
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cu(III)–bis-thiolato complex forms an unusual mono-thiolato Cu(III)–peroxido adduct†
Jane M.
Donnelly
 a,
Frederik
Lermyte
b,
Juliusz A.
Wolny
c,
Marc
Walker
d,
Ben G.
Breeze
d,
Russell J.
Needham
e,
Christina S.
Müller
c,
Peter B.
O’Connor
e,
Volker
Schünemann
c,
Joanna F.
Collingwood
a and
Peter J.
Sadler
*e
a,
Frederik
Lermyte
b,
Juliusz A.
Wolny
c,
Marc
Walker
d,
Ben G.
Breeze
d,
Russell J.
Needham
e,
Christina S.
Müller
c,
Peter B.
O’Connor
e,
Volker
Schünemann
c,
Joanna F.
Collingwood
a and
Peter J.
Sadler
*e
aSchool of Engineering, University of Warwick, Gibbet Hill Road, Coventry CV4 7Al, UK
bDepartment of Chemistry, Technical University of Darmstadt, Alarich-Weiss-Strasse 4, 64287 Darmstadt, Germany
cDepartment of Physics, Technische Universität Kaiserslautern, Erwin-Schrödinger-Straße 46, 67663 Kaiserslautern, Germany
dDepartment of Physics, University of Warwick, Gibbet Hill Road, Coventry CV4 7Al, UK
eDepartment of Chemistry, University of Warwick, Gibbet Hill Road, Coventry CV4 7AL, UK. E-mail: P.J.Sadler@warwick.ac.uk
First published on 18th December 2020
Abstract
The stable complex [bis(toluene-3,4-dithiolato)copper(III)][NEt3H] has been synthesised and characterised as a square-planar Cu(III) complex by X-ray photoelectron spectroscopy, cyclic voltammetry and DFT calculations. Intriguingly, when fragmented in FTICR-MS, an unusual [(toluene-3,4-dithiolate)Cu(III)(peroxide)]− complex is formed by reaction with oxygen. Natural 1,2-dithiolenes known to bind molybdenum might stabilise Cu(III) in vivo.
Cuproenzymes play important roles in regulating oxygen species in both mammalian and microbial biochemistry, and contribute to innumerable biological processes such as immune regulation, mitochondrial function, and neurotransmission.1 The dysregulation of such reactivity can result in oxidative damage that characterises multiple disease pathologies. In the study of biological Cu–oxygen reactivity, the Cu(III) oxidation state remains an important yet elusive target of study. Although there is no direct experimental evidence for the existence of the Cu(III) state in biological systems, there are numerous computationally- and synthetically-based proposed Cu(III) intermediate states in monooxygenases and oxidases, two categories of enzymes that participate in some of the most critical biological redox processes.2,3
Mononuclear dopamine-β-hydroxylase (DβH) and peptidylglycine α-hydroxylating monooxygenase (PHM) are involved in generating the critical neurotransmitter norepinephrine and bioactive C-terminal amidated peptides, respectively.4 The binuclear Cu enzyme tyrosinase is responsible for the hydroxylation that converts L-tyrosine to L-DOPA, which has been proposed to proceed through a Cu(III) intermediate.2 Tyrosinase has recently gained increasing interest, due to its role in neurodegeneration and other critical neurochemistry.5 Given the ubiquity and essential functions of oxidative cuproenzymes, it is important to gain a fuller understanding of their chemistry, including possible Cu(III) intermediate states.
Due to the transient nature of Cu(III) intermediates, they are difficult to characterise directly in biological samples.2 Consequently, biologically plausible synthetic Cu(III) complexes have produced the most information about the ability of Cu(III) to bind and activate O2, forming Cu(III)–peroxide adducts.1,2,6 Very few examples of mononuclear Cu(III)–peroxides have been reported and interest remains in synthesizing mononuclear Cu(III)–peroxide complexes capable of C–H or O–H bond activation.4,7,8
Sometimes metal–O2 adducts can be generated in the gas phase using mass spectrometry (MS). Examples include bipyridyl complexes of Co, Ni, Cr, Ru, and Os, and carbonyl complexes of Fe.9,10 However, there has been no report of gas-phase Cu reactivity with molecular O2.
