
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The primary dipole of flipper probes†
José
García-Calvo
,
Javier
López-Andarias
,
Naomi
Sakai
and
Stefan
Matile
 *
*
Department of Organic Chemistry, University of Geneva, Geneva, Switzerland. E-mail: stefan.matile@unige.ch; Web: http://www.unige.ch/sciences/chiorg/matile/
First published on 23rd March 2021
Abstract
Despite their growing popularity in biology to image membrane tension, central design principles of flipper probes have never been validated. Here we report that upon deletion of their primary dipole, from electron-poor and electron-rich dithienothiophenes, absorptions blue-shift, lifetimes shorten dramatically, and mechanosensitivity in cells vanishes not partially, but completely.
The conversion of sulfides into sulfoxides and sulfones is arguably one of the most useful transformations in supramolecular chemistry, also because it can be reversed.1–9 Examples include conformational switching in peptides, proteins and polymers,1,2 turn-on/off of binding,3 fluorophores,4–7 chromophores,8a chirality,8a,b self-sorting,8a hydrogelation,2 voltage-gated ion channels,8c transporters8a and anion–π catalysts.8b “Sulfur switches” are also instrumental in fluorescent flipper probes,9a which are bioinspired small-molecule mechanophores for the imaging of membrane tension changes in live cells.9,10 The mechanosensitive part consists of an electron-donating dithienothiophene (DTT) and an electron-accepting DTT S,S-dioxide (DTTO2) with internal formal sulfide and sulfone bridges, respectively,4,7,11 connected by a single bond (Fig. 1a).9 In fluid media, the two DTTs are twisted out of co-planarity by repulsion between the methyl groups and σ holes next to the twistable bond,7 thus the primary push–pull dipole is weak.9 Mechanical planarization should then establish conjugation, turn on the push–pull dipole, red shift the absorption and increase fluorescence lifetime.9 Unlike most membrane probes operating off-equilibrium in the excited state,12,13 flipper probes respond in equilibrium in the ground-state.9b,14
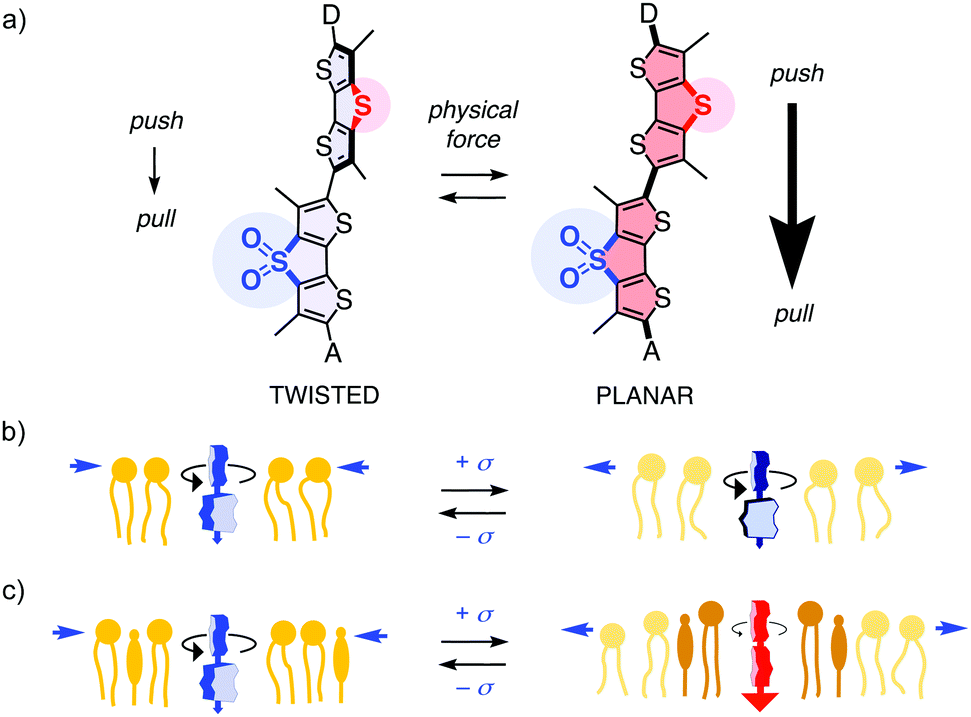 | ||
| Fig. 1 (a) The primary push–pull dipole from sulfide to sulfone bridges, supported by exocyclic donors D and acceptors A, is designed to turn on upon mechanical planarization of fluorescent flipper probes, shift absorption to the red and increase fluorescence lifetimes. (b) In uniform membranes, flippers respond to membrane tension by decreasing lifetime due to lipid decompression. (c) In mixed membranes, flipper lifetimes increase due to tension-induced membrane reorganization, particularly phase separation. | ||
