
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBase-catalysed 18F-labelling of trifluoromethyl ketones. Application to the synthesis of 18F-labelled neutrophil elastase inhibitors†
Denise N.
Meyer‡
 a,
Miguel A.
Cortés González‡
a,
Xingguo
Jiang‡
a,
Linus
Johansson-Holm
a,
Monireh
Pourghasemi Lati
a,
Mathias
Elgland
b,
Patrik
Nordeman
b,
Gunnar
Antoni
b and
Kálmán J.
Szabó
*a
a,
Miguel A.
Cortés González‡
a,
Xingguo
Jiang‡
a,
Linus
Johansson-Holm
a,
Monireh
Pourghasemi Lati
a,
Mathias
Elgland
b,
Patrik
Nordeman
b,
Gunnar
Antoni
b and
Kálmán J.
Szabó
*a
aDepartment of Organic Chemistry, Stockholm University, SE-106 91, Sweden. E-mail: kalman.j.szabo@su.se; Web: http://www.organ.su.se/ks/
bDepartment of Medicinal Chemistry, Uppsala University, SE-751 23, Sweden
First published on 30th July 2021
Abstract
A new method for the fluorine-18 labelling of trifluoromethyl ketones has been developed. This method is based on the conversion of a–COCF3 functional group to a difluoro enol silyl ether followed by halogenation and fluorine-18 labelling. The utility of this new method was demonstrated by the synthesis of fluorine-18 labelled neutrophil elastase inhibitors, which are potentially useful for detection of inflammatory disorders.
Introduction of fluorine into organic compounds can be used to control the physicochemical properties and the bioactivity of small molecules.1 Typical application areas of organofluorine compounds are the pharmaceutical industry,1c,2 agrochemistry3 and medical diagnostics.4 The high metabolic stability of the organofluorine species is also an attractive feature for applications in life-science related areas.1c Although the C–F bond is the strongest single bond that carbon may form, neighbouring functional groups may induce cleavage of the C–F bond via, for example, anomeric effects or hyperconjugation.1b Another possible complication can be the effects of the biological environment (such as high calcium or magnesium ion concentrations), which may lead to degradation of organofluorines.1c In drug substances and imaging tracers for Positron Emission Tomography (PET), mono-fluorination mainly occurs in aromatic species4c This is because the C(sp2)–F bond is very strong preventing both oxidative degradation and C–F bond cleavage via CaF2 formation or related processes.1c In the case of alkyl fluorides, application of CF3 or perfluoroalkyl groups are preferred due to the increased C–F bond strength. Installation of fluorine-18 labelled CF3 groups is still a formidable challenge in the synthesis of PET imaging tracers.5 The main difficulty is associated with the downscaling of the synthetic methodologies used for the construction of CF3 groups using nanomolar amounts of fluorine-18 precursors available for the radiosynthesis. Trifluoromethyl ketones (and analogues) are very important pharmacophores in many enzyme inhibitors (Fig. 1).6
 | ||
| Fig. 1 Bioactive molecules with fluoroalkyl ketone pharmacophores. NE = neutrophil elastase. | ||
A particularly important serine protease, neutrophil elastase (NE), can be efficiently inhibited by COCF3-containing drugs (1a–c). The main action of this functional group is forming covalent hemiketal type adducts with the hydroxy group of the serine moieties in the active site of the enzyme.6a,7 This ability of the carbonyl group arises from the strong electron-withdrawing character of the CF3 moiety.8 NE is an important biomarker of serious inflammatory disorders, which are causing fibrosis and organ failure. Examples of such disorders are chronic obstructive pulmonary disease9 (COPD) and abdominal aortic aneurysm10 (AAA). In addition, neutrophil elastase (NE) is a key enzyme in the formation of so-called neutrophil extracellular traps (NETs).11 Excessive formation of NETs have been identified as the major cause of acute respiratory distress syndrome (ARDS),12 which is the leading death cause in case of COVID-19.