Here we report the first observation of the formation of a gas-phase Cu–O2 adduct. For this, we synthesized a Cu(III)–thiolate complex with a bis(toluene-3,4-dithiolate) ligand system. Thiolate coordination is well documented in many Cu chaperone proteins, and square planar dithiolate coordination, as observed here, is characteristic of the molybdenum cofactor Moco.11,12
The synthesis of [bis(toluene-3,4-dithiolato)copper(III)][NEt3H] (NEt3H·1) was based on reported procedures for similar ligand systems.13,14 In the present case it was carried out in air, with O2 present as a likely electron acceptor.13,14 The formation of 1 was confirmed by 1H NMR, COSY, NOESY, CHN elemental analysis, and ultrahigh resolution (UHR)-MS (m/z = 370.91233) (Fig. S2–S6 and Table S1, ESI†). Negative ion mode UHR-MS indicates that 1 (MW 370.91) has an overall charge of −1, indicating a Cu(III) centre bound to two deprotonated dithiolato ligands. The sharp 1H NMR peaks are consistent with complex 1 being diamagnetic, containing low-spin d8 Cu(III). Cyclic voltammetry of NEt3H·1 in MeCN showed two irreversible redox processes, and a low positive reduction potential for the Cu(III)–Cu(II) redox couple (Ered = 0.437 V vs. NHE) (Fig. S7, ESI†).
X-ray photoelectron spectroscopy (XPS) analysis of the Cu 2p3/2 region further confirmed the Cu(III) oxidation state, as the feature at 934.9 eV falls within the previously established range of 934–935 eV for the Cu 2p3/2 feature of Cu(III) complexes (Fig. 1 and Fig. S9, ESI†).15–17 Correspondingly, the features at 163.9 eV and 164.9 eV are assigned to the S 2p3/2 and S 2p1/2 of NEt3H·1, respectively.
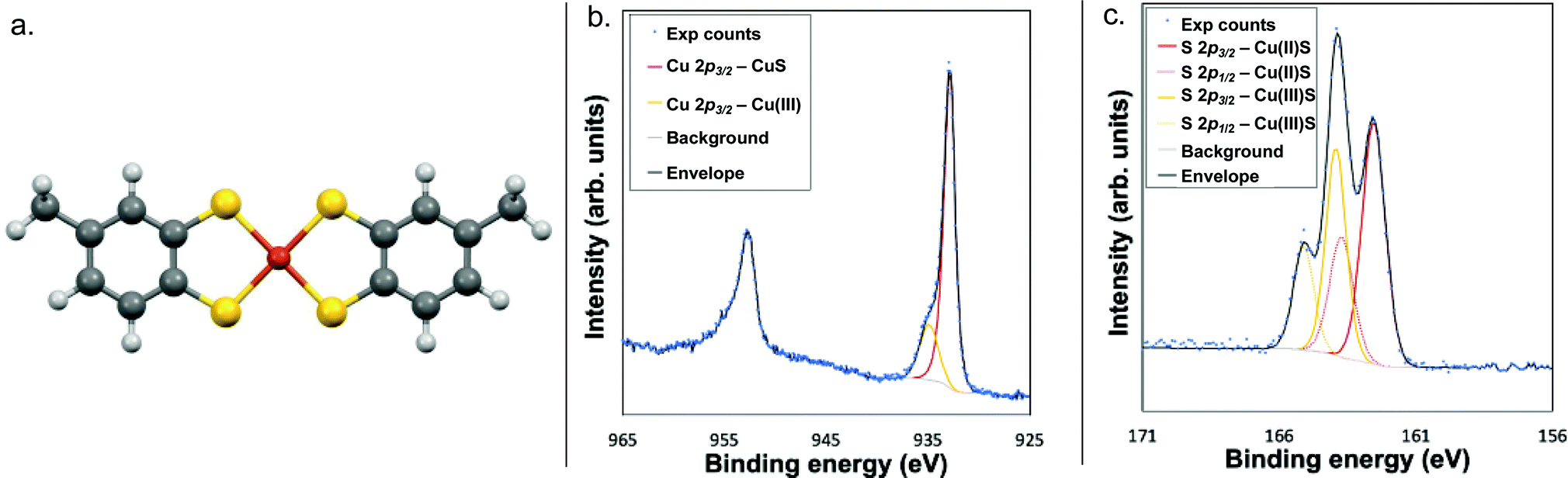 | ||
| Fig. 1 (a) DFT-calculated structure of ion 1; the Cu–S bond length is 2.208 Å. (b) Copper XPS spectrum, and (c) sulphur XPS spectrum of compound NEt3H·1. | ||
Additional features in both the Cu and S spectra indicate the presence of a separate Cu- and S-containing impurity, which was assigned as a Cu(II) analogue of NEt3H·1 with radical character on the ligand. The lack of significant shake-up features in the Cu 2p region and lack of signal around 530 eV in the O 1s region ruled out any copper oxides or hydroxides, while the feature at 932.8 eV in the Cu 2p3/2 region matches that previously reported for Cu(II)–S complexes (Fig. 1).18–21 This finding was corroborated by the existence of a S 2p doublet at 162.6 eV and 163.7 eV characteristic of Cu(II)–S complexes.18–21
