
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
pH-Dependent peptide bond formation by the selective coupling of α-amino acids in water†
Long-Fei
Wu
 *,
Ziwei
Liu
and
John D.
Sutherland
*
*,
Ziwei
Liu
and
John D.
Sutherland
*
MRC Laboratory of Molecular Biology, Cambridge Biomedical Campus, Cambridge, CB2 0QH, UK. E-mail: lfwu@mrc-lmb.cam.ac.uk; johns@mrc-lmb.cam.ac.uk
First published on 20th November 2020
Abstract
A novel mechanism enabling selective peptide elongation by coupling α-amino acids over other potentially competing prebiotic amines under acidic aqueous condition is suggested. It proceeds via the generation of a carboxylic acid anhydride intermediate with subsequent intramolecular formation of the amide bond.
The widespread importance of the synthesis of amides lies not only in synthetic organic chemistry,1–3 but also in long-standing efforts aimed at understanding the formation of peptides composed of α-amino acids in the origin of life.4–7 Although the “RNA world” hypothesis is a popular working model for studying the origin of life,8,9 peptides could have coexisted with RNA at the outset of life. This is suggested by the fact that α-amino acids and nucleotide building blocks might have had a common chemical origin.10–12 We have previously shown that the chemical energy inherent in methyl isonitrile 1 – a potentially prebiotic activating reagent13 – can be harnessed to activate and join phosphate and carboxylate building blocks simultaneously under acidic aqueous conditions.14 We have now investigated peptide formation chemistry mediated by methyl isonitrile 1 in a systems chemistry scenario. We considered various potentially prebiotic amine nucleophiles which could have been present together in a common pool on early Earth. We found that amide bond formation shows very limited selectivity for all amines examined at pH 5, regardless of the fact that the pKa values of their conjugate acids differ by up to 5.4 units. When the pH of the reactions was lowered to 3, however, α-amino acids had the highest reactivity amongst all the tested substrates, suggesting a unique mechanism and a potential means of selecting α-amino acids for peptide elongation.
A prebiotically plausible mixture would most likely have contained a range of different amine nucleophiles, regardless of whether it was produced endogenously15,16 or exogenously.17,18 Thus, a model peptide, N-acetyl L-alanine 2 (Ac-Ala, 100 mM), methyl isonitrile 1 (∼130 mM) and one of the different amine nucleophiles 3 (50 mM, the concentration of 2 was higher than that of the amine nucleophiles to minimise multiple