
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceManganese-catalyzed selective C–H activation and deuteration by means of a catalytic transient directing group strategy†
Sara
Kopf
 ,
Helfried
Neumann
* and
Matthias
Beller
*
,
Helfried
Neumann
* and
Matthias
Beller
*
Leibniz-Institut für Katalyse, Albert-Einstein-Straße 29a, Rostock 18059, Germany. E-mail: matthias.beller@catalysis.de
First published on 7th January 2021
Abstract
A novel manganese-catalyzed C–H activation methodology for selective hydrogen isotope exchange of benzaldehydes is presented. Using D2O as a cheap and convenient source of deuterium, the reaction proceeds with excellent functional group tolerance. High ortho-selectivity is achieved in the presence of catalytic amounts of specific amines, which in situ form a transient directing group.
Not only since the first deuterated drug was accepted by the U.S. Food and Drug Administration (FDA) in 2017, an increasing interest in the development of methodologies for the selective late-stage incorporation of deuterium into organic molecules has manifested itself within the field of organic synthesis.1 As a result, the current toolbox for labeling reactions comprises a number of established techniques for diverse substrates. As an example, deuteration of arenes predominantly relies on iridium-catalyzed hydrogen isotope exchange (HIE) (Scheme 1a). Here, the most important representative, the NHC-ligated Kerr catalyst, affects deuteration under deuterium gas atmosphere ortho to a number of pre-installed directing groups, including N-heterocycles, carbonyl groups and sulfur-containing functional groups.2 Most recently, base metal-catalyzed methodologies have expanded the field by presenting new selectivity concepts based on steric and/or electronic control. In this context, the deuteration of the least sterically hindered positions in arenes and hetarenes has been achieved under catalysis with iron pincer complexes (Scheme 1b),3 whereas a diamine nickel complex preferentially enabled HIE on electron-deficient positions of pyridine derivatives.4 However, the precision of a directed approach has so far been unmet by alternative strategies. In this respect, the development of metal catalyzed directed HIE methodologies,5 combining inexpensive and available metals with predictable selectivity, is desirable.
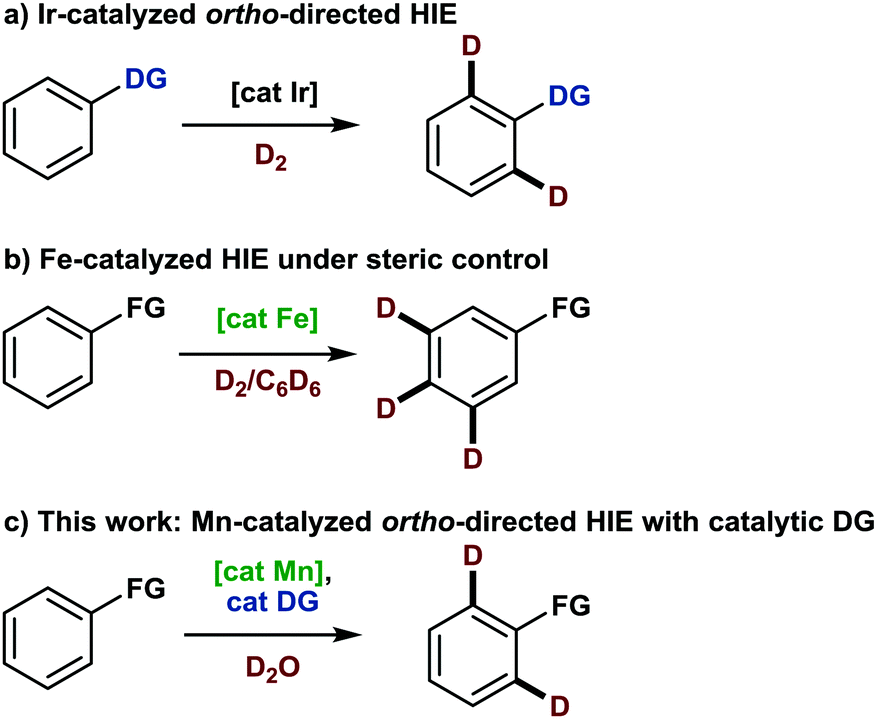 | ||
| Scheme 1 Selectivity control of deuteration methodologies. | ||
