π* transition of the heme group buried in the hydrophobic core of the peptide chain.22 The weak Q-bands at 529 nm and 549 nm indicate the reduced form of heme.55 A decrease in the absorption of the soret band in reverse micro-pools of W/HIL μEs as compared to that observed in buffer (Fig. 5A) indicates the alteration of enzyme structure at the BAIL–water interface. Cyt-c from horse heart at the investigated pH is positively charged.56,57 It is well documented that Cyt-c generally interacts with the negatively charged moieties via electrostatic interactions while positively charged moieties arrange themselves around the heme cleft present inside the enzyme.29 Hence, the binding of AOT anions with amino acid residues of the peptide chain that encapsulates the heme cleft could result in the unfolding of the hydrophobic core.58 This results in exposing the heme cleft towards a relatively polar environment and thus results in lowering the energy of the π* orbital.22 Such conformational changes around the heme cleft result in the exposure of the active sites of the enzymes, which results in an increased catalytic activity in the case of μE-1. On the other hand, ester functionalized BAIL-2 acquires varying orientations that result in enhancing the interactions with Cyt-c as also observed in reported SAIL-protein colloidal complexes.59 In μE-2, the H-bond acceptor ester moiety60 of BAIL-2 synergistically assists the interactions of BAIL-2 with Cyt-c along with the electrostatic and hydrophobic interactions. Such enhanced H-bonding interactions of Cyt-c with the ester moiety of the imidazolium cation may lead to complete denaturation of Cyt-c than that of BAIL-1 and hence a decrease in enzyme activity is observed in the case of μE-2.
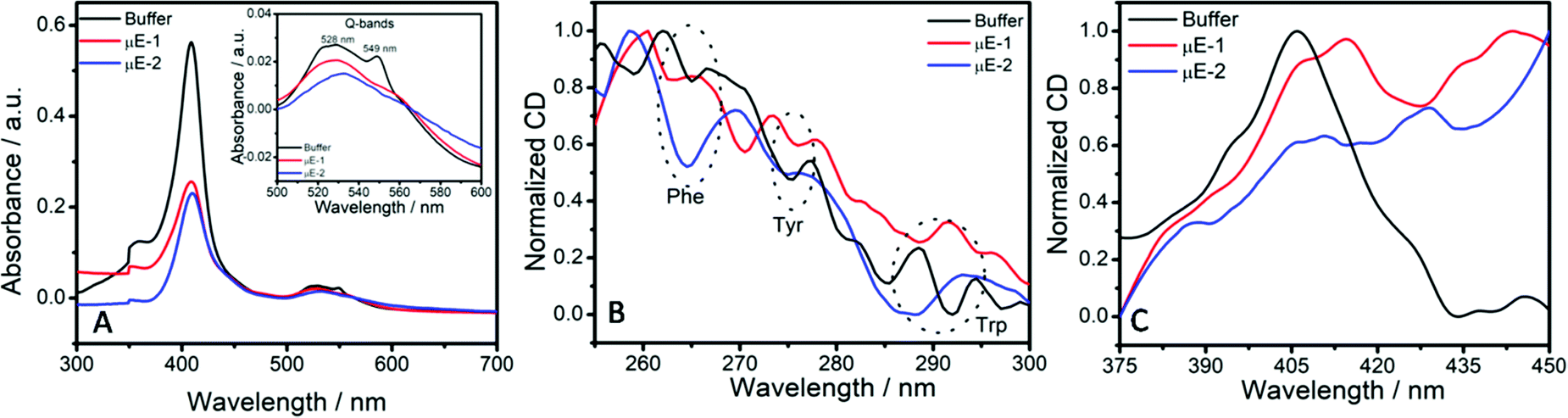 | ||
| Fig. 5 (A) Absorption spectra of Cyt-c; and (B) and (C) near UV-CD spectra of Cyt-c in buffer, μE-1 and μE-2. | ||
Due to the presence of the imidazolium cation in BAILs and HIL in the formed μEs, it is difficult to monitor the far-UV CD spectra of Cyt-c in μEs. However, the tertiary structure of the enzyme encapsulated in μEs is elucidated using the near-UV CD spectra. Near-UV CD spectra of Cyt-c exhibit bands at 265, 276 and 292 nm due to phenylalanine (Phe), tyrosine (Tyro) and tryptophan (Trp) (Fig. 5B) residues, respectively.61 Altered near-UV CD spectra in μEs when compared to that of native Cyt-c in buffer indicate the loss of the tertiary structure of Cyt-c due to unfolding of the peptide chain around the heme cleft. This is also supported by the decreased absorption values of the soret band in UV-Vis measurements. Alterations in the CD spectra are assigned to the electrostatic interactions of positively charged lysine (Lys) residues present in the vicinity of Trp and Tyr on the adjacent sites around the heme cleft with the negatively charged [AOT]− anions of the BAIL.