This design strategy has afforded operational mechanophores that image membrane tension.10 Increasing tension in uniform membranes caused the fluorescence lifetime of flipper probes to decrease, consistent with lipid decompression (Fig. 1b). Tension applied to heterogeneous membranes, including biomembranes, caused the fluorescence lifetimes to increase, consistent with tension-induced membrane reorganization to form highly ordered microdomains with planarized flippers (Fig. 1c). The observed linear correlations between lifetime changes in fluorescence lifetime imaging microscopy (FLIM) images and forces applied with micropipettes or osmotic stress demonstrate the applicability of flipper probes to image membrane tension. Strategies have been developed to target flipper probes to the membrane of interest inside living cells.15 The resulting probes have enabled the studies on how membrane mechanics regulate genome protection,16 HIV-1 fusion,17 cell extrusion,18 cell migration,19 mitochondrial fission, endocytosis, secretory pathway, signal transduction, and so on.15
The primary dipole of flipper probes is further supported by exocyclic donors and acceptors (Fig. 1a). These secondary push–pull systems have been varied extensively.14,20 However, the role of the primary push–pull system was never verified, except for a non-conclusive testing early on.9a Considering the importance of flipper probes in biology, we felt this fundamental design principle needs to be substantiated. In the following, we report design, synthesis and evaluation of the full “sulfur switching cycle” required for a comprehensive study of the primary dipole in flipper probes.
We selected the most advanced flipper 1 to assess the primary push–pull dipole (Fig. 2).20 Exocyclic triazole donors and ketone acceptors operate as chalcogen-bonding cascade switches triggered by mechanical planarization.14 Reversible dynamic covalent conversion of the trifluoromethyl ketone 1 acceptor into the non-fluorescent hydrate 1d affords the blinking needed for super-resolution microscopy (Fig. 2).20
 | ||
| Fig. 2 Amphiphilic flippers 1–3 made to assess the importance of the primary dipole, their hydrophobic analogues 1′–3′ with their color in CDCl3 (left), and their “dark” hydrates 1d–3d. | ||
Flippers 2 and 3 were the obvious choices to evaluate the importance of the primary dipole in push–pull flipper 1 (Fig. 2). Push–push flipper 2 is composed of two electron-rich sulfide-bridged DTTs, pull–pull flipper 3 has two electron-poor DTTO2s. This complementary removal of the primary dipole already affected flipper synthesis significantly. Both DTTs 4 and 5, prepared from tetrabromothiophene as described,20 were readily oxidized into the DTTO2s 6 and 7 (Scheme 1). Stille coupling of the electron-rich DTTs 4 and 5 gave flipper 8 in 25% yield. The yield of the original coupling of DTT 4 and DTTO2 7 to flipper 9 is poorly reproducible, can reach 23% but averages around 8%. Coupling of the two DTTO2s 6 and 7 gave a complex mixture. Pull–pull flipper 10 was, however, readily accessible by oxidation of the push–push flipper 8.