Among the different earth-abundant 3d metals, manganese is a most promising candidate, haven proven its efficacy for catalytic directed C(sp2)–H functionalization reactions by enabling a plethora of transformations.6 Moreover, although manganese-catalyzed deuteration of arenes has only been scarcely investigated yet,7 some mechanistic investigations already hint at the potential success of such an endeavor.8 However, despite these advances, manganese-catalyzed C–H activation is still limited to substrates comprising nitrogen-containing directing groups9 and thus often demands additional synthetic steps. For instance, a recently disclosed allylation reaction of arenes required the pre-formation of an imine as a directing group, which subsequently needed to be hydrolyzed in a separate step.10 A methodology that allows this imine formation to occur in situ with a catalytic amount of amine and thus reduces time and waste would certainly be more appealing. Interestingly, such a transient directing group strategy has been utilized in noble metal catalysis,11 but has thus far not been applied to either manganese-catalyzed C–H activation or directed HIE. We believed that both fields could be merged thus report herein on the first manganese-catalyzed ortho-selective C–H deuteration of aromatic aldehydes through transient formation of a catalytic directing group (Scheme 1c). We started our investigations by studying the deuteration of para-anisaldehyde under typical conditions for base-assisted manganese-catalyzed C–H activation. More specifically, we used 5 mol%12 manganese pentacarbonyl bromide along with D2O as the cheapest source of deuterium in the presence of catalytic amounts of different amines (Scheme 2).
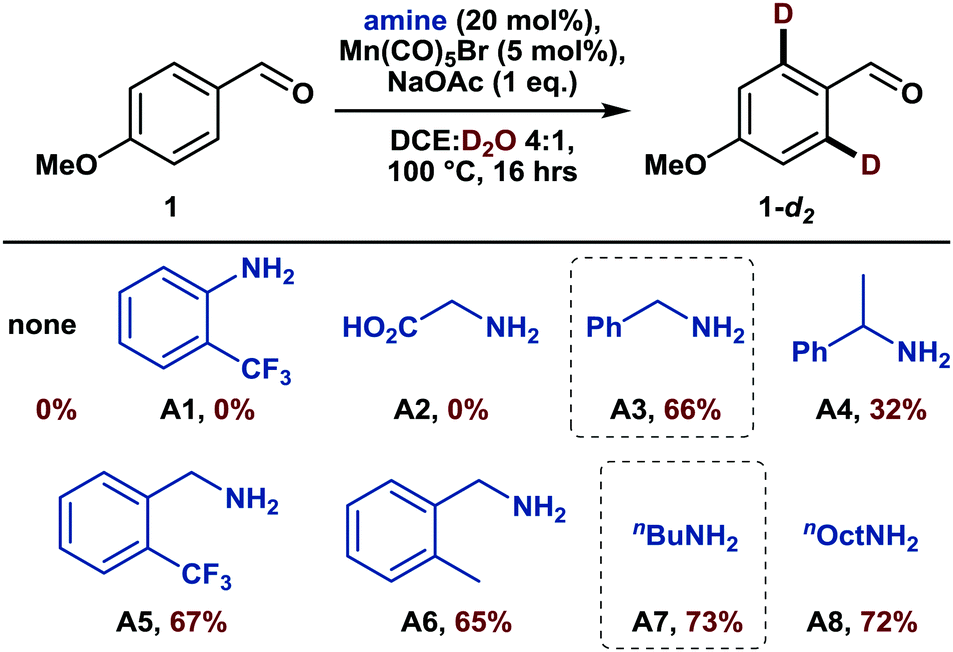 | ||
| Scheme 2 Mn-catalyzed o-deuteration of p-anisaldehyde in the presence of catalytic amine additives for the in situ formation of transient directing groups. Scale: 0.5 mmol 1. Deuterium incorporation was determined by 1H NMR through the decrease of the doublet at 7.81 ppm while referencing on the protons in meta position. | ||
Firstly, in the absence of amine additives, no deuteration was observed, confirming that the aldehyde moiety is not a feasible directing group for such C–H activation. Furthermore, established transient directing groups such as anilines and glycine11 could not afford any deuteration either. Surprisingly, the addition of 20 mol% of benzylamine to the reaction mixture enabled selective deuteration in the ortho positions of the aldehyde with a deuterium incorporation of 66% (A3). The use of sterically more hindered 1-phenylethylamine led to a drop in deuterium incorporation whereas substituents on the aromatic ring did not have a major influence on the reactivity (A5 and A6). A slightly increased deuteration was observed with aliphatic amines such as n-butylamine (A7) so that both benzylic and aliphatic amines can be considered efficient catalytic additives for transient directing group formation.