The presence of a Cu(II) trace compound was further confirmed by a signal in the powder EPR spectrum with axial four-line splitting characteristic of Cu(II) and a sharp radical signal suggesting significant organic-based spin density (Fig. S8, ESI†). Multiple examples have been reported of Cu(II) complexes with semiquinone-based ligands similar to the structure of the dithiolate used here, which produce similar EPR spectra.22,23 These complexes arise from the oxidation of the aryl ring. Quantitation of peaks in the EPR spectrum suggests that the paramagnetic Cu(II) complex is present only in trace amounts (ca. 0.3% of sample).
DFT calculations provide structural information about 1 and further evidence for the Cu(III) oxidation state (Fig. S10, S11 and Table S5, ESI†). The calculations indicate a perfectly square-planar coordination sphere, with Cu–S bond distances of 2.208 Å, within the 2.15–2.38 Å range of Cu–S distances reported for four-coordinate Cu(III)–thiolate complexes in the Cambridge Structural Database (CSD).24
The O2 reactivity of 1 was studied by UHR-MS, which provided evidence of a Cu–O2 adduct as a fragmentation product ion of 1. Collision-induced dissociation of the isolated ion of 1 (m/z = 370.91233; [C14H12S4Cu]−) results in the observation of the mono-dithiolato complex [(toluene-3,4-dithiolato)copper(I)]− (2) (m/z = 216.92123; [C7H6S2Cu]−) and a [(toluene-3,4-dithiolato)Cu–O2]− adduct (3) (m/z = 248.91107; [C7H6S2CuO2]−) after being passed through an Ar-filled chamber (Fig. 2 and Tables S2–S4, ESI†). The fine structure of the peak for 3 matched the calculated isotopic fine structure for [C7H6S2CuO2]−. The charge state determination is unambiguous and the formula assignment for 3 is definitive. The formation of 3 in the Ar-filled chamber, which contains only trace amounts of O2, indicates the strong affinity of 1 or 2 for O2 under the high energy conditions of the collision chamber. Ultraviolet photodissociation (UVPD) MS of 1, in which ions are activated by absorption of photons under ultrahigh-vacuum conditions, produces a signal at 216.92124 m/z (2), but not at 248.91107 m/z (3), further confirming that the observed behaviour is due to reactions with traces of O2 in the collision chamber.