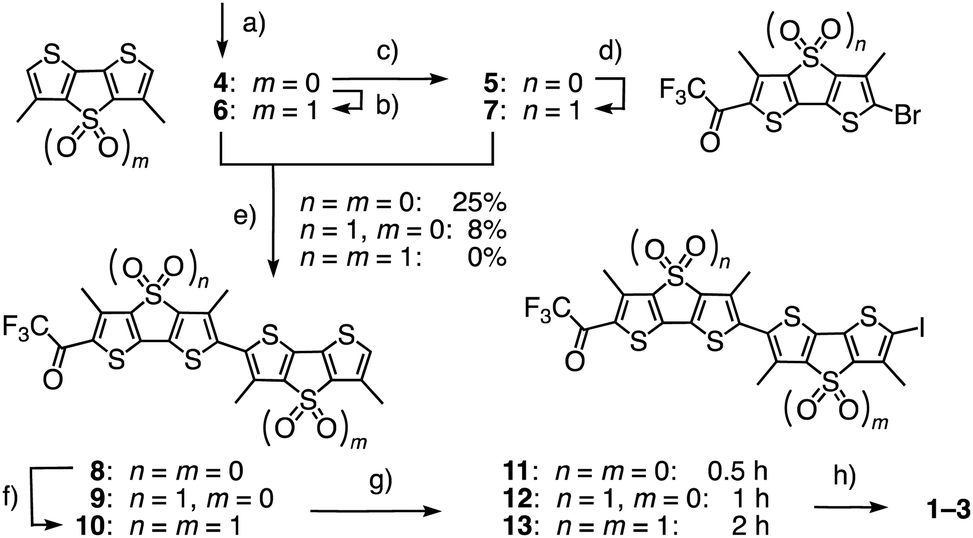 | ||
| Scheme 1 (a) Five steps from tetrabromothiophene, 20–40%;20 (b) mCPBA, CHCl3, rt, 4 h, 56%; (c) 2 steps, 70%;20 (d) mCPBA, CH2Cl2, rt, 8 h, 75%; (e) (1) nBuLi, THF, −78 °C, 15 min; (2) Bu3SnCl, −78 °C to rt, 30 min; (3) 5 or 7, Pd(PPh3)4, toluene/DMF, 80 °C, 48 h, 4 + 5 → 8, 25%; 4 + 7 → 9, ∼8% (max: 23%); 6 + 7 → 10, 0%; (f) mCPBA, CH2Cl2, rt, 12 h, 40%; (g) NIS, AcOH, CHCl3, 0 °C, 0.5 h, then rt, 0.5 h (8 → 11), 1 h (9 → 12), or 3 h (10 → 13), quantitative; (h) 3 steps, see ESI.† | ||
The different nature of flippers 8–10 also affected iodination into 11–13, being fast with the reactive push–push system 8 and slow with the electron-poor pull–pull counterpart 10. The following Sonogashira coupling and completion of the synthesis of amphiphiles 2 and 3 proceeded as described for original 1. The hydrophobic analogues 1′–3′ were prepared similarly for spectroscopic studies in apolar solvents (Fig. S1 and Schemes S1–S3, ESI†).
The effect of the missing primary dipole in flippers 2′ and 3′ was already visible by the naked eye, changing the orange-red appearance of original 1′ into a deep yellow (Fig. 2). The excitation and absorption maxima blue-shifted accordingly, from 450 nm for the push–pull 1′ to 436 nm for pull–pull 3′ and 402 nm for push–push 2′ (Fig. 3a and Table 1, entry 2). Thus, apparently, the polarization of push–pull 1 is significant even in the twisted form with a weak primary dipole. In emission, similar blue shifts occurred from 642 nm for 1′ to 586 nm for 3′ and 548 nm for 2′ (Fig. 3b and Table 1, entry 7). The fluorescence quantum yields of the twisted probes in solution decreased also upon deleting the primary push–pull dipole, from 27% for 1′ to 20% for 2′ and 10% for 3′ in dioxane (Table 1, entry 8). The poor fluorescence of 3′ was interesting because, as Barbarella and coworkers reported, quantum yields of monomers jump from <1% to 77% upon oxidation of DTTs like 4 to DTTO2s like 6.4 However, decreasing fluorescence with increasing numbers of DTTO2s in oligomers and polymers has been observed previously.5
 | ||
Fig. 3 (a) Normalized absorption and (b) emission spectra of flippers 1′ (red, λex 435 nm), 2′ (black, λex 380 nm) and 3′ (blue, λex 425 nm) in dioxane. (c) Normalized excitation (dashed) and emission spectra (solid) of 2′ in dioxane (black) or dioxane/water 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (grey). (d) Normalized fluorescence emission intensity of 1′ (red, 1 − I620) or ratio of 2′ (black, I450/I520) and 3′ (blue, I525/I580) in dioxane with increasing water concentration. (e) Normalized excitation spectra of flippers 1 (red), 2 (black) and 3 (blue, all 0.1 μM) in DOPC (dashed) and SM/CL (7:3) (solid) LUVs (75 μM lipid, 10 mM Tris, 100 mM NaCl, pH 7.4). (f) Same for 2 without normalization. :1 (grey). (d) Normalized fluorescence emission intensity of 1′ (red, 1 − I620) or ratio of 2′ (black, I450/I520) and 3′ (blue, I525/I580) in dioxane with increasing water concentration. (e) Normalized excitation spectra of flippers 1 (red), 2 (black) and 3 (blue, all 0.1 μM) in DOPC (dashed) and SM/CL (7:3) (solid) LUVs (75 μM lipid, 10 mM Tris, 100 mM NaCl, pH 7.4). (f) Same for 2 without normalization. | ||
| Entry | Parameter | 2/2′ | 1/1′ | 3/3′ | Unit |