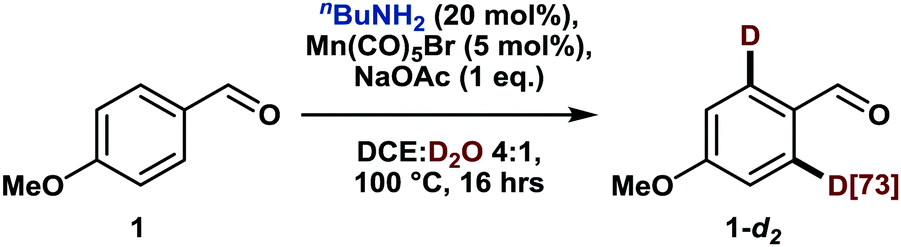
Notably, the deuterium incorporation can be simply increased by re-submitting the isolated, partially deuterated compound to the reaction conditions (Table 1, entry 1). Interestingly, the reaction does not benefit from elevated temperature (120 °C) but works equally well at 80 °C (Table 1, entries 2–4). Finally, in the absence of a manganese catalyst, no deuteration took place (Table 1, entry 5).
|
|
||
|---|---|---|
| Entry | Variation | D [%] |
| Scale: 0.5 mmol 1. Deuterium incorporation was determined by 1H NMR through the decrease of the doublet at 7.81 ppm while referencing on the protons in meta position. | ||
| 1 | 1-d2 (73% D) instead of 1 | 88 |
| 2 | 120 °C | 79 |
| 3 | 80 °C | 76 |
| 4 | 60 °C | 49 |
| 5 | No [Mn] | 0 |
Having gained insights into the influence of various parameters on the deuteration efficiency, we moved on to explore the substrate scope of this novel deuteration methodology. For this purpose, we conducted reactions at a temperature of 100 °C to ensure that less reactive substrates are also deuterated efficiently. To our delight, a variety of benzaldehyde derivatives proved to be amenable substrates, being selectively deuterated in ortho position without any side product formation (Scheme 3). In this context, halogenated benzaldehydes proved to be particularly viable substrates and did not undergo competing deuterodehalogenation (2–7 and 17).13 Only remaining amounts of the intermediary imine or the volatility of the substrates sometimes slightly impacted the recovery of the aldehyde. Further, substituents in ortho, meta and para positions as well as multi-substituted benzaldehydes are all well tolerated while guidance by Lewis-basic substituents is observed in the presence of two non-identical ortho positions, overriding potential steric effects (14, 16, 20).14