As a part of our synthetic organofluorine chemistry program5b,13 we decided to develop a new methodology for the construction of fluorine-18 labelled COCF3 groups targeting new potential PET imaging tracers for the detection of NE. Our approach (Fig. 2) is based on the defluorination of the natural isotope (1) of the trifluoromethyl ketone to be labelled affording its difluoro enol silyl ether. This conversion can be carried out by using Mg and TMSCl under dry conditions.14 Difluoro enol silyl ethers are highly reactive nucleophilic species, which can be used for the synthesis of various organohalogen and other species.15 Bromination or iodination of these compounds14b,15a leads to stable halodifluoromethyl ketones (2), which can be used as precursors for fluorine-18 labelling5a,b (Fig. 2). A potential advantage of this circular defluorination–fluorination sequence, 1 → [18F]1, is the easy access to suitable precursors for tracer development for bioactive small molecules, such as 1a–c. Difluoro enol silyl ethers have been used as precursors for fluorine-18 labelling using [18F]F2 with F2 carrier gas by Prakash, Olah and their co-workers.16 By the above method (Fig. 2) based on bromination/iodination of 2 the cumbersome handling of [18F]F2 and application of F2 as carrier gas can be avoided.
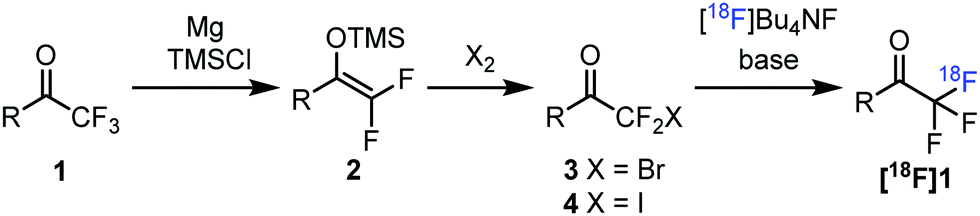 | ||
| Fig. 2 Circular defluorination–fluorination sequence for fluorine-18 labelling of trifluoromethyl ketones. | ||
We started the development of the above-mentioned circular approach by fluorination of 2-bromo-2,2-difluoro acetophenone 3g with [18F]Bu4NF targeting fluorine-18 labelled 2,2,2-trifluoro acetophenone [18F]1g (Table 1). The reaction proceeded with a low but encouraging radiochemical yield (RCY) of 5% at 100 °C in DMF (entry 1). Our earlier studies for the construction of fluorine-18 labelled trifluoromethyl compounds showed that nitrogen-containing bases facilitate the halogen exchange of the CF2X groups.5b When we performed the fluorine-18 labelling reaction in the presence of DBU, [18F]1g was obtained in 65% RCY (entry 2). Our previous results5b indicated an excellent performance of guanidine-like bases in these type of labelling studies. Indeed, application of TBD in place of DBU afforded [18F]1g in 92% RCY at 100 °C (entry 3). Furthermore, the temperature could be reduced to 75 °C without significant decrease in the radiochemical yield (90%, entry 4). When [18F]KF/K222 was used instead of [18F]Bu4NF, we found the reaction to proceed with similar RCY (entry 5). The use of MTBD, which is structurally similar to TBD (entry 6) or further reduction of the reaction temperature (entries 7 and 8) led to a lower radiochemical yield of [18F]1g. In most of the further studies we employed the optimal conditions, which are given in entry 4 of Table 1.