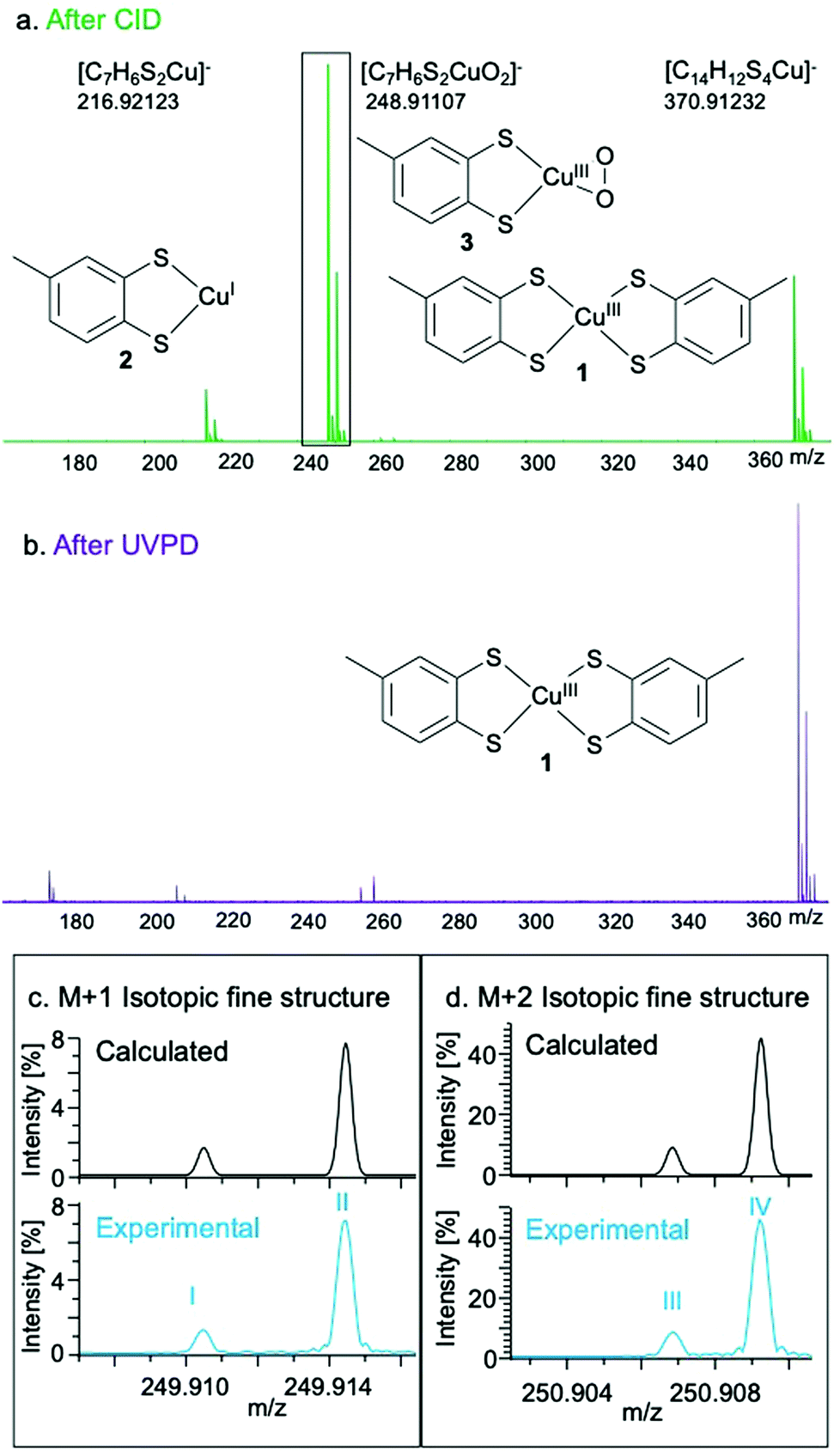 | ||
| Fig. 2 Experimental mass spectra of 1 after fragmentation by CID (a) and UVPD (b). Isotopic fine structure of the M + 1 (c) and M + 2 (d) peaks corresponding to 3. The fine structure peaks were assigned as: I – [63Cu32S33S12C71H616O2]−; II – [63Cu32S212C613C1H616O2]−; III – [63Cu32S34S1H612C716O2]−; IV – [65Cu32S21H612C716O2]−. Further fine structure assignments, including relative intensities of each peak, are reported in Table S4 (ESI†). | ||
DFT calculations for 3 indicate that it is best described as a Cu(III)–peroxide (Fig. 3). Initial geometry optimisations of singlet and triplet states of [C7H6S2CuO2]− revealed that the singlet ground state lies 30 kJ mol−1 below the triplet state, suggesting either a {Cu(III)–O2}− complex with no unpaired electrons, or a superoxide complex {Cu(II)–O2}− with the unpaired electron on the Cu antiferromagnetically coupled to the electron on the O2− ligand. The calculated geometry of the singlet complex favours {Cu(III)–O2}−, as the O–O bond distance of 1.408 Å lies closer to observed distances for O22− (i.e. 1.49 Å in BaO2) than for O2− (i.e. 1.28 in KO2) (Table S6, ESI†).25,26 It is also close to the experimental values of 1.392(3) and 1.392(12) Å reported for Cu(III)–peroxides (Table S6, ESI†). Moreover, the broken symmetry calculations for the optimised geometry of the triplet state reveal that even for this structure, the diamagnetic ground state is stabilised at ca. 6 kJ mol−1.27 Thus, the combination of UHR-MS and DFT indicates the ability of 1 to react with trace O2 in the gas phase to produce a mononuclear Cu(III)–peroxide.
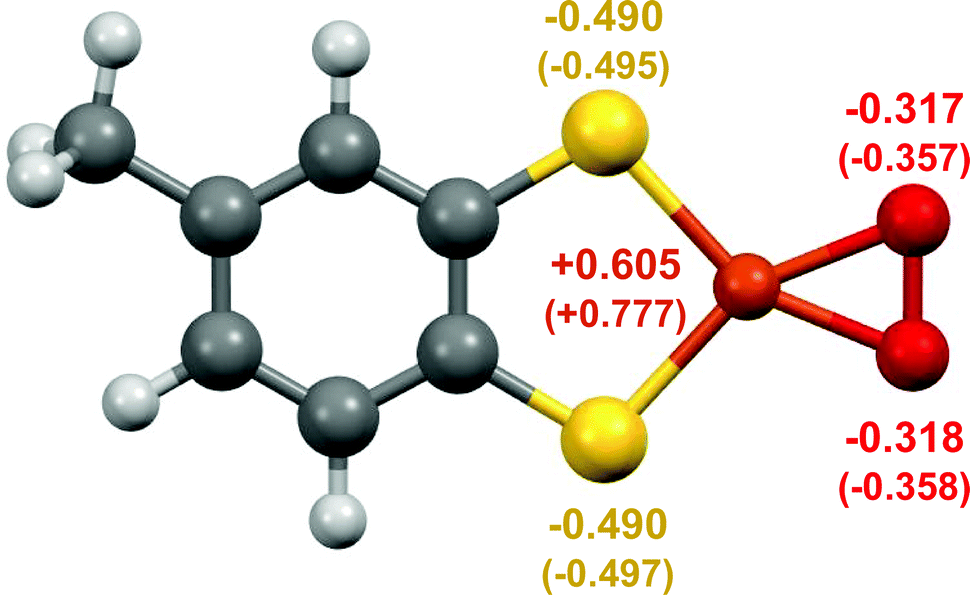 | ||
| Fig. 3 DFT calculated structure of 3 showing the planar coordination of the thiolate and O2 ligands, with the Cu, S, and O atoms labelled with the calculated electrostatic potential-derived (and in brackets Mulliken) charges. For comparison see Tables S7–S9, and Table S5, ESI,† for the respective data for 1. | ||
