
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Infrared action spectroscopy of nitrous oxide on cationic gold and cobalt clusters†
Ethan M.
Cunningham
 a,
Alice E.
Green
a,
Gabriele
Meizyte
a,
Alexander S.
Gentleman
a,
Peter W.
Beardsmore
a,
Sascha
Schaller
b,
Kai M.
Pollow
c,
Karim
Saroukh
c,
Marko
Förstel
c,
Otto
Dopfer
*c,
Wieland
Schöllkopf
b,
André
Fielicke
*bc and
Stuart R.
Mackenzie
*a
a,
Alice E.
Green
a,
Gabriele
Meizyte
a,
Alexander S.
Gentleman
a,
Peter W.
Beardsmore
a,
Sascha
Schaller
b,
Kai M.
Pollow
c,
Karim
Saroukh
c,
Marko
Förstel
c,
Otto
Dopfer
*c,
Wieland
Schöllkopf
b,
André
Fielicke
*bc and
Stuart R.
Mackenzie
*a
aDepartment of Chemistry, University of Oxford, Physical and Theoretical Chemistry Laboratory, South Parks Road, Oxford OX1 3QZ, UK. E-mail: stuart.mackenzie@chem.ox.ac.uk
bFritz-Haber-Institut der Max-Planck-Gesellschaft, Faradayweg 4-6, 14195 Berlin, Germany. E-mail: fielicke@fhi-berlin.mpg.de
cInstitut für Optik und Atomare Physik, Technische Universität Berlin, Hardenbergstr. 36, 10623 Berlin, Germany. E-mail: dopfer@physik.tu-berlin.de
First published on 8th December 2020
Abstract
Understanding the catalytic decomposition of nitrous oxide on finely divided transition metals is an important environmental issue. In this study, we present the results of a combined infrared action spectroscopy and quantum chemical investigation of molecular N2O binding to isolated Aun+ (n ≤ 7) and Con+ (n ≤ 5) clusters. Infrared multiple-photon dissociation spectra have been recorded in the regions of both the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (1000–1400 cm−1) and NN (2100–2450 cm−1) stretching modes of nitrous oxide. In the case of Aun+ clusters only the ground electronic state plays a role, while the involvement of energetically low-lying excited states in binding to the Con+ clusters cannot be ruled out. There is a clear preference for N-binding to clusters of both metals but some O-bound isomers are observed in the case of smaller Con(N2O)+ clusters.
O (1000–1400 cm−1) and NN (2100–2450 cm−1) stretching modes of nitrous oxide. In the case of Aun+ clusters only the ground electronic state plays a role, while the involvement of energetically low-lying excited states in binding to the Con+ clusters cannot be ruled out. There is a clear preference for N-binding to clusters of both metals but some O-bound isomers are observed in the case of smaller Con(N2O)+ clusters.
1. Introduction
Nitrous oxide (N2O) is a potent greenhouse gas1,2 accounting for around 5% of anthropogenic emissions and has a warming potential almost 300 times greater than carbon dioxide.3 As a result, there is considerable interest in reducing N2O emissions, especially from automobile combustion engines. To this end, modern vehicles employ metal-catalysed nitrogen oxide reduction in their three-way catalytic converters.4,5 The transition metals used are usually found in a highly dispersed form such as nanoparticles and the study of small isolated metal clusters is thus a vibrant and active research area.6 Understanding the physical and chemical properties of transition metal clusters has wider importance in the fields of heterogeneous catalysis, solid-state physics, surface chemistry, and organometallic chemistry.7–11 In this context, gas-phase metal clusters represent tractable model systems for developing molecular level insight into the thermodynamics and kinetics of catalytic reactions. Decades of work have shown that this activity depends on a complex interplay of various factors, including size, geometry, local charge density, dimensionality, electronic structure, and the presence as well as properties of defect sites.12–21Many experimental studies have investigated the structural evolution of isolated gold clusters with atom number.22–24 Early investigations utilised ion mobility mass spectrometry, which, combined with quantum chemical calculations, is a powerful tool for structure determination of charged metal clusters.25 Kappes and coworkers have performed such studies on charged gold clusters up to n = 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 26,27 and determined planar ground-state structures for Aun+ (n = 3–7) and Aun− (n = 3–11), respectively. Anionic gold clusters were investigated further by electron diffraction confirming these structural motifs, revealing a possible chiral structure for Au34−.28,29 Building on previous spectroscopic studies of Aun+ (n ≤ 5) clusters,30–33 Ferrari et al. have recently reported far-infrared multiple photon dissociation spectroscopy (IR-MPD) studies of Aun+ (n ≤ 9), identifying both 3D and planar isomers for n = 8.34 IR-MPD studies have also been performed on neutral gold clusters up to 20 gold atoms, revealing two-dimensional, planar structures up to Au10, with the onset of 3D structures for Au11 at 100 K.35–37 At room temperature, however, planar and non-planar isomers coexist for Au8, Au9, and Au10. The largest cluster studied, Au20, was shown to have a pyramidal structure.38
26,27 and determined planar ground-state structures for Aun+ (n = 3–7) and Aun− (n = 3–11), respectively. Anionic gold clusters were investigated further by electron diffraction confirming these structural motifs, revealing a possible chiral structure for Au34−.28,29 Building on previous spectroscopic studies of Aun+ (n ≤ 5) clusters,30–33 Ferrari et al. have recently reported far-infrared multiple photon dissociation spectroscopy (IR-MPD) studies of Aun+ (n ≤ 9), identifying both 3D and planar isomers for n = 8.34 IR-MPD studies have also been performed on neutral gold clusters up to 20 gold atoms, revealing two-dimensional, planar structures up to Au10, with the onset of 3D structures for Au11 at 100 K.35–37 At room temperature, however, planar and non-planar isomers coexist for Au8, Au9, and Au10. The largest cluster studied, Au20, was shown to have a pyramidal structure.38
Reactions between cationic gold clusters, Aun+ (n = 1–4), and N2O have been studied by Dietrich et al. combining a Penning trap with time-of-flight (ToF) mass spectrometry.39 Dissociative charge transfer yielding AunN and NO+ was the major reaction channel for n = 1 and 2, but a range of other products, including AunO+, AunN+ and AunNO+, were also detected, with n = 1 and 3 clusters leading preferentially to NO bond rupture and the n = 2 cluster breaking the NN bond. Relevant structural information on nitrogen oxides adsorbed to gold clusters is limited to the IR-MPD study of Aun(NO)+ by Fielicke and coworkers.40 Marked nitric oxide activation was observed in binding to the odd-electron, even-n, Aun+ clusters, reflecting efficient unpaired electron donation into the NO π* orbital. NO was found to bind non-linearly in atop arrangements, tending to favour low-coordinated gold atoms.
Gehrke et al. have employed IR-MPD to identify the structures of Con+–Ar (n = 4–8) clusters.41 All clusters observed exhibit high spin states and significant Jahn–Teller distortion and, unusually, the vibrational spectra of Con+ were shown to be strongly dependent on the number of adsorbed Ar messenger atoms. More recently, an IR-MPD study by Jia et al. on Con+–Arm (n = 3–5, m = 3, 4) confirmed these structural findings,42 and the high spin states of isolated Con+ (n = 4–9) clusters have been measured by X-ray absorption and X-ray magnetic circular dichroism spectroscopy.43 The structures of neutral Con clusters (n = 4–10, 13) have also been studied using two-colour IR-UV spectroscopy.44
Reactions of Con+ (n = 4–30) clusters with NO and N2O have been investigated using Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometry under single-collision conditions.45 Reactions with N2O typically proceed much more slowly than the collision rate and result in the formation of simple cluster oxides, ConOm+. By contrast, NO chemisorption rates exceed calculated collision rates. In both reactions there is evidence for cluster fragmentation, characterised typically by Co atom evaporation, and multiple collisions result in N2O decomposition leading to N2 loss. No molecularly adsorbed, i.e. Con(N2O)+, clusters were observed under single-collision conditions.
Theoretical studies by Castro and coworkers, utilising density functional theory methods, investigated the catalytic N2O reduction by Rh6− and Rh6+ clusters46 as well as the low-symmetry Pt8 cluster.47 We have previously reported IR-MPD studies of N2O binding on Rhn+ (n = 4–8)48–50 and Ptn+ clusters (n = 1–8).51 Extensive IR-induced reactivity was observed in both cases, with excitation of infrared modes resulting in N2O decomposition. By way of comparison with these studies, we report here an IR-MPD study of N2O adsorption on isolated Aun+ (n ≤ 7) and Con+ (n ≤ 5) clusters in which we investigate the nature of the molecular binding and its dependence on cluster size, electronic and geometrical structure.