elongation) were mixed in water and left to incubate at pH 3, 4 or 5 (Fig. 1A). The reaction mixture was monitored by 1H-NMR spectroscopy at 23 °C until most of the methyl isonitrile has been consumed. After 15 days at pH 5, 62% of glycine 3a (pKaH = 9.6) was incorporated into amide products, Ac-Ala-Gly 4a and Ac-Ala-Gly-Gly 4c (Fig. 1B and Fig. S1, ESI†). Using β-alanine 3b (β-Ala, pKaH = 10.2), glycylglycine 3c (Gly-Gly, pKaH = 8.1), glycinamide 3d (pKaH = 8.2), glycine nitrile 3e (pKaH = 5.2) and methylamine 3f (pKaH = 10.6), we observed reaction with Ac-Ala 2 to give the elongated amide products, Ac-Ala-β-Ala 4b, Ac-Ala-Gly-Gly 4c (and minor amount of Ac-Ala-Gly-Gly-Gly-Gly), Ac-Ala-Gly-NH24d, Ac-Ala-CN 4e, and Ac-Ala-NHCH3 4f, respectively, in comparable yield to the combined yield of products from 3a (as determined by 1H-NMR spectroscopy, Fig. 1B and Fig. S2–S6, ESI†). However, Ac-Ala-NH24g was only formed in 17% yield when ammonia 3g (pKaH = 9.3) was used as the nucleophile (Fig. S7, ESI†), this is consistent with the lower nucleophilicity of ammonia compared to some amines with similar pKaH values in water.19,20 At pH 4, the rate of reaction was significantly higher than it was at pH 5 with consumption of methyl isonitrile 1 in 48 hours.14,21,22 The yield of incorporation of glycine 3a into peptide product 4a was only slightly lower (58%) than the analogous reaction run at pH 5 (Fig. 1B and Fig. S8, ESI†), and the yield of 4b decreased to 47% when using 3b (Fig. 1B and Fig. S9, ESI†). However, the conversion of 3c or 3d proceeded in significantly lower yields (approximately 20%, Fig. 1B and Fig. S10, S11, ESI†), and no reaction of 3f and 3g with 2 was detected by 1H-NMR spectroscopy (Fig. 1B and Fig. S12, S13, ESI†). In contrast, quantitative conversion to 4e was obtained when glycine nitrile 3e was used as the nucleophile (Fig. 1B and Fig. S14, ESI†). We argued that it might be related to more of the unprotonated form of glycine nitrile being available because of its low pKaH.6 When reactions were run at pH 3, methyl isonitrile was consumed in only 6 hours. However, only glycine 3a, β-alanine 3b, glycine nitrile 3d underwent coupling with 2 to give 4a, 4b and 4d in yields of 46%, 5% and 30%, respectively (Fig. 1B and Fig. S15, 17, ESI†). The best yield when using glycine 3a as amine nucleophile suggests there must be a unique mechanism related to glycine 3a in amide bond formation mediated by methyl isonitirle 1 in low pH solution. To further evaluate if this behaviour also applied to other α-amino acids, we tested arginine, serine, valine and proline with Ac-Ala 2 at pH 4. Yields of incorporation of these amino acids into peptides ranging from 44% to 66% (comparable yields to glycine 3a) were observed in 48 hours as expected (Fig. S18–S21, ESI†).