|
|
|||
|---|---|---|---|
| Entry | Additiveb | Temperature [°C] | RCYc [%] |
| a Unless otherwise stated, a solution of the precursor (60 μmol, 14 mg) and the additive (60 μmol) in DMF (150 μL) was mixed with a solution of [18F]Bu4NF in DMF (150 μL). The reaction was stirred at the indicated temperature for 10 min. b DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene; TBD = 1,5,7-triazabicyclo-[4.4.0]dec-5-ene; MTBD = 7-methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene. c RCY was estimated by radio-HPLC analysis of the crude reaction mixture. d Using [18F]KF/K222. | |||
| 1 | — | 100 | 5 |
| 2 | DBU | 100 | 65 ± 9 (n = 2) |
| 3 | TBD | 100 | 92 ± 3 (n = 2) |
| 4 | TBD | 75 | 90 ± 1 (n = 2) |
| 5d | TBD | 75 | 90 ± 1 (n = 2) |
| 6 | MTBD | 75 | 73 ± 13 (n = 2) |
| 7 | TBD | 50 | 84 ± 1 (n = 2) |
| 8 | TBD | RT | 74 ± 1 (n = 2) |

With the optimized reaction conditions in hand, we explored the substrate scope of this transformation (Fig. 3). Similarly, to [18F]1g, para-phenyl substituted analogue [18F]1h was obtained in 90% RCY. The radiochemical yield of 1-naphthyl derivative [18F]1i was similarly high (92%), whereas 2-naphthyl substituted [18F]1j gave a somewhat lower RCY of 84%. The radiosynthesis could easily be extended to [18F]trifluoroacetophenones bearing electron-donating substituents, such as 4-methyl ([18F]1k, 71% RCY) and 4-methoxy ([18F]1l, 93% RCY) derivatives. Similarly, [18F]trifluoroacetophenones bearing electron-withdrawing substituents were obtained in high radiochemical yields. Thus, 4-fluoro ([18F]1m) and 3,5-difluoro ([18F]1n) substituted [18F]trifluoroacetophenones were labelled in 90% and 92% radiochemical yields, respectively. In