2. Experimental and computational methodology
The instrument at the Fritz-Haber Institute along with the infrared multiple photon dissociation (IR-MPD) technique employed have been described previously and only brief details are given here.52,53 Gas-phase Mn(N2O)+ (M = Co, Au) clusters are formed by entraining metal atoms/ions produced by pulsed laser ablation of a rotating metal rod within an intense pulse of helium carrier gas (10 bar backing pressure), with nitrous oxide introduced downstream via a late mixing valve. Efficient clustering and thermalisation occurs via collisions within an aggregation channel before expansion into vacuum creating the molecular beam. The beam is skimmed for collimation and positively-charged clusters are detected by orthogonal extraction into a reflectron time-of-flight mass spectrometer.54 Experimental parameters such as the total backing pressure behind the pulsed valve, ablation laser power and nitrous oxide partial pressure are optimised for the generation of Mn(N2O)+ (M = Co, Au; n ≤ 8) species of interest. For these experiments the source was operated at room temperature and the cluster distribution is assumed to be in thermal equilibrium with this.Intense tunable pulsed infrared radiation is provided by the free electron laser (FEL) at the Fritz-Haber Institute (FHI),53 operating in the regions of 1000–1400 cm−1 and 2100–2450 cm−1 and covering the ν1(NO) and ν3(NN) stretching modes of N2O, respectively.55 The IR beam is collinear with and counter-propagates the molecular beam, and IR-MPD spectra are recorded by monitoring the loss of N2O from the parent Mn(N2O)+ cluster as a function of wavenumber.56–62 In this way, photofragmentation provides a signature of IR absorption. To account for photon flux fluctuations, all experimental spectra were corrected for laser power.
To interpret the experimental vibrational spectra, quantum chemical calculations were performed, yielding structures and simulated infrared spectra of energetically low-lying isomers. Density functional theory (DFT) calculations were performed using the hybrid meta-GGA functional TPSSh, which includes 10% exact Hartree–Fock exchange,63 together with the Def2-TZVP basis set, a combination which has worked well in previous studies.64,65 Relativistic effects, essential in describing gold chemistry,66–68 were incorporated by means of the ECP-60 effective core potential (ECP)69 for all but the 19 valence electrons of each gold atom. Def2-TZVP was also shown to accurately predict the IR spectra of Rhn(CO)m+ clusters.70 The quadratically convergent SCF procedure was used along with 1.00 × 10−6 hartree convergence criterion along with the Gaussian 09 “very tight” geometry optimisation convergence.71 Plausible N2O binding at atop, bridge, and three-body binding sites were investigated with N2O found to bind exclusively atop, favouring low coordinated gold/cobalt atoms, with μ2 and μ3 arrangements converging to atop structures upon geometry optimisation. The lowest energy structures found typically involve dissociatively-adsorbed nitrous oxide, with molecularly-bound structures representing entrance-channel complexes trapped behind an activation barrier. There is no evidence for dissociative structures in the experimental spectra; N2 almost certainly desorbs as a result of the exoergicity of the dissociation and subsequent O-atom binding. To better compare with experimental spectra, all simulated IR spectra reported here have been scaled by a factor 0.955, determined from the simulated vibrational wavenumbers of isolated N2O.55 All calculations were performed using the Gaussian09 suite of programs,71 and relative energies of structural isomers are given in electron volts (eV), inclusive of zero-point energy. To accurately determine the multiplicity of each cluster structure, together with the relative energy, for each calculated structure the DFT wavefunction was stabilised and tested.
3. Results and discussion
3.1 Aun(N2O)+ clusters
Fig. 1 presents the ToF mass spectrum showing the production of Aun+ (n = 1–8) clusters along with attached adsorbates. Besides the naked Aun+ cluster cations, the most intense signals in the spectrum are the target Aun(N2O)+ clusters as well as additional peaks attributable to the simple cluster oxides, AunOm+ and, uniquely in the case of n = 1, AuO(N2O)+. Other species are observed, which we believe arise from reactions with trace hydrocarbons in the source, possibly introduced by glue used for sticking the rod to the holder. For the smallest, n = 1 and 2 clusters, the species produced here are different to those observed by Dietrich et al. in their single-collision reaction study, reflecting the stabilising effect of collisions in the source employed here and the possible effects of fragmentation of larger clusters.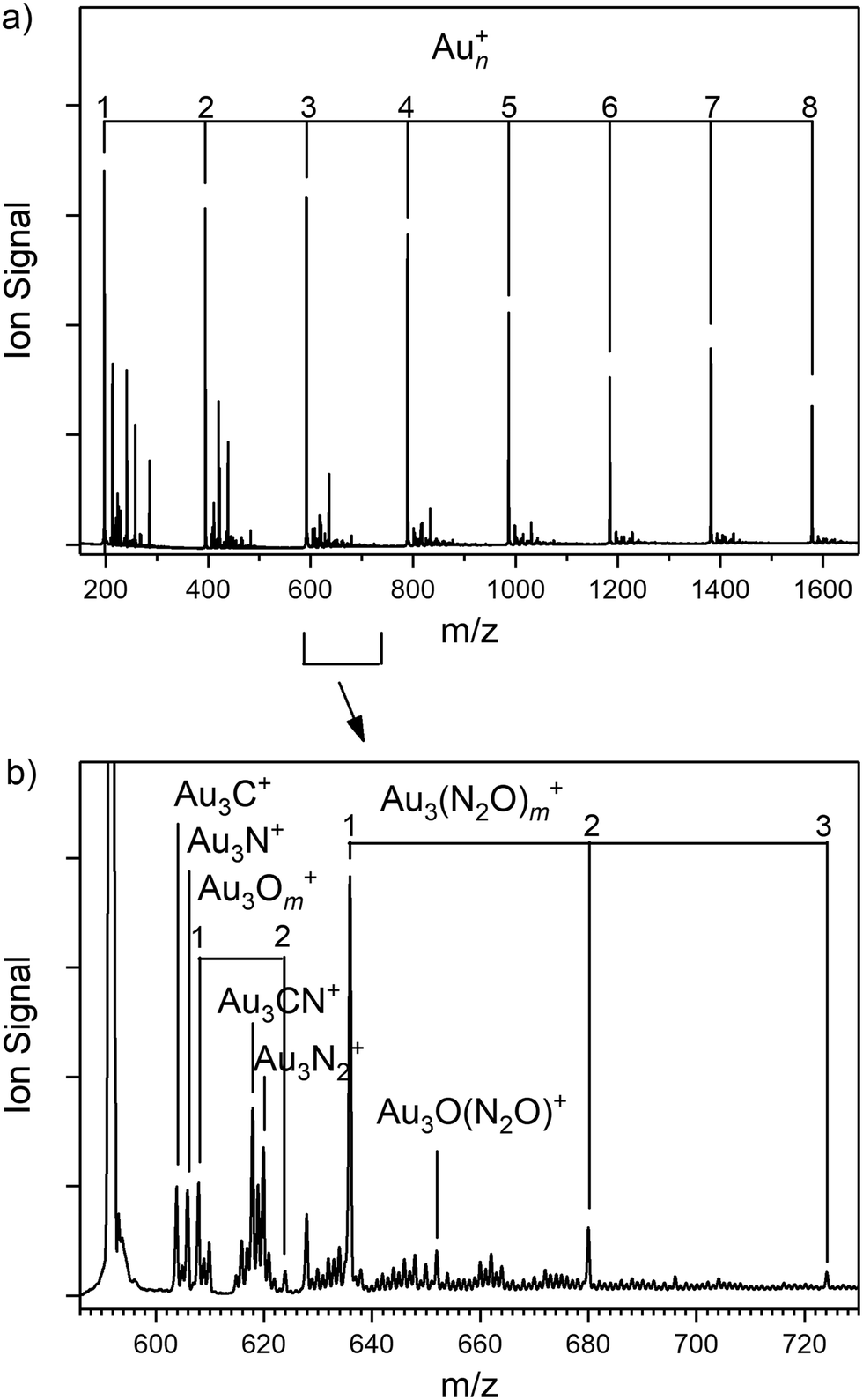 | ||
| Fig. 1 (a and b) Time-of-flight mass spectrum of Aun+ clusters produced by laser ablation of a gold rod in the presence of a helium carrier gas (10 bar backing pressure) and N2O added downstream in the cluster channel. | ||
Fig. 2 shows an overview of the IR-MPD spectra for Aun(N2O)+ (n = 3–7) species recorded in the spectral region of both the NO (1000–1400 cm−1) and NN (2100–2450 cm−1) stretches and presented as depletion in the parent ion signal. The presence of intense depletion bands (in some cases exceeding 60% of the parent ion signal) close to the vibrational bands in free nitrous oxide confirms the molecular nature of the N2O binding. In this respect, Aun(N2O)+ clusters are similar to the isoelectronic Aun(OCS)+ species whose spectra we have reported recently.72 The spectra of the n = 1 and 2 species unfortunately suffer from substantial distortions arising from dissociation of larger species (see Fig. S4 and S5, ESI†).
 | ||
| Fig. 2 IR-MPD depletion spectra of Aun(N2O)+ (n = 3–7) clusters with depletion given as a percentage of ion signal. The vertical dashed lines at 1285 and 2224 cm−1 indicate the wavenumber of the ν1(NO) and ν3(NN) modes in isolated N2O, respectively.55 | ||
In Fig. 2, a single band is observed in the NO stretch region around 1340 cm−1, blue-shifted up to 60 cm−1 from the ν1 mode of free N2O at 1285 cm−1.55 The blue-shift reduces smoothly with increasing cluster size from 1345 cm−1 for n = 3, to 1334 cm−1 for n = 7. A similar trend is observed in the NN stretch region as the main band moves from 2278 cm−1 for n = 3 to 2266 cm−1 for n = 7, again slightly blue-shifted from the ν3 mode of N2O at 2224 cm−1.55 In the latter spectral region a weak shoulder to the main spectral feature is observed for clusters n = 3 and 4, close to the free N2O stretch which is not present for the larger species.