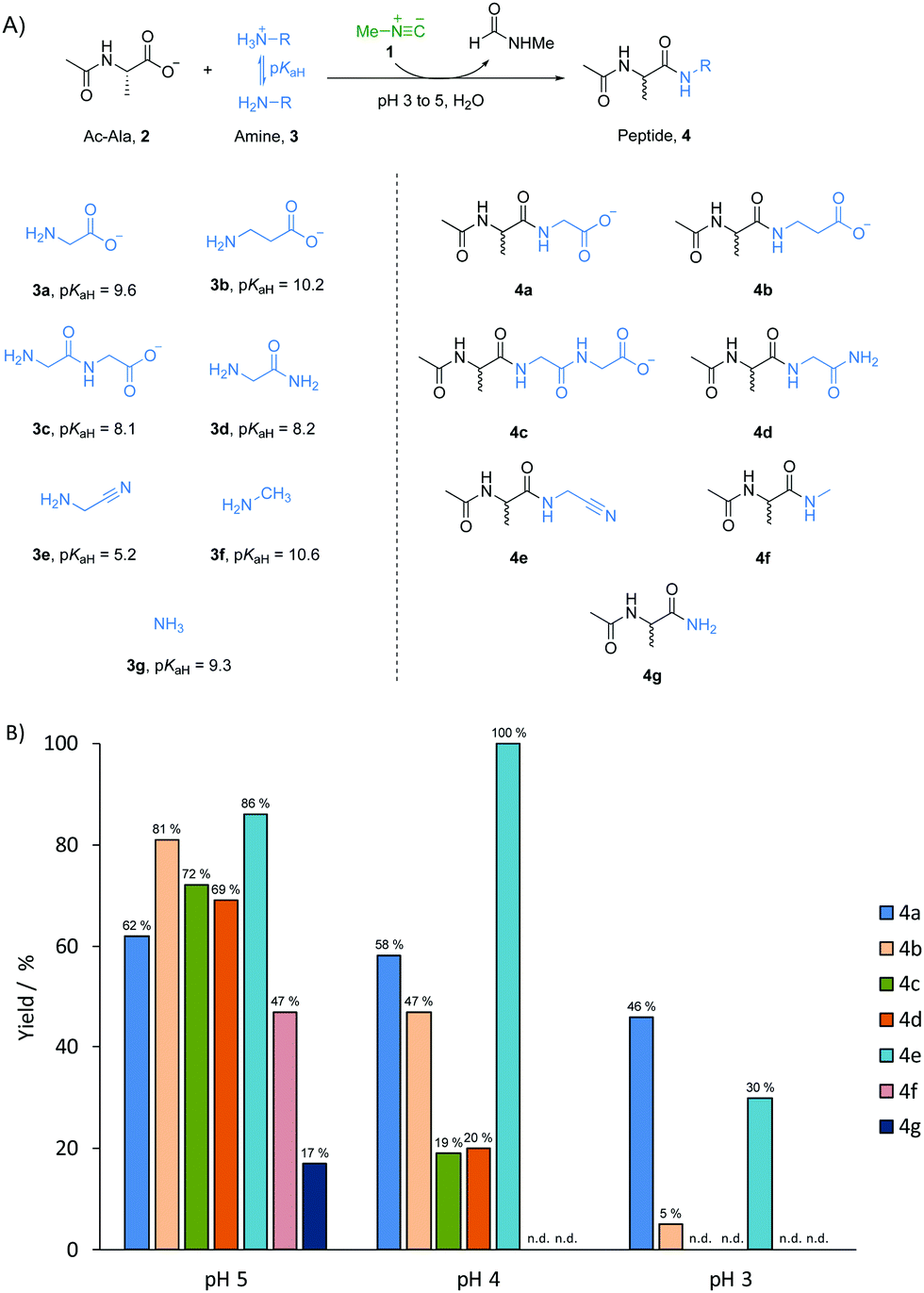 | ||
| Fig. 1 (A) Formation of amide bond mediated by methyl isonitrile with different amine nucleophiles in water at acidic pH values. (B) Yields based on the incorporation of the amine nucleophiles 3 into peptide products 4 at pH 5, 4 and 3 after 15 days, 48 hours and 6 hours, respectively. Reactions were run in duplicate and average yields are given. n.d.: amide product not detected. | ||
Accordingly, we wondered if the selectivity we had observed could play out in the sort of mixed system that might have existed on early Earth. A reaction mixture with Ac-Ala 2 (100 mM), glycine 3a (25 mM), β-alanine 3b (25 mM), glycylglycine 3c (25 mM), glycinamide 3d (25 mM), glycinenitrile 3e (25 mM), methylamine 3f (25 mM), ammonia 3g (25 mM) and methyl isonitrile 1 (∼130 mM) was incubated at pH 3 and monitored by 1H-NMR spectroscopy at 23 °C (Fig. 2A). Peptides 4a and 4e were the two major products with yields of 55% and 42%, respectively, based on 1H-NMR spectroscopy (Fig. S12, ESI†), and this is in line with the results of the separate experiments described above. Interestingly, 4b was not observed by 1H-NMR spectroscopy (Fig. S18, ESI†), which suggests that the reactivity of 3b was suppressed under a competing scenario.
Considering that progressive peptide elongation is needed to make potentially functional peptides, a reaction with Ac-Ala 2 (20 mM), glycine 3a (100 mM), and methyl isonitrile 1 (∼110 mM) was incubated at pH 3, 4 or 5 to evaluate multiple rounds of peptide elongation (Fig. 2B, other amine nucleophiles were excluded to simplify interpretation of NMR spectra). At pH 3, dipeptide Ac-Ala-Gly 4a and tripeptide Ac-Ala-Gly-Gly 4c were observed by 1H-NMR spectroscopy in yields of 46% and 14%, respectively, after 6 hours (Fig. S23, ESI†), which clearly showed that progressive peptide elongation can be achieved under α-amino acid-selective conditions. At pH 4, the yields of 4a and 4c were 54% and 24%, respectively, after 48 hours (Fig. S24, ESI†). Longer peptides could be observed in minor amounts based on 1H-NMR spectroscopy. At pH 5, after 15 days, the yields of 4a and 4c were 34% and 24% respectively, and tetrapeptide Ac-Ala-Gly-Gly-Gly 4h was also observed in a yield of 13% (Fig. 2B and Fig. S25, ESI†). Although higher yields and longer peptides could be achieved at pH 5, other amine nucleophiles would start to compete with α-amino acids to form amide bonds as shown in Fig. 1.