|---|---|---|---|---|---|
| a Flippers 1–3 for entries 3–6 and 1′–3′ for entries 1, 2 and 7–10, see Fig. 2. b Extinction coefficient in dioxane (= Lb, bulk liquid membrane, Table S2, ESI). c Excitation maximum in dioxane (Fig. 3a). d Ld membranes (liquid-disordered, DOPC LUVs, Fig. 3e). e Lo membranes (liquid-ordered, SM/CL LUVs, Fig. 3e). f Fluorescence lifetime τav in HeLa cells under isosmotic conditions (Fig. 4b, e). g Fluorescence lifetime τav in HeLa cells under hyperosmotic conditions (Fig. 4c, e). h Emission maximum in dioxane (Fig. 3b). i Fluorescence quantum yield in dioxane (Fig. S6, S7). j Time required for half maximal conversion of ketones 1′–3′ into hydrates 1d′–3d′ in dioxane with water (1.25 M, Fig. 3c and d). k Half maximal effective concentration of water in dioxane for the formation of hydrates 1d′–3d′ (Fig. 3c and d). l Data for 1 are partially reproduced from ref. 20. m Maximum of hydrate 1d. n Very weak fluorescence. | |||||
| 1 | ε (Lb)b | 47 | 37 | 32 | mM−1 cm−1 |
| 2 | λ ex (Lb)c | 402 | 450 | 436 | nm |
| 3 | λ ex (Ld)d | 432 | 418m | (422)n | nm |
| 4 | λ ex (Lo)e | 502 | 560 | (422)n | nm |
| 5 | τ av (iso)f | 1.27 | 4.03 | 0.49 | ns |
| 6 | τ av (hyper)g | 1.20 | 3.74 | 0.55 | ns |
| 7 | λ em (Lb)h | 548 | 642 | 586 | nm |
| 8 | ϕ f (Lb)i | 22 | 27 | 10 | % |
| 9 | t 1/2 (hydr)j | >16 | 1.6 | 0.38 | h |
| 10 | EC50 (hydr)k | 2400 | 120 | 106 | mM |
The addition of water shifted the equilibrium from ketones 1′–3′ to the less fluorescent hydrates 1d′–3d′ with blue-shifted absorption and emission (Fig. 2 and 3c, d). EC50 and t1/2 decreased with the number of electron-accepting DTTO2s (Fig. 3d, Fig. S8–S13 (ESI†), Table 1, entries 9 and 10). The remote DTTO2 in 3′ also increased the electrophilicity of the carbonyl compared to 1′, despite the twisted conformation of relaxed flippers in solution. Nucleophilic addition in polar protic solvents complicated the analysis of positive solvatochromism, which was overall preserved for all flippers (Fig. S2–S5 and Table S2, ESI†).
As reported previously,20 mechanical planarization of push–pull flipper 1 in the liquid-ordered (Lo) sphingomyelin/cholesterol (SM/CL) 7:3 large unilamellar vesicles (LUVs) results in a red shift of the excitation maximum to 560 nm (Fig. 3e, red solid, Table 1, entry 4). Deletion of the primary dipole in the new push–push flipper 2 weakened this shift by −58 nm to 502 nm, while the vibrational finestructure was well preserved (Fig. 3e, black solid; Fig. S14–S19, ESI†). Contrary to 1 and 2, pull–pull flipper 3 was fluorescent in buffer, perhaps because hydration hinders self-assembly into dark micelles. This fluorescence in buffer was weakened, but not shifted, by the presence of Lo membranes, suggesting that 3 does not align well along the highly ordered hydrophobic lipid tails (Fig. 3e, blue solid; Fig. S14–S19, ESI†). Overcompeting hydration could further hinder penetration into the membrane core (Table 1, entries 9 and 10), an interpretation that would be consistent with short fluorescence lifetime, mechano-insensitivity and rapid internalization in cells (Table 1, entries 5 and 6; Fig. S20–S22, vide infra, ESI†). The changing environment upon binding at the membrane surface would then account for the shift-less decrease of fluorescence of 3.