Interpretation of the IR-MPD spectra is greatly assisted by simulated infrared spectra of energetically low-lying isomers identified using DFT. By way of example, the observed spectra of Au3(N2O)+ and Au4(N2O)+ are presented in Fig. 3 and 4, respectively, along with simulated spectra of low-lying structural isomers.
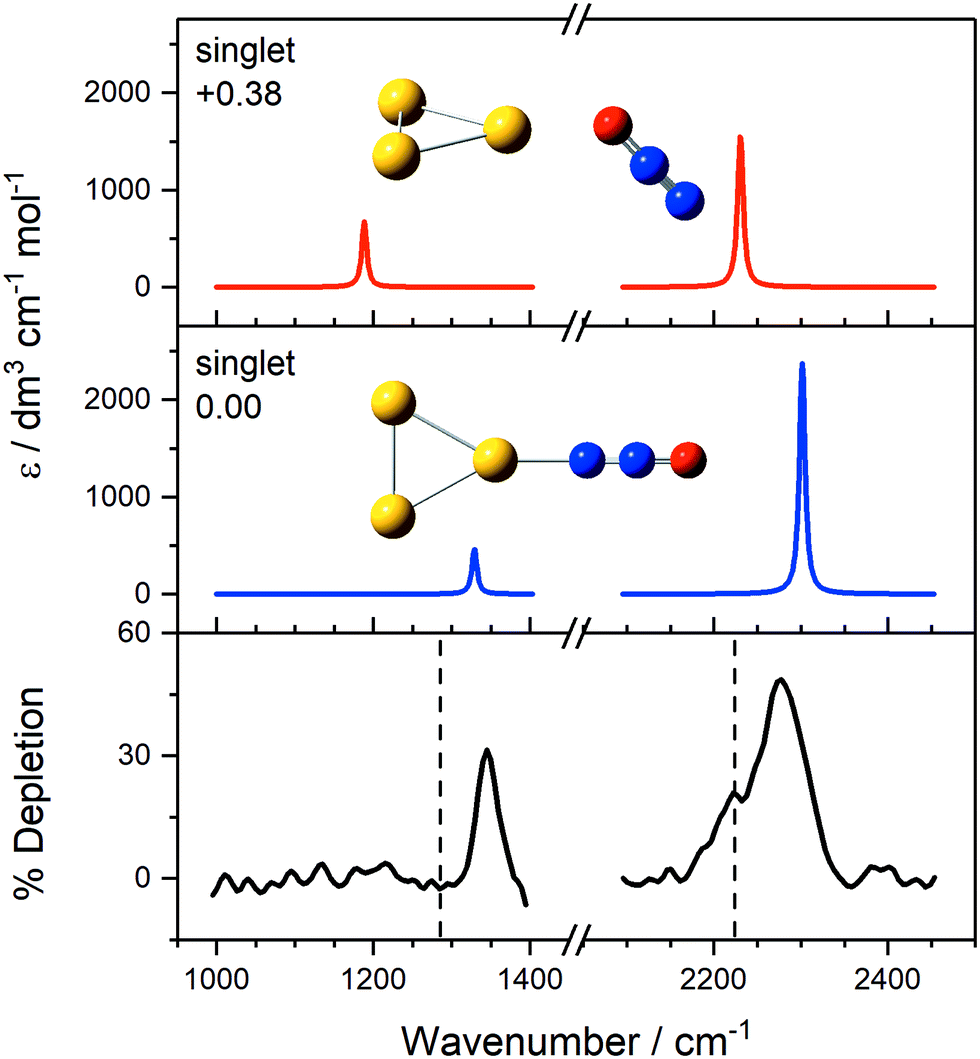 | ||
| Fig. 3 IR-MPD spectrum of Au3(N2O)+, along with simulated IR spectra of energetically low-lying isomers. The only low-lying electronic state is a singlet state and simulated IR bands corresponding to N-bound and O-bound isomers are indicated in blue and red, respectively. Energies are given in eV relative to the lowest molecularly bound isomer. The vertical dashed lines at 1285 and 2224 cm−1 indicate the wavenumber of the ν1(NO) and ν3(NN) modes in isolated N2O, respectively.55 | ||
 | ||
| Fig. 4 IR-MPD depletion spectrum of Au4(N2O)+, along with simulated IR spectra of low-lying isomers in the region of the N2O NO and NN stretches. The low-lying structures have doublet multiplicity. Simulated IR bands corresponding to N-bound and O-bound isomers are indicated in blue and red, respectively, with relative energies given in eV. The vertical dashed lines at 1285 and 2224 cm−1 indicate the wavenumber of the ν1(NO) and ν3(NN) modes in isolated N2O, respectively.55 | ||
For Au3(N2O)+ (Fig. 3), the observed spectrum agrees very well with the simulated spectrum of the N-bound, lowest energy molecularly-bound structure with both spectral features blue-shifted from the free N2O bands. With fractional depletion of >50% observed, it is clear that this structure dominates the distribution. It is tempting to interpret the shoulder at 2223 cm−1 as the higher energy O-bound isomer but there is little evidence for the equivalent red-shifted band in the NO stretch region. By contrast, both N- and O-bound isomers were observed in our recent study of Au(N2O)x+.73 The origin of this shoulder remains unclear but could represent the presence of another N-bound isomer. Alternatively, it could result from spectral power broadening due to the increased FEL power in the NN stretching region and the fact that fewer higher energy photons are required for IR-MPD in this region.
In the case of Au4(N2O)+ (Fig. 4), a richer distribution of low-lying structural isomers is predicted. However, it is clear that the vibrational spectrum in this region is diagnostic only of the nitrous oxide binding motif and not of the metal cluster structure. Again, the spectrum is dominated by N-bound structures with limited evidence for O-binding.
In both spectral regions, the weak blue shift observed in the N-bound isomers relative to the free N2O stretches can be explained by σ-donation from the 7σ (HOMO−1) orbital in N2O to the metal cluster, which is antibonding with respect to the NN bond. By contrast, O-binding, if present, would be signified by negligible shifts in the NN stretch region but a marked red shift in the NO band, reflecting an increased effective reduced mass. The nature of N2O binding to metal centres has been discussed extensively previously.4,73–75
The above interpretation of Aun(N2O)+ binding is readily extended to the larger, n = 5–7 clusters, in which single blue-shifted bands in both spectral regions are again assigned to N-bound isomers.