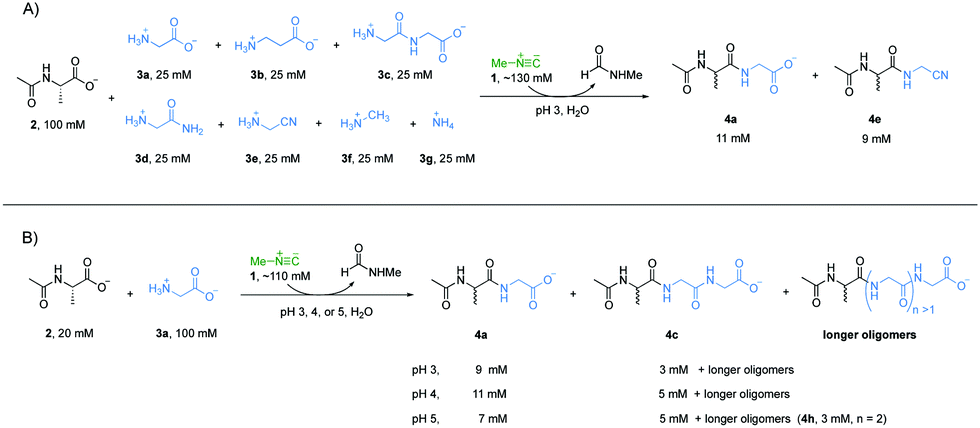 | ||
| Fig. 2 (A) Model peptide elongation by coupling a mixture of various amine nucleophiles at pH 3 mediated by methyl isonitrile 1. Concentrations of the two dominating products 4a and 4d were calculated based on proton signal integration on 1H-NMR spectroscopy. (B) Multiple rounds of peptide elongation by coupling an α-amino acid with model peptide 2 mediated by methyl isonitrile at pH 3, 4 or 5. Concentrations of the products were calculated based on proton signal integration on 1H-NMR spectroscopy. | ||
Next, we investigated the underlying mechanism of the unique property of α-amino acids in the amide bond formation chemistry mediated by methyl isonitrile 1. Considering the structural features of all the amine nucleophiles, 3a to 3g, we reasoned that the carboxylate group of glycine 3a might be implicated in the special reactivity of α-amino acids. We suggest that the carboxylate group of Ac-Ala 2 was activated by methyl isonitrile 1 to give intermediate imidoyl acetyl alanine 5via acid catalysis.14 Imidoyl acetyl alanine 5 can give oxazolone 6via intramolecular cyclization.14,23,24 Both 5 and 6 can be attacked by free amines, if they are sufficiently abundant, to give the amide products intermolecularly (Fig. 3A, direct pathway). Under more acidic condition, more protonation of the amines will inhibit their reactivities as nucleophiles. However, we assumed that the carboxylate/carboxylic acid group of glycine 3a (pKaH = 2.3) also acts as a nucleophile to attack intermediates 5 and/or 6 to give mixed anhydride 7. 7 could afford the desired peptide elongation product 4a by an O → N acyl transfer intramolecularly via a 5-membered ring transition state (Fig. 3A, indirect pathway). The pKaH of 7 is presumably lower than that of glycine 3a and this coupled with the lower entropic penalty of an intramolecular reaction25 is why this reaction can occur whilst direct intermolecular amide formation cannot (e.g.3aversus3d in amide bond formation at pH 3 in Fig. 1). To test this hypothesis, we devised an experiment to probe the putative formation of mixed anhydride intermediate 7a. Ac-Ala 2 (100 mM), methyl isonitrile 1 (∼130 mM) and glycolic acid 8 (50 mM) were mixed at pH 3 or 4, respectively, and the reactions were monitored by 1H-NMR spectroscopy at 23 °C (Fig. 3B). The ester product 9 was observed in good yields at both pH values (Fig. S26 and S27, ESI†). In a negative control reaction, Ac-Ala 2 (100 mM), methyl isonitrile 1 (∼130 mM) and methyl glycolate 11 (50 mM) were mixed at pH 4 (Fig. 3B), but ester 12 was not observed by 1H-NMR spectroscopy after all the methyl isonitrile had been consumed (Fig. S28, ESI†). These results strongly suggest that the formation of ester 9 proceeds via the generation of mixed anhydride 10 followed by an