The alterations in the CD spectra of the heme-absorption range (Fig. 5C) also indicate the modification of the tertiary structure of Cyt-c in μEs. The presence of an unusual positive peak at 420 nm in the case of μE-2 supports the observations made from UV-Vis absorption measurements that Cyt-c denatures to a greater extent in μE-2 as compared to μE-1. This supports the relatively lower catalytic activity of Cyt-c in μE-2. Such conformational changes of Cyt-c in μEs indicate that the nature of the interfaces offered by BAILs and water in reverse μEs plays an important role in the stabilization/destabilization of enzyme conformation resulting in varying enzymatic activity. The role of the varying nature of water molecules present at such interfaces in governing the conformational changes and hence enzymatic activity cannot be ruled out at this stage. The observation in the investigated system is in line with the reported IL based μEs where the nano-interfaces offered by μEs act as a platform for the storage of lysozyme and influence its antimicrobial activity.28
Therefore, the present study along with previous studies on encapsulation of enzymes in an aqueous pool of reverse μEs comprising conventional surfactants and organic solvents,62,63 conventional surfactants and HILs,32–35,64–67 as well as SAILs and HILs28,44 would provide a new platform for the materialization of new μEs based on BAILs for various biological applications.
5. Conclusion
Three new μEs based on BAILs and HIL using water as the polar medium are prepared and characterized using different techniques. In comparison to conventional sodium AOT surfactant, the investigated BAIL based on [AOT]− and imidazolium as the cation stabilizes the μEs effectively in the absence of any co-surfactant. The effect of functionalization of BAIL on the phase behavior of μEs has been explored for the first time. Different studies indicate the influence of functionalization of the alkyl chain of the imidazolium cation on the nano-interfaces created by BAIL and water in reverse μEs, where relatively stronger interactions between BAIL-2 and water are observed. Such nano-interfaces in reverse micelles are utilized as an efficient platform for biocatalysis using Cyt-c. Cyt-c in μEs exhibits more than 5-fold peroxidase activity as compared to that observed in buffer. Moreover, Cyt-c shows greater catalytic activity in μE-1 as compared to that observed in μE-2. This is due to the H-bonding interactions between the amino acid residues of Cyt-c and the ester-moiety of BAIL-2, which result in complete denaturation of Cyt-c in μE-2. Hence, the nano-interfaces play an important role in the storage of enzymes in μEs. It is anticipated that the present work along with the reported IL based μEs28,32–35,62–65 could be useful in providing a suitable scaffold for storing enzymes and for biocatalysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the DST, Govt. of India wide project number SB/FT/CS-057/2013. We are grateful to UGC, India, for the UGC-CAS program awarded to the Department of Chemistry, Guru Nanak Dev University, Amritsar. The infrastructure facility provided for this work under the UPE grant is highly acknowledged. M. K., H. K. and M. S. are thankful to CSIR and UGC, Govt. of India, for the fellowship.References
- P. Wasserscheid and T. Welton, Ionic Liquids in Synthesis, Wiley, New York, 2003 Search PubMed.
- T. Welton, Chem. Rev., 1999, 99, 2071–2084 CrossRef CAS PubMed.
- M. Watanabe, M. L. Thomas, S. Zhang, K. Ueno, T. Yasuda and K. Dokko, Chem. Rev., 2017, 117, 7190–7239 CrossRef CAS PubMed.
- S. Hisamitsu, N. Yanai and N. Kimizuka, Angew. Chem., Int. Ed., 2015, 54, 11550–11554 CrossRef CAS PubMed.
- T. V. Hoogerstraete, S. Wellens, K. Verachtert and K. Binnemansa, Green Chem., 2013, 15, 919–927 RSC.
- M. J. Earle, J. M. S. S. Esperança, M. A. Gilea, J. N. C. Lopes, L. P. N. Rebelo, J. W. Magee, K. R. Seddon and J. A. Widegren, Nature, 2006, 439, 831–834 CrossRef CAS PubMed.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4975 CrossRef CAS PubMed.
- J. Dupont, R. F. de Souza and P. A. Z. Suarez, Chem. Rev., 2002, 102, 3667–3692 CrossRef CAS PubMed.
- S. Zhang, N. Sun, X. He, X. Lu and X. Zhang, J. Phys. Chem. Ref. Data, 2006, 35, 1475–1517 CrossRef CAS.
- M. Deetlefs, K. R. Seddon and M. Shara, Phys. Chem. Chem. Phys., 2006, 8, 642–649 RSC.
- K. Fujita, D. R. MacFarlane and M. Forsyth, Chem. Commun., 2005, 4804–4806 RSC.