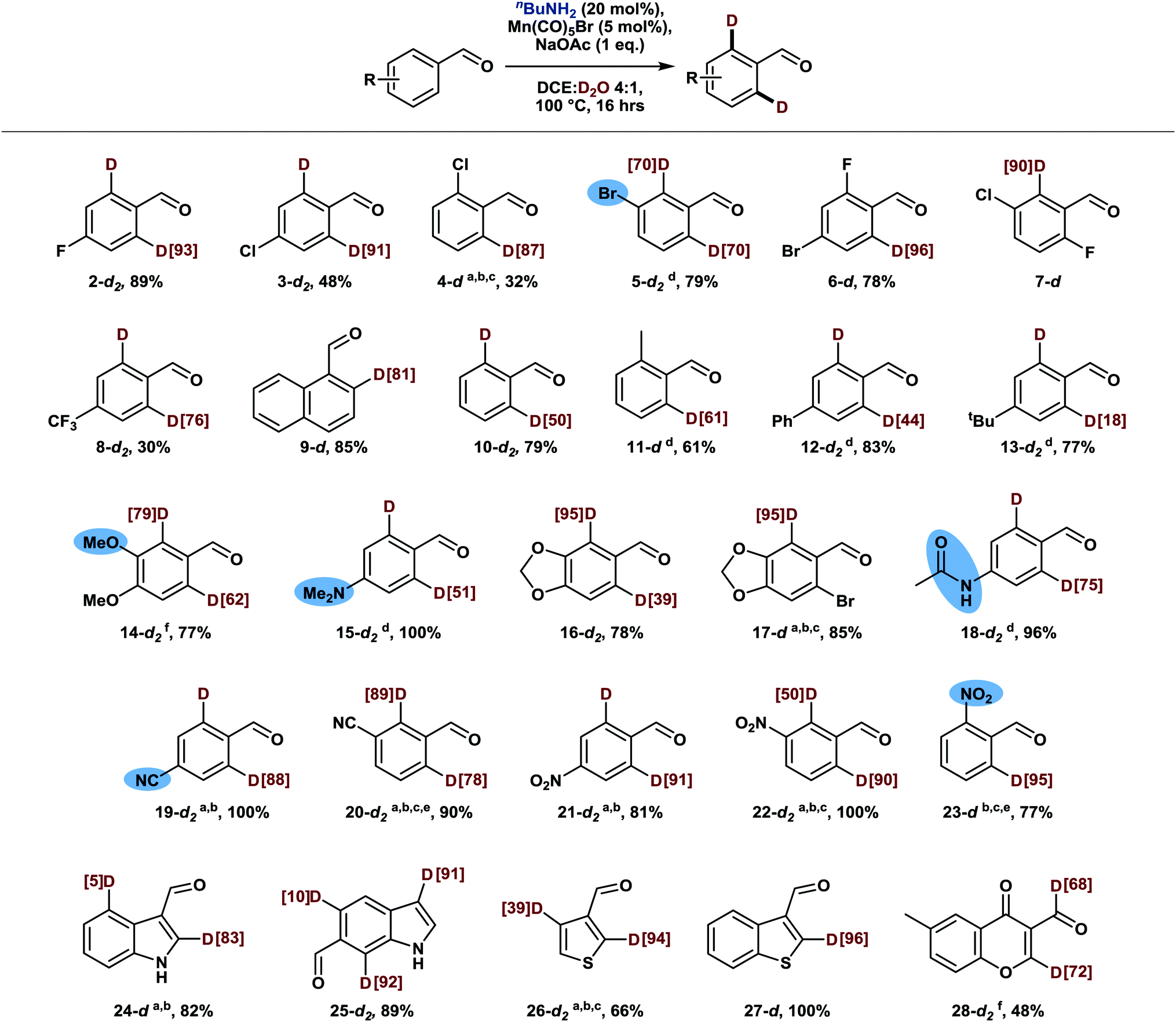 | ||
Scheme 3 Mn-catalyzed o-deuteration of various benzaldehyde derivatives. Scale: 0.5 mmol. Deuterium incorporation was determined by 1H NMR by referencing on the formyl proton and/or protons in m or p positions and is indicated in square brackets (red). Percentage values below the structures refer to isolated yields. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) BnNH2 instead of nBuNH2. b50 mol% p-chlorobenzoic acid instead of 1 eq. NaOAc. c2.5 mol% Mn(CO)5Br. d72 h. e8 h. f10 mol% Mn(CO)5Br. BnNH2 instead of nBuNH2. b50 mol% p-chlorobenzoic acid instead of 1 eq. NaOAc. c2.5 mol% Mn(CO)5Br. d72 h. e8 h. f10 mol% Mn(CO)5Br. | ||
Strongly electron-deficient benzaldehydes comprising nitro or cyano substituents (19–23) required slightly modified reaction conditions: due to the increased stability of the intermediary imine,15 a switch from base to acid catalysis facilitated turnover of the catalytic amine, thus yielding the corresponding aldehydes with high deuterium incorporation even at a low catalyst loading of 2.5 mol%. Similarly, good results were achieved with heterocyclic aldehydes (24–28), demonstrating the broad applicability of this methodology. Only substrates containing heteroatoms in α-position to the aldehyde moiety as well as pyridine derivatives appeared to form unreactive manganese complexes instead. Notably, chromone derivative 28 undergoes formyl deuteration additionally, presumably aided by coordination of the proximal keto group.