O → O acyl transfer. Taken together, these results are an indication that under the most acidic conditions amide 4a is still able to form in good yield because the reaction proceeds through the mixed anhydride intermediate 7a followed by an O → N acyl transfer (Fig. 3A, indirect pathway). A similar mechanism would be also expected to apply to β-alanine 3b, but the lesser amount of carboxylate anion of β-alanine 3b relative to that of glycine 3a because of the higher pKa of the carboxylic acid group of 3b (pKa = 3.6) compared to 3a (pKa = 2.3), the greater degree of protonation of the amino group of β-alanine because of its higher pKaH than glycine 3a, and different ring-size of the transition states of intramolecular acyl transfer (6-membered ring versus 5-membered ring) might contribute to its lower yield of amide product at pH 3 (Fig. 1B). It is worth mentioning that side products were observed (Fig. S1, S8 and S15, ESI†), and were assigned as cis- and trans-N-imidoyl glycine, 13-cis and 13-trans, based on NMR spectroscopy (Fig. 3C and Fig. S29–S33, ESI†). We suggest that the formation of 13-cis and 13-trans is due to the imidoylation of the amino group presumably via intermediate 14 followed by an O → N imidoyl transfer. 13 can hydrolyse to give N-formyl glycine.26 And N-formyl alanine was also shown to elongate effectively by coupling glycine 3a to give N-formylated peptides (Fig. S34, ESI†). These products are reminiscent of the formylated N-terminal residue in extant protein biosynthesis.
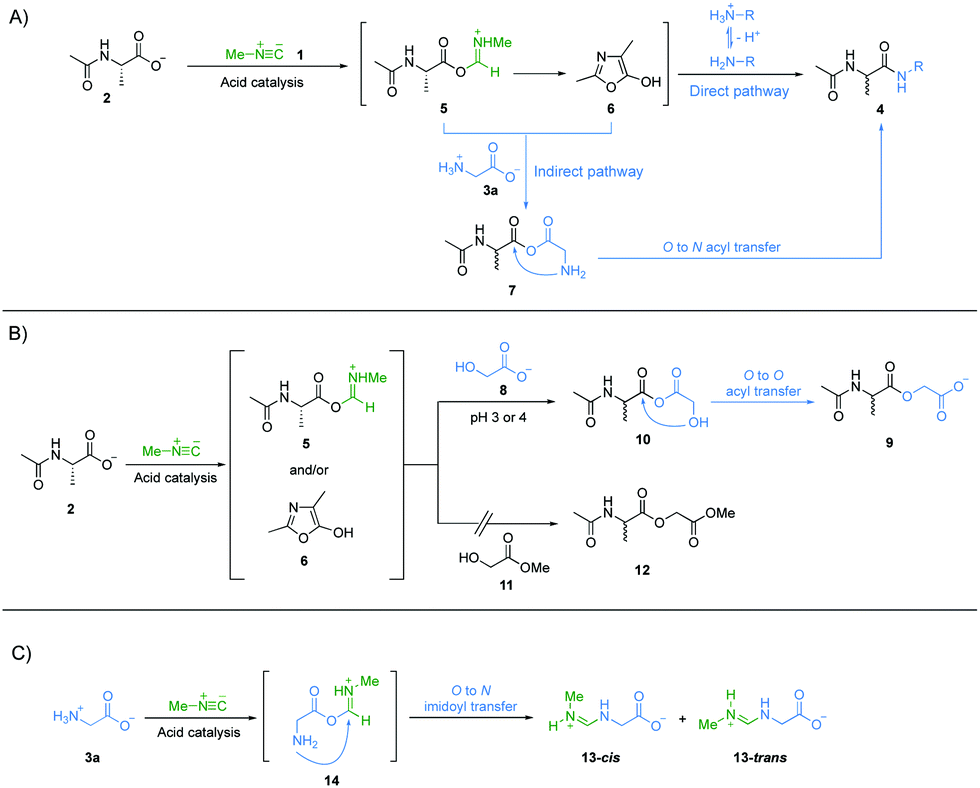 | ||
| Fig. 3 (A) Proposed mechanistic scheme for methyl isonitrile mediated model peptide elongation by coupling α-amino acids under acidic conditions. The direct pathway represents the mechanism of direct amide bond formation by attacking of the amine nucleophile on intermediates 5 or 6 intermolecularly, and the indirect pathway represents the mechanism of amide bond formation intramolecularly following the mixed anhydride intermediate 7. (B) Supporting evidence for the indirect pathway proposed in (A). (C) Possible mechanism for the formation of side product N-imidoyl glycine, 13-cis and 13-trans. | ||
Prebiotic chemistry is most likely to give a mixed pool of various plausible species on early Earth.10–12,15–18 Exploring whether there is selectivity among those mixed species in the process of forming oligomers is of great importance to understand the transition from prebiotic chemistry to biochemistry. Poor selectivity among various amine nucleophiles was observed when amide bond formation was mediated by methyl isonitrile 1 at only slightly acidic aqueous solution (e.g. pH 5). Under conditions of pH 6 or above, peptide elongation will be very slow because of a lack of acid catalysis. But, a unique chemical property inherent to α-amino acids could