Compared to those in Lo LUVs, both flippers 1 and 2 in liquid-disordered (Ld) dioleoyl phosphatidylcholine (DOPC) LUVs showed blue-shifted excitation maxima and weaker fluorescence, as expected for flipper deplanarization with decreasing membrane order (Fig. 3e, dashed, Table 1, entry 3; Fig. S14–S19, ESI†). Both changes were less pronounced without primary dipole in 2 because low reactivity of the ketone disfavors the formation of hydrate 2d also in the more hydrated Ld membranes (Fig. 3f).
In confocal laser scanning microscopy (CLSM) images of HeLa Kyoto cells, flippers 2 and 3 without primary dipole were found to label the plasma membrane selectively like original 1, but they were much less bright (Fig. 4a and Fig. S20–S22, ESI†). In FLIM images of the HeLa Kyoto cells, the poorly fluorescent flippers 2 and 3 without primary dipole showed much shorter fluorescence lifetime than the τav = 4.03 ns of the original push–pull flipper 1 (Fig. 4b and Table 1, entry 5). Triexponential deconvolution of the fluorescence decay of 1 revealed a large, long-lived population in the FLIM histogram that can be assigned to the planarized ketone 1 together with a small, short-lived population that could originate from hydrate 1d and/or mispositioned 1/1d, located near the interface rather than aligned along the lipid tails (Fig. 4d). Studies on the possibility to correlate changes in their distribution with lifetime changes and the imaging of membrane tension are ongoing.
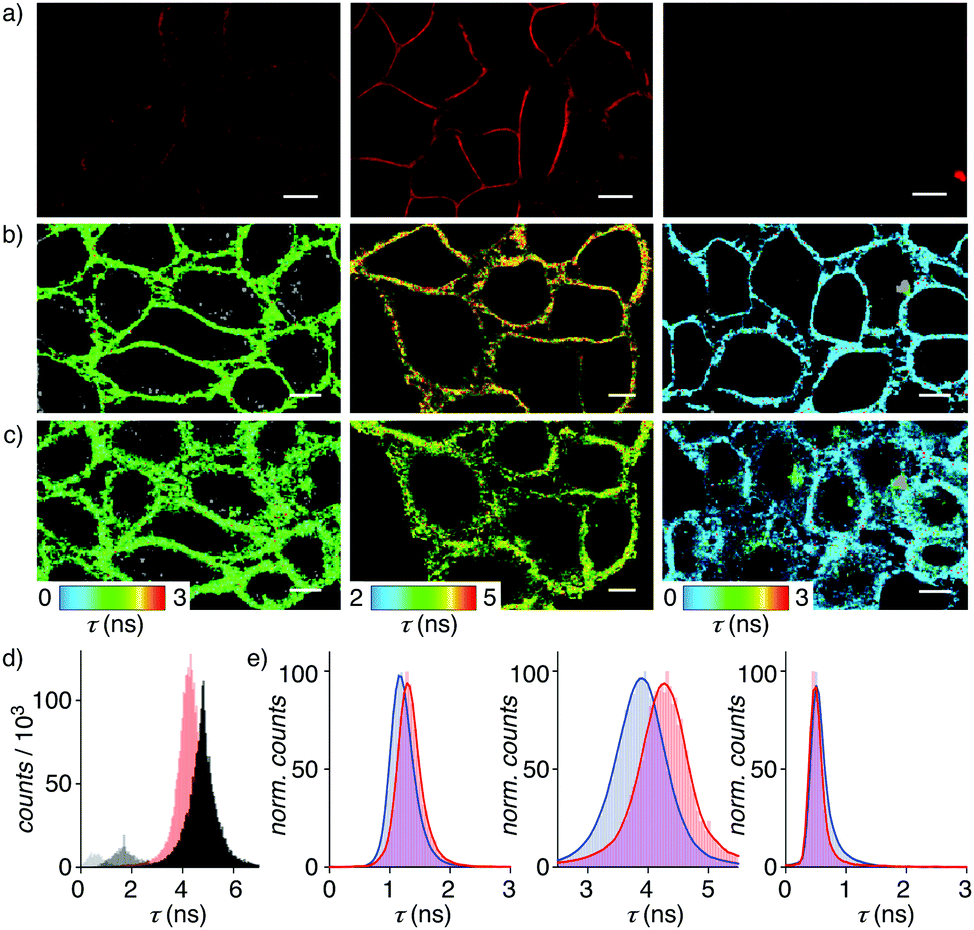 | ||
| Fig. 4 (a) CLSM images of 2, 1 and 3 (4 μM, left to right) in HeLa Kyoto cells after 3 min incubations and brief washes. (b) FLIM images of 2, 1 and 3 (4 μM) in HeLa Kyoto cells before and (c) after hyperosmotic shock. (d) Average (red) and deconvoluted (grey) lifetime histograms for 1. (e) Average lifetime histograms with Gaussian fits for 2, 1, and 3 (left to right) before (red) and after the hyperosmotic shock (blue). Scale bars: 10 μm. | ||
The very short lifetime τav = 0.49 ns of pull–pull 3 was consistent with the dysfunctional behavior identified in model membranes (Fig. 4b, right). The not so much longer lifetime τav = 1.27 ns of push–push 2 was unlikely to originate from hydrate 2d because hydration is less favored than for 1 (Fig. 4b, left, 3d, Table 1, entries 5, 9 and 10). Decreasing membrane tension upon hyperosmotic stress was reported by the original flipper 1 as decreasing lifetime from τav = 4.03 ns to τav = 3.74 ns (Fig. 4b and c middle; Table 1, entries 5 and 6). Concentration independent, this responsiveness of the lifetime is the essence of membrane tension imaging with flipper probes. Removal of the primary dipole in flipper 2 and 3 practically annihilated this mechanosensitivity (Fig. 4b, c, e and Table 1, entries 5 and 6).