3.2 Con(N2O)+ clusters
Fig. 5 shows the ToF mass spectrum of Con+-based clusters generated, exhibiting the production of Con(N2O)+ species as far as n = 5. Considerably more ConOm+ (m = 1–4) oxidation products are observed than for the gold clusters (see Fig. 5c). However, care needs to be taken in ascribing these to reactions with particular Con+ clusters. Under single-collision conditions, the Con+ + N2O reaction has previously been shown to result in efficient fragmentation of metal clusters in this size regime, notably as Con+ + N2O → Con−1O+ + [Co,N2].45 Hence, the observation of species such as Co5O4+ almost certainly reflects reactions of larger clusters as well as Co5+ itself.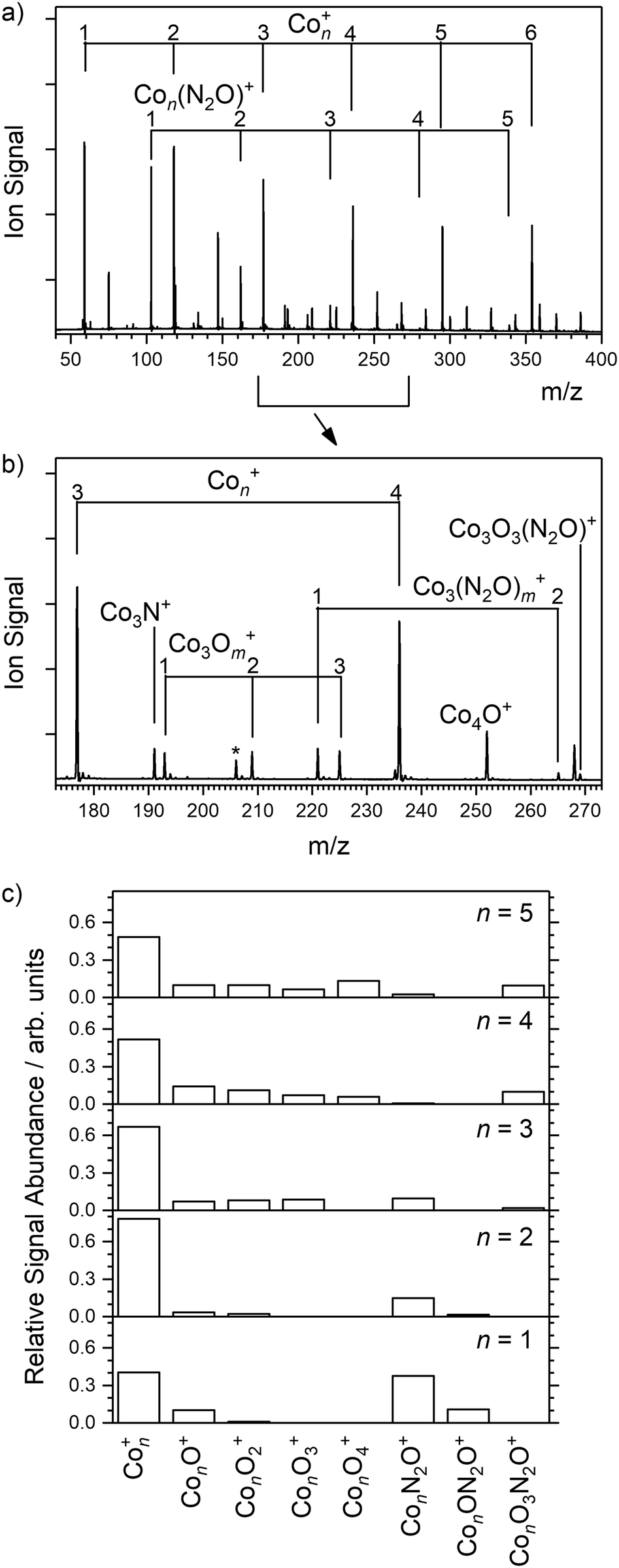 | ||
| Fig. 5 (a and b) Mass spectrum of ions observed following ablation of a cobalt rod along with N2O added downstream in the cluster channel. For clarity, the mass signal for Co2(N2O)2+ is marked with an asterisk (*). (c) Histogram showing relative abundance of major species observed relative to the naked Con+ mass signal. The larger clusters react readily with N2O forming simple oxides. | ||
Despite the abundance of oxides observed, sufficient Con(N2O)+ signals were produced to permit IR-MPD spectra of Con(N2O)+ clusters (n = 1–5) to be recorded, as shown in Fig. 6. The spectra of the very smallest (n = 1, 2) species again suffer enhancements (observed as negative depletions) due to fragmentation of larger and/or multiply decorated clusters, especially Con(N2O)2+, at the same wavenumber. Nevertheless, the observation of multiple spectral features in this region, including peaks red-shifted from the free N2O band, signifies the presence of both O-bound and N-bound N2O cluster structures.
 | ||
| Fig. 6 IR-MPD depletion spectra of Con(N2O)+ (n = 1–5) clusters in the NO and NN regions of N2O. The vertical dashed lines at 1285 and 2224 cm−1 indicate the wavenumber of the ν1(NO) and ν3(NN) modes in isolated N2O, respectively.55 | ||
A mix of N- and O-bound isomers was observed in related IR spectra of Co(N2O)x+ complexes in which the barrier for N2O free internal rotation in the Co(N2O)+ complex was calculated to be ca. 1.3 eV (0.8 eV from the O-bound minimum).76 Given the rate of collisions in the cluster formation channel, it is thus likely that a fraction of complexes become trapped in low-lying excited isomeric forms behind barriers of this magnitude. The larger clusters, however, represent more effective thermal heat baths facilitating a more efficient annealing of structures. Hence, the spectra of the larger n species are dominated by the lower energy N-bound isomers.
The strongest feature in the NO stretch region (ranging from 1345 cm−1 for n = 1 to 1323 cm−1 for n = 5) is characteristic of an N-bound structure. For n = 5 this band exceeds 80% depletion suggesting this is the dominant structure present. The lower fractional depletions observed for smaller cluster sizes reflect the presence of other isomers in the beam. In the NN stretch region, by contrast, all spectra exhibit one main band slightly blue-shifted from the ν3 band of N2O with a shoulder observed very close to the free ν3 stretch. Similarly to the NO region, the main band is slightly more strongly blue-shifted for the smallest cluster, n = 1, at 2280 cm−1 compared with 2271 cm−1 for n = 5.
Fig. 7 compares the IR-MPD spectrum of Co3(N2O)+ together with simulated spectra for low-lying calculated triplet (lowest energy) and quintet (1st excited state) isomeric forms, confirming the assignments above. The strong bands at 1337 cm−1 and 2275 cm−1, each blue-shifted from the free N2O bands, are readily assigned as N-bound structures (most likely the lowest-lying triplet state; IR-MPD is comparatively insensitive to cluster electronic state). The broad feature at 1219 cm−1 and the shoulder near 2220 cm−1, by contrast, agree well with simulated bands of the triplet O-bound isomer.
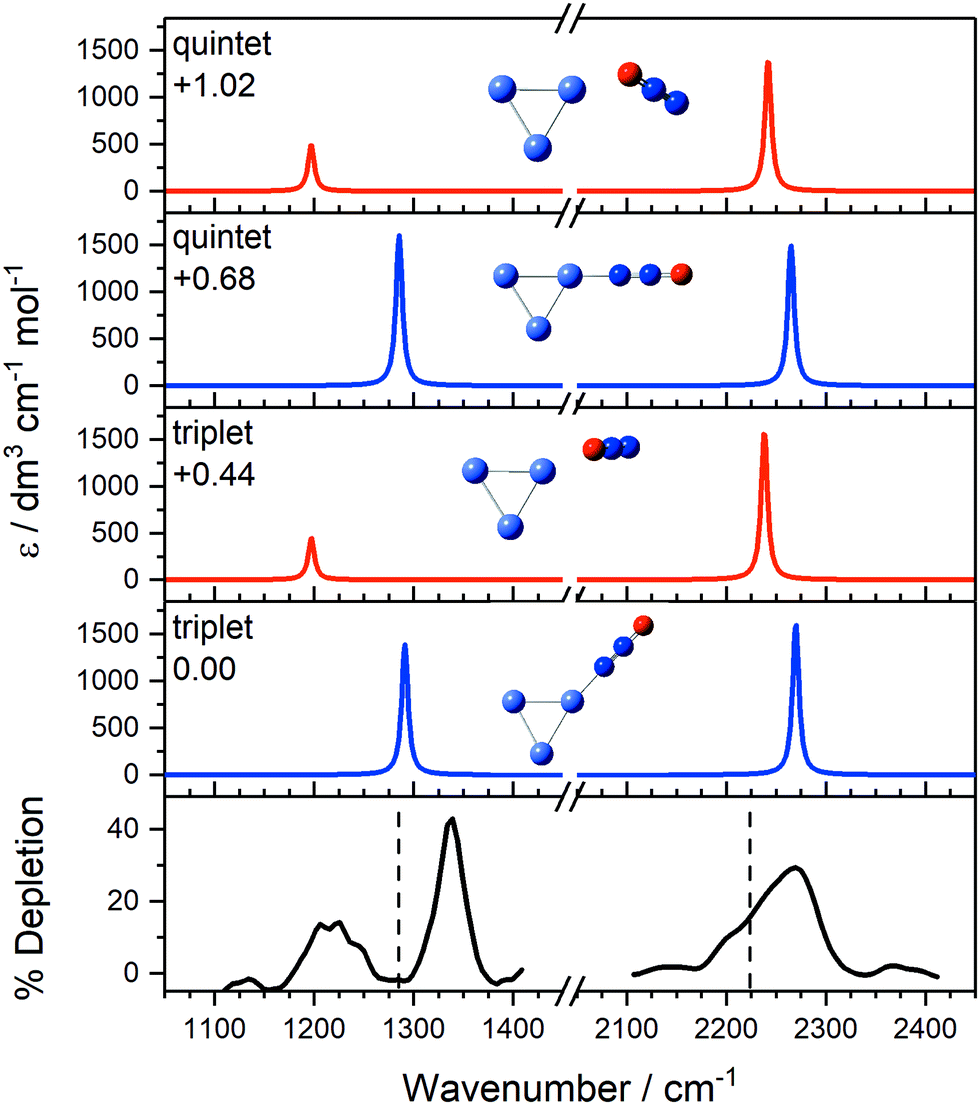 | ||
| Fig. 7 IR-MPD depletion spectrum of Co3(N2O)+, along with simulated IR spectra of low-lying isomers. Simulated IR spectra corresponding to N-bound and O-bound ligands are indicated in blue and red, respectively, for the two lowest energy calculated electronic states. The relative energies to the lowest molecularly bound isomer are given in eV. The vertical dashed lines at 1285 and 2224 cm−1 indicate the wavenumber of the ν1(NO) and ν3(NN) modes in isolated N2O, respectively.55 | ||
In addition to multiple isomers, Con(N2O)+ clusters with n = 4 and 5 are predicted to have several more low-lying electronic states, each with qualitatively similar IR spectra in this region (see ESI†).