enable the incorporation of α-amino acids selectively into peptides at pH 3 in the presence of other amine nucleophiles. Also, progressive peptide elongation by incorporating α-amino acids can still be achieved in the same set of conditions. A mixed carboxylic acid anhydride intermediate (e.g.7a, Fig. 3A) was suggested to be the reason contributing to the highest activity of α-amino acids at pH 3. As α-aminonitriles are easily hydrolysed to α-aminoamides (e.g. by organocatalysis by aldehydes27), then to α-aminoacids on a longer time scale (geologically plausible), one can imagine a prebiotic scenario in which α-amino acids existed alongside various other amines, but in the absence of α-aminonitriles. In such a scenario, peptides comprising only α-amino acids could have been produced at low pH by the chemistry described herein. Alternatively, if the hydrolysis of α-aminonitriles to α-amino acids was incomplete, peptides based on α-amino acids could still have been produced, but they would occasionally be terminated by α-aminonitriles. Interestingly, Powner and co-workers recently described a three-steps process in coupling each α-aminonitrile as α-amino acid equivalent in aqueous solution.6 The selective coupling of α-aminonitrile in their work resulted from the corresponding much lower pKaH values of α-aminonitrile than other amino nucleophiles, which was also partially exemplified here. Hud and co-workers have explored the ester product 9 analogues as key intermediates in amide bond formation driven by wet-dry cycles without using activating reagent.28 We anticipate that, in solution phase as described here, if the pH of the solution were brought up by environmental fluctuation, amines might react with ester 9 slowly and low selectivity would be expected.29 Regarding the low pH conditions, although models predict that the pH of the ocean of early Earth was near neutral,30,31 regional lakes/ponds with high acidity would have been possible.32,33
This work was supported by the Medical Research Council (no. MC_UP_A024_1009 to J. D. S.) and the Simons Foundation (no. 290362 to J. D. S.). The authors thank all J. D. S. group members for fruitful discussion.
Conflicts of interest
There are no conflicts to declare.Notes and references
- A. El-Faham and F. Albericio, Chem. Rev., 2011, 111, 6557–6602 CrossRef CAS.
- R. M. de Figueiredo, J.-S. Supp and J.-M. Campagne, Chem. Rev., 2016, 116, 12029–12122 CrossRef CAS.
- M. T. Sabatini, L. T. Boulton, H. F. Sneddon and T. D. Sheppard, Nat. Catal., 2019, 2, 10–17 CrossRef CAS.
- G. Danger, R. Plasson and R. Pascal, Chem. Soc. Rev., 2012, 41, 5416–5429 RSC.
- M. Frenkel-Pinter, M. Samanta, G. Ashkenasy and L. Leman, Chem. Rev., 2020, 120, 4707–4765 CrossRef CAS.
- P. Canavelli, S. Islam and M. Powner, Nature, 2019, 571, 546–549 CrossRef CAS.