Taken together, this evaluation of the central design paradigm reveals that the primary dipole of flipper probe is essential, arguably even more important than expected. Deletion of the primary dipole by formal sulfur redox switching1–9 – removing or adding only two oxygen atoms with, however, substantial synthetic effort – annihilates the flipper's mechanosensitivity in live cells. Other consequences of the removal of the primary dipole are significant losses of fluorescence lifetime, more than two-third, quantum yield and red shifts in absorption and emission maxima. These findings validate the original design and thus the fundamental understanding of flippers as mechanosensitive planarizable push–pull probes. The importance of these results cannot be overestimated with regard to future probe design as well as the interpretation of mechanobiological FLIM images.
We thank the NMR, MS and Bioimaging platforms for services, A. Roux for access to FLIM, and the University of Geneva, the National Centre for Competence in Research (NCCR) Chemical Biology, the NCCR Molecular Systems Engineering and the Swiss NSF for financial support.
Conflicts of interest
The authors declare the following competing financial interest: The University of Geneva has licensed four Flipper-TR® probes to Spirochrome for commercialization.Notes and references
- (a) G. P. Dado and S. H. Gellman, J. Am. Chem. Soc., 1993, 115, 12609–12610 CrossRef CAS; (b) J. R. Kramer and T. J. Deming, J. Am. Chem. Soc., 2014, 136, 5547–5550 CrossRef CAS; (c) C. Wolschner, A. Giese, H. A. Kretzschmar, R. Huber, L. Moroder and N. Budisa, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 7756–7761 CrossRef CAS; (d) H. Xu, W. Cao and X. Zhang, Acc. Chem. Res., 2013, 46, 1647–1658 CrossRef CAS PubMed.
- T. J. Deming, Bioconjugate Chem., 2017, 28, 691–700 CrossRef CAS PubMed.
- (a) N. N. Andersen, K. Eriksen, M. Lisbjerg, M. E. Ottesen, B. O. Milhøj, S. P. A. Sauer and M. Pittelkow, J. Org. Chem., 2019, 84, 2577–2584 CrossRef CAS PubMed; (b) M. A. E. Rebhan, A. Brunschweiger and J. Hall, ChemBioChem, 2013, 14, 2091–2094 CrossRef CAS PubMed; (c) Y. Cheng, A.-T. Pham, T. Kato, B. Lim, D. Moreau, J. López-Andarias, L. Zong, N. Sakai and S. Matile, Chem. Sci., 2021, 12, 626–631 RSC; (d) R. B. Ferreira, M. E. Law, S. C. Jahn, B. J. Davis, C. D. Heldermon, M. Reinhard, R. K. Castellano and B. K. Law, Oncotarget, 2015, 6, 10445–10459 CrossRef PubMed.
- G. Barbarella, L. Favaretto, G. Sotgiu, L. Antolini, G. Gigli, R. Cingolani and A. Bongini, Chem. Mater., 2001, 13, 4112–4122 CrossRef CAS.