The relative energies of the lowest N- and O-bound isomers of Mn(N2O)+ (M = Co, Au; n = 1–5), are shown in Fig. 8 for the lowest energy electronic state in each case. For all cluster sizes, the N-bound isomers (blue) represent the lowest energy molecularly-bound structures, however the relative energies of the higher-lying O-bound structures (red) vary with cluster size, especially in the case of cobalt clusters.
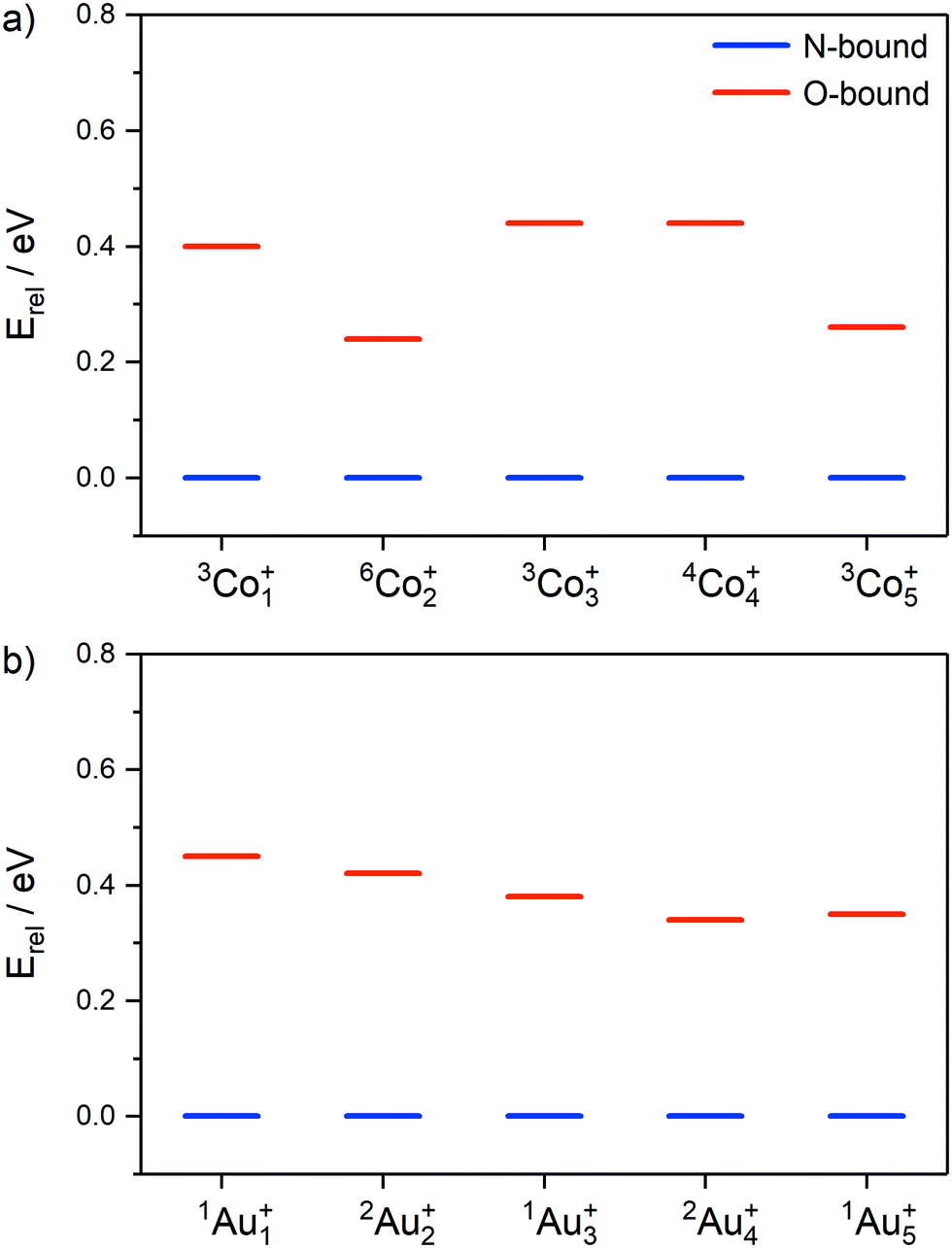 | ||
| Fig. 8 Relative energies of lowest energy molecularly-bound N- (shown in blue) and O-bound isomers (red) for (a) Con(N2O)+ and (b) Aun(N2O)+ (n = 1–5). | ||
In the case of Aun(N2O)+, the energy difference between N- and O-bound isomers reduces smoothly with cluster size between n = 1 (0.45 eV) and n = 5 (0.35 eV). For the Con(N2O)+ clusters the picture is more complicated. Many more low-lying electronic states exist for the Con(N2O)+ clusters (see ESI†), especially for the even n, odd electron clusters for which doublet, quartet and sextet states all lie within 1.3 eV of the lowest energy structures. Co2(N2O)+ has an anomalously low-lying O-bound state (ca. 0.24 eV in both doublet and sextet states). We have not calculated the barrier to internal N2O rotation for each cluster (which interchanges N- and O-bound isomers) but evidence of O-bound structures in the spectra of Con(N2O)+ clusters suggests a significant barrier behind which the higher energy structures can be trapped. As discussed above, such structures have been observed in the spectra of M(N2O)m+ for several individual metal ions73,76,77 but IR-MPD studies of Rhn(N2O)+ and, recently, Ptn(N2O)+ (n > 2) clusters concluded that N2O binds exclusively via the terminal N-atom.48,49,51 This suggests that, as well as larger metal clusters providing better annealing, the barriers to N2O rotation on Con+ are unusually large.
The calculated ligand binding energies of the lowest energy molecularly-bound N- and O-bound isomers for Con(N2O)+ and Aun(N2O)+ (n = 1–5) are presented in Fig. 9 illustrating the higher binding energy of N-bound structures relative to O-bound. The overall trend is for weaker binding with increased cluster size on both metals, though the N-bound isomer of Co2(N2O)+ is anomalously weakly bound.