The square-planar Cu(III) complex NEt3H·1 has chelated dithiolato ligands analogous to the chelated dithiolene in the natural molybdenum cofactor (Moco). Like copper, molybdenum also plays important roles in neurochemistry. A deficiency of Moco in the sulfite and xanthine oxidase families of enzymes is thought to be significant in severe neurologic abnormalities and dysmorphic features of the brain and head in some babies.28 The CSD contains over 20 structures of dithiolene complexes reminiscent of Moco; the S–S distance (3.152 Å) and SC–CS bond length (1.408 Å) of 1 fall in the middle of the ranges reported for these distances in the CSD (S–S: 3.02–3.22 Å; SC–CS: 1.32–1.45 Å), indicating that the geometry of 1 is comparable with reported models of Moco.24 XPS analysis confirms the presence of a Cu(III)–thiolate, with evidence of a Cu(II)-containing impurity. EPR suggests a trace amount of Cu(II)–thiolate with a ligand-based radical. While the XPS data indicate a higher intensity of the Cu(II) complex compared to Cu(III), calculations based on EPR data suggest that only a low concentration of the Cu(II) complex is present (ca. 0.3%), and the combination of UHR-MS, 1H NMR, COSY, NOESY, and CHN elemental analysis establish the high purity for NEt3H·1. Over time, very slow reduction of the Cu(III) centre and oxidation of the ligand may occur in air. This may explain the apparent accumulation of Cu(II) observed in XPS, a surface-sensitive technique. MS showed that complex 1 readily forms a Cu(III)–peroxide in the gas-phase. Notably, complex NEt3H·1 has a remarkably low reduction potential (+0.437 V vs. NHE in MeCN). Although numerous mononuclear Cu(III) complexes have been reported, the vast majority of these have positive reduction potentials of >+1 V vs. NHE (in water).2,24 Such positive potentials are not favourable for them to exhibit biologically relevant reactivity, as they would readily oxidise proteins and other biomolecules. The anionic nature of the thiolate ligation in 1 stabilizes the Cu(III) centre and decreases the reduction potential, moving it into a biologically relevant range.29–31 Based on this low reduction potential, complex 1 presents a system relevant to studies of biological processes that involve Cu(III) redox intermediates, such as enzymatic O2 activation.1,2