- (a) F. Di Maria, A. Zanelli, A. Liscio, A. Kovtun, E. Salatelli, R. Mazzaro, V. Morandi, G. Bergamini, A. Shaffer and S. Rozen, ACS Nano, 2017, 11, 1991–1999 CrossRef CAS; (b) F. Di Maria, M. Zangoli, I. E. Palamá, E. Fabiano, A. Zanelli, M. Monari, A. Perinot, M. Caironi, V. Maiorano, A. Maggiore, M. Pugliese, E. Salatelli, G. Gigli, I. Viola and G. Barbarella, Adv. Funct. Mater., 2016, 26, 6970–6984 CrossRef CAS.
- (a) E. L. Dane, S. B. King and T. M. Swager, J. Am. Chem. Soc., 2010, 132, 7758–7768 CrossRef CAS; (b) M. Taki, K. Kajiwara, E. Yamaguchi, Y. Sato and S. Yamaguchi, ACS Mater. Lett., 2021, 3, 42–49 CrossRef CAS; (c) M. M. Oliva, J. Casado, J. T. L. Navarrete, S. Patchkovskii, T. Goodson, M. R. Harpham, J. S. Seixas de Melo, E. Amir and S. Rozen, J. Am. Chem. Soc., 2010, 132, 6231–6242 CrossRef CAS; (d) J. Kosai, Y. Masuda, Y. Chikayasu, Y. Takahashi, H. Sasabe, T. Chiba, J. Kido and H. Mori, ACS Appl. Polym. Mater., 2020, 2, 3310–3318 CrossRef CAS; (e) E. Caron and M. O. Wolf, Macromolecules, 2017, 50, 7543–7549 CrossRef CAS; (f) C. Romero-Nieto and T. Baumgartner, Synlett, 2013, 920–937 CrossRef CAS; (g) V. M. Blas-Ferrando, J. Ortiz, K. Ohkubo, S. Fukuzumi, F. Fernández-Lázaro and Á. Sastre-Santos, Chem. Sci., 2014, 5, 4785–4793 RSC; (h) Y. Zhou, B. Xue, C. Wu, S. Chen, H. Liu, T. Jiu, Z. Li and Y. Zhao, Chem. Commun., 2019, 55, 13570–13573 RSC.
- K. Strakova, L. Assies, A. Goujon, F. Piazzolla, H. V. Humeniuk and S. Matile, Chem. Rev., 2019, 119, 10977–11005 CrossRef PubMed.
- (a) N.-T. Lin, A. Vargas Jentzsch, L. Guénée, J.-M. Neudörfl, S. Aziz, A. Berkessel, E. Orentas, N. Sakai and S. Matile, Chem. Sci., 2012, 3, 1121–1127 RSC; (b) Y. Zhao, Y. Cotelle, A.-J. Avestro, N. Sakai and S. Matile, J. Am. Chem. Soc., 2015, 137, 11582–11585 CrossRef CAS PubMed; (c) N. Sakai, D. Houdebert and S. Matile, Chem. – Eur. J., 2003, 9, 223–232 CrossRef CAS PubMed.
- (a) M. Dal Molin, Q. Verolet, A. Colom, R. Letrun, E. Derivery, M. Gonzalez-Gaitan, E. Vauthey, A. Roux, N. Sakai and S. Matile, J. Am. Chem. Soc., 2015, 137, 568–571 CrossRef CAS PubMed; (b) T. Kato, K. Strakova, J. García-Calvo, N. Sakai and S. Matile, Bull. Chem. Soc. Jpn., 2020, 93, 1401–1411 CrossRef CAS.
- A. Colom, E. Derivery, S. Soleimanpour, C. Tomba, M. D. Molin, N. Sakai, M. González-Gaitán, S. Matile and A. Roux, Nat. Chem., 2018, 10, 1118–1125 CrossRef CAS PubMed.
- M. E. Cinar and T. Ozturk, Chem. Rev., 2015, 115, 3036–3140 CrossRef CAS.
- (a) A. S. Klymchenko, Acc. Chem. Res., 2017, 50, 366–375 CrossRef CAS PubMed; (b) M. Páez-Pérez, I. López-Duarte, A. Vyšniauskas, N. J. Brooks and M. K. Kuimova, Chem. Sci., 2021, 16, 2604–2613 RSC; (c) C.-H. Wu, Y. Chen, K. A. Pyrshev, Y.-T. Chen, Z. Zhang, K.-H. Chang, S. O. Yesylevskyy, A. P. Demchenko and P.-T. Chou, ACS Chem. Biol., 2020, 15, 1862–1873 CrossRef CAS PubMed; (d) D. I. Danylchuk, P.-H. Jouard and A. S. Klymchenko, J. Am. Chem. Soc., 2021, 143, 912–924 CrossRef CAS PubMed; (e) J. E. Chambers, M. Kubánková, R. G. Huber, I. López-Duarte, E. Avezov, P. J. Bond, S. J. Marciniak and M. K. Kuimova, ACS Nano, 2018, 12, 4398–4407 CrossRef CAS PubMed.