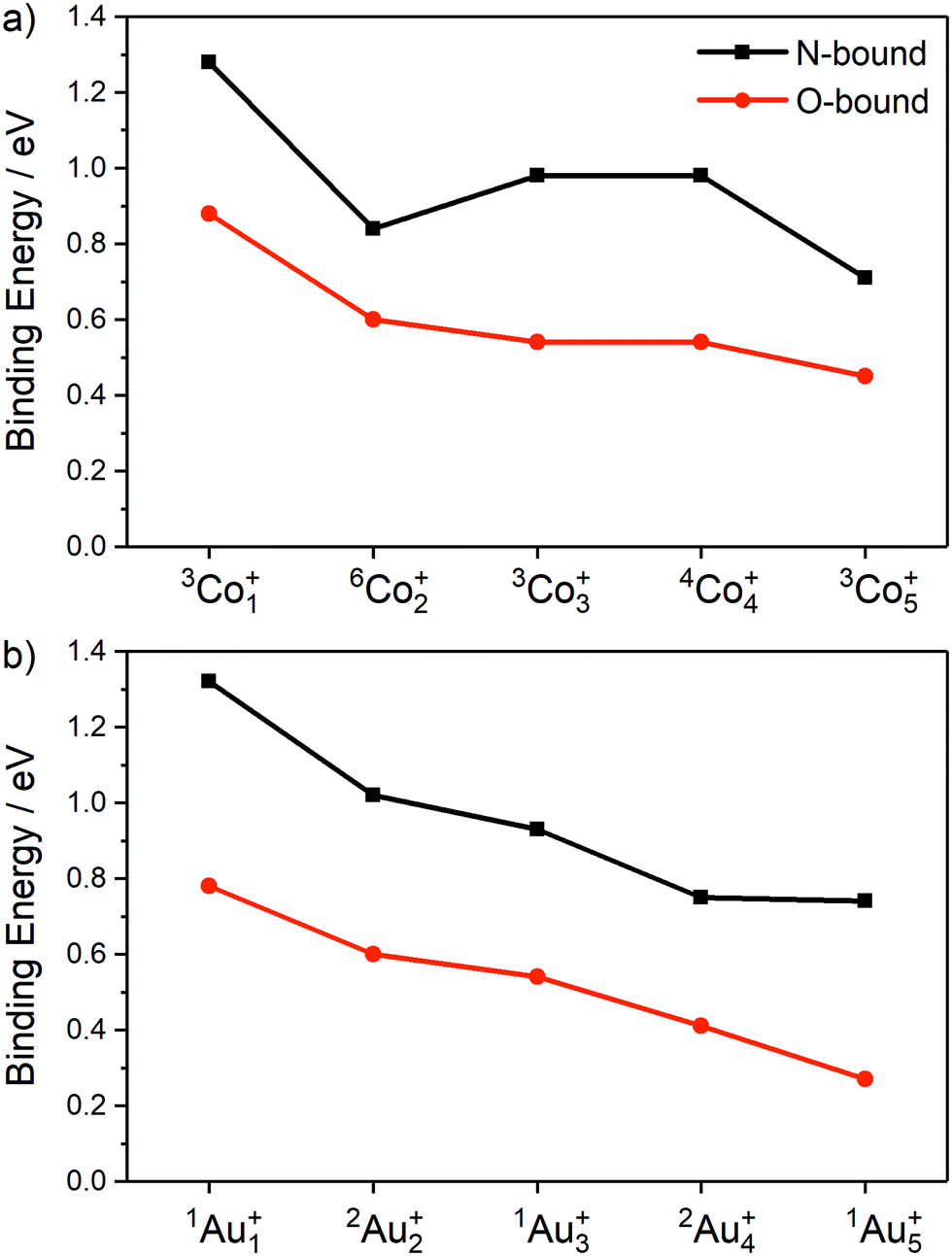 | ||
| Fig. 9 Relative ligand binding energies (given as absolute values) of lowest energy molecularly-bound N- and O-bound isomers for (a) Con(N2O)+ (n = 1–5), and (b) Aun(N2O)+ (n = 1–5), calculated as E[Mn(N2O)+] − E[Mn+] − E[N2O], including zero-point correction. | ||
4. Conclusions
The binding of nitrous oxide to small gas-phase Aun+ and Con+ clusters has been investigated using IR-MPD spectroscopy in conjunction with density functional theory calculations of energetically low-lying molecularly bound structures. In the case of Aun(N2O)+, the lowest energy N-bound isomers dominate the observed spectra as indicated by the slight blue-shift of vibrational bands relative to free N2O. For Con(N2O)+, a richer distribution of structural isomers is observed with clear evidence of both N- and O-bound isomers in the distributions produced for the smaller clusters. Calculations suggest the presence of more low-lying electronic states in the case of Con+ but these are imperceptible in the vibrational spectra. In all cases investigated here, the only significant dissociation channel observed is the simple N2O ligand loss with little or no clear evidence for the type of photoinitiated intra-complex reactivity previously characterised in the case of analogous Rhn(N2O)+ and Ptn(N2O)+ clusters48–51 and, more widely, in Aun(OCS)+ and PtnOm(CO)+.72,78 This suggests that the transition states for the N2O decomposition reaction on Con+ and Aun+ lie significantly higher in energy than the Mn+ + N2O dissociation asymptote.Statement of author contributions
This work was conceived of by SRM, OD and AF and funded under their Oxford-Berlin Research Partnership grant designed to establish new collaborations between Oxford and Berlin. WS is responsible for the free electron laser at the Fritz-Haber Institute where all experiments were run. EMC, AEG, GM, PWB, and ASG visited Berlin for the experimental runs at which SS, KMP, KS and MF were also present. The computational studies were performed by EMC, AEG, GM and PWB. The manuscript was written in Oxford with contributions from all authors.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was funded partly by EPSRC under Programme Grants EP/L005913 and EP/T021675/1. EMC is also grateful to the EPSRC for his graduate studentship in Oxford. GM and AEG thank Worcester and Magdalen Colleges, Oxford, respectively for their graduate studentships. AF thanks the Deutsche Forschungsgemeinschaft for his Heisenberg Grant (FI 893/5). Financial support is also gratefully acknowledged from the Oxford-Berlin Research Partnership (Ref. OXBER_STEM5, “A Collaborative Approach to Understanding Nitrogen Oxide Reduction at Metal Centres”). The authors acknowledge the use of the University of Oxford Advanced Research Computing facility (https://doi.org/10.5281/zenodo.22558) and the HPC infrastructure LEO of the University of Innsbruck in carrying out the calculations presented in this work. Open Access funding provided by the Max Planck Society.References
- M. J. Prather, Time Scales in Atmospheric Chemistry: Coupled Perturbations to N2O, NOy, and O3, Science, 1998, 279, 1339–1341 CrossRef CAS PubMed.
- A. R. Ravishankara, J. S. Daniel and R. W. Portmann, Nitrous Oxide (N2O): The Dominant Ozone-Depleting Substance Emitted in the 21st Century, Science, 2009, 326, 123–125 CrossRef CAS PubMed.
- R. K. Pachauri, M. R. Allen, V. R. Barros, J. Broome, W. Cramer, R. Christ, J. A. Church, L. Clarke, Q. Dahe, P. Dasgupta, N. K. Dubash, O. Edenhofer, I. Elgizouli, C. B. Field, P. Forster, P. Friedlingstein, J. Fuglestvedt, L. Gomez-Echeverri, S. Hallegatte, G. Hegerl, M. Howden, K. Jiang, B. Jimenez Cisneroz, V. Kattsov, H. Lee, K. J. Mach, J. Marotzke, M. D. Mastrandrea, L. Meyer, J. Minx, Y. Mulugetta, K. O’Brien, M. Oppenheimer, J. J. Pereira, R. Pichs-Madruga, G.-K. Plattner, H.-O. Pörtner, S. B. Power, B. Preston, N. H. Ravindranath, A. Reisinger, K. Riahi, M. Rusticucci, R. Scholes, K. Seyboth, Y. Sokona, R. Stavins, T. F. Stocker, P. Tschakert, D. van Vuuren and J.-P. van Ypserle, Climate Change 2014: Synthesis Report. Contribution of Working Groups I, II and III to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, IPCC, Geneva, Switzerland, 2014.
- W. B. Tolman, Binding and Activation of N2O at Transition-Metal Centers: Recent Mechanistic Insights, Angew. Chem., Int. Ed., 2010, 49, 1018–1024 CrossRef CAS PubMed.
- P. Chen, S. I. Gorelsky, S. Ghosh and E. I. Solomon, N2O Reduction by the μ4-Sulfide-Bridged Tetranuclear CuZ Cluster Active Site, Angew. Chem., Int. Ed., 2004, 43, 4132–4140 CrossRef CAS PubMed.
- V. Rosca, M. Duca, M. T. de Groot and M. T. M. Koper, Nitrogen Cycle Electrocatalysis, Chem. Rev., 2009, 109, 2209–2244 CrossRef CAS PubMed.
- E. L. Muetterties, T. N. Rhodin, E. Band, C. F. Brucker and W. R. Pretzer, Clusters and surfaces, Chem. Rev., 1979, 79, 91–137 CrossRef CAS.
- Z. Luo, A. W. Castleman and S. N. Khanna, Reactivity of Metal Clusters, Chem. Rev., 2016, 116, 14456–14492 CrossRef CAS PubMed.
- A. W. Castleman and K. H. Bowen, Clusters: Structure, Energetics, and Dynamics of Intermediate States of Matter, J. Phys. Chem., 1996, 100, 12911–12944 CrossRef CAS.
- A. W. Castleman and S. N. Khanna, Clusters, Superatoms, and Building Blocks of New Materials, J. Phys. Chem. C, 2009, 113, 2664–2675 CrossRef CAS.
- Z. Luo and A. W. Castleman, Special and General Superatoms, Acc. Chem. Res., 2014, 47, 2931–2940 CrossRef CAS PubMed.
- A. Sanchez, S. Abbet, U. Heiz, W.-D. Schneider, H. Häkkinen, R. N. Barnett and U. Landman, When Gold Is Not Noble: Nanoscale Gold Catalysts, J. Phys. Chem. A, 1999, 103, 9573–9578 CrossRef CAS.