To test the functional group compatibility of this reaction beyond the groups already mentioned in the text and highlighted in Scheme 3, further deuteration experiments were conducted in the presence of equimolar amounts of a terminal olefin, a boronic acid, a sulfonamide, and phenol. All these additives were well-tolerated and did not affect the deuterium incorporation in the aldehyde substrate. On the other hand, hydroxy-substituted aldehydes as well as substrates comprising terminal alkynes were not deuterated.
From a synthetic point of view it is interesting that the reaction is selective for aldehydes, leaving aromatic ketones untouched despite their ability to form ketimines. Moreover, other carbonyl groups which had previously been employed as directing groups for manganese catalyzed C–H functionalization16 did not induce ortho-deuteration under our conditions, whereas heterocyclic directing groups could potentially enable additional deuteration patterns in more complex substrates.
While a variety of methods exists for the labeling of arenes (vide supra), the deuteration of olefinic substrates is much less developed.17 Notably, current methodologies for the labeling of α,β-unsaturated carbonyl compounds either exchange all olefinic protons alike or are slightly selective for the β position.17c,e,f Likewise, a previously reported manganese-catalyzed olefinic C–H activation also led to exclusive functionalization in β position.18 In this context, we explored the deuteration of α,β-unsaturated aldehydes and ketones. Indeed, as shown in Scheme 4, labelling proceeded smoothly with high selectivity for the α-position. While this work was in progress, an amine-mediated α-selective deuteration of α,β-unsaturated aldehydes and ketones was published.19 Accordingly, substrates 29–31 can be also deuterated in the absence of the manganese catalyst, whereby the amine acts as a nucleophilic catalyst rather than a transient directing group.
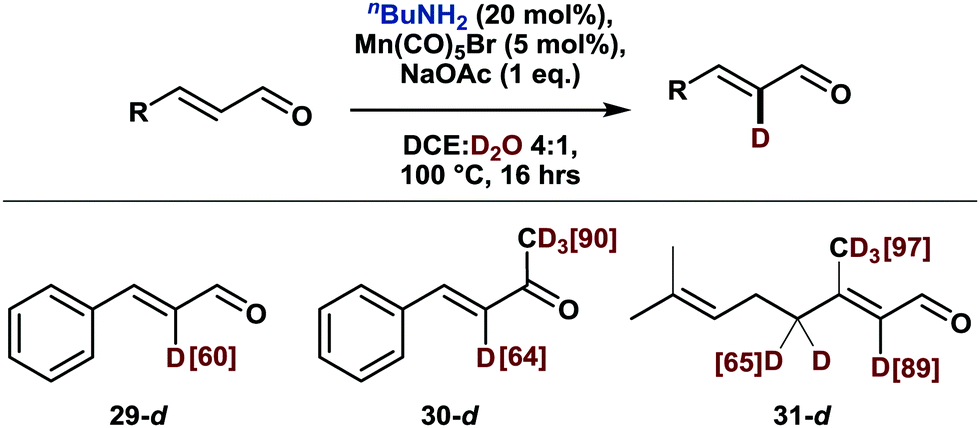 | ||
| Scheme 4 α-Deuteration of α,β-unsaturated aldehydes and ketones. Scale: 0.5 mmol. Deuterium incorporation was determined by 1H NMR. | ||
In conclusion, we describe a new base metal-catalyzed ortho-selective deuteration methodology using the most practical labelling reagent (D2O). This convenient alternative to current iridium-catalyzed directed hydrogen isotope exchange reactions shows broad scope of (hetero)aromatic aldehydes and versatile functional group compatibility. Moreover, we have introduced the transient directing group concept to the field of manganese-catalyzed C(sp2)–H activation which will undoubtedly improve the step economy of future transformations by alleviating the need for pre-installed directing groups.