Gas-phase formation of adducts with O2 after fragmentation in MS has been reported for organic radicals and complexes of first-row transition metals.9,10,32 This report appears to be the first example of an analogous process for a Cu complex. Such gas-phase reactivity offers insights into Cu–O2 chemistry. Historically, Cu(III) intermediate states have been proposed for multiple cuproenzymes, but the lack of experimental characterization of mononuclear Cu(III)–peroxides precludes extensive study of their involvement in biologically relevant processes.1,2 To date, only two such complexes have been isolated (one with a β-diketiminate ligand and one with an anilido imide), which were characterized spectroscopically and crystallographically.4,7,8
However, the thermodynamic stability of these complexes, which allowed their characterisation, also attenuated their reactivity. Interest has been expressed in changing the ligand systems of such complexes to increase the peroxo character of the O2 ligand, thus increasing their oxidative power and reactivity with organic substrates.8 Importantly, previous mechanistic studies of Cu(III)–peroxide formation indicate that both the ligands and Cu(I) contribute to the reduction of O2; electron density shifted from the formally −1 β-diketiminate and anilido imide ligands onto O2. Thus, the Cu(III)–peroxide complexes expected to have the most potent reactivity are those with electron-dense ligands and significant peroxo character.
The DFT calculations on complex 3 indicate that the bound O2 has strong peroxo character. Both the calculated electrostatic potential-derived charges and the Mulliken charge populations of 3 indicate significant electron density on the O2 ligand, to a greater extent than the calculated Mulliken charge populations of previously reported Cu(III)–peroxides (Tables S7–S9, ESI†). Such increased peroxo character may be partially attributable to the electron density of the dithiolate ligand, which has a formal −2 charge; if the ligand partially contributes to the reduction of O2, then a more electron-dense ligand may facilitate O2 reduction.