the presence of electron-withdrawing substituents on the aromatic ring, the air-stability of the difluoro enol silyl ethers (2) were somewhat lowered. Thioether-containing [18F]1o was obtained in only 20% radiochemical yield. A possible explanation is instability under the applied reaction conditions due to the presence of the SEt group. Labelled conjugated vinyl ketones [18F]1p and [18F]1q could also be obtained, albeit the RCY (51% and 67%) were lower than for aryl derivatives [18F]1g–n. The RCY for thiophene derivative [18F]1p could be increased to 67% by elevating the temperature to 100 °C. However, the increase of the reaction temperature led to a drop of RCY for the naphthyl derivative [18F]1q. The labelling was not limited to C(sp2)–COCF3 derivatives. Aliphatic derivative [18F]1r could be prepared with 51% RCY. This is an important finding indicating that the reaction proceeds with satisfactory yield even in the presence of an α-proton in the precursor. When TBD was replaced with DIPEA the RCY was slightly increased to 64%.
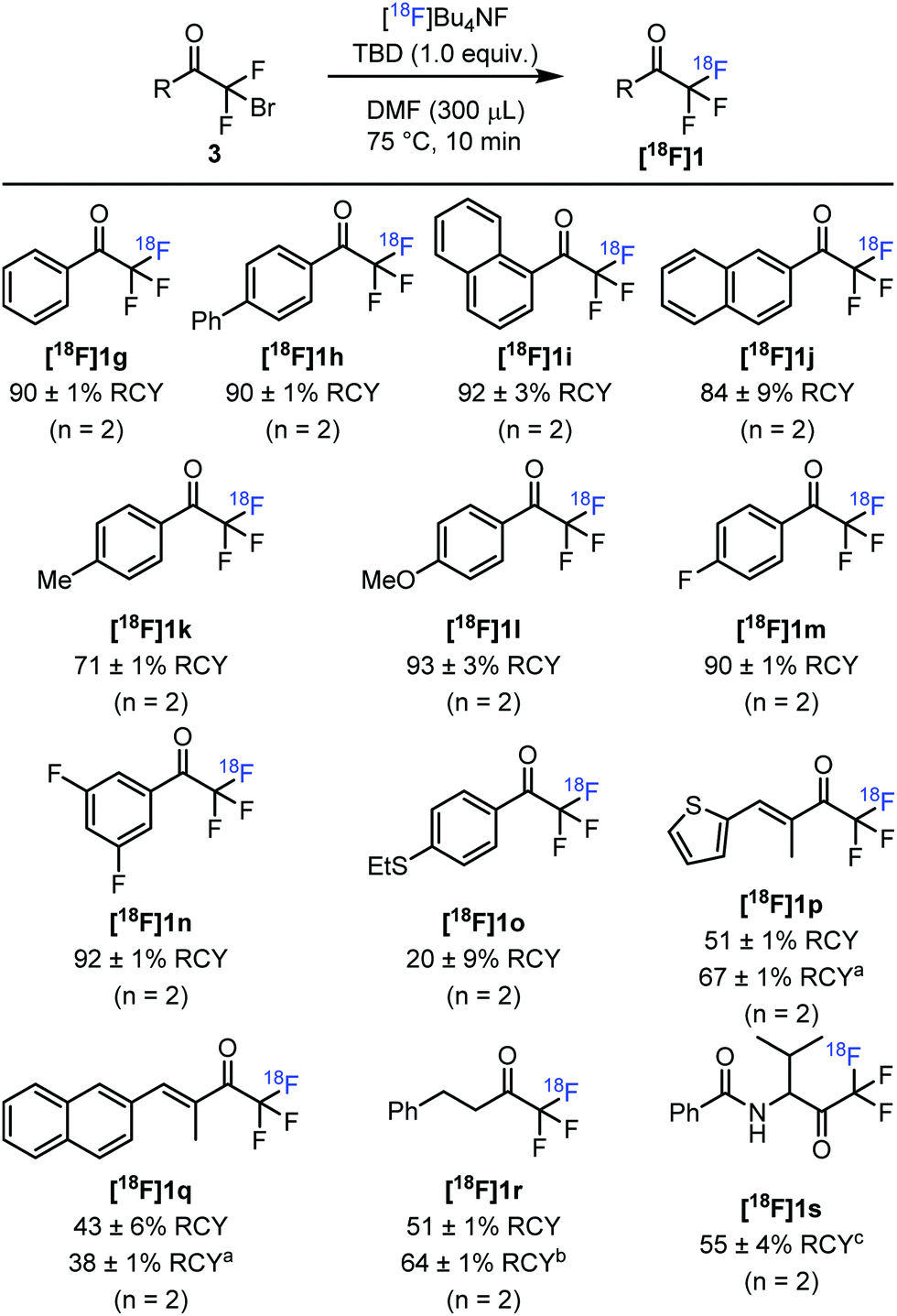 | ||
| Fig. 3 Substrate scope in the 18F-labelling of trifluoromethyl ketones. RCY was estimated by radio-HPLC or radio-TLC analysis of the crude reaction mixture. aThe reaction was performed at 100 °C. bDIPEA (1.0 equiv.) was used instead of TBD. cWith [18F]KF/K222. No additive was used. | ||
We have also studied the labelling of an analogue of 1a with a valine unit attached to the COCF3 functionality. The attempts using [18F]Bu4NF failed to give labelled product [18F]1s. However, when the fluorine source was changed to [18F]KF/K222 in the absence of base, [18F]1s was obtained in 55% RCY.