We acknowledge the financial support from the European Union (Flow Chemistry for Isotope Exchange, FLIX, 862179), the BMBF and the State of Mecklenburg–Western Pommerania. We thank the analytical team of LIKAT for their kind support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) J. Atzrodt, V. Derdau, W. J. Kerr and M. Reid, Angew. Chem., Int. Ed., 2018, 57, 1758 CrossRef CAS; (b) J. Atzrodt, V. Derdau, W. J. Kerr and M. Reid, Angew. Chem., Int. Ed., 2018, 57, 3022 CrossRef CAS.
- (a) J. A. Brown, A. R. Cochrane, R. Irvine, W. J. Kerr, B. Mondal, J. A. Parkinson, L. C. Paterson, M. Reid, T. Tuttle, S. Andersson and G. N. Nilsson, Adv. Synth. Catal., 2014, 356, 3551 CrossRef CAS; (b) W. J. Kerr, M. Reid and T. Tuttle, ACS Catal., 2015, 5, 402 CrossRef CAS; (c) M. Valero, T. Kruissink, J. Blass, R. Weck, S. Güssregen, A. T. Plowright and V. Derdau, Angew. Chem., Int. Ed., 2020, 59, 5626 CrossRef CAS; (d) W. J. Kerr, G. J. Knox, M. Reid, T. Tuttle, J. Bergare and R. A. Bragg, ACS Catal., 2020, 10, 11120 CrossRef CAS.
- (a) R. P. Yu, D. Hesk, N. Rivera, I. Pelczer and P. J. Chirik, Nature, 2016, 529, 195 CrossRef; (b) J. Corpas, P. Viereck and P. J. Chirik, ACS Catal., 2020, 10, 8640 CrossRef CAS; (c) S. Garhwal, A. Kaushansky, N. Fridman, L. J. W. Shimon and G. de Ruiter, J. Am. Chem. Soc., 2020, 142, 17131 CrossRef CAS.
- (a) H. Yang, C. Zarate, W. N. Palmer, N. Rivera, D. Hesk and P. J. Chirik, ACS Catal., 2018, 8, 10210 CrossRef CAS; (b) C. Zarate, H. Yang, M. J. Bezdek, D. Hesk and P. J. Chirik, J. Am. Chem. Soc., 2019, 141, 5034 CrossRef CAS.
- J. Zhang, S. Zhang, T. Gogula and H. Zou, ACS Catal., 2020, 10, 7486 CrossRef CAS.
- (a) W. Liu and L. Ackermann, ACS Catal., 2016, 6, 3743 CrossRef CAS; (b) P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192 CrossRef CAS; (c) R. Cano, K. Mackey and G. P. McGlacken, Catal. Sci. Technol., 2018, 8, 1251 RSC.
- There is a report on manganese catalyzed aliphatic HIE, though: S. Kar, A. Goeppert, R. Sen, J. Kothandaraman and G. K. S. Prakash, Green Chem., 2018, 20, 2706 RSC.
- (a) B. Zhou, P. Ma, H. Chen and C. Wang, Chem. Commun., 2014, 50, 14558 RSC; (b) S. Wu, Q. Yang, Q. Hu, Y. Wang, L. Chen, H. Zhang, L. Wu and J. Li, Org. Chem. Front., 2018, 5, 2852 RSC; (c) C. Zhu, R. Kuniyil and L. Ackermann, Angew. Chem., Int. Ed., 2019, 58, 5338 CrossRef CAS.