In summary, the Cu(III) complex reported here has a square-planar bis(dithiolato) coordination environment reminiscent of the biological cofactor Moco. When fragmented in the presence of trace O2 in the collision cell of a quadrupole/FTICR mass spectrometer, this Cu(III)–thiolato complex produces a Cu–O2 adduct, which DFT calculations describe as a Cu(III)–peroxide. The significant peroxide character of 3 suggests that it may be reactive towards organic substrates, thus indicating that dithiolene ligand systems could be valuable for future studies of O2 activation by Cu. That possibility, paired with the biologically relevant geometry and reduction potential of NEt3H·1, the precursor of 3, makes this system attractive for providing insight into biological processes thought to proceed through Cu(III) intermediates.
Data availability: The data that support the findings in this study are available in the Warwick Research Archive Portal (WRAP) repository, http://wrap.warwick.ac.uk/145605.
We thank the EPSRC (Grant no. EP/N033191/1 for J. F. C.; EP/P030572/1 for P. J. S.; EP/F034210/1, EP/J000302/1, and EP/N021630/1 for P. B. O.) and the US-UK Fulbright Commission (for J. M. D.). P. B. O. acknowledges support from University of Warwick Research Development Fund (RD16003); Horizon 2020 EU FTICR MS network (project 731077); BBSRC (BB/R022399, BB/P021875). V. S. acknowledges the support of the research initiative NANOKAT. J. A. W. and V. S. are grateful to Allianz für Hochleistungsrechnen Rheinland-Pfalz (AHRP) for providing CPU-time within the project TUK-SPINPLUSVIB.
Conflicts of interest
There are no conflicts to declare.Notes and references
- E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. Kieber-Emmons, C. H. Kjaergaard, R. G. Hadt and L. Tian, Chem. Rev., 2014, 114, 3659–3853 CrossRef PubMed.
- W. Keown, J. B. Gary and T. D. Stack, J. Biol. Inorg. Chem., 2017, 22, 289–305 CrossRef PubMed.
- C. Citek, J. B. Gary, E. C. Wasinger and T. D. P. Stack, J. Am. Chem. Soc., 2015, 137, 6991–6994 CrossRef PubMed.
- N. W. Aboelella, S. V. Kryatov, B. F. Gherman, W. W. Brennessel, V. G. Young, Jr., R. Sarangi, E. V. Rybak-Akimova, K. O. Hodgson, B. Hedman, E. I. Solomon, C. J. Cramer and W. B. Tolman, J. Am. Chem. Soc., 2004, 126, 16896–16911 CrossRef PubMed.
- I. Carballo-Carbajal, A. Laguna, J. Romero-Giménez, T. Cuadros, J. Bové, M. Martinez-Vicente, A. Parent, M. Gonzalez-Sepulveda, N. Peñuelas, A. Torra, B. Rodríguez-Galván, A. Ballabio, T. Hasegawa, A. Bortolozzi, E. Gelpi and M. Vila, Nat. Commun., 2019, 10, 973 CrossRef PubMed.
- D. A. Quist, M. A. Ehudin and K. D. Karlin, Inorg. Chim. Acta, 2019, 485, 155–161 CrossRef PubMed.
- N. W. Aboelella, E. A. Lewis, A. M. Reynolds, W. W. Brennessel, C. J. Cramer and W. B. Tolman, J. Am. Chem. Soc., 2002, 124, 10660–10661 CrossRef PubMed.
- A. M. Reynolds, B. F. Gherman, C. J. Cramer and W. B. Tolman, Inorg. Chem., 2005, 44, 6989–6997 CrossRef CAS PubMed.
- H. Molina-Svendsen, G. Bojesen and C. J. McKenzie, Inorg. Chem., 1998, 37, 1981–1983 CrossRef CAS.