To demonstrate the synthetic utility of the above-described method we prepared a NE PET imaging tracer candidate based on the known6a NE inhibitor 1a (Fig. 4). We started the synthesis by defluorination of NE inhibitor 1a6a to the corresponding difluoro enol silyl ether 2a under dry conditions. Halogenation of 2a was performed without further purification because of the poor air and moisture stability of this reaction intermediate. Using Br2, we could obtain the corresponding COCF2Br derivative from this enol silyl ether. However, this bromodifluoromethylated precursor could not be converted to the targeted [18F]1a. The low reactivity of the COCF2Br derivative was somewhat surprising as the valine analogue [18F]1s could be obtained from the corresponding bromo derivative 3s. A possible explanation was that the fluorine-18 reagent was deactivated under the applied reaction conditions.
 | ||
| Fig. 4 18F-Labelling of NE inhibitor 1a to obtain fluorine-18 NE ligand [18F]1a. | ||
After extensive studies (see ESI†) we have found that the iodo derivative 4a is sufficiently reactive for the synthesis of [18F]1a using [18F]Bu4NF without any additive. Thus, we obtained [18F]1a in 15 ± 12% RCY (Fig. 4). In addition, a preparative run starting from 3.6 GBq [18F]Bu4NF afforded an activity yield (AY) of 2% and a molar activity of 0.41 GBq μmol−1 70 minutes after the end of the bombardment. The obtained molar activity was relatively low probably due to fluorine-18–fluorine-19 isotopic exchange, which is a well-known problem for labelling trifluoroalkyl groups.5a
The pharmacological properties of these NE inhibitors can be varied without substantial alteration of the inhibitory constant (Ki) by changing the substituent on the pyridone group.6a This can be exploited for the development of radiosynthesis of fluorine-18 labelled NE inhibitors analogous to [18F]1a. We have found that the carbamate moiety of 4a can be hydrolysed by trifluoromethanesulfonic acid in the presence of anisole, affording 4t (Fig. 5). Subsequently, the amino group could be sulfonated by methanesulfonyl chloride in the presence of base and DMAP. This resulted in a new precursor, 4b, for fluorine-18 labelling. Fluorine-18 labelled NE inhibitor [18F]1b could be isolated with 30% RCY, 0.5% AY and a molar activity of 1.3 GBq μmol−1 (Fig. 5). Starting from 4a, this synthesis sequence can be exploited for a modular approach for radiosynthesis of a broad variety of fluorine-18 labelled NE inhibitors.
 | ||
| Fig. 5 Radiosynthesis of [18F]1b based on two step transformation of 4a. | ||
In summary, we have developed a new method for the fluorine-18 labelling of trifluoromethyl ketones based on a circular defluorination–fluorination sequence. The reactions took place in a short reaction time at 75–100 °C, affording radiochemical yields of up to 93%. We have demonstrated the radiosynthetic utility of this fluorine-18 labelling method for the preparation of [18F]1a–b, which are based on NE inhibitors 1a–b. The methodology can be extended to a modular approach, for synthesis of a large variety of fluorine-18 labelled NE inhibitors.
The authors would like to thank Dominik Baran for his valuable help, the Knut och Alice Wallenbergs Foundation (Dnr: 2018.0066), and Swedish Research Council (VR) for financial support and Carl Tryggers Foundation (CTS 17:458) for a postdoctoral fellowship to X. J.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) D. O’Hagan, Chem. Soc. Rev., 2008, 37, 308 RSC; (b) D. O’Hagan, Chem. – Eur. J., 2020, 26, 7981 CrossRef; (c) B. M. Johnson, Y.-Z. Shu, X. Zhuo and N. A. Meanwell, J. Med. Chem., 2020, 63, 6315 CrossRef CAS PubMed.
- (a) Y. Zhu, J. Han, J. Wang, N. Shibata, M. Sodeoka, V. A. Soloshonok, J. A. S. Coelho and F. D. Toste, Chem. Rev., 2018, 118, 3887 CrossRef CAS PubMed; (b) H. Mei, J. Han, S. Fustero, M. Medio-Simon, D. M. Sedgwick, C. Santi, R. Ruzziconi and V. A. Soloshonok, Chem. – Eur. J., 2019, 25, 11797 CrossRef CAS PubMed.
- P. Jeschke, in Organofluorine Chemistry: Synthesis, Modeling, and Applications, ed. K. J. Szabó and N. Selander, Wiley-VCH, 2021, ch. 11 Search PubMed.