- A few exceptions exist but need bimetallic C–H activation systems: (a) S. Sueki, Z. Wang and Y. Kuninobu, Org. Lett., 2016, 18, 304 CrossRef CAS; (b) B. Zhou, Y. Hu, T. Liu and C. Wang, Nat. Commun., 2017, 8, 1169 CrossRef; (c) Y. Hu, B. Zhou, H. Chen and C. Wang, Angew. Chem., Int. Ed., 2018, 57, 12071 CrossRef CAS; (d) X. Kong and B. Xu, Org. Lett., 2018, 20, 4495 CrossRef CAS; (e) S. Ali, J. Huo and C. Wang, Org. Lett., 2019, 21, 6961 CrossRef CAS.
- W. Liu, S. C. Richter, Y. Zhang and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 7747 CrossRef CAS.
- (a) P. Gandeepan and L. Ackermann, Chemistry, 2018, 4, 199 CrossRef CAS; (b) S. John-Campbell and J. A. Bull, Org. Biomol. Chem., 2018, 16, 4582 RSC; (c) B. Niu, K. Yang, B. Lawrence and H. Ge, ChemSusChem, 2019, 12, 2955 CrossRef CAS; (d) J. I. Higham and J. A. Bull, Org. Biomol. Chem., 2020, 18, 7291 RSC.
- Catalyst loadings of 10 mol% are most common for manganese-catalyzed C–H activation procedures. Cf. ref. 7, 8b, e.
- Recent examples of deuterodehalogenation: (a) Z.-Z. Zhou, J.-H. Zhao, X.-Y. Guo, X.-M. Chen and Y.-M. Liang, Org. Chem. Front., 2019, 6, 1649 RSC; (b) J. J. Gair, R. L. Grey, S. Giroux and M. A. Brodney, Org. Lett., 2019, 21, 2482 CrossRef CAS; (c) M. Kuriyama, G. Yano, H. Kiba, T. Morimoto, K. Yamamoto, Y. Demizu and O. Onomura, Org. Process Res. Dev., 2019, 23, 1552 CrossRef CAS.
- This effect has previously been observed in manganese catalyzed C–H activation: cf.ref. 9 and D. Zell, U. Dhawa, V. Müller, M. Bursch, S. Grimme and L. Ackermann, ACS Catal., 2017, 7, 4209 CrossRef CAS.
- R. W. Layer, Chem. Rev., 1963, 63, 489 CrossRef CAS.
- T. S. Amide, T. Yoshida, H. H. A. Mamari, L. Ilies and E. Nakamura, Org. Lett., 2017, 19, 5458 CrossRef ; ester: ref. 8a.
- Examples for olefinic deuteration: (a) S. K. S. Tse, P. Xue, Z. Lin and G. Jia, Adv. Synth. Catal., 2010, 352, 1512 CrossRef CAS; (b) A. Di Giuseppe, R. Castarlenas, J. J. Pérez-Torrente, F. L. Lahoz, V. Polo and L. A. Oro, Angew. Chem., Int. Ed., 2011, 50, 3938 CrossRef CAS; (c) W. J. Kerr, R. J. Mudd, L. C. Paterson and J. A. Brown, Chem. – Eur. J., 2014, 20, 14604 CrossRef CAS; (d) M. Hatano, T. Nishimura and H. Yorimitsu, Org. Lett., 2016, 18, 3674 CrossRef CAS; (e) A. L. Garreau, H. Zhou and M. C. Young, Org. Lett., 2019, 21, 7044 CrossRef CAS; (f) A. Bechtold and L. Ackermann, ChemCatChem, 2019, 11, 435 CrossRef.
- B. Zhou, Y. Hu and C. Wang, Angew. Chem., Int. Ed., 2015, 54, 13659 CrossRef CAS.
- V. G. Landge, K. K. Shrestha, A. J. Grant and M. C. Young, Org. Lett., 2020, 22, 9745 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc07675a |
| This journal is © The Royal Society of Chemistry 2021 |