- S. M. Lang, T. M. Bernhardt, R. N. Barnett, B. Yoon and U. Landman, Hydrogen-Promoted Oxygen Activation by Free Gold Cluster Cations, J. Am. Chem. Soc., 2009, 131, 8939–8951 CrossRef CAS PubMed.
- U. Landman, B. Yoon, C. Zhang, U. Heiz and M. Arenz, Factors in gold nanocatalysis: oxidation of CO in the non-scalable size regime, Top. Catal., 2007, 44, 145–158 CrossRef CAS.
- B. Yoon, H. Häkkinen, U. Landman, A. S. Wörz, J.-M. Antonietti, S. Abbet, K. Judai and U. Heiz, Charging Effects on Bonding and Catalyzed Oxidation of CO on Au8 Clusters on MgO, Science, 2005, 307, 403–407 CrossRef CAS PubMed.
- U. Landman, Materials by numbers: Computations as tools of discovery, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 6671–6678 CrossRef CAS PubMed.
- H. Häkkinen, S. Abbet, A. Sanchez, U. Heiz and U. Landman, Structural, Electronic, and Impurity-Doping Effects in Nanoscale Chemistry: Supported Gold Nanoclusters, Angew. Chem., Int. Ed., 2003, 42, 1297–1300 CrossRef PubMed.
- M. Haruta, Size- and support-dependency in the catalysis of gold, Catal. Today, 1997, 36, 153–166 CrossRef CAS.
- M. Valden, X. Lai and D. W. Goodman, Onset of Catalytic Activity of Gold Clusters on Titania with the Appearance of Nonmetallic Properties, Science, 1998, 281, 1647–1650 CrossRef CAS PubMed.
- G. C. Bond and D. T. Thompson, Catalysis by Gold, Catal. Rev., 1999, 41, 319–388 CrossRef CAS.
- L. D. Socaciu, J. Hagen, T. M. Bernhardt, L. Wöste, U. Heiz, H. Häkkinen and U. Landman, Catalytic CO Oxidation by Free Au2−: Experiment and Theory, J. Am. Chem. Soc., 2003, 125, 10437–10445 CrossRef CAS PubMed.
- D. Schooss, P. Weis, O. Hampe and M. M. Kappes, Determining the size-dependent structure of ligand-free gold-cluster ions, Philos. Trans. R. Soc., A, 2010, 368, 1211–1243 CrossRef CAS PubMed.
- A. P. Woodham and A. Fielicke, Gold Clusters, Colloids and Nano-Particles I, Springer International Publishing, 2014 Search PubMed.
- A. Zavras, G. N. Khairallah and R. A. J. O’Hair, Gold Clusters, Colloids and Nano-Particles II, Springer International Publishing, 2014 Search PubMed.
- P. R. Kemper and M. T. Bowers, Electronic-state chromatography: application to first-row transition-metal ions, J. Phys. Chem., 1991, 95, 5134–5146 CrossRef CAS.
- S. Gilb, P. Weis, F. Furche, R. Ahlrichs and M. M. Kappes, Structures of small gold cluster cations (Aun+, n < 14): ion mobility measurements versus density functional calculations, J. Chem. Phys., 2002, 116, 4094–4101 CrossRef CAS.
- F. Furche, R. Ahlrichs, P. Weis, C. Jacob, S. Gilb, T. Bierweiler and M. M. Kappes, The structures of small gold cluster anions as determined by a combination of ion mobility measurements and density functional calculations, J. Chem. Phys., 2002, 117, 6982–6990 CrossRef CAS.
- A. Lechtken, D. Schooss, J. R. Stairs, M. N. Blom, F. Furche, N. Morgner, O. Kostko, B. von Issendorff and M. M. Kappes, Au34−: A Chiral Gold Cluster?, Angew. Chem., Int. Ed., 2007, 46, 2944–2948 CrossRef CAS PubMed.
- X. Xing, B. Yoon, U. Landman and J. H. Parks, Structural evolution of Au nanoclusters: from planar to cage to tubular motifs, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 165423 CrossRef.
- A. Shayeghi, R. Schäfer, D. M. Rayner, R. L. Johnston and A. Fielicke, Charge-induced dipole vs. relativistically enhanced covalent interactions in Ar-tagged Au-Ag tetramers and pentamers, J. Chem. Phys., 2015, 143, 024310 CrossRef CAS PubMed.
- A. Shayeghi, R. L. Johnston, D. M. Rayner, R. Schäfer and A. Fielicke, The Nature of Bonding between Argon and Mixed Gold–Silver Trimers, Angew. Chem., Int. Ed., 2015, 54, 10675–10680 CrossRef CAS PubMed.
- A. N. Gloess, H. Schneider, J. M. Weber and M. M. Kappes, Electronically excited states and visible region photodissociation spectroscopy of Aum+·Arn clusters (m = 7–9): Molecular dimensionality transition?, J. Chem. Phys., 2008, 128, 114312 CrossRef PubMed.
- M. Förstel, W. Schewe and O. Dopfer, Optical Spectroscopy of the Au4+ Cluster: The Resolved Vibronic Structure Indicates an Unexpected Isomer, Angew. Chem., Int. Ed., 2019, 58, 3356–3360 CrossRef PubMed.
- P. Ferrari, G.-L. Hou, O. V. Lushchikova, F. Calvo, J. M. Bakker and E. Janssens, The structures of cationic gold clusters probed by far-infrared spectroscopy, Phys. Chem. Chem. Phys., 2020, 22, 11572–11577 RSC.
- L. M. Ghiringhelli, P. Gruene, J. T. Lyon, D. M. Rayner, G. Meijer, A. Fielicke and M. Scheffler, Not so loosely bound rare gas atoms: finite-temperature vibrational fingerprints of neutral gold-cluster complexes, New J. Phys., 2013, 15, 083003 CrossRef CAS.
- P. Gruene, B. Butschke, J. T. Lyon, D. M. Rayner and A. Fielicke, Far-IR Spectra of Small Neutral Gold Clusters in the Gas Phase, Z. Phys. Chem., 2014, 228, 337–350 CrossRef CAS.
- B. R. Goldsmith, J. Florian, J.-X. Liu, P. Gruene, J. T. Lyon, D. M. Rayner, A. Fielicke, M. Scheffler and L. M. Ghiringhelli, Two-to-three dimensional transition in neutral gold clusters: the crucial role of van der Waals interactions and temperature, Phys. Rev. Mater., 2019, 3, 016002 CrossRef CAS.
- P. Gruene, D. M. Rayner, B. Redlich, A. F. G. van der Meer, J. T. Lyon, G. Meijer and A. Fielicke, Structures of Neutral Au7, Au19, and Au20 Clusters in the Gas Phase, Science, 2008, 321, 674–676 CrossRef CAS PubMed.
- G. Dietrich, K. Lützenkirchen, S. Becker, H.-U. Hasse, H.-J. Kluge, M. Lindinger, L. Schweikhard, J. Ziegler and S. Kuznetsov, Au-induced decomposition of N2O, Ber. Bunsen-Ges., 1994, 98, 1608–1612 CrossRef CAS.
- A. Fielicke, G. von Helden, G. Meijer, B. Simard and D. M. Rayner, Direct observation of size dependent activation of NO on gold clusters, Phys. Chem. Chem. Phys., 2005, 7, 3906–3909 RSC.
- R. Gehrke, P. Gruene, A. Fielicke, G. Meijer and K. Reuter, Nature of Ar bonding to small Con+ clusters and its effect on the structure determination by far-infrared absorption spectroscopy, J. Chem. Phys., 2009, 130, 034306 CrossRef PubMed.
- M. Jia, J. van der Tol, Y. Li, V. Chernyy, J. M. Bakker, L. N. Pham, M. T. Nguyen and E. Janssens, Structures and magnetic properties of small Co+n and Con-1Cr+ (n = 3–5) clusters, J. Phys.: Condens. Matter, 2018, 30, 474002 CrossRef.
- V. Zamudio-Bayer, K. Hirsch, A. Langenberg, A. Lawicki, A. Terasaki, B. von Issendorff and J. T. Lau, Large orbital magnetic moments of small, free cobalt cluster ions Con+ with n ≤ 9, J. Phys.: Condens. Matter, 2018, 30, 464002 CrossRef CAS PubMed.
- J. M. Bakker, J. Jalink, D. Dieleman and A. Kirilyuk, Structural determination of neutral Con clusters (n = 4–10, 13) through IR–UV two-color vibrational spectroscopy and DFT calculations, J. Phys.: Condens. Matter, 2018, 30, 494003 CrossRef CAS PubMed.