- L. Leman, L. E. Orgel and M. R. Ghadiri, Science, 2004, 306, 283–286 CrossRef CAS.
- L. E. Orgel, Crit. Rev. Biochem. Mol. Biol., 2004, 39, 99–123 CrossRef CAS.
- G. F. Joyce and J. W. Szostak, Cold Spring Harbor Perspect. Biol., 2018, 10, a034801 CrossRef.
- A. Eschenmoser, Tetrahedron, 2007, 63, 12821–12844 CrossRef CAS.
- J. D. Sutherland, Angew. Chem., Int. Ed., 2016, 55, 104–121 CrossRef CAS.
- L.-F. Wu and J. D. Sutherland, Emerging Top. Life Sci., 2019, 3, 459–468 CrossRef CAS.
- A. Mariani, D. A. Russell, T. Javelle and J. D. Sutherland, J. Am. Chem. Soc., 2018, 140, 8657–8661 CrossRef CAS.
- Z. Liu, L.-F. Wu, J. Xu, C. Bonfio, D. A. Russell and J. D. Sutherland, Nat. Chem., 2020, 12, 1023–1028 CrossRef.
- S. L. Miller and L. E. Orgel, The Origin of Life on the Earth, Prentice-Hall, 1974 Search PubMed.
- B. H. Patel, C. Percivalle, D. Riston, C. D. Duffy and J. D. Sutherland, Nat. Chem., 2015, 7, 301–307 CrossRef CAS.
- C. Chyba and C. Sagan, Nature, 1992, 355, 125–132 CrossRef CAS.
- S. Pizzarello, Acc. Chem. Res., 2006, 39, 231–237 CrossRef CAS.
- F. Brotzel, Y. C. Chu and H. Mayr, J. Org. Chem., 2007, 72, 3679–3688 CrossRef CAS.
- F. Brotzel and H. Mayr, Org. Biochem. Chem., 2007, 5, 3814–3820 RSC.
- K. Sung and C.-C. Chen, Tetrahedron Lett., 2001, 42, 4845–4848 CrossRef CAS.
- Y.-Y. Lim and A. R. Stein, Can. J. Chem., 1971, 49, 2455–2459 CrossRef.
- G. Danger, A. Michaut, M. Bucchi, L. Boiteau, J. Canal, R. Plasson and R. Pascal, Angew. Chem., Int. Ed., 2013, 52, 611–614 CrossRef CAS.
- Z. Liu, D. Beaufils, J.-C. Rossi and R. Pascal, Sci. Rep., 2014, 4, 7440 CrossRef CAS.
- A. Kirby, J. Adv. Phys. Org. Chem., 1980, 17, 183 CAS.
- S. Vincent, C. Mioskowski and L. Lebeau, J. Org. Chem., 1999, 64, 99–997 Search PubMed.
- J. Taillades, I. Beuzelin, G. Laurence, V. Tabacik, C. Bied and A. Commeyras, Origins Life Evol. Biospheres, 1998, 28, 61–77 CrossRef CAS.
- J. G. Forsythe, S.-S. Yu, I. Mamajanov, M. A. Grover, R. Krishnamurthy, F. M. Fernández and N. V. Hud, Angew. Chem., Int. Ed., 2015, 54, 9871–9875 CrossRef CAS.
- A. C. Satterthwait and W. P. Jencks, J. Am. Chem. Soc., 1974, 96, 7018–7031 CrossRef CAS.
- I. Halevy and A. Bachan, Science, 2017, 355, 1069–1071 CrossRef CAS.
- J. Krissansen-Totton, G. N. Arney and D. C. Catling, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 4015–4110 CrossRef.
- R. W. Renaut and B. Jones, in Encyclopedia of Geobiology, ed. J. Reitner and V. Thiel, Springer, Berlin, Dordrecht. 2011, pp. 467–479 Search PubMed.
- R. W. Henley, in Volcanic Lakes, ed. D. Rouwet, B. Christenson, F. Tassi and J. Vandemeulebrouck, Springer, Berlin, Heidelberg, 2015, pp. 155–178 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc06042a |
| This journal is © The Royal Society of Chemistry 2021 |