- D. Constatinescu, C. Herrmann and H. Weingärtner, Phys. Chem. Chem. Phys., 2010, 12, 1756–1763 RSC.
- J. L. Karr, A. M. Jesionowski, J. A. Berberich, R. Moulton and A. J. Russel, J. Am. Chem. Soc., 2003, 125, 4125–4131 CrossRef PubMed.
- Z. Yang and W. Pan, Enzyme Microb. Technol., 2005, 37, 19–28 CrossRef CAS.
- R. A. Sheldon, R. M. Lau, M. J. Sorgedrager, F. Van Rantwijk and K. R. Seddon, Green Chem., 2002, 4, 147–151 RSC.
- F. Van Rantwijk and R. A. Sheldon, Chem. Rev., 2007, 107, 2757–2785 CrossRef CAS PubMed.
- M. Moniruzzaman, N. Kamiya and M. Goto, Org. Biomol. Chem., 2010, 8, 2887–2899 RSC.
- H. Ohno, C. Suzuki, K. Fukumoto, M. Yoshizawa and K. Fujita, Chem. Lett., 2003, 32, 450–451 CrossRef CAS.
- K. Nakashima, T. Maruyama, N. Kamiya and M. Goto, Chem. Commun., 2005, 4297–4299 RSC.
- M. B. Turner, S. K. Spear, J. G. Huddleston, J. D. Holbrey and R. D. Rogers, Green Chem., 2003, 5, 443–447 RSC.
- A. J. Walker and N. C. Bruce, Chem. Commun., 2004, 2570–2571 RSC.
- P. Bharmoria, T. J. Trivedi, A. Pabbathi, A. Samanta and A. Kumar, Phys. Chem. Chem. Phys., 2015, 17, 10189–10199 RSC.
- S. P. M. Ventura, L. D. F. Santos, J. A. Saraiva and J. A. P. Coutinho, Green Chem., 2012, 14, 1620–1625 RSC.
- P. L. Luisi and B. E. Straub, Reverse Micelles: Biological and Technological Relevance of Amphiphilic Structures in Apolar Media, Plenum, New York, 1984 Search PubMed.
- C. C. Müller-Goymann, Eur. J. Pharm. Biopharm., 2004, 58, 343–356 CrossRef PubMed.
- K. L. Kadam, Enzyme Microb. Technol., 1986, 8, 266–273 CrossRef CAS.
- C. R. Vestal and Z. J. Zhang, Chem. Mater., 2002, 14, 3817–3822 CrossRef CAS.
- M. Kaur, G. Singh, A. Kaur, P. K. Sharma and T. S. Kang, Langmuir, 2019, 35, 4085–4093 CrossRef CAS PubMed.
- P. Brochette, C. Petit and P. Pileni, J. Phys. Chem., 1988, 92, 3505–3511 CrossRef CAS.
- M. Bisht, D. Mondal, M. M. Pereira, M. G. Freire, P. Venkatesu and J. A. P. Coutinho, Green Chem., 2017, 19, 4900–4911 RSC.
- D. Das, A. Dasgupta and P. K. Das, Tetrahedron Lett., 2007, 48, 5635–5639 CrossRef CAS.
- M. Moniruzzaman, N. Kamiya, K. Nakashima and M. Goto, Green Chem., 2008, 10, 497–500 RSC.
- I. V. Pavlidis, D. Gournis, G. K. Papadopoulos and H. Stamatis, J. Mol. Catal. B: Enzym., 2009, 60, 50–56 CrossRef CAS.
- M. Moniruzzaman, N. Kamiya and M. Goto, Langmuir, 2009, 25, 977–982 CrossRef CAS PubMed.
- X. Yu, Q. Li, M. Wang, N. Du and X. Huang, Soft Matter, 2016, 12, 1713–1720 RSC.
- M. Zoumpanioti, M. Karali, A. Xenakis and H. Stamatis, Enzyme Microb. Technol., 2006, 39, 531–539 CrossRef CAS.
- G. Singh, Komal, M. Singh, O. Singh and T. S. Kang, J. Phys. Chem. B, 2018, 122, 12227–12239 CrossRef CAS PubMed.
- T. K. De and A. Maitra, Adv. Colloid Interface Sci., 1995, 59, 95–193 CrossRef CAS.
- M. Moniruzzaman, N. Kamiya, K. Nakashima and M. Goto, ChemPhysChem, 2008, 9, 689–692 CrossRef CAS PubMed.
- C. P. Fredlake, J. M. Crosthwaite, D. G. Hert, S. N. V. K. Aki and J. F. Brennecke, J. Chem. Eng. Data, 2004, 49, 954–964 CrossRef CAS.