- (a) P. Liu and E. W. Miller, Acc. Chem. Res., 2020, 53, 11–19 CrossRef CAS PubMed; (b) Y. Xiong, A. Vargas Jentzsch, J. W. M. Osterrieth, E. Sezgin, I. V. Sazanovich, K. Reglinski, S. Galiani, A. W. Parker, C. Eggeling and H. L. Anderson, Chem. Sci., 2018, 9, 3029–3040 RSC; (c) I. E. Steinmark, A. L. James, P.-H. Chung, P. E. Morton, M. Parsons, C. A. Dreiss, C. D. Lorenz, G. Yahioglu and K. Suhling, PLoS One, 2019, 14, e0211165 CrossRef CAS PubMed; (d) R. Guo, J. Yin, Y. Ma, Q. Wang and W. Lin, J. Mater. Chem. B, 2018, 6, 2894–2900 RSC; (e) L. Yu, J. F. Zhang, M. Li, D. Jiang, Y. Zhou, P. Verwilst and J. S. Kim, Chem. Commun., 2020, 56, 6684–6687 RSC; (f) P. E. Deal, P. Liu, S. H. Al-Abdullatif, V. R. Muller, K. Shamardani, H. Adesnik and E. W. Miller, J. Am. Chem. Soc., 2020, 142, 614–622 CrossRef CAS PubMed; (g) M. A. Haidekker and E. A. Theodorakis, J. Mater. Chem. C, 2016, 4, 2707–2718 RSC; (h) J. A. Robson, M. Kubánková, T. Bond, R. A. Hendley, A. J. P. White, M. K. Kuimova and J. D. E. T. Wilton-Ely, Angew. Chem., Int. Ed., 2020, 59, 21431–21435 CrossRef CAS PubMed; (i) D. I. Danylchuk, S. Moon, K. Xu and A. S. Klymchenko, Angew. Chem., Int. Ed., 2019, 58, 14920–14924 CrossRef CAS PubMed.
- M. Macchione, A. Goujon, K. Strakova, H. V. Humeniuk, G. Licari, E. Tajkhorshid, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2019, 58, 15752–15756 CrossRef CAS.
- J. López-Andarias, K. Straková, R. Martinent, N. Jiménez-Rojo, H. Riezman, N. Sakai and S. Matile, JACS Au, 2021, 1, 221–232 CrossRef.
- M. M. Nava, Y. A. Miroshnikova, L. C. Biggs, D. B. Whitefield, F. Metge, J. Boucas, H. Vihinen, E. Jokitalo, X. Li, J. M. García Arcos, B. Hoffmann, R. Merkel, C. M. Niessen, K. N. Dahl and S. A. Wickström, Cell, 2020, 181, 800–817 CrossRef CAS PubMed.
- C. A. Coomer, I. Carlon-Andres, M. Iliopoulou, M. L. Dustin, E. B. Compeer, A. A. Compton and S. Padilla-Parra, PLoS Pathog., 2020, 16, e1008359 CrossRef CAS PubMed.
- A. Villars and R. Levayer, Curr. Biol., 2020, 30, R168–R171 CrossRef CAS PubMed.
- J. H. R. Hetmanski, H. de Belly, I. Busnelli, T. Waring, R. V. Nair, V. Sokleva, O. Dobre, A. Cameron, N. Gauthier, C. Lamaze, J. Swift, A. del Campo, T. Starborg, T. Zech, J. G. Goetz, E. K. Paluch, J.-M. Schwartz and P. T. Caswell, Dev. Cell, 2019, 51, 460–475 CrossRef CAS PubMed.
- J. García-Calvo, J. Maillard, I. Fureraj, K. Strakova, A. Colom, V. Mercier, A. Roux, E. Vauthey, N. Sakai, A. Fürstenberg and S. Matile, J. Am. Chem. Soc., 2020, 142, 12034–12038 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cc00860a |
| This journal is © The Royal Society of Chemistry 2021 |