- (a) J. Rong, A. Haider and S. Liang, in Organofluorine Chemistry: Synthesis, Modeling, and Applications, ed. K. J. Szabó and N. Selander, Wiley-VCH, 2021, ch. 12 Search PubMed; (b) X. Deng, J. Rong, L. Wang, N. Vasdev, L. Zhang, L. Josephson and S. H. Liang, Angew. Chem., Int. Ed., 2019, 58, 2580 CrossRef CAS; (c) S. Preshlock, M. Tredwell and V. Gouverneur, Chem. Rev., 2016, 116, 719 CrossRef CAS PubMed; (d) D. van der Born, A. Pees, A. J. Poot, R. V. A. Orru, A. D. Windhorst and D. J. Vugts, Chem. Soc. Rev., 2017, 46, 4709 RSC.
- (a) V. T. Lien and P. J. Riss, BioMed Res. Int., 2014, 10 Search PubMed; (b) A. Bermejo Gomez, M. Cortes, M. Lübcke, M. Johansson, C. Halldin, K. J. Szabo and M. Schou, Chem. Commun., 2016, 52, 13963 RSC; (c) C. W. Kee, O. Tack, F. Guibbal, T. C. Wilson, P. G. Isenegger, M. Imiołek, S. Verhoog, M. Tilby, G. Boscutti, S. Ashworth, J. Chupin, R. Kashani, A. W. J. Poh, J. K. Sosabowski, S. Macholl, C. Plisson, B. Cornelissen, M. C. Willis, J. Passchier, B. G. Davis and V. Gouverneur, J. Am. Chem. Soc., 2020, 142, 1180 CrossRef CAS; (d) S. Verhoog, C. W. Kee, Y. Wang, T. Khotavivattana, T. C. Wilson, V. Kersemans, S. Smart, M. Tredwell, B. G. Davis and V. E. Gouverneur, J. Am. Chem. Soc., 2018, 140, 1572 CrossRef CAS PubMed; (e) T. Khotavivattana, S. Verhoog, M. Tredwell, L. Pfeifer, S. Calderwood, K. Wheelhouse, T. Lee Collier and V. Gouverneur, Angew. Chem., Int. Ed., 2015, 54, 9991 CrossRef CAS; (f) P. J. Riss, T. Ruehl, W. Rafique and V. T. Lien, Chem. Commun., 2014, 50, 6056 RSC; (g) A. Pees, M. J. W. D. Vosjan, N. Vasdev, A. D. Windhorst and D. J. Vugts, Chem. Commun., 2021, 57, 5286 RSC.
- (a) P. R. Bernstein, D. Andisik, P. K. Bradley, C. B. Bryant, C. Ceccarelli, J. R. Damewood, R. Earley, P. D. Edwards and S. Feeney, J. Med. Chem., 1994, 37, 3313 CrossRef CAS PubMed; (b) F. von Nussbaum and V. M. J. Li, Bioorg. Med. Chem. Lett., 2015, 25, 4370 CrossRef CAS PubMed; (c) D. Schirlin, C. Tarnus, S. Baltzer and J. M. Rémy, Bioorg. Med. Chem. Lett., 1992, 2, 651 CrossRef CAS; (d) G. B. Dreyer and B. W. Metcalf, Tetrahedron Lett., 1988, 29, 6885 CrossRef CAS; (e) A. Nikolaou, M. G. Kokotou, S. Vasilakaki and G. Kokotos, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2019, 1864, 941 CrossRef CAS PubMed.
- M. H. Gelb, J. P. Svaren and R. H. Abeles, Biochemistry, 1985, 24, 1813 CrossRef CAS PubMed.
- J.-P. Bégué and D. Bonnet-Delpon, Bioorganic and Medicinal Chemistry of Fluorine, Wiley, 2008, p. 223 Search PubMed.