- V. G. Rao, S. Ghosh, C. Ghatak, S. Mandal, U. Brahmachari and N. Sarkar, J. Phys. Chem. B, 2012, 116, 2850–2855 CrossRef CAS PubMed.
- V. G. Rao, S. Mandal, S. Ghosh, C. Banerjee and N. Sarkar, J. Phys. Chem. B, 2012, 116, 8210–8221 CrossRef CAS PubMed.
- K. Damarla, P. Bharmoria, K. S. Rao, P. S. Gehlot and A. Kumar, Chem. Commun., 2016, 52, 6320–6323 RSC.
- M. Kaur, G. Singh, S. Kumar, Navnidhi and T. S. Kang, J. Colloid Interface Sci., 2018, 511, 344–354 CrossRef CAS PubMed.
- C. M. O. Lépori, N. M. Correa, J. J. Silber and R. D. Falcone, Soft Matter, 2016, 12, 830–844 RSC.
- J. Piekart and J. Łuczak, Soft Matter, 2015, 11, 8992–9008 RSC.
- M. Olla and M. Monduzzi, Langmuir, 2000, 16, 6141–6147 CrossRef CAS.
- R. Wang, Z. Feng, W. Jin and X. Huang, Ind. Eng. Chem. Res., 2018, 57, 14846–14853 CrossRef CAS.
- T. S. Kang, K. Ishiba, M.-A. Morikawa and N. Kimizuka, Langmuir, 2014, 30, 2376–2384 CrossRef CAS PubMed.
- H. Zhang, K. Li, H. Liang and J. Wang, Colloids Surf., A, 2008, 329, 75–81 CrossRef CAS.
- J. K. Gregory, D. C. Clary, K. Liu, M. G. Brown and R. J. Saykally, Science, 1997, 275, 814–817 CrossRef CAS PubMed.
- E. I. Izgorodina, M. Forsyth and D. R. MacFarlane, Phys. Chem. Chem. Phys., 2009, 11, 2452–2458 RSC.
- T. Singh and A. Kumar, J. Phys. Chem. B, 2008, 112, 12968–12972 CrossRef CAS PubMed.
- P. Bharmoria and A. Kumar, Chem. Commun., 2016, 52, 497–500 RSC.
- H. Tajima, S. Ikeda, M. Matsuda, N. Hanasaki, J.-W. Oh and H. Akiyama, Solid State Commun., 2003, 126, 579–581 CrossRef CAS.
- D. Flöck and V. Helms, Biophys. J., 2004, 87, 65–74 CrossRef PubMed.
- C. Kawai, F. S. Pessoto, T. Rodrigues, K. C. U. Mugnol, V. Tórtora, L. Castro, V. A. Milícchio, I. L. S. Tersariol, P. D. Mascio, R. Radi, A. M. Carmona-Ribeiro and I. L. Nantes, Biochemistry, 2009, 48, 8335–8342 CrossRef CAS PubMed.
- S. Abel, M. Waks and M. Marchi, Eur. Phys. J. E: Soft Matter Biol. Phys., 2010, 32, 399–409 CrossRef CAS PubMed.
- G. Singh, G. Singh, S. Kancharla and T. S. Kang, J. Phys. Chem. B, 2019, 123, 2169–2181 CrossRef CAS PubMed.
- R. B. Barlow, J. B. Bremner and K. S. Soh, Br. J. Pharmacol., 1978, 62, 39–50 CrossRef CAS PubMed.
- L. S. Kaminsky, F. C. Yong and T. E. King, J. Biol. Chem., 1972, 247, 1354–1359 CrossRef CAS.
- C. Shah, S. Sellappan and D. Madamwar, Process Biochem., 2000, 35, 971–975 CrossRef CAS.
- K. Carlile, G. D. Rees, B. H. Robinson, T. D. Steer and M. Svensson, J. Chem. Soc., Faraday Trans., 1996, 92, 4701–4708 RSC.
- G.-P. Zhou, Y. Zhang, X.-R. Huang, C.-H. Shi, W.-F. Liu, Y.-Z. Li, Y.-B. Qu and P.-J. Gao, Colloids Surf., B, 2008, 66, 146–149 CrossRef CAS PubMed.
- F. Xu, L. Chen, A. Wang and Z. Yan, Bioresour. Technol., 2016, 208, 19–23 CrossRef CAS PubMed.
- L. Xue, H. Qiu, Y. Li, L. Lu, X. Huang and Y. Qu, Colloids Surf., B, 2011, 82, 432–437 CrossRef CAS PubMed.
- L. Xue, Y. Li, F. Zou, L. Lu, Y. Zhao, X. Huang and Y. Qu, Colloids Surf., B, 2012, 92, 360–366 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cp04513f |
| This journal is © the Owner Societies 2021 |