- M. L. Anderson, A. Lacz, T. Drewello, P. J. Derrick, D. P. Woodruff and S. R. Mackenzie, The chemistry of nitrogen oxides on small size-selected cobalt clusters, Con+, J. Chem. Phys., 2009, 130, 064305 CrossRef PubMed.
- H. Francisco, V. Bertin, J. R. Soto and M. Castro, Charge and Geometrical Effects on the Catalytic N2O Reduction by Rh6− and Rh6+ Clusters, J. Phys. Chem. C, 2016, 120, 23648–23659 CrossRef CAS.
- E. Hernández, V. Bertin, J. Soto, A. Miralrio and M. Castro, Catalytic Reduction of Nitrous Oxide by the Low-Symmetry Pt8 Cluster, J. Phys. Chem. A, 2018, 122, 2209–2220 CrossRef.
- S. M. Hamilton, W. S. Hopkins, D. J. Harding, T. R. Walsh, P. Gruene, M. Haertelt, A. Fielicke, G. Meijer and S. R. Mackenzie, Infrared Induced Reactivity on the Surface of Isolated Size-Selected Clusters: Dissociation of N2O on Rhodium Clusters, J. Am. Chem. Soc., 2010, 132, 1448–1449 CrossRef CAS PubMed.
- S. M. Hamilton, W. S. Hopkins, D. J. Harding, T. R. Walsh, M. Haertelt, C. Kerpal, P. Gruene, G. Meijer, A. Fielicke and S. R. Mackenzie, Infrared-Induced Reactivity of N2O on Small Gas-Phase Rhodium Clusters, J. Phys. Chem. A, 2011, 115, 2489–2497 CrossRef CAS PubMed.
- A. C. Hermes, S. M. Hamilton, W. S. Hopkins, D. J. Harding, C. Kerpal, G. Meijer, A. Fielicke and S. R. Mackenzie, Effects of Coadsorbed Oxygen on the Infrared Driven Decomposition of N2O on Isolated Rh5+ Clusters, J. Phys. Chem. Lett., 2011, 2, 3053–3057 CrossRef CAS.
- G. Meizyte, A. E. Green, A. S. Gentleman, S. Schaller, W. Schöllkopf, A. Fielicke and S. R. Mackenzie, Free electron laser infrared action spectroscopy of nitrous oxide binding to platinum clusters, Ptn(N2O)m+, Phys. Chem. Chem. Phys., 2020, 22, 18606–18613 RSC.
- A. Fielicke, G. von Helden and G. Meijer, Far-Infrared spectroscopy of isolated transition metal clusters, Eur. Phys. J. D, 2005, 34, 83–88 CrossRef CAS.
- W. Schöllkopf, S. Gewinner, H. Junkes, A. Paarmann, G. von Helden, H. P. Bluem and A. M. M. Todd, Advances in X-ray Free-Electron Lasers Instrumentation III, International Society for Optics and Photonics, 2015, vol. 9512, p. 95121L Search PubMed.
- B. A. Mamyrin, V. I. Karataev, D. V. Shmikk and V. A. Zagulin, The mass-reflectron, a new nonmagnetic time-of-flight mass spectrometer with high resolution, J. Exp. Theor. Phys., 1973, 37, 45 Search PubMed.
- G. Herzberg, Molecular Spectra and Molecular Structure: II Infrared and Raman Spectra of Polyatomic Molecules, Krieger, Malabar, Florida, 1991, vol. 2 Search PubMed.
- O. Rodriguez and J. M. Lisy, Infrared Spectroscopy of Li+(CH4)1Arn, n = 1–6, Clusters, J. Phys. Chem. A, 2011, 115, 1228–1233 CrossRef CAS PubMed.
- O. Rodriguez and J. M. Lisy, Infrared spectroscopy of Li+(CH4)n, n = 1–9, clusters, Chem. Phys. Lett., 2011, 502, 145–149 CrossRef CAS.
- P. Ayotte, G. H. Weddle, J. Kim and M. A. Johnson, Vibrational Spectroscopy of the Ionic Hydrogen Bond: Fermi Resonances and Ion–Molecule Stretching Frequencies in the Binary X−·H2O (X = Cl, Br, I) Complexes via Argon Predissociation Spectroscopy, J. Am. Chem. Soc., 1998, 120, 12361–12362 CrossRef CAS.
- M. Okumura, L. I. Yeh, J. D. Myers and Y. T. Lee, Infrared spectra of the solvated hydronium ion: vibrational predissociation spectroscopy of mass-selected H3O+·(H2O)n(H2)m, J. Phys. Chem., 1990, 94, 3416–3427 CrossRef CAS.
- M. Okumura, L. I. Yeh, J. D. Myers and Y. T. Lee, Infrared spectra of the cluster ions H7O3+·H2 and H9O4+·H2, J. Chem. Phys., 1986, 85, 2328–2329 CrossRef CAS.
- M. B. Knickelbein and W. J. C. Menezes, Metal cluster-rare gas van der Waals complexes: physisorption on a microscopic scale, J. Phys. Chem., 1992, 96, 6611–6616 CrossRef CAS.
- W. Herschel, XIV. Experiments on the refrangibility of the invisible rays of the sun, Philos. Trans. R. Soc. London, 1800, 90, 284–292 CrossRef.
- V. N. Staroverov, G. E. Scuseria, J. Tao and J. P. Perdew, Comparative assessment of a new nonempirical density functional: molecules and hydrogen-bonded complexes, J. Chem. Phys., 2003, 119, 12129–12137 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Weigend, Accurate Coulomb-fitting basis sets for H to Rn, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- P. Pyykkö, Theoretical Chemistry of Gold, Angew. Chem., Int. Ed., 2004, 43, 4412–4456 CrossRef PubMed.
- P. Pyykkö, Theoretical chemistry of gold. II, Inorg. Chim. Acta, 2005, 358, 4113–4130 CrossRef.
- P. Pyykkö, Theoretical chemistry of gold. III, Chem. Soc. Rev., 2008, 37, 1967–1997 RSC.
- D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Energy-adjusted ab initio pseudopotentials for the second and third row transition elements, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- I. Swart, F. M. F. de Groot, B. M. Weckhuysen, D. M. Rayner, G. Meijer and A. Fielicke, The Effect of Charge on CO Binding in Rhodium Carbonyls: From Bridging to Terminal CO, J. Am. Chem. Soc., 2008, 130, 2126–2127 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- A. E. Green, S. Schaller, G. Meizyte, B. J. Rhodes, S. P. Kealy, A. S. Gentleman, W. Schöllkopf, A. Fielicke and S. R. Mackenzie, Infrared Study of OCS Binding and Size-Selective Reactivity with Gold Clusters, Aun+ (n = 1–10), J. Phys. Chem. A, 2020, 124, 5389–5401 CrossRef CAS PubMed.
- E. M. Cunningham, A. S. Gentleman, P. W. Beardsmore, A. Iskra and S. R. Mackenzie, Infrared Signature of Structural Isomers of Gas–Phase M+(N2O)n (M = Cu, Ag, Au) Ion–Molecule Complexes, J. Phys. Chem. A, 2017, 121, 7565–7571 CrossRef CAS PubMed.
- X. Jin, G. Wang and M. Zhou, Matrix Isolation Infrared Spectroscopic and Theoretical Study of Nickel, Palladium, and Platinum Nitrous Oxide Complexes, J. Phys. Chem. A, 2006, 110, 8017–8022 CrossRef CAS PubMed.
- G. Wang, X. Jin, M. Chen and M. Zhou, Matrix isolation infrared spectroscopic and theoretical study of the copper (I) and silver (I)–nitrous oxide complexes, Chem. Phys. Lett., 2006, 420, 130–134 CrossRef CAS.
- E. M. Cunningham, A. S. Gentleman, P. W. Beardsmore and S. R. Mackenzie, Structural isomers and low-lying electronic states of gas-phase M+(N2O)n (M = Co, Rh, Ir) ion–molecule complexes, Phys. Chem. Chem. Phys., 2019, 21, 13959–13967 RSC.
- E. M. Cunningham, A. S. Gentleman, P. W. Beardsmore and S. R. Mackenzie, Infrared spectroscopy of closed s-shell gas-phase M+(N2O)n (M = Li, Al) ion-molecule complexes, Mol. Phys., 2019, 117, 2990–3000 CrossRef CAS.
- A. C. Hermes, S. M. Hamilton, G. A. Cooper, C. Kerpal, D. J. Harding, G. Meijer, A. Fielicke and S. R. Mackenzie, Infrared driven CO oxidation reactions on isolated platinum cluster oxides, PtnOm+, Faraday Discuss., 2012, 157, 213–225 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cp05195k |
| This journal is © the Owner Societies 2021 |