- D. Schroeder, A. Fiedler, J. Schwarz and H. Schwarz, Inorg. Chem., 1994, 33, 5094–5100 CrossRef CAS.
- P. Palumaa, FEBS Lett., 2013, 587, 1902–1910 CrossRef CAS.
- D. Collison, C. D. Garner and J. A. Joule, Chem. Soc. Rev., 1996, 25, 25–32 RSC.
- P. Amo-Ochoa, O. Castillo, E. Delgado, A. Gallut, E. Hernández, J. Perles and F. Zamora, CrystEngComm, 2019, 21, 957–963 RSC.
- S. Banerjee, A. Omlor, J. A. Wolny, Y. Han, F. Lermyte, A. E. Godfrey, P. B. O'Connor, V. Schünemann, M. Danaie and P. J. Sadler, Dalton Trans., 2019, 48, 9564–9569 RSC.
- R. R. Mondal, S. Khamarui and D. K. Maiti, ACS Omega, 2016, 1, 251–263 CrossRef CAS.
- E. Sacher, J. E. Klemberg-Sapieha, A. Cambron, A. Okoniewski and A. Yelon, J. Electron Spectrosc. Relat. Phenom., 1989, 48, C7–C12 CrossRef CAS.
- M. S. Kim, J. B. Yang, J. Medvedeva, W. B. Yelon, P. E. Parris and W. J. James, J. Phys.: Condens. Matter, 2008, 20, 255228 CrossRef.
- F. Navarro-Pardo, L. Jin, R. Adhikari, A. Tong, D. Benetti, A. Basu, A. Vanka, H. Zhao, A. Mi, C. Sun, V. Castaño, A. Vomiero and F. Rosei, Mater. Chem. Front., 2016, 1, 65–72 RSC.
- M. Panthakkal Abdul Muthalif, C. Sunesh and Y. Choe, Appl. Surf. Sci., 2018, 440, 1022–1026 CrossRef CAS.
- N. Karikalan, R. Karthik, S.-M. Chen, C. Karuppiah and A. Elangovan, Sci. Rep., 2017, 7, 2494 CrossRef.
- C. Sunesh, C. Gopi, M. Panthakkal Abdul Muthalif, H.-J. Kim and Y. Choe, Appl. Surf. Sci., 2017, 416, 446–453 CrossRef CAS.
- L. Najder-Kozdrowska, B. Pilawa, A. Więckowski, E. Buszman and D. Wrześniok, Appl. Magn. Reson., 2009, 36, 81–88 CrossRef CAS.
- S. K. Dey and A. Mukherjee, J. Mol. Catal. A: Chem., 2015, 407, 93–101 CrossRef CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- W. Wong-Ng and R. S. Roth, Phys. C, 1994, 233, 97–101 CrossRef CAS.
- S. C. Abrahams and J. Kalnajs, Acta Crystallogr., 1955, 8, 503–506 CrossRef CAS.
- L. Noodleman, J. Chem. Phys., 1981, 74, 5737–5743 CrossRef CAS.
- R. R. Mendel, Dalton Trans., 2005, 21, 3404–3409 RSC.
- F. P. Bossu, K. L. Chellappa and D. W. Margerum, J. Am. Chem. Soc., 1977, 99, 2195–2203 CrossRef CAS PubMed.
- M. R. McDonald, W. M. Scheper, H. D. Lee and D. W. Margerum, Inorg. Chem., 1995, 34, 229–237 CrossRef CAS.
- J. Hanss, A. Beckmann and H.-J. Krüger, Eur. J. Inorg. Chem., 1999, 163–172 CrossRef CAS.
- Y. Xia, P. A. Chrisman, S. J. Pitteri, D. E. Erickson and S. A. McLuckey, J. Am. Chem. Soc., 2006, 128, 11792–11798 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc06921c |
| This journal is © The Royal Society of Chemistry 2021 |