- P. J. Barnes, Nat. Rev. Drug Discovery, 2013, 12, 543 CrossRef PubMed.
- R. W. Thomson, J. A. Curci, T. L. Ennis, D. Mao, M. B. Pagano and C. T. N. Pham, Ann. N. Y. Acad. Sci., 2006, 1085, 59 CrossRef PubMed.
- M. Jiménez-Alcázar, C. Rangaswamy, R. Panda, J. Bitterling, Y. J. Simsek, A. T. Long, R. Bilyy, V. Krenn, C. Renné, T. Renné, S. Kluge, U. Panzer, R. Mizuta, H. G. Mannherz, D. Kitamura, M. Herrmann, M. Napirei and T. A. Fuchs, Science, 2017, 358, 1202 CrossRef PubMed.
- (a) B. J. Barnes, J. M. Adrover, A. Baxter-Stoltzfus, A. Borczuk, J. Cools-Lartigue, J. M. Crawford, J. Daßler-Plenker, P. Guerci, C. Huynh, J. S. Knight, M. Loda, M. R. Looney, F. McAllister, R. Rayes, S. Renaud, S. Rousseau, S. Salvatore, R. E. Schwartz, J. D. Spicer, C. C. Yost, A. Weber, Y. Zuo and M. Egeblad, J. Exp. Med., 2020, 217 Search PubMed; (b) P. Mehta, D. F. McAuley, M. Brown, E. Sanchez, R. S. Tattersall and J. J. Manson, Lancet, 2020, 395, 1033 CrossRef CAS PubMed.
- (a) N. O. Ilchenko, B. O. A. Tasch and K. J. Szabó, Angew. Chem., Int. Ed., 2014, 53, 12897 CrossRef CAS; (b) W. Yuan, L. Eriksson and K. J. Szabó, Angew. Chem., Int. Ed., 2016, 55, 8410 CrossRef CAS PubMed; (c) W. Yuan and K. J. Szabó, Angew. Chem., Int. Ed., 2015, 54, 8533 CrossRef CAS PubMed; (d) Q. Wang, M. Lübcke, M. Biosca, M. Hedberg, L. Eriksson, F. Himo and K. J. Szabó, J. Am. Chem. Soc., 2020, 142, 20048 CrossRef CAS PubMed; (e) M. A. Cortes Gonzalez, P. Nordeman, A. Bermejo Gomez, D. N. Meyer, G. Antoni, M. Schou and K. J. Szabó, Chem. Commun., 2018, 54, 4286 RSC; (f) M. A. Cortés González, X. Jiang, P. Nordeman, G. Antoni and K. J. Szabo, Chem. Commun., 2019, 55, 13358 RSC.
- (a) H. Amii, T. Kobayashi, Y. Hatamoto and K. Uneyama, Chem. Commun., 1999, 1323 RSC; (b) G. K. Surya Prakash, J. Hu and G. A. Olah, J. Fluorine Chem., 2001, 112, 355 CrossRef.
- (a) X.-S. Hu, J.-S. Yu and J. Zhou, Chem. Commun., 2019, 55, 13638 RSC; (b) M. Decostanzi, J. M. Campagne and E. Leclerc, Org. Biomol. Chem., 2015, 13, 7351 RSC; (c) X. Jiang, D. Meyer, D. Baran, M. A. Cortés González and K. J. Szabo, J. Org. Chem., 2020, 85, 8311 CrossRef CAS PubMed.
- (a) G. K. Surya Prakash, M. M. Alauddin, J. Hu, P. S. Conti and G. A. Olah, J. Labelled Compd. Radiopharm., 2003, 46, 1087 CrossRef; (b) G. K. S. Prakash, M. M. Alauddin, J. Hu, P. S. Conti and G. A. Olah, US Pat., US 6872855, 2005 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data and radiochemistry. See DOI: 10.1039/d1cc03624f |
| ‡ Contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |